

Mapping enhancer–target gene regulatory interactions and analyzing enhancer mutations at the level of their target genes and pathways reveal convergence of recurrent enhancer mutations on biological processes involved in tumorigenesis and prognosis.
Abstract
Enhancers are noncoding regulatory DNA regions that modulate the transcription of target genes, often over large distances along with the genomic sequence. Enhancer alterations have been associated with various pathological conditions, including cancer. However, the identification and characterization of somatic mutations in noncoding regulatory regions with a functional effect on tumorigenesis and prognosis remain a major challenge. Here, we present a strategy for detecting and characterizing enhancer mutations in a genome-wide analysis of patient cohorts, across three lung cancer subtypes. Lung tissue–specific enhancers were defined by integrating experimental data and public epigenomic profiles, and the genome-wide enhancer–target gene regulatory network of lung cells was constructed by integrating chromatin three-dimensional architecture data. Lung cancers possessed a similar mutation burden at tissue-specific enhancers and exons but with differences in their mutation signatures. Functionally relevant alterations were prioritized on the basis of the pathway-level integration of the effect of a mutation and the frequency of mutations on individual enhancers. The genes enriched for mutated enhancers converged on the regulation of key biological processes and pathways relevant to tumor biology. Recurrent mutations in individual enhancers also affected the expression of target genes, with potential relevance for patient prognosis. Together, these findings show that noncoding regulatory mutations have a potential relevance for cancer pathogenesis and can be exploited for patient classification.
Significance:
Mapping enhancer–target gene regulatory interactions and analyzing enhancer mutations at the level of their target genes and pathways reveal convergence of recurrent enhancer mutations on biological processes involved in tumorigenesis and prognosis.
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide (1), with the majority of lung cancers associated with long-term tobacco smoking. Furthermore, lung cancer is the tumor type with the second highest reported mutation burden amounting to 12.9 single-nucleotide variants (SNV) per megabase for smokers (2).
In this landscape of overall high mutation rate, driver somatic mutations with transforming potential in lung cancers have been reported for several oncogenes such as EGFR, ALK, ERBB2, BRAF, ROS1, MET, RET, NTRK1, NRG1, KRAS, as well as tumor-suppressor genes TP53 and STK11 (3–5). Despite the advances in targeted therapies against driver genes, lung cancers are still associated with poor survival and a high death-to-incidence rate (1). Therefore, it would be crucial to identify additional alterations beyond the commonly mutated genes to further characterize lung cancer biology and possibly open new avenues for treatment.
Cancer genomics studies predominantly focused on characterizing mutations in protein coding sequence (CDS) regions. However, noncoding regulatory elements such as enhancers and promoters play a pivotal role in transcription regulation (6) and are enriched for transcription factor–binding sites (TFBS; ref. 7). An ever-increasing amount of evidence suggests that alterations in noncoding regulatory elements can drive pathological phenotypes in various diseases (8–10). Moreover, genome-wide association studies reported a large fraction of disease-associated SNPs in distal noncoding regulatory elements (enhancers; refs. 11, 12), including SNPs associated to increased cancer risk. Furthermore, accumulating evidence corroborates the functional consequences for tumorigenesis of mutations in regulatory elements (13). In this context, the genetic or epigenetic mis-regulation of enhancers emerged as a potential pathogenic mechanism in cancer (14). Moreover, mutations in noncoding regulatory regions may exert a functional effect through multiple mechanisms, including alterations in transcription regulation, disruption of chromatin domain structure, changes in mRNA stability, and creation of de novo TFBSs (13).
However, characterizing genome-wide functional relevance of noncoding regulatory regions mutations in cancer is still a significant challenge, despite some scoring methods proposed in the literature (15–18). In this context, enhancers stand out as especially critical noncoding regulatory features. Indeed, on the one hand, enhancers are the most cell type–specific players among the epigenetic determinants of cell identity (19) and several chromatin remodeling factors acting on enhancers are often mutated in tumors (20), thus attesting to their crucial functional relevance for cell homeostasis. However, on the other hand, studying genetic or epigenetic alterations of enhancers is specifically challenging due again to their intrinsic cell-type specificity and the difficulties in the genome-wide definition of the enhancer–target genes (ETG) regulatory network.
Indeed, even though a specific gene may be active in multiple cell types, its activation can be regulated by different enhancers in different contexts (21). Active enhancers are generally defined using functional genomics data, thus providing a cell type–specific definition (22, 23). Therefore, to assess the impact of noncoding mutations in putative enhancer regions, it is necessary to estimate the cell-specific activity of enhancers within the tissue of origin of the cancer being studied.
Enhancers are generally distant from their target promoters along with the linear sequence of the genome (24). Moreover, a gene can be regulated by more than one enhancer, and an enhancer can regulate multiple genes (25). To this concern, we recently showed that chromatin three-dimensional (3D) architecture could help to disentangle the complexity of these interactions (26). Namely, we leveraged the biological consensus on enhancer-gene regulatory contacts occurring in the 3D context of topologically associated domains (TAD; ref. 27). TADs are dynamically formed by chromatin loop extrusion in interphase cells (28, 29), thus, determining the stochasticity and dynamics of enhancer–promoter interactions within a hierarchy of insulated domains. This is a change of perspective with respect to identifying ETG pairs just based on interaction frequencies inferred from chromosome conformation capture data, and it is in line with the latest evidence, indicating that the non-linear relationship between enhancer contact frequency and transcription output (30, 31) can be explained by the dynamic nature of TADs and loop extrusion (32).
Given the complexity of the ETG regulatory interactions and the overall high mutational burden in lung cancers, in this context, it is crucial to develop strategies to identify and prioritize the mutations in noncoding regulatory elements with the potential to affect tumor biology. Here, we present complementary approaches to identify and characterize the functional role of noncoding somatic mutations in lung cancer patient genomes. We leverage public and novel epigenomic profiles to define lung tissue-specific enhancers (Fig. 1A and B). We use state-of-the-art techniques to reconstruct the ETG regulatory interactions by leveraging the three-dimensional architecture of chromatin (Fig. 1C). We examine mutational burden and mutational signature differences across distinct classes of coding and noncoding regulatory elements. We highlight the aggregation of enhancer mutations in biologically relevant pathways and gene sets (Fig. 1D and E). We show that recurrent mutations of individual enhancers may affect the target gene with a potential clinical relevance (Fig. 1F).
Figure 1.

Methodological framework overview. Schematic illustration of our workflow for enhancer mutation characterization. A, Lung-specific enhancer definition from eight different lung cell and tissue types. ChIP-seq for open chromatin (H3K27ac) and chromatin accessibility (DNase-seq or ATAC-seq) from each sample are intersected to obtain cell-specific putative enhancers. The union of the regions from each cell creates the master list after removing regions overlapping with promoters and exons. *, in the cell line, indicates the in-house data. B, Somatic mutation calling. Whole-genome sequencing data of tumor and corresponding normal blood of three lung cancer cohorts viz., LUAD, LUSC, and SCLC obtained from public resources are processed using an ensemble mutation calling approach to identify somatic mutations. C, Enhancer–target gene prediction using canonical correlation of functional genomics data to investigate the synchronized activity of enhancer–promoter pairs across multiple cell types. Implementation of the 3D colocalization information encoded in hierarchical contact score to control for FDR in multiple testing hypotheses. D, Reconstructed ETG network with somatic mutations in enhancers. Lung-specific enhancer regulatory network reconstructed with somatic mutations at enhancers obtained through steps A, B, and C. Dark cyan circles represent enhancers, red lightning marks represent mutations, and colored ovals represent genes. E, Aggregation of enhancer mutations at the pathway level. Pathway level enrichment of TGEM is performed using three approaches, that is, over-representation analysis to determine biological pathways with TGEM enrichment, direct heat diffusion to determine significantly affected sub-networks, and a global test to assess the effect of TGEM on gene expression at the pathway level. F, Functional analysis to characterize recurrently mutated enhancers. Recurrently mutated enhancer cores are determined for the functional characterization of a relevant enhancer mutation. The effect of the enhancer mutation on the target gene expression is assessed by stratifying patients based on the presence of the enhancer mutation. Similarly, survival probability is estimated in patients stratified on the basis of the presence of the enhancer mutation. TFBS alteration in the enhancer core upon somatic mutation is assessed to determine the mechanistic effect of enhancer mutation. (Created with BioRender.com.)
Materials and Methods
Cell cultures
Human NCI-H460 cells were obtained from the ATCC and were cultured in RPMI-1640 medium (catalog no. BE12–167F, Lonza) containing 10% FBS (catalog no. 10270–106 Life Technologies) and 2 mmol/L glutamine (catalog no. LOBE17605F, Euroclone).
HBEC3-KT (human bronchial epithelial cells immortalized with CDK4 and hTERT) cells were obtained from Voden and also donated to co-author (Luca Roz) by Prof. John Minna (UT Southwestern, Dallas, TX) and were cultured in Keratinocyte-SFM with l-glutamine (catalog no. 17005034 Thermo Fisher Scientific) with Keratinocyte-SFM supplements: 0.025 μg/mL of human recombinant EGF (1–53) and 62.5 μg/mL of bovine pituitary extract (catalog no. 37000015 Thermo Fisher Scientific).
Cell line authentication was performed using GenePrint 10 System (Promega). Each cell line is tested any time we have to prepare a new stock to refill our bank. All cells were cultured at 37°C in 5% CO2 and regularly tested for Mycoplasma contamination using MycoAlert (Lonza) in addition to the PCR method.
Chromatin immunoprecipitation sequencing experiments
Cells were cross-linked in 1% fixing solution (50 mmol/L Hepes KOH pH7.5, 100 mmol/L NaCl, 1 mmol/L EDTA, 0.5 mmol/L EGTA, 11% formaldehyde in water) for 10 minutes at room temperature, followed by lyses and chromatin shearing. 5% of chromatin was saved as input. Immunoprecipitation (IP) was performed overnight on a wheel at 4°C with H3K27ac antibody (Abcam, catalog no. ab4729, RRID:AB_2118291) or control IgG (Abcam, catalog no. ab37415, RRID:AB_2631996) with a dilution of 2.5 ng/μL. The following day, antibody–chromatin immune complexes were loaded onto Dynabeads Protein G (Invitrogen, catalog no. 10004D).
The bound complexes were washed twice in IP buffer (10 mmol/L Tris-HCl pH 8.0, 140 mmol/L NaCl, 1 mmol/L EDTA, 0.1% SDS, 0.1% DOC, 1% Triton X-100, 1X PMSF, 1X protease inhibitors), twice in High Salt Solution (10 mmol/L Tris-HCl pH 8.0), 500 mmol/L NaCl, 1 mmol/L EDTA, 0.1% SDS, 0.1% DOC, 1% Triton X-100, 1X PMSF, 1X protease inhibitors) followed twice by RIPA-LiCl buffer (10 mmol/L Tris-HCl pH 8.0, 1 mmol/L EDTA, 250 mmol/L LiCl, 0.5% DOC, 0.5% NP-40, 1X PMSF, 1X protease inhibitors) and once in 10 mmol/L Tris-HCl (pH 8.0). Crosslinking was reversed at 65°C overnight in elution buffer (10 mmol/L Tris-HCl pH 8.0, 5 mmol/L EDTA, 300 mmol/L NaCl, 0.4% SDS), DNA was purified by standard phenol/chloroform [Phenol:Chloroform:Isoamyl Alcohol 25:24:1 (Sigma), saturated with 10 mmol/L Tris, pH 8.0, 1 mmol/L EDTA (catalog no. P3803)] extracted, precipitated, and resuspended in 30 μL of 10 mmol/L Tris-HCl (pH 8). Chromatin immunoprecipitation (ChIP) efficiency was tested by qPCR reactions, performed in triplicate using the SYBR Select Master Mix (Invitrogen, 4472908) on a StepOnePlus Real-Time PCR System (Applied Biosystems) on CDH13 promoter (positive control) and on the gene body of RARRES2P9 (negative control; Supplementary Table S1 for primers ETG-p1 and ETG-p2, respectively). Relative enrichment was calculated as the IP/Input ratio. Library preparation was performed starting from 5 ng of Input or IP-DNA, using Kapa HyperPrep kit from Kapa Biosystems. A dual index barcoded adapter was ligated to each library, followed by size selection using Ampure XP beads; libraries undergo 12 cycles of PCR amplification and after additional purification, were checked on Agilent Bioanalyzer 2100 for size and quantitated on Qubit 4.0 fluorometer. Equimolar amounts of indexed libraries were pooled and loaded on Illumina HighOutput flowcell on NextSeq550 for sequencing in 2×75nt read mode. Approximately 80 million paired-end sequencing reads were generated for each library.
Assay for transposase-accessible chromatin with sequencing experiments
Assay for transposase-accessible chromatin with sequencing (ATAC-seq) library preparation was carried out as previously described in ref. 33. Briefly, 50,000 cells from each sample were centrifuged at 500 × g at 4°C, then resuspended in ATAC-seq resuspension buffer (RSB; 10 mmol/L Tris-HCl, 10 mmol/L NaCl, 3 mmol/L MgCl2) supplemented with 0.1% NP-40, 0.1% Tween-20 and 0.01 digitonin. Samples were incubated on ice for 15 minutes and washed twice with 300 μL of RSB supplemented with 0.1% Tween-20. Nuclei were pelleted at 500 × g for 10 minutes at 4°C. The nuclei pellet was resuspended in 50 μL transposition mix [25 μL 2X TD buffer, 2.5μL transposase (Illumina), 16.5 μL PBS, 0.5 μL 1% digitonin, 0.5 μL 10% Tween-20, and 5 μL H20] and incubated for 30 minutes at 37°C in a thermal-mixer at 1000 rpm. Samples were purified using the Qiagen Mini elute PCR Purification kit according to the manufacturer's protocol (elution in 21 μL of elution buffer). Libraries were PCR-amplified using the NEBNext High-Fidelity PCR Master Mix and amplified for 5 cycles using NEBNext 2x MasterMix and custom primers, as previously described in ref. 34. Libraries were sufficiently amplified individually in addition to 5 cycles of PCR as computed from qPCR fluorescence curves. Libraries were purified using Zymo DNA Clean and Concentrator. The libraries were then size selected using AMPure XP Beads.
After 12 cycles of PCR amplification and additional purification, libraries were checked on Agilent Bioanalyzer 2100 for size and quantitated on Qubit 4.0 fluorometer. Equimolar amounts of indexed libraries were pooled and loaded on Illumina HighOutput flowcell on NextSeq550 for sequencing in 2×75nt read mode. Approximately 80 million paired-end sequencing reads were generated for each library.
ChIP-seq and ATAC-seq data analysis
Paired-end raw reads were filtered on the basis of the quality value obtained from FastQC (RRID:SCR_014583) v0.11.9 (-q 10 and -p 30; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) using the Trim Galore! (RRID:SCR_011847) software v0.6.4_dev (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The filtered reads were aligned to the hg19 reference genome using BWA (RRID:SCR_010910) v0.7.17-r1188 (https://sourceforge.net/projects/bio-bwa/files/) to produce the alignment file (BAM). The PCR duplicates were removed from the BAM files using PICARD (RRID:SCR_006525) tools v2.23.1 (http://broadinstitute.github.io/picard/). The BAM files were sorted and indexed for peak calling using SAMtools (RRID:SCR_002105; https://sourceforge.net/projects/samtools/files/samtools/). The BedGraph files were generated by comparing BAM files of IP and input (IP read coverage/input read coverage), resulting in a ratio for every base across the whole-genome using bamCompare from deepTools (RRID:SCR_016366) v3.4.3 (https://deeptools.readthedocs.io/en/develop/content/tools/plotProfile.html). To call the peaks MACS2 (RRID:SCR_013291) v2.2.7.1 (https://github.com/macs3-project/MACS) was used. This framework was implemented using Nextflow (RRID:SCR_024135) nfcore ChIP-seq (https://nf-co.re/chipseq) v1.2.1 or ATAC-seq (https://nf-co.re/atacseq) v1.2.1 pipeline for ChIP and ATAC sequencing data, respectively. The quality assessment of the ChIP-seq and ATAC-seq profile are provided in Supplementary Fig. S1A and S1B. The bed and bedgraph files obtained from the analysis were visualized using the IGV (RRID:SCR_011793) browser and further processed using custom made R and Python scripts.
Lung-specific enhancers’ definition
For the definition of lung-specific enhancers across the genome, we leveraged two epigenetic markers of open chromatin, that is, H3K27ac and DNase or transposase (through ATAC-seq) sensitivity. We downloaded uniformly processed H3K27ac ChIP-seq and DNAse-seq files in bigbed format for six lung tissue/cell types (lung, IMR-90, PC-9, A549, AG04450, lung fibroblasts) with replicates from ENCODE3 (RRID:SCR_015482; https://www.encodeproject.org/matrix/?type=Experiment&replicates.library.biosample.donor.organism.scientific_name=Homo+sapiens&biosample_ontology.organ_slims=lung). In addition, we performed H3K27ac ChIP-seq and ATAC-seq on two lung cell lines viz., HBEC3-KT and NCI-H460.
For each of the cell or tissue type, the corresponding peak files were first filtered to retain only peaks with strong significant enrichment, that is, (adjusted P value ≤0.01). Following which, we merged peak genomic coordinates across replicates and defined consensus peaks as merged peaks overlapping individual replicate peaks in more than 50% of replicates.
To obtain a comprehensive list of cis-regulatory elements active in lung cells and tissue types, we conducted a two-step procedure. First, for each of the eight-lung cell/tissue types (viz., lung, IMR-90, PC-9, A549, AG04450, fibroblast, NCI-H460 and HBEC3-KT), the intersection between H3K27ac and chromatin accessibility (ATAC or DNase-seq–based) peaks with overlapping regions (≥6 bps) were used to define cell-specific regulatory regions.
Additional filters were applied ex post, with respect to the transcription start site (TSS) to separate promoter-proximal (within 3.5 kb upstream and 1.5 kb downstream of TSS) or distal regulatory regions, and only the promoter-distal ones were retained as putative cell-type–specific enhancers for the following steps. Second, cell-type–specific enhancers with overlapping intervals across different cell types were merged (union) together to define a consensus set of enhancer regions. This set was filtered on the basis of size to remove intervals larger than 2.5 kb. Noncanonical and Y chromosomes were excluded. The merged set of genomic regions was also filtered on the basis of position to retain only noncoding promoter-distal regions, similarly to the previous step, to obtain the reference list of lung-specific enhancers (N = 187,206). This was used as a comprehensive reference set of enhancer regions, which are active in at least one of the lung cell types considered (Supplementary Fig. S1C and S1F).
Enhancer core definition
We defined enhancer cores, as the region of overlap between lung-specific enhancers and DNase footprinting peaks from the IMR-90 cell line. For this purpose, we obtained the footprinting peaks from the Roadmap Epigenomics consortium (https://egg2.wustl.edu/roadmap/data/byDataType/dgfootprints/). The obtained enhancer cores (N = 335,955) were in the size range 6 to 40 bp with an average of 13bps.
Promoter definition
We defined reference promoters as 2 kb regions (1.5 kb upstream and 0.5 kb downstream) around the TSS of annotated protein-coding genes, based on RefSeq (RRID:SCR_003496) annotations in (hg19.ncbiRefseq.gtf.gz; May 2019 download, hg19 reference genome assembly). Noncanonical and Y chromosomes were excluded. To create a more comprehensive list of promoters, in case of multiple alternative transcripts for the same gene, the promoter for each transcript was considered barring overlapping regions with exons and 5′UTR of another transcript.
Somatic mutations calling and mapping
High-coverage (average coverage for each pair of tumor and matched normal ≥35x) whole-genome sequencing (WGS) data of 55 lung adenocarcinomas (LUAD; ref. 4), 50 lung squamous cell carcinoma (LUSC; ref. 5), and 54 small-cell lung cancer (SCLC; ref. 35) samples were downloaded from The Cancer Genome Atlas (TCGA; for LUAD and LUSC cohorts) and European Genome Archive (EGA; for the SCLC cohort) in the form of tumor and matched normal BAM files (Supplementary Table S2; Supplementary Fig. S2A–S2C). For the uniform processing of the samples, the sequencing data were realigned on hg19 reference genome using BWA following the GATK best practices (RRID:SCR_001876). Mutations (SNVs and small indels) were called across the whole-genome using FreeBayes (RRID:SCR_010761), MuTect (RRID:SCR_000559), Scalpel (RRID:SCR_012107), VarDict (RRID:SCR_023658), and Varscan (RRID:SCR_006849). FreeBayes, Varscan, and VarDict are indel and SNV callers whereas Scalpel is an indel-only caller and Mutect is an SNV-only caller, thus, altogether resulting in 4 variant callers in the ensemble. Mutations present in the low-complexity regions as defined in ref. 36 were removed. Finally, for determining a somatic variant, we used the concordance of a variant call by at least two tools. A custom pipeline based on BC-BIO/bcbio-nextgen (RRID:SCR_004316; https://github.com/bcbio/bcbio-nextgen) was used to perform all the operations on WGS data above Supplementary Fig. S2D–S2F. The mutation list of each sample was then mapped on the lung-specific enhancers and promoters using pybedtools (RRID:SCR_021018; https://daler.github.io/pybedtools/; Fig. 2A and B).
Figure 2.

Mutational landscape of lung cancer cohort. A, Circos plot of the global landscape of mutations in patients with lung cancer. Chromosomes are shown on the outermost circle. The following circle is a bar graph of gene density obtained by binning the genome in 1 Mbp windows (dark blue). The next circles from the periphery to the center are the bar graphs representing the number of enhancers (dark cyan), promoters (salmon pink), and exons (powder blue) mutated (log-scale). The scale of each bar graph is represented at the start of chromosome1. Mutations in the noncanonical chromosome (chromosome Y) were removed from the analysis. B, Sample-wise mutation distribution. The bottom shows the line plot representing the mean somatic mutation per Mb in the lung cancer sample. The middle shows the relative proportions in the percentage of the six possible base-pair substitutions, as indicated in the legend on the right. The top shows the stacked bar plot depicting the number of mutated genomic elements. Each bar represents the total number of enhancer mutations (dark cyan), promoter mutations (salmon pink), and exon mutations (powder blue) for a patient. Samples are sorted on the basis of the total number of mutations in exons (x-axis). C, Mutation burden comparisons. Scatter plots showing the mutation burden per Mb in enhancers (x-axis) and exons (y-axis). D, Scatter plots showing the mutation burden per Mb in enhancers (x-axis) and the rest of the noncoding region (RNCR; y-axis). Each blue dot in scatter plots represents a sample, the gray line represents the bisectors, the blue line represents the line of regression, and the slope of the regression is mentioned in the plot.
Region-specific mutation burden
To identify somatic mutation enrichment of various regions of the genome, we computed the burden of somatic mutations in exons, enhancers, promoters, and the rest of noncoding regions (RNCR) for each sample. RNCR was defined as the whole-genome devoid of exons, enhancers, and promoters.
where xi is any genomic region of type x, with x ∈{exons, enhancers, promoters, RNCR}.
The mutation burden of each sample was reported in the results section as scatter plots in various comparison scenarios and the slopes of each linear regression were estimated (Fig. 2C and D; Supplementary Fig. S2G and S2H).
Mutation signatures
To obtain an approximate estimate of the contribution of different known mutational signatures to each sample, we used the MutationalPatterns v3.17 package from Bioconductor (RRID:SCR_006442). As a reference set of mutational signatures, we used a table with the relative frequency of each of the 96 trinucleotide substitutions across 30 known mutation signatures from COSMIC (RRID:SCR_002260; https://cancer.sanger.ac.uk/signatures/signatures_v2/) database version 2. Mutation signatures were estimated for the whole-genome and the relative frequency of each signature was plotted in a heat map (Fig. 3A).
Figure 3.

Mutation signatures comparison. A, Mutation signatures in lung cancer cohort. Heat map of the relative contribution of each COSMIC single-base substitutions (SBS) signature for each sample. The samples are grouped on the basis of the lung cancer subtype indicated by the color band (orange, SCLC; purple, LUSC; and green, LUAD). B, Mutation signature difference between coding and the noncoding genome. Comparison of underlying signature distribution between coding and noncoding regions in LUAD, LUSC, and SCLC for a subset of COSMIC SBS signatures. For a given signature, the color of the marker corresponds to the difference between the mean contribution in coding and the noncoding region. The size represents the P value (Wilcoxon rank-sum test). Only the subset of signatures with significant contribution differences in at least one of the cohorts (P < 0.05) are reported. C, Mutation signature associated with the enhancers, promoters, and exons. Box and whiskers plots of the relative contribution of mutation signature in enhancers (dark cyan), promoters (salmon pink), and exons (powder blue). The statistical significance of comparisons (P value < 0.05) is presented as star marks. Each point in the boxplot represents a sample.
To assess the variations in mutation signature between coding and noncoding regions in LUAD, LUSC, and SCLC samples, we computed the relative contribution of the 30 COSMIC mutation signatures for coding regions and noncoding regions for all the samples (Supplementary Fig. S3A). To compute the difference in the relative contribution of the frequency (Fig. 3B), the Wilcoxon rank-sum test using the scipy.stats.ranksums function in SciPy (RRID:SCR_008058) was used for each signature individually in each lung cancer subtype (LUAD, LUSC, and SCLC). A mutation signature was deemed significantly different between the two categories with a P value lower than 0.01, and we reported in Fig. 3B all signatures significantly different in at least one of the cancer subtypes. For assessing the difference, (noncoding average relative contribution)–(coding average relative contribution) was computed, for each lung cancer subtype.
Furthermore, the mutation signatures prevalent in enhancers, promoters, and exons were computed (Supplementary Fig. S3B) and the relative contribution of the signatures across samples was used to plot box and whiskers plot. Significance was determined by one-way ANOVA and followed by multiple comparison of means with a post hoc test (Tukey test; Fig. 3C) to define which group means are driving the differences, using the scipy.stats.multicomp function in SciPy.
ETG pairing
ETG pairs were identified starting with an input of 180,852 lung-specific enhancers and 18,027 promoters as described in ref. 26. Briefly, we computed the maximum enrichment signal of DNase-seq and H3K27ac ChIP-seq over enhancer regions, and then we used DNase-seq, H3K27ac, and H3K4me3 over promoter regions. As in the original published procedure, consolidated fold-change enrichment signal tracks in bigwig format from the Roadmap Epigenomics consortium for 44 cell and tissue types were used as the reference compendium of epigenomic profiles. Canonical correlation was adopted to investigate the inter-set correlation patterns to quantify the strength of each enhancer–promoter pair and a P value was computed for the overall dependence between each promoter and enhancer. We selected 1,809,529 enhancer–promoter pairs with canonical correlation P value of <0.05. To control the number of false discoveries due to multiple hypothesis testing, we adopted the Adaptive P value thresholding procedure (AdaPT; ref. 37) by considering relevant contextual three-dimensional colocalization information in the form of the Hierarchical Contact (HC) score. We used the same HC score that was computed as described in ref. 26 based on 11 Hi-C datasets (38–43) covering multiple cell lines and primary tissues: Lung (n = 3), pancreas (n = 2), breast (n = 2), ovary, and B cells (n = 2). The HC score was computed as described in (26). Finally, we identified 48,829 enhancer–promoter pairs with adjusted P value thresholding based on AdaPT <0.01 (Fig. 4; Supplementary Fig. S4A–S4C).
Figure 4.

Enhancer–target gene pairing. A, Distance between enhancer and predicted target gene. The x-axis denotes the distance (in Kb) between the enhancer and the predicted target gene, and the y-axis denotes the number of ETG pairs in the distance range. Each bar represents the total number of ETG pairs (in thousands) within the distance range per chromosome, as indicated in the legend on the right. B, Hierarchical contact score and the distance between ETG pairs. Bubble plot representing the number of ETG pairs (color) with HC score (y-axis) and the distance between the enhancer and the predicted target gene in Kb (x-axis). C, Number of enhancers versus number of mutated samples. Heat map showing the number of enhancers predicted for a gene (x-axis) compared with the number of enhancers mutated (y-axis). The color of the square indicates the number of genes with x number of enhancers and y number of mutated samples. D, Differential gene expression between genes with enhancer mutations. The volcano plot displays the log2-fold change expression (x-axis) between samples grouped by the specific enhancer mutation status of each gene (with vs. without enhancer mutations). Transcripts with significant difference and log2-fold change >1 are highlighted in pink, or log2-fold change < −1 are highlighted in violet. The y-axis is the −log10(P value) of the coefficient for enhancer mutations in the linear regression model. The horizontal red line marks the P value of <0.05 significance threshold. The size of the circles for significantly altered genes indicates the number of associated enhancers mutated. E, Transcription factor–binding sites at enhancer cores. Scatter plot showing the TF motif score computed by FIMO at enhancer core with the reference sequence (x-axis) and altered sequence (y-axis).
Enrichment of mutations at enhancer cores
To compute the enrichment of mutations at the enhancer core level, we use the Poisson Binomial Distribution (PBD), as proposed by (44). The PBD allows us to calculate the probability of observing a certain number of samples with at least one mutation in a specific enhancer core, considering the sample-specific background mutation rates.
Let Xi ∈[0, N] be a random variable representing the number of samples with at least one mutation in the i-th enhancer core, where N is the number of samples in the cohort. Then Xi follows a PDB with a k-dimensional vector of probability pi = [1-(1-pk)ni]k, where ni is the size of the i-th enhancer core in base pairs, and pk is the background mutation rate for the k-th sample. Namely, we computed the k-th sample background mutation frequency as follows:
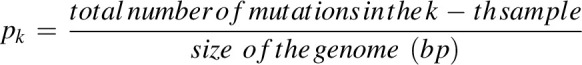 |
Finally, we calculated the probability of having at least si samples with at least one mutation in the i-th enhancer core as P(Xi≥si), using the poibin v1.5 R package (http://cran.nexr.com/web/packages/poibin/index.html). P values were computed for all the enhancer cores and adjusted for multiplicity using the Bonferroni method (Supplementary Fig. S5).
CIEN-Ins detection
For detecting the presence of CIEN-Ins in the WGS data, we leveraged the Compact Idiosyncratic Gapped Alignment Report strings from the BAM file. Using custom scripts, we identified the soft clipped sequences at the genomic loci of interest (chr16:82672414–82672441, hg19), along with the number of reads associated with the insert. We then computed the proportion of CIEN-Ins reads to the total reads at the loci to determine the samples with CIEN-Ins variation (≥ 25%) in the TCGA lung and breast cancer cohorts (Supplementary Fig. S6A–S6D).
Cell line screening for experimental validation
For the experimental validation of the role of the CIEN in regulating its gene expression, 10 lung cancer cell lines were screened for (i) sequence of CIEN-core, (ii) expression of CDH13 gene, and (iii) copy number of CDH13 gene (Supplementary Fig. S7A–S7C).
CIEN-core sequence determination
DNA was isolated using the Qiagen AllPrep DNA/RNA Mini Kit following the manufacturer's protocols. CIEN-core was amplified (Primers Supplementary Table S1) and run on a 1.5% agarose gel. All the bands were purified with the Qiagen PCR purification kit, according to the manufacturer's instructions. Samples were eluted in 30 μL and sequenced by the Sanger method.
CDH13 expression quantification
RNA was isolated using the Qiagen AllPrep DNA/RNA Mini Kit following the manufacturer's protocols. For qRT-PCR, 500 ng of RNA was reverse-transcribed using superscript III (Thermo Fisher Scientific) following the manufacturer's protocol. qRT-PCR was performed with TB Green Premix Ex Taq (Tli RNase H Plus) using Roche LightCycler 96. PCR amplification parameters were 98°C (30s), and 35 cycles of 98°C (10s), 65°C (30s), 72°C (10s), and 72°C (2 min).
The expression of CDH13 gene was quantified for all the isoforms of the gene in 10 lung cancer cell lines in comparison with WI38 (normal lung fibroblast cell line) and BJ (normal skin fibroblast cell line).
Copy-number assessment
Copy number of CDH13 gene in the tested lung cell lines was determined by quantitative PCR, by comparing the cycle threshold (Ct) values of CDH13 and GAPDH loci. As a reference, the WI38 cell line was used, because its GAPDH copy number is already known. All primer details are provided in Supplementary Table S1.
Candidate enhancer validation
Clones bearing a deletion of CIEN-core were generated using the CRISPR/Cas9 technology. To this purpose, oligonucleotides corresponding to three different protospacer sequences (named T1, T2, and T3) located 5′ (T1 and T2) or 3′ (T3) to the CIEN-Ins sequence were cloned in the PX459 vector (Addgene, plasmid #62988). Two plasmid pairs (T1+T3 and T2+T3) were used to induce double-strand breaks (DSB) and trigger deletion of the intervening sequence. Briefly, a total of 1×106 NCI-H460 cells (2 μg/100 μL) were electroporated in A69 buffer (30 mmol/L sodium phosphate buffer, 5 mmol/L potassium chloride, 10 mmol/L magnesium chloride, 20 mmol/L HEPES, 11 mmol/L glucose, 100 mmol/L NaCl, pH6.9) using the Amaxa Nucleofector II Device (program T-020). Subsequently, after 24 hours of electroporation transfected cells were positively selected for 3 days with 1.5 μg/mL puromycin. Single-cell clones were obtained by limiting dilution and screened by PCR using primers ETG-p3, flanking the region to be deleted. Amplicons of different sizes allowed to identify clones homozygous and heterozygous for the deleted targeted region. The characteristics of the designed sgRNA are detailed in Supplementary Table S1 and Supplementary Fig. S7D).
Screening-enhancer deletion clones
The genomic locus was amplified with the phosphorylated primers ETG-p4 using the Phusion polymerase. The amplicons were gel purified and blunt cloned into EcoRV-linearized and dephosphorylated pCRII-delta18 vector. Transformation was followed by sequencing using SP6.
CDH13 expression was quantified using primer ETG-p5. Assays were done in triplicate and relative levels of gene expression were normalized to beta-actin (ETG-p6).
Number of mutations versus number of predicted enhancers
The upper and lower right quadrant was defined as genes with more than 14 associated enhancers with more than 20 mutated samples and more than 14 associated enhancers with less than 10 mutated samples, respectively. The upper-left quadrant was defined as genes with 14 or less associated enhancers and 20 or less mutations. The genes from the respective quadrants were then individually assessed for gene set enrichment with MSigDB-curated gene sets. Gene sets with FDR < 0.05 are reported in Supplementary Table S3.
Gene expression analysis
RNA sequencing data were obtained for patients with high coverage WGS data from the TCGA (105 samples) and EGA (30 samples). The quantification of the transcripts was obtained using kallisto (RRID:SCR_016582; https://pachterlab.github.io/kallisto/) as Transcripts per Kilobase Million (TPM) based on the hg19 reference genome. To assess the impact of enhancer mutation on the gene expression, a regression model was applied on the normalized gene expression level e as a function of l, that is the mutation status of its enhancers [1, mutated; 0, wild-type (WT)], controlling for the impact of CNA status c (0, WT; positive value, amplification; negative value, deletion), DNA methylation m (mean beta value), promoter mutation p (1, mutated; 0, WT), and exon mutation x (1, mutated; 0, WT).
The R function model used for estimation:
Mod.lm← lm[log2(Expression(, x)+1) ∼ as.factor(enhancer(, x)) + CNA(, x) + Methylation(, x) + as.factor(promoter(, x))+ as.factor(exon(, x)].
Samples were stratified into mutated and non-mutated on the basis of the presence of enhancer mutation (at least five), and their corresponding gene expression as TPM values were compared, the P value obtained from the regression model for the coefficient enhancer mutation was then used for significant thresholding (P < 0.05). The genes with significant difference between the mutated and non-mutated samples were assessed for gene set enrichment with MSigDB-curated gene sets. Gene sets with FDR < 0.05 are reported in Supplementary Table S3.
For the quantification of transcription factor expression in the NCI-H460 cell line, RNA-sequencing data were obtained in FASTQ file format from the GEO (Gene Expression Omnibus) database (GSM2072563). The reads were processed to quantify transcripts using kallisto as described above.
Promoter methylation
Methylation data for the TCGA samples were obtained as Methylation Beta Value from HumanMethylation450 (HM450) arrays (45). To assess the methylation status of a promoter, mean methylation beta values of the probes present in the promoter (2 kb around TSS) were computed.
Survival analysis
Clinical features such as sex, vital status, TNM stage (tumor, lymph node, metastasis), and smoke exposure were also obtained from the TCGA (46) for the patients. Event-free survival probabilities were calculated by using the Kaplan–Meier method (survminer v0.4.9 R package https://cran.r-project.org/web/packages/survminer/index.html; RRID:SCR_021094). The Log-rank test was used to assess the statistical significance of the different groups.
Regression analysis
To assess the association of genomic features and clinical features with the CIEN-Ins, generalized linear model (GLM) was used to compute the P value through tidy verse R package (https://cran.r-project.org/web/packages/tidyverse/index.html). The following function was used for the estimation:
mod.glm←glm(mut∼Methylation-beta-values+as.factor(Tumor-stage) + as.factor(sex) + log2(CDH13exp+1) + as.factor(exonic_mut) + as.factor(copynumberalteration), data = TCGA-lung_data, family = “binomial”).
TFBS analysis
To calculate the presence of TFBS motifs in enhancer cores, we used FIMO (https://meme-suite.org/meme/tools/fimo) Version 5.4.1 from the MEME suite (RRID:SCR_001783) with a custom library of all TRANSFAC (RRID:SCR_005620) and JASPAR motifs (RRID:SCR_003030; https://jaspar.genereg.net/downloads/) at a q value threshold (FDR—Benjamini–Hochberg multiple testing correction) of 0.05. TF motif analysis was performed on the reference genome sequence of the enhancer cores and the altered sequence of enhancer cores resulting from somatic mutations in patients. The predicted motif scores for the reference and altered sequence was plotted in a scatter plot.
Tissue-specific expression quantification
For studying the tissue-specific expression levels, we obtained the gene TPMs from the Genotype-Tissue Expression (GTEx; RRID:SCR_013042) project database v8, (GTEx_Analysis_2017–06–05_v8_RNASeQCv1.1.9_gene_tpm.gct.gz). We compared the expression levels of genes with more than 25 enhancers (arbitrary cutoff value) with the genes with fewer lung-specific enhancers. The test gene set is composed of genes with at least 25 enhancers, (n = 130). For the background gene set, we bootstrapped 10 sets from the genes with less than 25 enhancers with approximately the same size. The mean expression levels of the genes in each group were quantified and log2 fold change was computed. The Mann–Whitney U test was implemented to assess the significance, and Bonferroni correction for multiple hypothesis testing was used to obtain the adjusted P value. Gene expression values for all the tissues were represented in box plots.
Gene set enrichment analysis
Target genes with enhancer mutations (TGEM) with at least 12 mutations (n = 466) were used for the gene set enrichment analysis (GSEA). Two different datasets of the MSigDB resources (http://www.gsea-msigdb.org/gsea/msigdb/index.jsp) C2 (literature-curated gene sets) and C6 (oncogenic gene sets) were used through the GSEA (RRID:SCR_003199). Gene sets with a P value of <0.01 were considered significantly enriched and the results were plotted using custom Python scripts.
For the gene ontology and pathway enrichment analyses, TGEM with (n > 3) were used. The list of target genes (n = 7,102) were then used to obtain gene ontology–biological process and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment through the g:profiler tool (RRID:SCR_006809; https://biit.cs.ut.ee/gprofiler/gost) with a P value of <0.01.
Global test for groups of genes
We implemented a global test for groups of genes (47) using the R package (https://bioconductor.org/packages/release/bioc/html/globaltest.html) to identify the impact of TGEM. Explanatory variable is the matrix of gene expression for genes within a gene set (X), and the response variable is the number of mutated enhancers that are associated to genes within the gene set (Y).
As input gene sets, we used MSigDB C2–curated (n = 6366) and the Hallmark gene sets (n = 50; RRID:SCR_016863) for the analysis. Poisson regression was used to model count response. Custom R scripts were used to identify significantly affected gene sets. A gene set was considered significantly affected, when the P value was <0.05.
Network diffusion for sub-network identification
Hotnet2 (https://github.com/raphael-group/hotnet2) was applied to identify sub-networks of protein–protein interaction network with more mutations than expected. We used the HINT database (RRID:SCR_002762) for the protein interaction information, with a beta of 0.4 and 100 network permutations and 1,000 heat permutations.
Data availability
The data generated in this study are publicly available in GEO at GSE228832, including H3K27ac ChIP-Seq and ATAC-seq data on HBEC3-KT and NCI-H460 cell lines. The cancer genomics WGS and RNA-seq data analyzed in this study were obtained from The database of Genotypes and Phenotypes at phs000178.v11.p8 and the European Genome–Phenome Archive (EGA) at EGAS00001000925. The COSMIC mutation signatures analyzed in this study were obtained from the COSMIC database version 2 at https://cancer.sanger.ac.uk/signatures/signatures_v2/. The ENCODE3 ChIP-seq and DNase-seq data analyzed in this study were obtained from https://www.encodeproject.org. All other raw data are available upon request from the corresponding author.
Results
Lung-specific enhancers’ identification
To achieve a comprehensive and accurate genome-wide definition of lung-specific enhancers, we created a repertoire of epigenomics profiles for eight different lung cell types and tissue samples, including bronchial epithelial cells, primary lung tissue, lung fibroblasts (n = 3), LUAD (n = 2), and large-cell lung cancer (Supplementary Table S4). This repertoire builds on similar solutions adopted in recent reference literature in the field (e.g., compared with lung cancer cell lines matching in ref. 48), but is purposely expanded to include normal epithelial cells. The rationale of this approach for the comprehensive definition of lung-specific enhancers stems from three factors: (i) The exact cellular origin of lung cancers is unclear (49), but most likely related to different cells within the epithelial compartment, potentially including also neuroendocrine cells for SCLCs (50, 51); (ii) lung cancers have relevant intratumoral heterogeneity and intricate cellular communications within the tumor microenvironment (52–54); (iii) the intent to examine noncoding mutations considering multiple lung cancer subtypes and highlight differences between them. To this aim, we primarily relied on ENCODE consortium data as they cover a broad range of cell and tissue types; standard data quality control and pre-processing procedures are adopted. To define enhancers in six lung cell and tissue types, we obtained ENCODE 3 epigenomics data for ChIP-seq (chromatin IP followed by high-throughput sequencing) enrichment peaks of histone H3 lysine 27 acetylation (H3K27ac), that is a chromatin mark associated to active enhancers (23), and chromatin accessibility peaks based on DNase-seq, a general feature of active regulatory elements (55). Although H3K4me1 is also found at enhancers, it is often reported to be present also in poised or weak enhancers (23), thus we did not use it.
When studying mutations connected to chromatin features, it is crucial to have a reference epigenomic profile for the cell type of origin of the tumor. To this concern, an earlier study in ref. 56 specifically remarked that the lack of reference normal lung epithelium was confounding the match of lung cancer mutations with epigenetic features. Hence, to address this gap, we ensured the inclusion of immortalized HBEC3-KT data in our repertoire by performing in-house experiments for H3K27ac ChIP-seq and ATAC-seq, an alternative genome-wide method to probe chromatin accessibility (57). To our knowledge, this is the first genome-wide epigenomic profile for active enhancers chromatin marks for immortalized normal lung epithelial cells, thus achieving crucial complementation of publicly available datasets. In addition, we also enriched our repertoire with data from the NCI-H460 cell line, a large-cell lung carcinoma line commonly used in standard experimental validations.
We combined H3K27ac and chromatin accessibility epigenomic profiles to identify active enhancers in each cell type (see Materials and Methods): Average number 49,017 and average size 402 bp (Supplementary Fig. S1C). We observe that 49% of the enhancers derived from the lung normal tissue sample are cell type–specific, as they are not found in the other samples that always have a lower portion of cell-type–specific enhancers (Supplementary Fig. S1D). Their pairwise comparison showed, on average, 31% similarity (Jaccard Index, JI): This reflects, on the one hand, the cell-specific nature of enhancers and, on the other hand, the partial conservation observed at the tissue level (Supplementary Fig. S1E). The JI between the three fibroblasts is higher and more similar between themselves than cancer cell lines, possibly indicating that the latter are more heterogenous in activating different epigenetic features. Nevertheless, A549 and NCI-H460 have considerable overlap. HBEC3-KT cell have good overlap (0.28 JI) with both A549 and PC9 cancer cell lines, thus confirming the importance of considering HBEC3-KT to define enhancers relevant for lung tissues and lung cancers. To define a comprehensive list, we considered the union of the cell-specific enhancers resulting in 180,852 enhancers (Supplementary Table S5) with an average size of 456 bp (Supplementary Fig. S1F). The number of enhancers per chromosome (chr) ranged between 2,213 in chr21 and 16,710 in chr1, in line with the size of each chromosome and its gene density. We considered this the reference list of “lung-specific enhancers” throughout our analyses.
Genomic landscape of noncoding mutations in lung cancer
To understand the effect of noncoding mutations in lung cancers, we used high-coverage WGS data from three different lung cancer cohorts, including 55 patients with LUAD (4), 50 patients with LUSC (5), and 54 patients with SCLC (35). Samples in the cohorts were selected on the basis of the availability of high-coverage WGS data from paired tumor and normal samples (average coverage for each pair ≥35x), in addition to transcriptomic data (RNA-seq) for functional characterization (Supplementary Fig. S2A–S2C).
In the analysis of somatic mutations inferred from WGS data, the standard bioinformatic pipelines adopt a single-variant caller. We aimed to go beyond the limitations of a single bioinformatic tool by adopting an ensemble approach that combines the results of four complementary algorithms (calling both indels and SNVs) to balance sensitivity and specificity (see Materials and Methods). Although individual tools have shown similar performances with 80%–90% concordance (58), the ensemble can reduce the differences attributed to individual callers. To ensure sensitivity and specificity, we obtained high-confidence somatic SNVs and indels retaining only the ones called by at least two somatic mutation calling tools for each tumor sample against the matched normal (Supplementary Fig. S2D).
With this approach, in total, we observe 6,937,213 somatic variations (SNVs and small indels) in the lung cancer cohort, where the number of variations per sample ranges from 2,926 to 288,853 (mean = 52,956; median = 48,292). The mean mutation density per megabase (Mb) per sample ranges between 0.88 and 89.73 (mean = 16.33; median = 14.99). We observe that the average mutation density in LUAD, LUSC, and SCLC samples are 14.34, 15.76, and 18.89, respectively (Supplementary Fig. S2E).
We observe that noncoding regulatory mutations (enhancer and promoters) and coding mutations are spread across the genome, with chr1 being the most mutated and chr21 the least, in line with being the largest and smallest chromosomes, respectively (Fig. 2A). Meanwhile, we identify that, on average, 863 enhancers, 620 promoters, and 2128 exons are mutated (at least once) per sample (Fig. 2B). Furthermore, among the SNVs, the C > A and C > T are the most prevalent single base-pair substitutions (SBS) across the cohorts (Fig. 2B). Overall, the SBS rates across the considered lung cancer subtypes are similar (Supplementary Fig. S2F).
With the knowledge that lung cancer is a highly mutated cancer type, we sought to understand whether the noncoding regulatory elements were mutated at the same rate as the rest of the noncoding region. Thus, we computed the region-specific somatic mutation burden in exons, promoters, enhancers, and the rest of the noncoding regions devoid of enhancers and promoters (the latter referred to as RNCR from now on). We observe that for all the samples, enhancers have a similar mutation burden with respect to exons and promoters (Fig. 2C; Supplementary Fig. S2G and S2H). In contrast, the mutation burden of the RNCR is higher compared with the enhancer regions (Fig. 2D). The similar propensity for mutation burden in regulatory and coding regions of the genome can be suggestive of a functional relevance.
Mutation signature differences across regulatory and coding regions
To further investigate the mechanisms determining the emergence of mutations in coding and noncoding regulatory regions, we examined the mutational signatures across different portions of the genome. First, to understand the general mutational processes at play, we computed the mutational profiles from the WGS data. We then compared them with the COSMIC SBS profiles (version 2) to identify the prevailing signatures and their relative contribution in each sample. We observe that the most prevalent signature in our cohort is associated with smoking (signature 4), as expected for lung cancers, together with three signatures of unknown etiology (signatures 5, 8, and 16; Fig. 3A).
We then examined whether any difference is detected in mutational signatures between the coding and the noncoding portions of the genome (Fig. 3B). After computing the relative frequency of mutational signatures in the coding and noncoding portions of the genome, we compared them for each lung cancer type. We identified a significant difference in at least one cancer type for 18 out of 30 signatures. Among them, signatures 5, 8, and 16 were the most prevalent in coding regions compared with noncoding regions, with 8 and 16 significantly different in all the three tumor types. On the contrary, signatures associated with defective DNA mismatch repair (signature 6), likely UV exposure (signature 7), aflatoxin exposure (signature 24), and APOBEC activity (signature 1) were significantly higher in noncoding regions in all three tumor types (Fig. 3B; Supplementary Fig. S3A).
These differences in the mutational processes associated with coding and noncoding portions of the genome prompted us to further explore the mutational profile in specific functional regions. Thus, we compared the relative contribution of mutation signatures between enhancer, promoter, and exon regions: In this analysis, we considered the three tumor types together as in most cases the differences between coding and noncoding mutational signatures reported above were concordant (Fig. 3B). When comparing enhancer, promoter, and exon regions, we detected significant differences across many signatures, including 1, 3, 6, 10, 12, 18, 24, and 25, although with different trends across the genomic features considered (Fig. 3C). Signatures 5, 7, 16, and 19 also show significant variations, but they are driven by the pairwise differences of “enhancers versus promoters” and “promoters versus exons,” as confirmed by the ANOVA post hoc test. Interestingly, the signature associated with defective DNA mismatch repair (signature 6; ref. 59) is higher in promoters than enhancers and exons. In contrast, the signature associated with failure of DSB repair is higher in enhancers (signature 3) compared with promoters and exons. Signature 3 is also characteristic of insertions and deletions with overlapping microhomology at breakpoint junctions (59). We also observed a significant difference in the signature associated with the activity of error-prone polymerase POLE (signature 10; ref. 59; Fig. 3C; Supplementary Fig. S3B). Even though the mutation burden in the selected regions is similar, the differences in signatures indicate that they are differently affected by mutagenic processes. Moreover, the presence of signatures associated with failures in DSB repair at enhancer regions is in line with previous literature reporting this type of DNA damage occurring at enhancers (60–62).
Overall, these results highlight the mechanistic differences in the mutational processes at the coding and noncoding regulatory regions, with specific differences involving regulatory regions.
Mutations in enhancers significantly affect the target gene expression
For the functional characterization of mutations in enhancers, it is crucial to interpret their effect in the context of the regulatory network linking enhancers and their target genes. In this context, relying on a comprehensive yet accurate reconstruction of lung-specific ETG pairs is essential. For this purpose, we adapted the statistical framework recently developed in our laboratory (26), incorporating information on chromatin 3D architecture and our reference set of lung-specific enhancers obtained as described above. In our statistical framework, we incorporate TADs hierarchical structure inferred from Hi-C datasets and encoded in the HC score, which is then used as side information in the AdaPT procedure to adjust the P value for each ETG pair. As a result, we obtained 48,829 ETG pairs (AdaPT FDR adjusted P value ≤ 0.01) with distances ranging from <5kb to >500kb (Fig. 4A and B; Supplementary Table S6). This is a crucial advancement over previous cancer genomics studies that attempt to link noncoding regulatory mutations to genes without accounting for the TADs hierarchical structure that drives their interactions.
Our ETG pairing resulted in 10,709 genes with at least one associated lung-specific enhancer (total of 33,797 enhancers out of the initial set). Our ETG approach has paired one gene with five enhancers (on average) and one target gene per enhancer (median). These results align with previous literature, indicating that multiple enhancers can regulate the same gene, whereas only one gene is the preferred target of each enhancer (Supplementary Fig. S4A and S4B). Incidentally, genes associated with many lung-specific enhancers (≥25) are highly transcribed specifically in normal lung tissue compared with genes with fewer enhancers (Supplementary Fig. S4C). Upon intersecting enhancer mutations and their target genes, we observed that 10,425 genes had at least one enhancer mutated in at least one tumor sample. As it may be expected, we noticed that some genes with a large number of associated enhancers tend to have a large number of samples with a mutation in any of their enhancers (Fig. 4C). However, in Fig. 4C, we also see several genes with few associated enhancers but many mutations (upper-left quadrant) and genes with many associated enhancers but with few mutations (lower-right quadrant), suggesting that regulatory mutations for these genes may undergo a positive or negative selection, respectively.
Genes with few enhancers and higher number of mutations (upper-left quadrant genes n = 17) have an overlap with developmental biology-related gene sets. On the other hand, the genes with many enhancers and higher mutations (upper-right quadrant n = 74) are enriched in gene sets associated with invasiveness, epithelial-to-mesenchymal transition and extracellular matrix organization (ECM). Genes with many enhancers and few mutations (lower-right quadrant n = 262) are enriched in gene sets associated with cytokines, natural killer cells, and NOTCH signaling pathway (FDR < 0.05 in MsigDB-curated gene sets; Supplementary Table S3).
To assess the impact of mutated enhancers, we applied a regression model of the normalized gene expression level as a function of the mutation status of its enhancers, controlling for the impact of copy-number alterations (CNA), DNA methylation, promoter and exon mutation status. The regression analysis was performed on a total of 1,322 genes with at least 5 mutations in the enhancers associated to a gene. We found that 72 genes were specifically affected by enhancer mutations (P <0.05 on the enhancer mutation status coefficient in the regression model; Fig. 4D; Supplementary Table S3). As a confirmation of their relevance in cancer, we observe that 30 out of 65 significantly enriched gene sets (FDR < 0.05 in MsigDB-curated gene sets) are indeed derived from studies on 14 different cancer types (Supplementary Table S3). The genes with large differential expression (at least two-fold change) are highlighted in (Fig. 4D).
We also examined the impact of enhancer mutations on TFBS. For this purpose, we focused on the DNase-seq–footprinting calls available from high-coverage DNase seq data of the IMR-90 cell line to identify regions of active TF binding within the selected lung-specific enhancers. DNase I–footprinting regions were used to narrow down the genomic regions of interest to the most likely location of TFBS that we named enhancer cores. To identify the extent of TFBS sequence alterations at enhancer cores, we first determined the TFBS sequence motifs in these regions using the reference genome sequence. We then examined how the motifs would change with the sequence alterations induced by somatic mutations detected in our lung cancer patient cohort. We observed that the changes in the sequence of enhancer cores might result in both gain- or loss-of-TFBS, depending on the specific instance (Fig. 4E). In several cases, the somatic mutations do not change the TFBS motifs composition of the enhancer core. However, there is a higher number of TFBS losses rather than gain of such motifs.
Overall, these data suggest that a loss of function may be more frequent than a gain of function as a consequence of enhancer mutations. Moreover, these data confirm the potential functional impact of enhancer mutations with respect to their target gene expression.
Enhancer mutations aggregate at pathway level and participate in the same biological process
According to the literature and our data as well, a gene can be regulated by multiple enhancers, and in principle, an individual enhancer can regulate multiple genes. As such, enhancers are, in fact, the constituents of the broader gene regulatory network comprising distal enhancers and their target genes. Thus, to interpret perturbations of the enhancer regulatory activity, we should analyze these events at the pathway and regulatory network level. For the prioritization of enhancer mutations at pathway level, we explored and proposed three approaches, including gene sets enrichment analyses, network diffusion analysis to identify mutated sub-networks (63) and a custom global-test–based analysis (47) to determine whether enhancer-mutated genes are differentially expressed.
First, we performed a GSEA. We mapped mutations in lung-specific enhancers and linked them to their target genes, thus defining a list of TGEMs. Then we used these genes in KEGG functional annotations to test for the over-representation of terms associated with specific pathways or functions enriched in the list of TGEM. We found that 10 out of the 20 significantly enriched pathways (FDR adjusted P value < 0.01) are directly involved in cancer or cancer-related properties, such as the PI3K–AKT signaling, focal adhesion and regulation of actin cytoskeleton pathways (Fig. 5A). Other 6 out of the 20 pathways are related to immunity and inflammation. The remaining enriched pathways have 70% of genes from our list overlapping with the cancer-related pathways. As such, there's a striking majority of association to cancer-related biological processes. As the PI3K-AKT signaling is one of the cancer driver pathways targeted by therapies, we further explored its mutational landscape in our cancer cohort. To this aim, we mapped the mutations in exons, enhancers, and promoters of therapeutically targeted genes obtained from DrugBank database (64) belonging to the PI3K–AKT signaling pathway. We observed that mutations in exons, enhancers, and promoters show a non-overlapping occurrence pattern in patients, that is, an individual gene of the pathway is targeted by either of the three categories of mutations in any given patient, whereas combination of two is rare and all the three types of mutations are never observed together in the therapeutically targeted genes (Fig. 5B).
Figure 5.

Pathway level enrichment of enhancer mutations. A, Scatter plot shows the over-representation of genes with enhancer mutations in the KEGG pathway. The x-axis represents the ratio of the overlapping genes to the total number of genes in the pathway. The size of the circle denotes the number of genes in overlap, and the color shows the negative logarithmic adjusted P value. B, Mutational landscape of the PI3K–AKT pathway. Co-mutation plot showing druggable PI3K–AKT signaling pathway genes (y-axis) affected in lung cancer samples (x-axis) by mutations in enhancers, promoters, and exons as indicated in the legend on the right. The top stacked bar plot shows the number of mutations in each sample, and the gene-wise mutation rate is displayed on the right. C, Network view of protein interactions among two sub-networks of regulation of transcription by RNA Pol II identified by HotNet2. Interactions between proteins in the sub-network from each interaction network are colored on the basis of a P value. D, Significantly altered gene sets (MSigDB-C2 curated). The violin plot shows the gene sets that are affected by TGEM. E, Hierarchical clustering graph of the WAKASUGI_HAVE_ZNF143_BINDING_SITES gene set from MSigDB C2 gene set. The gene plot shows the P value associated with the impact of the enhancer mutation on the gene expression. The part of the clustering graph with a significant P value is plotted in black.
We also performed a similar analysis using a curated list of gene sets derived from the literature (MSigDB database- C2 gene sets). We observed a significant overlap (FDR adjusted P value < 0.01) with gene sets associated with invasive tumor features, stemness, ECM organization, and focal adhesion (Supplementary Fig. S5A). We observed that the TGEM converge on Gene ontology biological processes such as positive regulation of kinase activity, regulation of protein phosphorylation, positive regulation of MAPK cascade, mononuclear cell differentiation, negative regulation of transcription by RNA pol II, and angiogenesis (Supplementary Table S7). These results confirm, using independent and complementary definitions of functional pathways, that mutations in enhancers recurrently target genes participating in biological processes with known relevance in cancer biology.
As mentioned above, the analysis of enhancer alterations integrated within the context of the broader gene regulatory network is mainly motivated by biological reasons related to their mechanism of action. However, there are also statistical reasons because we observe heterogeneity of mutations at enhancers, whereby at the level of individual enhancers, the recurrence of mutations is generally low (no. of enhancers with <5 mutations: 14386; Supplementary Fig. S5B). Thus, we applied the direct heat-diffusion method implemented in HotNet2 (63), originally designed for coding regions meant to overcome the long-tail phenomenon (extensive heterogeneity of mutations leading to low recurrence on individual features). We used the TGEM information as input to HotNet2 and identified 11 significantly mutated sub-networks (Supplementary Table S7). Interestingly, two sub-networks associated with Regulation of transcription by RNA Pol II (Fig. 5C) were identified as significantly enriched on the basis of mutations in its associated enhancers. These results suggest that the dysregulation of enhancers due to somatic mutations may have a cascade effect on the dysregulation of transcription also through their target genes.
A crucial issue with the interpretation of enhancer mutations at the pathway level is the ability to understand whether their combinatorial effect results in a disruption of transcriptional regulation. For this purpose, to understand whether the mutations in enhancers can affect gene expression at the pathway level, we adopted a custom approach based on the global test (47). We used this statistical test to determine whether the global expression of genes in a gene set (explanatory variables) is related to the number of mutated enhancers associated with genes within the gene set itself (response variable). We used the global test on TGEM with MSigDB-curated gene sets (n = 6,366) and observed that the enhancer-mutated genes impact 434 gene sets (P < 0.05; Fig. 5D). The top-most significant gene set (P = 0.0002) was derived from a publication by (65) as a set of DNA repair genes with a putative ZNF143-binding site in their promoter (MsigDB gene set “WAKASUGI_HAVE_ZNF143_BINDING_SITES”). Upon further exploring the specific gene set (Fig. 5E) using a hierarchical clustering graph, we observe that the positively associated genes have a significant impact on the response variable. We also used the MSigDB Hallmark gene set (n = 50) and found the DNA repair gene set to be most significantly affected (Supplementary Fig. S5C). Overall, these results indicate that indeed mutations in enhancers can accumulate in specific pathways with known biological relevance for tumor biology. The integrated analysis of regulatory mutations occurrence and gene expression differences with the global test approach also confirms a potential disruption of the target genes regulation. However, more focused experimental work is needed to confirm and dissect the functional role of individual enhancers.
Recurrently mutated intronic enhancer affects CDH13 expression
To prioritize individual noncoding regulatory regions affected by mutations for further experimental investigation, we quantified the recurrence of mutations at the level of individual enhancers, which may seem in line with approaches also adopted previously (66). However, a crucial difference with prior publications is that we narrowed down the selection of SNVs and small indels occurring within the enhancer core regions only (defined by DNase footprinting peaks), as discussed above.
We looked for the recurrence of mutations focusing at the enhancer core levels for biological and statistical reasons (Supplementary Fig. S5D). From a biological point of view, the effect of an enhancer core mutation is more likely to affect the TFBS directly, thereby leading to alterations in the regulation of its target gene. Furthermore, focusing on more functionally relevant regions may increase the statistical power (66). Thus, within each enhancer, we considered only mutations falling within any of its cores, defined as described above. We identified 9,151 enhancer cores to be mutated in at least one sample, of which, 57 have a significantly higher number of mutations compared with the background mutation rate of the samples (adjusted P value < 0.01; Supplementary Table S8).
The top most significantly mutated enhancer cores, resides within an intronic enhancer hosted in the cadherin 13 (CDH13) gene (Fig. 6A). Indeed, CDH13 is an atypical (without a transmembrane domain) member of the cadherin family, often known to be downregulated in cancerous cells (67, 68). CDH13 downregulation is associated with poor prognosis (69). The CDH13 intronic enhancer (chr16: 82671674–82672964, hg19; CIEN from now on) is present in the first intron of the gene but distant from the TSS (>11Kb). On the basis of our ETG pairing method (26), we observed that the CIEN and CDH13 promoter have highly correlated activity (0.826 canonical correlation from), they both reside within the same TAD across multiple hierarchies (HC score, 75.32), thus predicting a regulatory interaction with high confidence (adjusted P value = 0.001). Moreover, among pathways significantly affected by enhancers mutations based on the global test analysis reported in the previous paragraph, we found two pathways, both related to lung, that also contains the CDH13 gene: Namely, the MSigDB pathways MCDOWELL_ACUTE_LUNG_INJURY_DN and SCHLESINGER_METHYLATED_DE_NOVO_IN_CANCER.
Figure 6.
![Figure 6. CDH13 insertion variation. A, CDH13 enhancer loci at chromosome 16. The top shows the hierarchical TAD boundaries in the lung tissue within the locus 82.6 Mb to 83.3 Mb in chromosome 16. The next is the zoomed-in version between chr16: 82.6 Mb to 82.8 Mb, showing the location of the CDH13 promoter (salmon) and enhancers associated with CDH13 (dark cyan). The next shows the enhancer of interest (chr16:82.6718Mb–82.6728Mb) with enhancer mutations (red lollipop)—the height of the lollipop reflects the number of mutations found across the patient cohort. The last shows the enhancer cores in blue, with the most mutated enhancer core (CIEN-core) in dark blue. B, CIEN-Ins and patient clinical information. Co-mutation plot shows CDH13 expression [log2(TPM+1)], CIEN-Ins, presence of CIEN-Ins in tumor tissue, presence of CIEN-Ins in matched normal, source of normal (blood or tissue), copy-number variation (CNV; as neutral N, loss L, and gain G), exon mutation (number of mutations), promoter methylation (beta values), TNM staging of cancer, sex of the patient, and the lung cancer subtype are represented by indicated colors in the legend on the right. C, CDH13 expression upon CIEN-Ins deletion. CDH13 gene expression relative to β-actin in wild-type and homozygous deletion of CIEN-Ins in NCI-H460 cell line. The dots represent biological replicates (n = 5). D, Progression-free survival interval probability in lung cancer samples (LUAD+LUSC). E, Disease-free survival interval (DFI) probability in lung cancer samples. F, DFI probability in LUSC samples. For the survival analysis, patients were stratified on the basis of the presence of CIEN-Ins in the tumor, and Kaplan–Meier curves were plotted for the two groups. The risk table with the number of patients is described at the bottom. Differences between the two groups were evaluated using a log‐rank test.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10758689_133fig6.jpg)
CDH13 insertion variation. A, CDH13 enhancer loci at chromosome 16. The top shows the hierarchical TAD boundaries in the lung tissue within the locus 82.6 Mb to 83.3 Mb in chromosome 16. The next is the zoomed-in version between chr16: 82.6 Mb to 82.8 Mb, showing the location of the CDH13 promoter (salmon) and enhancers associated with CDH13 (dark cyan). The next shows the enhancer of interest (chr16:82.6718Mb–82.6728Mb) with enhancer mutations (red lollipop)—the height of the lollipop reflects the number of mutations found across the patient cohort. The last shows the enhancer cores in blue, with the most mutated enhancer core (CIEN-core) in dark blue. B, CIEN-Ins and patient clinical information. Co-mutation plot shows CDH13 expression [log2(TPM+1)], CIEN-Ins, presence of CIEN-Ins in tumor tissue, presence of CIEN-Ins in matched normal, source of normal (blood or tissue), copy-number variation (CNV; as neutral N, loss L, and gain G), exon mutation (number of mutations), promoter methylation (beta values), TNM staging of cancer, sex of the patient, and the lung cancer subtype are represented by indicated colors in the legend on the right. C,CDH13 expression upon CIEN-Ins deletion. CDH13 gene expression relative to β-actin in wild-type and homozygous deletion of CIEN-Ins in NCI-H460 cell line. The dots represent biological replicates (n = 5). D, Progression-free survival interval probability in lung cancer samples (LUAD+LUSC). E, Disease-free survival interval (DFI) probability in lung cancer samples. F, DFI probability in LUSC samples. For the survival analysis, patients were stratified on the basis of the presence of CIEN-Ins in the tumor, and Kaplan–Meier curves were plotted for the two groups. The risk table with the number of patients is described at the bottom. Differences between the two groups were evaluated using a log‐rank test.
CIEN comprises 16 enhancer cores with an average length of 14bps, among which, only two cores (chr16:82672417–82672441 and chr16:82672305–82672343) are mutated in 8 and 1 samples, respectively. The enhancer core (chr16:82672417–82672441, CIEN-core) is mutated in 7 samples with an SBS from C to T at locus chr16:82,672,430 (hg19) and in one sample with an insertion at chr16:82,672,428. Incidentally, this insertion is a reported germline sequence variant in dbSNP (rs139451683), comprising of a stretch of TG dinucleotides followed by a tail of CG bases [ins (TG)n(CG)4, referred to as CIEN-Ins from now on]. A visual inspection of sequencing reads in the 7 samples with a C to T SNV highlighted the presence of clipped reads that may suggest that a germline CIEN-Ins was lost in the tumor and erroneously annotated as an SBS by the mutation calling algorithms.
Indeed, as CIEN-Ins is rich in GT and GC bases, the reads partially overlapping the insertion may be clipped by the sequence aligner and erroneously interpreted by the mutation calling algorithm as a putative sequencing artefact. Upon careful reprocessing of all WGS data specifically for this region (see Materials and Methods), we confirmed the presence of CIEN-Ins in different combinations across patients: (i) presence of CIEN-Ins in tumor and normal samples; (ii) presence in tumor samples and absence in their matched normal; (iii) vice versa (Fig. 6B; Supplementary Fig. S6A) resulting in a total of 51 out of 105 samples with CIEN-Ins (somatic + germline combined) in the TCGA lung cancer cohorts. The number and percentage of reads supporting the CIEN-Ins variant across samples, mostly around 50% (Supplementary Fig. S6A), support the conclusion that this is not a random sequencing error, but a real sequence variant in the samples. Nevertheless, we adopted a conservative approach by not considering samples with less than 25% of reads supporting the CIEN-Ins presence.
Incidentally, genomic features, including hypermethylation of the promoter, CNA, gene expression, and exon mutation and clinical features such as stage of the tumor, sex of the patients, and the lung cancer subtype (LUAD and LUSC), are not significantly associated with CIEN-Ins occurrence in the TCGA lung cancer samples (GLM; Fig. 6B; Supplementary Table S9). However, stratifying patients with respect to CIEN-Ins and hypermethylation of promoter, show that the samples with hypermethylation and CIEN-Ins have higher CDH13 expression than samples with either of the alterations (Supplementary Fig. S6B).
To functionally validate the impact of CIEN-Ins on the regulation of CDH13 gene in the most appropriate cellular model, we first screened 10 lung cancer cell lines and two fibroblast cell lines used as reference (see Materials and Methods and Supplementary Fig. S7A–S7C). On the basis of (i) the presence of CIEN-Ins, (ii) expression of the CDH13 gene, and (iii) a diploid copy of the CDH13 gene, we chose the NCI-H460 lung cancer cell line. We then used CRISPR-Cas9 to delete CIEN-Ins in the NCI-H460. The successful deletion of the regulatory region was confirmed by Sanger sequencing. Upon homozygous deletion of CIEN-Ins, CDH13 is significantly downregulated (P value = 0.002 independent t test in 5 biological replicates, that is, 5 different clones; Fig. 6C).
Intronic enhancers are known to regulate their harboring gene expression by establishing direct or indirect enhancer–promoter contacts, which may be achieved by recruiting and clustering multiple TFs (70). To this concern we must note that the CIEN-core reference genome sequence is 23bp long and houses putative TFBS motifs for three transcription factors (EGR1, KLF9, and ZSCAN4), whereas the CIEN-Ins sequence has additional putative motifs for seven transcription factors (HES1, HES2, ZBTB14, EGR4, TCFL5, NRF1, and RREB1; Supplementary Fig. S6C).
We predicted the survival impact of CIEN-Ins by calculating the disease-free interval (DFI) and progression-free interval probability for lung cancer subtypes where these data are available (LUAD and LUSC, n = 102). Strikingly, we found that patients with CIEN-Ins had a better disease-free survival in LUSC (Fig. 6D). When considering together the LUAD and LUSC samples with disease-free survival annotations, we observed that the patients with CIEN-Ins had better DFI (Fig. 6E), but they did not pass statistical significance thresholds. Similarly, we observed the progression-free survival to be better in patients with insertion than those without, but not passing statistical thresholds (Fig. 6F).
We asked whether the presence of CIEN-Ins may be a general phenomenon observed also in other cancer types. As the downregulation of CDH13 was also reported in breast cancer (71), we examined a cohort of the TCGA high-coverage WGS samples (n = 112) for this tumor type. Surprisingly, we found that only 4% of the breast cancer samples had the insertion sequence variant in either tumor or normal tissue, in contrast with 48.6% in patients with lung cancer (Supplementary Fig. S6D). This low occurrence of CIEN-Ins in patients with breast cancer suggests that it is not a general phenomenon.
Overall, these results attest to the importance of combining the tissue-specific definition of enhancers and tailored refinement of sequence variants analysis over enhancer core regions to dissect the disruption potential of regulatory noncoding mutations recurrent at individual loci.
Discussion
The coding genome has been extensively studied in cancer to identify potential driver mutations and therapeutic targets. Despite these efforts, there is a sizeable heterogeneity in terms of cancer biology and clinical behavior of patients that is not explained by known coding mutations. We hypothesized that noncoding mutations in regulatory regions, such as enhancers and promoters, could contribute to cancer development or progression and can be exploited to develop novel strategies for the molecular classification of patients. Without a consensus on solutions to assess whether mutations in regulatory regions can be driver of tumorigenesis, it may be safer to assume they are passenger. However, they may still have an impact on tumor biology and specific strategies for their identification and prioritization are needed.
We worked on various challenges connected to this goal to go beyond the limitations of the most commonly adopted solutions in the field, in particular with regard to defining the reference set of lung-specific enhancers, pairing enhancers with their target genes, identifying mutations in distal regulatory regions and characterizing their functional effects.
Defining enhancers has been a major challenge due to the lack of an exhaustive reference list for all cell types. Enhancers are cell-type–specific; hence, a list of enhancers from one cell type does not represent the whole-lung tissue. Moreover, the cell of origin of lung cancers is not well defined. Hence, we opted for a comprehensive list of lung-specific enhancers defined from a collection of eight distinct lung cell lines and primary tissue samples, including epithelial cells, primary lung tissue, and fibroblasts, as well as a few cancer cell lines. Our goal was to cover primarily enhancers active in the tissue of origin of the tumor. Thus, in our compendium, we included only a couple of lung cancer cell lines (A549 and PC9) because they were previously adopted by reference literature in the field exploring noncoding regulatory mutations (48). As such, we aimed to consider also this information to be as comprehensive as previous literature. We then added also NCI-H460 as in the subsequent screening was identified as a good candidate for experimental validations on the CDH13 locus (Supplementary Fig. S7).
This may seem a counterintuitive solution as opposed to directly using only one cell type for enhancer definition. However, as lung tumors are highly heterogeneous and our cohort of samples consisted of a mix of different lung cancer subtypes, stages, and other clinical features, we applied a comprehensive approach to map all active lung enhancers. Although this rationale is in line with similar solutions chosen in recent literature, we adopted a broader set of samples with respect to reference articles in this field (e.g., compared with lung cancer cell lines matching in ref. 48). In particular, we included novel epigenomics profiles generated for this project, most notably for HBEC3-KT so far not available from lung cancer genomics studies despite their possible role as the cell of origin for a good fraction of these tumors. For identifying active enhancers in the lung, we used a combination of H3K27ac and chromatin accessibility (DNase-seq or ATAC-seq) profiles.
Somatic mutations calling on WGS data is challenging due to the hurdles posed by various factors, including tumor heterogeneity, mutations clonality, and tumor ploidy. In addition, somatic mutation calling can be highly affected by the sensitivity of the adopted algorithm. To overcome these concerns, we selected only high-coverage WGS datasets with tumor and matched normal genomes from three lung cancer subtypes cohorts. Then we applied a tailored bioinformatic pipeline implementing an ensemble approach using a total of five algorithms (four tools for calling indels and four tools for calling SNVs) and used the concordance of at least two variant callers to achieve a comprehensive yet reliable set of somatic sequence variants. This solution was aimed to go beyond the limits of the standard approaches based on a single algorithm. Moreover, as lung cancer has a high mutation burden, our priority was ensuring the control of false positives arising from individual mutation calling algorithms.
Lung cancer is reported to have a high mutation burden genome-wide (72). We observed that the mutation burden at enhancers is lower than the rest of the noncoding genome and that enhancers, promoters, and exons have a comparable mutation burden. This observation is in line with previous literature showing across multiple tumor types that promoter and enhancer regions are mutated at a rate similar to the transcribed genic regions, whereas intergenic regions carry a higher mutational burden (73). We speculate that this lower and comparable mutation burden across such functionally relevant regions could be attributed to a combination of negative selection and transcription-coupled DNA repair mechanisms. However, more specific experiments on tumor evolution would be required to properly investigate this hypothesis.
Nevertheless, we compared the mutation signatures observed across these genomic features to shed light on the process of mutagenesis affecting coding and noncoding regulatory regions. Signature 4, associated with smoking, was prevalent in all of the considered genomic regions, in concordance with its association with lung cancer. We also identify and show different prevalence for specific mutation signatures across cancer subtypes and distinct genomic features (enhancers, promoters, and exons) that were not previously reported. The differential prevalence of signatures between these coding and noncoding regulatory elements is also a novel result, to the best of our knowledge. When looking at the differential mutation signatures across genomic regions, we observe mutation signatures associated with defective DNA mismatch repair and DNA DSB repair to be more prevalent in regulatory regions compared with coding regions. These results corroborate and extend beyond recent data on the accumulation of single and DSBs at regulatory regions (60–62). Moreover, the role of the mismatch repair system in maintaining genome stability is well characterized, and its role in regulating gene enhancer activity in cancer is emerging (74).
For connecting enhancer mutations to their putative target genes, we adopted our framework integrating multi-scale hierarchical chromatin 3D architecture organization in TADs to reconstruct ETG pairs. This approach accounts for the most updated biological knowledge on ETG interactions occurring in the context of structural domains and their dynamic and hierarchical structure. Then, a novel strategy and a crucial change of perspective in comparison with previous studies has been adopted in the integration and interpretation of the composite effect of enhancers mutations at the level of pathways connecting their target genes. Analyzing enhancers’ mutations in terms of their target gene or pathway is meant to solve, at the same time, the problem of focusing the interpretation of effects on the downstream biological targets and aggregates mutations that may have a complementary effect.
Hence, we first aggregated the enhancer mutations at the pathway level and identified them as frequently harboring enhancer mutations in several pathways, including PI3K–AKT signaling, focal adhesion, and ECM organization. Interestingly, initial observations may suggest that only half of the enriched pathways are directly involved in cancer, but a more detailed examination revealed that other pathways are associated to relevant processes for cancer, such as immunity and inflammation. Moreover, the remaining enriched pathways have several genes in common with cancer pathways, thus confirming a general association of enhancers’ mutations with pathways potentially affecting tumorigenesis.
Furthermore, we also observed enhancer mutations affecting gene expression in pathways regulating DNA repair through a custom approach based on the global test. This approach has the conceptual and practical advantage that the effect on target genes and pathways expression can be interpreted by a statistical test summing up the combinatorial effect of multiple sparse variations across all enhancers regulating those genes and pathways. Overall, these results highlight the role of enhancers in the gene regulatory networks that affect critical pathways relevant to cancer progression.
These results also confirm that the aggregation and interpretation of enhancer mutations at the level of their target genes and pathways are the keys to highlighting their convergence over biological processes that can affect cancer development and progression. Indeed, these approaches allow identifying recurrent alterations affecting pathways with known relations to cancer biology, whereas highly recurrent mutations at individual enhancers are generally less frequent. The combinatorial effect of mutations spread across multiple enhancers targeting the same gene or pathway can explain why the field has been struggling to identify recurrent mutations at individual regulatory elements. Indeed, we can speculate that regulatory mutations at enhancers may contribute to the dysregulation of specific functions and pathways related to tumorigenesis. Their combinatorial effects would distribute the selective pressure across multiple genes and their even larger set of enhancers rather than on individual regulatory elements.
The identification of biologically relevant mutations in CDS has primarily relied on assessing their recurrence across cancer patients. The sheer size of the noncoding genome lowers the chance of recurrence of mutations in any specific regulatory element. However, we explored the recurrence at individual enhancer cores as a possible strategy to prioritize biologically relevant mutations. We found the enhancer core of CIEN (CDH13 intronic enhancer) associated with the CDH13 gene to be recurrently mutated with a characteristic insertion variation [ins(TG)n(CG)4]. We hypothesized that CIEN-Ins affect CDH13 expression by expansion of existing or generation of new TFBS. We also observe the presence of TFBS motifs at multiple adjacent locations within the region. This result is interesting, especially in the light of literature reports that at gene regulatory regions, there can be an accumulation of potential TFBSs, and the presence of multiple degenerate or weakly competing binding sites could accelerate the TF search for its target gene (75). Using CRISPR/Cas9-based genome editing, we confirmed that the deletion of the enhancer core results in the downregulation of the CDH13 gene in a cellular model. We found the presence of CIEN-Ins associated with higher disease-free survival in a cohort of LUSC samples, although the number of patients with complete clinical follow-up data was limited. Previous literature indicated a tumor-suppressor function for CDH13 and its downregulation associated to poor prognosis. Thus, we conclude that the CIEN-Ins may also act as a tumor suppressor by promoting CDH13 expression.
In conclusion, here we present a combination of strategies to identify functionally relevant noncoding mutations. Furthermore, we have shown that enhancer mutations can affect the expression of target genes and that recurrent mutations can influence survival probability of patients. Finally, we also highlight how mutations in enhancers can affect key cancer pathways. These results confirm the potential relevance of noncoding regulatory mutations with respect to cancer pathogenesis and how they can be exploited for patient molecular classification and to understand more details of tumor biology. We expect that extending this approach to other tumor types may further elucidate tumor-specific programs of biological alterations.
Supplementary Material
Supplementary Figures S1-S7
Primer details
Lung cancer cohort used in the study
Number of enhancers associated with genes and the number of mutations in the lower right quadrant of figure 4c Overlap with Msigdb C2 dataset
Epigenomics data from lung cell and tissue type used for Enhancer definition
Genomic co-ordinates of lung specific enhancers (hg19)
Enhancer target gene prediction results
Gene set enrichment analysis on C6 oncogenic dataset from MsigDB
Mutation frequency analysis at enhancer cores
CDH13 enhancer regression: Generalised linear model
Acknowledgments
We thank the support provided to us by the IFOM cell culture unit, IFOM genomics unit, IFOM genome editing unit, and the IFOM IT Team. We thank Federica Mainoldi (IFOM) for her suggestions in the experimental set-ups. We thank Maria Vivo (UniSA) for help in the ChIP-seq experiments handling. We thank Marco Cosentino Lagomarisno and Martin Schaefer for critical feedback on earlier versions of the article. We thank CINECA-Regione Lombardia LISA 2016–2018 call for high-performance computing resources and technical support to optimize the WGS mutation calling pipeline. The funding for this research was provided by Italian Association for Cancer Research (AIRC) Start-up grant 2015 n.16841 and FRRB INTERSTRAT-CAD (grant #CP2_14/2018 to F. Ferrari), AIRC IG grant #23747 (to S. Casola), AIRC Fellowship no. 22416 (to J.M. Hariprakash), AIRC fellowship No. 22351 (to E. Salviato), AIRC Fellowship no. 22538 (to F. La Mastra), AIRC iCARE Fellowship no. 800924 (to R.S. Silva). The results published here are in whole or part based upon data generated by The Cancer Genome Atlas (TCGA) managed by the NCI and NHGRI. Information about the TCGA can be found at http://cancergenome.nih.gov.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
R.S. Silva reports grants from AIRC during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
J.M. Hariprakash: Data curation, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. E. Salviato: Software, formal analysis. F. La Mastra: Data curation, validation. E. Sebestyén: Data curation, software. I. Tagliaferri: Data curation, software. R.S. Silva: Funding acquisition, validation, investigation, writing–review and editing. F. Lucini: Data curation, validation, writing–review and editing. L. Farina: Data curation, software. M. Cinquanta: Conceptualization, data curation, validation, investigation, writing–original draft, writing–review and editing. I. Rancati: Conceptualization, data curation, supervision, validation, investigation, writing–original draft, writing–review and editing. M. Riboni: Data curation, supervision, validation, investigation, writing–review and editing. S.P. Minardi: Investigation. L. Roz: Investigation. F. Gorini: Data curation, supervision, validation, investigation, writing–review and editing. C. Lanzuolo: Data curation, supervision, validation, investigation, writing–original draft, writing–review and editing. S. Casola: Conceptualization, supervision, investigation, writing–review and editing. F. Ferrari: Conceptualization, supervision, funding acquisition, writing–original draft, writing–review and editing.
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frankell AM, Dietzen M, Al Bakir M, Lim EL, Karasaki T, Ward S, et al. The evolution of lung cancer and impact of subclonal selection in TRACERx. Nature 2023;616:525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tippens ND, Vihervaara A, Lis JT. Enhancer transcription: what, where, when, and why? Genes Dev 2018;32:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 2012;13:613–26. [DOI] [PubMed] [Google Scholar]
- 8. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 2014;46:989–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weedon MN, Cebola I, Patch A-M, Flanagan SE, De Franco E, Caswell R, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet 2014;46:61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015;161:1012–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elliott K, Larsson E. Non-coding driver mutations in human cancer. Nat Rev Cancer 2021;21:500–9. [DOI] [PubMed] [Google Scholar]
- 14. Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer 2016;16:483–93. [DOI] [PubMed] [Google Scholar]
- 15. Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, et al. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 2014;15:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lochovsky L, Zhang J, Fu Y, Khurana E, Gerstein M. LARVA: an integrative framework for large-scale analysis of recurrent variants in noncoding annotations. Nucleic Acids Res 2015;43:8123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mularoni L, Sabarinathan R, Deu-Pons J, Gonzalez-Perez A, López-Bigas N. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol 2016;17:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Umer HM, Smolinska K, Komorowski J, Wadelius C. Functional annotation of noncoding mutations in cancer. Life Sci Alliance 2021;4:e201900523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith E, Shilatifard A. Enhancer biology and enhanceropathies. Nat Struct Mol Biol 2014;21:210–9. [DOI] [PubMed] [Google Scholar]
- 21. Yen A, Kheradpour P, Zhang Z, Heravi-moussavi A, Liu Y, Amin V, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Plajzer-Frick I, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2010;457:854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nat Rev Genet 2013;14:288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature 2012;489:109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salviato E, Djordjilović V, Hariprakash JM, Tagliaferri I, Pal K, Ferrari F. Leveraging three-dimensional chromatin architecture for effective reconstruction of enhancer–target gene regulatory interactions. Nucleic Acids Res 2021;49:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci USA 2015;112:10995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of chromosomal domains by loop extrusion. Cell Rep 2016;15:2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao JY, Hafner A, Boettiger AN. How subtle changes in 3D structure can create large changes in transcription. eLife 2021;10:e64320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuin J, Roth G, Zhan Y, Cramard J, Redolfi J, Piskadlo E, et al. Nonlinear control of transcription through enhancer–promoter interactions. Nature 2022;604:571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gabriele M, Brandão HB, Grosse-Holz S, Jha A, Dailey GM, Cattoglio C, et al. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 2022;376:496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science 2018;362:eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 2015;109:21.29.1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. George J, Lim JS, Jang SJ, Cun Y, Ozretia L, Kong G, et al. Comprehensive genomic profiles of small-cell lung cancer. Nature 2015;524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H, Wren J. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014;30:2843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lei L, Fithian W. AdaPT: an interactive procedure for multiple testing with side information. J R Stat Soc Ser B Statistical Methodol 2018;80:649–79. [Google Scholar]
- 38. Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 2013;503:290–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lawlor N, Márquez EJ, Orchard P, Narisu N, Shamim MS, Thibodeau A, et al. Multiomic profiling identifies cis-regulatory networks underlying human pancreatic β-cell identity and function. Cell Rep 2019;26:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barutcu AR, Lajoie BR, McCord RP, Tye CE, Hong D, Messier TL, et al. Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol 2015;16:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bunting KL, Soong TD, Singh R, Jiang Y, Béguelin W, Poloway DW, et al. Multi-tiered reorganization of the genome during B cell affinity maturation anchored by a germinal center-specific locus control region. Immunity 2016;45:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmitt AD, Hu M, Jung I, Lin Y, Barr CL. Resource a compendium of chromatin contact maps reveals spatially active regions in the human genome resource a compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep 2016;17:2042–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corona RI, Seo J-H, Lin X, Hazelett DJ, Reddy J, Fonseca MAS, et al. Non-coding somatic mutations converge on the PAX8 pathway in ovarian cancer. Nat Commun 2020;11:2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou W, Laird PW, Shen H. Comprehensive characterization, annotation, and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 2017;45:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018;173:400–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goeman JJ, Van de Geer S, De Kort F, van Houwellingen HC. A global test for groups fo genes: testing association with a clinical outcome. Bioinformatics 2004;20:93–9. [DOI] [PubMed] [Google Scholar]
- 48. Zhang J, Lee D, Dhiman V, Jiang P, Xu J, McGillivray P, et al. An integrative ENCODE resource for cancer genomics. Nat Commun 2020;11:3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sutherland KD, Berns A. Cell of origin of lung cancer. Mol Oncol 2010;4:397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sutherland KD, Proost N, Brouns I, Adriaensen D, Song J-Y, Berns A. Cell of origin of small-cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011;19:754–64. [DOI] [PubMed] [Google Scholar]
- 51. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non–small cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 2015;16:e165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu F, Fan J, He Y, Xiong A, Yu J, Li Y, et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non–small cell lung cancer. Nat Commun 2021;12:2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong K-K. Non–small cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 2014;14:535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24:1277–89. [DOI] [PubMed] [Google Scholar]
- 55. Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008;132:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence MS, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015;518:360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Koboldt DC. Best practices for variant calling in clinical sequencing. Genome Med 2020;12:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004;91:355–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pessina F, Giavazzi F, Yin Y, Gioia U, Vitelli V, Galbiati A, et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat Cell Biol 2019;21:1286–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of oncogenic super-enhancers is coupled with DNA Repair by RAD51. Cell Rep 2019;29:560–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Melton C, Reuter JA, Spacek DV, Snyder M. Recurrent somatic mutations in regulatory regions of human cancer genomes. Nat Genet 2016;47:710–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge JV, Thomas JL, et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 2015;47:106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wakasugi T, Izumi H, Uchiumi T, Suzuki H, Arao T, Nishio K, et al. ZNF143 interacts with p73 and is involved in cisplatin resistance through the transcriptional regulation of DNA repair genes. Oncogene 2007;26:5194–203. [DOI] [PubMed] [Google Scholar]
- 66. Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, Tiao G, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020;578:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Alexandra AV, Kutuzov MA. Cadherin 13 in Cancer. Genes Chromosomes Cancer 2010;49:775–90. [DOI] [PubMed] [Google Scholar]
- 68. Sato M, Mori Y, Sakurada A, Fujimura S, Horii A. The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum Genet 1998;103:96–101. [DOI] [PubMed] [Google Scholar]
- 69. Kim DS, Kim MJ, Lee JY, Kim YZ, Kim EJ, Park JY. Aberrant methylation of E-cadherin and H-cadherin genes in non–small cell lung cancer and its relation to clinicopathologic features. Cancer 2007;110:2785–92. [DOI] [PubMed] [Google Scholar]
- 70. Panigrahi A, O'Malley BW. Mechanisms of enhancer action: the known and the unknown. Genome Biol 2021;22:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, et al. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res 2001;61:4556–60. [PubMed] [Google Scholar]
- 72. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science 2015;349:1483–9. [DOI] [PubMed] [Google Scholar]
- 73. Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet 2014;46:1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hung S, Saiakhova A, Faber ZJ, Bartels CF, Neu D, Bayles I, et al. Mismatch repair-signature mutations activate gene enhancers across human colorectal cancer epigenomes. eLife 2019;8:e40760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rao S, Ahmad K, Ramachandran S. Cooperative binding between distant transcription factors is a hallmark of active enhancers. Mol Cell 2021;81:1651–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1-S7
Primer details
Lung cancer cohort used in the study
Number of enhancers associated with genes and the number of mutations in the lower right quadrant of figure 4c Overlap with Msigdb C2 dataset
Epigenomics data from lung cell and tissue type used for Enhancer definition
Genomic co-ordinates of lung specific enhancers (hg19)
Enhancer target gene prediction results
Gene set enrichment analysis on C6 oncogenic dataset from MsigDB
Mutation frequency analysis at enhancer cores
CDH13 enhancer regression: Generalised linear model
Data Availability Statement
The data generated in this study are publicly available in GEO at GSE228832, including H3K27ac ChIP-Seq and ATAC-seq data on HBEC3-KT and NCI-H460 cell lines. The cancer genomics WGS and RNA-seq data analyzed in this study were obtained from The database of Genotypes and Phenotypes at phs000178.v11.p8 and the European Genome–Phenome Archive (EGA) at EGAS00001000925. The COSMIC mutation signatures analyzed in this study were obtained from the COSMIC database version 2 at https://cancer.sanger.ac.uk/signatures/signatures_v2/. The ENCODE3 ChIP-seq and DNase-seq data analyzed in this study were obtained from https://www.encodeproject.org. All other raw data are available upon request from the corresponding author.