

ABSTRACT
Voltage-gated sodium channels initiate action potentials in nerve and muscle, and voltage-gated calcium channels couple depolarization of the plasma membrane to intracellular events such as secretion, contraction, synaptic transmission, and gene expression. In this Review and Perspective article, I summarize early work that led to identification, purification, functional reconstitution, and determination of the amino acid sequence of the protein subunits of sodium and calcium channels and showed that their pore-forming subunits are closely related. Decades of study by antibody mapping, site-directed mutagenesis, and electrophysiological recording led to detailed two-dimensional structure-function maps of the amino acid residues involved in voltage-dependent activation and inactivation, ion permeation and selectivity, and pharmacological modulation. Most recently, high-resolution three-dimensional structure determination by X-ray crystallography and cryogenic electron microscopy has revealed the structural basis for sodium and calcium channel function and pharmacological modulation at the atomic level. These studies now define the chemical basis for electrical signaling and provide templates for future development of new therapeutic agents for a range of neurological and cardiovascular diseases.
KEYWORDS: sodium channel, calcium channel, protein structure, cryogenic electron microscopy, X-ray crystallography
Sodium channels initiate action potentials
Voltage-gated sodium (NaV) channels are found in both kingdoms of prokaryotes and throughout the four kingdoms of eukaryotes. In the majority of these physiological settings, sodium channels generate conducted action potentials in response to small membrane depolarizations. In their classic work on sodium channels in nerve axons, Hodgkin and Huxley showed that small membrane depolarizations activate voltage-gated sodium channels, which generate action potentials that are conducted along the length of the nerve axons of the squid [1]. Sodium channels play a similar role in skeletal muscle and cardiac muscle fibers [2,3]. In addition, in endocrine cells and many other cell types that are not cylindrical, sodium channels initiate action potentials that are conducted over the cell body [4].
Calcium channels couple membrane depolarization to calcium entry
In some excitable cells, such as Paramecium and invertebrate skeletal muscle, voltage-gated calcium channels initiate and conduct calcium-dependent action potentials similarly to sodium channels [5,6]. Moreover, in repetitively firing cells of the sinoatrial node in the heart and the thalamus in the brain, calcium channels participate in rhythmic firing of action potentials [7–9]. However, the primary role of voltage-gated calcium channels is to couple depolarization of the cell surface membrane to calcium entry that initiates and regulates intracellular events such as contraction, secretion, neurotransmission, and gene expression [10–13]. Compared to sodium channels, the physiological roles of calcium channels are more varied and multifaceted, and their structures and functions are similarly more diverse.
Sodium and calcium channels as toxin targets
Voltage-gated sodium and calcium channels are the molecular targets for a broad array of natural toxins that are used in defense and in attacking prey [14,15]. They are produced, stored, and released by species ranging from single-celled dinoflagellates and corals to complex organisms such as spiders, frogs, and snakes. All of these chemical agents take advantage of the essential role of sodium and calcium channels in nerve conduction and synaptic transmission to stun, immobilize, and kill prey and predators, typically by binding specifically with high affinity to target receptor sites in the ion channel structure. These naturally occurring ion channel ligands act as gating modifiers by altering voltage-dependent gating or as pore-blockers by physically occluding the pore [14,15].
Finding sodium channels
Discovery of the sodium channel protein was directly dependent upon use of neurotoxins that bind to them with high affinity as molecular probes. α-Scorpion gating-modifier toxins from Leiurus quinquestriatus were used to photoaffinity label the protein subunits of sodium channels in mammalian brain with a photoreactive arylazide toxin derivative (Figure 1(a) [16]). This method identified two protein subunits, a large α-subunit with a molecular weight of 260 kilodaltons (kDa) and a smaller β-subunit of 33–36 kDa [17]. A complex of the large α subunit with two closely related β1 and β2 subunits was solubilized and purified from rat brain using specific binding of the pore blocker saxitoxin as a molecular probe (Figure 1(b) [18–20]) and shown to function as a voltage-gated sodium channel when reconstituted into phospholipid vesicles (Figure 1(c) [21,22]) or planar phospholipid bilayers (Figure 1(d)) [23]. All three subunits were found to be hydrophobic membrane glycoproteins. A similarly large, highly glycosylated sodium channel α subunit was identified in purified sodium channel preparations from electric eel electroplax and mammalian skeletal muscle [24–26]. These results led to formulation of a biochemical model for the subunit structure of sodium channels (Figure 1(c) [27]).
Figure 1.
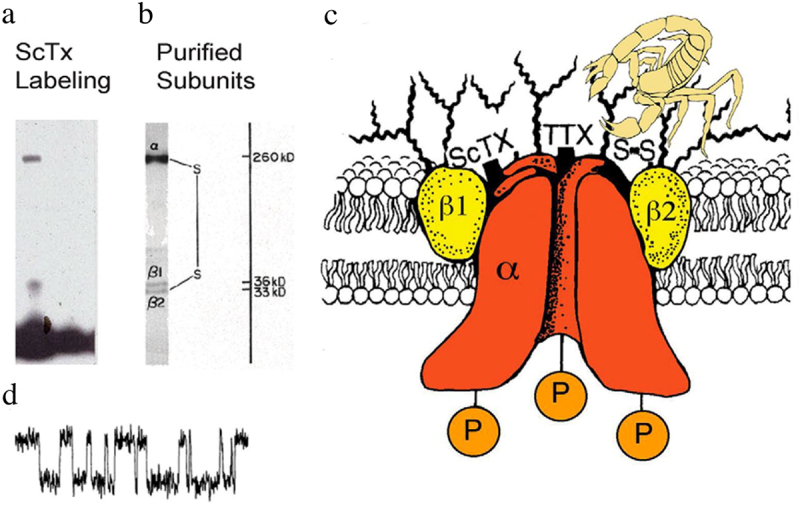
Subunit structure of voltage-gated sodium channels. a) SDS polyacrylamide gel electrophoresis illustrating the α and β subunits of rat brain sodium channels covalently labeled with125I-labeled leiurus quinquestriatus scorpion toxin (ScTx) and imaged by autoradiography. Adapted from Beneski and Catterall, 1980 [16]. b) SDS polyacrylamide gel electrophoresis patterns illustrating the α and β subunits of the brain Na+ channels. Sodium channel purified from rat brain showing the α, β1, and β2 subunits and their molecular weights. Adapted from Hartshorne et al., 1982 [19]. As illustrated, the α and β2 subunits are linked by a disulfide bond. Tetrodotoxin (TTX) and scorpion toxins (ScTx) bind to the α subunits of Na+ channels as indicated and were used as molecular tags to identify and purify the sodium channel protein from brain. c) drawing of the subunit structure of the brain Na+ channel based on biochemical data. Ψ, sites of N-linked glycosylation. Adapted from Catterall, 1984 [27]. d) single channel currents conducted by a single purified Na+ channel incorporated into a planar bilayer [23].
A key advance in studies of sodium channels was cloning and sequencing cDNA encoding the α subunits based on amino acid sequence and antibodies derived from biochemical studies of purified sodium channels – first from electric eel electroplax [28] and then from mammalian brain [29–31], skeletal muscle [32], and heart [33]. These results provided the key insights into the primary structures of sodium channels, which were used in extensive structure-function studies for more than three decades (reviewed in [34–36]). Some of the highlights of these studies are summarized in the form of a transmembrane folding model for the sodium channel (Figure 2). The ~ 2000 amino acid residues of the sodium channel α subunit are arranged in 24 transmembrane segments, which are organized in four homologous domains, DI to DIV (Figure 2). The six transmembrane segments in each domain were numbered S1-S6. The amino acid sequence is approximately 50% identical in the conserved transmembrane domains, but the large intracellular loops that connect the four homologous domains differ greatly from each other. The intracellular linker connecting Domains III and IV was found to serve as the inactivation gate (Figure 2, inset). The smaller β1 and β2 subunits were also cloned and sequenced using primary structure information and antibodies derived from biochemical studies [37,38]. These subunits are closely related single membrane-spanning cell adhesion molecules having a large extracellular N-terminal domain with an immunoglobulin-like folding pattern and a short intracellular C-terminal domain [39]. Related β3 and β4 subunits were cloned and analyzed many years later [40,41].
Figure 2.
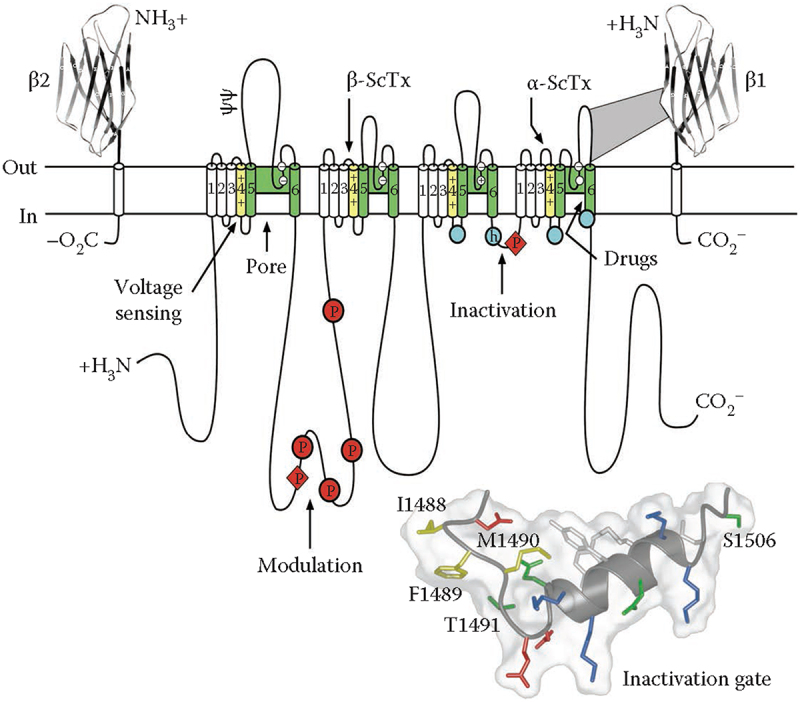
The primary structures of the subunits of the voltage-gated sodium channels. Cylinders represent alpha helical segments. Bold lines represent the polypeptide chains of each subunit with length approximately proportional to the number of amino acid residues in the brain sodium channel subtypes. The extracellular domains of the β1 and β2 subunits are shown as immunoglobulin-like folds. Ψ, sites of N-linked glycosylation; P in red circles, sites of demonstrated protein phosphorylation by PKA (circles) and PKC (diamonds); green, pore-lining segments; white circles, the outer and inner (DEKA) rings of amino residues that form the ion selectivity filter and the tetrodotoxin binding site; yellow, S4 voltage sensors; h in blue circle, inactivation particle in the inactivation gate loop; blue circles, sites implicated in forming the inactivation gate receptor. Sites of binding of α- and β-scorpion toxins and a site of interaction between α and β1 subunits are also shown. Tetrodotoxin is a specific blocker of the pore of Na+ channels, whereas the α- and β-scorpion toxins block fast inactivation and enhance activation, respectively, and thereby generate persistent Na+ current that causes hyperexcitability and depolarization block of nerve conduction. Adapted from Catterall, 2000 [34]. Inset. Structure of the fast inactivation gate in solution determined by NMR. Adapted from Rohl et al. [86].
Finding calcium channels
The protein subunits of calcium channels were discovered using similar ligand binding and biochemical purification methods as for sodium channels, but the primary ligands were radiolabeled calcium antagonist drugs in the dihydropyridine family, including nifedipine and isradipine [42,43]. Initial purification studies revealed α, β, and γ subunits from skeletal muscle, an abundant calcium channel protein source [44–47]. However, more detailed biochemical analysis, covalent-labeling, lectin-binding studies, and subunit-specific antibodies led to a surprisingly complex subunit structure with five protein subunits organized as in Figure 3(a) [48,50,51]. The large transmembrane α1 subunit was found to be associated with an intracellular transmembrane β subunit, a glycosylated transmembrane γ subunit, and a glycosylated transmembrane complex of disulfide-linked α2 and δ subunits [48,52–55]. This protein complex was functional in voltage-gated calcium conductance when reconstituted into phospholipid vesicles and planar phospholipid bilayers [45,47,56].
Figure 3.
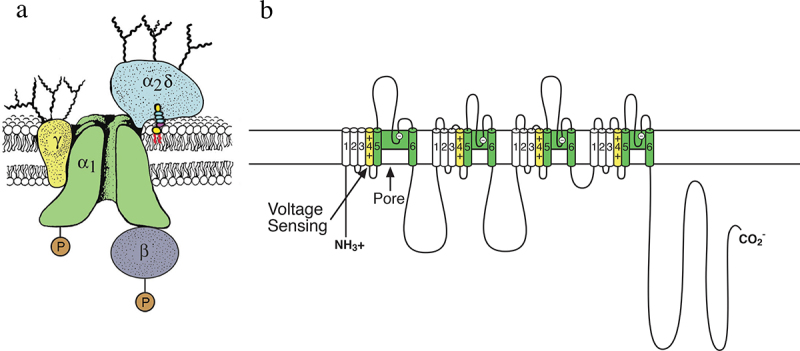
The subunit structure of calcium channels purified from skeletal muscle. a) a biochemical model of the skeletal muscle calcium channel taken from the original description of the subunit structure of skeletal muscle Ca2+ channels but with the mature α2δ subunit depicted following proteolytic processing, disulfide bond formation and attachment of a glycosylphosphatidylinositol membrane anchor. Adapted from Takahashi et al., 1987 [48]. P, sites of phosphorylation by cAMP-dependent protein kinase and protein kinase C. Ψ, sites of N-linked glycosylation. b) transmembrane folding models of the CaV1.1 subunits. Predicted alpha helices are depicted as cylinders. The lengths of lines correspond approximately to the lengths of the polypeptide segments represented. Adapted from Catterall, 1991 [49].
Cloning and sequencing the cDNA encoding the large α1 subunit of the skeletal muscle calcium channel revealed a protein of 2000 amino acid residues, closely resembling the sodium channel α subunit in transmembrane architecture, but only 25% identical in amino acid sequence (Figure 3(b) [57]). Further cloning and sequencing studies confirmed that the β subunit is an intracellular subunit, while the γ subunit has four transmembrane segments and multiple glycosylation sites, as expected from biochemical data [53,54]. Surprisingly, cloning, sequencing, and biochemical studies showed that the disulfide-linked α2 and δ subunits are encoded by a single gene [58], proteolytically processed to yield the disulfide-linked α2 and δ polypeptides [59,60], and further processed to cleave and replace the apparent transmembrane segment of the δ subunit with a glycosylphosphatidylinositol membrane anchor (Figure 3(a)) [61]). These complex processing and protein interactions of the α2δ subunits may be involved in regulation of synaptic plasticity [62].
Structure/Function studies of sodium and calcium channels
The structure and function of sodium and calcium channels have been studied extensively by site-directed mutagenesis and by mapping with site-directed antibodies (reviewed in [34–36]). Some of the highlights of these extensive studies are presented in the following sections.
Voltage dependent activation
In their classic work on voltage clamp analysis of sodium channels in the squid giant axon, published in 1952 long before any molecular studies of sodium channels, Hodgkin and Huxley proposed based on physical principles that voltage-dependent activation must involve the outward movement of three positively charged “gating particles” across the membrane upon depolarization, which led to opening of sodium channels [1]. This prophetic proposal has guided most subsequent work on the mechanism of activation of sodium channels. The predicted outward movement of the gating particles, now called “gating charges,” was detected by high-resolution voltage clamp studies of the squid giant axon in the absence of sodium and other permeant ions [63,64]. Armstrong and Bezanilla detected tiny capacitative “gating currents” produced by outward movement of the gating charges upon sodium channel activation and their subsequent inward movement upon repolarization and deactivation [63,64]. Detailed measurements with more modern methods have detected the movement of 12 to 16 gating charges per sodium channel voltage sensor [65,66]. But where are the gating charges located and how do they work? The S4 segments in each domain contain four to seven repeats of a highly conserved three-residue motif of a positively charged amino acid residue (usually Arg) flanked by two hydrophobic residues [35,67]. This structure creates a ladder of positive charges across the membrane. The “sliding-helix” model of voltage sensing [67–69] posited that the positively charged residues in the S4 segment were neutralized and stabilized in their transmembrane position by ion pair interactions with negatively charged amino acid residues in the neighboring transmembrane segments. In the resting state, these positive charges are pulled toward the cytosol by the negative membrane potential. Upon depolarization, this electrostatic force is released, and the S4 gating charges move outward along a spiral pathway by exchanging ion pair partners [67–69]. In support of this mechanism, mutations that neutralize the gating charges alter the voltage dependence of activation [70–73], toxin labeling studies show that the S4 segment remains in a transmembrane position in both resting and activated states [74,75], and substituted Cys labeling and crosslinking experiments show that the gating charges do indeed exchange ion pair partners and become accessible at the cell surface [76–78]. A consensus article by several leading investigators supports the sliding-helix model of voltage sensing [79], and the structural basis for the sliding helix model of voltage sensing and activation has now been further established as described below.
Fast inactivation
As shown by Hodgkin and Huxley [1], sodium channels inactivate within a few milliseconds after opening. Fast inactivation can be prevented by proteolytic treatment of the internal compartment of the squid axon, placing the inactivation process on the intracellular surface of the membrane [80]. Studies with sequence-direct antibodies targeted to each of the intracellular linkers showed that the short, highly conserved linker between Domains III and IV of the α subunit can impair fast inactivation of the sodium current and can induce re-openings of single sodium channels due to block of fast inactivation (Figure 2 [81,82]). Similarly, expression of the sodium channel as two separate proteins cut between Domains III and IV prevents fast inactivation [70]. Mutations in a conserved hydrophobic motif (Ile-Phe-Met, IFM) in the linker between Domains III and IV can completely prevent fast inactivation [83], which can be restored by intracellular perfusion of a short peptide containing the IFM motif (KIFMK [84,85]). The structure of the inactivation particle was first elucidated by NMR analysis of the intracellular linker between Domains III and IV expressed as a separate protein (Figure 2, inset [86]). Altogether, these studies led to a model of fast inactivation in which the IFM motif folds into the intracellular surface of the channel and inhibits ion conductance through the pore [83].
Slow inactivation
Voltage-gated sodium channels have a second, much slower inactivation process [87], which is engaged in tens to hundreds of milliseconds and regulates action potential generation during long trains of impulses [88]. It is not affected by treatment of the intracellular surface of the channel with proteases, suggesting that conformational changes in the transmembrane part of the sodium channel protein may be involved [89]. Extensive mutagenesis studies indicate that slow inactivation involves conformational changes in the outer half of the pore, involving the S5 and S6 segments as well as their extracellular connecting loops [90–92].
Ion Conductance and selectivity
The high affinity neurotoxins tetrodotoxin and saxitoxin block sodium channels from the extracellular solution in a manner that is consistent with direct occlusion of the pore [4]. The first evidence for the location of the pore came from identification of the amino acid residues in the P loop connecting the extracellular ends of the S5 and S6 segments as components of the receptor site for the pore blocker tetrodotoxin [93]. Mutagenesis studies revealed that sodium channels could be converted to calcium selectivity by mutation of a single amino acid residue in each of the four P loops to the negatively charged residue Glu, placing these residues in position to form the ion selectivity filter [94]. These studies led to a model in which the extracellular end of the pore of the sodium channel is lined by the P loops in Domains I -IV, which are surrounded by the S5 and S6 transmembrane segments [95].
Three dimensional structure of an ancestor of sodium and calcium channels
The α subunits of sodium channels are among the largest and most hydrophobic membrane proteins, which greatly slowed their study by structural biology methods. However, the unexpected discovery of prokaryotic voltage-gated sodium channels [96] provided an experimental model that allowed structural studies by X-ray crystallography. These prokaryotic channels are formed from a homotetramer of subunits that are similar in structure to one domain of a mammalian sodium channel, and biochemical and structural studies are facilitated by the lack of large intracellular and extracellular linkers [96]. The voltage-gated sodium channel from Arcobacter butzleri is a protein of 285 amino acid residues that can be expressed at high levels in insect cells driven by the powerful baculovirus promoter [97]. This channel protein could be expressed, purified, and crystallized in good yield and analyzed by X-ray crystallography [97]. The resulting structure at a resolution of 2.7 Å revealed the three-dimensional structure of a voltage-gated sodium channel for the first time [97]. As viewed from the extracellular side, the central pore is surrounded by four pore-forming domains composed of the S5 and S6 segments and the P loops between them (Figure 4(a), blue). Four voltage-sensing modules composed of the S1-S4 transmembrane segments are located on the periphery of the structure in a nearly symmetrical square array (Figure 4(a), green). As viewed from the membrane side, the S4-S5 linkers in each domain are seen as alpha-helical segments lying along the inner surface of the membrane (Figure 4(b), red). The four domains are linked in a domain-swapped organization, such that each voltage-sensing module is covalently connected to the pore module of its neighbor [97]. This organization may enforce concerted activation and opening of all four subunits simultaneously, thereby contributing in an important way to the rapid rate of increase of sodium current and the beginning of an action potential.
Figure 4.
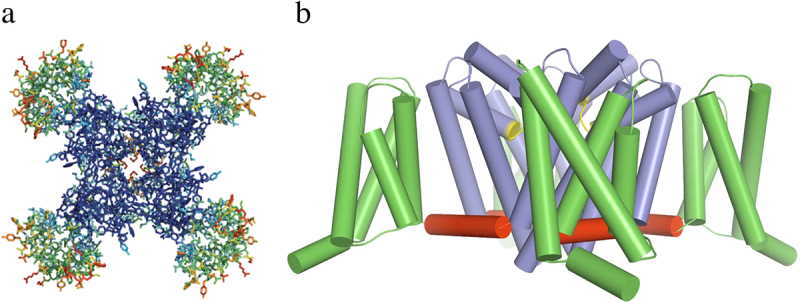
Structure of the bacterial sodium channel NaVAb. a) top view of NaVAb channels colored according to crystallographic temperature factors of the main-chain (blue <50 Å2 to red > 150 Å2). The four pore modules in the center are rigid in the crystal structure and therefore are blue. The four voltage-sensing modules surround the pore and are more mobile, as illustrated by warmer colors. b) side view of NaVAb. Voltage sensing module (S1-S4), green; pore module (S5, S6, and P loop), blue; selectivity filter, yellow; S4-S5 linker, red. Adapted from Payandeh et al.,2011 [97].
The original structure of NaVAb revealed the overall design of the voltage sensor and pore module (Figure 5 [97]). The voltage sensor is a V-shaped array of the S1-S4 segments, with S1-S2 forming one helical pair and S3-S4 forming a second helical pair. The two helical bundles are separated by an extracellular aqueous cleft that penetrates approximately half of the depth of the membrane (Figure 5(a) [97]). The key Arg gating charges, labeled R1-R4, are arrayed across the membrane in the S4 segment. The positively charged side chains of R1-R3 protrude into the extracellular aqueous cleft, and they make ion pair interactions with the negatively charged side chains of Glu residues in the Extracellular Negative Cluster (ENC; Figure 5(a), red [97]). In contrast, the positively charged side chain of R4 is located below the inner end of the extracellular aqueous cleft and makes interactions with the negatively charged side chains of the Intracellular Negative Cluster (INC, red). In between the ENC and INC is a hydrophobic barrier, the Hydrophobic Constriction Site (HCS, green), which seals the voltage sensor against penetration by water and ions. The structure of the voltage sensor suggests that is designed to move the gating charges in the S4 segment outward and inward in response to changes in the electrical field and trigger conformational changes to open and close the pore [97]. The voltage sensors of NaVAb were in an activated conformation in this structure.
Figure 5.
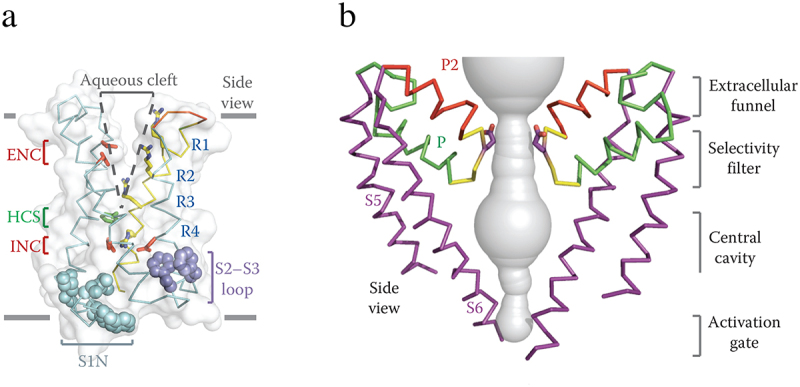
Architecture of the NaVAb voltage sensing module and pore module. a) side view of the voltage-sensing module of NaVAb illustrating the conformations of the S1-S4 helices and the size of the extracellular aqueous cleft with the R1-R4 gating charges (blue), extracellular negative cluster (ENC, red), intracellular negative cluster (INC, red), and hydrophobic constriction site (HCS, green). b) side view of the pore-forming module of two subunits of NaVAb. S5 and S6 transmembrane helices, purple: P helix loop, green; P2 helix green; water-filled space revealed by MOLE, gray. Note that the two S6 segments are crossed at their intracellular ends, forming the closed conformation of the activation gate. Adapted from Payandeh et al., 2011 [97].
The pore is formed by the S5 and S6 segments and the connecting P loop (Figure 5(b)). As a sodium ion approaches the pore from the extracellular solution, it enters a wide vestibule followed by the narrow ion selectivity filter. It then exits into the large water-filled central cavity and eventually moves into the cytosol through the activation gate formed by the intracellular ends of the four S6 segments (Figure 5(b)). In this first structure of NaVAb, the pore was in the closed conformation, with the four S6 segments binding tightly to each other. Remarkably, sodium ions complete this transit through the open pore at a rate of ~ 107 per second.
Structural basis for voltage dependent activation and pore opening in NaVAb
The structural basis for activation of the voltage sensor was revealed by comparison of high-resolution structures of NaVAb in resting and activated states [97,98]. The resting state was captured by introducing mutations that positively shifted the voltage dependence of activation by 250 mV, which stabilized the resting state at 0 mV, and by forming a disulfide bond between two substituted Cys residues to lock the voltage sensor in its resting conformation. Successful trapping of the functional resting state was confirmed by reducing the disulfide bonds, releasing the voltage sensor from its inactivated trapped position, and allowing it to respond to membrane potential with full recovery of sodium channel function [98]. The structures of NaVAb in resting and pre-open states are very similar, but there is a remarkable conformational change in the voltage sensor (Figure 6 [98]). When viewed from the side, the S4 segment is pulled toward the cytosol by 11.5 Å, which results in positioning the R2-R4 gating charges on the intracellular side of the HCS (Figure 6(a) [98]). This movement forms an elbow at the intracellular end of S4 where it joins the S4-S5 linker (Figure 6(b) [98]). In essence, this striking conformational change captures the energy of the electric field in the inward elbow conformation of the S4-S5 linker. This conformation serves as a cocked gun, ready to shoot the gating charges outward upon depolarization of the membrane (Figure 6(b) [98]). Looking outward toward the intracellular side of the voltage sensor, the four S4-S5 linkers form a nearly square corral (Figure 6(c), blue) surrounding the intracellular ends of the S6 segments (Figure 6(c), left, red). In the activated state, the four S4-S5 linkers move ~ 45° in a clockwise direction (Figure 6(c), blue). In contrast, the movement of the S6 helical backbone of the S6 segments is much more subtle (Figure 6(c), left, red/green). However, a space-filling representation shows a dramatic change. In the resting state, the orifice of the activation gate formed by the intracellular ends of the S6 segments is completely closed by the side chains of the four I217 residues (Figure 6(d), left, red shading), whereas these side chains are rotated out of the orifice of the pore in the open state leaving a pathway of 10.5 Å diameter for ion permeation (Figure 6(d), right, white). This orifice is sufficient for rapid permeation of hydrated sodium ions, as illustrated in the Movies in Reference [98].
Figure 6.
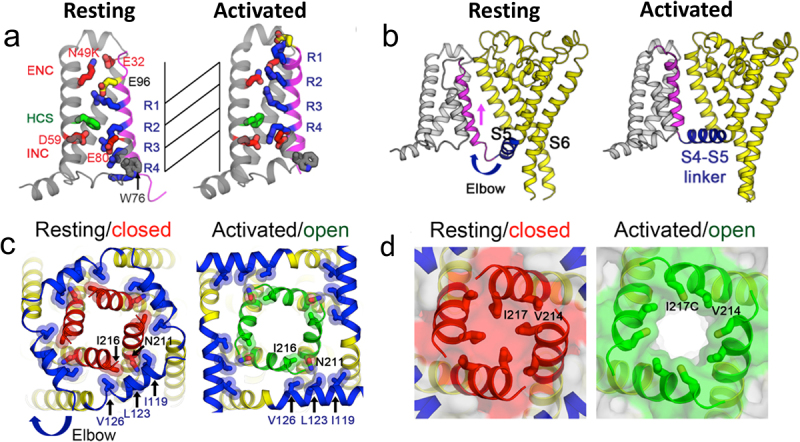
Sodium channel activation and pore gating mechanism of NaVAb. a) gating charge movement. Four Arg gating charges, R1–R4 (blue); the extracellular negative charge (ENC) cluster of E32 and N49(K) and the intracellular negative charge (INC) cluster of E59 and E80 (red); Phe in the hydrophobic constriction site (HCS) (green); and conserved W76 (gray) and E96 (yellow) are shown as sticks. S4 (magenta) moves outward by 11.5 Å, passing two gating charges through the HCS. Part of S3 is omitted for clarity. b) sideview of the structures focusing on S4 (magenta) and the S4-S5 linker (blue), with the S0–S3 segments shown in gray and the pore module in yellow. The S4 segment moves outward across the membrane from the resting to the activated states, whereas the S1–S3 segments remain relatively unchanged with respect to the membrane. The S4-S5 linker acts as an elbow that connects the S4 movement to modulate the pore. c) bottom (intracellular) view of the structures in (b), with S0–S4 omitted for clarity. The S4-S5 linker (blue) undergoes a large conformational change that tightens the collar around the S5 (yellow) and S6 segments (red or green) of the PM in the resting state and loosens the collar in the activated state. d) space-filling model of the structures in (c) at high magnification. Adapted from Wisedchaisri et al., 2019 [98].
Structural analysis of ion conductance and selectivity in NaVAb
As a sodium ion approaches the outer edge of the ion selectivity filter, it passes through the high field-strength site (HFS) formed by the side chains of four negatively charged Glu residues (E177, Figure 7(a) [97]). Interaction with the high field-strength site requires the sodium ion to release some of its waters of hydration, which are replaced by Glu side chains as ligands. The sodium ion then moves through the central and inner ion binding sites, which are formed by backbone carbonyls of L176 and T175 (Figure 7(b) [97]). Although the helical backbone that forms the walls of the ion selectivity filter is rigid, molecular dynamics analysis shows that the side chains of E177 “dunk” with the sodium ion and catalyze its inward movement (Figure 7(c) [99]). This rapid dunking movement requires only rotation around a single torsion angle, allowing it to take place on the time scale of ion conductance–107 ions/sec.
Figure 7.
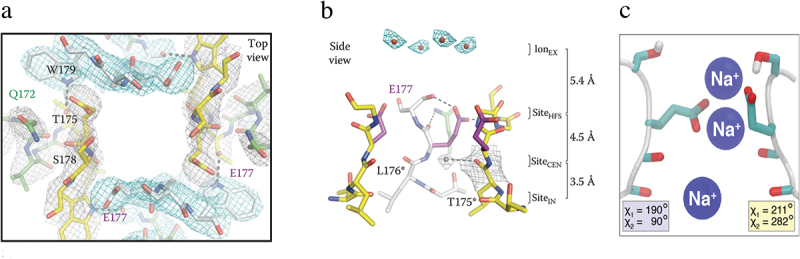
Mechanism of sodium conductance and selectivity. a) top view of the ion selectivity filter illustrating the high field strength site formed by four E177 residues. Hydrogen bonds between T175 and W179 are indicated by gray dashes. b) side view of the ion selectivity filter. E177 (purple) interactions with Q172, S178 and the backbone of S180 are shown for one subunit; putative cations or water molecules (red spheres). Electron-density around L176 (gray) and a bound water molecule are shown in gray mesh. Na+-coordination sites: SiteHFS, SiteCEN and SiteIN. Adapted from (Payandeh et al. 2011) [97]. c) E177 dunking. Movement of Na+ through the ion selectivity filter catalyzed by inward movement (dunking) of the side chains of E177 via a single torsion angle bend. Adapted from Chakrabarti et al., 2013 [99].
Multi-phase slow inactivation and drug block in NaVAb at the atomic level
Homotetrameric prokaryotic sodium channels do not have the structural equivalent of the fast inactivation gate of metazoan sodium channels, but they do have a process similar to slow inactivation that is mediated by the pore module [100]. Structure-function studies show that NaVAb has three kinetic phases of inactivation, which result in a highly stable inactivated state [101]. The structure of this stable inactivated state was determined by X-ray crystallography [102]. The overall structure is very similar to NaVAb in the pre-open state, but there are striking conformational changes in the pore [102]. Two of the S6 segments that line the pore have moved toward the central axis and the other two have moved away, resulting in a structure with two-fold symmetry rather than four-fold symmetry (Figure 8 [102]). The ion selectivity filter is in the shape of a parallelogram, rather than square (Figure 8(a)). A similar conformational change is observed in the central cavity (Figure 8(b)). Finally, the intracellular ends of the S6 segments where they form the activation gate have an oval configuration rather than circular (Figure 8(c)). Extensive structure-function studies implicate both the outer pore and the S6 segments in slow inactivation, consistent with this structural change [91,92,103].
Figure 8.
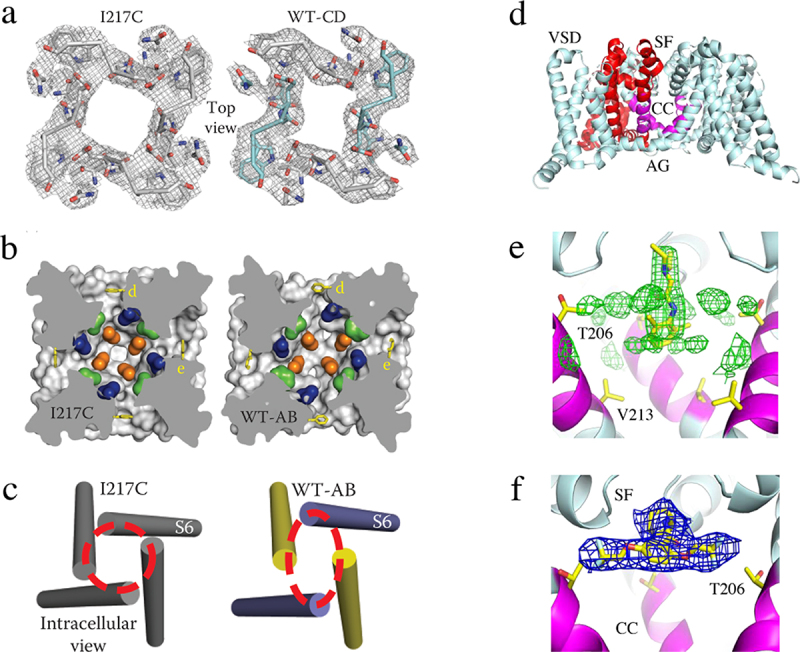
Slow inactivation and drug block of Na+ channels. a) top view of the collapse of the pore during slow inactivation of NaVAb. Two S6 segments move inward the central axis of the pore and two move outward to produce an asymmetric, partially collapsed conformation. The selectivity filter structure has changed from nearly square in the pre-open state of NaVAb/I217C to a partially collapsed parallelogram in the inactivated state of NaVAb/WT-CD. b) the central cavity is partially collapsed. c) the activation gate is tightly closed, but collapsed into a two-fold symmetric conformation. Adapted from Payandeh et al, 2012 [102]. d) structure of NaVAb with the selectivity filter (SF), central cavity (CC), and activation gate (AG) highlighted. e) lidocaine bound in the central cavity at the base of the selectivity filter. f) flecainide bound at the local anesthetic/antiarrhythmic receptor site in the central cavity at the base of the selectivity Filter.
Local anesthetics and Class I antiarrhythmic drugs inhibit sodium channels in a frequency- and voltage-dependent manner, which allows them to preferentially reduce sodium current and action potential generation in rapidly firing neurons that signal pain and in damaged cardiomyocytes that generate arrhythmias [104,105]. Both the local anesthetic lidocaine and the antiarrhythmic drug flecainide bind in the central cavity of NaVAb in a position to block sodium exit from the ion selectivity filter (Figure 8(d–f) [106]). Remarkably, access to this receptor site is controlled both by entry through the intracellular mouth of the pore in the open state and through fenestrations in the sides of the pore that lead to the into the central cavity from lipid bilayer in the resting state [106]. The rate of drug entry through the pore in the transient open state compared to the entry through fenestrations in the long-lasting resting state determines the extent of voltage- and frequency-dependent block of the pore, which is a critical element of controlling drug action [106].
Structural basis for calcium selectivity
The prokaryotic sodium channel NaVAb is a member of a large protein family whose members are as similar in amino acid sequence to mammalian calcium channels as to sodium channels and likely served as evolutionary precursors to both sodium and calcium channels [107,108]. These ancestral channels provide an exceptional opportunity to mimic evolution and build calcium channel features in their ancestral setting, which is amenable to high-resolution structural studies. Calcium channels must conduct calcium rapidly and selectively, yet the tight calcium binding needed for high selectivity would block the pore and prevent high conductance. Biophysical models resolved this paradox by combining high affinity binding to multiple calcium binding sites in sequence in the pore in order to induce electrical repulsion between bound calcium ions [109–112]. In this situation, calcium binds tightly in the pore and prevents other ions from permeating, but approaching calcium ions can “knock off” resident calcium ions in the pore and generate high conductance. Substitution of only three amino acid residues in the vestibule and selectivity filter of NaVAb is sufficient to mimic vertebrate calcium channel structure and form CaVAb, a calcium channel construct whose calcium selectivity is comparable to mammalian cardiac calcium channels with PCa/PNa ~400 (Figure 9(a), top [113]). Structural analysis shows that these amino acid substitutions do not alter the backbone fold of the voltage sensor ([113]); however, high-resolution views from X-ray crystallographic analysis reveals a series of calcium binding sites (Figure 9(a), green balls with mesh) that lead from the outer vestibule through to the central cavity. In favorable samples, one layer of water molecules is observed between the bound calcium ion and the walls of the selectivity filter (Figure 9(a), bottom [113]). The spacing of these calcium binding sites is ideal for electrostatic repulsion (Figure 9(a) [113]), which would knock off the resident calcium ion and allow rapid conductance. Consistent with this model, binding of a single blocking divalent cation, such as cadmium or nickel, would generate pore block rather than high conductance by producing high affinity binding without the knock-off effect [113]. These results provide the structural basis for the paradox of simultaneous high selectivity and high conductance that are essential for calcium channel function.
Figure 9.
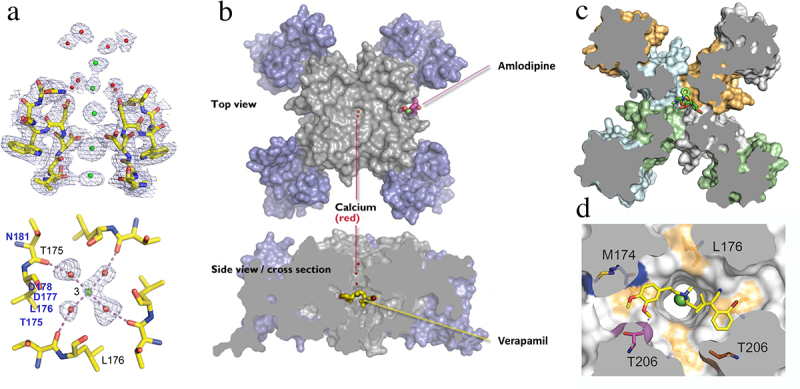
Calcium and drugs binding to the pore module of CaVAb. a) side view of the ion selectivity filter of CaVAb from X-ray crystallography. a) the ion selectivity filter of CaVAb at high resolution. Green balls, calcium ions; red balls, water; mesh, electron density. Top. side view; bottom, bottom view illustrating a single calcium ion with a square array of four waters of hydration bound. Adapted from Tang et al., 2014 [113]. b) structure of CaVAb in top and side views by X-ray crystallography. Blue, voltage sensor; gray, pore module. Bound amlodipine and verapamil, yellow sticks. c) top view of a cross-section of CaVAb with diltiazem bound in its receptor site as indicated (green sticks). Adapted from Tang et al., 2018 [138]. d) bottom X-ray crystallographic view of a cross-section in high resolution with verapamil (yellow sticks) bound in its receptor site and a calcium ion (green) bound in the pore. Adapted from Tang et al., 2014 [113].
Pharmacological modulation of the model calcium channel CaVAb
Voltage-gated calcium channels are the molecular targets for calcium antagonist drugs that are used in treatment of cardiac arrhythmia, hypertension, and angina pectoris [114,115]. Phenylalkylamines like verapamil and benzothiazepines like diltiazem are primarily used to treat atrial arrhythmias, whereas dihydropyridines like nifedipine and amlodipine are primarily used for treatment of hypertension and angina pectoris [116]. The sites of action of these important therapeutic agents have been progressively resolved by ligand binding, photoaffinity labeling, site-directed mutagenesis, and high-resolution structure determination, and they give key insights into the mechanisms of action of these important drugs [117–119]. Ligand binding studies showed that these three chemical classes of drugs have three allosterically coupled receptor sites, a distinct receptor site for the dihydropyridines and two separate but overlapping sites for verapamil and diltiazem [42,43]. Binding of verapamil and diltiazem and their derivatives is frequency-dependent, similar to the pore-blocking local anesthetics that inhibit sodium channels [120], suggesting that these calcium antagonist drugs are also pore-blockers [105,121]. In contrast, binding of dihydropyridines is voltage-dependent but not frequency-dependent, suggesting that they may interact allosterically with the voltage sensor rather than block the pore [105,122]. Photoaffinity labeling and antibody mapping of the labeled receptor sites showed that phenylalkylamines and benzothiazepines label the S6 segments in Domains III and IV that line the pore [123,124]. Photoaffinity labeling and antibody mapping also showed that dihydropyridines bind to the S5 and S6 segments in Domain III and the S6 segment in Domain IV [125,126]. These findings were greatly amplified by extensive site-directed mutagenesis studies of the S5 and S6 segments of Domains III and IV [127–136]. Functional dihydropyridine receptor sites were constructed by substitution of only nine amino acid residues in the IIIS5, IIIS6, and IVS6 segments, confirming that all of the important molecular interactions had been identified [133,135,136]. Altogether, this work on ligand binding, photoaffinity labeling, and site-directed mutagenesis indicated that phenylalkylamines and benzothiazepines bind to overlapping sites in the pore and block it, whereas dihydropyridines bind to a separate site on the membrane-facing aspect of the pore module and inhibit ion conductance through the pore in an indirect allosteric manner [117–119].
Structural studies have further resolved these receptor sites. In the prokaryotic model calcium channel CaVAb (Figure 9(b) [137,138]), amlodipine and other benzodiazepines bind to a receptor site on the lipid-facing surface of the pore module, between two voltage sensing domains, through interactions with the membrane-facing surface of the S5 and S6 segments (Figure 9(b) [137]). In contrast, verapamil binds to a site in the central cavity of the pore and physically occludes it (Figure 9(b) [137]). Similarly, diltiazem binds to an overlapping site and blocks the pore (Figure 9(c) [138]). High-resolution views of bound verapamil reveal that its tertiary amino group is lodged at the exit from the ion selectivity filter into the central cavity, while its two hydrophobic rings bind tightly to two sides of the pore and adhere tightly like a band-aid (Figure 9(d)). These two distinct types of receptor sites, which have been characterized at the atomic level, reveal the structural basis for frequency-dependent block of the pore by verapamil and diltiazem versus allosteric, voltage-dependent inhibition of calcium channels by dihydropyridines.
Key insights into the binding and action of these drugs have also come from high resolution cryogenic electron microscopy studies of the mammalian skeletal muscle calcium channel by Professor Nieng Yan and her colleagues [139,140], as summarized in the third article in this Special Issue. This remarkable work gives dramatic new insights into the subunit architecture, pore, and drug receptor sites of mammalian voltage-gated calcium channels at near-atomic resolution.
Structure of the cardiac sodium channel
Studies of mammalian sodium and calcium channels have benefited greatly from the revolution in high-resolution cryo-EM (Nieng Yan, Articles 2 and 3 in this Special Issue [139,141]). In addition to the structure of the skeletal muscle CaV1.1 channel noted above, these powerful methods have opened the way to new high-resolution structures of sodium channels from nerve [142–146], skeletal muscle [141], and heart [147,148], as well as calcium channels from peripheral nerve and brain [149,150]. Some highlights of the structure of the cardiac sodium channel as revealed by cryo-EM studies in our laboratory [147] are summarized here, and the structures and pharmacology of nerve and skeletal muscle sodium channels are summarized in more detail in Article 2 of this Special Issue by Professor Nieng Yan [141,142].
The structure of the cardiac sodium channel has been captured in both closed/inactivated and open states [147,151]. The structure of the closed/inactivated state of the cardiac sodium channel was determined using a construct in which the large intracellular linkers and C-terminal domain, which are predicted to be unstructured, were deleted by site-directed mutagenesis [152]. The resulting NaV1.5c construct is fully functional and retains its characteristic sensitivity to toxins and antiarrhythmic drugs [147]. The alpha-helical backbone of the transmembrane core of NaV1.5c (Figure 10(a), center [147]) is nearly identical to NaVAb, with root mean square deviation (RMSD) of 3.2 Å, well within the level of uncertainty expected due to the limits of resolution of the cryo-EM structure (3–4 Å). The four voltage sensors are in different activated conformations with 2–4 gating charge Arg residues positioned on the extracellular side of the HCS (Figure 10(a), center [147]). The pore is closed by the intracellular activation gate formed the four S6 segments. The fast inactivation gate is tightly bound to a receptor site adjacent to the IVS6 segments and the closed activation gate (Figure 10(a), lower left [147]). Thus, this structure has all of the features expected for a closed/inactivated state of the sodium channel.
Figure 10.
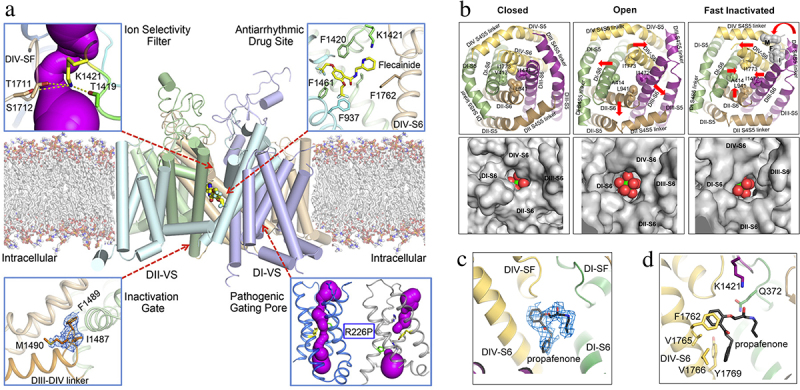
The cardiac sodium channel NaV1.5 at high resolution. a) graphical abstract. Center. NaV1.5 structure. Upper left, sodium permeation pathway in the pore module, purple. Upper right, bound flecainide. Lower right, arrhythmia mutation R227P in the voltage sensor. The pathogenic gating pore caused by this mutation creates a complete water-filled pathway through the voltage sensor, as revealed with the program MOLE (purple). Lower left. IFM motif of the fast inactivation gate. b) bottom view of the structure of the activation gate formed by the inner ends of the alpha-helical S6 segments. Top row, backbone structure in closed, open, and fast inactivated states. Bottom row. A sodium ion (yellow) with waters of hydration (red). Adapted from Jiang et al., 2020 [147]. c) propafenone binding site in side view. d) propafenone binding site in top view.
Structural basis for pathogenic gating pores
The voltage sensors of sodium channels are tightly packed, such that the movement of the S4 segment through the hydrophobic constriction site takes place without significant leakage of water and ions (Figure 6). However, mutation of one of the Arg gating charges to a smaller, hydrophilic residue can cause pathogenic ion leakage, termed “gating pore current,” through the hydrophobic constriction site when the small amino acid side chain occupies the hydrophobic constriction site of the sodium channel without fully blocking it [153]. This persistent ionic leak is the cause of periodic paralysis [154–156], cardiac arrhythmia [157], and autism [158]. Structural studies of a pathogenic gating pore inserted in the ancestral sodium channel NaVAb introduces a hole of ~ 3 Å diameter in the voltage sensor, just large enough for permeation of a sodium ion [159]. The pathogenic gating pore mutation R226P in rNaV1.5, which causes cardiac arrhythmia [157], is illustrated in Figure 10(a), lower right. The resulting complete water-filled pathway is illustrated in purple for the mutant, whereas the water-filled pathway is interrupted by the presence of Arg226 in wild-type (Figure 10(a), lower right). These pathogenic gating pores allow persistent leak of sodium into nerve and muscle cells, which causes persistent depolarization, increased resting membrane conductance, and impairment of action potential firing.
Structural basis for sodium conductance and selectivity in the cardiac sodium channel
The pore of NaV1.5c is asymmetric and tortuous (Figure 10(a), upper left [147]). The high field-strength site of the ion selectivity filter is surrounded by four different amino acid residues – Asp-Glu-Ala-Lys (DEKA). It is surprising to find a positively charged Lys residue in a sodium-specific ion selectivity filter, but Lys in this position is absolutely required for sodium selectivity [160]. Careful analysis of the structure of the selectivity filter in NaV1.5c revealed that the positive charge of the ε-amino group of Lys1421 is delocalized by formation of three hydrogen bonds with uniquely placed backbone carbonyls, which points the partially negatively charged electron pair of the ε-amino group into the lumen of the pore, where it is available for interaction with conducted sodium ions [147]. These structural findings led to the ε-electron-pair hypothesis of sodium selectivity, which posits that this unique chemical interaction is essential for sodium selectivity [147].
Subunit structure of the cardiac sodium channel
Vertebrate sodium channels typically have one or two NaVβ subunits associated with their pore-forming α subunit [34,36]. However, even when the NaVβ1 and NaVβ2 subunits were over-expressed with NaV1.5c, neither subunit was resolved in the structure, suggesting that these subunits have low affinity for cardiac sodium channels. Consistent with this hypothesis, an N-linked carbohydrate chain was found in an overlapping position with the expected interaction site of the NaVβ1 subunit, and a Cys residue that forms a disulfide linkage with the NaVβ2 subunit was replaced by Leu [147].
Structural basis for pore opening and fast inactivation of cardiac sodium channels
The fast Inactivation gate is formed by the short intracellular loop connecting Domains III and IV of mammalian sodium channels [70,81–83], and the hydrophobic motif Ile-Phe-Met (IFM) serves as the inactivation particle, binds to the intracellular surface of the pore module, and inhibits sodium current [83,84]. In contrast to the closed/inactivated state, NaV1.5c trapped in the open state by mutation of the IFM inactivation particle in the inactivation gate to the hydrophilic amino acid residues QQQ revealed a similar structure for the voltage sensors, with RMDS of 1.04 Å; however, many conformational changes were observed in the inner pore region near the activation gate and inactivation gate receptor site (Figure 10(b) [151]). In particular, the IFM motif is observed bound in its receptor site in the closed/inactivated structure, but it has dissociated and is not observed in the structure in the open state (Figure 10(b) [147,151]). The red arrows in Figure 10(b) show the motion of the amino acid residues in the activation gate in closed, open, and inactivated structures. The pore-opening mechanism is nearly identical to NaVAb, but the configuration of the open activation gate is slightly asymmetric with dimensions of 10.6 Å x 9.7 Å. The open activation gate is just wide enough to accommodate a fully hydrated sodium ion (Figure 10(b) [151]), indicating that sodium conductance out of the central cavity occurs in single file, even though the total flux of sodium is very rapid at ~ 107 ions/sec.
Receptor site for antiarrhythmic drugs in the cardiac sodium channel
The antiarrhythmic drug flecainide bound in the central cavity of NaV1.5c, in position to block ion conductance by preventing movement of sodium ions from the selectivity filter into the central cavity (Figure 10(a), upper right [147]), as previously observed in NaVAb (Figure 8(d–f) [106]). It makes prominent contacts with F937, F1420, K1421, and F1461, but the key residue F1762 is not close enough for a strong interaction (Figure 10(a), upper right [147]). In the open state of NaV1.5c, the antiarrhythmic drug propafenone also binds in a pore-blocking position in the central cavity (Figure 10(c,d)). Its interaction site is overlapping, yet distinct from flecainide (Figure 10(c,d) [151]). It makes a strong π-π interaction with the aromatic side chain of F1762 and additional interactions with K1421, V1760, V1765, and Y1769. These open-state interactions seem likely to provide higher affinity binding than the interactions observed for flecainide in the closed/inactivated state (Figure 10(c,d) [147,151]). Both propafenone and flecainide are Class IC antiarrhythmic drugs, which are thought to bind with high affinity to the open state of sodium channels [161,162]. Therefore, it is likely that these structures capture a low-affinity binding pose for flecainide to a closed/inactivated state (Figure 10(b)) and a high-affinity binding pose for propafenone to the open state (Figure 10(c,d)). The classical Class I antiarrhythmic drug quinidine also binds to this receptor site [148]. In addition, the atypical antiarrhythmic drug ranolazine binds to the core of the antiarrhythmic drug receptor site, but projects its atypical second aromatic ring beyond this classical drug receptor site to make new interactions with the IS6 segment that may be responsible for its unique channel-blocking properties [163]. Further studies may reveal the structural and chemical basis for state-dependent binding and block of sodium channels by these life-saving antiarrhythmic drugs.
Receptor site for a gating modifier toxin on the cardiac sodium channel
Polypeptide gating modifier toxins bind to the extracellular surface of the voltage sensors of sodium channels and alter voltage sensor function [14,164]. For example, α-scorpion toxins bind in a voltage-dependent manner and greatly slow fast inactivation of sodium channels, leading to pathologic depolarization and repetitive firing in nerve and muscle and to life-threatening arrhythmias in the heart [164]. The receptor site for α-scorpion toxins was identified by photoaffinity labeling [16] and mapped by site-directed antibodies and site-directed mutagenesis [74,165], leading to the conclusion that these toxins bind to a receptor site formed primarily by the S3-S4 extracellular linker in Domain IV, with secondary contributions by the S1-S2 linker in Domain IV and the SS2-S6 linker in the adjacent region of Domain I. The deathstalker α-scorpion toxin LqhIII has high affinity for the cardiac sodium channel and generates lethal arrhythmias in the heart [166,167]. The structure of this toxin bound to its receptor site on cardiac sodium channels has been solved at high resolution for both closed/inactivated and open sodium channels using the NaV1.5c construct ([168]; Jiang et al., submitted). The toxin binds to a receptor site on the Domain IV voltage sensor (Figure 11(a)). It wedges its flexible β2-β3 loop and C-terminal region into the aqueous cleft in the voltage sensor, making strong interactions with the S1-S2 linker and S3-S4 linker (Figure 11(b) [168]), consistent with previous structure-function studies [74,165]. The voltage sensor is locked in a partially activated conformation (Figure 11(c) [168]), suggesting how binding of the toxin can allow sodium channel activation and also prevent fast inactivation at the same time. Strong interactions are observed with Asp1612 via a pincers complex with His15 and His43 (Figure 11(d)). An analogous negatively charged residue is present in this position in the binding sites of many different scorpion toxins acting on a range of sodium channel types, suggesting that this interaction is central to toxin action. The bound toxin grips an arc of amino acid residues bridging the S1-S2 and S3-S4 linkers, in position to control the movements of these two helical hairpins in the gating process (Figure 11(d)). LqhIII is bound tightly through interaction of residue K64 with the negatively charged residues of the ENC, which further stabilizes the toxin-channel complex and prevents outward movement of gating charges R1 and R2 into this position during activation of the channel (Figure 11(c,d)). This charge-charge interaction of K64 with the ENC may be responsible for the characteristic slow, voltage-dependent dissociation of LqhIII and other α-scorpion toxins, as the toxin is slowly forced off its receptor site by electrostatic repulsion from the R1 and R2 gating charges during the outward movement of the S4 segment. Altogether, these structural features explain the hallmark features of scorpion toxin action – high affinity binding to sodium channels in resting states, strong effects to prevent fast inactivation, and slow voltage-dependent dissociation.
Figure 11.
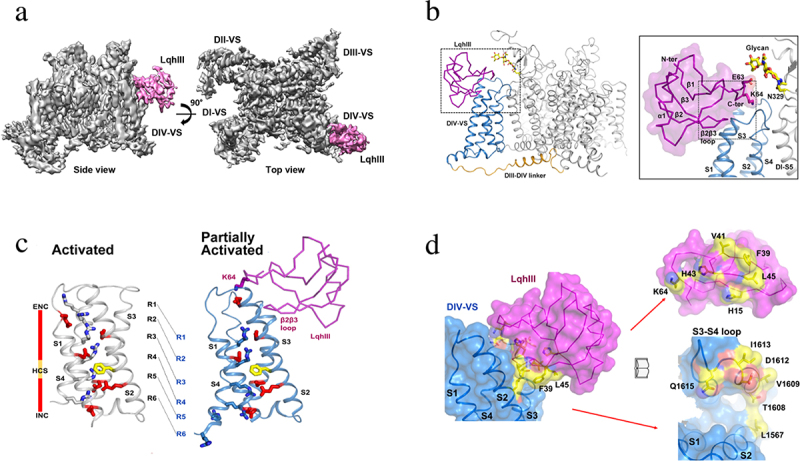
Receptor site for the deathstalker scorpion toxin LqhIII on NaV1.5. a) spacefilling side view and top view of NaV1.5 (gray) with LqhIII bound (purple). b) close-up view of bound LqhIII (purple) to its receptor site on the NaV1.5 backbone structure. Inset. higher resolution image of the interface between LqhIII and NaV1.5. c) superposition of the voltage sensor in the unmodified activated state and the toxin-modified partially activated state. d) mutational map of the interface residues in the LqhIII receptor site illustrated with “open book” format. LqhIII, purple; NaV1.5, blue. Amino acid residues highlighted in yellow and orange are required for high-affinity binding. Adapted from Jiang et al., 2021 [168].
Conclusion
Biochemical studies in the 1980’s led to identification of the protein components of sodium and calcium channels and defined their subunit structures [27,48], and cDNA cloning and sequencing determined their primary amino acid sequences [28,29,57]. Based on these early results, extensive structure-function studies in the 1990’s and 2000’s led to key insights into the mechanisms of ion conductance and selectivity [94,160,169], voltage-dependent activation and inactivation [68,70,81,83], and pharmacological modulation [119,170]. Beginning in 2011, high-resolution structures of ancestral sodium channels [97] followed by mammalian sodium and calcium channels [139,171] have provided increasingly precise views of their mechanisms of action at atomic resolution. We look forward to deeper understanding of the fundamental mechanisms of action of these ion channels and to development of higher affinity, more efficacious, and safer drugs for use as local anesthetics, analgesics, and antiarrhythmics in the future.
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Because this is a review article, no new experimental data are presented. Interested individuals should write to the authors of the papers that are cited in each figure legend to obtain the data presented in that figure.
References
- [1].Hodgkin AL, Huxley AF.. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117(4):500–23. doi: 10.1113/jphysiol.1952.sp004764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hall AE, Hutter OF, Noble D. Current-voltage relations of Purkinje fibres in sodium-deficient solutions. J Physiol. 1963;166:2225–2240. doi: 10.1113/jphysiol.1963.sp007102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Adrian RH, Chandler WK, Hodgkin AL. Voltage clamp experiments in striated muscle fibres. J Physiol. 1970;208(3):607–644. doi: 10.1113/jphysiol.1970.sp009139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hille B. Ionic channels of excitable membranes. 3rd ed. Sunderland (MA): Sinauer Associates Inc.; 2001. [Google Scholar]
- [5].Eckert R, Brehm P. Ionic mechanisms of excitation in Paramecium. Annu Rev Biophys Bioeng. 1979;8(1):353–383. doi: 10.1146/annurev.bb.08.060179.002033 [DOI] [PubMed] [Google Scholar]
- [6].Ashcroft FM, Stanfield PR. Calcium and potassium currents in muscle fibres of an insect (carausius morosus). J Physiol. 1982;323(1):93–115. doi: 10.1113/jphysiol.1982.sp014063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Torrente AG, Mesirca P, Bidaud I, et al. Channelopathies of voltage-gated L-type Cav1.3 channels. Pflugers Arch. 2020;472(7):817–830. doi: 10.1007/s00424-020-02421-1 [DOI] [PubMed] [Google Scholar]
- [8].Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83(1):117–161. doi: 10.1152/physrev.00018.2002 [DOI] [PubMed] [Google Scholar]
- [9].Deleuze C, David F, Behuret S, et al. T-type calcium channels consolidate tonic action potential output of thalamic neurons to neocortex. J Neurosci. 2012;32(35):12228–12236. doi: 10.1523/JNEUROSCI.1362-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Reuter H. The dependence of slow inward current in Purkinje fibres on the extracellular calcium-concentration. J Physiol (Lond). 1967;192(2):479–492. doi: 10.1113/jphysiol.1967.sp008310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Reuter H. Properties of two inward membrane currents in the heart. Annu Rev Physiol. 1979;41(1):413–424. doi: 10.1146/annurev.ph.41.030179.002213 [DOI] [PubMed] [Google Scholar]
- [12].Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium Science. Science. 1978;202(4373):1203–1206. doi: 10.1126/science.103199 [DOI] [PubMed] [Google Scholar]
- [13].Tsien RW, Elinor PT, Horne WA. Molecular diversity of voltage-dependent calcium channels. Trends Neurosci. 1991;12:349–354. doi: 10.1016/0165-6147(91)90595-J [DOI] [PubMed] [Google Scholar]
- [14].Catterall WA. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu Rev Pharmacol Toxicol. 1980;20(1):15–43. doi: 10.1146/annurev.pa.20.040180.000311 [DOI] [PubMed] [Google Scholar]
- [15].Olivera BM, Miljanich GP, Ramachandran J, et al. Calcium channel diversity and neurotransmitter release: The omega-conotoxins and omega-agatoxins. Annu Rev Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135 [DOI] [PubMed] [Google Scholar]
- [16].Beneski DA, Catterall WA. Covalent labeling of protein components of the sodium channel with a photoactivable derivative of scorpion toxin. Proc Natl Acad Sci, USA. 1980;77(1):639–643. doi: 10.1073/pnas.77.1.639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hartshorne RP, Coppersmith J, Catterall WA. Size characteristics of the solubilized saxitoxin receptor of the voltage-sensitive sodium channel from rat brain. J Biol Chem. 1981;255:10572–10575. doi: 10.1016/S0021-9258(19)70342-1 [DOI] [PubMed] [Google Scholar]
- [18].Hartshorne RP, Catterall WA. Purification of the saxitoxin receptor of the sodium channel from rat brain. Proc Natl Acad Sci, USA. 1981;78(7):4620–4624. doi: 10.1073/pnas.78.7.4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hartshorne RP, Messner DJ, Coppersmith JC, et al. The saxitoxin receptor of the sodium channel from rat brain evidence for two nonidentical beta subunits. J Biol Chem. 1982;257:13888–13891. doi: 10.1016/S0021-9258(19)45312-X [DOI] [PubMed] [Google Scholar]
- [20].Hartshorne RP, Catterall WA. The sodium channel from rat brain. Purification and subunit composition. J Biol Chem. 1984;259(3):1667–1675. doi: 10.1016/S0021-9258(17)43460-0 [DOI] [PubMed] [Google Scholar]
- [21].Talvenheimo JA, Tamkun MM, Catterall WA. Reconstitution of neurotoxin-stimulated sodium transport by the voltage-sensitive sodium channel purified from rat brain. J Biol Chem. 1982;257(20):11868–11871. doi: 10.1016/S0021-9258(18)33644-5 [DOI] [PubMed] [Google Scholar]
- [22].Tamkun MM, Talvenheimo JA, Catterall WA. The sodium channel from rat brain. Reconstitution of neurotoxin-activated ion flux and scorpion toxin binding from purified components. J Biol Chem. 1984;259(3):1676–1688. doi: 10.1016/S0021-9258(17)43461-2 [DOI] [PubMed] [Google Scholar]
- [23].Hartshorne RP, Keller BU, Talvenheimo JA, et al. Functional reconstitution of the purified brain sodium channel in planar lipid bilayers. Proc Natl Acad Sci, USA. 1985;82(1):240–244. doi: 10.1073/pnas.82.1.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Agnew WS, Moore AC, Levinson SR, et al. Identification of a large molecular weight peptide associated with a tetrodotoxin binding proteins from the electroplax of electrophorus electricus. Biochem Biophys Res Commun. 1980;92:860–866. doi: 10.1016/0006-291X(80)90782-2 [DOI] [PubMed] [Google Scholar]
- [25].Miller JA, Agnew WS, Levinson SR. Principal glycopeptide of the tetrodotoxin/saxitoxin binding protein from electrophorus electricus: isolation and partial chemical and physical characterization. Biochemistry. 1983;22:462–470. doi: 10.1021/bi00271a032 [DOI] [PubMed] [Google Scholar]
- [26].Barchi RL. Protein components of the purified sodium channel from rat skeletal sarcolemma. J Neurochem. 1983;36:1377–1385. doi: 10.1111/j.1471-4159.1983.tb13580.x [DOI] [PubMed] [Google Scholar]
- [27].Catterall WA. The molecular basis of neuronal excitability. Science. 1984;223(4637):653–661. doi: 10.1126/science.6320365 [DOI] [PubMed] [Google Scholar]
- [28].Noda M, Shimizu S, Tanabe T, et al. Primary structure of electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312(5990):121–127. doi: 10.1038/312121a0 [DOI] [PubMed] [Google Scholar]
- [29].Noda M, Ikeda T, Kayano T, et al. Existence of distinct sodium channel messenger RNAs in rat brain. Nature. 1986;320(6058):188–192. doi: 10.1038/320188a0 [DOI] [PubMed] [Google Scholar]
- [30].Goldin AL, Snutch T, Lubbert H, et al. Messenger RNA coding for only the α subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes. Proc Natl Acad Sci, USA. 1986;83:7503–7507. doi: 10.1073/pnas.83.19.7503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Auld VJ, Goldin AL, Krafte DS, et al. A rat brain sodium channel α subunit with novel gating properties. Neuron. 1988;1:449–461. doi: 10.1016/0896-6273(88)90176-6 [DOI] [PubMed] [Google Scholar]
- [32].Trimmer JS, Cooperman SS, Tomiko SA, et al. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3(1):33–49. doi: 10.1016/0896-6273(89)90113-X [DOI] [PubMed] [Google Scholar]
- [33].Rogart RB, Cribbs LL, Muglia LK, et al. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc Natl Acad Sci, USA. 1989;86(20):8170–8174. doi: 10.1073/pnas.86.20.8170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. doi: 10.1016/S0896-6273(00)81133-2 [DOI] [PubMed] [Google Scholar]
- [35].Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80(2):555–592. doi: 10.1152/physrev.2000.80.2.555 [DOI] [PubMed] [Google Scholar]
- [36].Ahern CA, Payandeh J, Bosmans F, et al. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J Gen Physiol. 2016;147(1):1–24. doi: 10.1085/jgp.201511492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Isom LL, De Jongh KS, Patton DE, et al. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256(5058):839–842. doi: 10.1126/science.256.5058.839 [DOI] [PubMed] [Google Scholar]
- [38].Isom LL, Ragsdale DS, De Jongh KS, et al. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83(3):433–442. doi: 10.1016/0092-8674(95)90121-3 [DOI] [PubMed] [Google Scholar]
- [39].Isom LL, Catterall WA. Na+ channel subunits and Igg domains. Nature. 1996;383(6598):307–308. doi: 10.1038/383307b0 [DOI] [PubMed] [Google Scholar]
- [40].Morgan K, Stevens EB, Shah B, et al. Beta-3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci, USA. 2000;97(5):2308–2313. doi: 10.1073/pnas.030362197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yu FH, Westenbroek RE, Silos-Santiago I, et al. Sodium channel beta-4, a new disulfide-linked auxiliary subunit with similarity to beta-2. J Neurosci. 2003;23(20):7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Murphy KMM, Gould RJ, Largent BL, et al. A unitary mechanism of calcium antagonist drug action. Proc Natl Acad Sci, USA. 1983;80(3):860–864. doi: 10.1073/pnas.80.3.860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Glossmann H, Ferry R, Goll A, et al. Calcium channels: basic properties as revealed by radioligand binding studies. J Cardiovasc Pharmacol. 1985;7:520–530. doi: 10.1097/00005344-198500076-00005 [DOI] [PubMed] [Google Scholar]
- [44].Curtis BM, Catterall WA. Purification of the calcium antagonist receptor of the voltage-sensitive calcium channel from skeletal muscle transverse tubules. Biochem. 1984;23(10):2113–2118. doi: 10.1021/bi00305a001 [DOI] [PubMed] [Google Scholar]
- [45].Curtis BM, Catterall WA. Reconstitution of the voltage-sensitive calcium channel purified from skeletal muscle transverse tubules. Biochemistry. 1986;25(11):3077–3083. doi: 10.1021/bi00359a002 [DOI] [PubMed] [Google Scholar]
- [46].Sieber M, Nastainczyk W, Zubor V, et al. The 165-kDa peptide of the purified skeletal muscle dihydropyridine receptor contains the known regulatory sites of the calcium channel. Eur J Biochem. 1987;167(1):117–122. doi: 10.1111/j.1432-1033.1987.tb13311.x [DOI] [PubMed] [Google Scholar]
- [47].Flockerzi V, Oeken HJ, Hofmann F, et al. Purified dihydropyridine-binding site from skeletal muscle t-tubules is a functional calcium channel. Nature. 1986;323(6083):66–68. doi: 10.1038/323066a0 [DOI] [PubMed] [Google Scholar]
- [48].Takahashi M, Seagar MJ, Jones JF, et al. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc Natl Acad Sci, USA. 1987;84(15):5478–5482. doi: 10.1073/pnas.84.15.5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Catterall WA. Structure and function of voltage-gated sodium and calcium channels. Curr Opin Neurobiol. 1991. Jun;1(1):5–13. doi: 10.1016/0959-4388(91)90004-Q [DOI] [PubMed] [Google Scholar]
- [50].Sharp AH, Imagawa T, Leung AT, et al. Identification and characterization of the dihydropyridine-binding subunit of the skeletal muscle dihydropyridine receptor. J Biol Chem. 1987;262(25):12309–12315. doi: 10.1016/S0021-9258(18)45353-7 [DOI] [PubMed] [Google Scholar]
- [51].Striessnig J, Knaus HG, Grabner M, et al. Photoaffinity labelling of the phenylalkylamine receptor of the skeletal muscle transverse-tubule calcium channel. FEBS Lett. 1987;212(2):247–253. doi: 10.1016/0014-5793(87)81354-6 [DOI] [PubMed] [Google Scholar]
- [52].Leung AT, Imagawa T, Campbell KP. Structural characterization of the 1,4-dihydropyridine receptor of the voltage-dependent Ca2+ channel from rabbit skeletal muscle. Evidence for two distinct high molecular weight subunits. J Biol Chem. 1987;262(17):7943–7946. doi: 10.1016/S0021-9258(18)47507-2 [DOI] [PubMed] [Google Scholar]
- [53].Jay SD, Ellis SB, McCue AF, et al. Primary structure of the gamma subunit of the DHP-sensitive calcium channel from skeletal muscle. Science. 1990;248:490–492. doi: 10.1126/science.2158672 [DOI] [PubMed] [Google Scholar]
- [54].Ruth P, Röhrkasten A, Biel M, et al. Primary structure of the beta subunit of the DHP-sensitive calcium channel from skeletal muscle. Science. 1989;245:1115–1118. doi: 10.1126/science.2549640 [DOI] [PubMed] [Google Scholar]
- [55].Vaghy PL, Striessnig J, Miwa K, et al. Identification of a novel 1,4-dihydropyridine- and phenylalkylamine-binding polypeptide in calcium channel preparations. J Biol Chem. 1987;262(29):14337–14342. doi: 10.1016/S0021-9258(18)47943-4 [DOI] [PubMed] [Google Scholar]
- [56].Nunoki K, Florio V, Catterall WA. Activation of purified calcium channels by stoichiometric protein phosphorylation. Proc Natl Acad Sci, USA. 1989;86(17):6816–6820. doi: 10.1073/pnas.86.17.6816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tanabe T, Takeshima H, Mikami A, et al. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328(6128):313–318. doi: 10.1038/328313a0 [DOI] [PubMed] [Google Scholar]
- [58].Ellis SB, Williams ME, Ways NR, et al. Sequence and expression of mRNAs encoding the α1 and α2 subunits of a DHP-sensitive calcium channel. Science. 1988;241(4873):1661–1664. doi: 10.1126/science.2458626 [DOI] [PubMed] [Google Scholar]
- [59].De Jongh KS, Warner C, Catterall WA. Subunits of purified calcium channels. α2 and δ are encoded by the same gene. J Biol Chem. 1990;265:14738–14741. doi: 10.1016/S0021-9258(18)77174-3 [DOI] [PubMed] [Google Scholar]
- [60].Jay SD, Sharp AH, Kahl SD, et al. Structural characterization of the dihydropyridine-sensitive calcium channel alpha-2 subunit and the associated delta peptides. J Biol Chem. 1991;266:3287–3293. doi: 10.1016/S0021-9258(18)49986-3 [DOI] [PubMed] [Google Scholar]
- [61].Davies A, Kadurin I, Alvarez-Laviada A, et al. The α2δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci, USA. 2010;107(4):1255–1690. doi: 10.1073/pnas.0908735107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hoppa MB, Lana B, Margas W, et al. α 2 δ expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486(7401):122–125. doi: 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242(5398):459–461. doi: 10.1038/242459a0 [DOI] [PubMed] [Google Scholar]
- [64].Armstrong CM, Benzanilla F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J Gen Physiol. 1974;63:533–552. doi: 10.1085/jgp.63.5.533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kuzmenkin A, Bezanilla F, Correa AM. Gating of the bacterial sodium channel, NaChBac: voltage-dependent charge movement and gating currents. J Gen Physiol. 2004;124(4):349–356. doi: 10.1085/jgp.200409139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hirschberg B, Rovner A, Lieberman M, et al. Transfer of twelve charges is needed to open skeletal muscle Na+ channels. J Gen Physiol. 1995;106(6):1053–1068. doi: 10.1085/jgp.106.6.1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67(6):915–928. doi: 10.1016/j.neuron.2010.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Catterall WA. Voltage-dependent gating of sodium channels: correlating structure and function. Trends Neurosci. 1986;9:7–10. doi: 10.1016/0166-2236(86)90004-4 [DOI] [Google Scholar]
- [69].Catterall WA. Molecular properties of voltage-sensitive sodium channels. Annu Rev Biochem. 1986;55(1):953–985. doi: 10.1146/annurev.bi.55.070186.004513 [DOI] [PubMed] [Google Scholar]
- [70].Stuhmer W, Conti F, Suzuki H, et al. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339(6226):597–603. doi: 10.1038/339597a0 [DOI] [PubMed] [Google Scholar]
- [71].Papazian DM, Timpe LC, Jan YN, et al. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349(6307):305–310. doi: 10.1038/349305a0 [DOI] [PubMed] [Google Scholar]
- [72].Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J Gen Physiol. 1997;110(4):391–401. doi: 10.1085/jgp.110.4.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aggarwal SK, MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker potassium channel. Neuron. 1996;16:1169–1177. doi: 10.1016/S0896-6273(00)80143-9 [DOI] [PubMed] [Google Scholar]
- [74].Rogers JC, Qu Y, Tanada TN, et al. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950 [DOI] [PubMed] [Google Scholar]
- [75].Cestèle S, Qu Y, Rogers JC, et al. Voltage sensor-trapping: enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron. 1998;21:919–931. doi: 10.1016/S0896-6273(00)80606-6 [DOI] [PubMed] [Google Scholar]
- [76].Yang N, George AL, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16(1):113–122. doi: 10.1016/S0896-6273(00)80028-8 [DOI] [PubMed] [Google Scholar]
- [77].Yang N, George AL, Horn R. Probing the outer vestibule of a sodium channel voltage sensor. Biophys J. 1997;73(5):2260–2268. doi: 10.1016/S0006-3495(97)78258-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].DeCaen PG, Yarov-Yarovoy V, Zhao Y, et al. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci, USA. 2008;105(39):15142–15147. doi: 10.1073/pnas.0806486105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Vargas E, Yarov-Yarovoy V, Khalili-Araghi F, et al. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J Gen Physiol. 2012;140(6):587–594. doi: 10.1085/jgp.201210873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Armstrong CM, Bezanilla F, Rojas E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol. 1973;62(4):375–391. doi: 10.1085/jgp.62.4.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241(4873):1658–1661. doi: 10.1126/science.2458625 [DOI] [PubMed] [Google Scholar]
- [82].Vassilev P, Scheuer T, Catterall WA. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci, USA. 1989;86(20):8147–8151. doi: 10.1073/pnas.86.20.8147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].West JW, Patton DE, Scheuer T, et al. A cluster of hydrophobic amino acid residues required for fast Na + channel inactivation. Proc Natl Acad Sci, USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12(5):1041–1048. doi: 10.1016/0896-6273(94)90312-3 [DOI] [PubMed] [Google Scholar]
- [85].Eaholtz G, Zagotta WN, Catterall WA. Kinetic analysis of block of open sodium channels by a peptide containing the isoleucine, phenylalanine, and methionine (IFM) motif from the inactivation gate. J Gen Physiol. 1998;111(1):75–82. doi: 10.1085/jgp.111.1.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rohl CA, Boeckman FA, Baker C, et al. Solution structure of the sodium channel inactivation gate. Biochemistry. 1999;38(3):855–861. doi: 10.1021/bi9823380 [DOI] [PubMed] [Google Scholar]
- [87].Adelman WJ, Palti Y. The effects of external potassium and long duration voltage conditioning on the amplitude of sodium currents in the giant axon of the squid Loligo pealei. J Gen Physiol. 1968;54:589–606. doi: 10.1085/jgp.54.5.589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Vilin YY, Ruben PC. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys. 2001;35(2):171–90. doi: 10.1385/CBB:35:2:171. PMID: 11892790. [DOI] [PubMed] [Google Scholar]
- [89].Rudy B. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. Journal of Physiology. 1978;283(1):1–21. doi: 10.1113/jphysiol.1978.sp012485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Balser JR, Nuss HB, Chiamvimonvat N, et al. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J Physiol (Lond). 1996;494:431–442. doi: 10.1113/jphysiol.1996.sp021503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Balser JR, Nuss HB, Chiamvimonvat N, et al. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J Physiol. 1996;494(Pt 2):431–442. doi: 10.1113/jphysiol.1996.sp021503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Xiong W, Farukhi YZ, Tian Y, et al. A conserved ring of charge in mammalian Na+ channels: a molecular regulator of the outer pore conformation during slow inactivation. J Physiol. 2006;576(Pt 3):739–754. doi: 10.1113/jphysiol.2006.115105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Noda M, Suzuki H, Numa S, et al. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989;259(1):213–216. doi: 10.1016/0014-5793(89)81531-5 [DOI] [PubMed] [Google Scholar]
- [94].Heinemann SH, Terlau H, Stühmer W, et al. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356(6368):441–443. doi: 10.1038/356441a0 [DOI] [PubMed] [Google Scholar]
- [95].Schlief T, Schönherr R, Imoto K, et al. Pore properties of rat brain II sodium channels mutated in the selectivity filter domain. Eur Biophys J. 1996;25(2):75–91. doi: 10.1007/s002490050020 [DOI] [PubMed] [Google Scholar]
- [96].Ren D, Navarro B, Xu H, et al. A prokaryotic voltage-gated sodium channel. Science. 2001;294(5550):2372–2375. doi: 10.1126/science.1065635 [DOI] [PubMed] [Google Scholar]
- [97].Payandeh J, Scheuer T, Zheng N, et al. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475(7356):353–358. doi: 10.1038/nature10238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wisedchaisri G, Tonggu L, McCord E, et al. Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell. 2019;178(4):993–1003.e12. doi: 10.1016/j.cell.2019.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chakrabarti N, Ing C, Payandeh J, et al. Catalysis of Na+ permeation in the bacterial sodium channel NaVAb. Proc Natl Acad Sci, USA. 2013;110(28):11331–11336. doi: 10.1073/pnas.1309452110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Pavlov E, Bladen C, Winkfein R, et al. The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys J. 2005;89(1):232–242. doi: 10.1529/biophysj.104.056994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].El-Din TM G, Lenaeus MJ, Ramanadane K, et al. Molecular dissection of multiphase inactivation of the bacterial sodium channel NaVAb. J Gen Physiol. 2019;151(2):174–185. doi: 10.1085/jgp.201711884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Payandeh J, Gamal El-Din TM, Scheuer T, et al. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486(7401):135–139. doi: 10.1038/nature11077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ong BH, Tomaselli GF, Balser JR. A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J Gen Physiol. 2000;116(5):653–662. doi: 10.1085/jgp.116.5.653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69(4):497–515. doi: 10.1085/jgp.69.4.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hondeghem LM, Katzung BG. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel blocking drugs. Annu Rev Pharmacol Toxicol. 1984;24:387–423. doi: 10.1146/annurev.pa.24.040184.002131 [DOI] [PubMed] [Google Scholar]
- [106].El-Din TM G, Lenaeus MJ, Zheng N, et al. Fenestrations control resting-state block of a voltage-gated sodium channel. Proc Natl Acad Sci, USA. 2018;115:13111–13116. doi: 10.1073/pnas.1814928115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Koishi R, Xu H, Ren D, et al. A superfamily of voltage-gated sodium channels in bacteria. J Biol Chem. 2004;279(10):9532–9538. doi: 10.1074/jbc.M313100200 [DOI] [PubMed] [Google Scholar]
- [108].Payandeh J, Minor DL Jr.. Bacterial voltage-gated sodium channels (BacNavs) from the soil, sea, and salt lakes enlighten molecular mechanisms of electrical signaling and pharmacology in the brain and heart. J Mol Biol. 2015;427(1):3–30. doi: 10.1016/j.jmb.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Almers W, McCleskey EW, Palade PT. A nonselective cation conductance in frog muscle membrane blocked by micromolar external Ca++. J Physiol. 1984;353:565–583. doi: 10.1113/jphysiol.1984.sp015351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Almers W, McCleskey EW. The nonselective conductance due to calcium channels in frog muscle: calcium-selectivity in a single file pore. J Physiol. 1984;353:585–608. doi: 10.1113/jphysiol.1984.sp015352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Hess P, Tsien RW. Mechanism of ion permeation through calcium channels. Nature. 1984;309(5967):453–456. doi: 10.1038/309453a0 [DOI] [PubMed] [Google Scholar]
- [112].Sather WA, McCleskey EW. Permeation and selectivity in calcium channels. Annu Rev Physiol. 2003;65(1):133–159. doi: 10.1146/annurev.physiol.65.092101.142345 [DOI] [PubMed] [Google Scholar]
- [113].Tang L, Gamal El-Din TM, Payandeh J, et al. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature. 2014;505(7481):56–61. doi: 10.1038/nature12775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Glossmann H, Striessnig J. Calcium channels. Vitam Horm. 1988;44:155–328. [DOI] [PubMed] [Google Scholar]
- [115].Godfraind T. Discovery and development of calcium channel blockers. Front Pharmacol. 2017;8:286. doi: 10.3389/fphar.2017.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Sampson KJ, Kass RK. Antiarrhythmic drugs. Goodman & Gilman’s pharmacological basis of therapeutics. 12th ed. McGraw Hill, New York. 2011. pp. 815–848. [Google Scholar]
- [117].Striessnig J. Pharmacology, structure and function of cardiac L-type calcium channels. Cell Physiol Biochem. 1999;9(4–5):242–269. doi: 10.1159/000016320 [DOI] [PubMed] [Google Scholar]
- [118].Hofmann F, Lacinová L, Klugbauer N. Voltage-dependent calcium channels: from structure to function. Rev Physiol Biochem Pharmacol. 1999;139:33–87. [DOI] [PubMed] [Google Scholar]
- [119].Hockerman GH, Peterson BZ, Johnson BD, et al. Molecular determinants of drug binding and action on L-type calcium channels. Annu Rev Pharmacol Toxicol. 1997;37(1):361–396. doi: 10.1146/annurev.pharmtox.37.1.361 [DOI] [PubMed] [Google Scholar]
- [120].Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol. 1983;81(5):613–642. doi: 10.1085/jgp.81.5.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hondeghem LM, Katzung BG. Timed- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-X [DOI] [PubMed] [Google Scholar]
- [122].Bean BP. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc Natl Acad Sci, USA. 1984;81(20):6388–6392. doi: 10.1073/pnas.81.20.6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Striessnig J, Glossmann H, Catterall WA. Identification of a phenylalkylamine binding region within the α1 subunit of skeletal muscle Ca channels. Proc Natl Acad Sci, USA. 1990;87:9108–9112. doi: 10.1073/pnas.87.23.9108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Kraus R, Reichl B, Kimball SD, et al. Identification of benz(othi)azepine-binding regions within L-type calcium channel α1 subunits. J Biol Chem. 1996;271(33):20113–20118. doi: 10.1074/jbc.271.33.20113 [DOI] [PubMed] [Google Scholar]
- [125].Striessnig J, Murphy BJ, Catterall WA. The dihydropyridine receptor of L-type Ca2+ channels: identification of binding domains for (+)-[3H]PN200-110 and [3H]azidopine within the alpha-1 subunit. Proc Natl Acad Sci, USA. 1991;88:10769–10773. doi: 10.1073/pnas.88.23.10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Nakayama H, Taki M, Striessnig J, et al. Identification of 1,4-dihydropyridine binding regions within the α1 subunit of skeletal muscle Ca channels by photoaffinity labeling with diazipine. Proc Natl Acad Sci, USA. 1991;88:9203–9207. doi: 10.1073/pnas.88.20.9203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Hockerman GH, Johnson BD, Scheuer T, et al. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels. J Biol Chem. 1995;270:22119–22122. doi: 10.1074/jbc.270.38.22119 [DOI] [PubMed] [Google Scholar]
- [128].Hockerman GH, Johnson BD, Abbott MR, et al. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels in transmembrane segment IIIS6 and the pore region of the a1 subunit. J Biol Chem. 1997;272:18759–18765. doi: 10.1074/jbc.272.30.18759 [DOI] [PubMed] [Google Scholar]
- [129].Peterson BZ, Hockerman GH, Abbot MR, et al. Analysis of the dihydropyridine receptor site of L-type calcium channels by alanine-scanning mutagenesis. J Biol Chem. 1997;272:18752–18758. doi: 10.1074/jbc.272.30.18752 [DOI] [PubMed] [Google Scholar]
- [130].Hockerman GH, Dilmac N, Scheuer T. Molecular determinants of diltiazem block in domains III S6 and IVS6 of L-type Ca channels. Mol Pharmacol. 2000;58:1264–1270. doi: 10.1124/mol.58.6.1264 [DOI] [PubMed] [Google Scholar]
- [131].Mitterdorfer J, Wang ZY, Sinnegger MJ, et al. Two amino acid residues in the IIIS5 segment of L-type calcium channels differentially contribute to 1,4-dihydropyridine sensitivity. J Biol Chem. 1996;271(48):30330–30335. doi: 10.1074/jbc.271.48.30330 [DOI] [PubMed] [Google Scholar]
- [132].Schuster A, Lacinová L, Klugbauer N, et al. The IVS6 segment of the L-type calcium channel is critical for the action of dihydropyridines and phenylalkylamines. EMBO J. 1996;15(10):2365–2370. doi: 10.1002/j.1460-2075.1996.tb00592.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Ito H, Klugbauer N, Hofmann F. Transfer of the high affinity dihydropyridine sensitivity from L-type to non-L-type calcium channel. Mol Pharmacol. 1997;52(4):735–740. doi: 10.1124/mol.52.4.735 [DOI] [PubMed] [Google Scholar]
- [134].Qian H, Patriarchi T, Price JL, et al. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the beta2-adrenergic receptor in neurons. Sci Signal. 2017 Jan 24;10(463). doi: 10.1126/scisignal.aaf9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hockerman GH, Peterson BZ, Sharp E, et al. Construction of a high-affinity receptor site for dihydropyridine agonists and antagonists by single amino acid substitutions in a non-L-type Ca2+ channel. Proc Natl Acad Sci, USA. 1997;94(26):14906–14911. doi: 10.1073/pnas.94.26.14906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Sinnegger MJ, Wang ZY, Grabner M, et al. Nine L-type amino acid residues confer full 1,4-dihydropyridine sensitivity to the neuronal calcium channel α1A subunit. J Biol Chem. 1997;272(44):27686–27693. doi: 10.1074/jbc.272.44.27686 [DOI] [PubMed] [Google Scholar]
- [137].Tang L, Gamal El-Din TM, Swanson TM, et al. Structural basis for inhibition of a voltage-gated Ca2+ channel by Ca2+ antagonist drugs. Nature. 2016;537(7618):117–121. doi: 10.1038/nature19102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Tang L, Gamal El-Din TM, Lenaeus MJ, et al. Structural basis for diltiazem block of a voltage-gated Ca2+ channel. Mol Pharmacol. 2019;96(4):485–492. doi: 10.1124/mol.119.117531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wu J, Yan Z, Li Z, et al. Structure of the voltage-gated calcium channel CaV.1 at 3.6 a resolution. Nature. 2016;537(7619):191–196. doi: 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- [140].Zhao Y, Huang G, Wu J, et al. Molecular basis for ligand modulation of a mammalian voltage-gated calcium channel. Cell. 2019;177(6):1495–1506.e12. doi: 10.1016/j.cell.2019.04.043 [DOI] [PubMed] [Google Scholar]
- [141].Pan X, Li Z, Zhou Q, et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science. 2018;362(6412). doi: 10.1126/science.aau2486 [DOI] [PubMed] [Google Scholar]
- [142].Shen H, Liu D, Wu K, et al. Structures of human NaV.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363(6433):1303–1308. doi: 10.1126/science.aaw2493 [DOI] [PubMed] [Google Scholar]
- [143].Pan X, Li Z, Huang X, et al. Molecular basis for pore blockade of human Na+ channel Nav.2 by the μ-conotoxin KIIIA. Science. 2019;363(6433):1309–1313. doi: 10.1126/science.aaw2999 [DOI] [PubMed] [Google Scholar]
- [144].Fan X, Huang J, Jin X, et al. Cryo-EM structure of human voltage-gated sodium channel Nav.6. Proc Natl Acad Sci U S A. 2023;120(5):e2220578120. doi: 10.1073/pnas.2220578120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Zhang J, Shi Y, Huang Z, et al. Structural basis for NaV.7 inhibition by pore blockers Nat Struct Mol Biol. Nat Struct Mol Biol. 2022;29(12):1208–1216. doi: 10.1038/s41594-022-00860-1 [DOI] [PubMed] [Google Scholar]
- [146].Li Y, Yuan T, Huang B, et al. Structure of human NaV.6 channel reveals Na+ selectivity and pore blockade by 4,9-anhydro-tetrodotoxin. Nat Commun. 2023;14(1):1030. doi: 10.1038/s41467-023-36766-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Jiang D, Shi H, Tonggu L, et al. Structure of the cardiac sodium channel. Cell. 2020;180(1):122–134.e10. doi: 10.1016/j.cell.2019.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Li Z, Jin X, Wu T, et al. Structural basis for pore blockade of the human cardiac sodium channel Nav1.5. Angew Chem Int Ed Engl. 2021;60(20):11474–11480. doi: 10.1002/anie.202102196 [DOI] [PubMed] [Google Scholar]
- [149].Zhao Y, Huang G, Wu Q, et al. Cryo-EM structures of apo and antagonist-bound human CaV.1. Nature. 2019;576(7787):492–497. doi: 10.1038/s41586-019-1801-3 [DOI] [PubMed] [Google Scholar]
- [150].Gao S, Yao X, Yan N. Structure of human Cav2.2 channel blocked by the painkiller ziconotide. Nature. 2021;596(7870):143–147. doi: 10.1038/s41586-021-03699-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Jiang D, Banh R, Gamal El-Din TM, et al. Open-state structure and pore gating mechanism of the cardiac sodium channel. Cell. 2021;184(20):5151–5162.e11. doi: 10.1016/j.cell.2021.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Jiang D, Gamal El-Din T, Zheng N, et al. Expression and purification of the cardiac sodium channel Nav1.5. Methods Enzymol. 2021;653:89–101. [DOI] [PubMed] [Google Scholar]
- [153].Sokolov S, Scheuer T, Catterall WA. Ion permeation through a voltage-sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron. 2005;47(2):183–189. doi: 10.1016/j.neuron.2005.06.012 [DOI] [PubMed] [Google Scholar]
- [154].Sokolov S, Scheuer T, Catterall WA. Gating pore current in an inherited ion channelopathy. Nature. 2007;446(7131):76–78. doi: 10.1038/nature05598 [DOI] [PubMed] [Google Scholar]
- [155].Struyk AF, Cannon SC. A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol. 2007;130(1):11–20. doi: 10.1085/jgp.200709755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Sokolov S, Scheuer T, Catterall WA. Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci U S A. 2008;105(50):19980–19985. doi: 10.1073/pnas.0810562105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Moreau A, Gosselin-Badaroudine P, Boutjdir M, et al. Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front Pharmacol. 2015;6:301. doi: 10.3389/fphar.2015.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].El-Din TM G, Lantin T, Tschumi CW, et al. Autism-associated mutations in Kv7/KCNQ channels induce gating pore current. Proc Natl Acad Sci, USA. 2021;118(45). doi: 10.1073/pnas.2112666118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Jiang D, Gamal El-Din TM, Ing C, et al. Structural basis for gating pore current in periodic paralysis. Nature. 2018;557(7706):590–594. doi: 10.1038/s41586-018-0120-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Favre I, Moczydlowski E, Schild L. On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys J. 1996;71(6):3110–3125. doi: 10.1016/S0006-3495(96)79505-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Benndorf K. Properties of single cardiac Na channels at 35 degrees C. J Gen Physiol. 1994;104(5):801–820. doi: 10.1085/jgp.104.5.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ramos E, O’Leary ME. State-dependent trapping of flecainide in the cardiac sodium channel. J Physiol. 2004;560(Pt 1):37–49. doi: 10.1113/jphysiol.2004.065003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Lenaeus ML, Gamal El-Din TM, Tonggu L, et al. Structural basis for inhibition of the cardiac sodium channels by the atypical antiarrhythmic drug ranolazine. Nat Cardiovasc Res. 2023;2(6):587–594. doi: 10.1038/s44161-023-00271-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Catterall WA, Cestele S, Yarov-Yarovoy V, et al. Voltage-gated ion channels and gating modifier toxins. Toxicon. 2007;49(2):124–141. doi: 10.1016/j.toxicon.2006.09.022 [DOI] [PubMed] [Google Scholar]
- [165].Wang J, Yarov-Yarovoy V, Kahn R, et al. Mapping the receptor site for alpha-scorpion toxins on a Na+ channel voltage sensor. Proc Natl Acad Sci, USA. 2011;108(37):15426–15431. doi: 10.1073/pnas.1112320108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Leipold E, Lu S, Gordon D, et al. Combinatorial interaction of scorpion toxins lqh-2, lqh-3, and LqhalphaIT with sodium channel receptor sites-3. Mol Pharmacol. 2004;65(3):685–691. doi: 10.1124/mol.65.3.685 [DOI] [PubMed] [Google Scholar]
- [167].Chen H, Heinemann SH. Interaction of scorpion alpha-toxins with cardiac sodium channels: binding properties and enhancement of slow inactivation. J Gen Physiol. 2001;117(6):505–518. doi: 10.1085/jgp.117.6.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Jiang D, Tonggu L, Gamal El-Din TM, et al. Structural basis for voltage-sensor trapping of the cardiac sodium channel by a deathstalker scorpion toxin. Nat Commun. 2021;12(1):128. doi: 10.1038/s41467-020-20078-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Sather WA, Yang J, Tsien RW. Structural basis of ion channel permeation and selectivity. Curr Opin Neurobiol. 1994;4:313–323. doi: 10.1016/0959-4388(94)90091-4 [DOI] [PubMed] [Google Scholar]
- [170].Ragsdale DS, McPhee JC, Scheuer T, et al. Molecular determinants of state-dependent block of sodium channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162 [DOI] [PubMed] [Google Scholar]
- [171].Yan Z, Zhou Q, Wang L, et al. Structure of the Nav1.4-beta1 complex from electric eel. Cell. 2017;170(3):470–482 e11. doi: 10.1016/j.cell.2017.06.039 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Because this is a review article, no new experimental data are presented. Interested individuals should write to the authors of the papers that are cited in each figure legend to obtain the data presented in that figure.