

Abstract
The flap endonuclease (FEN) of the hyperthermophilic archaeon Methanococcus jannaschii was expressed in Escherichia coli and purified to homogeneity. FEN retained activity after preincubation at 95°C for 15 min. A pseudo-Y-shaped substrate was formed by hybridization of two partially complementary oligonucleotides. FEN cleaved the strand with the free 5′ end adjacent to the single-strand–duplex junction. Deletion of the free 3′ end prevented cleavage. Hybridization of a complementary oligonucleotide to the free 3′ end moved the cleavage site by 1 to 2 nucleotides. Hybridization of excess complementary oligonucleotide to the free 5′ end failed to block cleavage, although this substrate was refractory to cleavage by the 5′-3′ exonuclease activity of Taq DNA polymerase. For verification, the free 5′ end was replaced by an internally labeled hairpin structure. This structure was a substrate for FEN but became a substrate for Taq DNA polymerase only after exonucleolytic cleavage had destabilized the hairpin. A circular duplex substrate with a 5′ single-stranded branch was formed by primer extension of a partially complementary oligonucleotide on virion φX174. This denaturation-resistant substrate was used to examine the effects of temperature and solution properties, such as pH, salt, and divalent ion concentration on the turnover number of the enzyme.
In a wide variety of biological processes, including DNA replication (1), recombination (21), and repair (5), DNA structures with single-stranded branches or “flaps” may be found in intermediates. For example, flaps may be produced during lagging-strand synthesis when the DNA polymerase displaces the primer RNA of the adjacent Okazaki fragment. While the 5′-3′ exonuclease activity of eubacterial DNA polymerases such as Escherichia coli DNA polymerase and Thermus aquaticus DNA polymerase will remove flaps endonucleolytically (18), all known eukaryotic DNA polymerases lack this activity.
While investigators have postulated the existence of eukaryotic enzymes with this activity, it was not until 1994 that the flap-cleaving activity was demonstrated in a pure protein (10). The acronym FEN-1, standing for flap endonuclease as well as 5′ exonuclease and thus referring to the dual properties of the enzyme, was proposed. Upon further analysis, the mammalian FEN-1 was found to be the same enzyme previously named DNase IV based on its DNase activity (17) and CCA exonuclease based on its activity in circle closing in an in vitro polyomavirus DNA replication assay (7). FEN-1 has become the prototype of a eukaryotic structure-specific nuclease family (16). Mammalian FEN-1 is most similar to Saccharomyces cerevisiae RAD27 (also known as YKL510 or RTH-1 [for RAD2 homolog]) and Schizosaccharomyces pombe RAD2. Other members of the family include a group of larger and similar proteins, human xeroderma pigmentosum protein XPG (8) (also known as rodent ERCC5), S. cerevisiae RAD2, and S. pombe RAD13. In addition, some bacteriophage-encoded structure-specific nucleases such as T5 5′ exonucleases apparently belong to the same family.
Many groups of investigators have characterized various properties of the enzymes in this family. The solvent parameters and substrates specificities, including the requirement for a free 5′ single-stranded end (19), have been defined for murine FEN-1 (10). S. cerevisiae RAD2 has similar substrate requirements (9). The T5 5′-exonuclease crystal structure suggests that the free 5′ end may thread through the enzyme (4).
FEN-1 (27) or XPG (6) will form a complex with human proliferating cell nuclear antigen (PCNA), which itself interacts not only with the DNA polymerase but also with DNA ligase (15), thus including many of the elements needed to synthesize and join Okazaki fragments. PCNA stimulates FEN-1 activity 10-fold. An analysis of the mutator phenotype of S. cerevisiae RAD27 mutants revealed a preponderance of insertion mutations with duplication of the sequence between direct repeats (25), consistent with recombination initiated by out-of-register single-strand annealing of uncleaved flap structures. The human FEN1 gene has been sequenced, and the chromosomal location has been determined (12). Site-directed mutagenesis studies have identified amino acids essential for enzymatic activity (24) as well as a region responsible for PCNA–FEN-1 complex formation (6), but detailed site-directed mutagenesis studies have not been reported.
When the archaea were first discovered in the 1970s, they were difficult to classify. While cytologically they were similar to eubacteria, at a molecular level they were more like eukaryotes (20). The archaeon Methanococcus jannaschii was first discovered in a deep-sea hydrothermal vent in 1983 (14). M. jannaschii was a methanogenic, extremely thermophilic (growing at 48 to 94°C), extremely piezophilic (growing at pressure of up to 500 atm) motile coccus. For these reasons, it was selected as the prototype archaeon for complete genomic sequencing, which was completed in 1996 (3). In this study, we report the cloning, expression, purification, substrate requirements, and biochemical activity of the FEN protein from the M. jannaschii.
MATERIALS AND METHODS
Bacterial strains and genomic, plasmid, and bacteriophage DNA.
For all DNA manipulations, standard techniques and procedures (22) were used. E. coli BL21(DE3)pLysS (Novagen, Inc.) was used to propagate the expression plasmid pET20b+ (Novagen). M. jannaschii cells were purchased from David R. Boone, Oregon Collection of Methanogens. Genomic DNA was extracted by a modification of the protocol described by Sandler et al. (23). In brief, the cells were pelleted and resuspended in 100 mM NaCl–10 mM Tris-HCl–1 mM EDTA (pH 8.0). One-tenth volume of 10% solution of sodium lauryl sarcosine was added before phenol extraction. The aqueous phase was diluted 1:1 with 5 M ammonium acetate and then 1:5 with ethanol. After microcentrifugation, the pellet was washed with 70% ethanol and resuspended in 10 mM Tris-HCl–1 mM EDTA (pH 8.0) (TE). Bacteriophage φX174 virion DNA was purchased from New England Biolabs, Inc. (NEB). Plasmid pSK101 was obtained from Soo Jung Kim (Mount Sinai School of Medicine). In it, the polylinker sequence in pUC19, 5′ GGGGATCCTCTAGAGTCGACct 3′, was replaced with 5′ TTGGGTCTTCCAGGGTAGATct 3′, thus deleting a BamHI site and introducing a BglII site (both underlined).
Nucleotides and enzymes.
Absorbance spectra and melting temperatures were determined by using a Hewlett-Packard diode array spectrophotometer equipped with a Peltier temperature controller. DNA and primer concentrations were determined by using 50 and 36 μg ml−1 A260−1, respectively, as conversion factors. Deoxynucleoside triphosphates were purchased from Boehringer Mannheim. [α-35S]dATP and [γ-32P]ATP were purchased from NEN/DuPont. Amplitaq DNA polymerase was purchased from Perkin-Elmer and used in the buffer supplied by the manufacturer. Restriction endonucleases, T4 polynucleotide kinase, and T4 DNA ligase were purchased from NEB and used as recommended by the manufacturer. Simultaneous reactions with two or more restriction endonucleases were carried out in NEB3 buffer (NEB).
Oligonucleotides.
All synthetic oligodeoxynucleotide primers for PCR and sequencing were synthesized on automated instruments using standard phosphoramidite chemistry. The sense primer 5′ GCGCATATGGGAGTGCAGTTTGGTGAT 3′ and the antisense primer 5′ GCGCTCGAGTTATTTAAACCATGCATCTAA 3′ were used to amplify the M. jannaschii fen gene. The primers contained GCG caps adjacent to NdeI and XhoI restriction endonuclease cleavage sites (underlined). The NdeI site contained the initiation codon (boldface). Additional sequencing primers lacking the GCG cap and restriction endonuclease sites were 5′ GGCAACCCCGCCAGCCTAGC 3′ (sense) and 5′ GAAAGGAGCGGGCGCTAGGG 3′ (antisense), corresponding to pET20b+ vector sequences, and 5′ GCTGAACTTAAGATGAAAGA 3′ (sense) and 5′ AAATATGGCTATATCTATCA 3′ (antisense), corresponding to internal FEN sequences. Two oligonucleotides capable of hybridization to form a forked FEN substrate were synthesized with the 5′ end of the sense oligonucleotide: FS (5′ CGAGTACCTAGCAAGGCAGTTAGCATGCTTAGGACTG 3′), complementary to the 3′ end of the antisense oligonucleotide, and FA (5′ AATTCAGTCCTAAGCATGCCATTCGCTTAAGCTATCAGGCC 3′). A truncated version of FA containing only the duplex-forming sequence, FA-T (5′ CAGTCCTAAGCATGC 3′), was synthesized. Oligonucleotides FS-C (5′ TAACTGCCTTGCTAGGTACTCG 3′) and FA-C (5′ GGCCTGATAGCTTAAGCGAATG 3′) were synthesized to be complementary to the single-stranded regions of sense and antisense strands, respectively, of the forked structure. Finally, the oligonucleotide FS-L (5′ TAACTGCCTTGCTAGG-TACTCGGATCGTTTTTCGATC 3′) was synthesized for converting the single-stranded portion of FS into a loop by ligation. We also synthesized three oligonucleotides with 3′ complementarity to φX174 virion DNA, P-15 (5′ CTTTGTTGGACTAATGCGGCGTTGACAGATGTATC 3′), P-4 (5′ TAATGCGGCGT-TGACAGATGTATC 3′), and P-6 (5′ CTGAATCCAGAAAACTGGCCTAAC 3′), with 15, 4, and 6 5′ unpaired nucleotides (nt), respectively. A BstXI site adjacent to the branch point is underlined.
DNA amplification, cloning, and sequence analysis.
PCR amplifications were carried out on an Ericomp Powerblock thermocycler or a Stratagene thermocycler gradient. The M. jannaschii FEN amplicon was digested with primer-specific restriction endonucleases, ligated into compatible sites on pET20b+, and transformed into E. coli BL21(DE3)pLysS. Inserts in the expression vectors were sequenced in both orientations by using insert-specific and vector-specific oligodeoxynucleotide primers and fluorescent dideoxy terminators and an ABI model 377 DNA sequencer. After expression and purification, the identity of the FEN protein was confirmed at the Protein Structure Core Facility at the University of Nebraska Medical Center by amino acid analysis and N-terminal sequencing. Nucleic acid and protein sequence analyses also were carried by using BLAST at the National Center for Biotechnology Information web site (www.ncbi.nlm.nih.gov) and the Genetics Computer Group programs (2).
Expression.
The BL21(DE3)pLysS clone containing the pET20b+ M. jannaschii FEN expression vector was propagated at 37°C in LB containing ampicillin (50 μg/ml) and chloramphenicol (25 μg/ml). Overnight cultures were diluted 1/100 into the same medium, grown to an A600 of ≈0.5, induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and grown for an additional 4 to 6 h. Cells were collected, resuspended in 10 mM EDTA–1 mM phenylmethylsulfonyl fluoride–1 mM dithiothreitol (DTT)–50 mM Tris-HCl (pH 9.0), frozen and thawed to disrupt the envelopes, sonicated to reduce the viscosity, and clarified by microcentrifugation. The supernatants were made 0.3 M (NH4)2SO4 by addition of 3 M stock, heated to 75°C for 15 min to denature thermolabile proteins, placed on ice for 30 min to aggregate the denatured proteins, and clarified by microcentrifugation for 15 min at 4°C.
Purification.
Crude FEN protein, approximately 1 ml per 250-ml culture, was diluted sixfold with 20 mM Tris-HCl (pH 9.0), loaded onto a 1-ml HiTrap Q anion-exchange column (Pharmacia), repeatedly washed with 20 mM Tris-HCl (pH 9.0), and eluted with 0.3 M NaCl in the same buffer. The eluate was diluted fivefold with 20 mM Tris-HCl (pH 9.0), loaded onto a 1-ml HiTrap SP ion-exchange column (Pharmacia), repeatedly washed with 20 mM Tris-HCl (pH 9.0), and eluted with 0.3 M NaCl in the same buffer. Following dialysis against 50 mM KCl–1 mM EDTA–10 mM Tris-HCl (pH 8) and concentration in a Centricon-30 filter, protein concentrations were determined and compared with complete absorbance spectra to determine an extinction coefficient and to verify removal of nucleic acids. The purified protein was stored at 4°C. Purification from other proteins was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in which overloaded gels were stained with Coomassie brilliant blue R.
FEN-dependent restriction endonuclease cleavage at a site containing a branch.
φX174 virion DNA was annealed to oligonucleotide P-6 containing a branch point adjacent to a BstXI recognition sequence and extended by 3′-5′ exonuclease-negative Pfu DNA polymerase (Stratagene) for 18 cycles of 55°C for 1 min and 65°C for 25 min. An aliquot was digested by HaeIII and analyzed by agarose gel electrophoresis to verify conversion to duplex DNA. Various concentrations of FEN and 500 ng of branched duplex φX174 were incubated in 50 mM KCl–10 mM Tris-HCl (pH 8.0)–2.5 mM MgCl2–1 mM DTT–10 μg of bovine serum albumin (BSA) per ml at 55°C for 1 h and digested with 1 U each of BstXI and BsiEI (NEB) at 55°C for 1 h. Products were separated on a 1.5% agarose gel, stained with ethidium bromide, and visualized by fluorography.
Pseudo-Y-shaped (pseudo-Y) oligonucleotide cleavage assays.
The oligonucleotide FS was phosphorylated with [γ-32P]ATP and purified by phenol extraction and ethanol precipitation. A typical reaction mixture contained 1 pmol of phosphorylated oligonucleotide FS, 2 pmol of oligonucleotide FA, and 2 pmol of FEN in 50 mM Tris-HCl (pH 8.0)–5 mM MgCl2–1 mM DTT–100 μg of BSA per ml. Incubation was carried out at 50°C for 45 min and stopped by the addition of 5 μl of the stop solution from the United States Biochemical reagent kit for DNA sequencing. The products were separated by 6% denaturing PAGE and analyzed by autoradiography.
Modified pseudo-Y oligonucleotide cleavage assays.
A 5′ or 3′ single-stranded arm was converted to a duplex by annealing to either 5 pmol of FS-C or 5 pmol of FA-C, respectively. The 3′ arm was truncated by replacement of oligonucleotide FA by oligonucleotide FA-T. A hairpin structure was formed on the 5′ arm by ligation of oligonucleotide FS-L onto the 5′-32P-labeled oligonucleotide FS. The same experimental protocol was used for cleavage and analysis of these modified pseudo-Y structures.
Quantitation of FEN activity.
5′-32P-labeled oligonucleotide P-15 was annealed to φX174 virion DNA and extended to form a branched duplex as described above. The duplex was freed from unincorporated oligonucleotide by filtration through a Millipore Ultrafree-MC unit and resuspended in TE. Incubations with FEN were carried out in 50 mM KCl–2.5 mM MgCl2–1 mM DTT–10 μg of BSA per ml, with 10 mM sodium acetate for pH 6 to 7 and 10 mM Tris-HCl for pH 7 to 9. For each set of conditions, aliquots were placed in stop solution as a function of time. The duplex structures and cleavage products were separated by filtration through a Millipore Ultrafree-MC unit. The radioactive counts in the filtrate were determined using a Beckman liquid scintillation counter.
Loop assays.
To produce a loop-containing heteroduplex (50% yield), equimolar pSK101 and pUC19 were linearized by digestion with ScaI, denatured at 97°C for 3 min, and renatured at 68°C for 1 h. Aliquots with 250 ng of this mixture were incubated for 70°C for 18 h with 2 pmol of FEN in 50 μl in 50 mM Tris-HCl (pH 8.0)–5 mM MgCl2–1 mM DTT–100 μg of BSA per ml.
RESULTS
Cloning, expression, and sequence analysis of the M. jannaschii fen gene.
The annotation of the M. jannaschii genome (3) assigned a translated sequence, C64480, the label “DNA repair protein RAD2 homolog.” A BLAST search of available databases revealed one highly related protein each from Archaeoglobus fulgidus (AE001087; P ≈ e−90) and Methanobacterium thermoautotrophicum (AE000922; P ≈ e−87), both archaea. The next group of related proteins included human FEN-1 (P39750; P ≈ e−50), mouse FEN-1 (P39749; P ≈ e−49), and S. cerevisiae RAD27 (P ≈ e−48). Much less related was S. cerevisiae RAD2 (P ≈ e−18). Because the M. jannaschii translated sequence was more closely related to sequences of mammalian FENs (36% identity) and yeast RAD27 (34% identity) than to that of yeast RAD2 (25% identity over fewer than two-thirds as many base pairs), and based on the enzymatic activity described in this work, the M. jannaschii protein was referred to as FEN.
The M. jannaschii fen coding sequence was amplified from genomic DNA and cloned into a pET20b+ expression vector. DNA sequence analysis revealed one difference that affected the amino acid sequence, T189I. This difference was verified by cycle sequencing genomic DNA.
Thermostable FEN was expressed and purified to homogeneity (Fig. 1). A heating step removed most of the contaminating, thermolabile E. coli proteins. SDS-PAGE analysis following induction of a control vector without insert revealed a less abundant thermostable protein band slightly larger than FEN (37 kDa). This contaminating protein did not bind to HiTrap Q. A HiTrap SP chromatography step was used to remove a major contaminant at 22 kDa as well as nucleic acids (lanes 3 to 10). After purification, the FEN was free of contaminating proteins, as shown by a single band on an overloaded SDS-polyacrylamide gel, and free of contaminating nucleic acids, as shown by an absorbance ratio, A280/A260, greater than 1.5.
FIG. 1.

SDS-PAGE analysis of M. jannaschii FEN protein on a HiTrap SP column. Lanes: 1, molecular weight markers; 2, fraction after heating at 70°C; 3 to 8, flowthrough and low-salt wash fractions; 9 and 10, 0.3 M NaCl elution fractions.
Purified FEN has the expected N-terminal amino acid sequence. The overall yield of the thermostable FEN from various preparations was approximately 0.2 mg/1011 cells, corresponding to approximately 2.5% of the initial protein content of the cells. All subsequent experiments were carried out with purified FEN.
Cleavage of a pseudo-Y structure.
A pseudo-Y structure was formed by using two partially complementary oligonucleotides with 15 nt of complementarity and 22 nt of 5′ (oligonucleotide FS) or 3′ (oligonucleotide FA) single-stranded extensions (Fig. 2A). Oligonucleotide FS, which was labeled with 32P, also contained a low concentration set of 5′-truncated sequences, providing a convenient internal size control. Oligonucleotide FA was present in twofold excess to ensure complete incorporation of the labeled oligonucleotide into the pseudo-Y structure. Aliquots of this structure were incubated with FEN protein or with Taq DNA polymerase as a positive control. The products were separated by denaturing PAGE and analyzed by autoradiography. The cleavage products resulting from both Taq DNA polymerase (Fig. 3, lane 5) and FEN protein (lane 6) were about 25 nt in length, indicating a cleavage site distal to the elbow by about 3 nt. Oligonucleotide FS was not cleaved in the absence of oligonucleotide FA (lane 7). The same result was observed with FEN. Thus, M. jannaschii FEN has the expected structure-dependent endonuclease activity.
FIG. 2.

Diagrams of FEN substrates. (A) Pseudo-Y structure formed by annealing oligonucleotides FS and FA; (B) structure formed by annealing a primer (P-4, P-6, or P-15) to φX174 virion DNA and extending; (C) substitution of oligonucleotide FA-T for FA; (D) annealing of oligonucleotide FA-C to FA; (E) annealing of oligonucleotide FS-C to FS; (F) ligation of oligonucleotide FS-L to FS.
FIG. 3.

Cleavage of pseudo-Y structure at 50°C. Lanes: 1 to 3, size markers (10, 15, and 21 nt, respectively); 4, oligonucleotide FS (37 nt) with truncated sequences; 5, FS after incubation of FS plus FA with Taq DNA polymerase; 6, FS after incubation of FS plus FA with FEN; 7, FS after incubation of FS alone with Taq DNA polymerase.
To test the thermostability of purified FEN, the protein was incubated at either 90 or 95°C for 15 min prior to measurement of its activity with the pseudo-Y substrate. Control lanes showed absence of cleavage when oligonucleotide FA was omitted (Fig. 4, lanes 4, 6, and 8). The cleavage product produced by unheated FEN is depicted in lane 5. M. jannaschii FEN is quite thermostable, losing only a small amount of enzymatic activity after incubation for 15 min at 90°C (lane 7) and even retaining activity after 15 min at 95°C (lane 3).
FIG. 4.

Thermostability of FEN protein. Lanes: 1, 21-nt marker; 2, oligonucleotide FS (37 nt); 3, FS after incubation of FS plus FA with FEN; 5, FS after incubation of FS plus FA with unheated FEN protein; 7, FS after incubation of FS plus FA with FEN protein preheated to 90°C for 15 min; lanes 4, 6, and 8, same as lanes 3, 5, and 7 but with omission of oligonucleotide FA.
Figure 5 depicts a time course for cleavage of a pseudo-Y structure, with increasing cleavage over the 1.5 h. Significant cleavage occurred between 45 min (lane 3) and 1.5 h (lane 4), suggesting that the pseudo-Y structure may not be the ideal substrate for this enzyme.
FIG. 5.
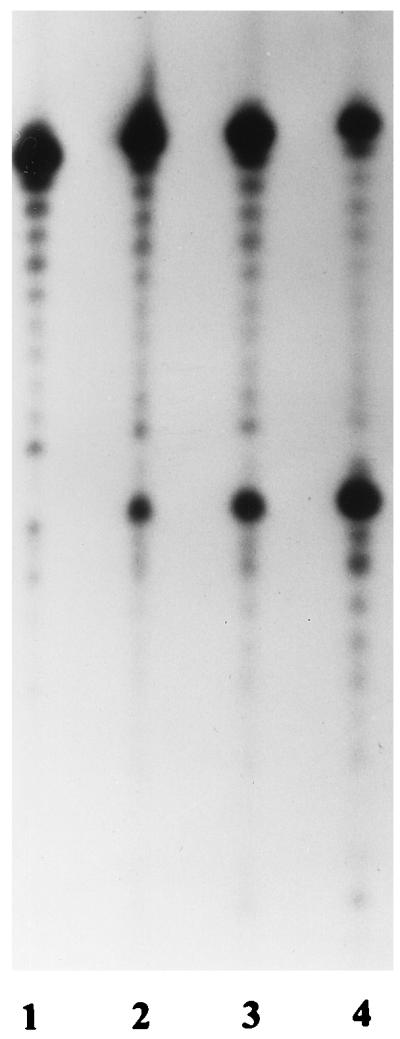
Time course of cleavage of a pseudo-Y structure at 50°C. Lanes 1 to 4 represent reactions stopped at 16, 32, 45, and 90 min, respectively.
FEN-dependent restriction endonuclease cleavage at a site containing a branch.
To provide a quick, nonradioactive semiquantitative alternative method for assessing the activity of FEN, the branched primer oligonucleotide P-6 was annealed to φX174 virion DNA and extended with 3′-5′ exonuclease-negative Pfu DNA polymerase to obtain a complete circle (Fig. 2B). The resultant product had a 6-nt flap which was adjacent to a BstXI restriction endonuclease recognition site. The flap protected this site from digestion (Fig. 6, lane 1, upper band), whereas following incubation as little as 50 pg of FEN in 20 μl (lane 6), the site was available for digestion (lanes 2 to 6).
FIG. 6.

Removal of a flap at a restriction endonuclease cleavage site at 55°C. Lanes: 1 to 6, φX174 primer extension product cut with BstXI and BsiEI; 1, no preincubation with FEN protein; 2 to 6, preincubation with a twofold serial dilution of FEN protein, with 50 pg (in 20 μl) in lane 6; 7, markers.
Quantitation of FEN activity.
Two substrates were produced for use in quantitation of the solvent and temperature dependence of FEN activity. A 32P-labeled branched primer with a 4-nt (P-4) or 15-nt (P-15) flap was annealed to φX174 virion DNA and extended as described above. The resultant structures were resistant to denaturation at temperatures above the maximum temperature for FEN activity. The flap cleavage products were generated as a function of time and separated from the duplex molecules by ultrafiltration. The cleavage rates with these substrates were similar to the rates observed with the pseudo-Y substrate. The 15- and 4-nt flaps were released at the same rate. Addition of magnesium was required for FEN activity, and magnesium could not be replaced by calcium, manganese, or zinc. The Mg2+ concentration dependence was weak, with an optimum of 2.5 mM and at least 50% activity between 1 and 10 mM. The monovalent salt effect was also broad, with equal activities observed in 50 and 100 mM KCl. Figure 7 depicts the effects of temperature and pH. The optimum temperature was 70°C, with significant activity observed from 50 to 80°C (Fig. 7A). The decreased enzymatic activity beginning at 80°C is a property of the enzyme and not the result of thermal instability of either the substrate, with a melting temperature of 91°C, or the enzyme. Surprisingly, the apparent optimum pH was 6 to 7, measured at 25°C, with activity decreasing rapidly from pH 7 to 9 (Fig. 7B). Because pH decreases with increasing temperature, apparent pH optima for proteins from thermophiles are usually higher than those of homologous proteins from mesophiles.
FIG. 7.

Dependence of FEN activity on temperature (A) and pH (B). All reaction mixtures contained 2 pmol of FEN protein in 50 μl.
FEN activity on modified pseudo-Y substrates.
Figure 8A depicts FEN and Taq DNA polymerase cleavage products with pseudo-Y (lanes 2 and 3) and modified pseudo-Y substrates. Oligonucleotide FA was replaced by truncated oligonucleotide FA-T, which was completely complementary to oligonucleotide FS and lacked the free 3′ end (Fig. 2C). Taq DNA polymerase acts on this substrate (Fig. 8A, lane 4), apparently cutting near the duplex–single-strand junction, but FEN appears to have little or no activity (lane 5). Oligonucleotide FA was hybridized to oligonucleotide FA-C, converting the 3′ single-stranded tail to a duplex (Fig. 2D). Both Taq DNA polymerase (lane 6) and FEN (lane 7) demonstrated activity on this substrate. In both cases, the cleavage products are at least one nucleotide nearer to the elbow than seen with the pseudo-Y structure controls (lanes 2 and 3), with FEN producing more of the smaller product. Figure 8B depicts FEN and Taq DNA polymerase cleavage products with pseudo-Y (lanes 3 and 5) and a pseudo-Y substrate with oligonucleotide FS-C hybridized to oligonucleotide FS, converting the 5′ single-stranded tail to a duplex (Fig. 2E). As expected, cleavage by Taq DNA polymerase was abolished (Fig. 8B, lane 4), but surprisingly, FEN was still active on this substrate (lane 6).
FIG. 8.

Activity of FEN on modified pseudo-Y substrates at 50°C. (A) Lanes: 1, marker (FS, 37 nt); 2 and 3, FS after incubation of FS plus FA with Taq DNA polymerase and FEN, respectively; 4 and 5, FS after incubation of FS plus FA-T with Taq DNA polymerase and FEN, respectively (see Fig. 2C); 6 and 7, FS after incubation of FS, FA, and FA-C (hybridized to FA) with Taq DNA polymerase and FEN, respectively (see Fig. 2D). (B) Lanes: 1, 21-nt marker; 2, oligonucleotide FS (37 nt); 3 and 5, FS after incubation of FS plus FA (unmodified substrate) with Taq DNA polymerase and FEN, respectively; 4 and 6, FS after incubation of FS, FA, and FS-C (hybridized to FS) with Taq DNA polymerase and FEN, respectively (see Fig. 2E). (C) Lanes: 1, marker formed by ligating oligonucleotide FS-L to 32P-labeled FS to form a hairpin loop (see Fig. 2F); 2 to 4, hairpin loop after incubation with Taq DNA polymerase for 5, 15, and 45 min, respectively; 5 to 7, hairpin loop after incubation with FEN for 5, 15, and 45 min, respectively.
To verify the activity of FEN on a branched structure with a duplex 5′ branch, the duplex 5′ branch was stabilized by lengthening the duplex from 22 to 27 bp while converting it from an intermolecular complex to an intramolecular hairpin (Fig. 2F). A hairpin oligonucleotide with an internal 32P label was formed by ligating oligonucleotide FS-L to 32P-labeled oligonucleotide FS. Figure 8C depicts a kinetic analysis of the cleavage product profiles produced by using FEN (lanes 5 to 7) or Taq DNA polymerase (lanes 2 to 4) digestion of the hairpin loop pseudo-Y substrate. The substrate alone is shown in lane 1. FEN was active, and cleavage increased with reaction time. The major cleavage product was shorter than the ligated oligonucleotide by the expected length. The action of Taq DNA polymerase on this substrate appears to more complex, involving first exonucleolytic and then endonucleolytic activities. Essentially, no endonucleolytic cleavage products were seen at 5 and 15 min, whereas well over half of the strands were cleaved by 45 min. The hairpin structures migrated anomalously fast due to retention of duplex character on denaturing PAGE. At 15 min, slower-migrating species became apparent, corresponding to shorter molecules with reduced duplex character. By 45 min, many of these intermediates were converted into endonucleolytic cleavage products.
Action of FEN on an internal loop.
The other members of the family of proteins that includes FEN exhibit a substrate requirement or preference for free 5′ ends on single-stranded branches. A heteroduplex substrate was produced by reassociation of equimolar concentrations of two linearized plasmids containing a 20-nt heterologous region, effectively forming two pseudo-Y substrates of opposite orientation but lacking free 5′ ends. The homoduplexes contained BamHI or BglII recognition sites that were lost with formation of the heteroduplex molecules. The homoduplexes and heteroduplexes migrated together. BamHI or BglII digestion cleaved the corresponding homoduplex molecules. Both enzymes cleaved all of the homoduplex molecules. Incubation for 18 h with excess FEN at 70°C produced less than a 5% yield of cleavage products of the same mobility (data not shown).
DISCUSSION
In this study, we have described the cloning into E. coli, expression, purification, biochemical properties, and substrate requirements of the FEN protein from M. jannaschii, a hyperthermophilic archaeon which evolved in a very unusual locale, a deep-sea hydrothermal vent at a pressure of 200 atm. M. jannaschii FEN was very thermostable, surviving incubation at 95°C for 15 min, consistent with the hyperthermophilic nature of the organism.
The substrate used to examine the biochemical properties of M. jannaschii FEN was a circular DNA duplex with a single flap, a substrate which would be formed during displacement synthesis. This substrate could be examined for cleavage of a radioactive flap or for exposing a restriction endonuclease cleavage site. Incubation of the substrate for 1 h with 1.3 fmol M. jannaschii FEN in 20 μl removed most of the flaps. However, the maximum velocity of the enzymatic reaction was rather slow, with approximately 1 h of incubation needed for completion of flap removal even with saturating levels of FEN. In vitro cleavage rates comparable to presumed in vivo rates may require interaction between FEN and DNA polymerase, PCNA, or both.
As was expected, the optimum temperature for activity of M. jannaschii FEN was relatively high (70°C), with activity occurring over a broad range (50 to 80°C). The pH optimum was 6 to 7, in contrast to 8 for murine FEN-1 (10). This result was somewhat surprising because proteins from thermophilic organisms often have higher pH optima than homologous proteins from mesophilic organisms. This shift in pH optimum is thought to reflect the temperature dependence of the isoelectric point. The enzymatic activity depended on magnesium, with a broad concentration range for optimal activity. Unlike the case for Taq DNA polymerase, which still retained activity (18), or murine FEN-1, in which activity was increased 10-fold (10), substitution of manganese for magnesium greatly reduced M. jannaschii FEN activity. Neither calcium nor zinc could substitute for magnesium. Unlike the case for murine FEN-1, addition of KCl increased M. jannaschii FEN activity.
The pseudo-Y substrate consisted of two complementary strands, resulting in a single-stranded 3′ tail. Various members of the FEN family of proteins act on similar substrates in different ways. Thus, while the substrate is efficiently cleaved by the S. cerevisiae RAD2 protein (9), it is not a substrate for murine FEN-1 (10) or S. cerevisiae YKL510 (FEN-1, RAD27) (25). The pseudo-Y substrate was efficiently cleaved by the M. jannaschii FEN protein. The low level of truncated sequences in the 5′-labeled oligonucleotide FS enabled precise determination of the point of cleavage. The cleavage site was three bases distal to the elbow with both the 5′-3′ exonuclease activity of Taq DNA polymerase and M. jannaschii FEN. Previous studies using related substrates with Taq DNA polymerase have shown that the cleavage site was determined by the G+C content at the 5′ end of the complementary region (13). The lower the G+C content, the greater is the instability of the complementary region. Thus, there is more likelihood of displacement by the cleaving enzyme, causing the site to be shifted away from the elbow. In our studies, both Taq DNA polymerase and M. jannaschii FEN cut the oligonucleotide after the elbow, FS between GCA and TGC.
Like murine FEN-1 (10), the length of the flap did not appear to influence the reaction rates of FEN, which cleaved both 15-flaps and 4 nt flaps with equal efficiency on the circular duplex substrate.
Several modified pseudo-Y substrates were examined. Substitution of the truncated oligonucleotide FA-T for FA resulted in almost complete suppression of cleavage activity. In this respect, M. jannaschii FEN was similar to murine FEN-1 (10). Annealing oligonucleotide FA-C to the free 3′ end of oligonucleotide FA did not dramatically increase its cleavage activity; rather, it caused a shift of the cleavage point closer to the elbow. This result differs from those for S. cerevisiae YKL510 (FEN-1, RAD27) and murine FEN-1, for which cleavage activities are increased about 100-fold (11).
The activity of M. jannaschii FEN on a substrate with a duplex 5′ flap was unexpected but was confirmed in assays using a substrate where this duplex was intramolecular rather than intermolecular. Formation of a duplex flap completely inhibits the activity of both human FEN-1, with or without the addition of PNCA (27) and murine FEN-1.
Among the family of structure-specific nucleases, the only crystal structure reported to date is that of bacteriophage T5 5′ exonuclease (4). An arch is formed by helix 4 (containing positively charged residues) and helix 5 (containing hydrophobic residues). The authors proposed that single-stranded DNA could be threaded through this arch prior to cleavage. Duplex DNA would not fit through this arch. One possible explanation for the activity of M. jannaschii FEN on a substrate with a duplex flap could be the presence of a larger arch. Based on the results with the intramolecular duplex flap, the arch would need to accommodate the loop as well as the duplex stem. Inferring a helical arch in M. jannaschii FEN based on the crystal structure of T5 5′ exonuclease may or may not be warranted. Sequence alignments reveal only a 24% identity over about two-thirds of the amino acid sequence between the two proteins. An alternative explanation for the activity on a substrate with a duplex flap would involve recognition of the elbow, perhaps in the context of additional topological constraints. We observed weak activity of M. jannaschii FEN on an internal loop. Among the structure-specific nuclease family, only XPG protein appears to cleave this efficiently (26), while RAD2 has a similar weak activity (9) and human FEN-1 has none at all (27). Elucidation of the crystal structure of M. jannaschii FEN, with a substrate, would provide a framework for understanding substrate limitations in the FEN family of proteins.
ACKNOWLEDGMENTS
We thank Soo-Jung Kim for plasmid pSK101.
This research was supported in part by grants from Roche Molecular Systems, Inc., and the National Institutes of Health (HG01356).
ADDENDUM IN PROOF
The crystal structure of M. jannaschii FEN has been reported by Hwang et al. (K. Y. Hwang, K. Baek, H.-Y. Kim, and Y. Cho, Nature Struct. Biol. 5:707–713, 1998).
REFERENCES
- 1.Bambara R A, Murante R S, Henricksen L A. Enzymes and reactions at the eukaryotic replication fork. J Biol Chem. 1997;272:4647–4650. doi: 10.1074/jbc.272.8.4647. [DOI] [PubMed] [Google Scholar]
- 2.Benson D, Lipman D J, Ostell J. Genbank. Nucleic Acids Res. 1993;21:2963–2965. doi: 10.1093/nar/21.13.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, et al. Complete genomic sequence of the methanogenic archaeon Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 4.Ceska T A, Sayers J R, Stier G, Suck D. A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature. 1996;382:90–93. doi: 10.1038/382090a0. [DOI] [PubMed] [Google Scholar]
- 5.DeMott M S, Shen B, Park M S, Bambara R A, Zigman S. Human RAD2 homolog 1 5′- to 3′-exo/endonuclease can efficiently excise a displaced DNA fragment containing a 5′-terminal abasic lesion by endonuclease activity. J Biol Chem. 1996;271:30068–30076. doi: 10.1074/jbc.271.47.30068. [DOI] [PubMed] [Google Scholar]
- 6.Gary R, Ludwig D L, Cornelius H L, MacInnes M A, Park M S. The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. J Biol Chem. 1997;272:24522–24529. doi: 10.1074/jbc.272.39.24522. [DOI] [PubMed] [Google Scholar]
- 7.Goulian M, Richards S H, Heard C J, Bigsby B M. Discontinuous DNA synthesis by purified mammalian proteins. J Biol Chem. 1990;265:18461–18471. [PubMed] [Google Scholar]
- 8.Habraken Y, Sung P, Prakash L, Prakash S. A conserved 5′ to 3′ exonuclease activity in the yeast and human nucleotide excision repair proteins RAD2 and XPG. J Biol Chem. 1994;269:31342–31345. [PubMed] [Google Scholar]
- 9.Habraken Y, Sung P, Prakash L, Prakash S. Structure-specific nuclease activity in yeast nucleotide excision repair protein Rad2. J Biol Chem. 1995;270:30194–30198. doi: 10.1074/jbc.270.50.30194. [DOI] [PubMed] [Google Scholar]
- 10.Harrington J J, Lieber M R. The characterization of a mammalian DNA structure specific endonuclease. EMBO J. 1994;13:1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrington J J, Lieber M R. Functional domains within FEN-1 and RAD2 define a family of structure specific endonucleases: implications for nucleotide excision repair. Genes Dev. 1994;8:1344–1355. doi: 10.1101/gad.8.11.1344. [DOI] [PubMed] [Google Scholar]
- 12.Hiraoka L R, Harrington J J, Gerhard D S, Lieber M R, Hsieh C-L. Sequence of human FEN-1, a structure specific endonuclease, and chromosomal localization of the gene (FEN1) in mouse and human. Genomics. 1995;25:220–225. doi: 10.1016/0888-7543(95)80129-a. [DOI] [PubMed] [Google Scholar]
- 13.Holland P M, Abramson R D, Watson R, Gelfand D H. Detection of specific polymerase chain reaction product by utilizing the 5′-3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci USA. 1991;88:7276–7282. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones W J, Leigh J A, Mayer F, Woese C R, Wolfe R S. Methanococcus jannaschii sp. nov.; an extremely thermophilic methanogen from a submarine hydrothermal vent. Arch Microbiol. 1983;136:254–261. [Google Scholar]
- 15.Levin D S, Bai W, Yao N, O’Donnell M O, Tomkinson A E. An interaction between DNA ligase I and proliferating cell nuclear antigen: implications for Okazaki fragment joining. Proc Natl Acad Sci USA. 1997;94:12863–12868. doi: 10.1073/pnas.94.24.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieber M R. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. BioEssays. 1996;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 17.Lindahl T, Gally J A, Edelman G M. Deoxyribonuclease IV: a new exonuclease from mammalian tissues. Proc Natl Acad Sci USA. 1969;62:597–603. doi: 10.1073/pnas.62.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyamichev V, Brow M A D, Dahlberg J E. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial DNA polymerases. Science. 1993;260:778–783. doi: 10.1126/science.7683443. [DOI] [PubMed] [Google Scholar]
- 19.Murante R S, Rust L, Bambara R A. Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J Biol Chem. 1995;270:30377–30383. doi: 10.1074/jbc.270.51.30377. [DOI] [PubMed] [Google Scholar]
- 20.Olsen G J, Woese C R. Lessons from the Archaeal genome: what are we learning from Methanococcus jannaschii? Trends Genet. 1996;12:337–339. doi: 10.1016/0168-9525(96)30092-9. [DOI] [PubMed] [Google Scholar]
- 21.Pont-Kingdon G, Dawson R J, Carroll D. Intermediates in extrachromosomal homologous recombination in Xenopus laevis oocytes: characterization by electron microscopy. EMBO J. 1993;12:23–34. doi: 10.1002/j.1460-2075.1993.tb05628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 23.Sandler S J, Satin L H, Samra H S, Clark A J. RecA-like genes from three archaean species with putative protein products similar to Rad51 and Dcm1 proteins of the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:2125–2132. doi: 10.1093/nar/24.11.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen B, Nolan J P, Sklar L A, Park M S. Functional analysis of point mutations in human flap endonuclease-1 active site. Nucleic Acids Res. 1997;25:3332–3338. doi: 10.1093/nar/25.16.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tishkoff D X, Filosi N, Gaida G M, Kolodner R D. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88:253–263. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- 26.Wakasugi M, Reardon J T, Sancar A. The non-catalytic function of XPG protein during dual incision in human nucleotide excision repair. J Biol Chem. 1997;272:16030–16034. doi: 10.1074/jbc.272.25.16030. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Li J, Li X, Hsieh C-L, Burgers P M J, Lieber M R. Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res. 1996;24:2036–2043. doi: 10.1093/nar/24.11.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]