

Abstract
Traumatic brain injury (TBI) is associated with mortality and morbidity worldwide. Accumulating pre-clinical and clinical data suggests TBI is the leading extrinsic cause of progressive neurodegeneration. Neurological deterioration after either a single moderate-severe TBI or repetitive mild TBI often resembles dementia in aged populations; however, no currently approved therapies adequately mitigate neurodegeneration. Inflammation correlates with neurodegenerative changes and cognitive dysfunction for years post-TBI, suggesting a potential association between immune activation and both age- and TBI-induced cognitive decline. Inflammaging, a chronic, low-grade sterile inflammation associated with natural aging, promotes cognitive decline. Cellular senescence and the subsequent development of a senescence associated secretory phenotype (SASP) promotes inflammaging and cognitive aging, although the functional association between senescent cells and neurodegeneration is poorly defined after TBI. In this mini-review, we provide an overview of the pre-clinical and clinical evidence linking cellular senescence with poor TBI outcomes. We also discuss the current knowledge and future potential for senotherapeutics, including senolytics and senomorphics, which kill and/or modulate senescent cells, as potential therapeutics after TBI.
Keywords: Inflammation, Neurotrauma, Aging, Senescence, Neurodegeneration, Immune
Traumatic brain injury (TBI) is a significant worldwide public health issue, killing or debilitating individuals regardless of age, gender, race, or socioeconomic status (Centers for Disease C, Prevention, 2009). TBI, which is broadly defined as a blow or jolt to the head that disrupts neurological function, has a heterogenous presentation that complicates the development of efficacious therapies to improve short and long-term quality of life. As a result, up to 2% of the population currently lives with the neurological consequences of a prior TBI. Research spanning several decades has focused on defining and treating the acute pathophysiology after a moderate-severe TBI, with significant effort directed toward the identification of acute cerebroprotectants that limit neuronal loss, including blockade of excitatory pathways and quenching of free radicals; however, despite widespread pre-clinical success, these approaches failed to translate into better long-term clinical outcomes. Whether due to poor target selection, incorrect drug choices, limited therapeutic windows, and/or inadequate pre-clinical modeling, these clinical failures illustrate the daunting challenge that remains to develop neuroprotective therapies after TBI. As such, pre-clinical and translational research must continue to mechanistically define TBI pathophysiology with the hope that this knowledge will advance the development of innovative therapeutic approaches.
The preponderance of research to date has targeted axonal pathology, neuronal loss, cerebral edema, and elevated intracranial pressure within the acute and sub-acute periods after TBI. Conversely, compelling epidemiological data indicate TBI increases the lifelong dementia risk by up to 50%, as compared to non-traumatic, age-matched control cohorts, making neurotrauma the leading external risk factor for dementia (Fleminger et al., 2003; Mortimer et al., 1991; Molgaard et al., 1990; Sullivan et al., 1987; Gedye et al., 1989; Nemetz et al., 1999; Ross, 2011; Tomaiuolo et al., 2004; Warner et al., 2010; Lye and Shores, 2000; Plassman et al., 2000; Schofield et al., 1997; Guo et al., 2000; Graves et al., 1990; Mortimer et al., 1985). Thus, elucidating how acute brain injuries drive subsequent progressive neurodegeneration, which often manifests decades after the initial TBI, is imperative for improving quality of life in neurotrauma patients. The goal of this mini-review is to provide an overview of the emerging evidence suggesting TBI accelerates the aging progress and promotes neurodegeneration. We also speculate on potential new therapeutic targets/approaches, including the unexplored possibility that inflammaging is a primary driver of premature cognitive aging after TBI. This knowledge will provide a foundation for future experimental and clinical research to improve chronic outcomes after TBI.
1. TBI accelerates cognitive aging and elevates the risk of dementia
The term ‘aging’ encompasses both chronological and biological aging. Chronological aging describes the linear changes over the lifespan while biological aging may produce nonlinear changes due to the influence of genetics and/or the environment. Ideally, chronological and biological age match, whereas deviations between chronological and biological age reflect accelerated aging, as compared to a “typical” individual. As aging is associated with an increased risk for developing chronic diseases and incident multi-morbidity, particularly during the final 25% of the lifespan, understanding the factors that influence accelerated aging may reduce societal burden of age-associated chronic illnesses (St Sauver et al., 2015; Kennedy et al., 2014).
Dementia encompasses pathological impairments in memory, language, cognition, and decision making that interferes with quality of life. Compelling epidemiological data, including prospective clinical studies, indicate TBI accelerates brain aging and is the leading extrinsic risk factor for later onset dementia (Fleminger et al., 2003; Mortimer et al., 1991; Molgaard et al., 1990; Sullivan et al., 1987; Gedye et al., 1989; Nemetz et al., 1999; Ross, 2011; Tomaiuolo et al., 2004; Warner et al., 2010; Lye and Shores, 2000; Plassman et al., 2000; Schofield et al., 1997; Guo et al., 2000; Graves et al., 1990; Mortimer et al., 1985; Faden and Loane, 2015; de Freitas Cardoso et al., 2019; Irimia et al., 2014). Toward this end, post-traumatic neurological deficits in young adults often resemble cognitive decline in elderly populations with no neurological history while a single moderate-severe TBI or repetitive mild TBI is a risk factor for the development of neurodegenerative diseases, including Alzheimer’s disease (AD) (Fann et al., 2018; Tolppanen et al., 2017; Mackay et al., 2019; Lehman et al., 2012), frontotemporal dementia (Rosso et al., 2003; Kalkonde et al., 2012; Deutsch et al., 2015), Parkinson’s disease (Mackay et al., 2019; Lehman et al., 2012; Gardner et al., 2015), and amyotrophic lateral sclerosis (Mackay et al., 2019; Lehman et al., 2012; Pupillo et al., 2020; Chio et al., 2005). Both observational and case-control studies found a “dose-dependent” effect of TBI, with more severe injury associated with higher risks of dementia (Tolppanen et al., 2017; Barnes et al., 2018; Nordstrom and Nordstrom, 2018), although a dissenting study found no clear association between TBI and AD risk (Sugarman et al., 2019). In addition, a history of TBI may be a risk factor for earlier onset AD, although this remains a topic of debate (Mortimer et al., 1991; Nemetz et al., 1999; Plassman et al., 2000; Guo et al., 2000; LoBue et al., 2017; Schaffert et al., 2018). Data primarily collected in military personnel and contact sport athletes further suggest repetitive (sub)concussive hits may lead to chronic traumatic encephalopathy (CTE), a progressive neurodegenerative disease associated with a unique pathology and both cognitive and psychiatric deficits (Katz et al., 2021; Bieniek et al., 2021). Together, these findings suggest a poorly understood relationship between neurotrauma and premature brain aging (Wood, 2017).
In line with the clinical association between TBI and subsequent neurodegeneration (Crane et al., 2016), histopathological features of TBI frequently mirror the classical features of AD. Using experimental models of TBI, we and others reported β-amyloid accumulation in the thalamus one year after moderate/severe TBI (Iwata et al., 2002; Chen et al., 2004; Zhang et al., 2012). Moderate/severe TBI also induces chronic widespread neurofibrillary tangles (NFT), temporally paralleling dementia onset (Corsellis and Brierley, 1959; Rudelli et al., 1982; Smith et al., 2003a), while amyloid precursor protein (APP) and APP processing enzymes, presenilin-1 and BACE1, accumulate at points of disrupted axonal transport, persisting for over six months post-TBI in large animals and humans (Iwata et al., 2002; Gentleman et al., 1993; Sherriff et al., 1994; Smith et al., 1999; Smith et al., 2003b; Chen et al., 2009). Diffuse amyloid pathology is acutely observed in ~30% of moderate/severe TBI patients, resembling the pathology in early AD, whereas an elevated fibrillar plaque burden presents at >1 year post-TBI, resembling established AD (Johnson et al., 2010; Roberts et al., 1991; Roberts et al., 1994; Ikonomovic et al., 2004; Johnson et al., 2012); however, widespread accumulation of extracellular amyloid-beta (Aβ) plaques and intracellular tau tangles often appear in postmortem brain tissue from patients with a history of TBI, but without a clinical diagnosis of AD (Johnson et al., 2012; Hyman et al., 2012; Agrawal et al., 2022). Similarly, cognitive and psychiatric symptoms may partially, but not entirely, overlap between TBI and dementia patients, possibility attributed to divergent etiologies (McDonald et al., 2002; Bray et al., 2021; Rao et al., 2010). Thus, despite overlapping neuropathology between TBI and AD, mechanistic and clinical differences may exist between the conditions.
In addition to neuropathological changes, white matter (WM) pathology, including extensive demyelination, contributes toward the clinical presentation of dementia (Sullivan et al., 1987; Gedye et al., 1989; Nemetz et al., 1999; Ross, 2011). Impact and/or coup-contrecoup injuries produce immediate diffuse axonal shearing and myelin degradation, yet most demyelination and WM loss develops for years after severe TBI in both rodents and humans (Bramlett and Dietrich, 2002; Pierce et al., 1998; Johnson et al., 2013a; Ramlackhansingh et al., 2011; Buki and Povlishock, 2006; Green et al., 2014; Gale et al., 1995; Ng et al., 1994; Berger et al., 2005; Wakade et al., 2010; Farook et al., 2013; Liu et al., 2006; Ottens et al., 2008; Ross et al., 1993; Su et al., 2012; Johnson et al., 2013b; Smith et al., 1997). WM lesions present within the corpus callosum of up to 76% of severe TBI patients while 96% of moderate/severe TBI patients exhibit cerebral atrophy, including reduced callosal volume (Green et al., 2014; Gentry et al., 1988). These structural changes manifest as memory loss, mood and concentration impairments, and psychiatric deficits (Kraus et al., 2007; Kumar et al., 2009; Pischiutta et al., 2018; Yoshita et al., 2006; Kovari et al., 2007; Bagi et al., 2022; Hase et al., 2018). While no FDA-approved therapies effectively prevent, delay, or reverse post-traumatic neurodegeneration, the degree of chronic inflammation correlates with progressive, bilateral thalamo-cortical WM tract damage, including reduced callosal thickness, for years after a moderate/severe TBI in rodents and humans (Johnson et al., 2013a; Pischiutta et al., 2018; Scott et al., 2015), supporting a poorly defined relationship between axonal injury, chronic inflammation, and progressive neurodegeneration after TBI.
2. Inflammaging as a contributing factor in delayed neurodegeneration
Inflammation is critical for host defense against infection and injury. Conversely, prolonged immune activation contributes toward a multitude of chronic, age-related ailments while chronological aging is associated with immune dysfunction, including elevated levels of inflammatory modulators that may be adaptive and/or detrimental for longevity (Fulop et al., 2017). While the CNS is traditionally regarded as immune privileged, meningeal lymphatic vessels that drain cerebrospinal fluid into deep cervical lymph nodes provide a conduit for the bidirectional movement of immune cells into and out of the brain (Louveau et al., 2015; Aspelund et al., 2015). A temporally and spatially coordinated series of immune responses develop within the CNS after experimental TBI while chronic neuroinflammation, the complex integration of CNS resident cells and infiltrating immune cells, correlates with WM loss, dementia, and premature cognitive aging for decades after moderate/severe TBI in humans (Wood, 2017; Johnson et al., 2013a; Scott et al., 2015). Despite the long-held assumption that immunosuppression may be beneficial after CNS injury (Braughler and Hall, 1985), the 10,008 patient CRASH trial was prematurely halted due to worse outcomes in the corticosteroid arm, as compared to the placebo arm (Roberts et al., 2004), supporting the notion that inflammation is a double-edged sword after injury (Bramlett and Dietrich, 2002; Pierce et al., 1998; Johnson et al., 2013a; Ramlackhansingh et al., 2011; Buki and Povlishock, 2006; Green et al., 2014; Gale et al., 1995; Johnson et al., 2013b; Gentry et al., 1988; Kraus et al., 2007; Kumar et al., 2009; Pischiutta et al., 2018; Scott et al., 2015). Thus, research to define the initiation and perpetuation of cerebral immunity is critical to spur targeted therapeutic development to minimize the deleterious consequences of unrestrained immune activation without impacting the protective actions of inflammation after TBI.
Inflammaging, an age-related increase in the chronic production of pro-inflammatory mediators, is a risk factor for accelerated aging, oncogenesis, cardiovascular and neurological disease, and premature death (Fulop et al., 2017; Ruparelia et al., 2017; Leonardi et al., 2018; Miller and Raison, 2016; Gorelick, 2010; Fabbri et al., 2015; Volpato et al., 2001; Soysal et al., 2016). Potential mechanisms underlying the initiation of inflammaging remain elusive, but genetic susceptibility, microbiome changes, oxidative stress, immune dysregulation, and infection are implicated. In addition, cellular senescence is a potent stimulus for inflammaging and chronic inflammation (Di Micco et al., 2021) while the presence of senescent cells correlates with cognitive aging and neurodegeneration (Cole et al., 2019; Franke and Gaser, 2019). Cellular senescence, first described over six decades ago by Hayflick and Moorehead, is characterized by irreversible cell cycle arrest, sustained viability with resistance to cell death, and increased metabolic activity (Hayflick and Moorhead, 1961; Tchkonia and Kirkland, 2018). Senescent cells functionally contribute to biochemical changes associated with neurodegeneration, including aberrant protein aggregation, metabolic changes, autophagy deficits, mitochondrial dysfunction, oxidative stress, and impaired neurogenesis (Chini et al., 2019; Xu et al., 2015a; Chapman et al., 2019; Nacarelli et al., 2019; Musi et al., 2018; Ogrodnik et al., 2019). Thus, cellular senescence may be an underexplored, yet critical factor in facilitating inflammaging and progressive neurodegeneration after TBI.
3. Does glial senescence promote post-traumatic neurodegeneration after TBI?
Moderate-severe TBI increased cell cycle markers and the number of senescence associated beta-galactosidase (SA-β-Gal) positive cells, a gold standard marker of cellular senescence, in the ipsilateral hemisphere for one-month post-injury (Tominaga et al., 2019; Schwab et al., 2021). SA-β-Gal was elevated throughout the cortex, hippocampus, and thalamus at one day and one month following a single or repeated moderate blast TBI in rats (Arun et al., 2020). Repeated mild TBI, induced by a concussive-like, closed head injury for three consecutive days, showed gene expression profiles consistent with activation of cell cycle markers, genotoxic stress responses, and inflammation over the first week post-injury (Schwab et al., 2021). With respect to cell type(s), post-mitotic neurons do not undergo senescence whereas glial senescence more readily occurs (Fielder et al., 2017; Moreno-Blas et al., 2019; Chinta et al., 2013; Cohen and Torres, 2019; Han et al., 2020). Moderate TBI elevated the expression of senescence markers in microglia, with more pronounced effects on microglial responses, leukocyte invasion, and senescence observed in aged mice, as compared to young mice (Ritzel et al., 2019). Interestingly, microglial proliferation in response to facial nerve axotomy exhibits age-dependent changes while aged microglia exhibit defects in phagocytosis, disrupted homeostatic functions, increased senescence, and potentiated inflammation (Koellhoffer et al., 2017; Conde and Streit, 2006). Moreover, senescent astrocytes are observed in both aged and Alzheimer’s brain tissue (Pertusa et al., 2007; Limbad et al., 2020; Bhat et al., 2012). Similarly, SA-β-gal activity, increased for over two weeks post-TBI while elevated expression of p16INK4a, a cyclin-dependent kinase inhibitor associated with senescence, was specifically elevated in astrocytes after TBI (Tominaga et al., 2019; LaPak and Burd, 2014). While temporally correlated with cognitive decline, it is unknown whether glial senescence causes neurodegeneration or reflects ongoing pathology. In support of the former possibility, genetic clearance of senescent glia slowed neurodegeneration and improved cognitive function in a murine tauopathy model (Bussian et al., 2018). In addition, aged astrocytes reduce neurotrophic support while astrocyte senescence is associated with an elevated risk of excitotoxicity, an important cause of neuronal loss and post-traumatic seizures, after TBI (Pertusa et al., 2007; Limbad et al., 2020). The mechanisms underlying the cell-type and age-dependent effects are unknown; however, a higher baseline of cellular senescence in the aged brain could explain why chronologically older individuals exhibit worse neurological outcomes after TBI (Thompson et al., 2006). Further analysis of cell-type specific mechanisms underlying the initiation of senescence may provide valuable insights into determining the wide range of outcomes after TBI.
4. Senescence associated secretory phenotype (SASP) and chronic inflammation
Depending on the cell type and initiating stimulus, approximately 30–70% of senescent cells undergo a phenotypic change to develop a senescence associated secretory phenotype (SASP). SASP consists of increased, cell-dependent secretion of interleukins, cytokines, chemokines, and matrix metalloproteinases to potentiate senescence and perpetuate inflammaging (Coppe et al., 2008; Kuilman and Peeper, 2009). Genotoxic stress, mTOR signaling, Jak2/STAT3 signaling, or mitochondrial dysfunction drive the expression of SASP-associated mediators, at least in part, via activation of the pro-inflammatory transcription factors, nuclear factor κB (NFκB) and CCAAT/enhancer-binding protein β (CEBP/β) (Hernandez-Segura et al., 2018).We and others reported elevated expression of SASP-associated factors, including interleukin-1β (IL-1β), IL-6, IL-8, tumor necrosis factor-α (TNF-α) and chemokines, such as C-X-C motif chemokine ligand 10 (CXCL10), C-C motif chemokine ligand 2 (CCL2), and CCL5 following TBI (Vaibhav et al., 2020; Woodcock and Morganti-Kossmann, 2013). These inflammatory mediators simultaneously enhance immune cell recruitment to eliminate senescent cells and perpetuate inflammation. Cells exhibiting the SASP also release matrix metalloproteinases (MMP), including MMP-3, MMP-9, MMP-12 and serpins, such as plasminogen activator inhibitor-1 (PAI-1) that modify the extracellular matrix and induce both tissue fibrosis and destruction (Jiang et al., 2017; Yamamoto et al., 2014). Interestingly, fibrosis and glial scarring persist for years after TBI (Di Giovanni et al., 2005; Burda et al., 2016) and contribute to chronic neurodegeneration (D’Ambrosi and Apolloni, 2020). Finally, release of exosomes and microRNA from senescent cells modulate stem cells and promote inflammation (Xu and Tahara, 2013). Thus, targeting the SASP may provide an innovative therapeutic approach to limit the long-term consequences of chronic inflammation after TBI.
5. Cellular senescence drives neurodegeneration
Cellular senescence drives tau aggregation (Musi et al., 2018) and is associated with Aβ pathology, the accumulation of the neurotoxic oligomer characteristic of AD and CTE (Stein et al., 2014). In a reciprocal manner, Aβ accelerated cellular senescence, as quantified by increased p16INK4a expression, and worsened cognitive function in a 5xFAD mouse model of AD (Wei et al., 2016). AD patient brains showed increased senescence in oligodendrocyte precursor cells while administration of senolytic drugs reduced cellular senescence, decreased neuroinflammation, prevented neurofibrillary tangles, decreased neurodegeneration, and improved cognitive outcomes in rodent AD models (Musi et al., 2018; Zhang et al., 2019; Mendelsohn and Larrick, 2018). Similarly, genetic elimination of senescent cells prevented tau pathology and slowed cognitive decline in transgenic mouse models of tau-dependent neurodegeneration (Bussian et al., 2018). Consistent with pre-clinical modeling, evidence of glial senescence and SASP phenotypes were observed in post-mortem brains from contact athletes with a history of brain trauma (Schwab et al., 2019); however, many classic neuropathological signs of dementia were absent in this cohort, despite clinical symptoms and neurological deficits during life (Schwab et al., 2019), raising the intriguing and largely unexplored possibility that senescent cells drive neurodegeneration.
6. Genotoxic stress: a initiating factor in cellular senescence after TBI?
Cellular senescence is triggered by diverse stimuli, including genotoxic stress, telomere attrition, oncogenes, stress, protein aggregation, reduced autophagy, reactive metabolites, oxidative stress, mechanical stress, danger associated molecular patterns, pathogens, and exposure to inflammatory mediators (Fig. 1). Of these inducers, oxidative stress, a metabolic state whereby the production of reactive oxygen species overwhelms endogenous antioxidant defense, develops immediately after acute brain injuries and is believed to induce senescence, at least in part, via DNA damage. Genotoxic stress is associated with the induction of senescence pathways, including elevated expression of the cyclin-dependent kinase inhibitors, p16INK4a and p21WAF1/Cip1 (Rayess et al., 2012; Yosef et al., 2017), or the tumor suppressor gene, p53, which maintain cell cycle arrest (Rufini et al., 2013). While the precise mechanisms remain poorly defined, reduced DNA repair pathways may confer susceptibility to chronic neurodegeneration (Jeppesen et al., 2011; Madabhushi et al., 2014; Pessina et al., 2021), providing a potential candidate risk factor for further exploration.
Fig. 1.
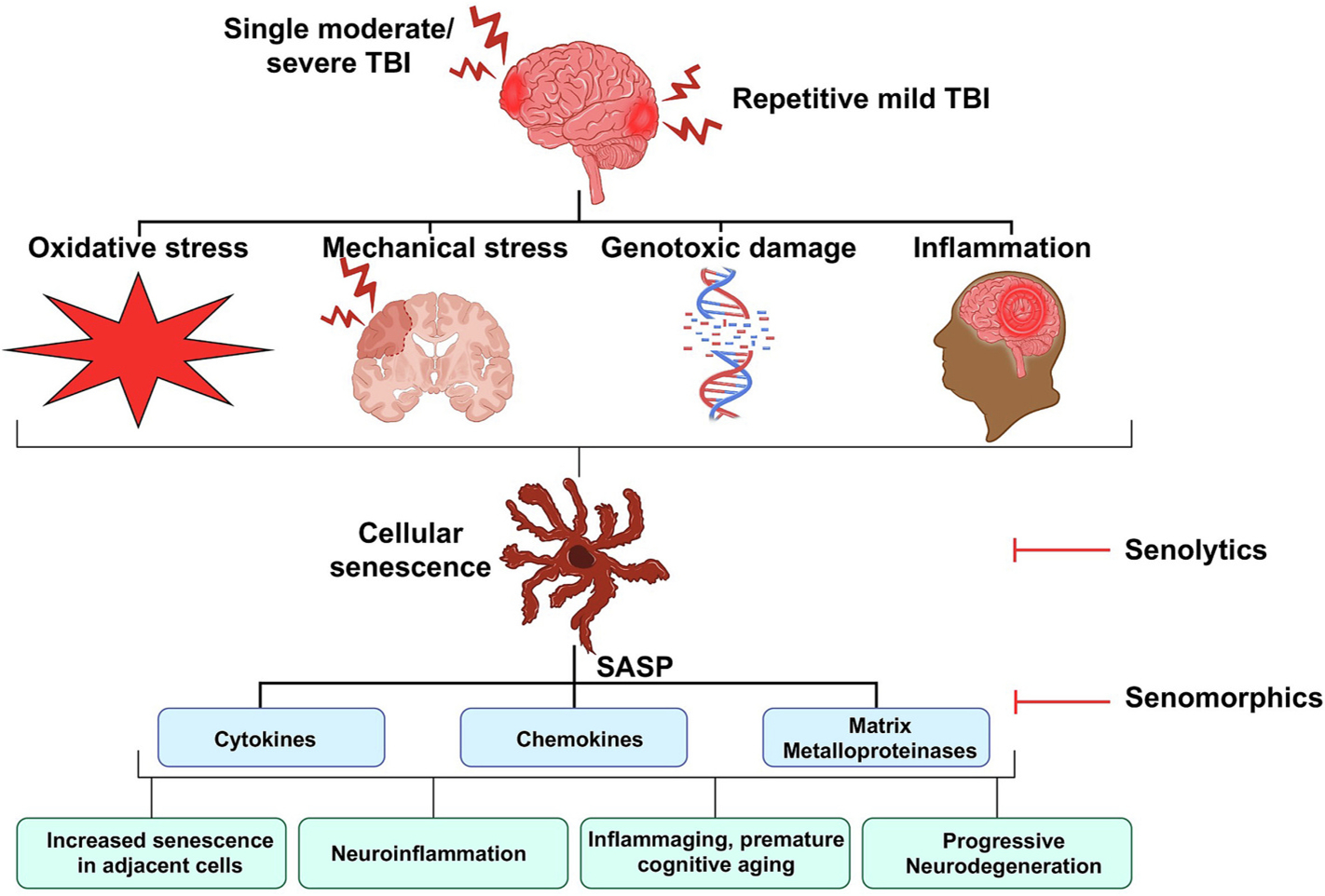
Proposed mechanisms of senescence following TBI. A single moderate-severe TBI or repetitive mild TBI produces oxidative and mechanical stress, inflammation, and genotoxic stress. These events contribute toward the development of cellular senescence and a SASP, which potentiate cellular senescence, advance cognitive aging and drive inflammaging to accelerate progressive neurodegeneration. Administration of senolytic and/or senomorphic therapies removes or modulates senescence cells, respectively, to improve cognitive outcomes.
Oxidative genotoxic stress is present in multiple models of TBI (Morita-Fujimura et al., 1999; Zhang et al., 2002; Wang et al., 2014). Toward this end, moderate controlled cortical impact increased both single and double strand DNA breaks predominantly within neurons of the ipsilateral cortex and hippocampus over the first days post-injury (Clark et al., 2001). In parallel, diffuse staining for 8-hydroxy-2’-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage, was elevated within both neurons and astrocytes of the ipsilateral cortex following severe TBI (Mendez et al., 2004). Moreover, double-strand DNA breaks were associated with early onset senescence and cognitive impairments after repeated mild TBI in mice (Schwab et al., 2021). In line with experimental data, a reduction in DNA repair pathways increased WM injury and axonal damage in professional athletes after mild TBI (Schwab et al., 2019) while genotoxic stress induced senescent oligodendrocytes, which reduced myelination and worsened axonal health, during chronological aging (Tse and Herrup, 2017). Consistent with these findings, mice with deficient DNA repair exhibited delayed neurological recovery after TBI (Tomasevic et al., 2012; Fujimura et al., 2000). Furthermore, elevated DNA damage markers in TBI patient blood correlated with injury severity while serum 8-OHdG levels associated with mortality after severe TBI (Shaked et al., 2014; Lorente et al., 2020).
Poly (ADP-ribose) polymerase (PARP) is a key DNA-binding enzyme that detects DNA strand breaks following genotoxic stress. Mice with genetic deletion of PARP exhibit increased genomic instability and elevated mortality after genotoxic stress (de Murcia et al., 1997). With respect to TBI, persistently elevated PARP expression is observed in brain mitochondria following controlled cortical impact, indicative of mitochondrial dysfunction and metabolic oxidative stress (Lai et al., 2008; Hill et al., 2017). Of note, PARP activation leads to energy depletion and activation of cell death pathways to exacerbate tissue damage (Andrabi et al., 2006). In support of this possibility, PARP inhibition attenuated microglial activation, reduced neurodegeneration, and improved functional outcomes after moderate TBI in mice (Stoica et al., 2014). Similarly, PARP inhibitors reduced injury volume after fluid percussion injury in rats (LaPlaca et al., 2001). Thus, early DNA damage responses may induce stress-induced cellular senescence to potentiate progressive neurodegeneration.
7. Senotherapeutics – the future of chronic TBI management
Senescent cells exhibit profound resistance to apoptosis, in part, due to upregulation in senescence cell associated anti-apoptotic pathways (SCAP) (Zhu et al., 2015). Interestingly, genetic elimination of as few as 30% of p16INK4a positive senescent cells delayed the onset of aging-associated phenotypes and extended the healthy lifespan of progeroid mice (Zhu et al., 2015; Baker et al., 2011; Farr et al., 2017). Based on these exciting studies, a push to identify and/or develop senotherapeutic agents, a class of drugs that selectively target and remove senescent cells (senolytics), emerged to combat the detrimental aspects of aging. Senolytic agents, broadly divided into classes based on mechanism of action, include kinase inhibitors, inhibitors of Bcl-2 family members, natural compounds, p53 inhibitors, heat shock protein 90 inhibitors, and histone deacetylase inhibitors (HDAC). As TBI appears to prematurely accelerate aging, these approaches may provide an innovative approach to reduce the long-term deleterious consequences of TBI. Table 1 summarizes research to date studying senolytics in the pre-clinical and clinical management of TBI.
Table 1.
Senolytic treatments after pre-clinical and clinical TBI.
| Drug | Dose/Route/Timing | Species | TBI Model | Main findings | Ref |
|---|---|---|---|---|---|
|
| |||||
| Pre-clinical | |||||
| Dasatinib | 25 mg/kg, i.p., daily beginning at 2h post-injury | Mice | Moderate lateral fluid percussion | Reduced inflammation, neurodegeneration, and improved motor and cognitive outcomes after TBI | (Yu et al., 2016) |
| Quercetin | 25 pmol/kg, i.p., 1h post-injury | Rats | Moderate anterior midline fluid percussion | Improved compound action potential amplitudes, reduced oxidative stress | (Schultke et al., 2005) |
| Quercetin | 30 mg/kg i.p., daily for 3 days post-injury | Rats | Weight drop | Improved cognitive, reduced cell death, improved antioxidant enzymes expression | Yang et al., 2014 |
| Quercetin | 50 mg/kg, i.p., 0.5h post-injury | Mice | Weight drop | Improved mitochondrial function, increased antioxidant defense, reduced cell death, decreased gliosis, improved neurobehavioral outcomes | (Li et al., 2016a)- (Kosari-Nasab et al., 2019) |
| Quercetin | 50 mg/kg, i.p., 0.5h, 12h, 24h post-injury | Rats | Weight drop | Reduced neuronal autophagy, decreased apoptosis, improved cognitive function | (Du et al., 2016)- (Du et al., 2018) |
| Quercetin | 5, 20, 50 mg/kg i.p., 0.5h, 12h, 24h post-injury | Rats | Weight drop | Reduced edema, microgliosis, inflammation, and oxidative stress | (Song et al., 2021) |
| Navitoclax (ABT- 263) | 1.5 mg/kg, i.p, 1 week post-injury | Mice | Repeated mild (controlled cortical impact) closed head injury | Reduced senescence markers, improved cognitive performance in males, but not females | (Schwab et al., 2022) |
| Fisetin | 25, 50 or 75 mg/kg, i.p, 0.5h post-injury | Mice | Weight drop | Improved neurological function, reduced oxidative stress, decreased neuronal apoptosis | (Zhang et al., 2018) |
| Curcumin | 500 ppm, p.o, 1 month pre-injury | Rats | Mild fluid percussion injury | Increased neurotrophin expression, reduced oxidative stress, improved cerebral energy homeostasis | (Sharma et al., 2009) |
| Curcumin | 75, 150 mg/kg, i.p. 15 minutes pre-injury | Mice | Controlled cortical impact | Reduced inflammation, improved neurological outcomes | (Khayatan et al., 2022)- (Sun et al., 2020) |
| Curcumin | 100 mg/kg, i.p., 5d pre-injury | Rats | Weight drop | Reduced oxidative stress, decreased lesion volume | (Samini et al., 2013) |
| Curcumin | 100 mg/kg, i.p. 15 minutes pre-injury | Mice | Weight drop | Decreased microglial/macrophage activation, reduced neuronal apoptosis | (Zhu et al., 2014) |
| Clinical | |||||
| Curcumin (Curcuminoids, C3 Complex) | 500 mg, via enteral nutrition, daily for 7d | Human | ICU-admitted adult TBI patients | Improved nutritional status and reduced serum levels of IL- 6, TNF-alpha, MCP-1 and CRP compared to baseline | (Zahedi et al., 2021) |
| Luteolin (PEALut, Glialia) | 700 mg palmitoyl ethanolamide+70 mg luteolin, oral, twice daily for 180d | Human | Moderate (GCS 9–13) TBI patients | Improved cognitive function assessed via mini mental state examination (MMSE) and brief neuropsychological cognitive examination (BNCE) and facilitated neural recovery on working memory | (Campolo et al., 2021) |
Dasatinib, an FDA-approved tyrosine kinase inhibitor used to treat newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia, promotes apoptosis in part, via inhibition of Src kinase while quercetin is a naturally occurring flavonoid that gives apple peels a bitter taste. Combination treatment with dasatinib and quercetin, but not treatment with either compound alone, induced apoptosis in senescent human primary adipocyte progenitor cells and in human umbilical vein endothelial cells (HUVEC), but not in non-senescent adipocyte progenitor cells or HUVEC (Zhu et al., 2015). Administration of dasatinib and quercetin also removed senescent cells in vivo, improving outcomes in fibrotic pulmonary disease, reducing age-associated bone loss, improving physical function in old age, and improving outcomes after diabetic kidney disease (Schafer et al., 2017; Xu et al., 2018; Hickson et al., 2019).
Astrocytes acquire a senescent phenotype in Alzheimer’s disease patients while genetic clearance of senescent glia reduced gliosis, tau accumulation, and preserved cognitive function in a pre-clinical model of tauopathy (Bhat et al., 2012; Bussian et al., 2018). Similarly, administration of dasatinib and quercetin ablated senescent astrocytes and reversed tau-mediated neurodegeneration in experimental models of Alzheimer’s disease (Musi et al., 2018). In line with these findings, post-mortem analysis of athletes with a history of neurotrauma revealed widespread astrocyte senescence and activation of SASP pathways, which paralleled the development of neurodegenerative changes (Schwab et al., 2019). While the functional significance of this association remains unclear, intraperitoneal administration of dasatinib decreased inflammation, reduced neurodegeneration, and improved neurological outcomes, as compared to placebo control (Yu et al., 2016). Quercetin also provided beneficial effects in rodent models of TBI, reducing oxidative stress after moderate fluid percussion injury (Schultke et al., 2005), improving mitochondrial function and cognitive function after weight drop injury (Yang et al., 2014; Li et al., 2016a; Li et al., 2016b), and reducing gliosis and anxious behavior after repetitive mild TBI in mice and rats (Zabenko and Pivneva, 1994; Kosari-Nasab et al., 2019; Du et al., 2016; Du et al., 2018; Song et al., 2021); however, it remains undetermined whether these beneficial effects are due to the direct free radical scavenging properties of quercetin or whether these changes are due to downstream senolytic activity. The clinical utility of dasatinib and quercetin remains unexplored in TBI patients; however, Enzogenol, a quercetin containing extract derived from Pinus radiata bark, was safe, well tolerated, and appeared to improve cognitive function, enhance sleep quality, and reduce mental fatigue for up to one year after mild TBI (Theadom et al., 2013; Walter et al., 2017). Furthermore, dasatinib plus quercetin is currently under investigation in an exploratory open-label clinical study for early-stage Alzheimer’s disease (Gonzales et al., 2022). These promising pre-clinical and early-stage clinical studies suggest a potential benefit of senolytic therapy after TBI.
Curcumin, the principal curcuminoid of the spice turmeric, is a multi-functional compound with anti-aging properties that exhibits potent senolytic activity (Beltzig et al., 2021). Similarly, a putative curcumin metabolite, o-vanillin, and a curcumin analog, EF24, exhibit senolytic activity (Li et al., 2019; Zanzer et al., 2019). We and others reported that administration of curcumin reduced glial activation, decreased inflammation, and improved cognitive outcomes in rodent models of both mild and moderate-severe TBI (Khayatan et al., 2022; Laird et al., 2010; Li et al., 2022; Sun et al., 2020; Zhu et al., 2014; Wu et al., 2011; Wu et al., 2006; Sharma et al., 2009; Samini et al., 2013). Curcumin also exhibits protective effects against neurodegeneration in animal models (Ojha et al., 2012; Kim et al., 2012; Sundaram et al., 2017; Abrahams et al., 2019; Ulamek-Koziol et al., 2020; Nebrisi, 2021; Pluta et al., 2022) while curcuminoid supplementation exhibits beneficial effects on inflammation and clinical outcomes in critically ill TBI patients (Zahedi et al., 2021). Similarly, a randomized, placebo-controlled, double-blind study found curcumin supplementation slowed cognitive decline in aged adults (Rainey-Smith et al., 2016). In line with these data, administration of a cocktail containing luteolin, another natural compound with predicted senolytic activity (Vazquez et al., 2022), improved cognitive and neuropsychological function in moderate TBI patients (Campolo et al., 2021). While these promising experimental and early-stage clinical studies suggest a potential therapeutic role for senolytics, additional pre-clinical and randomized controlled trials are needed to define the precise mechanism of action and establish the efficacy of senolytics to improve cognitive outcomes after TBI.
Fisetin (7, 3’, 4’-flavon-3-ol), a natural flavonol present in strawberries, apples, persimmons, onions, and cucumbers, exhibits senolytic activity, at least in part, by inhibiting anti-apoptotic Bcl-2 family members and other targets within SCAP networks (Yousefzadeh et al., 2018; Zhu et al., 2017). While fisetin has not been explored after clinical TBI, it is noteworthy that fisetin suppressed neuronal loss after experimental TBI (Zhang et al., 2018) and reduced neurodegeneration in experimental models of neurodegeneration (Hassan et al., 2022; Nabavi et al., 2016; Ahmad et al., 2017; Patel et al., 2012). In addition to natural senolytics that exhibit broad spectrum effects in vivo, newer compounds are under development to specifically target senescence inducing pathways. Navitoclax (ABT263), an experimental oral Bcl-2/Bcl-xL inhibitor, reduced the expression of senescence markers and improved cognitive function in male, but not female mice after repetitive mild TBI (Schwab et al., 2022), suggesting a possible sex-difference in the mechanisms of neurodegeneration after head injury; however, the impact of inhibitors of Bcl-2 family members after moderate-severe TBI or in clinical populations remain untested to date.
In addition to senolytics, which directly kill senescent cells, senomorphics modulate senescent cells to interfere with senoinflammation and the SASP to prevent inflammaging. Putative senomorphic therapies include telomerase activators, caloric restriction, sirtuin activators, mTOR activators, antioxidants, autophagy activators, and proteasome activators (Hubbard and Sinclair, 2014; Wei et al., 2017; Lamming et al., 2013; Chondrogianni et al., 2015; Tilstra et al., 2012; Xu et al., 2015b; Kang et al., 2017; Cao et al., 2011; Lee et al., 2016). While less characterized as compared to senolytic agents, caloric restriction, a nutritional, non-pharmaceutic intervention, increases longevity via activation of sirtuin 1 and modulation of mTOR, which affects cellular metabolism and autophagy. Caloric restriction of 50% of daily food intake for three months, suppressed microglial activation and reduced neurodegeneration in a cortical stab wound model of TBI in rats (Loncarevic-Vasiljkovic et al., 2012; Loncarevic-Vasiljkovic et al., 2016). Similarly, chronic dietary caloric restriction of 70% of normal food intake decreased lesion volume and improved spatial memory following weight drop injury in rat (Rich et al., 2010; Rubovitch et al., 2019). Whether the benefits of caloric restriction are due to senomodulation and/or other mechanisms of action remain untested and will be the subject of exciting future investigations.
8. Conclusions and future perspectives
TBI remains a worldwide public health issue that places a major burden on society. Compelling epidemiological data suggests TBI is a distinct neurodegenerative disorder that may be initiated in otherwise healthy individuals following a single moderate-severe TBI or repetitive mild head trauma; however, the mechanisms underlying the development of progressive neurological injury after TBI remain poorly defined, which contributes to the lack of efficacious therapies to improve long-term quality of life. Cellular senescence is a conserved mechanism of inflammaging and may represent a cause of premature cognitive aging after all types of TBI. Oxidative stress and genotoxic damage are key initiators of senescence. Consistent with this possibility, the polymorphic nature of DNA repair factors and sex-differences in genomic instability may be responsible for the clinical heterogeneity in outcomes after TBI (Fischer and Riddle, 2018; Leung and Hazrati, 2021). Thus, proactive targeting of the early genotoxic stress response may restrain the initiation of cellular senescence/SASP as a “first domino” that potentiates progressive neurodegeneration at all levels of TBI. As such, senolytics may provide an innovative therapeutic approach to improve long-term neurological outcomes after TBI. First generation senolytics are largely natural compounds with pleiotropic mechanisms of action, including the removal of senescent cells. Newer generation senolytics will optimize and more precisely target senescence to potentially limit or reverse post-traumatic cognitive aging. In addition to the selective killing of senescent cells by senolytic agents, senomorphic agents act by blocking SASP in senescent cells to control the deleterious aspects of chronic inflammation. Future translational studies are also needed in humans to further refine and optimize the timing, dosing, and routes of administration of senotherapeutics. These exciting topics offer great promise in the future of TBI management.
Funding
The authors are supported by grants from the National Institutes of Health (R01NS110378 to KMD and BB, R01NS117565 to KMD, and R01NS122724 to DWB).
Footnotes
CRediT authorship contribution statement
Yujiao Lu: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Visualization. Abbas Jarrahi: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. Nicholas Moore: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. Manuela Bartoli: Conceptualization, Writing – review & editing. Darrell W. Brann: Conceptualization, Writing – review & editing, Funding acquisition. Babak Baban: Conceptualization, Writing – review & editing. Krishnan M. Dhandapani: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Supervision, Project administration, Funding acquisition.
Data availability
No data was used for the research described in the article.
References
- Abrahams S, Haylett WL, Johnson G, Carr JA, Bardien S, 2019. Antioxidant effects of curcumin in models of neurodegeneration, aging, oxidative and nitrosative stress: a review. Neuroscience. 406, 1–21. 10.1016/j.neuroscience.2019.02.020. [DOI] [PubMed] [Google Scholar]
- Agrawal S, Leurgans SE, James BD, Barnes LL, Mehta RI, Dams-O’Connor K, Mez J, Bennett DA, Schneider JA, 2022. Association of traumatic brain injury with and without loss of consciousness with neuropathologic outcomes in community-dwelling older persons. JAMA Netw Open. 5, e229311 10.1001/jamanetworkopen.2022.9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A, Ali T, Park HY, Badshah H, Rehman SU, Kim MO, 2017. Neuroprotective effect of Fisetin against amyloid-beta-induced cognitive/synaptic dysfunction, neuroinflammation, and neurodegeneration in adult mice. Mol Neurobiol. 54, 2269–2285. 10.1007/s12035-016-9795-4. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, et al. , 2006. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 103, 18308–18313. 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Rossetti F, Wilder DM, Sajja S, Van Albert SA, Wang Y, Gist ID, Long JB, 2020. Blast exposure leads to accelerated cellular senescence in the rat brain. Front Neurol. 11, 438. 10.3389/fneur.2020.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K, 2015. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 212, 991–999. 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagi Z, Kroenke CD, Fopiano KA, Tian Y, Filosa JA, Sherman LS, Larson EB, Keene CD, Degener O’Brien K, Adeniyi PA, et al. , 2022. Association of cerebral microvascular dysfunction and white matter injury in Alzheimer’s disease. Geroscience. 10.1007/s11357-022-00585-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM, 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 479, 232–236. 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K, 2018. Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol. 75, 1055–1061. 10.1001/jamaneurol.2018.0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltzig L, Frumkina A, Schwarzenbach C, Kaina B, 2021. Cytotoxic, genotoxic and senolytic potential of native and micellar curcumin. Nutrients. 13. 10.3390/nu13072385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger RP, Adelson PD, Pierce MC, Dulani T, Cassidy LD, Kochanek PM, 2005. Serum neuron-specific enolase, S100B, and myelin basic protein concentrations after inflicted and noninflicted traumatic brain injury in children. J Neurosurg. 103, 61–68. 10.3171/ped.2005.103.1.0061. [DOI] [PubMed] [Google Scholar]
- Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C, 2012. Astrocyte senescence as a component of Alzheimer’s disease. PLoS One. 7, e45069 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniek KF, Cairns NJ, Crary JF, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, et al. , 2021. The second NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 80, 210–219. 10.1093/jnen/nlab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD, 2002. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 103, 607–614. 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- Braughler JM, Hall ED, 1985. Current application of “high-dose” steroid therapy for CNS injury. A pharmacological perspective. J Neurosurg. 62, 806–810. 10.3171/jns.1985.62.6.0806. [DOI] [PubMed] [Google Scholar]
- Bray MJC, Richey LN, Bryant BR, Krieg A, Jahed S, Tobolowsky W, LoBue C, Peters ME, 2021. Traumatic brain injury alters neuropsychiatric symptomatology in all-cause dementia. Alzheimers Dement. 17, 686–691. 10.1002/alz.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buki A, Povlishock JT, 2006. All roads lead to disconnection?–Traumatic axonal injury revisited. Acta Neurochir (Wien). 148, 181–193. Discussion 193–184. 10.1007/s00701-005-0674-4. [DOI] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, Sofroniew MV, 2016. Astrocyte roles in traumatic brain injury. Exp Neurol. 275 (Pt 3), 305–315. 10.1016/j.expneurol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ, 2018. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 562, 578–582. 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campolo M, Crupi R, Cordaro M, Cardali SM, Ardizzone A, Casili G, Scuderi SA, Siracusa R, Esposito E, Conti A, et al. , 2021. Co-Ultra PEALut enhances endogenous repair response following moderate traumatic brain injury. Int J Mol Sci. 22. 10.3390/ijms22168717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS, 2011. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 121, 2833–2844. 10.1172/JCI43578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease C, Prevention, 2009. Prevalence and most common causes of disability among adults–United States, 2005. MMWR Morb Mortal Wkly Rep. 58, 421–426. [PubMed] [Google Scholar]
- Chapman J, Fielder E, Passos JF, 2019. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett. 593, 1566–1579. 10.1002/1873-3468.13498. [DOI] [PubMed] [Google Scholar]
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH, 2004. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 165, 357–371. 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH, 2009. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 19, 214–223. 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini C, Hogan KA, Warner GM, Tarrago MG, Peclat TR, Tchkonia T, Kirkland JL, Chini E, 2019. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD(+) decline. Biochem Biophys Res Commun. 513, 486–493. 10.1016/j.bbrc.2019.03.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK, 2013. Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med. 273, 429–436. 10.1111/joim.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A, Benzi G, Dossena M, Mutani R, Mora G, 2005. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 128, 472–476. 10.1093/brain/awh373. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Voutetakis K, Kapetanou M, Delitsikou V, Papaevgeniou N, Sakellari M, Lefaki M, Filippopoulou K, Gonos ES, 2015. Proteasome activation: an innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res Rev. 23, 37–55. 10.1016/j.arr.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Clark RSB, Chen M, Kochanek PM, Watkins SC, Jin KL, Draviam R, Nathaniel PD, Pinto R, Marion DW, Graham SH, 2001. Detection of single- and double-strand DNA breaks after traumatic brain injury in rats: comparison of in situ labeling techniques using DNA polymerase I, the Klenow fragment of DNA polymerase I, and terminal deoxynucleotidyl transferase. J Neurotrauma. 18, 675–689. 10.1089/089771501750357627. [DOI] [PubMed] [Google Scholar]
- Cohen J, Torres C, 2019. Astrocyte senescence: evidence and significance. Aging Cell. 18, e12937 10.1111/acel.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JH, Marioni RE, Harris SE, Deary IJ, 2019. Brain age and other bodily ‘ages’: implications for neuropsychiatry. Mol Psychiatry. 24, 266–281. 10.1038/s41380-018-0098-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde JR, Streit WJ, 2006. Effect of aging on the microglial response to peripheral nerve injury. Neurobiol Aging. 27, 1451–1461. 10.1016/j.neurobiolaging.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J, 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsellis JA, Brierley JB, 1959. Observations on the pathology of insidious dementia following head injury. J Ment Sci. 105, 714–720. 10.1192/bjp.105.440.714. [DOI] [PubMed] [Google Scholar]
- Crane PK, Gibbons LE, Dams-O’Connor K, Trittschuh E, Leverenz JB, Keene CD, Sonnen J, Montine TJ, Bennett DA, Leurgans S, et al. , 2016. Association of traumatic brain injury with late-life neurodegenerative conditions and neuropathologic findings. JAMA Neurol. 73, 1062–1069. 10.1001/jamaneurol.2016.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosi N, Apolloni S, 2020. Fibrotic scar in neurodegenerative diseases. Front Immunol. 11, 1394. 10.3389/fimmu.2020.01394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Freitas Cardoso MG, Faleiro RM, de Paula JJ, Kummer A, Caramelli P, Teixeira AL, de Souza LC, Miranda AS, 2019. Cognitive impairment following acute mild traumatic brain injury. Front Neurol. 10, 198. 10.3389/fneur.2019.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, et al. , 1997. Requirement of poly (ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 94, 7303–7307. 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch MB, Mendez MF, Teng E, 2015. Interactions between traumatic brain injury and frontotemporal degeneration. Dement Geriatr Cogn Disord. 39, 143–153. 10.1159/000369787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI, 2005. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 102, 8333–8338. 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F, 2021. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 22, 75–95. 10.1038/s41580-020-00314-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Zhao Z, Chen Y, Li Z, Tian Y, Liu Z, Liu B, Song J, 2016. Quercetin attenuates neuronal autophagy and apoptosis in rat traumatic brain injury model via activation of PI3K/Akt signaling pathway. Neurol Res. 38, 1012–1019. 10.1080/01616412.2016.1240393. [DOI] [PubMed] [Google Scholar]
- Du G, Zhao Z, Chen Y, Li Z, Tian Y, Liu Z, Liu B, Song J, 2018. Quercetin protects rat cortical neurons against traumatic brain injury. Mol Med Rep. 17, 7859–7865. 10.3892/mmr.2018.8801. [DOI] [PubMed] [Google Scholar]
- Fabbri E, An Y, Zoli M, Simonsick EM, Guralnik JM, Bandinelli S, Boyd CM, Ferrucci L, 2015. Aging and the burden of multimorbidity: associations with inflammatory and anabolic hormonal biomarkers. J Gerontol A Biol Sci Med Sci. 70, 63–70. 10.1093/gerona/glu127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI, Loane DJ, 2015. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics. 12, 143–150. 10.1007/s13311-014-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann JR, Ribe AR, Pedersen HS, Fenger-Gron M, Christensen J, Benros ME, Vestergaard M, 2018. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. 5, 424–431. 10.1016/S2215-0366(18)30065-8. [DOI] [PubMed] [Google Scholar]
- Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N, 2013. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis. 4, e754 10.1038/cddis.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, et al. , 2017. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 23, 1072–1079. 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielder E, von Zglinicki T, Jurk D, 2017. The DNA damage response in neurons: Die by apoptosis or survive in a senescence-like state? J Alzheimers Dis. 60, S107–S131. 10.3233/JAD-161221. [DOI] [PubMed] [Google Scholar]
- Fischer KE, Riddle NC, 2018. Sex differences in aging: genomic instability. J Gerontol A Biol Sci Med Sci. 73, 166–174. 10.1093/gerona/glx105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A, 2003. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 74, 857–862. 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke K, Gaser C, 2019. Ten years of brain AGE as a neuroimaging biomarker of brain aging: What insights have we gained? Front Neurol. 10, 789. 10.3389/fneur.2019.00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Noshita N, Yoshimoto T, Chan PH, 2000. Reduction of the DNA base excision repair protein, XRCC1, may contribute to DNA fragmentation after cold injury-induced brain trauma in mice. Brain Res. 869, 105–111. 10.1016/s0006-8993(00)02375-1. [DOI] [PubMed] [Google Scholar]
- Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, Witkowski JM, Franceschi C, 2017. Immunosenescence and inflamm-aging as two sides of the same coin: Friends or Foes? Front Immunol. 8, 1960. 10.3389/fimmu.2017.01960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale SD, Johnson SC, Bigler ED, Blatter DD, 1995. Nonspecific white matter degeneration following traumatic brain injury. J Int Neuropsychol Soc. 1, 17–28. 10.1017/s1355617700000060. [DOI] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K, 2015. Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol. 77, 987–995. 10.1002/ana.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E, 1989. Severe head injury hastens age of onset of Alzheimer’s disease. J Am Geriatr Soc. 37, 970–973. 10.1111/j.1532-5415.1989.tb07283.x. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW, 1993. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 160, 139–144. 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Gentry LR, Thompson B, Godersky JC, 1988. Trauma to the corpus callosum: MR features. AJNR Am J Neuroradiol. 9, 1129–1138. [PMC free article] [PubMed] [Google Scholar]
- Gonzales MM, Garbarino VR, Marques Zilli E, Petersen RC, Kirkland JL, Tchkonia T, Musi N, Seshadri S, Craft S, Orr ME, 2022. Senolytic therapy to modulate the progression of Alzheimer’s Disease (SToMP-AD): a pilot clinical trial. J Prev Alzheimers Dis. 9, 22–29. 10.14283/jpad.2021.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick PB, 2010. Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann N Y Acad Sci. 1207, 155–162. 10.1111/j.1749-6632.2010.05726.x. [DOI] [PubMed] [Google Scholar]
- Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, Raskind M, 1990. The association between head trauma and Alzheimer’s disease. Am J Epidemiol. 131, 491–501. 10.1093/oxfordjournals.aje.a115523. [DOI] [PubMed] [Google Scholar]
- Green RE, Colella B, Maller JJ, Bayley M, Glazer J, Mikulis DJ, 2014. Scale and pattern of atrophy in the chronic stages of moderate-severe TBI. Front Hum Neurosci. 8, 67. 10.3389/fnhum.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, et al. , 2000. Head injury and the risk of AD in the MIRAGE study. Neurology. 54, 1316–1323. 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- Han X, Zhang T, Liu H, Mi Y, Gou X, 2020. Astrocyte senescence and Alzheimer’s disease: a review. Front Aging Neurosci. 12, 148. 10.3389/fnagi.2020.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase Y, Horsburgh K, Ihara M, Kalaria RN, 2018. White matter degeneration in vascular and other ageing-related dementias. J Neurochem. 144, 617–633. 10.1111/jnc.14271. [DOI] [PubMed] [Google Scholar]
- Hassan SSU, Samanta S, Dash R, Karpinski TM, Habibi E, Sadiq A, Ahmadi A, Bunagu S, 2022. The neuroprotective effects of fisetin, a natural flavonoid in neurodegenerative diseases: focus on the role of oxidative stress. Front Pharmacol. 13, 1015835. 10.3389/fphar.2022.1015835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS, 1961. The serial cultivation of human diploid cell strains. Exp Cell Res. 25, 585–621. 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Hernandez-Segura A, Nehme J, Demaria M, 2018. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453. 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, et al. , 2019. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 47, 446–456. 10.1016/j.ebiom.2019.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RL, Singh IN, Wang JA, Hall ED, 2017. Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem Int. 111, 45–56. 10.1016/j.neuint.2017.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Sinclair DA, 2014. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 35, 146–154. 10.1016/j.tips.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, et al. , 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST, 2004. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 190, 192–203. 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Irimia A, Goh SY, Torgerson CM, Vespa P, Van Horn JD, 2014. Structural and connectomic neuroimaging for the personalized study of longitudinal alterations in cortical shape, thickness and connectivity after traumatic brain injury. J Neurosurg Sci. 58, 129–144. [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH, 2002. Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 61, 1056–1068. 10.1093/jnen/61.12.1056. [DOI] [PubMed] [Google Scholar]
- Jeppesen DK, Bohr VA, Stevnsner T, 2011. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 94, 166–200. 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Liu G, Luckhardt T, Antony V, Zhou Y, Carter AB, Thannickal VJ, Liu RM, 2017. Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell. 16, 1114–1124. 10.1111/acel.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2010. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 11, 361–370. 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2012. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W, 2013a. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 136, 28–42. 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH, 2013b. Axonal pathology in traumatic brain injury. Exp Neurol. 246, 35–43. 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkonde YV, Jawaid A, Qureshi SU, Shirani P, Wheaton M, Pinto-Patarroyo GP, Schulz PE, 2012. Medical and environmental risk factors associated with frontotemporal dementia: a case-control study in a veteran population. Alzheimers Dement. 8, 204–210. 10.1016/j.jalz.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Kang HT, Park JT, Choi K, Kim Y, Choi HJC, Jung CW, Lee YS, Park SC, 2017. Chemical screening identifies ATM as a target for alleviating senescence. Nat Chem Biol. 13, 616–623. 10.1038/nchembio.2342. [DOI] [PubMed] [Google Scholar]
- Katz DI, Bernick C, Dodick DW, Mez J, Mariani ML, Adler CH, Alosco ML, Balcer LJ, Banks SJ, Barr WB, et al. , 2021. National Institute of Neurological Disorders and stroke consensus diagnostic criteria for traumatic encephalopathy syndrome. Neurology. 96, 848–863. 10.1212/WNL.0000000000011850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, et al. , 2014. Geroscience: linking aging to chronic disease. Cell. 159, 709–713. 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayatan D, Razavi SM, Arab ZN, Niknejad AH, Nouri K, Momtaz S, Gumpricht E, Jamialahmadi T, Abdolghaffari AH, Barreto GE, et al. , 2022. Protective effects of curcumin against traumatic brain injury. Biomed Pharmacother. 154, 113621 10.1016/j.biopha.2022.113621. [DOI] [PubMed] [Google Scholar]
- Kim DS, Kim JY, Han Y, 2012. Curcuminoids in neurodegenerative diseases. Recent Pat CNS Drug Discov. 7, 184–204. 10.2174/157488912803252032. [DOI] [PubMed] [Google Scholar]
- Koellhoffer EC, McCullough LD, Ritzel RM, 2017. Old maids: aging and its impact on microglia function. Int J Mol Sci. 18. 10.3390/ijms18040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosari-Nasab M, Shokouhi G, Ghorbanihaghjo A, Mesgari-Abbasi M, Salari AA, 2019. Quercetin mitigates anxiety-like behavior and normalizes hypothalamus-pituitary-adrenal axis function in a mouse model of mild traumatic brain injury. Behav Pharmacol. 30, 282–289. 10.1097/FBP.0000000000000480. [DOI] [PubMed] [Google Scholar]
- Kovari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, Giannakopoulos P, 2007. Cortical microinfarcts and demyelination affect cognition in cases at high risk for dementia. Neurology. 68, 927–931. 10.1212/01.wnl.0000257094.10655.9a. [DOI] [PubMed] [Google Scholar]
- Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM, 2007. White matter integrity and cognition in chronic traumatic brain injury: a diffusion tensor imaging study. Brain. 130, 2508–2519. 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS, 2009. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 9, 81–94. 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Kumar R, Husain M, Gupta RK, Hasan KM, Haris M, Agarwal AK, Pandey CM, Narayana PA, 2009. Serial changes in the white matter diffusion tensor imaging metrics in moderate traumatic brain injury and correlation with neuro-cognitive function. J Neurotrauma. 26, 481–495. 10.1089/neu.2008.0461. [DOI] [PubMed] [Google Scholar]
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, Jenkins LW, Szabo C, Clark RS, 2008. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 104, 1700–1711. 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- Laird MD, Sukumari-Ramesh S, Swift AE, Meiler SE, Vender JR, Dhandapani KM, 2010. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? J Neurochem. 113, 637–648. 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Sabatini DM, Baur JA, 2013. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 123, 980–989. 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPak KM, Burd CE, 2014. The molecular balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res. 12, 167–183. 10.1158/1541-7786.MCR-13-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK, 2001. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 18, 369–376. 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Jung YS, Yoon MH, Kang SM, Oh AY, Lee JH, Jun SY, Woo TG, Chun HY, Kim SK, et al. , 2016. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J Clin Invest. 126, 3879–3893. 10.1172/JCI84164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman EJ, Hein MJ, Baron SL, Gersic CM, 2012. Neurodegenerative causes of death among retired National Football League players. Neurology. 79, 1970–1974. 10.1212/WNL.0b013e31826daf50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi GC, Accardi G, Monastero R, Nicoletti F, Libra M, 2018. Ageing: from inflammation to cancer. Immun Ageing. 15, 1. 10.1186/s12979-017-0112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E, Hazrati LN, 2021. Breast cancer type 1 and neurodegeneration: consequences of deficient DNA repair. Brain Commun. 3, fcab117. 10.1093/braincomms/fcab117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang H, Gao Y, Li L, Tang C, Wen G, Yang Y, Zhuang Z, Zhou M, Mao L, et al. , 2016a. Quercetin induces mitochondrial biogenesis in experimental traumatic brain injury via the PGC-1alpha signaling pathway. Am J Transl Res. 8, 3558–3566. [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang H, Gao Y, Li L, Tang C, Wen G, Zhou Y, Zhou M, Mao L, Fan Y, 2016b. Protective effects of quercetin on mitochondrial biogenesis in experimental traumatic brain injury via the Nrf2 signaling pathway. PLoS One. 11, e0164237 10.1371/journal.pone.0164237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, He Y, Zhang R, Zheng G, Zhou D, 2019. The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY). 11, 771–782. 10.18632/aging.101787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Duan L, Yang F, Yang L, Deng Y, Yu Y, Xu Y, Zhang Y, 2022. Curcumin suppress inflammatory response in traumatic brain injury via p38/MAPK signaling pathway. Phytother Res. 36, 1326–1337. 10.1002/ptr.7391. [DOI] [PubMed] [Google Scholar]
- Limbad C, Oron TR, Alimirah F, Davalos AR, Tracy TE, Gan L, Desprez PY, Campisi J, 2020. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS One. 15, e0227887 10.1371/journal.pone.0227887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MC, Akle V, Zheng W, Kitlen J, O’Steen B, Larner SF, Dave JR, Tortella FC, Hayes RL, Wang KK, 2006. Extensive degradation of myelin basic protein isoforms by calpain following traumatic brain injury. J Neurochem. 98, 700–712. 10.1111/j.1471-4159.2006.03882.x. [DOI] [PubMed] [Google Scholar]
- LoBue C, Wadsworth H, Wilmoth K, Clem M, Hart J Jr., Womack KB, Didehbani N, Lacritz LH, Rossetti HC, Cullum CM, 2017. Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin Neuropsychol. 31, 85–98. 10.1080/13854046.2016.1257069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncarevic-Vasiljkovic N, Pesic V, Todorovic S, Popic J, Smiljanic K, Milanovic D, Ruzdijic S, Kanazir S, 2012. Caloric restriction suppresses microglial activation and prevents neuroapoptosis following cortical injury in rats. PLoS One. 7, e37215 10.1371/journal.pone.0037215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncarevic-Vasiljkovic N, Milanovic D, Pesic V, Tesic V, Brkic M, Lazic D, Avramovic V, Kanazir S, 2016. Dietary restriction suppresses apoptotic cell death, promotes Bcl-2 and Bcl-xl mRNA expression and increases the Bcl-2/Bax protein ratio in the rat cortex after cortical injury. Neurochem Int. 96, 69–76. 10.1016/j.neuint.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Lorente L, Martin MM, Gonzalez-Rivero AF, Perez-Cejas A, Abreu-Gonzalez P, Ramos L, Argueso M, Caceres JJ, Sole-Violan J, Alvarez-Castillo A, et al. , 2020. Association between DNA and RNA oxidative damage and mortality of patients with traumatic brain injury. Neurocrit Care. 32, 790–795. 10.1007/s12028-019-00800-w. [DOI] [PubMed] [Google Scholar]
- Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al. , 2015. Structural and functional features of central nervous system lymphatic vessels. Nature. 523, 337–341. 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye TC, Shores EA, 2000. Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol Rev. 10, 115–129. 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- Mackay DF, Russell ER, Stewart K, MacLean JA, Pell JP, Stewart W, 2019. Neurodegenerative disease mortality among former professional soccer players. N Engl J Med. 381, 1801–1808. 10.1056/NEJMoa1908483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH, 2014. DNA damage and its links to neurodegeneration. Neuron. 83, 266–282. 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BC, Flashman LA, Saykin AJ, 2002. Executive dysfunction following traumatic brain injury: neural substrates and treatment strategies. NeuroRehabilitation. 17, 333–344. [PubMed] [Google Scholar]
- Mendelsohn AR, Larrick JW, 2018. Cellular senescence as the key intermediate in Tau-mediated neurodegeneration. Rejuvenation Res. 21, 572–579. 10.1089/rej.2018.2155. [DOI] [PubMed] [Google Scholar]
- Mendez DR, Cherian L, Moore N, Arora T, Liu PK, Robertson CS, 2004. Oxidative DNA lesions in a rodent model of traumatic brain injury. J Trauma. 56, 1235–1240. 10.1097/01.ta.0000130759.62286.0e. [DOI] [PubMed] [Google Scholar]
- Miller AH, Raison CL, 2016. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 16, 22–34. 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgaard CA, Stanford EP, Morton DJ, Ryden LA, Schubert KR, Golbeck AL, 1990. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology. 9, 233–242. 10.1159/000110778. [DOI] [PubMed] [Google Scholar]
- Moreno-Blas D, Gorostieta-Salas E, Pommer-Alba A, Mucino-Hernandez G, Geronimo-Olvera C, Maciel-Baron LA, Konigsberg M, Massieu L, Castro-Obregon S, 2019. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging (Albany NY). 11, 6175–6198. 10.18632/aging.102181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita-Fujimura Y, Fujimura M, Kawase M, Chan PH, 1999. Early decrease in apurinic/apyrimidinic endonuclease is followed by DNA fragmentation after cold injury-induced brain trauma in mice. Neuroscience. 93, 1465–1473. 10.1016/s0306-4522(99)00231-6. [DOI] [PubMed] [Google Scholar]
- Mortimer JA, French LR, Hutton JT, Schuman LM, 1985. Head injury as a risk factor for Alzheimer’s disease. Neurology. 35, 264–267. 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA, et al. , 1991. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 20 (Suppl. 2), S28–S35. [DOI] [PubMed] [Google Scholar]
- Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, Orr ME, 2018. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 17, e12840 10.1111/acel.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi SF, Braidy N, Habtemariam S, Sureda A, Manayi A, Nabavi SM, 2016. Neuroprotective effects of fisetin in Alzheimer’s and Parkinson’s Diseases: from chemistry to medicine. Curr Top Med Chem. 16, 1910–1915. 10.2174/1568026616666160204121725. [DOI] [PubMed] [Google Scholar]
- Nacarelli T, Lau L, Fukumoto T, Zundell J, Fatkhutdinov N, Wu S, Aird KM, Iwasaki O, Kossenkov AV, Schultz D, et al. , 2019. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol. 21, 397–407. 10.1038/s41556-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebrisi EE, 2021. Neuroprotective activities of curcumin in Parkinson’s disease: a review of the literature. Int J Mol Sci. 22. 10.3390/ijms222011248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT, 1999. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. Am J Epidemiol. 149, 32–40. 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- Ng HK, Mahaliyana RD, Poon WS, 1994. The pathological spectrum of diffuse axonal injury in blunt head trauma: assessment with axon and myelin strains. Clin Neurol Neurosurg. 96, 24–31. 10.1016/0303-8467(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Nordstrom A, Nordstrom P, 2018. Traumatic brain injury and the risk of dementia diagnosis: a nationwide cohort study. PLoS Med. 15, e1002496 10.1371/journal.pmed.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Kruger P, Fielder E, Victorelli S, Ruswhandi RA, Giorgadze N, Pirtskhalava T, et al. , 2019. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 29 (1061–1077), e1068 10.1016/j.cmet.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha RP, Rastogi M, Devi BP, Agrawal A, Dubey GP, 2012. Neuroprotective effect of curcuminoids against inflammation-mediated dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J Neuroimmune Pharmacol. 7, 609–618. 10.1007/s11481-012-9363-2. [DOI] [PubMed] [Google Scholar]
- Ottens AK, Golden EC, Bustamante L, Hayes RL, Denslow ND, Wang KK, 2008. Proteolysis of multiple myelin basic protein isoforms after neurotrauma: characterization by mass spectrometry. J Neurochem. 104, 1404–1414. 10.1111/j.1471-4159.2007.05086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MY, Panchal HV, Ghribi O, Benzeroual KE, 2012. The neuroprotective effect of fisetin in the MPTP model of Parkinson’s disease. J Parkinsons Dis. 2, 287–302. 10.3233/JPD-012110. [DOI] [PubMed] [Google Scholar]
- Pertusa M, Garcia-Matas S, Rodriguez-Farre E, Sanfeliu C, Cristofol R, 2007. Astrocytes aged in vitro show a decreased neuroprotective capacity. J Neurochem. 101, 794–805. 10.1111/j.1471-4159.2006.04369.x. [DOI] [PubMed] [Google Scholar]
- Pessina F, Gioia U, Brandi O, Farina S, Ceccon M, Francia S, d’Adda di Fagagna F, 2021. DNA damage triggers a new phase in neurodegeneration. Trends Genet. 37, 337–354. 10.1016/j.tig.2020.09.006. [DOI] [PubMed] [Google Scholar]
- Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK, 1998. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience. 87, 359–369. 10.1016/s0306-4522(98)00142-0. [DOI] [PubMed] [Google Scholar]
- Pischiutta F, Micotti E, Hay JR, Marongiu I, Sammali E, Tolomeo D, Vegliante G, Stocchetti N, Forloni G, De Simoni MG, et al. , 2018. Single severe traumatic brain injury produces progressive pathology with ongoing contralateral white matter damage one year after injury. Exp Neurol. 300, 167–178. 10.1016/j.expneurol.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, et al. , 2000. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 55, 1158–1166. 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- Pluta R, Furmaga-Jablonska W, Januszewski S, Czuczwar SJ, 2022. Post-ischemic brain neurodegeneration in the form of Alzheimer’s Disease proteinopathy: possible therapeutic role of curcumin. Nutrients. 14. 10.3390/nu14020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupillo E, Bianchi E, Vanacore N, Montalto C, Ricca G, Robustelli Della Cuna FS, Fumagalli F, Castellani M, Poli F, Romeo F, et al. , 2020. Increased risk and early onset of ALS in professional players from Italian Soccer Teams. Amyotroph Lateral Scler Frontotemporal Degener. 21, 403–409. 10.1080/21678421.2020.1752250. [DOI] [PubMed] [Google Scholar]
- Rainey-Smith SR, Brown BM, Sohrabi HR, Shah T, Goozee KG, Gupta VB, Martins RN, 2016. Curcumin and cognition: a randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br J Nutr. 115, 2106–2113. 10.1017/S0007114516001203. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, et al. , 2011. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 70, 374–383. 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Rao V, Rosenberg P, Miles QS, Patadia D, Treiber K, Bertrand M, Norton M, Steinberg M, Tschanz J, Lyketsos C, 2010. Neuropsychiatric symptoms in dementia patients with and without a history of traumatic brain injury. J Neuropsychiatry Clin Neurosci. 22, 166–172. 10.1176/jnp.2010.22.2.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayess H, Wang MB, Srivatsan ES, 2012. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 130, 1715–1725. 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich NJ, Van Landingham JW, Figueiroa S, Seth R, Corniola RS, Levenson CW, 2010. Chronic caloric restriction reduces tissue damage and improves spatial memory in a rat model of traumatic brain injury. J Neurosci Res. 88, 2933–2939. 10.1002/jnr.22443. [DOI] [PubMed] [Google Scholar]
- Ritzel RM, Doran SJ, Glaser EP, Meadows VE, Faden AI, Stoica BA, Loane DJ, 2019. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging. 77, 194–206. 10.1016/j.neurobiolaging.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Graham DI, 1991. beta A4 amyloid protein deposition in brain after head trauma. Lancet. 338, 1422–1423. 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI, 1994. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 57, 419–425. 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, et al. , 2004. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet. 364, 1321–1328. 10.1016/S0140-6736(04)17188-2. [DOI] [PubMed] [Google Scholar]
- Ross DE, 2011. Review of longitudinal studies of MRI brain volumetry in patients with traumatic brain injury. Brain Inj. 25, 1271–1278. 10.3109/02699052.2011.624568. [DOI] [PubMed] [Google Scholar]
- Ross DT, Graham DI, Adams JH, 1993. Selective loss of neurons from the thalamic reticular nucleus following severe human head injury. J Neurotrauma. 10, 151–165. 10.1089/neu.1993.10.151. [DOI] [PubMed] [Google Scholar]
- Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, van Swieten JC, 2003. Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosurg Psychiatry. 74, 1574–1576. 10.1136/jnnp.74.11.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubovitch V, Pharayra A, Har-Even M, Dvir O, Mattson MP, Pick CG, 2019. Dietary energy restriction ameliorates cognitive impairment in a mouse model of traumatic brain injury. J Mol Neurosci. 67, 613–621. 10.1007/s12031-019-01271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudelli R, Strom JO, Welch PT, Ambler MW, 1982. Posttraumatic premature Alzheimer’s disease. Neuropathologic findings and pathogenetic considerations. Arch Neurol. 39, 570–575. 10.1001/archneur.1982.00510210040009. [DOI] [PubMed] [Google Scholar]
- Rufini A, Tucci P, Celardo I, Melino G, 2013. Senescence and aging: the critical roles of p53. Oncogene. 32, 5129–5143. 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- Ruparelia N, Chai JT, Fisher EA, Choudhury RP, 2017. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol. 14, 133–144. 10.1038/nrcardio.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samini F, Samarghandian S, Borji A, Mohammadi G, bakaian M, 2013. Curcumin pretreatment attenuates brain lesion size and improves neurological function following traumatic brain injury in the rat. Pharmacol Biochem Behav. 110, 238–244. 10.1016/j.pbb.2013.07.019. [DOI] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. , 2017. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 8, 14532. 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffert J, LoBue C, White CL, Chiang HS, Didehbani N, Lacritz L, Rossetti H, Dieppa M, Hart J, Cullum CM, 2018. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed Alzheimer’s disease. Neuropsychology. 32, 410–416. 10.1037/neu0000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, Sano M, Stern Y, Mayeux R, 1997. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 62, 119–124. 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultke E, Kamencic H, Zhao M, Tian GF, Baker AJ, Griebel RW, Juurlink BH, 2005. Neuroprotection following fluid percussion brain trauma: a pilot study using quercetin. J Neurotrauma. 22, 1475–1484. 10.1089/neu.2005.22.1475. [DOI] [PubMed] [Google Scholar]
- Schwab N, Grenier K, Hazrati LN, 2019. DNA repair deficiency and senescence in concussed professional athletes involved in contact sports. Acta Neuropathol Commun. 7, 182. 10.1186/s40478-019-0822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab N, Ju Y, Hazrati LN, 2021. Early onset senescence and cognitive impairment in a murine model of repeated mTBI. Acta Neuropathol Commun. 9, 82. 10.1186/s40478-021-01190-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab N, Taskina D, Leung E, Innes BT, Bader GD, Hazrati LN, 2022. Neurons and glial cells acquire a senescent signature after repeated mild traumatic brain injury in a sex-dependent manner. Front Neurosci. 16, 1027116. 10.3389/fnins.2022.1027116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott G, Hellyer PJ, Ramlackhansingh AF, Brooks DJ, Matthews PM, Sharp DJ, 2015. Thalamic inflammation after brain trauma is associated with thalamo-cortical white matter damage. J Neuroinflammation. 12, 224. 10.1186/s12974-015-0445-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked G, Douvdevani A, Yair S, Zlotnik A, Czeiger D, 2014. The role of cell-free DNA measured by a fluorescent test in the management of isolated traumatic head injuries. Scand J Trauma Resusc Emerg Med. 22, 21. 10.1186/1757-7241-22-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Zhuang Y, Ying Z, Wu A, Gomez-Pinilla F, 2009. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 161, 1037–1044. 10.1016/j.neuroscience.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Sivaloganathan S, 1994. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol. 87, 55–62. 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK, 1997. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 14, 715–727. 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF, 1999. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 58, 982–992. 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith C, Graham DI, Murray LS, Nicoll JA, 2003a. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. 29, 496–502. 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Iwata A, Graham DI, 2003b. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg. 98, 1072–1077. 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- Song J, Du G, Wu H, Gao X, Yang Z, Liu B, Cui S, 2021. Protective effects of quercetin on traumatic brain injury induced inflammation and oxidative stress in cortex through activating Nrf2/HO-1 pathway. Restor Neurol Neurosci. 39, 73–84. 10.3233/RNN-201119. [DOI] [PubMed] [Google Scholar]
- Soysal P, Stubbs B, Lucato P, Luchini C, Solmi M, Peluso R, Sergi G, Isik AT, Manzato E, Maggi S, et al. , 2016. Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res Rev. 31, 1–8. 10.1016/j.arr.2016.08.006. [DOI] [PubMed] [Google Scholar]
- St Sauver JL, Boyd CM, Grossardt BR, Bobo WV, Finney Rutten LJ, Roger VL, Ebbert JO, Therneau TM, Yawn BP, Rocca WA, 2015. Risk of developing multimorbidity across all ages in an historical cohort study: differences by sex and ethnicity. BMJ Open. 5, e006413 10.1136/bmjopen-2014-006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Alvarez VE, McKee AC, 2014. Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther. 6, 4. 10.1186/alzrt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, Kabadi SV, Hanscom M, Byrnes KR, Faden AI, 2014. PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma. 31, 758–772. 10.1089/neu.2013.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su E, Bell MJ, Kochanek PM, Wisniewski SR, Bayir H, Clark RS, Adelson PD, Tyler-Kabara EC, Janesko-Feldman KL, Berger RP, 2012. Increased CSF concentrations of myelin basic protein after TBI in infants and children: absence of significant effect of therapeutic hypothermia. Neurocrit Care. 17, 401–407. 10.1007/s12028-012-9767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman MA, McKee AC, Stein TD, Tripodis Y, Besser LM, Martin B, Palmisano JN, Steinberg EG, O’Connor MK, Au R, et al. , 2019. Failure to detect an association between self-reported traumatic brain injury and Alzheimer’s disease neuropathology and dementia. Alzheimers Dement. 15, 686–698. 10.1016/j.jalz.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P, Petitti D, Barbaccia J, 1987. Head trauma and age of onset of dementia of the Alzheimer type. JAMA. 257, 2289–2290. 10.1001/jama.1987.03390170045014. [DOI] [PubMed] [Google Scholar]
- Sun G, Miao Z, Ye Y, Zhao P, Fan L, Bao Z, Tu Y, Li C, Chao H, Xu X, et al. , 2020. Curcumin alleviates neuroinflammation, enhances hippocampal neurogenesis, and improves spatial memory after traumatic brain injury. Brain Res Bull. 162, 84–93. 10.1016/j.brainresbull.2020.05.009. [DOI] [PubMed] [Google Scholar]
- Sundaram JR, Poore CP, Sulaimee NHB, Pareek T, Cheong WF, Wenk MR, Pant HC, Frautschy SA, Low CM, Kesavapany S, 2017. Curcumin ameliorates neuroinflammation, neurodegeneration, and memory deficits in p25 transgenic mouse model that bears hallmarks of Alzheimer’s Disease. J Alzheimers Dis. 60, 1429–1442. 10.3233/JAD-170093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Kirkland JL, 2018. Aging, cell senescence, and chronic disease: emerging therapeutic strategies. JAMA. 320, 1319–1320. 10.1001/jama.2018.12440. [DOI] [PubMed] [Google Scholar]
- Theadom A, Mahon S, Barker-Collo S, McPherson K, Rush E, Vandal AC, Feigin VL, 2013. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 20, 1135–1144. 10.1111/ene.12099. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, McCormick WC, Kagan SH, 2006. Traumatic brain injury in older adults: epidemiology, outcomes, and future implications. J Am Geriatr Soc. 54, 1590–1595. 10.1111/j.1532-5415.2006.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al. , 2012. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 122, 2601–2612. 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolppanen AM, Taipale H, Hartikainen S, 2017. Head or brain injuries and Alzheimer’s disease: a nested case-control register study. Alzheimers Dement. 13, 1371–1379. 10.1016/j.jalz.2017.04.010. [DOI] [PubMed] [Google Scholar]
- Tomaiuolo F, Carlesimo GA, Di Paola M, Petrides M, Fera F, Bonanni R, Formisano R, Pasqualetti P, Caltagirone C, 2004. Gross morphology and morphometric sequelae in the hippocampus, fornix, and corpus callosum of patients with severe non-missile traumatic brain injury without macroscopically detectable lesions: a T1 weighted MRI study. J Neurol Neurosurg Psychiatry. 75, 1314–1322. 10.1136/jnnp.2003.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasevic G, Laurer HL, Mattiasson G, van Steeg H, Wieloch T, McIntosh TK, 2012. Delayed neuromotor recovery and increased memory acquisition dysfunction following experimental brain trauma in mice lacking the DNA repair gene XPA. J Neurosurg. 116, 1368–1378. 10.3171/2012.2.JNS11888. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Shimada R, Okada Y, Kawamata T, Kibayashi K, 2019. Senescence-associated-beta-galactosidase staining following traumatic brain injury in the mouse cerebrum. PLoS One. 14, e0213673 10.1371/journal.pone.0213673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse KH, Herrup K, 2017. DNA damage in the oligodendrocyte lineage and its role in brain aging. Mech Ageing Dev. 161, 37–50. 10.1016/j.mad.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulamek-Koziol M, Czuczwar SJ, Januszewski S, Pluta R, 2020. Substantiation for the use of curcumin during the development of neurodegeneration after brain ischemia. Int J Mol Sci. 21. 10.3390/ijms21020517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaibhav K, Braun M, Alverson K, Khodadadi H, Kutiyanawalla A, Ward A, Banerjee C, Sparks T, Malik A, Rashid MH, et al. , 2020. Neutrophil extracellular traps exacerbate neurological deficits after traumatic brain injury. Sci Adv. 6, eaax8847. 10.1126/sciadv.aax8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez OSB, Magos-Guerrero GA, Escobar Ramirez JL, Gomez-Verjan JC, 2022. Natural products as a major source of candidates for potential senolytic compounds obtained by in silico screening. Med Chem. 10.2174/1573406419666221019153537. [DOI] [PubMed] [Google Scholar]
- Volpato S, Guralnik JM, Ferrucci L, Balfour J, Chaves P, Fried LP, Harris TB, 2001. Cardiovascular disease, interleukin-6, and risk of mortality in older women: the women’s health and aging study. Circulation. 103, 947–953. 10.1161/01.cir.103.7.947. [DOI] [PubMed] [Google Scholar]
- Wakade C, Sukumari-Ramesh S, Laird MD, Dhandapani KM, Vender JR, 2010. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. J Neurosurg. 113, 1195–1201. 10.3171/2010.3.JNS091212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter A, Finelli K, Bai X, Arnett P, Bream T, Seidenberg P, Lynch S, Johnson B, Slobounov S, 2017. Effect of Enzogenol(R) supplementation on cognitive, executive, and vestibular/balance functioning in chronic phase of concussion. Dev Neuropsychol. 42, 93–103. 10.1080/87565641.2016.1256404. [DOI] [PubMed] [Google Scholar]
- Wang Y, Arun P, Wei Y, Oguntayo S, Gharavi R, Valiyaveettil M, Nambiar MP, Long JB, 2014. Repeated blast exposures cause brain DNA fragmentation in mice. J Neurotrauma. 31, 498–504. 10.1089/neu.2013.3074. [DOI] [PubMed] [Google Scholar]
- Warner MA, Marquez de la Plata C, Spence J, Wang JY, Harper C, Moore C, Devous M, Diaz-Arrastia R, 2010. Assessing spatial relationships between axonal integrity, regional brain volumes, and neuropsychological outcomes after traumatic axonal injury. J Neurotrauma. 27, 2121–2130. 10.1089/neu.2010.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Chen XC, Song Y, Pan XD, Dai XM, Zhang J, Cui XL, Wu XL, Zhu YG, 2016. Amyloid beta protein aggravates neuronal senescence and cognitive deficits in 5XFAD mouse model of Alzheimer’s Disease. Chin Med J (Engl). 129, 1835–1844. 10.4103/0366-6999.186646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, Groshen S, Mack WJ, Guen E, Di Biase S, et al. , 2017. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med. 9. 10.1126/scitranslmed.aai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RL, 2017. Accelerated cognitive aging following severe traumatic brain injury: a review. Brain Inj. 31, 1270–1278. 10.1080/02699052.2017.1332387. [DOI] [PubMed] [Google Scholar]
- Woodcock T, Morganti-Kossmann MC, 2013. The role of markers of inflammation in traumatic brain injury. Front Neurol. 4, 18. 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F, 2006. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp Neurol. 197, 309–317. 10.1016/j.expneurol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Schubert D, Gomez-Pinilla F, 2011. Brain and spinal cord interaction: a dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabil Neural Repair. 25, 332–342. 10.1177/1545968310397706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Tahara H, 2013. The role of exosomes and microRNAs in senescence and aging. Adv Drug Deliv Rev. 65, 368–375. 10.1016/j.addr.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N, et al. , 2015a. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 4, e12997 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, White TA, Johnson KO, Stout MB, Mezera V, et al. , 2015b. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 112, E6301–E6310. 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, et al. , 2018. Senolytics improve physical function and increase lifespan in old age. Nat Med. 24, 1246–1256. 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Takeshita K, Saito H, 2014. Plasminogen activator inhibitor-1 in aging. Semin Thromb Hemost. 40, 652–659. 10.1055/s-0034-1384635. [DOI] [PubMed] [Google Scholar]
- Yang T, Kong B, Gu JW, Kuang YQ, Cheng L, Yang WT, Xia X, Shu HF, 2014. Anti-apoptotic and anti-oxidative roles of quercetin after traumatic brain injury. Cell Mol Neurobiol. 34, 797–804. 10.1007/s10571-014-0070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben-Dor S, Krizhanovsky V, 2017. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 36, 2280–2295. 10.15252/embj.201695553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshita M, Fletcher E, Harvey D, Ortega M, Martinez O, Mungas DM, Reed BR, DeCarli CS, 2006. Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology. 67, 2192–2198. 10.1212/01.wnl.0000249119.95747.1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, et al. , 2018. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 36, 18–28. 10.1016/j.ebiom.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Parakramaweera R, Teng S, Gowda M, Sharad Y, Thakker-Varia S, Alder J, Sesti F, 2016. Oxidation of KCNB1 potassium channels causes neurotoxicity and cognitive impairment in a mouse model of traumatic brain injury. J Neurosci. 36, 11084–11096. 10.1523/JNEUROSCI.2273-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabenko YY, Pivneva TA, 1994. Flavonoid quercetin reduces gliosis after repetitive mild traumatic brain injury in mice. Fiziol Zh 2016 (62), 50–56. [PubMed] [Google Scholar]
- Zahedi H, Hosseinzadeh-Attar MJ, Shadnoush M, Sahebkar A, Barkhidarian B, Sadeghi O, Najafi A, Hosseini S, Qorbani M, Ahmadi A, et al. , 2021. Effects of curcuminoids on inflammatory and oxidative stress biomarkers and clinical outcomes in critically ill patients: a randomized double-blind placebo-controlled trial. Phytother Res. 35, 4605–4615. 10.1002/ptr.7179. [DOI] [PubMed] [Google Scholar]
- Zanzer YC, Batista AG, Dougkas A, Tovar J, Granfeldt Y, Ostman E, 2019. Difficulties in translating appetite sensations effect of turmeric-based beverage When given prior to isoenergetic medium- or high-fat meals in healthy subjects. Nutrients. 11. 10.3390/nu11040736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, et al. , 2002. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 82, 181–191. 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW, 2012. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 7, e34504 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang H, Zhou Y, Zhu Y, Fei M, 2018. Fisetin alleviates oxidative stress after traumatic brain injury via the Nrf2-ARE pathway. Neurochem Int. 118, 304–313. 10.1016/j.neuint.2018.05.011. [DOI] [PubMed] [Google Scholar]
- Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe M, et al. , 2019. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 22, 719–728. 10.1038/s41593-019-0372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK, 2014. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 11, 59. 10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. , 2015. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 14, 644–658. 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, Kirkland JL, 2017. New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463 . Aging (Albany NY). 9, 955–963. 10.18632/aging.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.