

Abstract
Sleep latency, the amount of time that it takes an individual to fall asleep, is a key indicator of sleep need. Sleep latency varies considerably both among and within species and is heritable, but lacks a comprehensive description of its underlying genetic network. Here we conduct a genome-wide association study of sleep latency. Using previously collected sleep and activity data on a wild-derived population of flies, we calculate sleep latency, confirming significant, heritable genetic variation for this complex trait. We identify 520 polymorphisms in 248 genes contributing to variability in sleep latency. Tests of mutations in 23 candidate genes and additional putative pan-neuronal knockdown of 9 of them implicated CG44153, Piezo, Proc-R and Rbp6 in sleep latency. Two large-effect mutations in the genes Proc-R and Piezo were further confirmed via genetic rescue. This work greatly enhances our understanding of the genetic factors that influence variation in sleep latency.
Subject terms: Genetics, Behavioural genetics, Genetic association study, Heritable quantitative trait
Introduction
Sleep onset latency in humans is the number of minutes that it takes to fall asleep once the attempt to sleep is made. It is the difference between the desired sleep time and the actual start of sleep. Short sleep latencies may reflect sleep deprivation or excessive sleepiness1. Long sleep latencies have a more nuanced interpretation; they may reflect a reduced sleep need, or they may reflect difficulty in falling asleep due to increased arousal, as occurs in individuals with insomnia2. Researchers use a combination of polysomnography, actigraphy, and self-report/questionnaires to assess sleep latency in humans1,3–7. Human mean sleep latencies range from 10 to 21 min3,4. Heritabilities for human sleep latency are moderate, from 0.18 to 0.321,4, suggesting that genes influence this trait. Accordingly, several human studies identified candidate genes for sleep latency. SNPs in the third intron of the CACNA1C gene were associated with sleep latency in individuals from the Australian Twin Registry4. A GWAS of sleep and activity parameters assessed with actigraphy found a SNP near DMRT1 associated with sleep latency3. A meta-analysis of GWAS found that sleep latency associated with three variants in an intron of RBFOX31. Also, a scan of 2000 candidate genes identified polymorphisms in DRD2 associated with sleep latency and sleep duration5. In addition, candidate gene studies identified the 5-HTR2A receptor6 as being associated with sleep latency, and MTNR1B with REM sleep latency7. Thus, sleep latency exhibits a partial genetic basis in humans, and several candidate genes have been identified for the trait.
Additional evidence indicates that sleep latency is a complex trait, modifiable by potentially large numbers of genes. Sleep latency is often measured after a subject has been deprived of sleep. The earliest use of sleep latency as an objective measure of sleep debt noted a roughly linear relationship between the amount sleep lost and the corresponding sleep latency in humans8. However, the linear relationship could be altered by sampling sleep latency at different timepoints, suggesting the involvement of the molecular circadian clock8, and the relationship between sleep latency and the circadian clock was later demonstrated in flies9. The correlation between sleep latency and prior wakefulness has been observed in rodent models as well10, but can be disrupted by exposing the animals to different types of sleep-depriving stimuli11. For example, sleep latency was reduced in mice deprived of sleep via gentle handling but increased when constant cage change was used to deprive the animals of sleep, despite the fact that the amount of sleep lost was the same among groups11. Broad-sense heritabilities for sleep latency in mice under two different handling conditions were estimated to be between 0.28 and 0.70 for Diversity Outcross founder strains, comparable to heritabilities found in humans; however, narrow-sense (i.e., additive) heritability estimates for the Diversity Outcross itself were low, indicating a significant influence of both dominance and epistasis on this trait12. The overall implication is that while sleep latency has a genetic component, it can be perturbed by the circadian factors, sensory experience, and arousal in addition to exhibiting epistasis and dominance.
Sleep latency in flies is typically calculated without additional experimental intervention. Night sleep latency (often simply referred to as sleep latency) is the number of minutes that it takes the flies to fall asleep after the lights are turned off. Day sleep latency has a similar definition—it is the number of minutes it takes the fly to fall asleep after the lights are turned on. These quantitative measures in flies reflect the endogenous need to sleep. Though many mutant screens and candidate gene approaches report the effects of mutations on sleep latency (for example wake9, Rdl13, amn14, and NPF and NPFR115), a systematic unbiased search for the genes that contribute to variability in night sleep latency in nature has not yet been conducted in flies. We therefore conducted a genome-wide association study of night sleep latency in the Drosophila Genetic Reference Panel (DGRP). The DGRP is a community resource created to identify genetic variants underlying complex traits like sleep latency16,17. The DGRP has been used to investigate over 61 complex traits18, including sleep, day-to-day fluctuations in sleep, and circadian behavior19–21. We found that sleep latency was not only variable in the DGRP, but heritable as well. Sleep latency is genetically correlated with many sleep traits, including a strong negative genetic correlation with night sleep duration. We mapped 520 variants tagging 248 genes to sleep latency, and verified candidate genes through mutational analysis and genetic rescue.
Results
Sleep latency is highly variable and heritable in the DGRP
Mean sleep latency was highly variable in the DGRP, ranging from 4.2 min ± 5.2 SD to 176.6 min ± 148.7 SD, with a population average of 41.4 min ± 2.0 SD (Fig. 1A; Supplementary Table S1). Similar to the male population mean of 42.08 min ± 0.52 SD and the female population mean of 40.45 min ± 0.66 SD, w1118; Canton-S B control males had a sleep latency of 30.91 min ± 1.12 SD, while control females had a sleep latency of 39.26 min ± 2.11 SD. The genetic component of variance was highly significant for both sexes combined or for each sex separately (PLine(Block) < 0.0001) (Table 1). Accordingly, the combined-sex broad-sense heritability H2 was 0.44, relatively high for a behavioral trait. Sleep latency was highly sexually dimorphic; consequently, the cross-sex genetic correlation was low (rmf = 0.32), suggesting that sex-specific differences in the genetic basis of sleep latency exist. When mean sleep latencies are ordered numerically for females and compared to males, large differences between the two sexes can be seen (Fig. 1B). The minimum sleep latency in females was 6.2 min while the maximum was 261.7 min, a greater range than males, whose sleep latencies ranged from 2.1 to 163.3 min. The high levels of genetic variance and heritability for sleep latency indicated that genome-wide association mapping would be fruitful.
Figure 1.
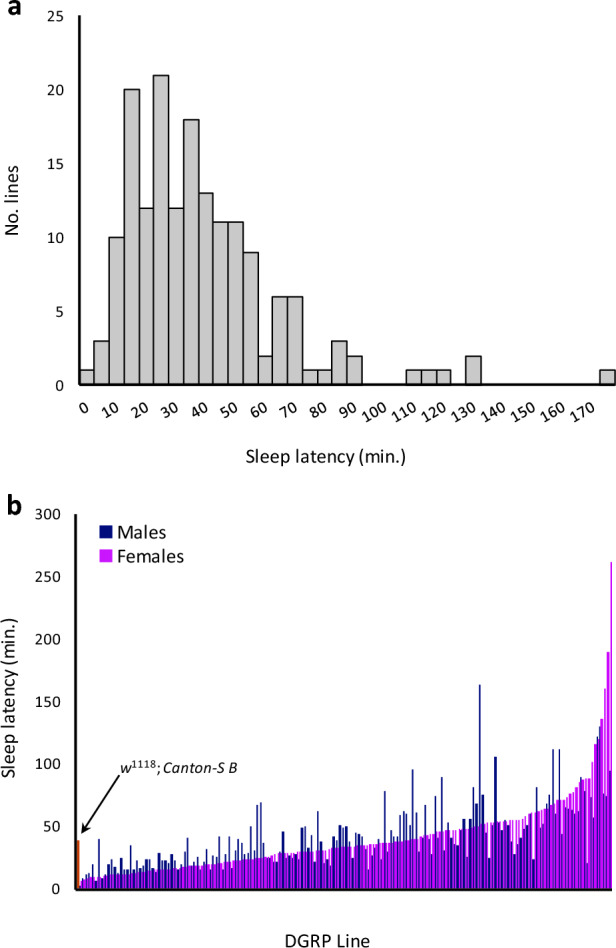
Sleep latency is highly variable and sexually dimorphic in the DGRP. (a), the histogram shows the distribution of sleep latency in the DGRP. (b), Mean sleep latency for each line/sex ordered numerically by sleep latency in females. w1118; Canton-S B control flies are plotted at the beginning of the distribution, with males shown by a light orange bar and females shown by a dark orange bar.
Table 1.
Quantitative genetic analysis of sleep latency.
| Sex | Source | d.f | M.S | F | P | σ2 | H2 | rmf |
|---|---|---|---|---|---|---|---|---|
| Combined | Block | 3 | 52,165.08 | 1.20 | 0.3133 | 2.46 | 0.44 | 0.32 |
| Sex | 1 | 6536.99 | 0.62 | 0.4338 | Fixed | |||
| Line (Block) | 164 | 40,502.69 | 3.99 | < 0.0001 | 510.44 | |||
| Sex × Line (Block) | 164 | 9963.45 | 7.75 | < 0.0001 | 289.05 | |||
| Rep (Block) | 12 | 4781.18 | 2.21 | 0.0803 | 4.37 | |||
| Sex × Rep (Block) | 12 | 1989.84 | 1.55 | 0.1038 | 1.86 | |||
| Line × Rep (Block) | 492 | 1459.89 | 1.13 | 0.0817 | 11.47 | |||
| Sex × Line × Rep (Block) | 490 | 1287.50 | 1.37 | < 0.0001 | 45.47 | |||
| Error | 8897 | 941.28 | – | – | 941.22 | |||
| Males | Block | 3 | 17,143.10 | 0.83 | 0.4810 | 0.00 | 0.45 | |
| Line (Block) | 164 | 19,799.61 | 17.60 | < 0.0001 | 617.69 | |||
| Rep (Block) | 12 | 2179.83 | 1.94 | 0.0281 | 3.35 | |||
| Line × Rep (Block) | 490 | 1126.91 | 1.58 | < 0.0001 | 54.59 | |||
| Error | 4462 | 711.18 | – | – | 711.01 | |||
| Females | Block | 3 | 42,328.98 | 1.27 | 0.2881 | 6.63 | 0.44 | |
| Line (Block) | 164 | 30,669.99 | 18.95 | < 0.0001 | 979.86 | |||
| Rep (Block) | 12 | 4545.23 | 2.81 | 0.0010 | 8.85 | |||
| Line × Rep (Block) | 492 | 1620.16 | 1.38 | < 0.0001 | 58.92 | |||
| Error | 4435 | 1172.77 | – | – | 1172.89 |
d.f., Degrees of freedom; M.S., mean sum of squares; F, F-value; P, P-value; σ2, variance estimated by restricted maximum likelihood; H2, broad-sense heritability; rmf, cross-sex correlation.
Genotype–phenotype associations identify candidate genes for sleep latency
The presence of genetic variance indicates that sleep latency can be mapped to the genome, but the low cross-sex genetic correlation suggests that different genes may contribute to sleep latency in males and females. Accordingly, we conducted the genome-wide association tests with male and female sleep latency separately and the difference between the line means of each sex (male–female) in addition to the analysis for both sexes combined. The DGRP2 webtool first determines whether Wolbachia pipientis infection and chromosomal inversions affect the phenotype of interest17, in this case sleep latency; however, neither Wolbachia nor inversion status had a significant effect. Using the DGRP2 webtool, we identified 520 unique single nucleotide polymorphisms (SNPs) that mapped to within ± 1000 bp of 248 genes (Fig. 2; Supplementary Table S2). For sexes combined, 132 polymorphisms mapped to 67 genes, with 168 polymorphisms mapping to intergenic regions. Most of the polymorphisms significant in the male-only analysis were unique to males; of the 95 polymorphisms mapping to male sleep latency, 17 overlapped with the sexes-combined analysis, only 2 overlapped with female sleep latency, and 2 overlapped with the sex difference analysis. In contrast, polymorphisms significant in the female-only analysis overlapped with 45% of the polymorphisms from the sexes-combined and sex difference analysis (146 out of 304). Minor allele frequencies were relatively low, with a mean 0.09 for all analyses (Fig. 2A). The relatively low minor allele frequencies of significant SNPs are typical of genome-wide association studies in sleep19,20 as well as other phenotypes in the DGRP16,22–25. A recent meta-analysis of phenotypes studied using the DGRP reported that lower-frequency alleles tend to have higher effect sizes and pleiotropic effects26. Little linkage disequilibrium existed among significant polymorphisms, also a typical characteristic of DGRP studies19,20,22,24,25. Combined-sex effect sizes, estimated as one-half the difference between the major and minor allele means, were large, ranging from − 24.8 to + 9.5 min per SNP, with an average of − 14.5 min. Interestingly, the presence of the minor allele was associated with an increase in sleep latency for all but one polymorphism, which mapped to an intergenic region of the X chromosome (Fig. 2C). The effects of the more common alleles in this population, therefore, were in the direction of reduced sleep latency. Thus, sleep latency is a typical complex trait influenced by many genes, some of which have sex-specific effects.
Figure 2.

Multiple sex-specific loci are associated with sleep latency. The 520 polymorphisms significantly associated with sleep latency (P ≤ 1 × 10–5) are plotted. Each point is color coded to indicate the most significant association from the female-only, male-only, combined sexes, or sex difference analysis. (a), plot of minor allele frequency (MAF) versus genomic location. (b), plot of -log10(P-value) versus genomic location. (c), plot of effect size a normalized by the genetic standard deviation σG.
Several of the 248 candidate genes were enriched for certain KEGG pathways and Gene Ontology categories. Seven Hippo pathway genes were implicated in sleep latency: app, CycE, dlg1, Dlg5, ed, fred, and scrib (FDR = 0.0139). Enrichment was also present for several biological process categories: synaptic target recognition (FDR = 0.0200), motor neuron axon guidance (FDR = 0.0228), homophilic cell–cell adhesion via plasma membrane adhesion molecules (FDR = 0.0013), and cell–cell adhesion (FDR = 0.0463) (Supplementary Table S3). The list was also enriched for plasma membrane proteins (FDR = 0.0013). We used DIOPT (DRSC Integrative Ortholog Prediction Tool) to identify human genes with high homology to the fly genes associated with sleep latency27. While 44 of the genes had no known human orthologs, 136 genes (54.8%) had high homology with at least one human gene, and 65 (26.2%) had more than one predicted human homolog (the remaining 3 genes were not found in the database) (Supplementary Table S4). Thus, the vast majority of the genes we identified, 81%, had human orthologs.
We also examined the phenotypic and genetic correlations between sleep latency and other sleep and circadian traits (Table 2). Notably, night sleep duration had a very strong negative correlation with sleep latency (rG = -1.0, P ≤ 0.0001), implying similar genetic architecture between night sleep duration and sleep latency. Overlapping the SNPs we previously identified for night sleep duration with sleep latency SNPs revealed three overlapping SNPs and two overlapping candidate genes, Rbp6 and alpha-Man-I (Supplementary Tables S5 and S6)19. In addition, seven genes (CG32103, CG34353, fz, kirre, mam, MsR1, and scrib) that we identified for night sleep duration from an artificial selection experiment also overlapped with sleep latency genes28. Tests of a mutation in fz in that study revealed a suggestive pleiotropic effect on sleep latency, increasing it in females28. Like night sleep duration, a high correlation [rG = 1.0 (P ≤ 0.0001)] existed between sleep latency and night sleep duration CVE (the coefficient of environmental variation). Night sleep duration CVE is a measure of the variability in sleep duration observed between flies with the same genotype19. Forty-seven SNPs overlapped between sleep latency and night sleep duration CVE, and twenty genes overlapped (Supplementary Tables S5 and S6)19. In addition, one intergenic SNP overlapped between sleep latency and night sleep σ, which measures the day-to-day fluctuations in night sleep in each fly20. Finally, one intergenic SNP overlapped between and sleep latency and rhythmicity index, the degree of similarity among daily activity patterns21. The shared genetic architecture between sleep latency and other sleep and circadian traits implies pleiotropic gene effects.
Table 2.
Phenotypic and genetic correlations between sleep latency and other sleep and circadian traits.
| Trait | rP | rG |
|---|---|---|
| Night sleep (min.) | − 0.7726 | − 1.0000 |
| Night avg. bout length (min.) | − 0.3733 | − 0.5914 |
| Day sleep (min.) | − 0.2270 | − 0.4251 |
| Day avg. bout length (min.) | − 0.1079 | − 0.2785 |
| Waking activity CVE | − 0.1486 | − 0.2655 |
| Night bout number CVE | − 0.1022 | − 0.2053 |
| Day bout number | − 0.0980 | − 0.1818 |
| Day avg. bout length CVE | − 0.0343 | − 0.1040 |
| Rhythmicity index | 0.0098 | 0.0005 |
| Χ2 period | 0.0182 | 0.0008 |
| MESA period | 0.0135 | 0.0009 |
| Waking activity (cts/min) | 0.0242 | 0.0013 |
| Night avg bout length CVE | 0.0692 | 0.1666 |
| Night bout number | 0.2153 | 0.3433 |
| Day bout number CVE | 0.1337 | 0.3488 |
| Day sleep CVE | 0.1913 | 0.4203 |
| Night sleep CVE | 0.7944 | 1.0000 |
rG, Genetic correlation; rP, phenotypic correlation; CVE, coefficient of environmental variation. Numbers in bold are statistically significant.
In addition, we tested 43 SNPs with moderate-to-high minor allele frequencies for potential SNP-SNP interactions (i.e., epistasis). We found 6 significant pairwise interactions in males, and 100 in females (Supplementary Table S7), providing evidence that epistasis contributes to sleep latency.
We combined the data from the epistasis analysis and the Gene Ontology analysis in the following way. We searched the BIOGRID29 database for evidence of genetic and protein–protein interactions among sleep latency candidate genes. We found 82 interactions among 69 sleep latency genes. We combined the BIOGRID data with our epistasis analysis, where the epistatic SNPs mapped to within ± 1000 bp of a gene. Figure 3 shows the resulting network, with Gene Ontology categories and gene pathways annotated (see Supplementary Table S8 for the list of genes and annotations). Interestingly, the epistatic network connects to the known genetic and protein–protein interaction network via three genes: RNA-binding protein 6 (Rbp6), terribly reduced optic lobes (trol), and roadkill (rdx).
Figure 3.

Interaction network of sleep latency genes. Known genetic and protein–protein interactions between genes via BIOGRID are plotted as solid gray lines. Epistatic interactions are plotted as blue dashed lines. Genes involved in synaptic target recognition are plotted in blue. Genes that have functions in motor neuron axon guidance are plotted in pink. Genes that are involved in cell–cell adhesion, including homophilic cell adhesion, are plotted in light orange. Gene names within octagons are part of the Hippo pathway. Tutl, which has known functions in synaptic target recognition and cell–cell adhesion, is plotted in green. Fra, which has known functions in motor neuron axon guidance and cell–cell adhesion, is plotted in dark orange. Only networks with connections among four or more genes were plotted; for the full list of genes, see Supplementary Table S8.
Mutant and RNAi knockdown tests verify candidate genes for sleep latency
Using the genome-wide association data, we chose candidate genes to verify. We selected the 100 polymorphisms having the largest effect sizes for both sexes combined that mapped to within ± 1000 bp of a gene. Twenty-three of these genes had Minos insertion lines available for testing. These transposable element insertions putatively disrupt gene function30. We measured sleep and activity in these mutants, and compared them with their isogenic controls. To analyze the mutant data, we considered both sexes-combined data and we also analyzed the data for each sex separately (Supplementary Table S9). For both sexes combined, sleep latency decreased significantly in comparison to the w1118 control for nine Minos mutations (Bonferroni P-value < 0.0031), including CG10713, CG32121, CG34353, CG8086, Gasp, Piezo, Proc-R, sns, and Syt β. Significant decreases in sleep latency were present in females for all nine mutations as well (Fig. 4A). In males, there was a significant decrease in sleep latency for Proc-R and sns only, although the same trend of decreased sleep latency relative to the control was observed for all mutations (Fig. 4A). To mitigate any effect of the w1118 control line genetic background we additionally tested whether these mutants were significantly different from their overall mean using Dunnett’s t-test (P < 0.05). CG43373, Piezo, Proc-R, and upSET were significantly different from the overall mean for both sexes combined; CG43373 was significantly different for both sexes separately; and Proc-R was significant for males (Supplementary Table S9). Both tests implicated Piezo and Proc-R, which had the most extreme sleep latencies.
Figure 4.

Minos insertions having a w1118 control affect sleep latency and exhibit pleiotropic effects on some sleep phenotypes. The plots show mean sleep phenotypes ± SEM in Minos insertion lines contrasted with the sleep phenotypes of the w1118 isogenic control. Light purple bars indicate female data, with the w1118 control shown in dark purple. Light blue bars indicate male data, with the w1118 control shown in dark blue. (a) Sleep latency. (b) Night avg. bout length. (c) Day sleep duration. (d) Day avg. bout length. Asterisks show the level of significance: **** P ≤ 0.0001; *** 0.0001 < P ≤ 0.001; ** 0.001 < P ≤ 0.0031. A pound sign (#) shows those mutants that are also significantly different from the overall mean of all mutant data combined.
Given the strong negative correlation between sleep latency and night sleep, night avg. bout length, day sleep, and day avg. bout length, one might anticipate pleiotropic effects on these traits. For females, we did observe increases in night avg. bout length, day sleep, and day avg. bout length in addition to the reduced sleep latency (Fig. 4B–D). For males, there was a corresponding increase in day sleep for Proc-R and sns (Fig. 4C). Little evidence of pleiotropy was present for night sleep or night bout number (Supplementary Fig. S2A,B) in either males or females. Likewise, few mutations had effects on day bout number or waking activity, as expected due to their low genetic correlation with sleep latency (Supplementary Fig. S2C,D).
Unlike the lines with the w1118 control, the direction of effects of Minos insertions in the y1w67c23 background were highly sex dimorphic. Four lines with insertions in the genes CG32206, CG43427, comm2, and teq had decreased sleep latency in females relative to the control (Fig. 5A; Bonferroni P-value < 0.0071; Supplementary Table S9). In contrast, all seven Minos insertions had significant changes in sleep latency in males—but sleep latency was increased rather than decreased relative to the control (Fig. 5A). In females, little pleiotropy was observed in the other sleep traits (Fig. 5B–H). A Minos insertion in one gene, comm2, exhibited the most pleiotropy, affecting not only sleep latency but night and day sleep duration, avg. bout length, and bout number (Fig. 5B–G). teq was highly pleiotropic in males, with changes occurring in night bout number, day sleep duration, day avg. bout length, and day bout number (Fig. 5D–G). For four of the Minos insertions, there was an increase in sleep latency in males, and a corresponding increase in night sleep, contrary to the expected negative correlation between these variables (Fig. 5A,B). Again we tested whether there were differences in sleep latency between these mutations and their overall mean using Dunnett’s t-test (P < 0.05) to mitigate any y1w67c23 control line effects. CG43427, CG44153, and Rbp6 were significant for sexes combined; CG44153 was significant for males, and Rbp6 was significant for females. Overall, 16 of the 23 Minos mutants tested had effects on sleep latency exceeding a strict Bonferroni correction in at least one sex, and the additional statistical tests supported Piezo, Proc-R, CG43427, CG44153, and Rbp6 as candidate genes for sleep latency.
Figure 5.

Minos insertion lines with y1w67c23 control affect sleep latency and exhibit pleiotropic effects on some sleep phenotypes. The plots show mean sleep phenotypes ± SEM in Minos insertion lines contrasted with the sleep phenotypes of the y1w67c23 isogenic control. Light purple bars indicate female data, with the y1w67c23 control shown in dark purple. Light blue bars indicate male data, with the w1118 control shown in dark blue. (a) Sleep latency. (b) Night sleep duration. (c) Night average bout length. (d) Night bout number. (e) Day sleep. (f) Day average bout length. (g) Day bout number. (h) Waking activity. Asterisks show the level of significance: **** P ≤ 0.0001; *** 0.0001 < P ≤ 0.001; ** 0.001 < P ≤ 0.0071. A pound sign (#) shows those mutants that are also significantly different from the overall mean of all mutant data combined.
We used an additional approach to test nine of the candidate genes having significant Minos insertion sleep latency phenotypes. We used RNAi to knock down the following genes pan-neuronally with an elav-GAL4 driver: CG32121, CG32206, CG34353, CG43427, CG8086, Proc-R, Rbp6, Syt-β, and teq. We compared each elav-GAL4 × RNAi cross with its respective RNAi and the elav-GAL4 driver line as controls. Sleep latency in each cross had a significant effect on genotype for sexes combined (Fig. 6A; Bonferroni-corrected P-value = 0.0055; Supplementary Table S10). We then examined the data for each sex separately (Fig. 6B,C). CG32121, CG32206, CG43427, Proc-R, and teq had significant genotypic effects in females, and CG8086, CG34353, CG43427, and Rbp6 had significant genotypic effects in males (Bonferroni-corrected P-value = 0.0055; Supplementary Table S10). Post-hoc Tukey analysis on the combined-sex data revealed that all but three of the elav-GAL4 × RNAi crosses (CG32121, CG34353, and Syt β) were significantly different from both the corresponding RNAi line as well as the elav-GAL4 driver. For females, CG32206, CG8086, Proc-R, Rbp6, and teq elav-GAL4 × RNAi crosses were significantly different from all controls; for males, only CG8086 crosses were significantly different (Fig. 6B,C). Inspection of Fig. 6 reveals that flies from the elav-GAL4 × RNAi cross have sleep latencies that are intermediate between the controls and could be due to the heterozygous background. We tested this formally using a t-test to determine whether the sleep phenotypes in each elav-GAL4 × RNAi cross were different from what would be expected under a strictly additive model. For females and both sexes combined, Proc-R, Rbp6, and teq crosses were both significantly different from controls and significantly different from an additive model, while males of the CG8086 cross were significantly different from controls and significantly different from an additive model (Supplementary Table S10). Thus, pan-neuronal RNAi knockdown corroborated the role of Proc-R and Rbp6 in sleep latency.
Figure 6.

Sleep latency in TriP RNAi knockdown lines. The plots show sleep latency for each RNAi line, the elav-GAL4 pan-neuronal driver, and the elav-GAL4 × RNAi cross. (a) Sexes combined; (b) Females; (c) Males. An asterisk (*) is plotted for each cross that is significantly different from both the elav-GAL4 driver and its respective RNAi line. A pound sign (#) shows those crosses that are also significantly different from strict additivity.
Overall, the combined mutational and RNAi analysis suggested that CG44153, Piezo, Proc-R, and Rbp6 are candidate genes for sleep latency.
Precise excisions rescue sleep latency phenotypes in Piezo and Proc-R mutants
We created precise excision lines for Piezo and Proc-R as the Minos insertions in these two genes had the most extreme decrease in sleep latency for both sexes combined compared to the w1118 control (Fig. 7A; Supplementary Table S9; Supplementary Figs. S3 and S4). Sleep in Proc-R and Piezo mutants occurred 55.3 and 52.3 min before that of the w1118 control, respectively. For each gene, we tested the sleep latency in the homozygous Minos insertion line, the w1118 control, and a heterozygous cross of the Minos insertion line and control against the precise excision line. The precise excision in Proc-R completely rescued the wildtype phenotype in both females and males (Fig. 7A). The precise excision line for Piezo partially rescued the wildtype phenotype in females and completely rescued the wildtype phenotype in males. For both sexes, sleep latency in the precise excision was indistinguishable from the w1118 control (Fig. 7B). However, for females, sleep latency in the precise excision was also not significantly different from the homozygous Piezo Minos insertion line while for males there was a clear distinction between the precise excision and the homozygous and heterozygous Piezo Minos insertion line. Sleep latency thus maps to both of these candidate genes. In addition, given its role in detecting mechanical stimuli31, we wondered whether Piezo mutants would respond to mechanical shaking during sleep. We measured the arousal threshold of Piezo mutants by stimulating them mechanically once per hour for three days. In comparison to w1118 control and precise excision flies, the proportion of Piezo mutant flies responding to the mechanical stimulus was the same, day or night (Supplementary Fig. S5). However, Piezo mutants slept longer prior to the stimulus, and were awake for less time after the stimulus during the night.
Figure 7.

Precise excision of Minos insertions in Piezo and Proc-R rescues wildtype sleep latency. The figures plot mean sleep latency for each line ± SEM. The rescued line (MB*) is marked in green. Means having the same letter are not significantly different by post-hoc Tukey test. (a) Piezo precise excision data for females and males. For females, sleep latency in the w1118 control was 59.39 ± 7.0 min (n = 32); PiezoMB* (precise excision) was 44.39 ± 5.2 min (n = 32); PiezoMB08675 heterozygote was 27.46 ± 3.4 min (n = 30); and PiezoMB08675 homozygous mutant was 33.45 ± 3.7 min (n = 31). For males, sleep latency in the w1118 control was 49.31 ± 7.0 min (n = 32); PiezoMB* precise excision was 57.54 ± 6.1 min (n = 32); PiezoMB08675 heterozygote was 29.68 ± 3.0 min (n = 30); and PiezoMB08675 homozygous mutant was 32.77 ± 2.9 min (n = 32). (b) Proc-R precise excision for females and males. For females, sleep latency in the w1118 control was 53.48 ± 7.9 min (n = 32); Proc-RMB* (precise excision) was 54.22 ± 4.7 min (n = 32); Proc-RMB00909 heterozygote was 38.02 ± 2.9 min (n = 31); and Proc-RMB00909 homozygous mutant was 22.39 ± 2.6 min (n = 32). For males, sleep latency in the w1118 control was 53.00 ± 4.7 min (n = 32); Proc-RMB* was 53.51 ± 5.2 min (n = 29); Proc-RMB00909 heterozygote was 33.48 ± 2.7 min (n = 31); and Proc-RMB00909 homozygous mutant was 25.74 ± 2.4 min (n = 31).
Discussion
Here we conducted a genome-wide association for sleep latency in flies, greatly extending the catalog of known variants and genes that modify this trait. Sleep latency was both genetically variable and heritable, and interestingly, had a greater range in females than males of the DGRP. We found a strong negative correlation between night sleep duration and sleep latency, consistent with previous artificial selection and candidate gene/mutagenesis experiments. Genetic mapping revealed 520 variants that mapped to 248 genes. By making use of the accessible Minos insertion lines, we effectively validated genes that were selected for testing their involvement in sleep latency. Moreover, our results were further confirmed by conducting experiments that utilized pan-neuronal knockdown through the elav-GAL4 driver. Notably, knockdown of CG44153, Piezo, Proc-R, and Rbp6 provided supplementary evidence of their impact on sleep latency. Of particular significance, the mutations in the Proc-R and Piezo genes exhibited the most substantial influence on sleep latency; interestingly, we achieved a successful restoration of normal effects for both of these genes by precisely eliminating the Minos element.
Variants from the combined-sex and female-specific GWAS tended to overlap, but most of the male-specific variants were unique. We chose genes with larger predicted effect sizes from the combined-sex analysis for verification via mutational analyses; most of these were implicated in the female-specific analysis as well. Interestingly, Minos insertion mutants and TRiP RNAi knockdown also had stronger effects on both sexes combined and on females in particular than on males, despite the differences in genetic background between the DGRP, the Minos insertion lines, and the TRiP RNAi lines. This observation coupled with the low cross-sex correlation for sleep latency implies that different sets of genes influence sleep latency in males and females.
Sleep latency genes were connected to one another in a network using previously reported genetic/protein–protein interactions and significant epistatic interactions from our own analysis. The epistasis analysis we conducted implies a genetic interaction network in addition to known gene interactions, with three genes connecting the two networks: Rbp6, trol, and rdx. Rbp6 is a RNA-binding protein implicated as a heterogeneous nuclear ribonucleoprotein with a putative role in the regulation of translation32,33. trol functions in many processes, including the regulation of cell signaling in hedgehog, FGF, and EGF pathways and the regulation of the stability of the extracellular matrix covering neurons33,34. rdx is part of the hedgehog signaling pathway, with a role in myriad developmental processes35,36. These three genes are highly pleiotropic, and the results from other genome-wide association studies using the DGRP support this observation. All three genes are implicated in olfactory avoidance and the resistance to oxidative stress25,37. Rbp6 is a candidate for activity in response to a rotating stimulus38, fitness traits including age-specific fecundity and the influence of the female nervous system on sperm competition39,40; aggression41, alcohol sensitivity42, and infection resistance43. Further, the three genes are candidate genes for sleep and activity phenotypes: Rbp6 for night sleep duration, night sleep CVE, and waking activity CVE; rdx for waking activity; and trol for day avg. bout length, night sleep CVE, and waking activity CVE19. Interestingly, Rbp6 was also implicated in a GWAS of circadian period21, and its expression is down-regulated in a DGRP line with a long circadian period44. The high degree of pleiotropy in these three genes supports the omnigenic model of inheritance45, which suggests that most of the genetic variation in a trait can be attributed to genes having indirect effects.
Sleep latency tests of Minos insertions were uniformly decreased in flies compared to the w1118 genetic background, while they were they were mostly uniformly decreased in females and increased in males compared to the y1w67c23 genetic background. As we demonstrate here, many genomic variants may impact sleep latency; thus, one possibility is that there are de novo mutations in addition to the focal gene segregating in the genetic backgrounds of the Minos lines that enhance the effect on sleep latency in either the insertion line or the control. However, several lines of evidence argue against the idea that de novo mutations enhance or exaggerate sleep phenotypes. First, standing genetic variation for sleep and activity traits in the DGRP is much lower than would be expected compared to a neutrally evolving model, suggesting that stabilizing selection acts against de novo mutations to favor intermediate phenotypes over extremes46. Similarly, a selective breeding experiment demonstrated that natural selection acts against extreme long and short sleep duration, shifting the allele frequencies of genomic modifiers to generate more moderate sleep47. In addition, long-standing stocks of Shaker mutant alleles did not exhibit a short-sleeping phenotype until outcrossed, suggesting that accumulated de novo mutations suppressed the expected short sleep duration phenotype48. In contrast, outcrossing a mutation in the gene Calreticulin to the DGRP both suppressed and enhanced the effects of the mutation on sleep duration49. These data would suggest that selective forces would tend to suppress the effects of de novo mutations accumulating in the Minos lines. To further support the Minos data, we conducted additional tests using RNAi knockdown lines that were available for nine of the genes we tested. Combined testing and analysis suggested that CG44153, Piezo, Proc-R, and Rbp6 impact sleep latency. We note that the effects of the Minos insertions are not an exact phenocopy of the effects observed using RNAi knockdown for Proc-R and Rbp6. The difference may be due to the location of the Minos insertion, or due to differences in genetic background50. A future direction would be to compare both types of knockdown in the same genetic background, including the Minos rescue experiments.
We conducted genetic rescue in Proc-R, suggesting that it has a role in sleep latency. Proc-R is the receptor for proctolin, a neuropeptide involved in the stimulation of muscle contraction in insects51. Proc-R expression was detected in glial and muscle cells using single-cell RNA-Seq52, while immunolabeling detected Proc-R in neurons of the brain and muscles of the hindgut53. Proc-R is expressed during all developmental stages and at the adult stage, with greater expression during early embryonic development54. Notably, Proc-R expression does not fluctuate in a circadian fashion; instead, it appears to be consistent across time and under different lighting conditions53. While a CRISPR/Cas9-induced knockout of Proc-R was assayed for sleep, no significant effect on day, night, or 24-h sleep was noted55, in contrast to the changes we observed in sleep duration. The identification of Proc-R as a candidate gene for sleep latency is intriguing given that one of the hallmarks of sleep is a relaxed muscle tone.
We also demonstrated that sleep latency maps to Piezo. Piezo is an ion channel expressed in sensory neurons that responds to both mechanical pressure and voltage52,56,57. Its expression is highest during embryonic development but it is expressed at all developmental stages, including the adult stage54. Piezo has many different functions in flies, including, for example, an intriguing role in axon regeneration after injury58. Knockouts of Piezo in larvae have a reduced response to strong mechanical stimuli mediated through ppk-positive neurons that line the larval body wall31. Responses to temperature change and gentle touch are normal in Piezo knockouts, suggesting that the channel detects only relatively strong mechanical stimuli in larvae31. Thus, the role of the Piezo polymorphism in sleep latency may involve an attenuation of arousal threshold during sleep. We tested this idea by measuring arousal threshold in Piezo mutants and their corresponding controls. While the proportion of sleeping flies responding to a repeated shaking stimulus during the night did not differ between Piezo mutants and controls, Piezo mutants slept more between each stimulus and stayed awake for less time after the stimulus, suggesting that they were less perturbed by the shaking. The reduced sleep latency of Piezo mutants may therefore reflect a dampened sensitivity to the environment, particularly mechanical stimuli in the environment. Piezo potentially influences sleep latency through satiety. Recent work demonstrates that dietary protein promotes sleep through peptidergic signaling in the posterior gut, specifically inhibiting the response to mechanical stimulation59. It would be interesting to determine whether Piezo, which functions to sense fullness in the anterior gut60, has a role in this signaling pathway.
The very high negative genetic correlation estimate between sleep duration and sleep latency that we calculated is consistent with much of the sleep literature in flies. For example, selective breeding for an insomnia-like phenotype and for short night sleep duration resulted in a correlated response for sleep latency, increasing it relative to control populations28,61. Similarly, selection for long night sleep had reduced sleep latency as a correlated response28. Likewise, mutational studies of genes affecting sleep duration reflect the negative correlation with sleep latency. For example, a reduction in night (and sometimes 24-h) sleep duration accompanied an increase in sleep latency for mutations in the following genes: 5-HT1A, 5-HT1B, aus, BomBc2, cv-c (also identified in this study), ltpr, Lztr1, Nlg4, rogdi, Shal, tara, Trh, and wake9,62–71. Overexpression of the genes Appl and fbx14 revealed the same pattern72,73. Mutations that increase night sleep duration, such as those in Ih, fbx14, Gabat, miR-276a, NPF, NPFR, para, and Rdl have a corresponding decrease in sleep latency13,15,72,74–77. Interestingly, increasing brp synaptic protein levels extended night sleep and decreased sleep latency in a dose-dependent manner78. Previous work also observed a similar pattern for sleep during the day: increased day sleep latency accompanies reduced day sleep duration, as observed in mutants of CG2277 and tau (also identified in this study)79,80. Mutants that increase day sleep duration have reduced sleep latency75,79,81. Only a few exceptions to this generalized result exist thus far: over-expression of miR-375 in tim-expressing neurons decreased both sleep and sleep latency; heterozygous mutations in conserved exons of wake increased 24-h sleep duration and day sleep latency in females; and changes in sleep latency emerged without a corresponding change in sleep duration in pan-neuronal knockdowns of CG32459 and CG181468,79,82. The negative relationship between sleep duration and sleep latency persists across brain structures and is present when genes are overexpressed pan-neuronally9,73 and in large ventral lateral neurons72; and when genes are reduced pan-neuronally65,79, in small and large ventral lateral neurons9,66,71,72,81, in R5 neurons83, and in glia63. Much of the data from mutational studies in flies, therefore, supports a negative genetic correlation between sleep duration and sleep latency, and the identification of a gene affecting sleep latency apart from sleep duration is rare. Very long sleep duration is necessarily coupled with short sleep latency, as the longer a fly sleeps during the day or night, the less time there is available to spend falling asleep. However, why very short sleep duration also correlates with very long sleep latency in flies is not clear. One possible explanation would be a strong genetic correlation between sleep latency and the genetic components of the circadian clock, which could alter the timing of sleep. We did not observe any correlation or connection between sleep latency and circadian period or rhythmicity index except for a single SNP in an intron of Pdp1 implicated for sleep latency in female flies.
Studying the genetics and environmental factors that influence sleep latency can offer valuable insights into the factors that contribute to insomnia and lead to the identification of novel therapeutic targets for the disorder. Here we have demonstrated significant genetic variation for sleep latency in a natural population of flies. In addition to sharing genetic architecture with night sleep duration, most of the genes mapping to this moderately heritable trait have human homologs. A long-term goal would be to determine whether the genes we have identified for sleep latency in flies are functionally conserved in humans.
Materials and methods
Drosophila stocks
This study re-analyzes sleep and activity data measured in a previous study of 167 lines of the Drosophila Genetic Reference Panel and w1118; Canton-S B19 in order to calculate the sleep latencies for each fly measured. We also tested 23 Minos insertion lines for potential effects on sleep latency (Bloomington Drosophila Stock Center, Bloomington, IN). Supplementary Table S9 lists the Minos lines, genotypes, Bloomington stock numbers, and control lines used. For the genetic rescue of Piezo and Proc-R mutants, we used w1118; snaSco/SM6a, P[w[+ mC] = hsILMiT]2.4 (Stock # 24613 from the Bloomington Drosophila Stock Center, Bloomington, IN) as the source of transposase. Finally, for nine genes, we crossed Transgenic RNAi Project (TriP) lines to an elav-GAL4 driver to reduce their expression pan-neuronally84–86. Supplementary Table S10 lists the TRiP RNAi lines, genotypes, Bloomington stock numbers, and elav-GAL4 line used.
Sleep and activity assays
Previously, sleep and activity was measured in flies from the Drosophila Genetic Reference Panel19. We briefly describe the original assay conditions here; the full details can be found in19. To measure sleep, flies were reared under standard conditions (cornmeal-molasses-agar food medium, 25 °C, 60–75% humidity, and a 12-h:12-h light:dark cycle). Both mating and social enrichment can affect sleep in flies87,88, thus virgin males and females were collected and held at 30 flies to a same-sex vial for four days to standardize the impact of these experimental parameters on sleep. We monitored the flies’ sleep and activity in DAM2 Drosophila Activity Monitors (Trikinetics, Waltham, MA) for seven days. Flies were allowed to recover from CO2 and acclimate to the monitor tubes during the first day, thus data from the first day was removed from further analysis. Data from flies not surviving the assay was also removed from further analysis. For sleep assays, the 167 DGRP lines were split into 4 blocks of 41–42 lines; each block was replicated four times. Each replicate contained 8 flies per sex per line. Eight male and eight female flies of w1118; Canton-S B were measured in each block and replicate as a contemporaneous control. 10,703 flies survived the assay. We used the raw sleep and activity data to calculate the sleep latency, which we defined as the number of minutes to the first sleep bout during the night period. We calculated the sleep latency per fly each day using a C# program (R. Sean Barnes). If the fly was already asleep when the lights were turned off in the incubator, we defined the sleep latency as zero.
For the verification tests, Minos lines were subjected to the same procedure, except for the following differences. Standard Drosophila medium was used (https://bdsc.indiana.edu/information/recipes/bloomfood/html), and flies were collected and held at 20 flies rather than 30 flies to a same-sex vial for four days. We replicated sleep and activity measures twice. Each replicate contained 8 flies per sex per line, for a total of 16 flies/sex/line tested.
Quantitative genetic analysis of sleep latency
We used an analysis of variance to partition the variance in sleep latency. The model was
where B is the random effect of block, S is Sex, L is the random effect of DGRP Line, and R is the random effect of replicate. We also analyzed the data for sexes separately using the reduced model
where the terms are defined as above. Broad-sense heritability (H2) was calculated for both sexes combined as H2 = σ2L + σ2S×L/(σ2L + σ2S×L + σ2E) and for sexes separately as H2 = σ2L/( σ2L + σ2E), where σ2L is the among-line variance, σ2L×S is the line-by-sex variance component, and σ2E is the sum of all other sources of variance. The cross-sex genetic correlation was computed as rmf = σ2L/√(σ2LM + σ2LF) where σ2L is the line variance component for both sexes combined, σ2LM is the line variance component for males, and σ2LF is the line variance component for females89. In addition, we calculated the genetic correlation between sleep latency and other sleep and circadian traits published previously. The genetic correlation was computed using rG = cov12/√(σL12 × σL22), where cov12 is the covariance between traits, and σL12 and σL22 are the among-line variance estimates for each trait. SAS (SAS Institute, Cary, NC) was used for all statistical analyses.
Genotype–phenotype associations
We used the DGRP Freeze 2.0 webtool for genome-wide association analysis (dgrp2.gnets.ncsu.edu). This tool tested sleep latency against 1,920,276 genome-wide polymorphisms segregating in the DGRP having minor allele frequencies ≥ 0.05 (9 or more lines having the minor allele)17. The webtool first adjusts phenotypes for cryptic genetic relatedness, the presence of chromosomal inversions, and Wolbachia pipientis infection status17. Neither the inversions nor Wolbachia infection status was significantly associated with sleep latency; however, adjustments were made to the sleep latency phenotype based on cryptic relatedness (Supplementary Table S1). Using the adjusted data, we then applied the following linear mixed model:
where y is the adjusted sleep latency means, X is the design matrix containing the fixed effect of each SNP, Z is the incidence matrix of random effects, and e is the random residual error17.
We defined our threshold P-value for each association as P ≤ 1 × 10–5, a threshold used in many other studies of the DGRP22,37,41,90–94 and supported by Q-Q plots (Supplementary Fig. S1). Variants meeting the threshold criterion were mapped to the Drosophila 6.0 genome. We used the DRSC Integrative Ortholog Prediction Tool (DIOPT) to identify Drosophila genes having human homologs27, and the Database for Annotation, Visualization, and Integrated Discovery (DAVID) to assess gene ontology and pathway enrichment27.
Pairwise epistasis and network analysis
We tested all significant polymorphisms with a minor allele frequency of 0.15 or greater (43 polymorphisms; 903 total pairwise tests) for possible pairwise epistatic interactions. Pairs were tested for high (r2 ≥ 0.8) linkage disequilibrium (LD) using PLINK95; none of the polymorphisms were in high LD. We applied the following model for males and females separately:
where M1 is the first marker tested, M2 is the second marker tested, and ε is error. We used a Bonferroni-corrected P-value to identify potential interactions (i.e., a significant M1 × M2 term).
In addition, we used BIOGRID to identify known genetic and protein–protein interactions among sleep latency candidate genes29. We visualized these interactions using Cytoscape96.
Candidate gene verification
To identify candidate genes for further testing we developed the following criteria: the polymorphism (1) must be located within ± 1000 bp of a gene; (2) within the top 100 polymorphisms with the largest predicted effect size for sexes combined; and (3) have a Minos insertion stock available for testing. We found 23 such genes (Supplementary Table S9). We measured the sleep latency in each mutant and compared it to its corresponding isogenic control line (w1118 or y1w67c23). We measured 8 flies per sex per line in two separate biological replicates. We analyzed the data using the ANOVA model
where µ is the overall population mean, G is genotype, S is Sex, R is replicate, and ε is the within-line variance. We used a Bonferroni-corrected P-value of 0.0031 as our criterion for significance for lines having the w1118 control (16 lines) and 0.0071 as the criterion for lines having the y1w67c23 control (7 lines). We additionally tested for differences among the mutants without the control lines using a Dunnett’s t-test.
We additionally used available RNAi lines to knock down gene expression in nine of the genes having significant Minos phenotypes. We used Transgenic RNAi Project (TriP) lines as the source of RNAi hairpins (Supplementary Table S10)84,85. We used an elav-GAL4 driver to reduce gene expression pan-neuronally86. The elav-GAL4 driver was crossed to each RNAi line, and sleep and activity were measured and analyzed as stated above. We used a Bonferroni-corrected P-value of 0.0056 as our criterion for significance. In addition, we tested whether sleep latency in the elav-GAL4 × RNAi cross progeny was different from what would be expected under a strict additive model (i.e., the mean of the sleep latency for the elav-GAL4 and corresponding RNAi line) using a t-test.
Genetic rescue of Piezo and Proc-R
We used a previously published procedure to produce precise excision lines from Piezo and Proc-R Minos insertion lines30. We crossed flies from a helper line, w1118; snaSco/SM6a, P[w[+ mC] = hsILMiT]2.4 to flies homozygous for the Mi[ET1] insertion in Piezo and in Proc-R. After two days, the parental flies were removed. We exposed larval progeny to heat shock using a 37 °C water bath for 1 h per day for 3 days. After eclosion, the adult progeny were screened for the presence of mosaic green fluorescent protein (GFP) in the eyes, an indication that the Minos element has been partially transposed. Individual mosaic males were backcrossed to females from their respective Minos insertion line as appropriate (PiezoMB08675 or Proc-RMB00909). The resulting heterozygous progeny were crossed and screened for the presence of GFP. GFP-negative flies were crossed to produce precise excision lines, PiezoMB* and Proc-RMB*. We verified precise excisions via DNA extraction with PCR amplification and sequencing.
PCR reactions were carried out in 0.2 mL PCR tube strips with strips of dome caps (USA Scientific, Inc., Ocala, FL) using Qiagen Taq PCR Master Mix containing 250 units Taq DNA Polymerase (Cat. No./ID: 201443 Qiagen). The PCR reactions were performed on an Eppendorf Mastercycler nexus PCR cycler (Eppendorf North America, Enfield, CT) using the manufacturer’s protocol. We designed primers using Primer-Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Primers were synthesized by IDT (Integrated DNA Technologies, Coralville, IA).
Primers to amplify and sequence a region of Piezo flanking the Minos insertion site in w1118 and PiezoMB* DNA were 1, TCCTGACCGAGGGATTTTTGG and 2, ACACAAGTGAATCCCCTGAAGC. To confirm the location of the Minos element Mi[ET1] in PiezoMB08675, a fragment spanning the junction between Piezo and Mi[ET1] was amplified and sequenced using primers 7, CCGAGGGATTTTTGGAACCG and 8, CTCATGTTTGACAGCTTATCATCG. A region flanking the Minos insertion site in Proc-R in w1118 and Proc-RMB* DNA was amplified using primers 20, TGTTCAGTATTTCCGCTACATTTGC and 21, TGATCTTATCTCCGTACGCTGC. The Proc-R fragments were sequenced using primers 30, GGAGAAAATTAAACTGCTCGGC, 32, TCATGAGATACAAAATGGCGGG, and 34, GGCGGGGAGATGAAGATTTTGG. The location of Mi[ET1] in Proc-RMB00909 was confirmed using primers 22, GATGGACTTGCTGCCATGACC, 23, CATGCTGGAGTTCTTCGCCC, and 30. See Supplementary Fig. S3 for a schematic of Minos element insertion and primer locations as well as the gel showing that the PCR fragments were of the expected size. Supplementary Fig. S4 presents confirmatory sequence data.
We tested the putative precise excision lines for sleep latency and compared them to the w1118 isogenic control, the PiezoMB08675 (Proc-RMB00909) Minos insertion line, as appropriate, and a heterozygous cross between w1118 and PiezoMB08675 (Proc-RMB00909). We measured 16 flies per sex per line, and the experiments were replicated twice. The same ANOVA model used for the verification tests was used for the rescue experiments, along with post-hoc Tukey tests to distinguish differences among individual genotypes.
Arousal threshold measures in Piezo mutant and rescue lines
We assessed the arousal threshold in the PiezoMB08675 Minos insertion line, the PiezoMB* rescue line, and the w1118 control. We perturbed male and female flies from each line for one second per hour for three days using the Trikinetics Vortexer mounting plate system (Trikinetics, Waltham, MA). The vortexer was set to the lowest setting, intensity 1. For each fly at each hour, we computed the amount of sleep in minutes prior to the stimulus and the number of minutes the fly was awake after the stimulus. We also calculated the proportion of flies responding to the stimulus each hour97,98. We analyzed the data using the ANOVA model Y = µ + G + S + G × S + ε, where G and S are as defined above.
Supplementary Information
Acknowledgements
This research was supported by the Intramural Research Program of the NIH, the National Heart Lung and Blood Institute (funding number HL006276 to STH).
Author contributions
M.E., S.K., and S.T.H. conceived of the experiment; M.E., S.K., Y.L.S.N. and T.R.T. acquired the data; M.E. and S.T.H analyzed the data; M.E., S.K., Y.S.L.N, and S.T.H. wrote the manuscript with input from all authors.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Data availability
The data generated for this manuscript are provided in the supplementary tables. We downloaded publicly available genome variant data for the DGRP from the DGRP2 website, dgrp2.gnets.ncsu.edu. Additional information on the DGRP, including the SRA accession numbers for the DNA sequences of each DGRP line, can be found in Huang et al.17.
Competing interests
Financial Disclosure: The author(s) declare no competing interests. Non-financial Disclosure: S.T.H. is on the Editorial Board of Scientific Reports. The review process for this manuscript was handled independently by other members of the editorial team. The remaining authors have no non-financial interests to declare.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Matthew N. Eiman and Shailesh Kumar.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-50552-z.
References
- 1.Amin N, et al. Genetic variants in RBFOX3 are associated with sleep latency. Eur. J. Hum. Genet. 2016;24:1488–1495. doi: 10.1038/ejhg.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riemann D, et al. Insomnia disorder: State of the science and challenges for the future. J. Sleep Res. 2022;31:e13604. doi: 10.1111/jsr.13604. [DOI] [PubMed] [Google Scholar]
- 3.Spada J, et al. Genome-wide association analysis of actigraphic sleep phenotypes in the LIFE adult study. J. Sleep Res. 2016;25:690–701. doi: 10.1111/jsr.12421. [DOI] [PubMed] [Google Scholar]
- 4.Byrne EM, et al. A genome-wide association study of sleep habits and insomnia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013;162B:439–451. doi: 10.1002/ajmg.b.32168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cade BE, et al. Common variants in DRD2 are associated with sleep duration: The CARe consortium. Hum. Mol. Genet. 2016;25:167–179. doi: 10.1093/hmg/ddv434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, et al. Effect of 5-HT2A receptor polymorphisms and occupational stress on self-reported sleep quality: a cross-sectional study in Xinjiang, China. Sleep Med. 2016;20:30–36. doi: 10.1016/j.sleep.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Chang AM, et al. Circadian gene variants influence sleep and the sleep electroencephalogram in humans. Chronobiol. Int. 2016;33:561–573. doi: 10.3109/07420528.2016.1167078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dement WC. The Promise of Sleep. New York: Random House Inc.; 1999. [Google Scholar]
- 9.Liu S, et al. WIDE AWAKE mediates the circadian timing of sleep onset. Neuron. 2014;82:151–166. doi: 10.1016/j.neuron.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veasey SC, Yeou-Jey H, Thayer P, Fenik P. Murine multiple sleep latency test: Phenotyping sleep propensity in mice. Sleep. 2004;27:388–393. doi: 10.1093/sleep/27.3.388. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki A, Sinton CM, Greene RW, Yanagisawa M. Behavioral and biochemical dissociation of arousal and homeostatic sleep need influenced by prior wakeful experience in mice. Proc. Natl. Acad. Sci. U. S. A. 2013;110:10288–10293. doi: 10.1073/pnas.1308295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keenan BT, et al. High-throughput sleep phenotyping produces robust and heritable traits in Diversity Outbred mice and their founder strains. Sleep. 2020;43:zsz278. doi: 10.1093/sleep/zsz278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agosto J, et al. Modulation of GABAA receptor desensitization uncouples sleep onset and maintenance in Drosophila. Nat. Neurosci. 2008;11:354–359. doi: 10.1038/nn2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu W, Guo F, Lu B, Guo A. amnesiac regulates sleep onset and maintenance in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2008;372:798–803. doi: 10.1016/j.bbrc.2008.05.119. [DOI] [PubMed] [Google Scholar]
- 15.He C, Yang Y, Zhang M, Price JL, Zhao Z. Regulation of sleep by neuropeptide Y-like system in Drosophila melanogaster. PLOS ONE. 2013;8:e74237. doi: 10.1371/journal.pone.0074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackay TF, et al. The Drosophila melanogaster genetic reference panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang W, et al. Natural variation in genome architecture among 205 Drosophila melanogaster genetic reference panel lines. Genome Res. 2014;24:1193–1208. doi: 10.1101/gr.171546.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackay TFC, Huang W. Charting the genotype-phenotype map: Lessons from the Drosophila melanogaster genetic reference panel. Wiley Interdiscip Rev. Dev. Biol. 2017 doi: 10.1002/wdev.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harbison ST, McCoy LJ, Mackay TF. Genome-wide association study of sleep in Drosophila melanogaster. BMC Genomics. 2013;14:281. doi: 10.1186/1471-2164-14-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu KJ, Kumar S, Serrano Negron YL, Harbison ST. Genotype influences day-to-day variability in sleep in Drosophila melanogaster. Sleep. 2018;41:zsx205. doi: 10.1093/sleep/zs1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harbison ST, et al. Genome-wide association study of circadian behavior in Drosophila melanogaster. Behav. Genet. 2019;49:60–82. doi: 10.1007/s10519-018-9932-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lobell AS, Kaspari RR, Serrano-Negron YL, Harbison ST. The genetic architecture of ovariole number in Drosophila melanogaster: Genes with major, quantitative, and pleiotropic effects. G3 Bethesda. 2017;7:2391–2403. doi: 10.1534/g3.117.042390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jordan KW, et al. Genome-wide association for sensitivity to chronic oxidative stress in Drosophila melanogaster. PLOS ONE. 2012;7:e38722. doi: 10.1371/journal.pone.0038722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montgomery SL, et al. Genome-wide association analysis of tolerance to methylmercury toxicity in Drosophila implicates myogenic and neuromuscular developmental pathways. PLOS ONE. 2015;9:e110375. doi: 10.1371/journal.pone.0110375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber AL, Khan GF, Magwire MM, Tabor CL, Mackay TF. Genome-wide association analysis of oxidative stress resistance in Drosophila melanogaster. PLOS ONE. 2012;7:e34745. doi: 10.1371/journal.pone.0034745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackay TFC, Huang W. Charting the genotype-phenotype map: Lessons from the Drosophila melanogaster genetic reference panel. Wiley Interdiscip Rev Dev Biol. 2018;7:e289. doi: 10.1002/wdev.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, et al. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harbison ST, Serrano Negron YL, Hansen NF, Lobell AS. Selection for long and short sleep duration in Drosophila melanogaster reveals the complex genetic network underlying natural variation in sleep. PLoS Genet. 2017;13:e10007098. doi: 10.1371/journal.pgen.1007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oughtred R, et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021;30:187–200. doi: 10.1002/pro.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metaxakis A, Oehler S, Klinakis A, Savakis C. Minos as a genetic and genomic tool in Drosophila melanogaster. Genetics. 2005;171:571–581. doi: 10.1534/genetics.105.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SE, Coste B, Chadha A, Cook B, Patapoutian A. The role of Drosophila Piezo in mechanical nociception. Nature. 2012;483:209–212. doi: 10.1038/nature10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim YJ, Baker BS. Isolation of RRM-type RNA-binding protein genes and the analysis of their relatedness by using a numerical approach. Mol. Cell. Biol. 1993;13:174–183. doi: 10.1128/mcb.13.1.174-183.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gramates LS, et al. FlyBase: A guided tour of highlighted features. Genetics. 2022;220:035. doi: 10.1093/genetics/iyac035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guss EJ, Akbergenova Y, Cunningham KL, Littleton JT. Loss of the extracellular matrix protein Perlecan disrupts axonal and synaptic stability during Drosophila development. Elife. 2023;12:RP88273. doi: 10.7554/eLife.88273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kent D, Bush EW, Hooper JE. Roadkill attenuates Hedgehog responses through degradation of Cubitus interruptus. Development. 2006;133:2001–2010. doi: 10.1242/dev.02370. [DOI] [PubMed] [Google Scholar]
- 36.Umberger PA, Ogden SK. SPOP and CUL3 modulate the sonic hedgehog signal response through controlled degradation of GLI family transcription factors. Front Cell Dev Biol. 2021;9:710295. doi: 10.3389/fcell.2021.710295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arya GH, et al. The genetic basis for variation in olfactory behavior in Drosophila melanogaster. Che. Senses. 2015;40:233–243. doi: 10.1093/chemse/bjv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riddle NC. Variation in the response to exercise stimulation in Drosophila: marathon runner versus sprinter genotypes. J Exp Biol. 2020;223:jeb229997. doi: 10.1242/jeb.229997. [DOI] [PubMed] [Google Scholar]
- 39.Durham MF, Magwire MM, Stone EA, Leips J. Genome-wide analysis in Drosophila reveals age-specific effects of SNPs on fitness traits. Nat. Commun. 2014;5:4338. doi: 10.1038/ncomms5338. [DOI] [PubMed] [Google Scholar]
- 40.Chen DS, et al. Female genetic contributions to sperm competition in Drosophila melanogaster. Genetics. 2019;212:789–800. doi: 10.1534/genetics.119.302284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shorter J, et al. Genetic architecture of natural variation in Drosophila melanogaster aggressive behavior. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E3555–E3563. doi: 10.1073/pnas.1510104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morozova TV, et al. Polymorphisms in early neurodevelopmental genes affect natural variation in alcohol sensitivity in adult drosophila. BMC Genomics. 2015;16:865. doi: 10.1186/s12864-015-2064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JB, Lu HL, St Leger RJ. The genetic basis for variation in resistance to infection in the Drosophila melanogaster genetic reference panel. PLoS Pathog. 2017;13:e1006260. doi: 10.1371/journal.ppat.1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar S, Tunc I, Tansey TR, Pirooznia M, Harbison ST. Identification of genes contributing to a long circadian period in Drosophila melanogaster. J. Biol. Rhythms. 2021;36:239–253. doi: 10.1177/0748730420975946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: From polygenic to omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang W, et al. Spontaneous mutations and the origin and maintenance of quantitative genetic variation. Elife. 2016;5:e14625. doi: 10.7554/eLife.14625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Souto-Maior C, Serrano Negron YL, Harbison ST. Natural selection on sleep duration in Drosophila melanogaster. Sci. Rep. 2020;10:20652. doi: 10.1038/s41598-020-77680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cirelli C, et al. Reduced sleep in Drosophila Shaker mutants. Nature. 2005;434:1087–1092. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- 49.Swarup S, et al. Extensive epistasis for olfactory behavior, sleep and waking activity in Drosophila melanogaster. Genet. Res. 2012;94:9–20. doi: 10.1017/S001667231200002X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandler CH, et al. How well do you know your mutation? Complex effects of genetic background on expressivity, complementation, and ordering of allelic effects. PLoS Genet. 2017;13:e1007075. doi: 10.1371/journal.pgen.1007075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Claeys I, et al. Insect neuropeptide and peptide hormone receptors: current knowledge and future directions. Vitam. Horm. 2005;73:217–282. doi: 10.1016/S0083-6729(05)73007-7. [DOI] [PubMed] [Google Scholar]
- 52.Li H, et al. Fly cell atlas: A single-nucleus transcriptomic atlas of the adult fruit fly. Science. 2022;375:eabk2432. doi: 10.1126/science.abk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson EC, et al. Identification and characterization of a G protein-coupled receptor for the neuropeptide proctolin in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6198–6203. doi: 10.1073/pnas.1030108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown JB, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014;512:393–399. doi: 10.1038/nature12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng B, et al. Chemoconnectomics: Mapping chemical transmission in Drosophila. Neuron. 2019;101:876–893. doi: 10.1016/j.neuron.2019.01.045. [DOI] [PubMed] [Google Scholar]
- 56.Coste B, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. 2012;483:176–181. doi: 10.1038/nature10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moroni M, Servin-Vences MR, Fleischer R, Sanchez-Carranza O, Lewin GR. Voltage gating of mechanosensitive PIEZO channels. Nat. Commun. 2018;9:1096. doi: 10.1038/s41467-018-03502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song Y, et al. The mechanosensitive ion channel piezo inhibits axon regeneration. Neuron. 2019;102:373–389. doi: 10.1016/j.neuron.2019.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Titos I, et al. A gut-secreted peptide suppresses arousability from sleep. Cell. 2023;186:1382–1397. doi: 10.1016/j.cell.2023.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Min S, et al. Control of feeding by Piezo-mediated gut mechanosensation in Drosophila. Elife. 2021;10:e63049. doi: 10.7554/eLife.63049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seugnet L, et al. Identifying sleep regulatory genes using a Drosophila model of insomnia. J. Neurosci. 2009;29:7148–7157. doi: 10.1523/JNEUROSCI.5629-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qian Y, et al. Sleep homeostasis regulated by 5HT2b receptor in a small subset of neurons in the dorsal fan-shaped body of drosophila. Elife. 2017;6:e26519. doi: 10.7554/eLife.26519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.You S, Yu AM, Roberts MA, Joseph IJ, Jackson FR. Circadian regulation of the Drosophila astrocyte transcriptome. PLoS Genetics. 2021;17:e1009790. doi: 10.1371/journal.pgen.1009790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bedont JL, et al. Short and long sleeping mutants reveal links between sleep and macroautophagy. Elife. 2021;10:e64140. doi: 10.7554/eLife.64140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maurer GW, et al. Analysis of genes within the schizophrenia-linked 22q11.2 deletion identifies interaction of night owl/LZTR1 and NF1 in GABAergic sleep control. PLOS Genet. 2020;16:e1008727. doi: 10.1371/journal.pgen.1008727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng G, et al. Control of sleep onset by Shal/Kv4 channels in drosophila circadian neurons. J Neurosci. 2018;38:9059–9071. doi: 10.1523/JNEUROSCI.0777-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim M, et al. Rogdi defines GABAergic control of a wake-promoting dopaminergic pathway to sustain sleep in Drosophila. Sci. Rep. 2017;7:11368. doi: 10.1038/s41598-017-11941-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang S, et al. Nmf9 encodes a highly conserved protein important to neurological function in mice and flies. PLOS Genet. 2015;11:e1005344. doi: 10.1371/journal.pgen.1005344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Afonso DJ, et al. TARANIS functions with cyclin A and Cdk1 in a novel arousal center to control sleep in Drosophila. Curr. Biol. 2015;25:1717–1726. doi: 10.1016/j.cub.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Donlea JM, Pimentel D, Miesenbock G. Neuronal machinery of sleep homeostasis in Drosophila. Neuron. 2014;81:860–872. doi: 10.1016/j.neuron.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y, et al. Drosophila neuroligin 4 regulates sleep through modulating GABA transmission. J. Neurosci. 2013;33:15545–15554. doi: 10.1523/JNEUROSCI.0819-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Q, et al. Fbxl4 serves as a clock output molecule that regulates sleep through promotion of rhythmic degradation of the GABAA receptor. Curr. Biol. 2017;27:3616–3625. doi: 10.1016/j.cub.2017.10.052. [DOI] [PubMed] [Google Scholar]
- 73.Shao L, Zhang Y, Hao Y, Ping Y. Upregulation of IP3 receptor mediates APP-induced defects in synaptic downscaling and sleep homeostasis. Cell Rep. 2022;38:110594. doi: 10.1016/j.celrep.2022.110594. [DOI] [PubMed] [Google Scholar]
- 74.Fernandez-Chiappe F, et al. High-frequency neuronal bursting is essential for circadian and sleep behaviors in Drosophila. J. Neurosci. 2021;41:689–710. doi: 10.1523/JNEUROSCI.2322-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang R, Zhao X, Du J, Wei L, Zhao Z. Regulatory mechanism of daily sleep by miR-276a. FASEB J. 2021;35:e21222. doi: 10.1096/fj.202001220R. [DOI] [PubMed] [Google Scholar]
- 76.Petruccelli E, Lansdon P, Kitamoto T. Exaggerated nighttime sleep and defective sleep homeostasis in a Drosophila knock-in model of human epilepsy. PLOS ONE. 2015;10:e0137758. doi: 10.1371/journal.pone.0137758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen WF, et al. A neuron-glia interaction involving GABA transaminase contributes to sleep loss in sleepless mutants. Mol. Psychiatry. 2015;20:240–251. doi: 10.1038/mp.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang S, Piao C, Beuschel CB, Gotz T, Sigrist SJ. Presynaptic active zone plasticity encodes sleep need in Drosophila. Curr. Biol. 2020;30:1077–1091. doi: 10.1016/j.cub.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 79.Singgih EL, et al. Investigating cytosolic 5'-nucleotidase II family genes as candidates for neuropsychiatric disorders in Drosophila (114/150 chr) Transl. Psychiatry. 2021;11:55. doi: 10.1038/s41398-020-01149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arnes M, et al. Role of tau protein in remodeling of circadian neuronal circuits and sleep. Front. Aging Neurosci. 2019;11:320. doi: 10.3389/fnagi.2019.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Z, Cao W, Edery I. The SR protein B52/SRp55 regulates splicing of the period thermosensitive intron and mid-day siesta in Drosophila. Sci. Rep. 2018;8:1872. doi: 10.1038/s41598-017-18167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xia X, et al. Regulation of circadian rhythm and sleep by miR-375-timeless interaction in Drosophila. FASEB J. 2020;34:16536–16551. doi: 10.1096/fj.202001107R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raccuglia D, et al. Network-specific synchronization of electrical slow-wave oscillations regulates sleep drive in Drosophila. Curr. Biol. 2019;29:3611–3621. doi: 10.1016/j.cub.2019.08.070. [DOI] [PubMed] [Google Scholar]
- 84.Perkins LA, et al. The transgenic RNAi project at harvard medical school: Resources and validation. Genetics. 2015;201:843–852. doi: 10.1534/genetics.115.180208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zirin J, et al. Large-scale transgenic drosophila resource collections for loss- and gain-of-function studies. Genetics. 2020;214:755–767. doi: 10.1534/genetics.119.302964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 87.Isaac RE, Li C, Leedale AE, Shirras AD. Drosophila male sex peptide inhibits siesta sleep and promotes locomotor activity in the post-mated female. Proc. R. Soc. B. 2010;277:65–70. doi: 10.1098/rspb.2009.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ganguly-Fitzgerald I, Donlea J, Shaw PJ. Waking experience affects sleep need in Drosophila. Science. 2006;313:1775–1781. doi: 10.1126/science.1130408. [DOI] [PubMed] [Google Scholar]
- 89.Falconer DS, Mackay TF. Introduction to Quantitative Genetics. 4. Boston: Addison Wesley Longman Limited; 1996. [Google Scholar]
- 90.Garlapow ME, Huang W, Yarboro MT, Peterson KR, Mackay TF. Quantitative genetics of food intake in Drosophila melanogaster. PLOS One. 2015;10:e0138129. doi: 10.1371/journal.pone.0138129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dembeck LM, et al. Genetic architecture of abdominal pigmentation in Drosophila melanogaster. PLOS Genet. 2015;11:e1005163. doi: 10.1371/journal.pgen.1005163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dembeck LM, et al. Genetic architecture of natural variation in cuticular hydrocarbon composition in Drosophila melanogaster. Elife. 2015;4:e09861. doi: 10.7554/eLife.09861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hunter CM, Huang W, Mackay TF, Singh ND. The genetic architecture of natural variation in recombination rate in Drosophila melanogaster. PLOS Genet. 2016;12:e1005951. doi: 10.1371/journal.pgen.1005951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zwarts L, et al. The genetic basis of natural variation in mushroom body size in Drosophila melanogaster. Nat. Commun. 2015;6:10115. doi: 10.1038/ncomms10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Purcell S, et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shannon P, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Faville R, Kottler B, Goodhill GJ, Shaw PJ, van Swinderen B. How deeply does your mutant sleep? Probing arousal to better understand sleep defects in Drosophila. Sci. Rep. 2015;5:8454. doi: 10.1038/srep08454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vienne J, Spann R, Guo F, Rosbash M. Age-related reduction of recovery sleep and arousal threshold in Drosophila. Sleep. 2016;39:1613–1624. doi: 10.5665/sleep.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated for this manuscript are provided in the supplementary tables. We downloaded publicly available genome variant data for the DGRP from the DGRP2 website, dgrp2.gnets.ncsu.edu. Additional information on the DGRP, including the SRA accession numbers for the DNA sequences of each DGRP line, can be found in Huang et al.17.