

Abstract
Influenza infection and vaccination impart strain-specific immunity that protects against neither seasonal antigenic variants nor the next pandemic. However, antibodies directed to conserved sites can confer broad protection. Here we identify and characterize a class of human antibodies that engage a previously undescribed, conserved epitope on the influenza hemagglutinin (HA) protein. Prototype antibody S8V1–157 binds at the normally occluded interface between the HA head and stem. Antibodies to this HA head-stem interface epitope are non-neutralizing in vitro but protect against lethal influenza infection in mice. Antibody isotypes that direct clearance of infected cells enhance this protection. Head-stem interface antibodies bind to most influenza A serotypes and seasonal human variants, and are present at low frequencies in the memory B cell populations of multiple human donors. Vaccines designed to elicit these antibodies might contribute to “universal” influenza immunity.
INTRODUCTION:
Influenza pandemics arise from antigenically novel zoonotic influenza A viruses transmitted to humans from animals. Historically, pandemic viruses generally have become endemic and have continued to circulate as seasonal viruses. Sustained viral circulation is enabled by on-going antigenic evolution that leads to escape from population-level immunity elicited by previous exposures 1. Although antibodies provide the strongest protection against infection, they also drive the antigenic evolution of the viral surface proteins 1. Therefore, neither infection nor the seasonal flu vaccine confers enduring immunity against future seasonal variants or new pandemic viruses. Nevertheless, broadly protective monoclonal antibodies that engage conserved influenza sites have been isolated from human donors 2. Passive transfer of these antibodies to animal models imparts broadly protective immunity. A next generation influenza vaccine that elicits similar antibodies would likely confer more durable protection than that offered by current seasonal vaccines 3–5.
The influenza hemagglutinin (HA) protein is the major target of protective antibodies 6. HA facilitates cell entry by attaching to cells via an interaction with its receptor, sialic acid, and by acting as a virus-cell membrane fusogen. HA is synthesized as a polyprotein, HA0, which forms homotrimers that are incapable of undergoing the full series of conformational rearrangements required for membrane fusion. Cellular proteases (often resident on the target cell) cleave the HA0 into HA1 and HA2 domains, resulting in a fusion-competent trimer 7–9. HA1 includes the globular HA head that contains the receptor binding site (RBS), while HA2 contains the helical stem regions that rearrange during endosomal acidification to drive fusion of viral and cellular membranes. The requirement for HA to transit through multiple conformations during fusion likely accounts for the intrinsic propensity of HA0 or HA1-HA2 to transiently explore states that deviate from its prefusion conformation, which reproducibly forms ordered protein crystal lattices that yield high-resolution structures determined by X-ray crystallography10–13.
Large genetic and antigenic differences separate influenza A HA serotypes, which are classified into group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16) and group 2 (H3, H4, H7, H10, H14, H15). Serotypes of a second glycoprotein, neuraminidase (NA), are denoted by a similar convention; consequently, influenza viruses are named by their HA and NA content (e.g., H2N2, H10N8). Currently, divergent H1N1 and H3N2 viruses circulate as seasonal human influenza viruses. Humans can produce antibodies that engage surfaces conserved between serotypes and thereby provide cross-serotype protection. These epitopic regions include the HA RBS, stem, anchor, head interface, the so-called "long alpha-helix", and the HA2 β-hairpin 2,14–19.
Here, we report a class of human antibodies directed to a previously unreported, widely conserved site at the base, or neck, of the HA head. In the “resting state” prefusion structure of the HA trimer, the epitope for prototype antibody S8V1–157 is occluded at the interface of the head and stem. Biochemical, cellular, and in vivo passive transfer experiments indicate that this HA head-stem interface epitope is sufficiently exposed to allow antibodies to bind and confer strong protection against lethal influenza virus infection in murine challenge models. Many humans harbor antibodies recognizing this epitope; some of these antibodies bind HAs from divergent influenza serotypes, groups, and >50 years of human seasonal virus antigenic variation. Immunogens that elicit such antibodies might be included in influenza vaccines intended to confer broader protection.
RESULTS:
A class of antibodies engages the interface of the HA head and stem regions
By culturing individual human memory B (Bmem) cells and then screening the culture supernatants containing secreted IgGs, we identified (as reported previously 20) 449 HA-reactive antibodies from four donors (S1, S5, S8 and S9). These antibodies represent the Bmem cells circulating in the blood at the time the donors were immunized with the TIV 2015–2016 seasonal influenza vaccine (visit 1; V1), or 7 days later (visit 2; V2). From donors S1 and S8, we found four IgGs (S1V2–17, S1V2–60, S1V2–65 and S8V1–157) that had a novel pattern of HA reactivity (Figure 1A). All bound seasonal H1s and H3s and an H5 HA; they also bound recombinant HA constructs comprising only the HA head, indicating the IgGs did not target stem epitopes (Figure 1A). These four antibodies competed with each other for HA-binding, implying overlapping epitopes; however, the four antibodies did not compete with other well-characterized antibodies that engage conserved, structure-verified, broadly protective epitopes spanning the HA molecule (Figure 1B).
Figure 1: Identification of a novel, broadly binding, HA head-directed antibody class.

A. Luminex screening of Bmem-cell Nojima culture supernatants identified four antibodies that broadly react with influenza A HA FLsEs and heads. The mean fluorescence intensity (MFI) values are colored according to the key. B. In a Luminex competitive binding assay, the four antibodies from (A) that share a pattern of reactivity did not compete with antibodies that engage known HA epitopes, but compete with each other for HA binding. Structures of Fab-HA complexes were aligned on an HA trimer from A/American black duck/New Brunswick/00464/2010(H4N6) (PDB: 5XL2)21. Fab structures include HC1932 (PDB 2VIR), S5V2–2920 (PDB 6E4X), HC4557 (PDB 1QFU), CR911458 (PDB 4FQY), CR802059 (PDB 3SDY) and FI6v347 (PDB 3ZTJ). SARS-CoV antibody CR302233 was used as an HA non-binding control. C. The cross-competing HA antibodies share genetic signatures.
All four new antibodies have light chains encoded by IGKV2–28*01 or IGKV2D-28*01 (Figure 1C), with identical germline coding sequences. The heavy chains are encoded by either IGHV1–24*01 or IGHV5–51*01 and have short (11–13 amino acid) third complementarity-determining regions (HCDR3s). S1V2–60 and S1V2–65 are clonally related, while S1V2–17 and S8V1–157 share a common IGHV1–24*01-IGKV2–28*01 pairing, despite their derivation from different donors.
We determined the structure of S8V1–157 complexed with a monomeric HA head construct. Crystals were only obtained using an A/American black duck/New Brunswick/00464/2010(H4N6) (H4-NB-2010) HA (Figure 2A and Table S1). S8V1–157 engages an epitope at the base of the head, just where it faces the stem 21. In the context of trimeric, full-length, prefusion HA, this epitope is occluded by the association of HA1 with HA2 (Figure 2A). Therefore, displacement of the HA1 head from the HA2 helical stem would be required for B cells or antibodies to recognize this site.
Figure 2: Human antibodies engage a recessed surface at the head-stem interface of the influenza HA molecule.
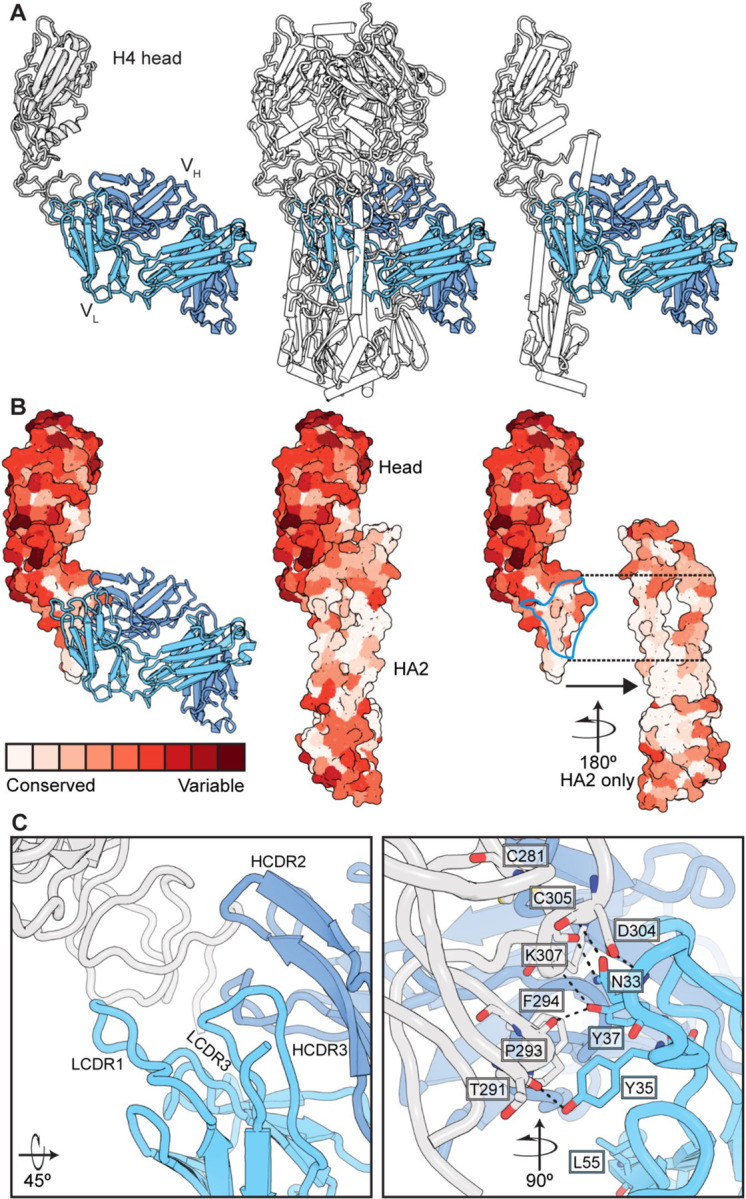
A. Structure of antibody S8V1–157 complexed with the HA head of A/American black duck/New Brunswick/00464/2010(H4N6) colored in gray. The heavy chain is colored darker blue and the light chain is lighter blue. Engagement of this site is incompatible with the defined prefusion H4 HA trimer21, colored in white (PDB: 5XL2) or with individual HA monomers. B. A surface projection showing the degree of amino acid conservation among HAs engaged by this antibody class (see Figure 3). The head-stem epitope is circumscribed in blue in the rightmost panel. Conservation scores were produced using ConSurf 60,61. C. Key S8V1–157 contacts. The orientation relative to panel A is indicated.
S8V1–157 contacts residues mediating the intra-protomer interaction, which are conserved across divergent HAs (Figures 2B and S1). Evolutionary constraints imposed by the requirement that HA1 and HA2 stably associate at neutral pH likely account for the conservation of the head-stem interface epitope, and by extension, the binding breadth of the corresponding antibodies.
The compact HCDR3 of S8V1–157 packs against the heavy chain CDR and framework regions (FR), HCDR1-FR1 and FR2-HCDR2, to produce a cleft between the heavy and light chains that accommodates the head-stem interface epitope (Figure 2C). Light chain CDR1 residues N33, Y35 and Y37 make extensive contacts with a conserved HA surface. The δ-amide of N33 donates and receives hydrogen bonds from the main chain of HA residue HA-C305. Its main chain amide also donates a hydrogen bond to the carboxyl group of HA-D304. Y35 donates a hydrogen bond to the main chain carbonyl of HA-T291 and participates in van der Waals interactions by stacking upon HA-P293. Y37 receives a hydrogen bond from the main chain residue HA-K307, donates a hydrogen bond to HA-P293, and participates in van der Waals contacts with the aryl group of HA-F294 and the aliphatic side chain of HA-K307.
The structure of the S8V1–157-HA complex explains the genetic features common to this class of head-stem interface antibodies. The LCDR1 NXYXY motif is germline-encoded by a small subset of IGKV-genes. Of these, only IGKV2–28/IGKV2D-28a also encode a hydrophobic residue at position 55. L55 participates in van der Waals contacts with HA-P293 (Figure 2C) and contributes to a hydrophobic surface on S8V1–157 that complements a hydrophobic patch within its epitope on the HA head (Figure S2). Polar residues at position 55 in otherwise similar light chains are predicted to be less favorable in this local environment. Similar sequence features are not present in the IGLV locus. Additionally, the short HCDR3 (common among these antibodies) creates shallow cavity that accommodates the hydrophobic surfaces that would normally pack against HA2 in the prefusion HA trimer.
Antibodies to the HA head-stem interface epitope are broadly binding.
We determined the binding of head-stem interface antibodies to divergent HA serotypes using recombinantly expressed soluble HA ectodomains and recombinantly expressed antibodies in enzyme-linked immunosorbent assays (ELISA). All HAs except H5 and H7 were expressed as HA0, which is incapable of transitioning to the postfusion conformation without first being processed by a trypsin-like protease8,22. H5 and H7 HAs contain polybasic cleavage sites that are processed by endogenous furin-like proteases in the expressing cell line, resulting in HA1-HA223–28.
The HA head-stem interface antibodies had very similar breadth: each bound all seasonal H1s (1977–2019) and H3s (1968–2020) assayed, and also bound several other HA subtypes within groups 1 and 2 (Figures 3A and S3). Typically, if an HA was bound by one head-stem epitope antibody, it was also bound by the other three (Figure 3). Affinities and breadth of binding were generally higher for group 2 HAs but extended to group 1 HAs, including seasonal H1, pandemic H2, and pre-pandemic H5 HAs. Overall, these antibodies engaged HAs from 11 of the 16 non-bat influenza A HA serotypes. Failure to engage specific HAs was not due to epitope inaccessibility, since the antibodies also did not bind matched, soluble, HA head that present the HA head-stem epitope without steric hindrance (Figures S4 and S5). None of the head-stem interface antibodies bound a truncated HA head lacking the S8V1–157 epitope. All four antibodies likely engage a common, discrete epitope comprising the terminus of the HA head.
Figure 3: Breadth of HA binding by HA head-stem epitope antibodies.

A. Equilibrium dissociation constants (Kd), determined by ELISA. Broadly binding influenza A HA antibody FI6v3 47 and influenza B HA antibody CR8071 58 served as binding controls. B. Phylogenetic relationships of HAs used in our panel. HAs bound by HA head-stem epitope antibodies are indicated. Binding data from Figure S4 are incorporated into panel B.
In a complementary flow cytometry assay (in the absence of exogenous trypsin19,29), HA head-stem epitope antibodies bound divergent HAs stably expressed in their native, transmembrane form on the cell surface (Figure 4). The binding pattern was consistent with our ELISA data. We used a curated panel of antibodies (largely overlapping with those in Figure 1), including K03.1230 and H5.331 which recognize the fully exposed receptor binding site. HA head-stem interface antibodies generally labeled cells less brightly than did control antibodies directed to solvent-exposed epitopes on the HA head (e.g., K03.12, H5.3, HC19); however, head-stem interface antibodies labeled cells comparably to the HA head-interface antibody S5V2–29 20. Together, these observations suggest that HA interface epitopes are not occupied as fully by antibody as are fully exposed epitopes. Epitope accessibility and/or differences in the number of IgG molecules bound per HA trimer may account for these differences.
Figure 4: HA head-stem epitope antibodies bind cell surface-anchored HA.

Flow cytometry histograms depict the fluorescence intensities of recombinant IgG binding to K530 cell lines expressing recombinant, native HA on the cell surface. K530 cells were labeled with 400 ng/ml of the four head-stem epitope antibodies or control antibodies targeting the HA receptor binding site (HC1932, K03.1230 and H5.331), the head interface (S5V2–2920), a lateral head epitope (HC45)57, stem (FI6v347), or SARS-CoV spike protein (CR302233).
To determine whether head-stem interface antibodies can engage native HA0 on the cell surface, 293F cells were transiently transfected with an expression vector encoding full-length, membrane-anchored A/Aichi/02/1968(H3N2)(X-31) HA. Western blotting analysis using an antibody against a linear epitope contained within HA1 confirmed that the transfected cells expressed only unprocessed, 83 kD HA0 (Fig. S5B). Processed HA1, which under reducing and denaturing conditions would be observed as a 55 kD band, was not detected. Flow cytometry analysis showed that S8V1–157 labeled the transfected cells (Fig. S5C), as did antibodies recognizing the RBS (HC19) and head interface epitope (S5V2–29). The head-stem interface epitope is therefore exposed, if only transiently, on native HA0 expressed on the cell surface.
Antibodies to the HA head-stem epitope protect against lethal influenza virus infection.
S8V1–157 failed to neutralize influenza virus in vitro, in an assay in which virus and antibody were co-incubated, added to cells, and neutralization scored four days post-infection (Figure S6). To determine if head-stem interface antibodies confer protection in vivo, we produced the antibodies as recombinant mouse IgG1 or IgG2c, then passively transferred the antibodies to mice and challenged them with a lethal dose of influenza virus. In mice, the IgG2c isotype potently directs protective, Fc-dependent, effector functions, including antibody-dependent cellular cytotoxicity (ADCC) and complement deposition (ADCD). In contrast, the IgG1 isotype does not appreciably stimulate these effector functions. Each mouse received 150 μg (~7.5 mg/kg) of the head-stem interface antibodies S8V1–157 or S5V2–65, HC1932 (which potently neutralizes A/Aichi/02/1968(H3N2)(X-31)), S5V2–2920 (an HA-head interface antibody that protects against lethal infection by Fc-dependent mechanisms), or CR302233 (SARS-CoV antibody). The ~7.5 mg/kg antibody dose is comparable or below commonly administered doses of antiviral monoclonal antibody therapeutics33–38.
HC19 protected mice from infection-induced weight loss, while the irrelevant antibody CR3022 offered no protection, requiring recipient mice to be ethically euthanized by day 10 post-infection (Figure 5A). All animals administered HA head-stem interface antibodies S8V1–157 or S1V2–65 recovered after experiencing mild to moderate weight loss; by day 10 post-infection, their body weights were similar to the HC19-treated group. IgG2c versions of S8V1–157 and S1V2–65 conferred slightly better protection than the corresponding IgG1s, indicating that Fc effector functions had a role in controlling the infection (Figure 5B-D). Similarly, head interface antibody S5V2–29 protected better against infection-induced weight loss when infused as an IgG2c. Thus, despite failing to neutralize the initial infection, HA head-stem interface antibodies protected influenza-infected mice against severe disease and mortality.
Figure 5: HA head-stem epitope antibodies protect against lethal influenza virus infection and severe disease.

C57BL/6 mice (n = 7 per group) were intraperitoneally injected with 150 μg of recombinant antibody via intraperitoneal injection three hours prior to intranasal challenge with 5xLD50 of A/Aichi/02/1968(H3N2)(X31). Mice were weighed daily and euthanized at a humane endpoint of 25% loss of body weight. Antibodies passively transferred included musinized IgG1 and IgG2c versions of HA head-stem epitope antibodies S8V1–157 and S1V2–65, neutralizing antibody HC1932, head interface antibody S5V2–2920 and SARS-CoV antibody CR302233. Mice injected with PBS were included as an additional control. A. Post-infection survival rate. B-D. Body weight curves for infected mice administered S8V1–157 (B), S1V2–65 (C), or S5V2–29 (D) antibodies, compared with controls. *p < 0.05 and ***p < 0.001 compared with isotype control CR3022. Not significant (n.s.), p 0.05; † p < 0.05, †† p < 0.01, and †††† p < 0.0001 IgG2c compared with IgG1.
The HA head-stem epitope is immunogenic in humans.
We screened additional human Bmem cell cultures for S8V1–157-competing antibodies (Figure 6). The samples were taken from seven donors, including the same subjects as before (S1, S5, S8, S9), but vaccinated and sampled during subsequent flu seasons (see Materials and Methods for details); subject S12, who was immunized and sampled at the same times; and subjects KEL01 and KEL03, who were sampled after receiving TIV in 2014–2015. From 528 clonal cultures that produced HA-binding IgG, we identified eight additional supernatants that inhibited S8V1–157 binding to HA by >90%. (Figure 6A). In a screen of antibody reactivity, five of the eight competing antibodies reacted with both group 1 and group 2 HAs.
Figure 6: The HA head-stem epitope epitope is immunogenic in humans.

A. An additional 528 Nojima culture supernatants from donors K01, K03, S1, S5, S8, S9 and S12 were screened for competition with a recombinant musinized S8V1–157 IgG1 for HA binding. Culture supernatants that inhibited S8V1–157 binding by >90% are colored and specified. B. HA reactivity of S8V1–157-competing Nojima culture supernatants, as determined by multiplex Luminex assay. N.D.: not determined.
Paired heavy and light chain sequences were recovered from seven of the eight S8V1–157-competing antibodies (Figure S7). Two antibodies, S1V4-P4-C5 and S12V6-P7-H4, use an IGKV2–28 light chain paired with IGHV1–24*01, like S8V1–157 and S1V2–17. All four antibodies have short, 11–12 amino acid HCDR3s. These features, shared with S8V1–157, likely define a public antibody class present in at least three human donors. The other newly identified S8V1–157 competitors use other IGHV and IGKV genes and have varying HCDR3 lengths (10–19 amino acids). These antibodies could either have footprints that overlap the head-stem interface epitope or that prevent its exposure.
DISCUSSION:
We identified and characterized a human antibody response directed to a previously unrecognized, widely conserved, cryptic epitope on the influenza HA head. These antibodies bind broadly to divergent HA serotypes found in animals and seasonal antigenic variants of human viruses. Prophylactic passive transfer of these antibodies to mice protects against lethal influenza virus disease. Approximately 1% of circulating, HA-reactive Bmem recognize the head-stem interface and are present in multiple donors both before and after seasonal influenza vaccination, though most such Bmem were isolated following vaccination. Head-stem interface antibodies have a bias, but not a restriction, for IGKV2–28 usage. Across the 12 known examples we find no additional constraints. Other humans are therefore likely to produce similar antibodies and to have the capacity to mount polyclonal, broadly protective antibody responses.
That HA head-stem interface antibodies confer robust protection against lethal influenza challenge demonstrates that the epitope is exposed on infected cells and/or virions, if only transiently. Exposure of the HA head-stem interface would be predicted to require a large-scale displacement of the HA head from HA2, and additional conformational rearrangements within the HA trimer might accompany such events. Nevertheless, our in vitro experiments, which used recombinant HA0 (either as soluble ectodomain or native, membrane-anchored HA on the surface of transiently-transfected cells), confirmed that head-stem interface antibodies must be able to recognize their epitope on some conformation of prefusion HA, because HA0 cannot transition to a stable postfusion structure8. Structural, biophysical, and computational approaches indicate that HA trimers transiently adopt conformations that expose epitopes normally occluded in the defined prefusion state10–13. Such transient fluctuations might explain how B cells and antibodies recognize ordinarily occluded epitopes16,17,20,39–41. In addition to binding HA0, head-stem interface antibodies might also directly recognize postfusion HA on the surface of infected cells, as has been proposed for some HA antibodies, including LAH3117, S1V2–7219 and m82641. It is plausible that, in vivo, HA0 is cleaved into HA1/HA2 by proteases and that some fraction of postfusion HA containing the exposed head-stem interface epitope accumulates on infected cell-surfaces and/or among the HAs presented to B cells in germinal centers.
Humans and mice mount antibody responses to several apparently occluded HA epitopes 14–17,19,20,39–43. Prevalence of such antibodies in immune repertoires indicates that these epitopes are immunogenic. That several of the human head-stem interface antibodies reported here had 5–11% IGHV gene mutation frequencies implies that the epitope was encountered repeatedly over recurrent influenza exposures (whether by infection or vaccination). The immunogenicity of the HA head-stem epitope poses a critical question for vaccinology: how does the antigen presented to a B cell in a germinal center reaction relate to its form on infectious virions or to what has been defined in the laboratory in considerable biochemical and structural detail? The existence of antibodies to interface/buried epitopes on other, unrelated viral glycoproteins demonstrates that our lack of understanding extends beyond HA44.
Several of the most conserved epitopes on HA are occluded. Adjacent protomers and/or membranes hinder access to some stem/anchor epitopes, and interface epitopes are normally hidden 2,14,15,19,45. Nevertheless, antibodies recognizing these epitopes protect against lethal influenza virus disease in small animal models. These antibodies are typically less potently neutralizing than antibodies targeting fully exposed sites, and their protection is often enhanced by or fully dependent on Fc-mediated effector functions 16,17,19,20,40,43,46,47. Their resistance to human influenza antigenic evolution and ability to engage emerging, pre-pandemic, and other animal viruses make their selective elicitation a potential strategy to improve current influenza vaccines. Given the characteristics of head-stem interface antibodies, a rigorous understanding of their potential to enhance human protective immunity and/or ameliorate disease is needed.
METHODS:
Human subjects.
Peripheral blood mononuclear cells (PBMCs) were obtained from human donors KEL01 (male, age 39) and KEL03 (female, age 39), under Duke Institutional Review Board committee guidelines. Written informed consent was obtained from both subjects. KEL01 and KEL03 received the trivalent inactivated seasonal influenza vaccine (TIV) 2014–2015 Fluvirin, which contained A/Christchurch/16/2010, NIB-74 (H1N1), A/Texas/50/2012, NYMC X-223 (H3N2), and B/Massachusetts/2/2012, NYMC BX-51B. Blood was drawn on day 14 post-vaccination, and PBMCs isolated by centrifugation over Ficoll density gradients (SepMate-50 tubes, StemCell Tech) were frozen and kept in liquid nitrogen until use.
PBMCs were also obtained from human donors S1 (female, age 51–55), S5 (male, age 21–25), S8 (female, age 26–30), S9 (female, age 51–55), and S12 (male, age 35–40), under Boston University Institutional Review Board committee guidelines. Written informed consent was obtained from all five subjects. Donors met all of the following inclusion criteria: between 18 and 65 years of age; in good health, as determined by vital signs [heart rate (<100 bpm), blood pressure (systolic ≤ 140 mm Hg and ≥ 90 mm Hg, diastolic ≤ 90 mm Hg), oral temperature (<100.0 °F)] and medical history to ensure existing medical diagnoses/conditions are not clinically significant; can understand and comply with study procedures, and; provided written informed consent prior to initiation of the study. Exclusion criteria included: 1) life-threating allergies, including an allergy to eggs; 2) have ever had a severe reaction after influenza vaccination; 3) a history of Guillain-Barre Syndrome; 4) a history of receiving immunoglobulin or other blood product within the 3 months prior to vaccination in this study; 5) received an experimental agent (vaccine, drug, biologic, device, blood product, or medication) within 1 month prior to vaccination in this study or expect to receive an experimental agent during this study; 6) have received any live licensed vaccines within 4 weeks or inactivated licensed vaccines within 2 weeks prior to the vaccination in this study or plan receipt of such vaccines within 2 weeks following the vaccination; 7) have an acute or chronic medical condition that might render vaccination unsafe, or interfere with the evaluation of humoral responses (includes, but is not limited to, known cardiac disease, chronic liver disease, significant renal disease, unstable or progressive neurological disorders, diabetes mellitus, autoimmune disorders and transplant recipients); 8) have an acute illness, including an oral temperature greater than 99.9°F, within 1 week of vaccination; 9) active HIV, hepatitis B, or hepatitis C infection; 10) a history of alcohol or drug abuse in the last 5 years; 11) a history of a coagulation disorder or receiving medications that affect coagulation. Subjects S1, S5, S8, S9, and S12 received seasonal influenza vaccination during three consecutive North American flu seasons (2015–2016, 2016–2017, 2017–2018), and had blood drawn on day 0 (pre-vaccination; visits 1, 3, and 5) and day 7 (post-vaccination, visits 2, 4, and 6) each year. During the 2015–2016 season (visits 1 and 2), the subjects received the TIV Fluvirin, which contained A/reassortant/NYMC X-181 (California/07/2009 x NYMC X-157) (H1N1), A/South Australia/55/2014 IVR-175 (H3N2), and B/Phuket/3073/2013. During the 2016–2017 season (visits 3 and 4), the subjects received the quadrivalent inactivated vaccine Flucelvax, containing A/Brisbane/10/2010 (H1N1), A/Hong Kong /4801/2014 (H3N2), B/Utah/9/2014, and B/Hong Kong/259/2010. During the 2017–2018 season (visits 5 and 6), the subjects received the quadrivalent inactivated vaccine Flucelvax, containing A/Singapore/GP1908/2015 IVR-180 (H1N1), A/Singapore/GP2050/2015 (H3N2), B/Utah/9/2014, and B/Hong Kong/259/2010.
Cell lines.
Human 293F cells were maintained at 37°C with 5–8% CO2 in FreeStyle 293 Expression Medium (ThermoFisher) supplemented with penicillin and streptomycin. HA-expressing K530 cell lines 29 (Homo sapiens) were cultured at 37°C with 5% CO2 in RPMI-1640 medium plus 10% FBS (Cytiva), 2-mercaptoethanol (55 μM; Gibco), penicillin, streptomycin, HEPES (10 mM; Gibco), sodium pyruvate (1 mM; Gibco), and MEM nonessential amino acids (Gibco). Madin-Darby canine kidney (MDCK) were maintained in Minimum Essential medium supplemented with 10% fetal bovine serum, 5 mM L-glutamine and 5 mM penicillin/streptomycin.
Recombinant Fab expression and purification.
The heavy and light chain variable domain genes for Fabs were cloned into a modified pVRC8400 expression vector, as previously described48–50. Fab fragments used in crystallization were produced with a noncleavable 6xhistidine (6xHis) tag on the heavy chain C-terminus. Fab fragments were produced by polyethylenimine (PEI) facilitated, transient transfection of 293F cells. Transfection complexes were prepared in Opti-MEM (Gibco) and added to cells. 5 days post transfection, cell supernatants were harvested and clarified by low-speed centrifugation. Fabs were purified by passage over TALON Metal Affinity Resin (Takara) followed by gel filtration chromatography on Superdex 200 (GE Healthcare) in 10 mM tris(hydroxymethyl)aminomethane (tris), 150 mM NaCl at pH 7.5 (buffer A).
Single B cell Nojima cultures.
Nojima cultures were previously performed18–20. Briefly, peripheral blood mononuclear cells (PBMCs) were obtained from four human subjects S1 (female, age range 51–55), S5 (male, age 21–25), S8 (female, age 26–30), and S9 (female, age 51–55). Single human Bmem cells were directly sorted into each well of 96-well plates and cultured with MS40L-low feeder cells in RPMI1640 (Invitrogen) containing 10% HyClone FBS (Thermo scientific), 2-mercaptoethanol (55 μM), penicillin (100 units/ml), streptomycin (100 μg/ml), HEPES (10 mM), sodium pyruvate (1 mM), and MEM nonessential amino acid (1X; all Invitrogen). Exogenous recombinant human IL-2 (50 ng/ml), IL-4 (10 ng/ml), IL-21 (10 ng/ml) and BAFF (10 ng/ml; all Peprotech) were added to cultures. Cultures were maintained at 37° degrees Celsius with 5% CO2. Half of the culture medium was replaced twice weekly with fresh medium (with fresh cytokines). Rearranged V(D)J gene sequences for human Bmem cells from single-cell cultures were obtained as described20,48,51. Specificity of clonal IgG antibodies in culture supernatants and of rIgG antibodies were determined in a multiplex bead Luminex assay (Luminex Corp.). Culture supernatants and rIgGs were serially diluted in 1 × PBS containing 1% BSA, 0.05% NaN3 and 0.05% Tween20 (assay buffer) with 1% milk and incubated for 2 hours at room temperature with the mixture of antigen-coupled microsphere beads in 96-well filter bottom plates (Millipore). After washing three times with assay buffer, beads were incubated for 1 hour at room temperature with Phycoerythrin-conjugated goat anti-human IgG antibodies (Southern Biotech). After three washes, the beads were re-suspended in assay buffer and the plates read on a Bio-Plex 3D Suspension Array System (Bio-Rad).
Recombinant HA expression and purification.
Recombinant HA head constructs and Full-length HA ectodomains (FLsE) were expressed by polyethylenimine (PEI) facilitated, transient transfection of 293F cells. To clone HA heads, synthetic DNA for the region was subcloned into a pVRC8400 vector encoding a C-terminal rhinovirus 3C protease site and a 6xHis tag. To produce FLsE constructs, synthetic DNA was subcloned into a pVRC8400 vector encoding a T4 fibritin (foldon) trimerization tag and a 6xHis tag. Transfection complexes were prepared in Opti-MEM (Gibco) and added to cells. 5 days post transfection, cell supernatants were harvested and clarified by low-speed centrifugation. HA was purified by passage over TALON Metal Affinity Resin (Takara) followed by gel filtration chromatography on Superdex 200 (GE Healthcare) in buffer A. HA heads used for crystallography underwent the following additional purification steps. HA heads were cleaved using the Pierce 3C HRV Protease Solution Kit (Ref 88947) and passed over TALON Metal Affinity Resin to capture cleaved tags. Cleaved HA heads were then further purified by gel filtration chromatography on Superdex 200 (GE Healthcare) in buffer A.
ELISA
Five hundred nanograms of rHA FLsE or HA head were adhered to high-capacity binding, 96 well-plates (Corning 9018) overnight in PBS pH 7.4 at 4°C. HA coated plates were washed with a PBS-Tween-20 (0.05%v/v) buffer (PBS-T) and then blocked with PBS-T containing 2% bovine serum albumin (BSA) for 1 hour at room temperature. Blocking solution was then removed, and 5-fold dilutions of IgGs (in blocking solution) were added to wells. Plates were then incubated for 1 hour at room temperature. Primary IgG solution was removed and plates were washed three times with PBS-T. Secondary antibody, anti-human IgG-HRP (Abcam ab97225) diluted 1:10,000 in blocking solution, was added to wells and incubated for 30 minutes at room temperature. Plates were then washed three times with PBS-T. Plates were developed using 150μl 1-Step ABTS substrate (ThermoFisher, Prod#37615). Following a brief incubation at room temperature, HRP reactions were stopped by the addition of 100μl of 1% sodium dodecyl sulfate (SDS) solution. Plates were read on a Molecular Devices SpectraMax 340PC384 Microplate Reader at 405 nm. KD values for ELISA were obtained as follows. All measurements were performed in technical triplicate. The average background signal (no primary antibody) was subtracted from all absorbance values. Values from multiple plates were normalized to the FI6v347 standard (FluA20 was used for the HA head assays) that was present on each ELISA plate. The average of the three measurements were then graphed using GraphPad Prism (v9.0). KD values were determined by applying a nonlinear fit (One site binding, hyperbola) to these data points. The constraint that Bmax must be greater than 0.1 absorbance units was applied to all KD analysis parameters.
Recombinant IgG expression and purification.
The heavy and light chain variable domains of selected antibodies were cloned into modified pVRC8400 expression vectors to produce full length human IgG1 heavy chains and human lambda or kappa light chains. IgGs were produced by transient transfection of 293F cells as specified above. Five days post-transfection supernatants were harvested, clarified by low-speed centrifugation, and incubated overnight with Protein A Agarose Resin (GoldBio) at 4°C. The resin was collected in a chromatography column and washed with one column volume of buffer A. IgGs were eluted in 0.1M Glycine (pH 2.5) which was immediately neutralized by 1M tris (pH 8.5). Antibodies were then dialyzed against phosphate buffered saline (PBS) pH 7.4.
Recombinant mIgG expression and endotoxin free purification.
The heavy and light chain variable domains of selected antibodies were cloned into respective modified pVRC8400 expression vector to produce full length murine IgG1 and IgG2C heavy chains and murine lambda or kappa light chains. IgGs were produced by transient transfection of 293F cells as specified above. Five days post-transfection supernatants were harvested, clarified by low-speed centrifugation. 20% volume of 1M 2-morpholin-4-ylethanesulfonic acid (MES) pH 5 and Immobilized Protein G resin (Thermo Scientific Prod#20397) were added to supernatant and sample was incubated overnight at 4°C. The resin was collected in a chromatography column and washed with one column volume of 10mM MES, 150 mM NaCl at pH 5. mIgGs were eluted in 0.1M Glycine (pH 2.5), which was immediately neutralized by 1M tris (pH 8.5). Antibodies were then dialyzed against phosphate buffered saline (PBS) pH 7.4. All mIgGs administered to mice were purified using buffers made with endotoxin-free water (HyPure Cell Culture Grade Water, Cytiva SH30529.03) and were dialyzed into endotoxin-free Dulbecco’s PBS (EMD Millipore, TMS-012-A). Following dialysis, endotoxins were removed using Pierce High Capacity Endotoxin Removal Spin Columns (Ref 88274).
Virus microneutralization assays.
Two-fold serial dilutions of 50 ug/mL of HC19, S8V1–157 or CR3022 were incubated with 103.3 TCID50 of A/Aichi/02/1968 H3N2 (X-31) influenza virus for 1 hour at room temperature with continuous rocking. Media with TPCK was added to 96-well plates with confluent MDCK cells before the virus:antibody mixture was added and left on the cells for the duration of the experiment. After 4 days, cytopathic effect was determined and the neutralizing antibody titer was expressed as the reciprocal of the highest dilution of serum required to completely neutralize the infectivity each virus on MDCK cells. The concentration of antibody required to neutralize 100 TCID50 of virus was calculated based on the neutralizing titer dilution divided by the initial dilution factor, multiplied by the antibody concentration.
Protection:
C57BL/6 female and male mice were obtained from the Jackson Laboratory. All mice were housed under pathogen-free conditions at Duke University Animal Care Facility. Eight to 10-week old mice were injected i.p. with 150 μg of recombinant antibody diluted to 200 μl in PBS. Three hours later, mice were anesthetized by i.p. injection of ketamine (85 mg/kg) and xylazine (10 mg/kg) and infected intranasally with 3×LD50 (1.5×104 PFU) of A/Aichi/2/1968 X-31 (H3N2) in 40 μL total volume (20 μL/nostril). Mice were monitored daily for survival and body weight loss until 14 days post-challenge. The humane endpoint was set at 20% body weight loss relative to the initial body weight at the time of infection. All animal experiments were approved by the standards and guidance set forth by Duke University IACUC.
Female C57BL/6J mice at 8 weeks old, purchased from Japan SLC, were i.p. injected with 150 μg of the antibodies in 150 μL PBS. Three hours later, the mice were intranasally infected with 5LD50 of A/Aichi/02/1968(H3N2)(X31), kindly gifted from Dr. Takeshi Tsubata (Tokyo Medical and Dental University), under anesthesia with medetomidine-midazolam-butorphanol. Survival and body weight were daily assessed for 14 days with a humane endpoint set as 25% weight loss from the initial body weight. The experimental procedures were approved by the Animal Ethics Committee of the National Institute of Infectious Diseases, Japan, and performed in accordance with the guidelines of the Institutional Animal Care and Use Committee. Statistical significance of body weight change and survival of mice after lethal influenza challenge was calculated by Two-way ANOVA test and Mantel-Cox test, respectively, using GraphPad Prism (v10.0) software
Competitive inhibition assay
Competitive binding inhibition was determined by a Luminex assay, essentially as described20,30. Briefly, serially diluted human rIgGs or diluted (1:10) Bmem cell culture supernatants were incubated with HA-conjugated Luminex microspheres for 2 h at room temperature or overnight at 4°C. S8V1–157 mouse IgG1 was then added at a fixed concentration (100 ng/ml final for competition with rIgGs, or 10 ng/ml final for competition with culture supernatants) to each well, and incubated with the competitor Abs and HA-microspheres for 2 h at room temperature. After washing, bound S8V1–157 was detected by incubating the microspheres with 2 μg/ml PE-conjugated rat anti-mouse IgG1 (SB77e, SouthernBiotech) for 1 hr at room temperature, followed by washing and data collection. Irrelevant rIgG or culture supernatants containing HA-nonbinding IgG were used as non-inhibiting controls.
Flow cytometry
Flow cytometry analysis of rIgG binding to HA-expressing K530 cell lines was performed essentially as described 29. Briefly, K530 cell lines were thawed from cryopreserved aliquots and expanded in culture for ≥3 days. Pooled K530 cells were incubated at 4°C for 30 min with 0.4 μg/ml recombinant human IgGs diluted in PBS plus 2% fetal bovine serum. After washing, cells were labeled with 2 μg/ml PE-conjugated goat anti-human IgG (Southern Biotech) for 30 min at 4°C. Cells were then washed and analyzed with a BD FACSymphony A5 flow cytometer. Flow cytometry data were analyzed with FlowJo software (BD).
Flow cytometry analysis of rIgG binding to HA-expressing 293F cells was performed as follows. 293F cells were transfected using PEI with either plasmid encoding full-length HA from A/Aichi/02/1968(H3N2)(X31) or with empty vector. 36 hours post-transfection, cells were incubated at 4°C for one hour with 0.4 μg/ml recombinant human IgGs diluted in PBS plus 2% fetal bovine serum. After washing, cells were labeled with BB515 mouse anti-human IgG (BD Biosciences) for 30 min at 4°C at the manufacturer’s recommended concentration, followed by washing and fixation in 2% paraformaldehyde. Cells were analyzed with a BD LSRFortessa flow cytometer. Flow cytometry data were analyzed with FlowJo software (BD).
Western blots
293F cells were transfected using PEI with either plasmid encoding full-length HA from A/Aichi/02/1968(H3N2)(X31) or with empty vector. 24 hours post-transfection, cells were lysed in RIPA buffer (25mM Tris pH7.6, 150mM NaCl, 1% NP40 alternative, 1% sodium deoxycholate, 0.1% SDS). Cell lysates were clarified of debris and boiled with Laemmli buffer with 2-mercaptoethanol. Samples were run on 4–20% acrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% milk in PBS with 0.05% Tween 20 and probed with anti-HA tag antibody (Thermo-Fisher A01244–100; 0.3 μg/ml), followed by IR800-conjugated goat anti-mouse immunoglobulin (LiCor 926–32210; 0.1 μg/ml) and DyLight680-conjugated mouse anti-rabbit GAPDH antibody (BioRad MCA4739D680; 0.3 μg/ml). Membranes were imaged using a LiCor Odyssey CLx imager.
Crystallization
S1V2–157 Fab fragments were co-concentrated with the HA-head of A/American black duck/New Brunswick/00464/2010(H4N6) at a molar ratio of ~1:1.3 (Fab to HA-head) to a final concentration of ~20 mg/ml. Crystals of Fab-head complexes were grown at 18°C in hanging drops over a reservoir solutions containing 0.1 M Lithium sulfate, 0.1 M Sodium chloride, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.5 and 30% (v/v) poly(ethylene glycol) (PEG) 400. Crystals were cryprotected with 30% (v/v) PEG 400, 0.12 M Lithium sulfate, 0.3 M Sodium chloride, and 0.06 M MES pH 6.5. Cryprotectant was added directly to the drop, crystals were harvested, and flash cooled in liquid nitrogen.
Structure determination and refinement
We recorded diffraction data from a single moderate to poorly diffracting crystal at the Advanced Photon Source on beamline 24-ID-C. The dataset was strongly anisotropic, variable mosaicity (average 1.4°) and comprised contributions from multiple lattices. Efforts to improve these crystals were unsuccessful. Data were processed and scaled (XSCALE) with XDS52. Molecular replacement was carried out with PHASER53, dividing each complex into four search models (HA-head, Vh, Vl and constant domain). Search models were 5XL2, 6EIK, 5BK5 and 6E56.We carried out refinement calculations with PHENIX54 and model modifications, with COOT55. Refinement of atomic positions and B factors was followed by translation-liberation-screw (TLS) parameterization. All placed residues were supported by electron density maps and subsequent rounds of refinement. Final coordinates were validated with the MolProbity server56. Data collection and refinement statistics are in Table S1. Figures were made with PyMOL (Schrödinger, New York, NY.
Supplementary Material
ACKNOWLEDGMENTS:
We thank the many members of our Program Project Consortium for advice and discussion. X-ray diffraction data were recorded at beamline ID-24-C (operated by the Northeast Collaborative Access team: NE-CAT) at the Advanced Photon Source (APS, Argonne National Laboratory). We thank NE-CAT staff members for advice and assistance in data collection. NE-CAT is funded by NIH grant P30 GM124165. APS is operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02–06CH11357. The research was supported by NIAID Program Project Grant P01 AI089618 (to G.H.K.), funds from the University of Pittsburgh Center for Vaccine Research (to K.R.M), and Japan Agency for Medical Research and Development grant JP22fk0108141 (to Y.A. and Y.T.)
DATA AND SOFTWARE AVAILABILITY:
Coordinates and diffraction data have been deposited at the PDB, accession number 8US0. Antibody sequences have been deposited in NCBI GenBank, accession numbers OR825693-OR825700.
REFERENCES:
- 1.Knossow M. & Skehel J. J. Variation and infectivity neutralization in influenza. Immunology 119, 1–7 (2006). 10.1111/j.1365-2567.2006.02421.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu N. C. & Wilson I. A. Influenza Hemagglutinin Structures and Antibody Recognition. Cold Spring Harb Perspect Med 10 (2020). 10.1101/cshperspect.a038778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krammer F., Palese P. & Steel J. Advances in universal influenza virus vaccine design and antibody mediated therapies based on conserved regions of the hemagglutinin. Curr Top Microbiol Immunol 386, 301–321 (2015). 10.1007/82_2014_408 [DOI] [PubMed] [Google Scholar]
- 4.Krammer F., Garcia-Sastre A. & Palese P. Is It Possible to Develop a "Universal" Influenza Virus Vaccine? Potential Target Antigens and Critical Aspects for a Universal Influenza Vaccine. Cold Spring Harb Perspect Biol 10 (2018). 10.1101/cshperspect.a028845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erbelding E. J. et al. A Universal Influenza Vaccine: The Strategic Plan for the National Institute of Allergy and Infectious Diseases. J Infect Dis 218, 347–354 (2018). 10.1093/infdis/jiy103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cox R. J. Correlates of protection to influenza virus, where do we go from here? Hum Vaccin Immunother 9, 405–408 (2013). 10.4161/hv.22908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skehel J. J. et al. Membrane fusion by influenza hemagglutinin. Cold Spring Harb Symp Quant Biol 60, 573–580 (1995). 10.1101/sqb.1995.060.01.061 [DOI] [PubMed] [Google Scholar]
- 8.Skehel J. J. & Wiley D. C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69, 531–569 (2000). 10.1146/annurev.biochem.69.1.531 [DOI] [PubMed] [Google Scholar]
- 9.Weissenhorn W. et al. Structural basis for membrane fusion by enveloped viruses. Mol Membr Biol 16, 3–9 (1999). 10.1080/096876899294706 [DOI] [PubMed] [Google Scholar]
- 10.Benton D. J., Gamblin S. J., Rosenthal P. B. & Skehel J. J. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 583, 150–153 (2020). 10.1038/s41586-020-2333-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das D. K. et al. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 174, 926–937 e912 (2018). 10.1016/j.cell.2018.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casalino L. et al. Breathing and Tilting: Mesoscale Simulations Illuminate Influenza Glycoprotein Vulnerabilities. ACS Cent Sci 8, 1646–1663 (2022). 10.1021/acscentsci.2c00981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia N. K. et al. Structural dynamics reveal subtype-specific activation and inhibition of influenza virus hemagglutinin. J Biol Chem 299, 104765 (2023). 10.1016/j.jbc.2023.104765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guthmiller J. J. et al. Broadly neutralizing antibodies target a haemagglutinin anchor epitope. Nature 602, 314–320 (2022). 10.1038/s41586-021-04356-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benton D. J. et al. Influenza hemagglutinin membrane anchor. Proc Natl Acad Sci U S A 115, 10112–10117 (2018). 10.1073/pnas.1810927115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adachi Y. et al. Exposure of an occluded hemagglutinin epitope drives selection of a class of cross-protective influenza antibodies. Nat Commun 10, 3883 (2019). 10.1038/s41467-019-11821-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tonouchi K. et al. Structural basis for cross-group recognition of an influenza virus hemagglutinin antibody that targets postfusion stabilized epitope. PLoS Pathog 19, e1011554 (2023). 10.1371/journal.ppat.1011554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmons H. C. et al. A new class of antibodies that overcomes a steric barrier to cross-group neutralization of influenza viruses. PLoS Biol 21, e3002415 (2023). 10.1371/journal.pbio.3002415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finney J. et al. Protective human antibodies against a conserved epitope in pre- and postfusion influenza hemagglutinin. Proc Natl Acad Sci U S A 121, e2316964120 (2024). 10.1073/pnas.2316964120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe A. et al. Antibodies to a Conserved Influenza Head Interface Epitope Protect by an IgG Subtype-Dependent Mechanism. Cell 177, 1124–1135 e1116 (2019). 10.1016/j.cell.2019.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song H. et al. Avian-to-Human Receptor-Binding Adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep 20, 1201–1214 (2017). 10.1016/j.celrep.2017.07.028 [DOI] [PubMed] [Google Scholar]
- 22.Bertram S., Glowacka I., Steffen I., Kuhl A. & Pohlmann S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 20, 298–310 (2010). 10.1002/rmv.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horimoto T. & Kawaoka Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J Virol 68, 3120–3128 (1994). 10.1128/JVI.68.5.3120-3128.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosch F. X., Garten W., Klenk H. D. & Rott R. Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of Avian influenza viruses. Virology 113, 725–735 (1981). 10.1016/0042-6822(81)90201-4 [DOI] [PubMed] [Google Scholar]
- 25.Webster R. G. & Rott R. Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell 50, 665–666 (1987). 10.1016/0092-8674(87)90321-7 [DOI] [PubMed] [Google Scholar]
- 26.Vey M. et al. Hemagglutinin activation of pathogenic avian influenza viruses of serotype H7 requires the protease recognition motif R-X-K/R-R. Virology 188, 408–413 (1992). 10.1016/0042-6822(92)90775-k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wood G. W., McCauley J. W., Bashiruddin J. B. & Alexander D. J. Deduced amino acid sequences at the haemagglutinin cleavage site of avian influenza A viruses of H5 and H7 subtypes. Arch Virol 130, 209–217 (1993). 10.1007/BF01319010 [DOI] [PubMed] [Google Scholar]
- 28.Stieneke-Grober A. et al. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J 11, 2407–2414 (1992). 10.1002/j.1460-2075.1992.tb05305.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song S. et al. A cell-based multiplex immunoassay platform using fluorescent protein-barcoded reporter cell lines. Commun Biol 4, 1338 (2021). 10.1038/s42003-021-02881-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy K. R. et al. Memory B Cells that Cross-React with Group 1 and Group 2 Influenza A Viruses Are Abundant in Adult Human Repertoires. Immunity 48, 174–184 e179 (2018). 10.1016/j.immuni.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winarski K. L. et al. Vaccine-elicited antibody that neutralizes H5N1 influenza and variants binds the receptor site and polymorphic sites. Proc Natl Acad Sci U S A 112, 9346–9351 (2015). 10.1073/pnas.1502762112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleury D., Wharton S. A., Skehel J. J., Knossow M. & Bizebard T. Antigen distortion allows influenza virus to escape neutralization. Nat Struct Biol 5, 119–123 (1998). 10.1038/nsb0298-119 [DOI] [PubMed] [Google Scholar]
- 33.ter Meulen J. et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med 3, e237 (2006). 10.1371/journal.pmed.0030237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta A. et al. Early Treatment for Covid-19 with SARS-CoV-2 Neutralizing Antibody Sotrovimab. N Engl J Med 385, 1941–1950 (2021). 10.1056/NEJMoa2107934 [DOI] [PubMed] [Google Scholar]
- 35.Weinreich D. M. et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N Engl J Med 384, 238–251 (2021). 10.1056/NEJMoa2035002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen P. et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med 384, 229–237 (2021). 10.1056/NEJMoa2029849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulangu S. et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N Engl J Med 381, 2293–2303 (2019). 10.1056/NEJMoa1910993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Group P. I. W. et al. A Randomized, Controlled Trial of ZMapp for Ebola Virus Infection. N Engl J Med 375, 1448–1456 (2016). 10.1056/NEJMoa1604330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zost S. J. et al. Canonical features of human antibodies recognizing the influenza hemagglutinin trimer interface. J Clin Invest 131 (2021). 10.1172/JCI146791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bangaru S. et al. A Site of Vulnerability on the Influenza Virus Hemagglutinin Head Domain Trimer Interface. Cell 177, 1136–1152 e1118 (2019). 10.1016/j.cell.2019.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu F. et al. A Potent Germline-like Human Monoclonal Antibody Targets a pH-Sensitive Epitope on H7N9 Influenza Hemagglutinin. Cell Host Microbe 22, 471–483 e475 (2017). 10.1016/j.chom.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCarthy K. R. et al. A Prevalent Focused Human Antibody Response to the Influenza Virus Hemagglutinin Head Interface. mBio 12, e0114421 (2021). 10.1128/mBio.01144-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bajic G. et al. Influenza Antigen Engineering Focuses Immune Responses to a Subdominant but Broadly Protective Viral Epitope. Cell Host Microbe 25, 827–835 e826 (2019). 10.1016/j.chom.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu Y. & Gao G. F. "Breathing" Hemagglutinin Reveals Cryptic Epitopes for Universal Influenza Vaccine Design. Cell 177, 1086–1088 (2019). 10.1016/j.cell.2019.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otterstrom J. J. et al. Relating influenza virus membrane fusion kinetics to stoichiometry of neutralizing antibodies at the single-particle level. Proc Natl Acad Sci U S A 111, E5143–5148 (2014). 10.1073/pnas.1411755111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiLillo D. J., Tan G. S., Palese P. & Ravetch J. V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med 20, 143–151 (2014). 10.1038/nm.3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corti D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011). 10.1126/science.1205669 [DOI] [PubMed] [Google Scholar]
- 48.McCarthy K. R., Raymond D. D., Do K. T., Schmidt A. G. & Harrison S. C. Affinity maturation in a human humoral response to influenza hemagglutinin. Proc Natl Acad Sci U S A (2019). 10.1073/pnas.1915620116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmidt A. G. et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci U S A 110, 264–269 (2013). 10.1073/pnas.1218256109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt A. G. et al. Viral receptor-binding site antibodies with diverse germline origins. Cell 161, 1026–1034 (2015). 10.1016/j.cell.2015.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuraoka M. et al. Complex Antigens Drive Permissive Clonal Selection in Germinal Centers. Immunity 44, 542–552 (2016). 10.1016/j.immuni.2016.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132 (2010). 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCoy A. J. et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221 (2010). 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004). 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 56.Chen V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 (2010). 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bizebard T. et al. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 376, 92–94 (1995). 10.1038/376092a0 [DOI] [PubMed] [Google Scholar]
- 58.Dreyfus C. et al. Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012). 10.1126/science.1222908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ekiert D. C. et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333, 843–850 (2011). 10.1126/science.1204839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashkenazy H. et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44, W344–350 (2016). 10.1093/nar/gkw408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Celniker G. et al. ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Israel Journal of Chemistry 53, 199–206 (2013). 10.1002/ijch.201200096 [DOI] [Google Scholar]
- 62.Eisenberg D., Schwarz E., Komaromy M. & Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179, 125–142 (1984). 10.1016/0022-2836(84)90309-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and diffraction data have been deposited at the PDB, accession number 8US0. Antibody sequences have been deposited in NCBI GenBank, accession numbers OR825693-OR825700.