

Summary
Skeletal muscle, the largest human organ by weight, is relevant to several polygenic metabolic traits and diseases including type 2 diabetes (T2D). Identifying genetic mechanisms underlying these traits requires pinpointing the relevant cell types, regulatory elements, target genes, and causal variants. Here, we used genetic multiplexing to generate population-scale single nucleus (sn) chromatin accessibility (snATAC-seq) and transcriptome (snRNA-seq) maps across 287 frozen human skeletal muscle biopsies representing 456,880 nuclei. We identified 13 cell types that collectively represented 983,155 ATAC summits. We integrated genetic variation to discover 6,866 expression quantitative trait loci (eQTL) and 100,928 chromatin accessibility QTL (caQTL) (5% FDR) across the five most abundant cell types, cataloging caQTL peaks that atlas-level snATAC maps often miss. We identified 1,973 eGenes colocalized with caQTL and used mediation analyses to construct causal directional maps for chromatin accessibility and gene expression. 3,378 genome-wide association study (GWAS) signals across 43 relevant traits colocalized with sn-e/caQTL, 52% in a cell-specific manner. 77% of GWAS signals colocalized with caQTL and not eQTL, highlighting the critical importance of population-scale chromatin profiling for GWAS functional studies. GWAS-caQTL colocalization showed distinct cell-specific regulatory paradigms. For example, a C2CD4A/B T2D GWAS signal colocalized with caQTL in muscle fibers and multiple chromatin loop models nominated VPS13C, a glucose uptake gene. Sequence of the caQTL peak overlapping caSNP rs7163757 showed allelic regulatory activity differences in a human myocyte cell line massively parallel reporter assay. These results illuminate the genetic regulatory architecture of human skeletal muscle at high-resolution epigenomic, transcriptomic, and cell state scales and serve as a template for population-scale multi-omic mapping in complex tissues and traits.
1. Introduction
Skeletal muscle, the largest organ in the adult human body by mass (>40%) (Frontera and Ochala, 2015), facilitates mobility, sustaining life functions, and influences quality of life. Beyond its mechanical functions, skeletal muscle plays a central role in metabolic processes, particularly in glucose uptake and insulin resistance (Frontera and Ochala, 2015; G. Kim and J. H. Kim, 2020; Morgan and Partridge, 2020; Musi et al., 2002; Sriwijitkamol et al., 2007). Metabolic diseases and traits, such as type 2 diabetes (T2D), fasting insulin, waist-to-hip ratio (WHR), and others are complex and polygenic, involving a multitude of genetic factors. Genome-wide association studies (GWAS) have identified thousands of genetic signals associated with these diseases and traits (J. Chen et al., 2021; Mahajan, Spracklen, et al., 2022; Mahajan, Taliun, et al., 2018; Pulit et al., 2019; Spracklen et al., 2020; Yengo et al., 2018). However, ~90% of these variants lie within non-coding regions (Maurano et al., 2012), are enriched to overlap tissue-specific enhancers, and are therefore expected to regulate gene expression (J. Chen et al., 2021; Parker et al., 2013; Quang et al., 2015; Thurner et al., 2018). Additionally, GWAS loci are often tagged by numerous variants in high linkage disequilibrium (LD), and can harbor multiple causal variants (Hormozdiari et al., 2016). For these reasons, identifying the biological mechanisms and pinpointing causal variants in GWAS loci remains challenging.
Information encoded in DNA, which is largely invariant across cells in the body, likely percolates through several molecular layers to influence disease. The mostly non-coding genetic variation identified through GWAS likely has the most proximal effect on the molecules bound to DNA (epigenome), which in turn can influence the expression of target genes (transcriptome), and then levels of proteins, all of which can vary by the cell type (Civelek and Lusis, 2014). This molecular cascade is not completely unidirectional and it is dynamic in nature. For example, changes in expression of a transcription factor (TF) can feed back to changes in the epigenome. The epigenome and the transcriptome layers are therefore valuable to gain insights about gene regulation. One approach to link these layers with GWAS is through identification of quantitative trait loci (QTL) for epigenomic modalities such as chromatin accessibility QTL (caQTL) and gene expression quantitative trait loci (eQTL) followed by testing whether common causal variants underlie the molecular QTL and GWAS signals (i.e. if the signals are formally colocalized) (Aygün, Elwell, et al., 2021; Aygün, Liang, et al., 2023; Barbeira et al., 2021; Currin et al., 2021; Hormozdiari et al., 2016; Khetan et al., 2018; Liang et al., 2021; Robertson et al., 2021; Soskic et al., 2021; Turner et al., 2022; Viñuela et al., 2020; Wallace, 2021).
Previous studies profiling the epigenome and transcriptome in bulk skeletal muscle across hundreds of samples identified expression and DNA methylation QTLs and provided valuable insights (Consortium, 2020; Scott et al., 2016; Taylor et al., 2019). However, bulk skeletal muscle profiles are dominated by the most prominent muscle fiber types, and other less abundant but relevant cell types are largely missed. Several resident cell types are essential for muscle function (Morgan and Partridge, 2020). For example, muscle fibro-adipogenic progenitors (FAPs) are resident interstitial stem cells involved in muscle homeostasis and along with muscle satellite cells, regulate muscle regeneration (Biferali et al., 2019; Contreras, Rossi, and Theret, 2021; Dumont et al., 2015; Molina, Fabre, and Dumont, 2021). Diabetes and obesity not only lead to structural and metabolic changes of the muscle fibers but also exert detrimental effects on these progenitor cells (Hilton et al., 2008; Teng and Huang, 2019; Travers et al., 2013). Endothelial cells and smooth muscle cells comprise the muscle vasculature which is another important component in diabetes-associated complications, involving insulin uptake (Kolka and Bergman, 2013). Immune cells are also critical, especially following injury (Pillon et al., 2013). Recent studies have generated reference epigenome and transcriptome maps in human skeletal muscle at a single-nucleus/single-cell resolution (De Micheli et al., 2020; Orchard et al., 2021; Rubenstein et al., 2020; K. Zhang et al., 2021). However, population-scale studies are imperative to identify e-and-caQTL within each cell type to enable exhaustive interrogation of mechanistic signatures underlying GWAS signals. To date, there is no single-nucleus/cell resolution population-scale study that maps e-and-caQTL in hundreds of samples.
We hypothesize that single-nucleus epigenome (snATAC-seq) and transcriptome (snRNA-seq) profiling across hundreds of genotyped samples will help identify the appropriate cell type, regulatory elements, target genes, and causal variants(s) in elucidating context-specific regulatory mechanisms within skeletal muscle. In this work, we perform snRNA-seq and snATAC-seq across skeletal muscle samples from 287 Finnish individuals (Scott et al., 2016). We integrate these molecular profiles with genetic variation to identify cell-specific eQTL and caQTL. We further integrate the e/caQTL signals with GWAS by testing for colocalization and infer the chain of causality between these modalities using mediation analyses, and highlight our findings with orthogonal methods at multiple example loci.
2. Results
2.1. snRNA and snATAC profiling and integration identifies 13 distinct cell type clusters
We generated a rich dataset of snRNA and snATAC across 287 frozen human skeletal muscle (vastus lateralis) biopsies from the FUSION study (Scott et al., 2016) (Figure. 1A), as part or a larger study with 408 total samples including three additional smaller cohorts. We processed the samples in ten batches of 40 or 41 samples multiplexed together using a randomized block study design to balance across experimental contrasts of interest (cohort, age, sex, BMI, oral glucose tolerance test (OGTT), Figures. S1A–S1E). We also included multiome data (snRNA and snATAC on the same nucleus) for one muscle sample to help assess the quality of our cross-modality clustering. We performed rigorous quality control (QC) of all nuclei and only included those deemed as high-quality (Methods). This led to a total of 188,337 pass-QC RNA nuclei and 268,543 pass-QC ATAC nuclei (Figures. S1F–S1I, Figures. S2A–S2D, Figures. S3A–S3E). As expected, there is a strong correlation across samples for the number of pass-QC RNA and ATAC nuclei (Figure. S3F), and nuclei counts correlate with the initial weights of the tissue samples (Figure. S3G), indicating that our genetic demultiplexing and QC recovered high-quality nuclei in expected proportions. Collectively, we generated total N = 625,722 high-quality RNA or ATAC nuclei from all 408 samples, and in this work we analyze N = 456,880 nuclei from the 287 FUSION and one multiome sample.
Figure 1: snRNA and snATAC -seq data generation and integration identifies 13 high quality cell-type clusters.
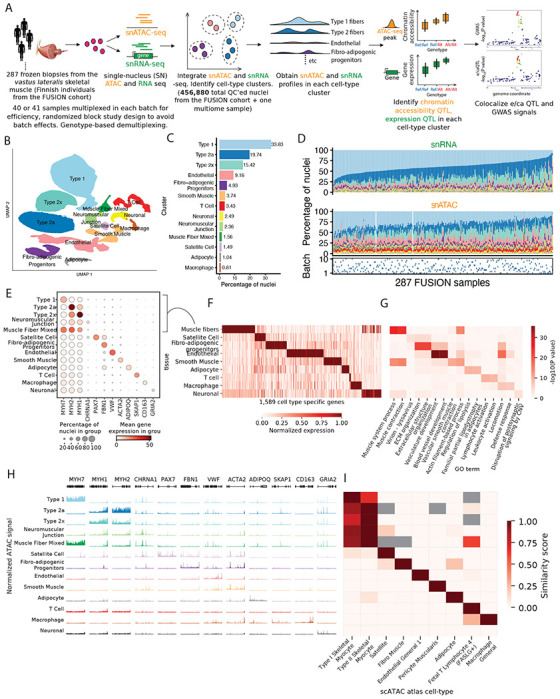
(A) Study design including sample processing, snRNA and snATAC -seq profiling, and analyses. (B) UMAP plot showing the 13 identified clusters after jointly clustering the snRNA and snATAC modalities. (C) Cluster abundance shown as percentage of total nuclei. (D) Cluster proportions across samples and modalities. (E) Gene expression (post ambient-RNA adjustment) in clusters for known marker genes for various cell-types. (F) Identification of cell-type-specific genes across clusters. Five related muscle fiber clusters (type 1, 2a, 2x, neuromuscular junction and muscle fiber mixed were taken together as a”muscle fiber” cell type). (G) GO term enrichment for cell-type-specific genes identified in (F), showing two GO terms for each cluster. (H) snATAC-seq profiles over known marker genes in clusters. (I) Comparison of snATAC-seq peaks identified for clusters in this study with reference data across various cell-types from the K. Zhang et al. (2021) scATAC-seq atlas.
We jointly clustered the snRNA and snATAC data, while avoiding batch and modality-specific effects using Liger (Gao et al., 2021; Welch et al., 2019) (Figure. S4A). We identified 13 distinct clusters representing diverse cell types (Figure. 1B) that ranged in abundance (Figure. 1C) from 34% (type 1 fiber) to <1% (macrophages). The aggregate cell-specific profiles provide clear evidence of muscle tissue heterogeneity (Figure. 1D). When treating the multiome RNA and ATAC modalities separate and integrating across them, we found that 82.8% of the non-muscle fiber multiome nuclei had the same RNA and ATAC cluster assignments (Figure. S4B). This is consistent with previous multiome studies (Duren et al., 2022; Xiong et al., 2023) (Supplementary note); for example, integrating 92 brain snATAC+snRNA samples (19 of which were multiome) obtained 79.5%-85% concordant cluster assignments depending on the clustering approach (Xiong et al., 2023).
The annotated clusters showed expected patterns of expression for known marker genes (Figure. 1E, Figure. S4C). We merged the five closely-related muscle fiber types together and annotated them as “muscle fiber” and identified 1,569 cell-specific genes using pair-wise differential gene expression analyses (Figure. 1F). Relevant gene ontology (GO) terms were enriched in these cell-specific genes (Figure. 1G), for example, muscle system process and muscle contraction terms for muscle fiber and regulation of lipolysis in adipocytes and familial partial lipodystrophy terms for the adipocyte cluster.
The ATAC modality also showed clear patterns of chromatin accessibility over known marker genes for various cell types (Figure. 1H). We optimized ATAC peak calls to be of similar statistical power, reproducible, and non-redundant across clusters to create a harmonized list of 983,155 consensus peak summits across the 13 cell types (Methods, Figures. S5A–S5D). We compared our snATAC profiles with reference snATAC data from 222 cell types from a previous study (K. Zhang et al., 2021). Our snATAC peaks were enriched to overlap peaks identified in related cell types (Figure. 1I), which reinforces the quality of our cluster labels using the independent ATAC modality. We identified 95,442 snATAC peaks that were specific for a cell type cluster (Figure. S5E).
DNA-binding motifs for cell type-relevant TFs were enriched in these cluster-specific peaks (Figure. S5F). For instance, motifs for the myocyte enhancer factor 2 (MEF2) family of TFs that are known regulators of skeletal muscle development and function (Anderson et al., 2015; N. Liu et al., 2014) were enriched for muscle fiber peaks; motifs for the SRY (Sex Determining Region Y)-related HMG box of DNA binding (SOX) TFs, implicated in endothelial differentiation and endothelial-mesenchymal cell transitions (Goveia et al., 2014; M. Liu et al., 2019; Y. Yao, J. Yao, and Boström, 2019) were enriched in endothelial-specific peaks. Collectively, these data demonstrate the high-quality of our snRNA and snATAC profiles and data integration.
2.2. Integrating genetic variation with snRNA and snATAC profiles identifies thousands of e/caQTL
We next identified genetic associations with gene expression and chromatin accessibility QTL (e/ca QTL) in the five most abundant cell type clusters (type 1, 2a, 2x muscle fibers, FAP, endothelial). Optimizing QTL discovery (Figure. S6A, Figure. S7A), we identified 6,866 eQTL and 100,928 caQTL across the five clusters (Figures. 2A–2B). The most abundant type 1, followed by type 2a and 2x fiber clusters (Figure. 1C) had higher power to detect QTL; consequently, a large number of e/caQTL are identified only in type 1, or shared across the three muscle fiber types (Figures. 2A–2B). 427 eQTL and 11,170 caQTL signals were identified specifically in FAP and endothelial clusters (Figures. 2A–2B). Despite differences in power to detect QTL, the e/caQTL effect sizes were highly concordant across the five clusters (Figure. S6B,Figure. S7B). Figure. 2C shows an example type 1 caQTL signal (P = 1.4x10−63) where the caQTL SNP (caSNP) rs12636284 lies within the caQTL peak (caPeak), and the C allele is associated with higher chromatin accessibility. This caQTL is also identified in FAPs (P = 2.4x10−34), and the peak is shared across multiple clusters (Figure. 2D). We identified cluster-specific caQTL even for peaks shared across cell types, indicating context-specific genetic effects on chromatin accessibility. For example, Figure. 2E shows a caQTL identified in FAPs (~5% ATAC nuclei) and not type 1 fibers (~30% ATAC nuclei), even when the overall peak was comparable between the two clusters (Figure. 2E, aggregate cluster snATAC tracks). Additionally, we identified cluster-specific peaks as caQTL (Figure. 2F). caPeaks in the five clusters were enriched to overlap TF motifs relevant to the corresponding cell type (Figure. S7C).
Figure 2: Thousands of e/ca QTLs identified in clusters.
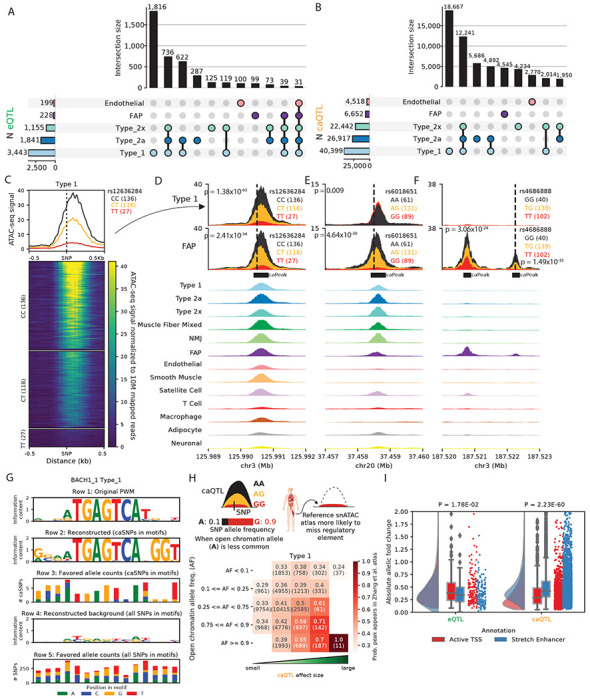
(A) UpSet plot showing the number of shared and specific eGenes, and (B) caPeaks across five clusters (C) An example caQTL is depicted in a heatmap of normalized snATAC-seq reads across samples in the type 1 cluster, separated by caSNP rs12336284 genotype classes. Aggregate profiles by genotype are shown on top. Examples of shared and cluster-specific caQTL are shown in (D) , (E) , and (F) . Top two rows show snATAC-seq profiles by the caSNP genotype classes in type 1 and FAPs, followed by aggregate snATAC profiles across all 13 clusters. (G) Reconstruction of the BACH_1 TF motif using caQTL data. From top, row 1: original motif PWM. Row 2: genetically reconstructed motif PWM. For all BACH_1 motifs occurring in type 1 snATAC-seq peaks (peak-motifs) that also overlapped type 1 caSNPs, alleles associated with higher chromatin accessibility (“favored alleles”) were quantified using the caQTL allelic fold change, followed by generating a PWM. Row 3: favored allele counts for caSNPs in BACH_1 peak-motifs. Row 4: PWM reconstructed using the nucleotide counts for all heterozygous SNPs overlapping the BACH_1 peak-motifs. Row 5: nucleotide counts for all heterozygous SNPs in the BACH_1 peak-motifs. (H) Comparison of caSNP effect size and MAF with the replication of snATAC-seq peaks in a reference scATAC dataset (K. Zhang et al., 2021). (I) Allelic fold change for type 1 e/caSNPs that overlap skeletal muscle active TSS or stretch enhancer chromatin states.
We next asked if the genetic regulatory signatures from our caQTL scans recapitulate patterns of TF binding. Most TFs bind accessible chromatin regions by recognizing specific DNA motifs. For genetic variants within bound activator motifs, the allele preferred by the TF should be preferentially associated with higher chromatin accessibility (Liang et al., 2021). In Figure. 2G, we show the known position weight matrix (PWM) for the TF motif BACH_1 (row 1). We considered all BACH_1 motif occurrences across snATAC peaks in type 1 fibers that also overlapped caSNPs, and used the caQTL allelic fold change (aFC) to quantify alleles associated with higher chromatin accessibility (“favored alleles”). We then used these favored alleles to genetically reconstruct the PWM (Figure. 2G, row 2) (Figure. 2G, row 3) and found it closely matches the canonical motif PWM (Figure. 2G, row 1), providing a caQTL-informed in vivo verification of the cognate PWM. To further verify that the caQTL-based genetically reconstructed PWM does not simply reflect the allelic composition of SNPs in motifs, we constructed the PWM using the allele count for all heterozygous SNPs observed in the BACH_1 motif occurrences in snATAC peaks (Figure. 2G, row 4,5). The resulting PWM had low information content and little similarity to the cognate motif (Figure. 2G, row 4,1). Other examples of caQTL-informed reconstructed motifs for MSC_1 and MEF2_known9 are shown in Figures. S7D–S7E. Overall, these results demonstrate how high quality snATAC and caQTL information can provide base-resolution insights into TF binding and regulation.
Given our deep caQTL results, we next compared caPeaks to snATAC peaks in the same cell types from reference atlas datasets. We reasoned that for caPeaks where the more commonly occurring caSNP allele is associated with lower chromatin accessibility, the caPeak is more likely to be missed in reference datasets that usually only include one or a few representative tissue samples and therefore do not capture population-scale genetic effects. We additionally reasoned that caPeak reproducibility in reference atlases will be lower for large effect-size caSNPs when the allele associated with high chromatin-accessibility occurs rarely in the population. Figure. 2H delineates this observation comparing type 1 fiber caPeaks with the K. Zhang et al. (2021) snATAC atlas type 1 fiber peaks. Even with moderate effect sizes and allele frequencies, the snATAC caPeak was missed in the snATAC atlas about equally as often as it was observed (Figure. 2H). Overall, this observation underscores the importance of population-scale snATAC studies to exhaustively identify regulatory elements in the human population.
To examine the local chromatin context, we compared chromatin state patterns at e/caQTL in muscle fibers. Type 1 caPeaks were enriched to overlap TSS and enhancer chromHMM states in skeletal muscle (Figure. S7E). We contrasted two classes of functional regulatory elements, the active TSS chromHMM state that constitutes shared and cell type-specific promoter elements and stretch enhancers that constitute cell identity enhancer elements (Parker et al., 2013; Varshney, Kyono, et al., 2021; Varshney, VanRenterghem, et al., 2019). Type 1 fiber eSNPs occurring in the skeletal muscle active TSS chromHMM state had higher eQTL absolute aFC than eSNPs occurring in stretch enhancers (Figure. 2I, P = 1.8x10−2), whereas, type 1 fiber caSNPs occurring in stretch enhancers had higher caQTL absolute aFC than caSNPs in active TSS states (Figure. 2I, P = 2.2x10−60). These results suggest that eQTL scans identify signals largely in proximal gene promoter regions, whereas caQTL scans are able to identify signals in distal and cell-specific regulatory elements, elucidating an important distinction in the two modalities. Collectively, these results reinforce the importance of joint snRNA and snATAC profiling along with e/caQTL analyses to gain mechanistic insights into the genetic regulation of gene expression and distal regulatory element accessibility.
2.3. e/caQTL finemapping, colocalization and causal inference informs cell-specific multi-omic genetic regulation
We performed genetic finemapping to identify independent e/caQTL signals and generate 95% credible sets using the sum of single effects (SuSiE) approach (J. Wang et al., 2020) (Figure. 3A). 266 out of 6,866 eQTL and 4,502 out of 100,928 caQTL signals could be finemapped to a single variant in the 95% credible set, whereas the remaining signals had more variants in their corresponding credible sets (Figures. 3B–3C). eSNPs occurring in snATAC peaks and caSNPs occurring in the corresponding caPeaks have higher posterior inclusion probability (PIP) in the e/caQTL signal credible sets, which reinforces the quality of our e/caQTL scans and the utility of finemapping to nominate causal e/caSNPs (Figures. 3B–3C). We next tested if the eQTL and caQTL signals shared causal variant(s), i.e. if the e/caQTL signals were colocalized using coloc v5 (Wallace, 2021) (Figure. 3D). We identified colocalized caQTL signals (coloc posterior probability for shared variant(s) (PPH4) > 0.5) across the five clusters for 1,973 eGenes; the majority (59.9%) of these e-caQTL colocalizations were cluster-specific (Figure. 3E). Several relevant TF motifs were enriched in caPeaks that colocalized with an eQTL signal relative to all caPeaks (Figure. 3F); for example, the motif for NKX2-5, a regulator of skeletal muscle differentiation (Riazi et al., 2005) is enriched in caPeaks colocalized with eQTL in muscle fibers. These results suggest that e-caQTL colocalizations nominate biologically relevant gene regulatory mechanisms and emphasizes the value of our sn-e/caQTL catalog.
Figure 3: e/ca QTL finemapping, colocalization and causal inference informs regulatory grammar in clusters.
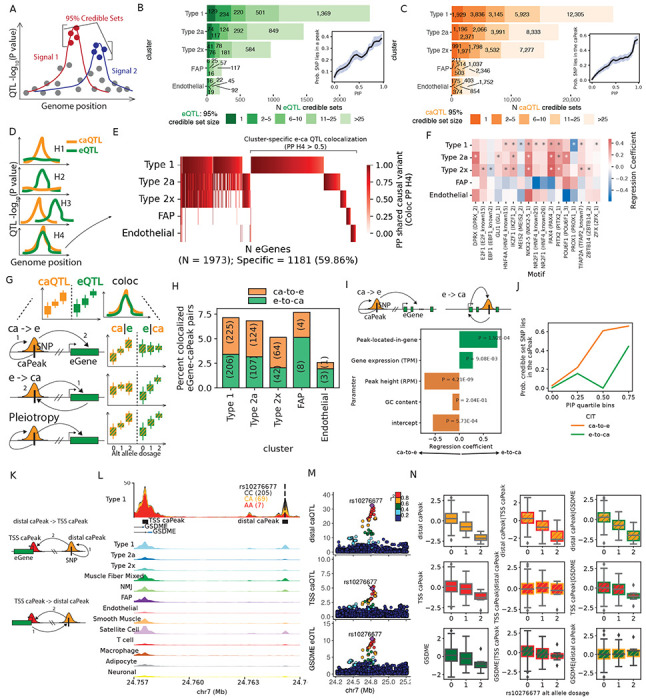
(A) Identification of independent signals and finemapping was performed using SuSiE for e,caQTL. (B) Number of eQTL signals and (C) caQTL signals classified by the number of variants in the finemapped 95% credible set. Insets show the probability of a eSNP overlapping a peak in (B) and caSNP overlapping the caPeak in (C) in a cluster relative to the PIP. (D) Testing for colocalization between e and ca QTL signals informs if the modalities share common causal variant(s). (E) Heatmap showing the posterior probability of shared causal variant (PP H4) from coloc for pairs of eGenes and caPeaks in the five clusters. (F) TF motif enrichment in caPeaks that colocalize with eGenes. “*” denotes significant logistic regression coefficient (5% FDR). (G) After identifying colocalizing e-and-ca QTL signals, causal inference tests (CIT) can inform the causal direction between the gene expression and chromatin accessibility modalities using e/ca SNPs as instrument variables. (H) (I) (J) (K) (L) (M) (N) continued on the next page. (H) Percentage of colocalizing eGene-caPeak pairs for which the putative causal direction could be determined (5% FDR) as chromatin accessibility over gene expression (ca-to-e) or vice versa from CIT. (I) Logistic regression modeling the causal direction between caPeak-eGene pairs with whether the caPeak lies within the eGene body, along with eGene expression (TPM,) caPeak height (RPM), and GC content. (J) Probability that a caSNP lies in the caPeak relative to caSNP PIP (binned into quartiles), classified by if the caPeak was inferred as ca-to-e or e-to-ca from CIT. (K) For colocalized caPeak and eGene pairs where a caPeak was also identified in the TSS+1kb upstream region of the eGene, causal direction can be estimated between the distal-caPeak and the TSS-caPeak. (L) Type 1 snATAC-seq signal track by rs10276677 genotype classes over the GDSME locus on chr7 showing a distal-caPeak, a TSS-caPeak and the GDSME gene TSS. Aggregate snATAC-seq in clusters are shown below. (M) Locus-zoom plots showing the distal-caQTL, TSS-caQTL and the GDSME eQTL. (N) Causal inference between the distal-caPeak, TSS-caPeak and the GDSME gene using rs10276677 as the instrument variable. Boxplots show inverse normalized chromatin accessibility or gene expression relative to the alternate allele dosages at rs10276677 before and after regressing out the corresponding modality.
For colocalized e/caQTL signals, we inferred the causal relationship between chromatin accessibility and gene expression using causal inference tests (CIT) and Mendelian randomization (MR) approaches (Hemani, Tilling, and Smith, 2017; Millstein, G. K. Chen, and Breton, 2016; Millstein, B. Zhang, et al., 2009) (Figure. 3G). We tested if chromatin accessibility mediates the effect of genetic variation on gene expression (Figure. 3G, row 1, “ca-to-e”), or if gene expression mediates the effect of genetic variation on chromatin accessibility (Figure. 3G, row 2, “e-to-ca”), compared to a model consistent with pleiotropic effects (Figure. 3G, row 3). In these analyses, “causal” implies that variance in the mediator determines some proportion of the variance in the outcome (Millstein, B. Zhang, et al., 2009). Since measurement errors in the molecular phenotypes can affect causal inference, we conservatively required consistent causal direction reported by both the CIT and the MR Steiger directionality test, and also performed sensitivity analyses that measured how consistent the inferred direction was over the estimated bounds of measurement error (Hemani, Tilling, and Smith, 2017) (Figure. S8A).
We discovered 787 colocalized e/caQTL signal pairs as ca-to-e or e-to-ca (consistent CIT and MR Steiger directionality test, 5% FDR Figure. 3H). The e-to-ca model may represent gene expression effects on chromatin accessibility for caPeaks within the body of the transcribed gene. To test this hypothesis, we modeled the inferred causal direction in a logistic regression coding e-to-ca as 1 and ca- to-e as 0, adjusting for caPeak height (reads per million mapped reads, RPM), eGene expression level (transcripts per million mapped reads, TPM), caPeak GC content and if the caPeak was located within the eGene body. We found that e-to-ca caPeaks occurred within the eGene body significantly more than ca-to-e caPeaks (regression coefficient = 0.8, P = 1.9x10−4; Figure. 3I), indicating that colocalized e/caQTL caPeaks in the gene body are more likely to be influenced by the act of transcription across the underlying DNA region. ca-to-e caPeaks were higher (CPM) than e-to-ca caPeaks (coefficient = −0.72, P = 4.2x10−9), whereas e-to-ca eGenes were more highly expressed than ca-to-e eGenes (coefficient = 0.28, P = 9.1x10−3).
High PIP caSNPs were more likely to occur within ca-to-e caPeaks than e-to-ca caPeaks (Figure. 3J), consistent with expectation for caPeaks that are causal on eGenes. For TSS-distal ca-to-e caPeaks where additional caPeaks were identified in TSS+1kb upstream region of the eGene (Figure. 3K), the distal caPeak was often causal on the TSS-caPeak as well (Figure. S8B), Fisher’s exact test P = 4.4x10−16). For example, a distal caPeak ~7.6 kb from the GSDME gene TSS is causal on both GSDME gene expression (CIT P = 5.4x10−5) and a TSS-caPeak accessibility (CIT P = 4.2x10−5) (Figures. 3L–3N). These analyses support an enhancer model for the ca-to-e caPeaks where the caSNP affects chromatin accessibility at the TSS-distal caPeak that then regulates gene expression.
We highlight a locus on chromosome 8 where two independent caQTL signals for a caPeak tagged by caSNPs rs700037 and rs1400506 (Figure. S8C), both of which lie within the caPeak (Figure. S8D) are colocalized with two independent eQTL signals for the lincRNA gene AC023095.1 (PPH4 0.99 and 0.76). This caPeak is specific for the type 1 and the mixed fiber cell-type clusters (Figure. S8D). Considering the independent signals as instruments, we identified the caPeak to be causal on the AC023095.1 gene expression (CIT P value 2.11x10−07) (Figure. S8E). Collectively, these results demonstrate how signal identification, finemapping, colocalization and causal inference analyses illuminate cell-specific causal event chains for the regulatory element, target gene and causal variant(s).
2.4. Cell-specific e/caQTL and GWAS signal integration to inform disease/trait regulatory mechanisms
To identify mechanisms underlying disease/trait associations, we integrated our e/caQTL signals with GWAS signals. We considered 374 publicly available disease/trait GWAS datasets from the UK Biobank (UKBB), along with 32 other GWAS datasets that included other skeletal muscle-relevant diseases/traits such as T2D, fasting insulin, WHR, body mass index (BMI), creatinine, and others. Computing the enrichment of these GWAS signals to overlap snATAC peaks across our 13 cell type clusters in an LD score regression (LDSC) joint model (B. K. Bulik-Sullivan et al., 2015; Finucane, B. Bulik-Sullivan, et al., 2015) systematically revealed the most relevant traits for the cell type clusters (Cano-Gamez and Trynka, 2020) (Figure. 4A, Figure. S9A). For example, muscle fiber snATAC peaks were enriched for diabetes, modified Stumvoll insulin sensitivity index (ISI), birth weight, sitting height, atrial fibrillation, creatinine, pulse rate, and others (LDSC coefficient Z score > 2.02, enrichment P value < 1.1x10−5; Figure. 4A, Figure. S9A). These results are highly consistent with previous K. Zhang et al. (2021) study where type 1 skeletal myocytes were enriched for height, pulse rate, atrial fibrillation, and creatinine. FAPs were enriched for waist-to-hip ratio, bone mineral density, height, and ocular trait signals (LDSC coefficient Z score > 3.69, enrichment P value < 6.9x10−11. Type 1 snATAc peaks that overlapped with a caSNP showed generally higher enrichment to overlap GWAS signals than peaks that overlapped eSNPs and all other peaks (Figure. 4B, Figure. S9B), indicating that trait-associated genetic variants are especially enriched in open chromatin peaks that are sensitive to genetic variation. Focusing on a shortlist of 43 relevant diseases/traits, we identified 3,378 GWAS signals colocalized with e/caQTL from our study (Figures. 5A–5B, Figure. S10A), the vast majority (2,616 signals, 77.4%) of which were GWAS-caQTL (not GWAS-eQTL) colocalizations (Figure. 5C). Since coloc results can be sensitive to the prior probability for the SNP being associated with both traits (p12), we performed sensitivity analyses relative to the p12 prior (Figures. S10B–S10D) and include the minimum p12 prior for PPH4>0.5 as a potential QC metric for colocalization analyses. We highlight GWAS signals for T2D, BMI, and fasting insulin that colocalize with e/caQTL across the five tested clusters, both in a shared and cell-specific manner (Figure. 5D, Figures. S10E–S10F).
Figure 4: Enrichment of GWAS traits in cluster snATAC peaks.
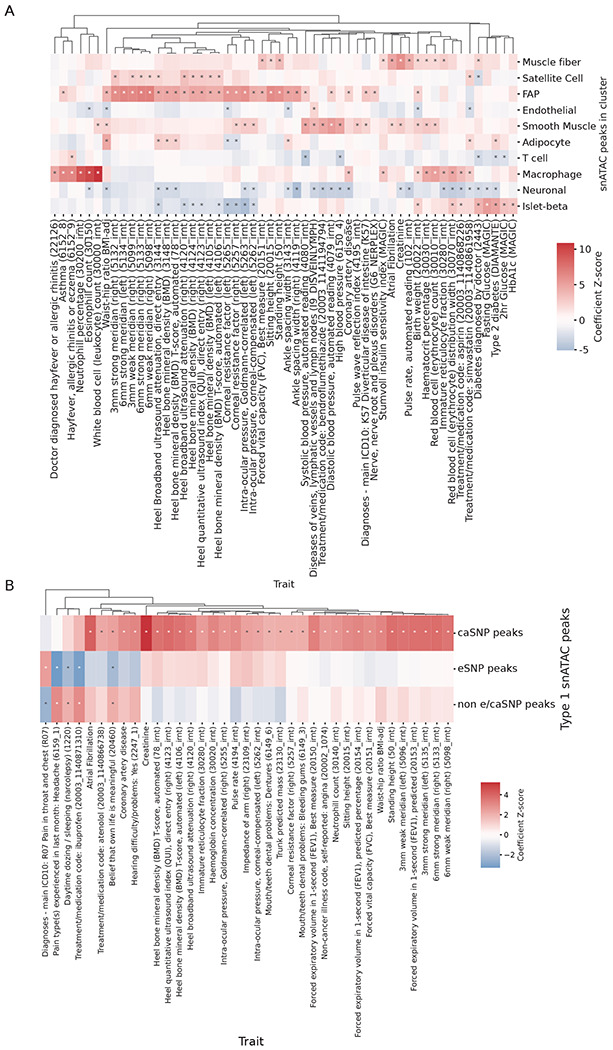
(A) GWAS enrichment in cluster snATAC peak features. Heatmap shows the LDSC regression coefficient Z scores from a joint model that included the cluster snATAC peaks, along with islet beta peaks (Rai et al., 2019) and a baseline model accounting for relevant cell-type agnostic features. (B) GWAS Enrichment for type 1 fiber snATAC peaks that contain a caSNP or eSNP versus all other Type 1 snATAC peaks. Both (A) and (B) show a pruned selection of traits (Methods), full results are shown in Figures. S9A–S9B. “*” = regression coefficient confidence intervals not overlapping zero.
Figure 5: Integrating e,ca QTL signals with GWAS inform disease/trait relevant regulatory mechanisms.
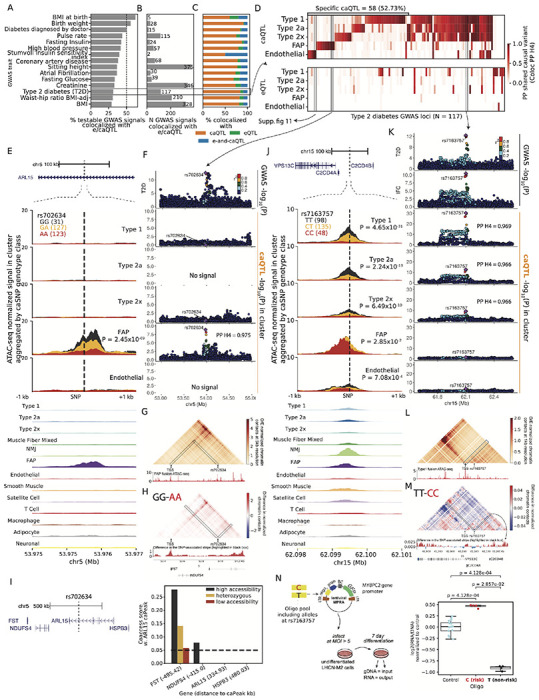
(A) Percentage of testable GWAS signals, and (B) Number of GWAS signals across traits that colocalize with e/ca QTL signals across the five clusters. (C) Proportion of colocalized GWAS signals (from B) that colocalize with only caQTL or only eQTL or both e-and-caQTL. (D) (E) (F) (G) (H) (I) (J) (K) (L) (M) (N) continued on the next page. (D) Heatmap showing T2D GWAS signal colocalization with e,ca QTL. (E) T2D GWAS signal at the ARL15 locus is colocalized with an FAP caQTL. The genomic locus is shown at the top, followed by zooming into a ±1kb neighborhood of the caSNP rs702634. snATAC-seq profiles in five clusters by the caSNP genotype are shown, followed by aggregate profiles in all 13 clusters. (F) Locuszoom plots showing the ARL15 GWAS signal (top) followed by the caQTL signal in the five clusters. The peak was not testable for caQTL in the type 2a and endothelial clusters due to low counts. (G) Hi-C chromatin contacts at 5kb resolution imputed by EPCOT using the FAP snATAC-seq data (shown below the heatmap) in a 1Mb region over rs702634. (H) Difference in the predicted normalized chromatin contacts using FAP ATAC-seq from samples with the high accessibility genotype (GG) and low accessibility genotype (AA) at rs702634. Interactions with rs702634 highlighted in black are shown as a signal track below the heatmap. (I) Genes in the 1Mb neighborhood of the ARL15 gene. Chromatin co-accessibility scores between the caPeak and TSS+1kb upstream peaks for the neighboring genes, classified by genotype classes at rs702634. Distance of the TSS peak to the caPeak in kb is shown in parentheses. (J) GWAS signals for T2D and insulin fold change (IFC) at the C2CD4A/B colocalize with a caQTL in type 1,2a and 2x fibers. Shown are the genomic locus, snATAC-seq profiles by the caSNP genotype and aggregated in clusters. (K) Locuszoom plots showing the C2CD4A/B GWAS and caQTL signals. (L) Micro-C chromatin contacts imputed at 1kb resolution by EPCOT using the type 1 snATAC-seq showing rs7163757 and the neighboring 500kb region. (M) Difference in the predicted normalized chromatin contacts by rs7163757 genotype. Interactions with rs7163757 highlighted in black are shown as a signal track below. (N) A massively parallel reporter assay in the muscle cell line LHCN-M2 tested a 198bp element centered on the caSNP rs7163757. Enhancer activity is measured as log2(RNA/DNA) normalized to controls.
The GLI2 locus T2D GWAS signal (P = 4.2x10−9) is colocalized (PPH4 = 1.0) with a caQTL idnetified specifically in the endothelial cells (P = 1.37x10−11, Figures. S11A–S11B), and the caSNP rs11688682 (PIP=1.0) occurs within the caPeak. We used a deep learning framework that can predict epigenome, chromatin organization and transcription (EPCOT) (Z. Zhang et al., 2023) to impute high-resolution 3D chromatin contacts (Micro-C) using the endothelial ATAC profile. This approach predicted high contacts of the caSNP-caPeak region with the INHBB gene TSS, nominating the gene as a target (Figure. S11C). Notably, we detected allelic differences in the predicted contacts, where the homozygous high accessibility genotype (GG) showed higher contacts with the INHBB gene than the homozygous low accessibility genotype (CC) (Figure. S11D).
The ARL15 locus T2D GWAS signal (P = 7.7x10−14) is colocalized (PPH4 = 0.975) with an FAP-specific caQTL (P = 2.5x10−9) (Figures. 5E–5F). EPCOT predicted high chromatin contact frequency of the caSNP rs702634 region with the FST gene TSS (Figure. 5G); the predicted contacts were higher with the homozygous high accessibility genotype (GG) compared to the homozygous low accessibility genotype (AA) at the caSNP (Figure. 5H). This FAP-specific caPeak is present in the analagous cell type at the orthologous region in the rat genome, and its allelic enhancer activity was validated in a luciferase assay in human mesenchymal stem cells (Orchard et al., 2021). The caPeak was highly co-accessible with the FST gene TSS peak in a genotype-specific manner (co-accessibility computed using Cicero (Pliner et al., 2018)) (Figure. 5I). The nominated target gene for this GWAS signal, FST, encodes follistatin, which is involved in increasing muscle growth and reducing fat mass and insulin resistance (Braga et al., 2014; Singh, Braga, et al., 2017; Singh, Pervin, et al., 2018; Zheng et al., 2017).
The C2CD4A/B locus T2D GWAS signal (P = 2.6x10−13) colocalizes (PPH4 = 0.969, 0.966, 0.966) with caQTL signals in the type 1, 2a and 2x fibers (P = 4.7x10−31, 2.2x10−13, 6.5x10−10) (Figures. 5J–5K). This GWAS signal also colocalizes with GWAS signals for fasting glucose, and insulin fold change (IFC) post 2 hour oral glucose tolerance test (OGTT), which is a measure of insulin sensitivity (Williamson et al., 2023). Notably, this GWAS-colocalized caPeak was not found as a type I skeletal myocyte cis regulatory element in the K. Zhang et al. (2021) snATAC atlas. The caSNP rs7163757 lies within the caPeak; the T (T2D non-risk) allele is associated with higher chromatin accessibility (Figure. 5J). While this caPeak is observed in the muscle fiber clusters, it appears strongest in the low-abundance neuro-muscular junction (NMJ) cluster (Figure. 5J, Figure. S11E). EPCOT predicted high chromatin contacts with the VPS13C gene TSS (Figure. 5L), which was higher for the high accessibility genotype (TT) compared to the low accessibility genotype (CC) (Figure. 5M). An siRNA-mediated knock-down of VPS13C in an adipocyte cell line affected the cell-surface-abundance of the glucose transporter GLUT4 upon insulin stimulation (Williamson et al., 2023), implicating the nominated target gene, VPS13C, in insulin resistance mechanisms (Mueckler, 2001). We validated the enhancer activity of the caPeak 198 bp distal regulatory element centered on caSNP rs7163757 in a massively parallel reporter assay (MPRA) framework in the LHCN-M2 human skeletal myoblast cell line (see Methods). The T2D risk allele C showed significantly higher activity relative to the empty vector control (P = 4.1x10−4) which was significantly higher than the activity of the non-risk T allele (P value = 2.9x10−2, Figure. 5N). Previously, Kycia et al. (2018) reported that rs7163757 occurred in accessible chromatin in pancreatic islets, the risk allele C showed higher enhancer activity in rodent islet model systems, and this allele was also associated with higher C2CD4A/B gene expression, thereby implicating this T2D GWAS signal in islet dysfunction, which was supported by an independent publication (Mehta et al., 2016). Our results highlight skeletal muscle fibers as another key cell type where this signal could modulate the genetic risk for T2D and insulin resistance through the VPS13C gene.
Collectively, these results demonstrate the importance of the snATAC modality and caQTL information in nominating mechanisms underlying GWAS associations and identifying causal variants in disease-relevant cell types.
3. Discussion
In this study, we present population-scale single-nucleus profiling of chromatin accessibility and gene expression on 287 frozen human skeletal muscle biopsies. We multiplexed 40 or 41 samples in each batch using a randomized block design to control for sample variables. Demultiplexing the data downstream using known genetic variation enabled reduced costs, helped protect against batch effects, allowed genetic detection of doublets, and overall increased rigor of the work. The integration and joint-clustering of multi-omic modalities provided a comprehensive view of the cell-specific molecular landscape within human skeletal muscle.
We identified 6,866 eQTL and 100,928 caQTL across the top five most abundant cell types. Concordant e/caQTL effects across clusters supported the high-quality of our e/caQTL scans. Chromatin accessibility directional allelic effects discovered from the caQTL scans mirrored the DNA-binding preferences of TF motifs which is a powerful demonstration of the depth of information snATAC and caQTL data capture. Notably, we identified 14-fold more caQTL compared to eQTL, which can be attributed to two factors: first, more peaks were tested for caQTL than genes for eQTL, and second, chromatin accessibility modality is likely an overall more proximal molecular trait to genetic variation than gene expression in the sequence of causal events, which likely contributes to the larger enhancer effects we observed and therefore results in higher power to detect caQTL with the same sample size.
The majority (77%) of GWAS signals colocalized with only caQTL rather than eQTL, in part because we detected many more caQTL than eQTL. As a corollary, we identified fewer triple GWAS-caQTL-eQTL colocalizations, which limited our efforts in using eQTL to identify target genes inferring the causal direction between omic modalities. It is becoming evident that eQTL alone fall short in fully elucidating the regulatory architecture of GWAS loci (Mostafavi et al., 2022; Umans, Battle, and Gilad, 2021). Our analyses revealed an intrinsic distinction between e- and caQTLs that may help reconcile these observations. Active TSS regions contained higher effect eSNPs compared to caSNPs whereas stretch enhancer regions, which are enriched for cell-type-relevant GWAS signals (J. Chen et al., 2021; Parker et al., 2013; Varshney, Scott, et al., 2017), contained higher effect caSNPs compared to eSNPs. Therefore, eQTL scans identify signals largely in gene TSS regions, whereas caQTL scans are able to identify strong effects in cell-specific distal enhancer elements enriched for GWAS signals.
Because complex traits are influenced by both genetic and environmental effects, examining gene expression in the conditions most relevant for disease could be more informative. The larger genetic effects on stretch enhancer chromatin accessibility could propagate to gene expression effects under specific environmental conditions. Alasoo et al. (2018) provided support for this hypothesis bulk RNA and ATAC data in a macrophage model system where ~60% of eQTL identified only under stimulatory conditions (response eQTL) were caQTL in the basal state. Another study found similar response eQTL overlapping basal caQTL results in a human neural progenitor system (Matoba et al., 2023). These studies, along with our data, suggest that chromatin in cell-identity stretch enhancers is primed to potentiate changes in gene expression under relevant conditions. Notably, recent sn-multiome studies observing lower cell-state resolution from chromatin accessibility compared to transcription also posited that cells could retain a primed or permissive chromatin landscape that can allow dynamic state transitions in response to relevant conditions (Sun et al., 2023; Xiong et al., 2023).
About half of GWAS-caQTL colocalizations were cluster-specific across traits, with many specific for the lower powered (due to nuclei abundance) Endothelial and FAP clusters, which adds to the importance of single nucleus chromatin accessibility profiling in identifying cell-specific genetic regulatory elements. Our snATAC caQTL data help delineate heterogeneity in the mechanistic pathways shaping T2D pathophysiology. We show the GLI2 signal is most relevant for endothelial cells and the ARL15 signal targets the FST gene in FAPs, implicating an interplay of fat and muscle mass regulation by these progenitor cells in T2D. We find evidence for the C2CD4A/B T2D GWAS signal, previously implicated in islet dysfunction through inflammatory cytokine-responsive C2CD4A/B genes, to also be involved in glucose uptake mechanisms in muscle fibers through the VPS13C gene. Cell types such as FAPs and endothelial occur in other T2D-relevant tissues such as adipose; comparing the snRNA/snATAC and e/caQTL profiles for these cell types from a wider array of tissues will help glean the similarities and differences in disease mechanisms in related cell type populations. Layering sn-e/caQTL colocalization information over GWAS signals across multiple relevant tissues will help generate a conceptual “signal scoreboard” that can help prioritize cell types, regulatory elements, target genes and causal variants(s) for each GWAS signal towards experimental validation.
While we identified 13 distinct cell type clusters, rare clusters had limited power to identify e/caQTL. In our single-nucleus study, most nuclei were identified as muscle fibers and relatively few nuclei as other cell types; this distribution of cell type proportions was especially skewed since muscle fibers are multi-nucleated. Approaches such as deeper sequencing, pre-selecting relevant cell types via fluorescence activated cell sorting (FACS) for non-frozen cell/tissue samples could allow for more power to identify QTLs in rare cell types. Cleaner nuclei preps with low ambient transcripts and better approaches to adjust for these would enable retrieving more quality nuclei from rare cell types. Signal upscaling via deep learning methods such as (Lal et al., 2021; Rai et al., 2019) is another possible avenue to enable caQTL scans in lower abundance cell types. Instead of identifying discrete clusters, identifying contiguous cell states through latent embedding and related approaches (Ahlmann-Eltze and Huber, 2023) could help mitigate the power issue and identify state-specific QTLs. The multiome protocol for profiling RNA and ATAC on the same nucleus was not available when our samples were processed. However, it is a powerful assay to link regulatory elements to genes, complementing eQTL analyses to nominate target genes.
To date, there have been some single cell/nucleus eQTL studies (Bryois et al., 2020; Cuomo et al., 2020; Jerber et al., 2020; Neavin et al., 2021; Wijst et al., 2018; Yazar et al., 2022), few sn-caQTL studies (Benaglio et al., 2023; Turner et al., 2022); however, these all had modest sample sizes, and were mainly in blood cell types or cell lines. There are no population-scale single cell/nucleus studies in skeletal muscle and none with both RNA and ATAC modality for hundreds of samples in any tissue. Our work bridges a large gap in knowledge in that it is the first study identifying both sn-eQTL and sn-caQTL across hundreds of samples in any tissue.
Overall, our study provides population-scale epigenomic and transcriptomic profiles across multiple cell types in human skeletal muscle. Our findings emphasize the need to consider chromatin accessibility in addition to gene expression when investigating the functional mechanisms underlying complex traits, and serves as a template for multi-omics maps in other tissue and disease contexts.
4. Methods
4.1. Sample collection
4.1.1. FUSION cohort
The Finland-United States Investigation of NIDDM Genetics (FUSION) study is a long-term project aimed at identifying genetic variants that contribute to the development of type 2 diabetes (T2D) or affect the variability of T2D-related quantitative traits. To conduct the FUSION Tissue Biopsy Study, we obtained vastus lateralis muscle biopsy samples from 331 individuals across the glucose tolerance spectrum, including 124 with normal glucose tolerance (NGT), 77 with impaired glucose tolerance (IGT), 44 with impaired fasting glucose (IFG), and 86 with newly-diagnosed T2D (Scott et al., 2016).
To ensure the validity of the study results, certain individuals were excluded from the study, including those receiving drug treatment for diabetes, those with conditions that could interfere with the analysis (such as cancer, inflammatory diseases, or skeletal muscle diseases), those with conditions that increase hemorrhage risk during biopsy (such as hemophilia, von Willebrand’s disease, or severe liver disease), those taking medications that increase hemorrhage risk during the biopsy (such as warfarin), those taking medications that could confound the analysis (for example oral corticosteroids, or other anti-inflimmatory drugs such as infliximab or methotrexate), and those under 18 years of age.
Clinical and muscle biopsy visits were conducted at three different study sites (Helsinki, Savitaipale, and Kuopio). The clinical visit included a 2-hour four-point oral glucose tolerance test (OGTT), BMI, waist-to-hip ratio (WHR), lipids, blood pressure, and other phenotypes measured after a 12-hour overnight fast, as well as health history, medication, and lifestyle questionnaires. The clinical visit was conducted an average of 14 days before the biopsy visit.
The muscle biopsies were performed using a standardized protocol. Participants were instructed to avoid strenuous exercise for at least 24 hours prior to the biopsy. After an overnight fast, approximately 250 mg of skeletal muscle from the vastus lateralis was obtained using a conchotome, under local anesthesia with 20 mg/mL lidocaine hydrochloride without epinephrine. A total of 331 muscle biopsies were collected by nine experienced and well-trained physicians at the three different study sites between 2009 and 2013, with three physicians performing the majority of the biopsies. All physicians were trained to perform the biopsy in an identical manner. The muscle samples were cleaned of blood, fat, and other non-muscle tissue by scalpel and forceps, rinsed with NaCl 0.9% solution, and frozen in liquid nitrogen within 30 seconds after sampling. Muscle samples were then stored at −80 degrees Celsius.
4.2. Sample preparation, snRNA-seq and ATAC profiling
The frozen tissue biopsy samples were processed in ten batches, each consisting of 40-41 samples. These batches were organized using a randomized block design to protect against experimental contrasts of interest including cohort, age, sex, BMI and stimulatory condition (for Texas cohort) (Figures. S1A–S1E). Samples in each batch were pulverized in four groups of 10 or 11 samples (each sample weighing between 6-9 mg) using a CP02 cryoPREP automated dry pulverizer (Covaris 500001) and resuspended in 1 mL of ice-cold PBS. Following, the material from all 40/41 samples was pooled together and nuclei were isolated. We developed a customized protocol (protocol S1, supplementary text) derived from the previously published ENCODE protocol https://www.encodeproject.org/experiments/ENCSR515CDW/ and used it to isolate nuclei, which is compatible with both snATAC-seq and snRNA-seq. The desired concentration of nuclei was achieved by re-suspending the appropriate number of nuclei in 1X diluted nuclei buffer (supplied by 10X genomics for snATAC, and RNA nuclei buffer (1% BSA in PBS containing 0.2U/uL of RNAse inhibitor) for snRNA). The nuclei at appropriate concentration for snATAC-seq and snRNA-seq were submitted to the University of Michigan Advanced Genomics core for all the snATAC-seq and snRNA-seq processing on the 10X Genomics Chromium platform (v. 3.1 chemistry for snRNA-seq). Nuclei to profile each modality from each batch were loaded onto 8 channels/wells of a 10X chip at 50k nuclei/channel concentration. For snRNA-seq, the libraries were single-ended, 50 bp, stranded. For snATAC-seq, the libraries were paired-ended, 50 bp. The sequencing for each modality and batch was performed on one NovaSeq S4 flowcell.
4.3. Muscle multiome sample
We obtained “multiome” data, i.e. snATAC-seq and snRNA-seq performed on the same nucleus for one muscle sample as part of newer ongoing projects in the lab. We used 70mg of pulverized human skeletal muscle tissue sample. The sample was pulverized using an automated dry cryo pulverizer (Covaris 500001). We developed a customized protocol (hybrid protocol with sucrose) from the previously published ENCODE protocol, and used it to isolate nuclei for single nuclei multiome ATAC and 3’GEX assay. The desired concentration of nuclei was achieved by re-suspending the appropriate number of nuclei in 1X diluted nuclei buffer (supplied by 10X genomics). The nuclei at the appropriate concentration for single nuclei multiome ATAC and 3’GEX assay was processed on the 10X genomics chromium platform. 20K nuclei were loaded on one well of the 8 well strip.
4.4. Genotyping and imputation
The FUSION cohort samples were genotyped using DNA extracted from blood on the HumanOmni2.5 4v1_H BeadChip array (Illumina, San Diego, CA, USA) during a previous study (Taylor et al., 2019). The Texas and Sapphire cohort samples were genotyped using DNA extracted from blood on the Infinium Multi-Ethnic Global-8 v1.0 kit. Probes were mapped to Build 37. We removed variants with multi mapping probes and updated the variant rsIDs using Illumina support files Multi-EthnicGlobal_D1 MappingComment.txt and annotated.txt downloaded from https://support.illumina.com/downloads/infinium-multi-ethnic-global-8-v1-support-files.html. We performed pre-imputation QC using the HRC-1000G-check-bim.pl script (v. 4.2.9) obtained from the Marc McCarthy lab website https://www.well.ox.ac.uk/~wrayner/tools/ to check for strand, alleles, position, Ref/Alt assignments and update the same based on the 1000G reference (https://www.well.ox.ac.uk/~wrayner/tools/1000GP_Phase3_combined.legend.gz). We did not conduct allele frequency checks at this step (i.e. used the -noexclude flag) since we had samples from mixed ancestries.
For all samples, we performed pre-phasing and imputation using the Michigan Imputation Server (Das et al., 2016). The standard pipeline (https://imputationserver.readthedocs.io/en/latest/pipeline/) included pre-phasing using Eagle2 (Loh et al., 2016) and genotype dosage imputation using Minimac4 (https://github.com/statgen/Minimac4) and the 1000g phase 3 v5 (build GRCh37/hg19) reference panel (The 1000 Genomes Project Consortium 2015). Post-imputation, we selected biallelic variants with estimated imputation accuracy (r2) > 0.3, variants not significantly deviating from Hardy Weinberg Equilibrium P>1e-6, MAF in 1000G European individuals > 0.05.
4.5. snRNA-seq data processing and quality control
snRNA: We mapped the reads to the human genome (hg38) using STARsolo https://github.com/alexdobin/STAR/blob/master/docs/STARsolo.md (v. 2.7.3a). We performed rigorous quality control (QC) to identify high-quality droplets containing single nuclei (Figures. S1F–S1G). We required the following criteria: 1) nUMI > 1000; 2) fraction of mitochondrial reads < 0.01; 3) identified as a “singlet” and assigned to a sample using Demuxlet (Kang et al., 2018) 4) identified as “non-empty”, i.e. where the RNA profile was statistically different from the background ambient RNA signal, using the testEmtpyDrops function from the Dropletutils package (Lun et al., 2019); and 5) passing the cluster-specific thresholds for the estimated ambient contamination from the DecontX package (Yang et al., 2020). This led to a total of 255,930 pass-QC RNA nuclei, 180,583 from the FUSION cohort. These individual qc steps are further described below.
4.6. snATAC-seq data processing and quality control
We made barcode corrections using the 10X Genomics whitelist using an approach implemented by the 10X Genomics Cell Ranger ATAC v. 1.0 software via a custom python script and counted the number of read pairs from each droplet barcode. We trimmed the adapter sequences using cta https://github.com/ParkerLab/cta and generated updated fastqs by replacing the cellular barcodes with the corrected cellular barcodes, while selecting reads corresponding to cellular barcodes that had at least 1000 pairs. Droplets with less than 1000 read pairs would not contain useful/high quality data from single nuclei and so were removed from processing. We mapped the reads to the human genome (hg38) using bwa mem (v. 0.7.15-r1140) (Li and Durbin, 2009) with flags “-I 200,200,5000 -M”. We performed rigorous quality control (QC) and retained high-quality droplets based on the following definitions (Figures. S1H–S1I): 1) 4,000 < high quality autosomal alignments (HQAA) < 300,000, 2) transcription start site (TSS) enrichment ≥ 2, 3) mitochondrial fraction < 0.2. For each snATAC-seq library bam file, we used the subset-bam tool (v. 1.0.0) https://github.com/10XGenomics/subset-bam to subset for the selected cellular barcodes, and used SAMtools to filter for high-quality, properly-paired autosomal read pairs (-f 3 -F 4 -F 8 -F 256 -F 1024 -F 2048 -q 30). To identify droplets containing a single nucleus “singlet” and determine the sample identity, we used the Demuxlet (Kang et al., 2018) tool. For each library (8 10X channels/wells in each of the 10 batches, N=80), we ran Demuxlet using default parameters providing the snATAC-seq library bam files the genotype vcf files containing all samples included in that batch and selected all the droplets assigned as singlets. This led to a total of 3,69,792 pass-QC ATAC nuclei, 2,68,543 from the FUSION cohort.
4.6.1. Two-stage Demuxlet pipeline
Multiplexing 40/41 samples in each batch in a randomized block study design helped protect against batch effects and it was cost-effective approach. To identify droplets containing a single nucleus “singlet” and determine the sample identity, we used the Demuxlet (Kang et al., 2018) tool. For each library (8 10X channels/wells in each of the 10 batches, N=80), we ran Demuxlet using default parameters providing the library bam files the genotype vcf files containing all samples included in that batch and selected all the droplets assigned as singlets. Background/ambient RNA contamination can influence singlet assignments, so we accounted for that next. We performed clustering of these pass-qc RNA droplets and annotated clusters using known marker genes. A large proportion of our data was muscle fiber nuclei, this is expected since muscle fibers are multi-nucleated. Therefore, a large proportion of ambient RNA would come from muscle fiber cells. Observing the barcode-nUMI rank plots (Figure. S1F), we considered droplets with less than 100 reads as unlikely to contain an intact nucleus and therefore representative of the ambient RNA profile. Top 100 genes contained top ~30% of ambient RNA reads (Figure. S2A). Most abundant genes in the ambient RNA were expectantly mitochondrial and muscle fiber genes such as MYH1, MYH7 etc (Figure. S2B). We reasoned that “masking” top n% of these top genes should reduce ambiguity arising due the ambient RNA, enabling more droplets to be assigned as a singlet. We would correct the gene counts for ambient RNA in the downstream steps. We tested masking to n% of genes from Demuxlet and observed that masking the top 30% of genes in the ambient RNA maximized singlet assignment (Figure. S2C). We therefore completed a second Demuxlet run masking top 30% genes, and any new droplets that were identified as singlets to the set of selected droplets. The singlet nuclei recovered from the masked stage 2 came mostly from lower abundance non-fiber clusters (Figure. S2D) (using cluster labels identified downstream).
4.6.2. Adjusting RNA counts for overlapping gene annotations
RNA mapping and gene quantification using STARsolo outputs a “GeneFull” matrix that quantifies intronic+exonic reads and a “Gene” matrix that quantifies only exonic reads. For our nuclear RNA expriment, we used the GeneFull matrices for all downstream applications. As of the STAR version 2.7.3a which was used in our analysis, in case of overlapping gene annotations, the program renders some read assignments ambiguous and therefore some genes receive less counts in the GeneFull matrix compared to the Gene matrix. We observed the distribution of count differences between the exon+intron (GeneFull) and exon (Gene) matrices for each gene across all 80 libraries and created a list of genes where this difference was consistently negative in at least 10 libraries. We then created custom counts matrices keeping the “Gene” counts for these 6,888 selected genes and kept the “GeneFull” counts for all other genes.
4.6.3. Ambient RNA adjustment
We used DecontX (celda v. 1.8.1, in R v. 4.1.1) (Yang et al., 2020) to adjust the nucleus x gene expression count matrices for ambient RNA. Taking all the qc’ed RNA nuclei up to this stage (N = 260,806), we identified cell type clusters using Liger (rliger R package v. 1.0.0) (Welch et al., 2019). Liger employs integrative non-negative matrix factorization (iNMF) to learn a low-dimensional space in which each nucleus is defined by both dataset-specific and shared factors called as metagenes. It then builds a graph in the resulting factor space, based on comparing neighborhoods of maximum factor loadings. We selected the top 2000 variable genes using the selectGene function and clustered with number of factors k=20 and regularization parameter lambda=5 along with other default parameters as it identified expected clusters (Figure. S3A). We then ran DecontX on a per-library basis using the decontX() function, passing our custom created RNA raw matrices (adjusted for overlapping gene annotations) for the QC’ed nuclei, barcodes with total UMIs < 100 for the background argument, cluster labels from liger, and set the delta parameter (prior for ambient RNA counts) as 30. This prior value was more stringent than the DecontX default of 10 and it was selected after exploring the parameter space and observing that delta=30 better reduced fiber type marker gene such as MYH7, MYH2 counts in rarer clusters such as Endothelial, Satellite Cell, while retaining respective marker gene VWF and PAX7 counts (Figure. S3B). Since the decontamination is sensitive to the provided cluster labels, we performed a second clustering using adjusted counts from the first DecontX run to obtain better optimized cluster labels. We also included the snATAC modality for this clustering. Liger’s online integrative non-negative matrix factorization (iNMF) algorithm was used at this step (Gao et al., 2021; Welch et al., 2019) which enabled efficient processing of this large snATAC+snRNA dataset by iteratively sampling a subset of nuclei at a time. We selected the clustering with liger k=19, lambda=5, epoch=5, batchsize=10,000 along with other default parameters (Figure. S3C). We then performed a second DecontX run using raw snRNA matrices (adjusted for overlapping gene annotations), droplets with UMIs < 100 as background, delta set to 30 while including the updated snRNA cluster labels.
DecontX also estimates fraction of ambient RNA per nucleus. We used this metric to further filter out RNA nuclei. We observed that this metric varied across clusters, and the immune cell, muscle fiber mixed and the smooth muscle clusters has a visible population of nuclei with high estimated ambient RNA fraction (Figure. S3D). We therefore fitted two Gaussians for these three clusters per batch and removed nuclei that obtained the probability of being from the high contamination population > probability of being from the low contamination population (Figure. S3E). For the rest of the clusters, we removed nuclei with estimated ambient RNA > 0.8. We retained all pass QC nuclei and used rounded decontaminated counts for the final joint clustering and all downstream analyses.
4.7. Joint clustering and cell type annotation
We jointly clustered snRNA and snATAC from the FUSION cohort and the one multiome muscle sample using Liger’s online iNMF algorithm (rliger v. 1.0.0) (Gao et al., 2021; Welch et al., 2019). The inputs to Liger are for snRNA, the adjusted gene by nucleus count matrices, and for snATAC, gene (gene body + 3kb promoter region) by nucleus fragment counts. We only considered protein coding, autosomal genes for Liger. We used the online iNMF Liger version (https://github.com/MacoskoLab/liger/tree/online), factorizing RNA nuclei first and then projecting ATAC nuclei using the following parameters: top 2000 variable genes, k=21, lambda=5, epoch=5, max iterations=4, batchsize=10,000, along with other default parameters. This process resulted in a joint clustering without batch or modality specific effects (Figure. S4A) We then used the Louvain community detection function in Liger with resolution = 0.04 to identify ‘coarse’ clusters that we annotated using known marker gene expression patterns (Figure. S4B). These cluster assignments were used in all downstream association analyses.
4.8. ATAC-seq peak calling and consensus peak feature definition
We created per-cluster snATAC-seq bam files by merging reads from all pass-QC ATAC nuclei for each cluster. We randomly subsampled bam files to 1 Billion reads and called narrow peaks using MACS2 (v. 2.1.1.20160309) (Y. Zhang et al., 2008). We used BEDTools bamToBed (Quinlan and Hall, 2010) to convert the bam files to the BED format, and then used that file as input to MACS2 callpeak (command “macs2 callpeak -t atac-$cluster.bed –outdir $cluster -f BED -n $cluster -g hs –nomodel –shift −100 – seed 762873 –extsize 200 -B –keep-dup all”) to call narrow peaks . We removed peaks overlapping the ENCODE blacklisted regions (Amemiya, Kundaje, and Boyle, 2019), and selected peaks passing 0.1% FDR from macs2. We then defined a set of consensus snATAC-seq peak summits across all 13 clusters. We considered the filtered narrow peak summits across all clusters and sorted by MACS2 q value. We sequentially collapsed summits across clusters within 150bp and retained the most significant one, identifying N=983,155 consensus summits (Figures. S5A–S5C). Aggregating ATAC-seq signal over broad peaks in a cluster while centering on the left-most summit showed the second summit usually occurred ~300bp away (Figure. S5D), in line with the nucleosome length being ~147 bp (Beshnova et al., 2014). We therefore considered each consensus summit extended by 150 bp on each side as the consensus peak-feature for all downstream analyses. To visualize the signal, we converted the bedGraph files output by MACS2 to bigWig files using bedGraphToBigWig (Kent et al., 2010).
4.9. Comparison to snATAC atlas
Per-cell type comparisons to the snATAC atlas from (K. Zhang et al., 2021) were performed using a modified version of the logistic regression-based technique described previously (Orchard et al., 2021). First, narrowPeaks from each cell type cluster were merged to produce a set of master peaks. Next, master peaks within 5kb upstream of a GENCODE TSS (GENCODE v40; (Frankish et al., 2021)) were dropped. Master peaks were annotated to muscle cell types according to whether or not they overlapped a narrowPeak in that cell type, and master peaks annotated to more than one cell type were dropped, resulting in a set of cell type-specific peaks. Next, for each of our cell types and each of the 222 cell types from (K. Zhang et al., 2021), we ran the logistic regression model: (master peak is specific to muscle cell type ~ + β0 + β1 *master peak overlaps peak from snATAC atlas cell type), where represents a model intercept. Within each of our cell types, we then produced a matching score for each of the snATAC atlas cell types by re-normalizing the resulting model coefficient β1 to range between 0 and 1 (by dividing the coefficients by the maximum coefficient, first setting coefficients to 0 if the model p-value was not significant after Bonferroni correction or the coefficient was negative). The snATAC atlas cell type with score = 1 was determined to be the best match.
4.10. Identification of cell type-specific genes and GO enrichments
Differential gene expression analyses between all pairs of cell types were performed to identify cell type-specific genes. Muscle fiber nuclei clusters (Type_1, Type_2a, Type_2x, Neuromuscular_junction, Muscle_Fiber_Mixed) were merged for this analysis due to their expected similarity. For each pair of cell types we used DESeq2 (Love, Huber, and Anders, 2014) to call differential genes between the cell types. Samples with less than 3,000 genes detected in either of the cell types were dropped, as were genes with less than 3 counts across all of the samples (when combining the cell types). The DESeq2 analysis was done in a paired sample fashion. A gene was considered to be a cell type-specific gene for cell type X if that gene was more highly expressed in cell type X than in all other cell types (5% FDR).
GO enrichments were performed using g:Profiler (python API, v. 1.0.0; (Raudvere et al., 2019)), using all genes with at least one count in one cell type as the background set.
4.11. Identification of cell type-specific open chromatin summits and motif enrichments
Using the per-cluster peak summit counts, we identified cell type-specific summits using the τ metric from (Yanai et al., 2005). As muscle fiber types show high gene expression similarity, we merged any nuclei assigned to muscle fibers (Type 1, Type 2a, Type 2x, NMJ, and Muscle fiber mixed clusters). Summits with τ > 0.8 were considered to be cell type-specific, and were assigned to the cell type showing greatest accessibility of that summit.
Motif enrichments were performed using the 540 non-redundant motifs from a previous study (D’Oliveira Albanus et al., 2021), with the logistic regression model (one model per motif per cell type): summit is specific to cell type ~ intersect + summit is TSS proximal + summit GC content + number of motif hits in summit where TSS proximal was defined as within 2kb upstream of a TSS, and the number of motif hits was determined using FIMO (v. 5.0.4, with default parameters and a 0-order Markov background model generated using fasta-get-markov (Grant, Bailey, and Noble, 2011)). We excluded two cell types (Neuronal and T_cell) with less than 500 cell type specific summits and excluded cases where the model didn’t converge. A motif was considered significantly enriched if the coefficient for the “number of motif hits in summit” term was significantly positive after Bonferroni correction within each cell type. The corresponding heatmap figure displays motifs that were amongst the top 5 significantly enriched motifs by either p-value or coefficient in at least one cell type.
4.12. snATAC-seq coaccessiblity
We ran CICERO (Pliner et al., 2018) (v. 1.4.0; R v. 4.0.1) on the narrow peak fragment counts in each cluster to score peak-peak co-accessibility. We used UMAP dimensions 1 and 2 (Figure. 1B) as the reduced coordinates and set window size to 1 Mb. A peak was considered to be a TSS peak for a gene if it overlapped the 1kb window upstream of that gene’s TSS. If multiple TSS peaks were present for a gene, the maximum co-accessibility score was considered.
4.13. QTL scan in clusters
We performed expression and chromatin accessibility QTL analysis in the top five most highly abundant cell-type clusters (clusters with the most number of nuclei) - Type 1, Type 2a, Type 2x, FAPs and Endothelial using QTLtools (v. 1.3.1-25-g6e49f85f20) (Delaneau et al., 2017). We removed three samples from out QTL analyses: one because it appeared to be of non-Finnish ancestry from PCA analysis, and two others which were found to be first degree related to other samples. We created a vcf file with imputed genotypes of all the selected FUSION samples, and filtered for autosomal, bi-allelic variants with MAF ≥ 5%, non-significant deviation from Hardy-Weinberg equilibrium P>1x10−6. We performed PCA using QTLtools pca with options –scale, –center and –distance 50,000.
4.14. eQTL scan
We selected the following gene biotypes (Gencode V30): protein_coding, lincRNA, 3prime_overlapping_ncRNA, antisense, bidirectional_promoter_lncRNA, macro_lncRNA, non_coding, sense_intronic, and sense_overlapping. For each cluster, we considered samples with at least 10 nuclei for the eQTL analysis. We generated RNA count matrices by summing up gene counts (post-ambient RNA decontamination) from nuclei for each sample in each cluster. We converted the gene counts into transcript per million (TPMs) and inverse-normalized across samples. TPM = RPK/factor, where RPK = counts/(length in kb) and factor = sum(RPK)/1M for each cluster. We used the top 10,000 genes based on median TPM to perform PCA using QTLtools. eQTL scans were performed considering variants within 250kb of gene TSSs. For each cluster, we ran test eQTL scans while considering the top 3 genotype PCs and a varying number of phenotype PCs to account for unknown biological and technical factors. We selected the number of phenotype PCs that maximized eQTL discovery as covariates Figure. S6A. We optimized within-cluster thresholds for minimum gene counts across minimum number of samples that defined our final set of testable genes that minimized the multiple testing burden. We performed the cis eQTL scans with 1,000 permutations, then applied an across-feature multiple testing correction using the qvalue Storey function on the beta distribution adjusted P values and reported eGenes at FDR ≤ 5%.
4.15. caQTL scan
For each cluster, we considered samples with at least 10 nuclei for the caQTL analysis. We quantified each consensus feature and obtained the sum of fragment counts across all nuclei from each samples in each cluster. For an initial lenient caQTL scan, we selected all consensus features in a cluster that had at least 2 counts in at least 10 samples to test for caQTL in each cluster. We used inverse-normalized counts per million (CPMs) as quantification for caQTL. CPM = RPK/factor, where RPK = counts/(feature length in kb) and factor = sum(RPK)/1M for each cluster. We performed PCA on the inverse-normalized CPMs and included the top n phenotype PCs that maximized QTL discovery in each cluster, along with the top 3 genotype PCs as covariates. We optimized within-cluster thresholds for minimum peak counts across minimum number of samples that defined our final set of testable peak that minimized the multiple testing burden. caQTL scans were performed considering variants within 10kb of the feature midpoint (peak summit). We performed the cis caQTL scans with 1,000 permutations, then applied an across-feature multiple testing correction using the qvalue Storey function on the beta distribution adjusted P values and reported caPeaks at FDR ≤ 5%.
4.16. QTL finemapping
We used the sum of single effects (SuSiE) (G. Wang et al., 2020) approach to identify independent e and caQTL signals and obtain 95% finemapped credible sets. We used QTLtools to adjust for the covariates optimized for e or caQTL scans and inverse-normalized the residuals. We used these adjusted phenotypes along with the sample genotype dosages for variants in a 250kb window in the susie function along with the following parameters: number of signals L=10, estimate_residual_variance=TRUE, estimate_prior_variance=TRUE, min_abs_cor=0.1.
4.17. Motif reconstruction using caQTL results
For a given cell type and motif, we identified all caQTL variants that sat within the corresponding caPeak and that overlapped a motif hit. Motif hits were determined by scanning the genomic sequence in a variant-aware manner using FIMO (v. 5.0.4, with default parameters and a 0-order Markov background model generated using fasta-get-markov (Grant, Bailey, and Noble, 2011)), i.e. scanning the genomic sequence containing the reference and the alternative allele. For each such overlapping caQTL, we calculated the caQTL allelic fold change (Mohammadi et al., 2017) using tensorQTL (Taylor-Weiner et al., 2019). To reconstruct the motif, for each of the four nucleotides and each position in the motif, we summed the absolute value of the allelic fold change for all caQTLs overlapping that position in the motif hit and having that nucleotide as the favored (open chromatin) allele. This was converted to a probability matrix (such that the four values at each motif position summed to one) for the final reconstructed motif. To demonstrate that the observed similarity between the original and reconstructed motif was not simply a result of the fact that a motif hit was called by FIMO, we additionally reconstructed motifs based on all variants that met filtering requirements for the caQTL scan, overlapped motif hits, and were in peaks tested in the caQTL scan. To do this, for each of the four nucleotides and each position in the motif, we counted the number of variants overlapping that position in the motif hit and having that nucleotide as either the ref or the alt allele, and then converted this to a probability matrix as before.
4.18. Relationship between caQTL effect size, caSNP MAF, and caQTL peak presence in scATAC atlas
Type 1 muscle fiber caPeaks were grouped based on the open chromatin allele frequency (calculated using the FUSION samples) and the caQTL effect size (absolute value of the slope, binned by 0-0.4, 0.4-0.8, 0.8-1.2, 1.2-1.6, and 1.6-2.0). We then calculated the fraction of the caPeaks within that bin that overlapped with a Type I Skeletal Myocyte peak from (K. Zhang et al., 2021).
4.19. caPeak chromatin state enrichments
CaPeak enrichment in chromatin states was computed using the Skeletal Muscle Female (E108) chromatin states (15-state model) from Roadmap Epigenomics (The Roadmap Epigenomics Consortium et al., 2015). First, muscle ATAC peaks were lifted from hg38 to hg19 using liftOver (kentUtils v. 343 (Hinrichs et al., 2006)). For each of the Type 1, Type 2a, and Type 2x cell types, we then ran the logistic regression:
where peak size was set as the average peak reads per million across samples. Only peaks tested for caQTL were included in the model. caPeaks were enriched for a state if the coefficient for the corresponding state term in the model was significantly positive after Bonferroni correction (Bonferroni correction performed within each cell type, across the 16 non-intercept terms).
4.20. Motif enrichment in caPeaks
Motif enrichments were performed using the 540 non-redundant motifs from (D’Oliveira Albanus et al., 2021), with the logistic regression model (one model per motif per cell type): peak is caPeak ~ intercept + peak is TSS proximal + peak GC content + peak size + number of motif hits in peak where TSS proximal was defined as within 2kb upstream of a TSS, peak size was set as the average peak reads per million across samples, and the number of motif hits was determined using FIMO (v. 5.0.4, with default parameters and a 0-order Markov background model generated using fasta-get-markov (Grant, Bailey, and Noble, 2011)). Only peaks tested the caQTL scans were included in each model. A motif was considered significantly enriched if the coefficient for the “number of motif hits in summit” term was significantly positive after Bonferroni correction within each cell type. The corresponding heatmap figure displays motifs that were amongst the top 3 significantly enriched motifs by either p-value or coefficient in at least one cell type.
4.21. eQTL and caQTL colocalization
We used coloc v5 (Wallace, 2021) to test for colocalization between e and ca QTL. We used the e and ca QTL finemapping output from SuSiE over the 250kb window as inputs to coloc v5. We considered colocalization between two signals if the PP H4 > 0.5.
4.22. Causal inferrence between chromatin accessibility and gene expression
For all pairs of colocalized eGenes and caPeaks, we inferred the causal chain between chromatin accessibility and gene expression using two orthogonal approaches - a mediation-based approach causal inference test (CIT, v2.3.1) (Millstein, G. K. Chen, and Breton, 2016; Millstein, B. Zhang, et al., 2009) and a Mendelian randomization approach MR Steiger directionality test (Hemani, Tilling, and Smith, 2017). We required consistent direction from both CIT and MR Steiger at 5% FDR to consider an inferred causal direction between an eGene and caPeak pair.
4.22.1. CIT
To test if an exposure mediates an effect on an outcome, CIT uses genetic instruments (eg SNPs) requiring a set of mathematical conditions to be met in a series of regressions under a formal hypothesis testing framework. If a SNP (L) is associated with an outcome (T) only through an exposure (G), outcome when conditioned on the exposure should be independent of the SNP. The conditions therefore are: (i) L is associated with T, (ii) L is associated with G conditional on T, (iii) T is associated with G conditional on L and (iv) T is independent of L conditional on G. For each pair of caPeak and eGene for which one or more independent caQTL and eQTL signal(s) colocalized, we ran four CIT models each returning an omnibus P value- a) eSNP(s) -> eGene -> caPeak (P e-to-ca-causal), b) eSNP(s) -> caPeak -> eGene (P e-to-ca-revCausal), c) caSNP(s) -> caPeak -> eGene (P ca-to-e-causal) and d) caSNP(s) -> eGene -> caPeak (P ca-to-e-revCausal). We included sample batch, age, sex, BMI and top 3 genotype PCs as covariates in the CIT model. For each model, we computed the omnibus FDR values using the fdr.cit function to account for multiple testing. To infer a caPeak causal on an eGene, we required q-ca-e-causal < 0.05, q-ca-e-revCausal > 0.05, q-e-ca-causal > 0.05 and q-e-ca-revCausal < 0.05, and vice versa to infer an eGene causal on a caPeak. We note that eGene-caPeak pairs without a putative causal CIT prediction could be truly independent or could have a causal relationship obscured by measurement error.
4.22.2. MR Steiger directionality test
In an MR-based approach, the genetic instrument (SNP) is used as a surrogate for the exposure to estimate its causal effect on an outcome, by scaling the association of SNP and outcome by the association between SNP and exposure. This approach is considered less susceptible to bias from measurement errors or confounding (Hemani, Tilling, and Smith, 2017). For each pair of caPeak and eGene for which one or more independent caQTL and eQTL signal(s) colocalized, we used the mr_steiger function (TwoSampleMR R package version 0.5.6) to test both caPeak and eGene as exposure over the other modality as outcome. To infer a caPeak causal on an eGene, we required ca-to-e “correct causal direction” as “True” at 5% FDR, and e-to-ca “correct causal direction” as “False” at 5% FDR, while estimating steiger test q values using the R qvalue package (http://github.com/jdstorey/qvalue). For each model, we provided the respective QTL scan sample sizes and set r_xxo = 1, r_yyo = 1, r_exp = NA and r_out = NA to estimate the sensitivity ratio - which computes over the bounds of measurement errors in the exposure and outcome, how much more often is one causal direction observed versus the other. The higher the sensitivity ratio, more robust is the inferred causal direction to measurement errors.
4.23. GWAS enrichment in ATAC-seq peak features
We computed enrichment of GWAS variants in ATAC-seq peak features using LD score regression (LDSC) (B. K. Bulik-Sullivan et al., 2015; Finucane, Reshef, et al., 2018). We downloaded GWAS summary statistics for 28 traits relevant for skeletal muscle such as T2D, glycemic traits, COVID19, exercise etc. Where required, we lifted over the summary stats onto hg38 using the UCSC liftOver tool. We formatted the summary stats according to LDSC requirements using the ldsc munge_sumstats.py script, which included keeping only the HapMap3 SNPs with minimum MAF of 0.01 (as recommended by the LDSC authors). We also downloaded several LDSC-formatted UKBB GWAS summary statistics from the Benjamin Neale lab website (Sudlow et al., 2015) https://nealelab.github.io/UKBB_ldsc/downloads.html. We selected selected primary GWASs on both sexes for high confidence traits with h2_significance > z7, following guidelines described on the Ben Neal lab blog https://nealelab.github.io/UKBB_ldsc/details.html. We created a baseline model with cell type agnostic annotations such as MAF, coding, conserved regions, etc. We set up LDSC in a joint model to test snATAC-seq peak features (consensus peak summit features that overlapped a peak summit called in that cluster) from all 13 snATAC-seq cell types, islet beta cell ATAC-seq peaks (Rai et al., 2019) since islets are relevant for several metabolic traits of interest, along with the baseline model. The various annotation files (regression weights, frequencies, etc.) required for running LDSC were downloaded from https://data.broadinstitute.org/alkesgroup/LDSCORE/GRCh38/. LD scores were calculated using the Phase 3 1000 Genomes data. For GWAS enrichment in cluster snATAC annotations (Figure. S9A) and type 1 snATAC annotations by e/caSNP overlap (Figure. S9B), we show traits that had a nominally significant regression coefficient in at least one annotation. We pruned out closely related traits by forming flat clusters on the trait hierarchical clustering at thresholds t=0.7 and t=1.0 respectively and retaining the trait with highest regression coefficient Z-score in Figures. 4A–4B.
4.24. eQTL and caQTL co-localization with GWAS
We considered the lead GWAS signals that if the individual study reported so; otherwise, we identified genome-wide significant (P < 5e-8) signals in 1Mb windows. We finemapped each GWAS signal using the available GWAS summary statistics along with 40,000 unrelated British individuals from the UKBB as the reference panel, over a 500kb window centered on the signal lead variant. We obatined pairwise r between variants using the cor() function in R on the genotype dosages for variants in the SuSiE window. We ran SuSiE using the following parameters: max number of signals L = 10; coverage = 0.95; r2.prune = 0.8; minimum absolute correlation = 0.1; maximum iterations = 10,000. For studies that provided lead signals (primary and or secondary), we selected the SuSiE credible sets that contained at least one variant with LD r2>0.8 with the lead variant for colocalization downstream. For other studies such as UKBB that did not provide lead signals, we selected all SuSiE detected signals for colocalization. We considered e/ca QTL signals where the lead variant was within 100kb of the GWAS lead variant to test for GWAS-QTL colocalization using the function coloc.susie from the coloc v5 package. We used the coloc sensitivity() function to assess sensitivity of findings to coloc’s priors. We considered two signals to be colocalized if the PP H4 > 0.5.
4.25. Imputing high-resolution 3D chromatin contact maps
We used EPCOT (Z. Zhang et al., 2023) to impute the high-resolution 3D chromatin contact maps. EPCOT is a computational framework that predicts multiple genomic modalities using chromatin accessibility profiles and the reference genome sequence as input. We predicted chromatin contacts in genomic neighborhoods of selected caPeaks of interest using snATAC-seq from the respective cluster - either Micro-C at 1kb resolution for a 500kb genomic region or Hi-C at 5kb resolution for a 1Mb genomic region. EPCOT was trained with existing Micro-C contact maps from H1 and HFF, or Hi-C contact maps from GM12878, H1, and HFF. Both the Micro-C and Hi-C contact maps are O/E normalized (i.e., the contact values present the ratio of the observed contact counts over the expected contact counts).
We then generated Micro-C maps by the genotype at the caSNP of interest. We created genotype-specific snATAC-seq profiles by aggregating samples with either homozygous reference or homozygous alternate genotypes at the caSNP of interest. We downsampled the data using Picard when required to make the two profiles have similar depth. We respectively incorporated the reference of alternate allele in the DNA sequence input to EPCOT. Subsequently, we subtracted the predicted contact values associated with the low chromatin accessibility genotype from the high accessibility genotype.
EPCOT’s input ATAC-seq (bigWig) processing:
bamCoverage –normalizeUsing RPGC –effectiveGenomeSize 2913022398
–Offset 1 –binSize 1 –blackListFileName ENCODE_black_list.bed
4.26. Massively parallel reporter assay for validation
4.26.1. Cloning
We ordered oligos as 230 bp sequences where 197 bp comprise the variant of interest flanked on both by 98 bp of genomic context, and the additional 33 bp are cloning adapters. Within this panel, we included a set of ~50 negative control sequences defined by a previous publication (Vanhille et al., 2015) We added 20 bp barcodes via a 2-step PCR amplification process then incorporated the barcoded oligos into a modified pMPRA1 vector (a gift from Tarjei Mikkelsen (Melnikov et al., 2014), Addgene #49349) upstream of the GFP reporter gene using Golden Gate assembly. After transforming and expanding in NEB 10-beta electrocompetent bacteria, we sequenced this version of the MPRA library to establish a barcode-oligo pairing dictionary. We performed a second Golden Gate assembly step to insert an ENCODE-annotated promoter for the human MYBPC2 gene in between the oligo and barcode. Finally, we used restriction cloning to port the assembled MPRA block (oligo, barcode, promoter, GFP) to a lentiviral transfer vector, which was used by the University of Michigan viral vector core to produce infectious lentiviral particles. Primer sequences used for cloning and sequencing library preparation along with the MYBPC2 promoter sequence are included in a separate table.
4.26.2. MPRA Experiment
For each replicate, we infected 4x106 LHCN-M2 human skeletal myoblasts with our MPRA library at an MOI of ~10. After infection, we passaged the cells for one week to remove any unincorporated virus or contaminating transfer plasmid, then differentiated the cells for one week. We isolated RNA and gDNA from each replicate using the Qiagen AllPrep DNA/RNA mini kit. We reverse transcribed RNA into cDNA with a GFP-specific primer, then constructed indexed sequencing libraries for both the cDNA and gDNA libraries using Illumina-compatible primers.
4.26.3. Data Analysis
After quality checks and filtering, we calculated the sum of barcode counts for each oligo within a replicate. We used DESeq2 v1.34.0 (Love, Huber, and Anders, 2014) to perform normalization and differential expression analysis. We used a nested model to identify oligos with significant activity (relative to plasmid input) and significant allelic bias (between reference and alternate alleles). All results were subject to a Benjamini-Hochberg FDR of 5%.
Supplementary Material
4.28. Acknowledgements
We thank Sarah Graham, Cristen Willer, Sarah Hanks, Marcel den Hoed, and Parker lab members for their help and feedback for this work. This work was supported by grants R01DK072193, UM1DK126185 to KLM, and R01DK117960, UM1DK126185, P30AR069620 to SCJP.
Footnotes
4.27 Data and code availability
The raw data is being submitted to dbGaP as an update over an existing repository with other FUSION molecular data phs001048.v2.p1. All data including full summary statistics and code used to run analyses in this work will be available upon publication.
4.30 Competing interests
SCJP has a research grant from Pfizer.
References
- Ahlmann-Eltze Constantin and Huber Wolfgang (Mar. 2023). Analysis of multi-condition single-cell data with latent embedding multivariate regression. en. Pages: 2023.03.06.531268 Section: New Results. DOI: 10.1101/2023.03.06.531268. [DOI]
- Alasoo Kaur et al. (Jan. 2018). “Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response”. En. In: Nature Genetics, p. 1. issn: 1546-1718. DOI: 10.1038/s41588-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya Haley M., Kundaje Anshul, and Boyle Alan P. (June 2019). “The ENCODE Blacklist: Identification of Problematic Regions of the Genome”. en. In: Scientific Reports 9.1. Number: 1 Publisher: Nature Publishing Group, p. 9354. ISSN: 2045-2322. DOI: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson Courtney M. et al. (Feb. 2015). “Myocyte enhancer factor 2C function in skeletal muscle is required for normal growth and glucose metabolism in mice”. In: Skeletal Muscle 5.1, p. 7. ISSN: 2044-5040. DOI: 10.1186/s13395-015-0031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygün Nil, Elwell Angela L., et al. (Sept. 2021). “Brain-trait-associated variants impact cell-type-specific gene regulation during neurogenesis”. eng. In: American Journal of Human Genetics 108.9, pp. 1647–1668. ISSN: 1537-6605. DOI: 10.1016/j.ajhg.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygün Nil, Liang Dan, et al. (May 2023). “Inferring cell-type-specific causal gene regulatory networks during human neurogenesis”. eng. In: Genome Biology 24.1, p. 130. ISSN: 1474-760X. DOI: 10.1186/s13059-023-02959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira Alvaro N. et al. (Jan. 2021). “Exploiting the GTEx resources to decipher the mechanisms at GWAS loci”. In: Genome Biology 22.1, p. 49. ISSN: 1474-760X. DOI: 10.1186/s13059-020-02252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaglio Paola et al. (June 2023). “Mapping genetic effects on cell type-specific chromatin accessibility and annotating complex immune trait variants using single nucleus ATAC-seq in peripheral blood”. In: PLOS Genetics 19.6, e1010759. ISSN: 1553-7390. DOI: 10.1371/journal.pgen.1010759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshnova Daria A. et al. (July 2014). “Regulation of the Nucleosome Repeat Length In Vivo by the DNA Sequence, Protein Concentrations and Long-Range Interactions”. In: PLoS Computational Biology 10.7, e1003698. ISSN: 1553-734X. DOI: 10.1371/journal.pcbi.1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biferali Beatrice et al. (2019). “Fibro-Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network”. In: Frontiers in Physiology 10. ISSN: 1664-042X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga Melissa et al. (Mar. 2014). “Follistatin promotes adipocyte differentiation, browning, and energy metabolism”. eng. In: Journal of Lipid Research 55.3, pp. 375–384. ISSN: 1539-7262. DOI: 10.1194/jlr.M039719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryois Julien et al. (Apr. 2020). “Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease”. en. In: Nature Genetics. Publisher: Nature Publishing Group, pp. 1–12. ISSN: 1546-1718. DOI: 10.1038/s41588-020-0610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan Brendan K. et al. (Mar. 2015). “LD Score regression distinguishes confounding from polygenicity in genome-wide association studies”. en. In: Nature Genetics 47.3, pp. 291–295. ISSN: 1546-1718. DOI: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Gamez Eddie and Trynka Gosia (2020). “From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases”. eng. In: Frontiers in Genetics 11, p. 424. ISSN: 1664-8021. DOI: 10.3389/fgene.2020.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Ji et al. (June 2021). “The trans-ancestral genomic architecture of glycemic traits”. en. In: Nature Genetics 53.6. Number: 6 Publisher: Nature Publishing Group, pp. 840–860. ISSN: 1546-1718. doi: 10.1038/s41588-021-00852-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek Mete and Lusis Aldons J. (Jan. 2014). “Systems genetics approaches to understand complex traits”. eng. In: Nature Reviews. Genetics 15.1, pp. 34–48. ISSN: 1471-0064. doi: 10.1038/nrg3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, The GTEx (Sept. 2020). “The GTEx Consortium atlas of genetic regulatory effects across human tissues”. en. In: Science 369.6509. Publisher: American Association for the Advancement of Science Section: Research Article, pp. 1318–1330. ISSN: 0036-8075, 1095-9203. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras Osvaldo, Rossi Fabio M. V., and Theret Marine (July 2021). “Origins, potency, and heterogeneity of skeletal muscle fibro-adipogenic progenitors—time for new definitions”. In: Skeletal Muscle 11.1, p. 16. ISSN: 2044-5040. doi: 10.1186/s13395-021-00265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo Anna S. E. et al. (Feb. 2020). “Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression”. en. In: Nature Communications 11.1. Number: 1 Publisher: Nature Publishing Group, p. 810. ISSN: 2041-1723. doi: 10.1038/s41467-020-14457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currin Kevin W. et al. (May 2021). “Genetic effects on liver chromatin accessibility identify disease regulatory variants”. en. In: The American Journal of Human Genetics. ISSN: 0002-9297. doi: 10.1016/j.ajhg.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Oliveira Albanus Ricardo et al. (Feb. 2021). “Chromatin information content landscapes inform transcription factor and DNA interactions”. en. In: Nature Communications 12.1. Number: 1 Publisher: Nature Publishing Group, p. 1307. ISSN: 2041-1723. doi: 10.1038/s41467-021-21534-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Sayantan et al. (2016). “Next-generation genotype imputation service and methods”. eng. In: Nature Genetics 48.10, pp. 1284–1287. ISSN: 1546-1718. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Micheli Andrea J. et al. (July 2020). “A reference single-cell transcriptomic atlas of human skeletal muscle tissue reveals bifurcated muscle stem cell populations”. In: Skeletal Muscle 10.1, p. 19. ISSN: 2044-5040. doi: 10.1186/s13395-020-00236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau Olivier et al. (May 2017). “A complete tool set for molecular QTL discovery and analysis”. En. In: Nature Communications 8, p. 15452. ISSN: 2041-1723. doi: 10.1038/ncomms15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont Nicolas A. et al. (July 2015). “Satellite Cells and Skeletal Muscle Regeneration”. eng. In: Comprehensive Physiology 5.3, pp. 1027–1059. ISSN: 2040-4603. doi: 10.1002/cphy.c140068. [DOI] [PubMed] [Google Scholar]
- Duren Zhana et al. (May 2022). “Regulatory analysis of single cell multiome gene expression and chromatin accessibility data with scREG”. In: Genome Biology 23.1, p. 114. ISSN: 1474-760X. doi: 10.1186/s13059-022-02682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane Hilary K., Bulik-Sullivan Brendan, et al. (Nov. 2015). “Partitioning heritability by functional annotation using genome-wide association summary statistics”. en. In: Nature Genetics 47.11, pp. 1228–1235. ISSN: 1546-1718. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane Hilary K., Reshef Yakir A., et al. (Apr. 2018). “Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types”. en. In: Nature Genetics 50.4, pp. 621–629. ISSN: 1546-1718. doi: 10.1038/s41588-018-0081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish Adam et al. (Jan. 2021). “GENCODE 2021”. In: Nucleic Acids Research 49.D1, pp. D916–D923. ISSN: 0305-1048. doi: 10.1093/nar/gkaa1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontera Walter R. and Ochala Julien (Mar. 2015). “Skeletal Muscle: A Brief Review of Structure and Function”. en. In: Calcified Tissue International 96.3, pp. 183–195. ISSN: 1432-0827. doi: 10.1007/s00223-014-9915-y. [DOI] [PubMed] [Google Scholar]
- Gao Chao et al. (Aug. 2021). “Iterative single-cell multi-omic integration using online learning”. In: Nature biotechnology 39.8, pp. 1000–1007. ISSN: 1087-0156. doi: 10.1038/s41587-021-00867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goveia Jermaine et al. (July 2014). “Endothelial Cell Differentiation by SOX17”. In: Circulation Research 115.2. Publisher: American Heart Association, pp. 205–207. doi: 10.1161/CIRCRESAHA.114.304234. [DOI] [PubMed] [Google Scholar]
- Grant Charles E., Bailey Timothy L., and Noble William Stafford (Apr. 2011). “FIMO: scanning for occurrences of a given motif”. en. In: Bioinformatics 27.7, pp. 1017–1018. ISSN: 1367-4803. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani Gibran, Tilling Kate, and Smith George Davey (Nov. 2017). “Orienting the causal relationship between imprecisely measured traits using GWAS summary data”. In: PLOS Genetics 13.11, e1007081. ISSN: 1553-7404. doi: 10.1371/journal.pgen.1007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton Tiffany N et al. (Nov. 2008). “Excessive Adipose Tissue Infiltration in Skeletal Muscle in Individuals With Obesity, Diabetes Mellitus, and Peripheral Neuropathy: Association With Performance and Function”. In: Physical Therapy 88.11, pp. 1336–1344. ISSN: 0031-9023. doi: 10.2522/ptj.20080079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs A. S. et al. (Jan. 2006). “The UCSC Genome Browser Database: update 2006”. eng. In: Nucleic Acids Research 34.Database issue, pp. D590–598. ISSN: 1362-4962. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari Farhad et al. (Dec. 2016). “Colocalization of GWAS and eQTL Signals Detects Target Genes”. In: The American Journal of Human Genetics 99.6, pp. 1245–1260. ISSN: 0002-9297. doi: 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerber J. et al. (May 2020). “Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation”. en. In: bioRxiv. Publisher: Cold Spring Harbor Laboratory Section: New Results, p. 2020.05.21.103820. DOI: 10.1101/2020.05.21.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Hyun Min et al. (Jan. 2018). “Multiplexed droplet single-cell RNA-sequencing using natural genetic variation”. en. In: Nature Biotechnology 36.1, pp. 89–94. ISSN: 1546-1696. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J. et al. (Sept. 2010). “BigWig and BigBed: enabling browsing of large distributed datasets”. In: Bioinformatics 26.17, pp. 2204–2207. ISSN: 1367-4803. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khetan Shubham et al. (Nov. 2018). “Type 2 Diabetes–Associated Genetic Variants Regulate Chromatin Accessibility in Human Islets”. en. In: Diabetes 67.11, pp. 2466–2477. ISSN: 0012-1797, 1939-327X. DOI: 10.2337/db18-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Gyuri and Kim Jae Hyeon (Mar. 2020). “Impact of Skeletal Muscle Mass on Metabolic Health”. In: Endocrinology and Metabolism 35.1, pp. 1–6. ISSN: 2093-596X. doi: 10.3803/EnM.2020.35.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolka Cathryn M and Bergman Richard N (Mar. 2013). “The Endothelium in Diabetes: its role in insulin access and diabetic complications”. In: Reviews in endocrine & metabolic disorders 14.1, pp. 13–19. ISSN: 1389-9155. DOI: 10.1007/s11154-012-9233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kycia Ina et al. (Apr. 2018). “A Common Type 2 Diabetes Risk Variant Potentiates Activity of an Evolutionarily Conserved Islet Stretch Enhancer and Increases C2CD4A and C2CD4B Expression”. In: The American Journal of Human Genetics 102.4, pp. 620–635. ISSN: 0002-9297. doi: 10.1016/j.ajhg.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal Avantika et al. (Mar. 2021). “Deep learning-based enhancement of epigenomics data with At-acWorks”. en. In: Nature Communications 12.1. Number: 1 Publisher: Nature Publishing Group, p. 1507. ISSN: 2041-1723. DOI: 10.1038/s41467-021-21765-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Heng and Durbin Richard (July 2009). “Fast and accurate short read alignment with Burrows-Wheeler transform”. eng. In: Bioinformatics (Oxford, England) 25.14, pp. 1754–1760. ISSN: 1367-4811. DOI: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Dan et al. (May 2021). “Cell-type-specific effects of genetic variation on chromatin accessibility during human neuronal differentiation”. en. In: Nature Neuroscience. Publisher: Nature Publishing Group, pp. 1–13. ISSN: 1546-1726. DOI: 10.1038/s41593-021-00858-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Menglin et al. (May 2019). “Sox17 is required for endothelial regeneration following inflammation-induced vascular injury”. en. In: Nature Communications 10.1. Number: 1 Publisher: Nature Publishing Group, p. 2126. ISSN: 2041-1723. DOI: 10.1038/s41467-019-10134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Ning et al. (Mar. 2014). “Requirement of MEF2A, C, and D for skeletal muscle regeneration”. In: Proceedings of the National Academy of Sciences 111.11. Publisher: Proceedings of the National Academy of Sciences, pp. 4109–4114. doi: 10.1073/pnas.1401732111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Po-Ru et al. (Nov. 2016). “Reference-based phasing using the Haplotype Reference Consortium panel”. en. In: Nature Genetics 48.11. Number: 11 Publisher: Nature Publishing Group, pp. 1443–1448. ISSN: 1546-1718. DOI: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love Michael I., Huber Wolfgang, and Anders Simon (Dec. 2014). “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2”. In: Genome Biology 15.12, p. 550. ISSN: 1474-760X. DOI: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun Aaron T. L. et al. (Dec. 2019). “EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data”. en. In: Genome Biology 20.1. Number: 1 Publisher: BioMed Central, pp. 1–9. ISSN: 1474-760X. doi: 10.1186/s13059-019-1662-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan Anubha, Spracklen Cassandra N., et al. (May 2022). “Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation”. en. In: Nature Genetics. Publisher: Nature Publishing Group, pp. 1–13. ISSN: 1546-1718. DOI: 10.1038/s41588-022-01058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan Anubha, Taliun Daniel, et al. (Nov. 2018). “Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps”. En. In: Nature Genetics 50.11, p. 1505. ISSN: 1546-1718. DOI: 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba Nana et al. (Apr. 2023). Wnt activity reveals context-specific genetic effects on gene regulation in neural progenitors. en. Pages: 2023.02.07.527357 Section: New Results. DOI: 10.1101/2023.02.07.527357. [DOI]
- Maurano Matthew T. et al. (Sept. 2012). “Systematic Localization of Common Disease-Associated Variation in Regulatory DNA”. In: Science (New York, N.Y.) 337.6099, pp. 1190–1195. ISSN: 0036-8075. DOI: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta Zenobia B. et al. (Aug. 2016). “Changes in the expression of the type 2 diabetes-associated gene VPS13C in the -cell are associated with glucose intolerance in humans and mice”. In: American Journal of Physiology - Endocrinology and Metabolism 311.2, E488–E507. ISSN: 0193-1849. doi: 10.1152/ajpendo.00074.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov Alexandre et al. (Aug. 2014). “Massively Parallel Reporter Assays in Cultured Mammalian Cells”. In: Journal of Visualized Experiments : JoVE 90, p. 51719. ISSN: 1940-087X. doi: 10.3791/51719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millstein Joshua, Chen Gary K., and Breton Carrie V. (Aug. 2016). “cit: hypothesis testing software for mediation analysis in genomic applications”. In: Bioinformatics 32.15, pp. 2364–2365. ISSN: 1367-4803. DOI: 10.1093/bioinformatics/btw135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millstein Joshua, Zhang Bin, et al. (May 2009). “Disentangling molecular relationships with a causal inference test”. In: BMC Genetics 10.1, p. 23. ISSN: 1471-2156. doi: 10.1186/1471-2156-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi Pejman et al. (Oct. 2017). “Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change”. en. In: Genome Research. ISSN: 1088-9051, 1549-5469. doi: 10.1101/gr.216747.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina Thomas, Fabre Paul, and Dumont Nicolas A. (Dec. 2021). “Fibro-adipogenic progenitors in skeletal muscle homeostasis, regeneration and diseases”. eng. In: Open Biology 11.12, p. 210110. ISSN: 2046-2441. DOI: 10.1098/rsob.210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan Jennifer and Partridge Terence (Feb. 2020). “Skeletal muscle in health and disease”. In: Disease Models & Mechanisms 13.2, p. dmm042192. ISSN: 1754-8403. doi: 10.1242/dmm.042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi Hakhamanesh et al. (May 2022). Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. en. Pages: 2022.05.07.491045 Section: New Results. doi: 10.1101/2022.05.07.491045. [DOI]
- Mueckler Mike (May 2001). “Insulin resistance and the disruption of Glut4 trafficking in skeletal muscle”. In: Journal of Clinical Investigation 107.10, pp. 1211–1213. ISSN: 0021-9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musi Nicolas et al. (July 2002). “Metformin Increases AMP-Activated Protein Kinase Activity in Skeletal Muscle of Subjects With Type 2 Diabetes”. In: Diabetes 51.7, pp. 2074–2081. ISSN: 0012-1797. DOI: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- Neavin Drew et al. (Mar. 2021). “Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells”. In: Genome Biology 22.1, p. 76. ISSN: 1474-760X. DOI: 10.1186/s13059-021-02293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard Peter et al. (Dec. 2021). “Human and rat skeletal muscle single-nuclei multi-omic integrative analyses nominate causal cell types, regulatory elements, and SNPs for complex traits”. en. In: Genome Research 31.12, pp. 2258–2275. ISSN: 1088-9051, 1549-5469. doi: 10.1101/gr.268482.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker Stephen C. J. et al. (Oct. 2013). “Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants”. en. In: Proceedings of the National Academy of Sciences 110.44, pp. 17921–17926. ISSN: 0027-8424, 1091-6490. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillon Nicolas J. et al. (Mar. 2013). “Cross-talk between skeletal muscle and immune cells: muscle-derived mediators and metabolic implications”. eng. In: American Journal of Physiology. Endocrinology and Metabolism 304.5, E453–465. ISSN: 1522-1555. DOI: 10.1152/ajpendo.00553.2012. [DOI] [PubMed] [Google Scholar]
- Pliner Hannah A. et al. (Sept. 2018). “Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data”. English. In: Molecular Cell 71.5, 858–871.e8. ISSN: 1097-2765. DOI: 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulit Sara L et al. (Jan. 2019). “Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry”. In: Human Molecular Genetics 28.1, pp. 166–174. ISSN: 0964-6906. DOI: 10.1093/hmg/ddy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang Daniel X. et al. (July 2015). “Motif signatures in stretch enhancers are enriched for disease-associated genetic variants”. In: Epigenetics & Chromatin 8, p. 23. ISSN: 1756-8935. DOI: 10.1186/s13072-015-0015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan Aaron R. and Hall Ira M. (Mar. 2010). “BEDTools: a flexible suite of utilities for comparing genomic features”. en. In: Bioinformatics 26.6, pp. 841–842. ISSN: 1367-4803. DOI: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai Vivek et al. (Dec. 2019). “Single cell ATAC-seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures”. en. In: Molecular Metabolism. ISSN: 2212-8778. DOI: 10.1016/j.molmet.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere Uku et al. (July 2019). “g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update)”. In: Nucleic Acids Research 47.W1, W191–W198. ISSN: 0305-1048. DOI: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazi Ali M. et al. (Mar. 2005). “CSX/Nkx2.5 modulates differentiation of skeletal myoblasts and promotes differentiation into neuronal cells in vitro”. eng. In: The Journal of Biological Chemistry 280.11, pp. 10716–10720. ISSN: 0021-9258. DOI: 10.1074/jbc.M500028200. [DOI] [PubMed] [Google Scholar]
- Robertson Catherine C. et al. (July 2021). “Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes”. en. In: Nature Genetics 53.7, pp. 962–971. ISSN: 1546-1718. DOI: 10.1038/s41588-021-00880-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein Aliza B. et al. (Jan. 2020). “Single-cell transcriptional profiles in human skeletal muscle”. en. In: Scientific Reports 10.1. Number: 1 Publisher: Nature Publishing Group, p. 229. ISSN: 2045-2322. DOI: 10.1038/s41598-019-57110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott Laura J. et al. (June 2016). “The genetic regulatory signature of type 2 diabetes in human skeletal muscle”. en. In: Nature Communications 7, ncomms11764. ISSN: 2041-1723. DOI: 10.1038/ncomms11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh Rajan, Braga Melissa, et al. (Jan. 2017). “Follistatin Targets Distinct Pathways To Promote Brown Adipocyte Characteristics in Brown and White Adipose Tissues”. In: Endocrinology 158.5, pp. 1217–1230. ISSN: 0013-7227. DOI: 10.1210/en.2016-1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh Rajan, Pervin Shehla, et al. (Mar. 2018). “Metabolic profiling of follistatin overexpression: a novel therapeutic strategy for metabolic diseases”. In: Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 11, pp. 65–84. ISSN: 1178-7007. DOI: 10.2147/DMSCI.S159315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskic Blagoje et al. (Dec. 2021). Immune disease risk variants regulate gene expression dynamics during CD4+ T cell activation. en. Tech. rep. Section: New Results Type: article. bioRxiv, p. 2021.12.06.470953. DOI: 10.1101/2021.12.06.470953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spracklen Cassandra N. et al. (June 2020). “Identification of type 2 diabetes loci in 433,540 East Asian individuals”. eng. In: Nature 582.7811, pp. 240–245. ISSN: 1476-4687. DOI: 10.1038/s41586-020-2263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriwijitkamol Apiradee et al. (Mar. 2007). “Effect of Acute Exercise on AMPK Signaling in Skeletal Muscle of Subjects With Type 2 Diabetes: A Time-Course and Dose-Response Study”. In: Diabetes 56.3, pp. 836–848. ISSN: 0012-1797. DOI: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow Cathie et al. (Mar. 2015). “UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age”. en. In: PLOS Medicine 12.3. Publisher: Public Library of Science, e1001779. ISSN: 1549-1676. DOI: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Na et al. (Sept. 2023). “Human microglial state dynamics in Alzheimer’s disease progression”. English. In: Cell 186.20. Publisher: Elsevier, 4386–4403.e29. ISSN: 0092-8674, 1097-4172. DOI: 10.1016/j.cell.2023.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. Leland et al. (May 2019). “Integrative analysis of gene expression, DNA methylation, physiological traits, and genetic variation in human skeletal muscle”. en. In: Proceedings of the National Academy of Sciences 116.22. Publisher: National Academy of Sciences Section: Biological Sciences, pp. 10883–10888. ISSN: 0027-8424, 1091-6490. doi: 10.1073/pnas.1814263116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Weiner Amaro et al. (Dec. 2019). “Scaling computational genomics to millions of individuals with GPUs”. en. In: Genome Biology 20.1. Number: 1 Publisher: BioMed Central, pp. 1–5. ISSN: 1474-760X. doi: 10.1186/s13059-019-1836-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Shuzhi and Huang Ping (Mar. 2019). “The effect of type 2 diabetes mellitus and obesity on muscle progenitor cell function”. In: Stem Cell Research & Therapy 10.1, p. 103. ISSN: 1757-6512. DOI: 10.1186/s13287-019-1186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Roadmap Epigenomics Consortium et al. (Feb. 2015). “Integrative analysis of 111 reference human epigenomes”. en. In: Nature 518.7539, pp. 317–330. ISSN: 1476-4687. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurner Matthias et al. (Feb. 2018). “Integration of human pancreatic islet genomic data refines regulatory mechanisms at Type 2 Diabetes susceptibility loci”. en. In: eLife 7, e31977. ISSN: 2050-084X. DOI: 10.7554/eLife.31977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers Mary E. et al. (Mar. 2013). “Insights into the molecular mechanism for type 2 diabetes susceptibility at the KCNQ1 locus from temporal changes in imprinting status in human islets”. eng. In: Diabetes 62.3, pp. 987–992. ISSN: 1939-327X. doi: 10.2337/db12-0819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner Adam W. et al. (May 2022). “Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk”. en. In: Nature Genetics. Publisher: Nature Publishing Group, pp. 1–13. ISSN: 1546-1718. DOI: 10.1038/s41588-022-01069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umans Benjamin D., Battle Alexis, and Gilad Yoav (Feb. 2021). “Where are the disease-associated eQTLs?” In: Trends in genetics : TIG 37.2, pp. 109–124. ISSN: 0168-9525. doi: 10.1016/j.tig.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhille Laurent et al. (Apr. 2015). “High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq”. In: Nature Communications 6, p. 6905. [DOI] [PubMed] [Google Scholar]
- Varshney Arushi, Kyono Yasuhiro, et al. (July 2021). “A Transcription Start Site Map in Human Pancreatic Islets Reveals Functional Regulatory Signatures”. en. In: Diabetes 70.7, pp. 1581–1591. ISSN: 0012-1797, 1939-327X. DOI: 10.2337/db20-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney Arushi, Scott Laura J., et al. (Feb. 2017). “Genetic regulatory signatures underlying islet gene expression and type 2 diabetes”. en. In: Proceedings of the National Academy of Sciences 114.9, pp. 2301–2306. ISSN: 0027-8424, 1091-6490. DOI: 10.1073/pnas.1621192114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney Arushi, VanRenterghem Hadley, et al. (Feb. 2019). “Cell Specificity of Human Regulatory Annotations and Their Genetic Effects on Gene Expression”. en. In: Genetics 211.2, pp. 549–562. ISSN: 0016-6731, 1943-2631. DOI: 10.1534/genetics.118.301525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñuela Ana et al. (Sept. 2020). “Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D”. en. In: Nature Communications 11.1. Number: 1 Publisher: Nature Publishing Group, p. 4912. ISSN: 2041-1723. doi: 10.1038/s41467-020-18581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace Chris (Sept. 2021). “A more accurate method for colocalisation analysis allowing for multiple causal variants”. en. In: PLOS Genetics 17.9. Publisher: Public Library of Science, e1009440. ISSN: 1553-7404. DOI: 10.1371/journal.pgen.1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Gao et al. (Dec. 2020). “A simple new approach to variable selection in regression, with application to genetic fine mapping”. en. In: Journal of the Royal Statistical Society: Series B (Statistical Methodology) 82.5, pp. 1273–1300. ISSN: 1369-7412, 1467-9868. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Jianhua et al. (Jan. 2020). “CAUSALdb: a database for disease/trait causal variants identified using summary statistics of genome-wide association studies”. en. In: Nucleic Acids Research 48.D1. Publisher: Oxford Academic, pp. D807–D816. ISSN: 0305-1048. doi: 10.1093/nar/gkz1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch Joshua D. et al. (June 2019). “Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity”. In: Cell 177.7, 1873–1887.e17. ISSN: 0092-8674. doi: 10.1016/j.cell.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijst Monique G. P. van der et al. (Apr. 2018). “Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs”. en. In: Nature Genetics 50.4, pp. 493–497. ISSN: 1546-1718. DOI: 10.1038/s41588-018-0089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson Alice et al. (June 2023). “Genome-wide association study and functional characterization identifies candidate genes for insulin-stimulated glucose uptake”. en. In: Nature Genetics 55.6. Number: 6 Publisher: Nature Publishing Group, pp. 973–983. ISSN: 1546-1718. doi: 10.1038/s41588-023-01408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Xushen et al. (Sept. 2023). “Epigenomic dissection of Alzheimer’s disease pinpoints causal variants and reveals epigenome erosion”. English. In: Cell 186.20. Publisher: Elsevier, 4422–4437.e21. ISSN: 0092-8674, 1097-4172. DOI: 10.1016/j.cell.2023.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai Itai et al. (Mar. 2005). “Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification”. eng. In: Bioinformatics (Oxford, England) 21.5, pp. 650–659. ISSN: 1367-4803. DOI: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yang Shiyi et al. (Dec. 2020). “Decontamination of ambient RNA in single-cell RNA-seq with De-contX”. en. In: Genome Biology 21.1. Number: 1 Publisher: BioMed Central, pp. 1–15. ISSN: 1474-760X. DOI: 10.1186/s13059-020-1950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Yucheng, Yao Jiayi, and Boström Kristina I. (Mar. 2019). “SOX Transcription Factors in Endothelial Differentiation and Endothelial-Mesenchymal Transitions”. In: Frontiers in Cardiovascular Medicine 6, p. 30. ISSN: 2297-055X. DOI: 10.3389/fcvm.2019.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazar Seyhan et al. (2022). “Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease”. In: Science 376.6589 (). Publisher: American Association for the Advancement of Science, eabf3041. doi: 10.1126/science.abf3041. [DOI] [PubMed] [Google Scholar]
- Yengo Loic et al. (Oct. 2018). “Meta-analysis of genome-wide association studies for height and body mass index in 700000 individuals of European ancestry”. In: Human Molecular Genetics 27.20, pp. 3641–3649. ISSN: 0964-6906. DOI: 10.1093/hmg/ddy271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Kai et al. (Nov. 2021). “A single-cell atlas of chromatin accessibility in the human genome”. English. In: Cell 184.24. Publisher: Elsevier, 5985–6001.e19. ISSN: 0092-8674, 1097-4172. doi: 10.1016/j.cell.2021.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Yong et al. (Sept. 2008). “Model-based Analysis of ChIP-Seq (MACS)”. In: Genome Biology 9.9, R137. ISSN: 1474-760X. DOI: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Zhenhao et al. (July 2023). “A generalizable framework to comprehensively predict epigenome, chromatin organization, and transcriptome”. eng. In: Nucleic Acids Research 51.12, pp. 5931–5947. ISSN: 1362-4962. DOI: 10.1093/nar/gkad436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Hui et al. (Sept. 2017). “Follistatin N terminus differentially regulates muscle size and fat in vivo”. en. In: Experimental & Molecular Medicine 49.9. Number: 9 Publisher: Nature Publishing Group, e377–e377. ISSN: 2092-6413. doi: 10.1038/emm.2017.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.