

NOS1AP variants impair actin remodeling in podocytes and cause glomerular disease in humans, kidney organoids, and mice.
Abstract
Nephrotic syndrome (NS) is a leading cause of chronic kidney disease. We found recessive NOS1AP variants in two families with early-onset NS by exome sequencing. Overexpression of wild-type (WT) NOS1AP, but not cDNA constructs bearing patient variants, increased active CDC42 and promoted filopodia and podosome formation. Pharmacologic inhibition of CDC42 or its effectors, formin proteins, reduced NOS1AP-induced filopodia formation. NOS1AP knockdown reduced podocyte migration rate (PMR), which was rescued by overexpression of WT Nos1ap but not by constructs bearing patient variants. PMR in NOS1AP knockdown podocytes was also rescued by constitutively active CDC42Q61L or the formin DIAPH3. Modeling a NOS1AP patient variant in knock-in human kidney organoids revealed malformed glomeruli with increased apoptosis. Nos1apEx3−/Ex3− mice recapitulated the human phenotype, exhibiting proteinuria, foot process effacement, and glomerulosclerosis. These findings demonstrate that recessive NOS1AP variants impair CDC42/DIAPH-dependent actin remodeling, cause aberrant organoid glomerulogenesis, and lead to a glomerulopathy in humans and mice.
INTRODUCTION
Nephrotic syndrome (NS) is the second leading cause of chronic kidney disease in the first three decades of life (1). NS manifests with proteinuria, edema, and hypoalbuminemia and arises from disruption of the glomerular filtration barrier, primarily the podocyte foot processes (2). Patients with steroid-resistant NS invariably progress to end-stage renal disease concomitant with podocyte loss (1, 2). The investigation of monogenic forms of NS has revealed >55 single-gene etiologies (3). A monogenic cause of NS has been identified in 11 to 45% of cases by gene panel or exome sequencing (ES) (4, 5). Monogenic NS genes are predominantly expressed in the glomerular podocyte, and variants in their encoded proteins impair podocyte structure and function (2, 6). These disease proteins have been shown to coalesce into molecular complexes and pathways that are essential for podocyte development or homeostasis (2, 7).
Variants in genes encoding dynamic regulators of the actin cytoskeleton have been shown to cause NS in humans and mice, including the Rho guanosine triphosphatase (GTPase) cell division control protein 42 (Cdc42), its guanine nucleotide exchange factors intersectin 1 and 2 (ITNS1 and ITSN2), and the CDC42 effector neural Wiskott-Aldrich syndrome protein (NWASP encoded by Wasl) (8–16). These genes encode actin remodeling proteins that, when mutated, lead to podocyte dysfunction marked by abnormal podocyte migration rate (PMR) (9, 16, 17). During cell migration, CDC42 stimulates actin assembly through downstream effectors such as diaphanous-related formin 3 (DIAPH3) during filopodia formation and NWASP during podosome and invadopodia formation (fig. S1) (18–24).
Human kidney organoids are an emerging system to investigate renal diseases. Recently, organoids harboring variants in known NS genes were shown to develop glomerular abnormalities (25–27). However, kidney organoids have not been used to investigate novel monogenic causes of NS.
Nitric oxide synthase 1 adaptor protein (NOS1AP) contains 506 amino acids residues with two protein interaction domains: the N-terminal phosphotyrosine binding domain (PTB) and the C-terminal PDZ binding domain (PDZ-BD) through which it interacts with nitric oxide synthase 1 (NOS1) (Fig. 1D) (28–33). There are multiple alternative transcripts described for NOS1AP (30, 31, 33). These alternative transcripts encode the same N-terminal domains, including the PTB domain, but distinct C-terminal domains lacking the PDZ-BD (30, 31, 33). At the cellular level, NOS1AP regulates actin dynamics, neuronal dendrite formation, and cancer cell migration (28–33). Nos1ap homozygous mutant mice have been reported (34). They were viable through adulthood and have no reported renal phenotypes. Homozygous mice were prone to cardiac arrhythmias and dysfunction when challenged with oxidative stress–inducing agents. This is consistent with literature demonstrating that NOS1AP common variants are associated with arrythmias in adult humans (35, 36). However, the role of rare variants in NOS1AP in human disease is unknown.
Fig. 1. Homozygous NOS1AP variants in individuals with steroid-resistant NS.
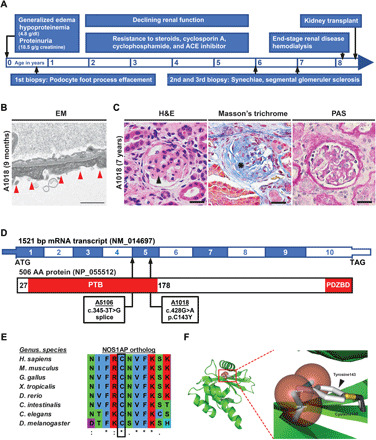
(A) Time bar outlining clinical course of patient A1018 with onset of disease at 4 days of age and progression to end-stage renal disease (ESRD) with subsequent kidney transplant at 8 years of age. (B) Renal biopsy in subject A1018 (9 months) revealed podocyte foot process effacement (red arrowheads) by electron microscopy (EM). Scale bar, 1 μm. (C) Renal biopsy in A1018 (7 years) showed glomerular synechiae formation (arrow) and capillary loop collapse [hematoxylin and eosin (H&E) staining], increased sclerosis (asterisk) (Masson’s trichrome staining), and thickened basement membrane (PAS staining). Scale bar, 50 μM. (D) Coding exon (top bar) and protein domain (bottom bar) structures of NOS1AP are shown with arrows indicating position of variants identified in A5106 and A1018. AA, amino acid. PDZBD, PDZ-binding domain. (E) Evolutionary conservation of primary amino acid sequence shows that cysteine 143 is conserved in NOS1AP orthologs through C. elegans. (F) Modeling of C143Y in the structure of the human NUMB-like protein PTB domain (Protein Data Bank: 3F0W) reveals that, relative to the Cys143 residue in black and gold, the Tyr143 substitution (gray aromatic ring) sterically clashes with neighboring amino acid residues (red spheres) (also see fig. S3H).
Because gene panel and ES have revealed a causative variant in one of the 55 published monogenic causes of NS in ~11 to 45% of families (4, 5), we hypothesized that novel single-gene etiologies may exist in the remaining subjects without a molecular diagnosis. By ES in an NS cohort of 300 families, we found recessive variants in the novel disease gene NOS1AP encoding nitric oxide synthase 1 adaptor protein in two unrelated families with early-onset NS. We demonstrate that NOS1AP is expressed in glomerular podocytes and localizes to actin-rich filopodia and podosomes in immortalized podocytes. We provide evidence that loss of NOS1AP through NS patient variants causes reduced CDC42 activation, defective podocyte actin remodeling, and aberrant glomeruli formation in kidney organoids. Furthermore, mice with biallelic variants in Nos1ap recapitulate the human phenotype, exhibiting severe proteinuria and podocyte foot process effacement. We, thereby, delineate a monogenic NS mechanism, in which recessive NOS1AP variants impair CDC42/DIAPH signaling and actin remodeling, cause aberrant glomerulogenesis in a knock-in human kidney organoids, and lead to proteinuric kidney disease in mice. Our findings propose a new standard for the functional genomic investigation of kidney disease.
RESULTS
Recessive variants in NOS1AP cause early onset NS in humans
Because many cases with NS remain unsolved, we hypothesized that novel monogenic causes and unknown disease pathways are yet to be found. We, therefore, performed ES (37) on 300 NS families with onset before age 25. In 146 families with prominent homozygosity by descent, we postulated that a causative homozygous recessive variant resides within a homozygous haplotype, which was inherited from a common ancestor. By this approach, we identified homozygous variants in NOS1AP in two unrelated subjects, A1018 and A5106, born from consanguineous unions (Fig. 1D, fig. S2A, and Table 1). Patient A1018 was a male born in Austria who presented at 4 days of life with generalized edema. Consistent with NS, his laboratory evaluation showed hypoproteinemia (total protein 4.8 g/dl), nephrotic-range proteinuria (18.5 g protein/g creatinine), and microscopic hematuria (Fig. 1A and Table 1). His proteinuria did not respond to corticosteroids, cyclophosphamide, cyclosporine A, or angiotensin-converting enzyme inhibitors. Renal biopsy at age 9 months showed podocyte foot process effacement by electron microscopy without noticeable changes by light microscopy (Fig. 1B). His renal function declined, and he had two subsequent biopsies at age 6 and 7. Both biopsies revealed glomerular sclerosis with collapse of capillary loops (Fig. 1C) and showed thickening of the glomerular basement membrane (Fig. 1C and fig. S2D). He, then, progressed to end-stage renal disease by age 8 and was transitioned to hemodialysis. He received a renal transplantation at age 8 and has not had recurrence of NS through age 21. Patient A5106 was an Egyptian male, who developed generalized edema at age 6 months. His laboratory evaluation, similarly, showed hypoproteinemia (total protein, 3.8 g/dl; albumin, 2.2 g/dl), nephrotic-range proteinuria (3 g protein/g creatinine), and microscopic hematuria (Table 1). He was diagnosed with NS but subsequently lost to follow-up.
Table 1. Recessive mutations in NOS1AP in two families with early-onset NS.
Cr, serum creatinine; DC, disease causing; DL, deleterious; ESRD, end-stage renal disease; ExAC v1.0 and gnomAD v2.11 and v3, summary of data within Exome and Genome Aggregation databases; H, homozygotes in ExAC and gnomAD; h, heterozygous alleles in ExAC and gnomAD; HD, hemodialysis; hom, homozygous; ME, MaxEnt splice prediction score; MT, “MutationTaster” prediction score; N/A, not available; NNS, NNSPLICE splice-site mutation prediction score; PC, parental consanguinity; PD, probably damaging; PP2, PolyPhen-2 prediction score; RUS, renal ultrasound; SGA, small for gestational age; SIFT, “Sorting Tolerant From Intolerant” prediction score; Seg, segregation; T, total alleles in ExAC and gnomAD; TP, total protein; TX, kidney transplantation; Zyg, zygosity.
| Family |
Nucleotide and
amino acid change |
Exon (Zyg and
Seg) |
In silico
severity scores |
Conservation |
ExAC and
gnomAD (H/h/T) |
Sex |
Ethnic
origin |
Renal disease |
| A1018 |
c.428G>A
p.Cys143Tyr |
5 (hom, mat) |
PP2 PD
SIFT DL MT DC |
C. elegans | Absent | M | N/A |
Initial onset:
4 days, SGA, edema Urinalysis: Microscopic hematuria; 18.5 g protein/g creatinine Serum studies: Cr 1.0 mg/dl, TP 4.8 g/dl. RUS: N/A Treatment: Resistant to corticosteroids, cyclophosphamide, and cyclosporin A. Biopsy: 75% podocyte foot effacement. ESRD: 7 years (HD and TX). |
| A5106 |
c.345-3T>G
Splice |
5 (hom) |
ME −66%
NNS −97% |
Not
applicable |
Absent | M | Egypt |
Initial onset:
6 months, edema Urinalysis: Microscopic hematuria; 3 g protein/g creatinine. Serum studies: Cr 0.6 mg/dl, TP 3.8 g/dl, Albumin 2.2 g/dl. RUS: Bilateral echogenicity |
In A1018, we detected a homozygous missense variant (c.428G>A; p.Cys143Tyr) in NOS1AP (Fig. 1D; fig. S2, A and B; and Table 1). NOS1AP is present within a region of homozygosity by descent in chromosome 1 (fig. S2A). The variant was deemed deleterious based on the following criteria: (i) It yielded strong in silico prediction scores (Table 1); (ii) the variant has extremely rare population prevalence, as it was not present in the homozygous or heterozygous state in the control Genome Aggregation Database (gnomAD) (Table 1); (iii) cysteine 143 is evolutionarily highly conserved down to the invertebrate ortholog in Caenorhabditis elegans (Fig. 1E); and (iv) the affected cysteine residue, which is within the PTB domain (Fig. 1D), is highly conserved across paralogous domains (fig. S3, A to C). We performed a multiple sequence alignment of 101 paralogous human PTB domains from the Simple Modular Architecture Research Tool (SMART) database (fig. S3B). The mutated cysteine residue, located within a β-pleated sheet, is highly conserved in 86 of 101 (85%) paralogous domains. Moreover, cysteine 143 is only one of four highly conserved amino acids (>75% of PTB domains) within the 147–amino acid NOS1AP PTB domain (fig. S3C). The effect of the human mutation p.Cys143Tyr was, furthermore, predicted structurally using Site Directed Mutator with five distinct structures, which are paralogous to the human NOS1AP PTB domain (fig. S3, D to H). When substituted into these structures, the mutation yielded consistently negative pseudo-ΔΔG scores, suggesting destabilization of the local structure (fig. S3F). The p.Cys143Tyr mutation was also visualized in PyMOL in each paralogous structure, demonstrating that the tyrosine mutation sterically clashes with adjacent amino acid residues in each case (Fig. 1F and fig. S3H).
In A5106, we detected a homozygous splice-site variant (c.345-3T>G) in NOS1AP, similarly residing within a homozygous peak region in chromosome 1 (Fig. 1D; fig.S2, A and C; and Table 1). This variant is absent from the gnomAD database of 120,000 control individuals (Table 1). Moreover, it is predicted in silico to suppress exon splicing (Table 1). Consistent with these prediction scores, >99.7% of 187,091 human transcripts contains cytidine, adenosine, or thymidine at the −3 acceptor splice-site position (38). However, guanosine—the mutated nucleoside observed in subject A5106—is extremely rare (<0.3%) across all human acceptor splice sites (38).
Evaluation of ES data from both subjects did not reveal causative variants in 60 established monogenic NS disease genes or 11 monogenic nephritis disease genes (3). We thereby identified homozygous recessive variants in NOS1AP as a potential novel cause of NS and subsequently evaluated their deleteriousness functionally (fig. S1).
NOS1AP is expressed by podocytes of mammalian glomeruli
By exploring single-cell mRNA sequencing data from adult mouse glomeruli and adult human kidney experiments (39, 40), we demonstrated that NOS1AP mRNA transcript is predominantly expressed in the mammalian podocyte relative to other kidney glomerular and renal epithelial cell types (Fig. 2A). We, furthermore, investigated endogenous NOS1AP localization in adult rat glomeruli by immunofluorescence (IF). First, we established that a NOS1AP antibody identifies tagged NOS1AP protein by immunoblotting and IF (fig. S4, A to C). Using this antibody, we performed IF microscopy in adult rat glomeruli (Fig. 2B) and detected Nos1ap staining in a linear pattern that showed partial colocalization with podocyte slit diaphragm marker nephrin, consistent with podocyte foot process localization. Nos1ap did not colocalize with endothelial cell marker CD31 nor with mesangial cell marker α-smooth muscle actin (αSMA), which marks the two other glomerular cell types (Fig. 2B). The Nos1ap signal was abrogated upon preabsorption of the antibody with its immunogen (fig. S4F).
Fig. 2. NOS1AP is expressed by podocytes of mammalian glomeruli and localizes to actin-rich filopodia and podosomes.
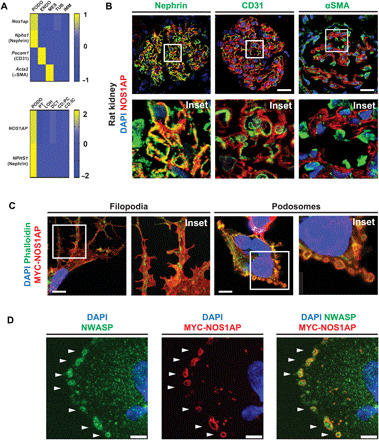
(A) NOS1AP mRNA (z-score) was predominantly expressed by podocytes from single-cell mRNA sequencing data (6, 40). Top: Nos1ap expression was highest in the podocyte cluster (Nphs1 and nephrin) relative to the endothelial cell cluster (Pecam1 and CD31), mesangial cell cluster (Acta2 and α-smooth muscle actin), and renal tubular and immune cells in murine glomeruli (6). Bottom: NOS1AP expression is similarly highest in the podocyte cluster relative to other nephron epithelial cell clusters from adult human kidney (40). PODO, podocytes; ENDO, endothelial cells; MES, mesangial cells; TUB, tubular epithelial cells; IMM, immune cells; PT, proximal tubular epithelial cells; LOH, Loop of Henle epithelial cells; DCT, distal convoluted tubular epithelial cells; CD:PC, collecting duct principal cells; CD:IC, collecting duct intercalated cells. (B) IF confocal microscopy imaging of rat kidney sections demonstrates NOS1AP colocalization (yellow) with podocyte slit diaphragm marker nephrin but not endothelial cell marker CD31 or mesangial cell marker αSMA. Insets are shown below each image. Scale bars, 25 μm. (C) In a human podocyte cell line, overexpression of MYC-tagged WT NOS1AP (MYC-NOS1AP) induced the formation of filopodia (left column) and peripheral ring structures (right column) in which NOS1AP colocalized with F-actin. (Images 26 hours after transfection.) Scale bars, 7.5 μm. (D) MYC-NOS1AP overexpression in a human podocyte cell line identifies peripheral ring structures as podosomes by colocalization of NOS1AP with NWASP (white arrows). Scale bars, 5 μm.
NOS1AP localizes to actin-rich filopodia and podosomes
To determine NOS1AP subcellular localization, we next overexpressed human NOS1AP cDNA in an immortalized human podocyte cell line. In 24% of cells, MYC-tagged NOS1AP localized to F-actin containing filopodia (Fig. 2C), which are regulated by multiple NS disease genes (8–11). In 44% of cells, it colocalized with F-actin in peripheral ring structures (Fig. 2C). We demonstrated that NOS1AP colocalizes with NWASP in these ring structures (Fig. 2D), thereby revealing them as the adhesive actin-containing structures known as podosomes (20). NOS1AP exhibited diffuse localization in the remaining cells (32%). Thus, consistent with its role in actin dynamics of neurons (30), we found that NOS1AP localizes to multiple actin-cytoskeletal structures: (i) filopodia, which mammalian NS disease proteins have been shown to regulate (8, 24), and (ii) podosomes, which have not been linked to NS pathogenesis previously.
We next postulated that the patient variants we found cause aberrant NOS1AP expression and/or localization. However, relative to wild-type (WT) MYC-tagged NOS1AP, MYC_NOS1AP_C143Y had comparable protein levels by Western blotting (WB) (fig. S4D). MYC_NOS1AP_C143Y localized to the cytoplasm and podosomes by IF in an indistinguishable fashion from the WT construct (fig. S4E). The splice variant in A5106 (c.345-3T>G) is predicted to reduce splicing that would cause exon 5 skipping and yield a truncated protein (I116Afs*4). MYC_NOS1AP_I116Afs*4 showed a lower protein level by WB (fig. S4D) and mislocalizes to the nucleus by IF despite lacking a predicted nuclear localization signal (fig. S4, E, G, and H).
WT NOS1AP, but not NS mutants, promotes filopodia and podosome formation
On the basis of its subcellular localization, we posited that WT NOS1AP, but not NS patient–derived mutants, would promote filopodia formation, as was shown for NS genes Cdc42, ITSN1, and ITSN2 (8, 24). Using fluorescent live-cell imaging, we found that green fluorescent protein (GFP)–tagged WT NOS1AP overexpression increased filopodia formation to 59% (range, 40 to 78%) of GFP-positive podocytes compared to only 1% of GFP mock overexpressing podocytes (Fig. 3, A and B). In contrast, constructs representing NS patient variants failed to induce filopodia (3 and 0%) (Fig. 3, A and B; fig. S1; and movie S1).
Fig. 3. NOS1AP induces filopodia formation and is essential for PMR, potentially via a CDC42/DIAPH pathway.
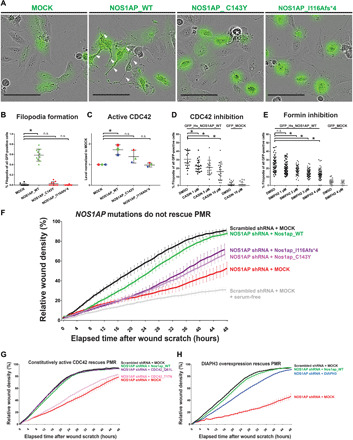
(A) Podocytes were transfected with GFP-plasmids (MOCK, WT NOS1AP, and NOS1AP patient mutation constructs). Representative images are shown with white arrows pointing to filopodia. Scale bars, 100 μm. (B) WT NOS1AP increased filopodia formation to 59% (range, 40 to 78%). NS mutant construct overexpression (C143Y and I116Afs*4) failed to induce filopodia (3 and 0%). [One-way analysis of variance (ANOVA), *P < 0.05; n.s., nonsignificant]. (C) Human embryonic kidney (HEK) 293T cells were transfected with NOS1AP WT or cDNA reflecting patient variants. WT NOS1AP overexpression caused a significant increase in active CDC42 levels, while NS patient variant constructs did not (one-way ANOVA; unique colors for independent experiments). (D) CDC42 inhibitor CASIN blocked filopodia formation in NOS1AP-transfected podocytes (one-way ANOVA, see fig. S6B for representative images). (E) Formin inhibitor SMIFH2 attenuated filopodia formation in NOS1AP-transfected podocytes (one-way ANOVA, see fig. S6C for representative images). DMSO, dimethyl sulfoxide. (F) Knockdown of NOS1AP (red) resulted in reduced PMR compared with scrambled shRNA control cells (black), which was rescued by overexpression of WT Nos1ap (green) but not NS patient variant constructs (purple and pink). (G) Reduced PMR upon knockdown of NOS1AP (red) was rescued by overexpression of WT Nos1ap (green) and a constitutively active human CDC42Q61L construct (purple) but not the hypomorphic CDC42T17N construct (pink). (H) Reduced PMR upon knockdown of NOS1AP (red) was rescued by overexpression of WT Nos1ap (green) and partially rescued by the formin DIAPH3 (blue).
We hypothesized that WT NOS1AP, but not NS mutants, would promote podosome formation, as described for murine NS genes Cdc42 and Wasl/NWASP (22, 41). Podocytes transfected with MYC MOCK, MYC-tagged WT NOS1AP, or the two mutants derived from NS patients were scored for the number of podosomes, defined as F-actin(+) peripheral ring structures using IF and confocal microscopy (fig. S5A). While only 6% of MOCK-transfected cells had >21 podosomes, 32% of WT NOS1AP overexpressing cells had >21 podosomes (fig. S5, B and C). In contrast, only 19 and 3% of immortalized podocytes transfected with the two mutant constructs p.C143Y and p.I116Afs*4, respectively, had >21 podosomes (fig. S5, B and C). We, thereby, showed that NOS1AP promotes filopodia and podosome formation, which is abrogated by NS patient variants (Fig. 3, A and B, and figs. S1 and S5).
NOS1AP promotes filopodia formation upstream of the GTPase CDC42
The finding that NOS1AP induced filopodia and podosome formation (Fig. 3, A and B, and fig. S5) led us to posit that it regulates actin dynamics upstream of CDC42 (fig. S1). We showed that overexpressed NOS1AP colocalizes with F-actin in a peripheral ring pattern in podosomes (Fig. 2C), which centrally contain two CDC42 effectors (18, 19, 23), NWASP (Fig. 2D), and DIAPH3 (fig. S5D). We therefore measured active guanosine triphosphate (GTP)–bound CDC42 levels using the G-LISA® assay in response to overexpression of WT NOS1AP versus overexpression of constructs reflecting variants from NS patients. We found that, relative to mock control, WT NOS1AP expression caused a significant increase in active CDC42 levels (1.72 mean fold change), while NS mutants C143Y and I116Afs*4 did not (Fig. 3C). As the Rho-like small GTPases may be coregulated, we additionally measured active levels of Rac family small GTPase 1 (RAC1) and ras homolog family member A (RHOA). WT NOS1AP overexpression did not significantly increase active RAC1 levels (1.33-fold change), while NS mutant constructs did (1.59- and 1.77-fold increases) (fig. S6A). In contrast, WT and mutant constructs had no effect on active RHOA levels (fig. S6A). This suggests that NOS1AP promotes active CDC42 levels, which is impaired by human NS patient variants, whereas these variants cause increased RAC1 activation, another established NS mechanism (fig. S1) (16).
If NOS1AP regulates actin cytoskeleton remodeling upstream of CDC42, then its effects (e.g., on filopodia formation) should be impaired by CDC42 inhibition. We tested this hypothesis by overexpressing GFP-tagged WT NOS1AP in a podocyte cell line in the presence of vehicle dimethyl sulfoxide control or increasing doses of CASIN, which blocks nucleotide exchange required for CDC42 activation (42). We found that CASIN inhibited filopodia formation in NOS1AP-treated cells in a dose-dependent manner (Fig. 3D and fig. S6B), further confirming that NOS1AP promotes filopodia formation by promoting CDC42 activation (fig. S1).
CDC42, in turn, binds and activates formin proteins, which mediate linear actin polymerization, filopodia formation, and cell migration (fig. S1) (18, 23). We hypothesized that formins act downstream of NOS1AP and CDC42 in filopodia formation (fig. S1). Consistent with this, we observed that filopodia formation upon overexpression of GFP-tagged WT NOS1AP was abrogated by the small molecule inhibitor of Formin Homology 2 domains (SMIFH2) (43) in a dose-dependent manner (Fig. 3E and fig. S6C).
We also tested whether NOS1AP-dependent filopodia formation required established NOS1AP-interacting factors NOS1 and YAP1 (32, 33), given Yes1 associated transcriptional regulator (YAP1) deficiency in mice causes NS (44). Knockdown of YAP1 did not alter filopodia formation, thereby excluding YAP1 from the pathogenic pathway (fig. S6, D and E). Likewise, the established NOS1 inhibitor L-nitroarginine methyl este (L-NAME) did not affect filopodia formation upon NOS1AP overexpression (fig. S6F).
WT Nos1ap, but not NS mutants, rescues podocyte migration
We hypothesized that NOS1AP also regulates PMR, which is modulated by multiple actin regulators encoded by monogenic NS disease genes (9, 16, 17). We therefore generated a NOS1AP-deficient human podocyte cell line by short hairpin RNA (shRNA) knockdown (fig. S6, G and H). We observed that, relative to scrambled shRNA expressing control cells, NOS1AP knockdown cells had reduced PMR (Fig. 3F), while cell proliferation and survival were not affected (fig. S6, I and J). NOS1AP knockdown PMR was rescued by overexpression of WT murine Nos1ap (Fig. 3F and figs. S1 and S6K). In contrast, overexpression of Nos1ap cDNA constructs reflecting the two NS patient variants (Fig. 1) failed to rescue PMR (Fig. 3F, figs. S1 and S6K, and movie S2).
NOS1AP promotes podocyte migration upstream of the GTPase CDC42
We have provided evidence that NOS1AP regulates the actin cytoskeleton upstream of CDC42 (Fig. 3, C and D ) and, therefore, hypothesized that it may similarly regulate PMR upstream of CDC42 (fig. S1). We found that, in NOS1AP shRNA knockdown cells, overexpression of the constitutively active CDC42Q61L variant did rescue PMR (Fig. 3G and fig. S6L) (45). However, overexpression of WT CDC42 or hypomorphic CDC42T17N failed to rescue PMR (Fig. 3G and fig. S6, L and N) (45).
In line with our observation that NOS1AP-induced filopodia formation requires formin proteins (Fig. 3E and fig. S6C), we postulated that formins would also act downstream of NOS1AP and CDC42 in podocyte migration (fig. S1). In podosomes, MYC-tagged NOS1AP encircled the classical formin and CDC42 effector DIAPH3 (fig. S5D). Overexpression of DIAPH3 rescued PMR in NOS1AP knockdown cells (Fig. 3H and fig. S6M). This strongly suggests that NOS1AP regulates PMR through activation of CDC42 and, in turn, DIAPH3 (fig. S1).
NOS1AP patient variant knock-in kidney organoids have aberrant glomeruli formation
Human kidney organoids differentiated from induced pluripotent stem cells (iPSCs) of patients with monogenic NS have demonstrated gene-specific abnormalities in glomeruli formation (25–27). We, therefore, hypothesized that kidney organoids harboring an NS patient–derived recessive NOS1AP variant would reveal aberrant glomerulogenesis consistent with podocyte dysfunction.
We determined that NOS1AP mRNA expression is enriched in organoid glomerular podocytes in single-cell mRNA sequencing datasets (46, 47) (fig. S7A) and is highest in three-dimensional organoid glomeruli relative to two-dimensional podocyte culture in bulk RNA sequencing datasets (fig. S7B) (25). Furthermore, by whole-mount IF, we determined that NOS1AP protein localizes to podocytes in organoid glomeruli adjacent to the podocyte marker synaptopodin and basement membrane protein laminin A5 (Fig. 4A and fig. S8A). These expression studies were consistent with our findings in mammalian kidneys (Fig. 2, A and B) and suggested that organoids provide a suitable system to interrogate NOS1AP function.
Fig. 4. Kidney organoids bearing the homozygous NOS1AP patient variant c.428G>A exhibit aberrantly formed glomeruli.
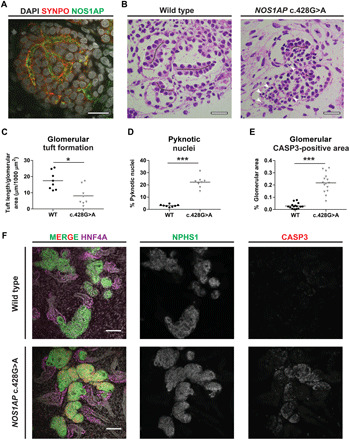
(A) IF imaging demonstrated NOS1AP localization to podocytes in organoid glomeruli adjacent to the podocyte marker SYNPO. Scale bar, 20 μm. (B) In WT and NOS1AP c.428G>A mutant organoids (PAS staining), glomerular tufts (within white dashed lines) were defined as linear podocyte monolayers organized bilaterally about established extracellular matrix (black lines) and were reduced in NOS1AP mutant organoids. NOS1AP mutant glomeruli also exhibited increased pyknotic nuclei (arrowheads), indicative of cell death. Scale bars, 20 μm. Also see fig. S8H for additional images of PAS staining. Scale bars, 20 μm. (C) Cumulative glomerular tuft length measurement is plotted from two to three independent fields (dots) from three independent organoid cultures. NOS1AP mutant organoids demonstrate significantly lower cumulative tuft length than WT organoids (Mann-Whitney U test, *P < 0.05). (D) Percentage of pyknotic nuclei is plotted from independent fields and organoids as in (B). NOS1AP mutant organoids exhibit significantly increased pyknotic nuclei relative to WT organoids (Mann-Whitney U test, ***P < 0.001). (E) Quantification of active caspase-3 (CASP3) staining in glomerular regions is shown from three paired differentiation experiments to substantiate increased cell death that was indicated by pyknotic nuclei in (B) and (D). NOS1AP mutant organoids demonstrate elevated apoptosis (Mann-Whitney U test, ***P < 0.001). (F) Whole mount IF of organoids for apoptotic marker cleaved CASP3 is shown. CASP3 staining is increased in glomeruli (NPHS1) of NOS1AP mutant organoid glomeruli, relative to WT organoids. CASP3 signal in tubular segments (HNF4A) is not increased. Scale bars, 100 μm. Also see fig. S8G for CASP3 staining in organoids derived from the second independent iPSC cell line PCS201010.
We introduced the recessive NOS1AP variant from subject A1018 (c.428G>A) into a healthy control iPSC line using CRISPR-Cas9 homology-directed repair (Table 1 and fig. S7, C and D). NOS1AP mutant and isogenic WT iPSC cell lines were differentiated into kidney organoids containing multisegmented nephrons (fig. S7, E and F). Knock-in organoids exhibited comparable NOS1AP mRNA level by quantitative reverse transcription polymerase chain reaction (RT-PCR) (fig. S8B) and NOS1AP protein levels by WB (fig. S8C). NOS1AP showed similar protein localization in WT and knock-in organoids by whole-mount IF using an antibody raised against a C-terminal region (amino acids 250 to 354) of the protein (fig. S8, D and F). An antibody raised against an immunogen (amino acids 149 to 230) adjacent to the mutated amino acid cysteine 143 did not detect the protein in knock-in organoids by whole-mount IF (fig. S8, D and E).
We measured organoid glomerular tuft formation by light microscopy as the cumulative length of linear podocyte monolayers organized bilaterally about established extracellular matrix. Tuft formation was significantly reduced in NOS1AP mutant organoids relative to WT organoid glomeruli (Fig. 4, B and C, and fig. S8H). NOS1AP mutant glomeruli also demonstrated increased pyknosis by periodic acid–Schiff (PAS) staining (Fig. 4, B and D, and fig. S8H). To characterize this cell death further, we performed cleaved caspase-3 (CASP3) staining, a marker of apoptosis. We found that CASP3 staining was increased in the knock-in organoids relative to the WT organoids (Fig. 4, E and F, and fig. S8G). Using cell type–specific markers, we observed apoptosis selectively in the glomerular regions of knock-in organoids as compared to tubular regions (Fig. 4E). This was confirmed in organoids derived from an independent iPSC cell line (fig. S8G).
Recessive variants in Nos1ap caused glomerulopathy in mice
On the basis of patient NOS1AP variants, we posited that recessive Nos1ap variants within the PTB domain would cause features of NS in mice. We evaluated mice bearing a deletion of the symmetrical third exon of Nos1ap (Nos1apEx3−) (Fig. 5A) (34). This exon encodes a highly conserved region of the PTB domain (fig. S9A).
Fig. 5. Nos1apEx3−/Ex3− mice develop glomerular proteinuria, foot process effacement, and mesangial matrix expansion.
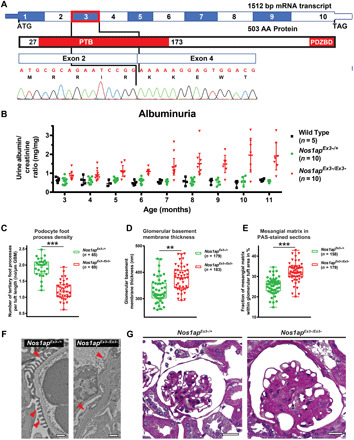
(A) Coding exon (top) and protein domain (bottom) structures of murine Nos1ap and Sanger sequencing trace for Nos1apEx3−/Ex3− cDNA are shown. Exon 3 deletion (red rectangle) causes an in-frame deletion within the PTB domain. (B) Urinary albumin/creatinine ratios were measured. Nos1apEx3−/Ex3− mice develop significant albuminuria. (C) Podocyte foot process density was quantified in transmission electron microscopy (TEM) images for Nos1apEx3−/+ and Nos1apEx3−/Ex3− mice [five animals per genotype, 11 months old (three) and 16 months old (two); 65 and 69 capillary loops per genotype, respectively]. Foot process effacement is observed in homozygous Nos1apEx3−/Ex3− mice. (Each dot represents one capillary loop, and bars represent minimum to maximum; Kruskal-Wallis test, ***P < 0.001). (D) Increased glomerular basement membrane (GBM) thickness was observed in TEM images of homozygous Nos1apEx3−/Ex3− mice. (Bars represent minimum to maximum; Kruskal-Wallis test, **P < 0.01). (E) Mesangial matrix area per glomerulus in PAS staining was increased in homozygous Nos1apEx3−/Ex3− mice. [Each dot represents one glomerulus, and bars represent minimum to maximum; five animals per genotype, 11 months old (three) and 16 months old (two); 158 and 179 images per genotype, respectively; Kruskal-Wallis test, ***P < 0.001]. (F) Representative glomerular TEM images for Nos1apEx3−/+ and Nos1apEx3−/Ex3− mice demonstrate podocyte foot process effacement and thickened basement membranes. Scale bars, 1 μm. (G) Representative PAS images show mesangial matrix expansion, partially collapsed capillary loops, and thickened basement membranes in Nos1apEx3−/Ex3− mice. Scale bars, 200 μm. Also see fig. S10.
Homozygous Nos1apEx3−/Ex3− mice developed albuminuria at 5 months of life, which worsened progressively through 11 months, while WT and heterozygous littermates did not (Fig. 5B and fig. S9, B and C). Male homozygotes had higher albuminuria than female homozygotes, consistent with prior literature (48), although the albuminuria in female Nos1apEx3−/Ex3− mice was, nonetheless, significantly higher than in control animals of either sex (fig. S9, E and F). Despite exhibiting proteinuria with full penetrance, Nos1apEx3−/Ex3− mice did not develop hypoalbuminemia, renal dysfunction, or early lethality (fig. S9D).
We next investigated whether there were histological and ultrastructural glomerular changes underlying the glomerular proteinuria in Nos1apEx3−/Ex3− mice. Electron microscopy of kidney sections from homozygotes demonstrated podocyte foot process effacement and thickening of the glomerular basement membrane (Fig. 5, C, D, and F), relative to heterozygote glomeruli exhibiting appropriate rhythmicity of foot process formation. Histologically, Nos1apEx3−/Ex3− animals exhibited mesangial matrix expansion (Fig. 5, E and G, and fig. S10A), increased glomerular size (fig. S10B), and glomerulosclerosis (fig. S10, C and D).
Notably, albuminuria in homozygotes was not ameliorated by corticosteroid treatment (fig. S9G). In contrast, albuminuria was increased in both heterozygote control and homozygote animals upon oral delivery of dexamethasone (fig. S9G), which is consistent with a previous report of the proteinuric effect of corticosteroids (49).
DISCUSSION
In summary, we delineated a pathogenesis for NOS1AP variants detected in NS patients (fig. S1). Our studies in human podocytes suggest that recessive NOS1AP variants (Fig. 1 and Table 1) impair CDC42 activation (Fig. 3C), which reduces formin function (Fig. 3E) and disrupt actin remodeling in podocyte filopodia and podosome formation (Fig. 3, A and B, and fig. S5). This was corroborated by defective PMR in NOS1AP-depleted podocytes, which is rescued by WT Nos1ap, active CDC42, and DIAPH3 but not by patient mutants (Fig. 3, F to H). These defects cause podocyte dysfunction, evidenced by aberrant glomerulogenesis in human renal organoids (Fig. 4 and figs. S7 and S8), proteinuria and glomerulosclerosis in mice (Fig. 5 and figs. S9 and S10), and NS in humans (Table 1).
Consistent with the role of NOS1AP in the actin cytoskeleton of neurons (30, 31), we show that NOS1AP NS patient variants abrogate filopodia and podosome formation in podocytes (Fig. 3, A and B, and figs. S1 and S5). Filopodia have been appreciated as an important cytoskeletal structure in podocytes and in the pathogenesis of other proteins that are deficient in monogenic NS such as Cdc42, ITSN1, and ITSN2 (8, 10, 12, 18, 21). Podosomes, on the other hand, are actin-based adhesive structure that play crucial roles in three-dimensional migration (20) but have not been previously linked to NS before this study.
For the small Rho-like GTPase CDC42, many downstream targets and effectors like p21 activated kinases (PAKs), WASPs (e.g., NWASP), or formins (e.g., DIAPH3) have been described (19, 23, 50). Our in vitro data suggest that formins act downstream of NOS1AP, as NOS1AP-induced filopodia formation was reduced by formin inhibition (Fig. 3E) and defective PMR in NOS1AP-deficient podocytes was rescued by DIAPH3 (Fig. 3H). However, given the multitude of CDC42 effectors, it will be important to determine whether other downstream CDC42 responses are affected by NOS1AP deficiency in podocytes.
In addition, it remains unclear how NOS1AP promotes CDC42 activity. NOS1AP may regulate CDC42 by activating CDC42 guanine exchange factors such as ITSN1 and ITSN2, whose genes are mutated in monogenic forms of NS (8). Alternatively, NOS1AP may inhibit factors, which down-regulate small Rho-like GTPases. This has been shown for ARHGDIA, which is mutated in human NS (9). These possibilities should be explored in future studies to determine what mediates the effects on NOS1AP on CDC42, podocyte actin remodeling, and NS pathogenesis.
It is known that RAC1 activation is linked to podocyte foot process effacement and other features of NS (12, 15, 16). While we observed increased levels of active RAC1 upon overexpression of NS patient–derived NOS1AP variants, our human genetics and mouse model data suggest that disease-causing recessive variants in NOS1AP lead to loss of function rather than gain of function as both the heterozygous mother of patient A1018 and heterozygous Nos1apEx3−/+ mice did not show a renal phenotype. NOS1AP loss of function may secondarily increase RAC1 activation due to dysregulated CDC42, given that CDC42 has been shown to regulate RAC1 previously (51). Further studies are needed to determine the exact molecular effects of NOS1AP loss of function on CDC42 and RAC1 activation.
Nos1apEx3−/Ex3− mice recapitulate the glomerular proteinuria, podocyte foot process effacement, and glomerulosclerosis (Fig. 5) observed in patients with NOS1AP variants, which similarly reside within the PTB domain. Moreover, these features of NS disease were also observed in Cdc42-deficient mice (10, 12), supporting our proposed pathogenic mechanism. While Nos1ap-deficient mice do not fully recapitulate the human phenotype (e.g., hypoalbuminemia and end-stage renal disease), this may be secondary to their C57BL/6 genetic background. It is well established that the severity of glomerular disease phenotypes in mice varies depending on genetic background. This has been associated with differences in glomeruli density and basement membrane protein expression (48, 52). For example, the Tns2-deficient monogenic NS mouse model exhibited strain-dependent severity with most severe features of NS when bred on the FVB/N background, while they demonstrated mild or no disease on C57BL/6J, 129/SvJ, and DBA/2 backgrounds (53). Therefore, it will be important to examine the impact of the Nos1apEx3− allele on potentially more disease-prone backgrounds.
Our findings provide the first example of kidney organoids to model a novel monogenic cause of NS. Consistent with its localization in primary rat glomeruli (Fig. 2B), NOS1AP was detected in organoid podocytes in colocalization with foot process marker synaptopodin (Fig. 4A and fig. S8A). In contrast, endogenous NOS1AP was not detectable in immortalized podocytes in two-dimensional culture by IF imaging with the same antibody (fig. S4C). This may be due to lower abundance of NOS1AP in two-dimensional cell culture compared to three-dimensional kidney organoids, which is supported by lower NOS1AP mRNA transcript levels in two-dimensional podocyte culture relative to three-dimensional kidney organoids (fig. S7B). Therefore, the NOS1AP antibody used may not be sufficiently sensitive to detect endogenous NOS1AP protein at the lower levels present in two-dimensional podocyte culture by IF imaging. These findings highlight the utility of three-dimensional human kidney organoids for interrogating NOS1AP function.
Kidney organoids bearing a NOS1AP NS patient variant demonstrate increased glomerular apoptosis (Fig. 4, D and F). This suggests that NOS1AP may regulate cell viability, consistent with the observed role for CDC42 signaling in podocyte survival in proteinuric disease (10, 12, 13). However, we found that loss of function of NOS1AP by shRNA knockdown in immortalized human podocytes did not affect cell survival or proliferation (fig. S6, I and J). Therefore, the apoptosis in organoid glomeruli may represent an intermediate phenotype of NOS1AP variant–induced podocytopathy that is unique to this system, in which podocytes are grown three-dimensionally without a vasculature and with growth factors withdrawn in the course of maturation.
In summary, we demonstrate that recessive variants in NOS1AP are a novel cause of glomerular proteinuric kidney disease in humans using a unique combination of cutting-edge functional genomic approaches including (i) multiple single-cell mRNA sequencing datasets, (ii) IF and high-resolution confocal microscopy imaging and live-cell imaging of two-dimensional podocyte culture, (iii) novel three-dimensional knock-in kidney organoids, and (iv) a mouse model bearing recessive in-frame deletion in Nos1ap. Overall, this work proposes a new standard for functional genomic studies of rare kidney diseases.
METHODS
Research subjects
We obtained blood samples and pedigrees following informed consent from individuals with NS or their legal guardians. The diagnosis of NS was based on clinical features of nephrotic-range proteinuria, hypoalbuminemia, and edema and supported by renal biopsy findings evaluated by renal pathologists. Clinical data were obtained using a standardized questionnaire (www.renalgenes.org).
ES, homozygosity mapping, and variant calling
ES and variant calling were performed as previously described (3, 54) to discover a novel genetic cause of NS using Agilent SureSelect human exome capture arrays (Thermo Fisher Scientific) with next-generation sequencing on an Illumina platform. Sequence reads were mapped against the human reference genome (National Center for Biotechnology Information build 37/hg19) using CLC Genomics Workbench (version 6.5.1) (CLC bio). Genetic location information is according to the February 2009 Human Genome Browser data, hg19 assembly (www.genome.ucsc.edu). Downstream processing of aligned BAM files were performed using Picard and samtools, and single nucleotide variant calling was performed using Genome Analysis Toolkit 5 (GATK5). Variant calling was performed as previously described (3, 54). The variants included were rare in the population with mean allele frequency < 1% in dbSNP147 and with only zero to one homozygote in the adult genome database gnomAD. In addition, variants were nonsynonymous and/or located within splice sites. On the basis of an autosomal homozygous recessive hypothesis, variants within homozygosity regions (see the next section) were included. Subsequently, variant severity was stratified on the basis of protein impact (truncating frameshift or nonsense variants, essential or extended splice-site variants, and missense variants). Splice-site variants were assessed by in silico tools MaxEnt and NNSPLICE splice-site variant prediction scores as well as conservation across human splice sites as described previously (3, 54). Missense variants were assessed on the basis of Sorting Intolerant From Tolerant (SIFT), MutationTaster, and PolyPhen 2.0 conservation prediction scores and evolutionary conservation based on manually derived multiple sequence alignments.
Homozygosity mapping
For subject A5106, the generated variant call format (VCF) file was subsequently used in homozygosity mapper to identify regions of homozygosity based on nonparametric lod (NPL) scores as previously described (54). In subject A1018, the GeneChip® Human Mapping 250K Sty Array from Affymetrix was used to generate homozygosity mapping plots as previously described (54). NPL scores were calculated using a modified version of the program GENEHUNTER 2.1 through stepwise use of a sliding window with sets of 110 single-nucleotide polymorphisms and the program ALLEGRO to identify regions of homozygosity using a disease allele frequency of 0.0001 and Caucasian marker allele frequencies. Genetic regions of homozygosity by descent were plotted across the genome as candidate regions for recessive genes as previously described (54).
Accession numbers
Human NOS1AP full-length protein (GenBank accession NP_055512) encoded by GenBank accession NM_014697.
Structural modeling for NOS1AP missense mutation
The NOS1AP PTB sequence (amino acids 16 to 74) was queried in the Protein Data Bank using Psi-BLAST with the BLOSUM62 matrix. In PyMOL, the structures with at least 30% identity were aligned, and calculations of the root mean square deviation and number of aligned amino acids were generated. The aligned homologous structures were visualized in PyMOL. The alignment was also confirmed using Expresso to highlight amino acids in close proximity to mutated NOS1AP residue Cys143 (55, 56). Site Directed Mutator (57) was used to assess the stability consequences of human NOS1AP mutations based on structural parameters (mainchain conformation, solvent accessibility, and hydrogen bonding class) to generate a pseudo-ΔΔG stability output. PyMOL was used to generate and visualize the mutations in paralogous structures with an algorithm that makes use of a rotamer library, using the rotamer that led to the loESt van der Waals strain.
cDNA cloning
cDNA clones were purchased from the following sources: human NOS1AP (Harvard PlasmID Database), mouse Nos1ap (OriGene), human DIAPH3 (GenScript Biotech Corporation), and human CDC42 (pRK5-Myc–tagged construct, gift from G. Bokoch, Addgene 12972). Mutagenesis was performed using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). Expression constructs (pRK5-N-Myc and pCDNA6.2-N-GFP) were produced using LR Clonase (Invitrogen, Thermo Fisher Scientific) following the manufacturer’s instructions.
Cell lines
The experiments described here were performed using human embryonic kidney (HEK) 293T cells and immortalized human podocytes. HEK293T cells were purchased from the American Type Culture Collection (ATCC) Biological Resource Center. Human immortalized podocytes were a gift of M. Saleem (University of Bristol, Bristol, UK) and were cultured as previously described (54).
Antibodies, reagents, and quantitative PCR reagents
The following primary antibodies were used: rabbit anti-NOS1AP (Novus Biologicals, NBP2-38758 and NBP2-38151), guinea anti-nephrin (Progen, GP-N2), mouse anti-SMA (Sigma-Aldrich, A2547), mouse anti-CD31 (MA3100, Thermo Fisher Scientific), mouse anti-NWASP (LSBio, LS-C133098-100), mouse anti-DIAPH3 (Proteintech, 14342-1-AP), rabbit anti-c-Myc (Sigma-Aldrich, C3956), mouse anti-myc (Santa Cruz Biotechnology, SC-40), mouse horseradish peroxidase (HRP)–linked anti–β-actin (Abcam, ab20272), rabbit anti-YAP1 (Cell Signaling Technology, 4912), anti-Golgin B1 (GOLGB1) (Sigma-Aldrich, HPA011008), anti-GRP78 Binding Immunoglobulin Protein (BiP) (Abcam, ab21685), anti-Nucleoporin 153 (NUP153) (Sigma-Aldrich, HPA027897), and anti–cleaved CASP3 (Abcam, ab2302). Donkey anti-mouse, anti-guinea, and anti-rabbit Alexa 488– and Alexa 594–conjugated secondary antibodies; 4′,6-diamidino-2-phenylindole (DAPI) staining reagents; and phalloidin–Alexa 488 were obtained from Invitrogen (Thermo Fisher Scientific). HRP-labeled secondary antibodies were purchased from Santa Cruz Biotechnology.
Filopodia quantification by live imaging
Human podocytes were plated and transfected with cDNA constructs after 24 hours. One hour later, transfection media was exchanged with regular culture media. Cells were then imaged hourly with light and fluorescence microscopy at 10× to 20× using the IncuCyte ZOOM System (Essen BioScience). For podocyte filopodia frequency, cells with two or more filopodia were quantified over all transfected cells in a field for 10 to 20 independent fields per experiment. Three independent experiments were performed for confirmation. This was evaluated at 20 hours after transfection for NOS1AP mutant evaluation when maximal effects were observed and at 10 to 14 hours for drug treatment experiments depending on drug half-life. For cell treatment, the following reagents were used: L-NAME (Cayman Chemical), CASIN (Millipore), and SMIFH2 (Millipore). For CASIN and L-NAME administration, the compound was added at time of media exchange at 1 hour after cDNA transfection, and representative images at 13 hours after transfection were captured for quantification. For SMIFH2 administration, the compound was added at 1 hour after cDNA transfection and changed every 2 hours throughout the experimental time course based on previous literature demonstrating its brief duration of effect (43, 58). Representative images at 14 hours after transfection were captured for quantification.
IF in tissue sections and cell lines
For immunostaining of frozen optimal cutting temperature (OCT) compound-embedded tissue sections (adult rat kidney), permeabilization was performed using 0.1% Triton X-100. After blocking, sections were incubated overnight at 4°C with primary antibodies. The cells were incubated in secondary antibodies for 90 min at room temperature, followed by mounting in hardening medium with DAPI. For cell IF, human immortalized podocytes were seeded on coverslips and grown at a permissive temperature. For overexpression studies, human podocytes were transiently transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Experiments were performed 24 to 48 hours after transfection. Cells were fixed for 15 min using 4% paraformaldehyde (PFA) and permeabilized as above. Staining was performed similarly to tissue sections. Confocal microscopy imaging was performed using the Leica SP5X system with an upright DM6000 microscope, and images were processed with the Leica AF software suite.
Single-cell mRNA sequencing data analysis
Heatmap results depicting differential mRNA expression levels (from z-scores) were based on single-cell transcriptomics data from either 8-week-old WT CD1 male mice (39) or the renal biopsy of a healthy male adult man (40). Processed data from each set were queried for percent expression in defined cell clusters (from 14,722 murine and 4259 human cells, respectively). Queried data were normalized using z-score calculation as previously described (6).
G-LISA Rho, Rac, and Cdc42 activation assays
Cells were transfected in six-well plates with WT constructs or mock using Lipofectamine 2000. Transfected cells were incubated in Dulbecco’s Modified Eagle Medium (HEK293T) with 10% fetal bovine serum for 24 hours and then in serum-free medium for 24 hours. RHOA, RAC1, or CDC42 activity was determined using a colorimetric G-LISA RHOA, RAC1, or CDC42 Activation Assay Biochem kit (Cytoskeleton), which is an enzyme-linked immunosorbent assay–based assay that detects GTP-bound small GTPases.
shRNA and siRNA transfection
shRNA against human NOS1AP was subcloned into pSIREN RetroQ for retroviral transduction using HEK293T cells. Two independent scramble shRNA and knockdown lines were generated and used in all podocyte migration studies. Forty-eight hours after transduction, puromycin at a final concentration of 2 to 4 μg/ml was added to the medium for selection of transduced cells. Podocytes were treated with 200 pmol ON TARGETplus small interfering RNA (siRNA) targeting YAP1 (GE Dharmacon) using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. Experiments were performed 20 hours after transfection for NS mutant filopodia assays and, subsequently, at 10 to 14 hours for drug treatment studies as function of compound stability.
Podocyte migration assay
Briefly, the podocyte migration assays were performed using the IncuCyte ZOOM System (Essen BioScience) as previously described (17). Upon scratch, wound was imaged hourly, and the degree of wound closure was measured as the relative wound density, defined as cell density in the wound area normalized to cell density outside of the wound area over time. This metric normalizes for changes in cell density caused by proliferation. Rescue experiments were performed with all constructs transfected at 2 μg/ml except MYC_CDC42_Q61L (0.5 μg/ml) as cellular toxicity was noted at higher doses.
Cell proliferation assay
XTT proliferation assay (ATCC) was performed according to the manufacturer’s instructions. Briefly, 1.5 ×104 cells were plated. Twenty-four hours later, media was changed, and complexed XTT reagent was added. Cells were incubated at 37°C, and absorbance was measured at 475 and 660 nm at set intervals. Full media without cells was used as a negative control. Six biological replicates were used per condition.
Generation of NOS1AP c.428G>A knock-in iPSCs
A single guide RNA (sgRNA) plasmid specific to exon 5 of the NOS1AP locus (pSMART-sgRNA-NOS1AP) was generated by annealing oligodeoxynucleotides CACCGTTACACCTGAAGATATTGC and AAACGCAATATCTTCAGGTGTAAC, followed by ligation into the BbsI sites of the pSMART-sgRNA vector (59). To generate the NOS1AP template for homology directed repair (HDR), a gblock (Integrated DNA Technologies) comprising 434 and 555 base pairs (bp) flanking the c.428G>A variant was ligated into the pSMART-HCKan plasmid vector (Lucigen) (fig. S7C). A 3-bp CAG>TTC synonymous change was also included just upstream of the c.428G>A variant to block Cas9 recutting and facilitate screening by allele-specific PCR. Plasmid DNA was propagated in DH5-alpha Escherichia coli (Bioline) and prepared for transfection using a Plasmid Maxi kit (QIAGEN). Transfections were performed using the Neon Transfection System (Thermo Fisher Scientific). iPSCs (derived from ATTC CRL-1502 and PCS201010 fibroblasts) were harvested with TrypLE (Thermo Fisher Scientific) 2 days after passaging and resuspended in buffer R at a final concentration of 1 × 107 cells/ml and added to a tube containing pSMART-sgRNA-NOS1AP, mRNA encoding Cas9-Gem (59, 60), and the NOS1AP template plasmid. Electroporation was performed in a 100-μl tip using 1100 V for 30 ms with one pulse. Electroporated iPSCs were plated on six-well Matrigel-coated plates containing Essential 8 medium with 5 μM Y-27632 (Tocris). Colonies containing the desired knock-in allele were screened by PCR, followed by Sanger sequencing to confirm homozygosity of the c.428G>A variant. Colonies containing successfully edited iPSCs were subjected to a subsequent round of subcloning and screening to ensure a clonal population of gene-edited iPSCs. iPSCs were maintained and expanded at 37°C, 5% CO2, and 5% O2 in Essential 8 medium (Thermo Fisher Scientific) on Matrigel-coated plates with daily media changes and passaged every 3 to 4 days with EDTA in 1× phosphate-buffered saline (PBS) as previously described (61).
Kidney organoid differentiation
Kidney organoids were differentiated by adapting a previously reported protocol (62, 63) depicted in fig. S7 (E and F). Briefly, 75,000 iPSCs were seeded in six-well plates on growth factor reduce Matrigel in Essential 8 media supplemented with Revitacell (Thermo Fisher Scientific). Cells were treated with 3 days of CHIR99021 (7 μM; R&D Systems, refreshed on day 2), followed by 4 days of fibroblast growth factor 9 (FGF9) (200 ng/ml) and heparin (1 μg/ml) in TeSR-E6 (STEMCELL Technologies) refreshed daily. Monolayers were dissociated to a single-cell suspension using TrypLE on day 7 and resuspended in 5 μl of TeSR-E6 media per million cells. This cell suspension was aspirated into a 100-μl Gastight syringe with a 21-guage removable needle (Hamilton) and loaded onto a NovoGen MMX Bioprinter™ (Organovo). Eight 4-mm lines of cells were printed onto 0.4-μm membrane six-well Transwell inserts (Corning). Transwells were treated with 5 μM CHIR99021 for 1 hour and then 5 days of FGF9/heparin, followed by 2 days of all-trans retinoic acid (2 μM) before withdrawal of all growth factors. TeSR-E6 was then refreshed to transwells three times weekly.
Organoid histology
Organoids were fixed in 4% PFA for 6 hours. They were set in 2% agar before tissue processing for standard paraffin wax histology. Organoid sections were cut at 3 μm and stained with PAS reagents. Tuft glomeruli formation was measured as the total length of podocyte monolayers that bilaterally lined established extracellular matrix divided by glomerular area per high-magnification field. Pyknotic nuclei were measured as a percentage of total glomerular nuclei per high-magnification field.
Organoid IF
Organoids were fixed in 2% PFA for 20 min on ice and washed with PBS. Whole organoids were cut from the Transwell filter and blocked/permeabilized for 3 hours in 10% donkey serum/0.3% Triton X-100/PBS. Both primary and secondary antibodies were incubated overnight at 4°C and washed with 0.3% Triton X/PBS. Whole mounts were cleared with 90% glycerol. Images were obtained with an Andor Dragonfly Spinning Disk confocal microscope. The following antibodies were used: synaptopodin (1:300; Santa Cruz Biotechnology, SC21537), Laminin subunit alpha-5 (LAMA5) (1:300; Abcam, AB77175), Hepatocyte Nuclear Factor 4-alpha (HNF4A) (1:500; Life Technologies, MA1-199), CASP3 (1:200; Cell Signaling Technology, 9661), NPHS1 (1:300; Bioscientific, AF4269), Lectin (LTL) (1:300; Vector Laboratories), E-cadherin (ECAD) (1:300; BD Biosciences, 610181), GATA binding protein 3 (GATA3) (1:300; Cell Signaling Technology, 5852S), podocin (NPHS2) (1:500; Sigma-Aldrich, P0372), streptavidin-AF405 (1:400; Life Technologies, S32351), donkey anti–mouse-AF488 (1:400; Life Technologies, A21202), donkey anti–rabbit-AF568 (1:400; Life Technologies, A10042), and donkey anti–sheep-AF647 (1:400; Life Technologies, A21448). In CASP3 studies, NPHS1 signal was digitally enhanced in all images from WT and knock-in groups to equivalent levels to identify glomerular regions between groups in a consistent fashion.
Organoid RNA and protein quantification
Quantitative RT-PCR was performed for NOS1AP mRNA transcript using the following primers: CACGTCCAGGCTTCCTAC (reverse) and CCTAGCAAAACCAAGTACAACC (forward). NOS1AP protein levels were measured using the SimplyWes capillary Western Blotting system.
Mouse breeding and maintenance
The animal experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the Boston Children’s Hospital (#18-12-3826R). All mice were handled in accordance with the Guidelines for the Care and Use of Laboratory Animals. Mice were housed under pathogen-free conditions with a light period from 7:00 a.m. to 7:00 p.m. and had ad libitum access to water and rodent chow. Nos1apEx3−/Ex3− mice were a gift from N. Kato from the National Center for Global Health and Medicine in Japan. Briefly, a linearized vector with targeted deletion of Nos1ap exon 3 was electroporated into ES cells, positive clones were selected by G418 on embryonic fibroblast feeder cells, and targeted ES cells were then microinjected into C57BL/6 blastocysts. Resulting male chimeras were backcrossed onto C57BL/6 mice, and heterozygous Nos1apEx3−/+ mice (N4 generations) were intercrossed to generate homozygous Nos1apEx3−/Ex3− mice. Genotyping was performed by multiplex PCR using the following primers: #1, CTTTGTCTTCTGCTTCGCC; #2, ACACTACCATTTGGTCTCC; #3,TCAAGACCGACCTGTCC; and #4, CAATAGCAGCCAGTCCC.
Urine analysis
Urine was collected by placing mice in collection cages with ad libitum access to water overnight (16 hours). Upon collection, samples were immediately frozen and stored at −80°C and only thawed on ice before urine albumin and creatinine measurements once. Urinary albumin levels were determined using the Albumin Blue Fluorescent Assay Kit (Active Motif) in combinations with standard dilutions prepared from mouse serum albumin (Equitech Bio Inc.). Urine creatinine was measured using the QuantiChrom Creatinine Assay Kit (BioAssay Systems). In-gel protein detection was performed with Coomassie blue dye.
Whole-blood analysis
Two hundred microliters of blood was collected once a month via facial vein bleeding method and collected in lithium heparin tubes. Blood samples were then immediately analyzed with the Vetscan VS2 Chemistry Analyzer using Comprehensive Diagnostic Profile rotors.
Histological analysis
The kidney tissues were fixed in 4% PFA, sectioned (3- to 5-μm thickness), and stained with PAS and Masson’s trichrome (MT) following standard protocols for histological examination. Per animal, 20 images were obtained by a blinded observer striving to only include well-oriented glomeruli. Semiquantitative analysis of PAS and MT stained sections was performed in a semiautomated manner using an ImageJ macro. Briefly, images were first subjected to histogram matching using Adobe Photoshop to account for slight differences in staining intensity and brightness. ImageJ was used to perform an automated Gaussian blur color thresholding to exclusively select strong pink-stained structures with high amounts of carbohydrates in PAS or strong blue-stained collagenous structures in MT, respectively. The glomerular tuft area was then outlined by the user, and the stained fraction per tuft area automatically was calculated.
Ultrastructural analysis
Kidney tissues were perfuse-fixed with 2.5% glutaraldehyde, 1.25% PFA in 0.1 M sodium cacodylate buffer (pH 7.4), and then fixed in 2.5% glutaraldehyde, 1.25% PFA, and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4) overnight at 4°C. Samples were then washed with 0.1 M phosphate buffer, postfixed with 1% OsO4 dissolved in 0.1 M PBS for 2 hours, dehydrated in ascending gradual series (50 to 100%) of ethanol, and infiltrated with propylene oxide. Samples were embedded using the Poly/Bed 812 kit (Polysciences) according to the manufacturer’s instructions. After pure fresh resin embedding and polymerization in a 65°C oven (TD-700, DOSAKA, Japan) for 24 hours, sections of approximately 200- to 250-nm thickness were cut and stained with toluidine blue for light microscopy. Sections of 70-nm thickness were double-stained with 6% uranyl acetate (Electron Microscopy Sciences, 22400) for 20 min and lead citrate (Thermo Fisher Scientific) for 10 min for contrast staining. The sections were cut using Reichert Ultracut S/Leica EM UC7 (Leica) with a diamond knife (Diatome) and transferred onto copper and nickel grids. Sections were evaluated by transmission electron microscopy (JEOL 1200EX) at an acceleration voltage of 80 kV. All steps, including image acquisition, were performed in a blinded manner by independent persons. For each animal [five per genotype (three 11 months old and two 16 months old)], ~20 images of two to four glomeruli were acquired, and for assessment of podocyte foot process density and glomerular basement membrane thickness, only perpendicularly cut capillary loops were evaluated. Podocyte foot process density quantification was performed for 65 or 69 capillary loops per genotype, respectively. Quantification of glomerular basement membrane thickness was performed on the basis of 158 and 179 images per genotype, respectively.
Statistics
Graphpad Prism 8.0.0 software was used to perform one-way analysis of variance (ANOVA) for statistical testing between groups. For organoid histological analysis, Mann-Whitney U statistical test and Welch’s test were used.
Study approval
Approval for human subjects research was obtained from Institutional Review Boards of the University of Michigan, Boston Children’s Hospital and local institutional review board equivalents.
Web resources
The following websites are used for this study: University of California Santa Cruz Genome Browser, genome.ucsc.edu; Ensembl Genome Browser, www.ensembl.org; gnomAD browser 2.0.3., gnomad.broadinstitute.org; Polyphen2, genetics.bwh.harvard.edu/pph2; SIFT, sift.jcvi.org; MutationTaster, www.mutationtaster.org; NNSPLICE splice-site mutation prediction, www.fruitfly.org/seq_tools/splice.html; MaxEnt splice prediction, hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html; SMART, smart.embl-heidelberg.de; Psi-BLAST, www.ncbi.nlm.nih.gov/blast/; Protein Data Bank, www.rcsb.org; Site Directed Mutator, marid.bioc.cam.ac.uk/sdm2.
Supplementary Material
Acknowledgments
Funding: F.H. is the William E. Harmon Professor of Pediatrics. This research is supported by a grant from the NIH to F.H. (DK-076683-13). A.J.M. was supported by an NIH Training Grant (T32DK-007726), by the 2017 Post-doctoral Fellowship Grant from the Harvard Stem Cell Institute, and by the American Society of Nephrology Lipps Research Program 2018 Polycystic Kidney Disease Foundation Jared J. Grantham Research Fellowship. F.B. was supported by a fellowship grant (404527522) from the German Research Foundation (DFG). V.K. is supported by a DFG fellowship grant (403877094). T.M.K. was supported by a Post-doctoral Fellowship award from the KRESCENT Program, a national kidney research training partnership of the Kidney Foundation of Canada, the Canadian Society of Nephrology, and the Canadian Institutes of Health Research. E.W. was supported by the Leopoldina Fellowship Program, German National Academy of Sciences Leopoldina (LPDS 2015-07). A.C.O.-W. acknowledges support from NIH T32 (DK007527) and F32 (DK122766) Ruth L. Kirschstein National Research Service Awards. J.D.F.-K. acknowledges funding from the Natural Sciences and Engineering Research Council of Canada (#RGPIN-2016-06718). Organoid work was funded by the National Health and Medical Research Council of Australia (NHMRC) (GNT1098654). T.A.F. is supported by an NHMRC Postgraduate Scholarship (GNT1114409) and a Royal Australasian College of Physicians Jacquot Award. M.H.L. is an NHMRC Senior Principal Research Fellow (GNT1136085). MCRI is supported by the Victorian Government’s Operational Infrastructure Support Program. The MCRI iPSC gene editing facility is supported by the Stafford Fox Foundation. We acknowledge the support of the Methuselah Foundation and Organovo Inc. for the Novogen MMX bioprinter. The Yale Center for Mendelian Genomics (UM1HG006504) is funded by the National Human Genome Research Institute. The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Competing interests: F.H. is a co-founder of Goldfinch Biopharma Inc. M.H.L. has received contract research support from Organovo Inc. The other authors declare that they have no competing interests. No part of this manuscript has been previously published. Author contributions: A.J.M., F.B., V.K., R.S., K.D., T.M.K., E.W., D.A.B., I.U., A.A., R.D., M.N., I.O., C.M.K., A.C.O.-W.., Y.M., N.M., S.M., R.P.L., S.S., and F.H. performed genetic analysis. M.K. and J.D.F.-K. performed structural modeling. A.J.M., F.B., V.K., R.S., K.D., E.W., D.A.B., I.U., A.A., A.K., K.E., C.H.C., D.S., R.D., M.N., I.O., and F.H. performed cell culture studies. A.J.M, F.B., V.K., R.S., K.D., T.M.K., E.W., D.A.B., I.U., A.A., R.D., M.N., I.O., C.M.K., A.C.O.-W.., Y.M., N.M., N.A.S., A.H., R.K., C.A., and F.H. recruited patients and gathered detailed clinical information for the study. M.H.L. planned, analyzed, and oversaw organoid studies. S.E.H. performed iPSC gene editing. T.A.F. and K.S.T. performed kidney organoids differentiation and analysis, and M.S. performed organoid histology. All authors critically reviewed the paper. F.H. conceived of and directed the project. A.J.M., F.B., and F.H. prepared the manuscript with contributions from T.A.F., S.E.H., and M.H.L. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabe1386/DC1
REFERENCES AND NOTES
- 1.W. Harmon, R. Fine, S. Alexander, B. Warady, M. Benfield, S. Goldstein, R. McDonald, K. Martz, D. Stablein, NAPRTCS 2008 Annual Report (2008).
- 2.Wiggins R.-C., The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 71, 1205–1214 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Connaughton D. M., Kennedy C., Shril S., Mann N., Murray S. L., Williams P. A., Conlon E., Nakayama M., van der Ven A. T., Ityel H., Kause F., Kolvenbach C. M., Dai R., Vivante A., Braun D. A., Schneider R., Kitzler T. M., Moloney B., Moran C. P., Smyth J. S., Kennedy A., Benson K., Stapleton C., Denton M., Magee C., O’Seaghdha C. M., Plant W. D., Griffin M. D., Awan A., Sweeney C., Mane S. M., Lifton R. P., Griffin B., Leavey S., Casserly L., de Freitas D. G., Holian J., Dorman A., Doyle B., Lavin P. J., Little M. A., Conlon P. J., Hildebrandt F., Monogenic causes of chronic kidney disease in adults. Kidney Int. 95, 914–928 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadowski C. E., Lovric S., Ashraf S., Pabst W. L., Gee H. Y., Kohl S., Engelmann S., Vega-Warner V., Fang H., Halbritter J., Somers M. J., Tan W., Shril S., Fessi I., Lifton R. P., Bockenhauer D., El-Desoky S., Kari J. A., Zenker M., Kemper M. J., Mueller D., Fathy H. M., Soliman N. A.; SRNS Study Group and Friedhelm Hildebrandt , A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 26, 1279–1289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warejko J. K., Tan W., Daga A., Schapiro D., Lawson J. A., Shril S., Lovric S., Ashraf S., Rao J., Hermle T., Jobst-Schwan T., Widmeier E., Majmundar A. J., Schneider R., Gee H. Y., Schmidt J. M., Vivante A., van der Ven A. T., Ityel H., Chen J., Sadowski C. E., Kohl S., Pabst W. L., Nakayama M., Somers M. J. G., Rodig N. M., Daouk G., Baum M., Stein D. R., Ferguson M. A., Traum A. Z., Soliman N. A., Kari J. A., Desoky S. E., Fathy H., Zenker M., Bakkaloglu S. A., Müller D., Noyan A., Ozaltin F., Cadnapaphornchai M. A., Hashmi S., Hopcian J., Kopp J. B., Benador N., Bockenhauer D., Bogdanovic R., Stajić N., Chernin G., Ettenger R., Fehrenbach H., Kemper M., Munarriz R. L., Podracka L., Büscher R., Serdaroglu E., Tasic V., Mane S., Lifton R. P., Braun D. A., Hildebrandt F., Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 13, 53–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park J., Shrestha R., Qiu C., Kondo A., Huang S., Werth M., Li M., Barasch J., Suszták K., Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lovric S., Ashraf S., Tan W., Hildebrandt F., Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol. Dial. Transplant. 31, 1802–1813 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashraf S., Kudo H., Rao J., Kikuchi A., Widmeier E., Lawson J. A., Tan W., Hermle T., Warejko J. K., Shril S., Airik M., Jobst-Schwan T., Lovric S., Braun D. A., Gee H. Y., Schapiro D., Majmundar A. J., Sadowski C. E., Pabst W. L., Daga A., van der Ven A. T., Schmidt J. M., Low B. C., Gupta A. B., Tripathi B. K., Wong J., Campbell K., Metcalfe K., Schanze D., Niihori T., Kaito H., Nozu K., Tsukaguchi H., Tanaka R., Hamahira K., Kobayashi Y., Takizawa T., Funayama R., Nakayama K., Aoki Y., Kumagai N., Iijima K., Fehrenbach H., Kari J. A., Desoky S. E., Jalalah S., Bogdanovic R., Stajić N., Zappel H., Rakhmetova A., Wassmer S.-R., Jungraithmayr T., Strehlau J., Kumar A. S., Bagga A., Soliman N. A., Mane S. M., Kaufman L., Lowy D. R., Jairajpuri M. A., Lifton R. P., Pei Y., Zenker M., Kure S., Hildebrandt F., Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat. Commun. 9, 1960 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gee H. Y., Saisawat P., Ashraf S., Hurd T. W., Vega-Warner V., Fang H., Beck B. B., Gribouval O., Zhou W., Diaz K. A., Natarajan S., Wiggins R. C., Lovric S., Chernin G., Schoeb D. S., Ovunc B., Frishberg Y., Soliman N. A., Fathy H. M., Goebel H., Hoefele J., Weber L. T., Innis J. W., Faul C., Han Z., Washburn J., Antignac C., Levy S., Otto E. A., Hildebrandt F., ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Invest. 123, 3243–3253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott R. P., Hawley S. P., Ruston J., Du J., Brakebusch C., Jones N., Pawson T., Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J. Am. Soc. Nephrol. JASN. 23, 1149–1154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibata S., Nagase M., Yoshida S., Kawarazaki W., Kurihara H., Tanaka H., Miyoshi J., Takai Y., Fujita T., Modification of mineralocorticoid receptor function by Rac1 GTPase: Implication in proteinuric kidney disease. Nat. Med. 14, 1370–1376 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Blattner S. M., Hodgin J. B., Nishio M., Wylie S. A., Saha J., Soofi A. A., Vining C., Randolph A., Herbach N., Wanke R., Atkins K. B., Kang H. G., Henger A., Brakebusch C., Holzman L. B., Kretzler M., Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 84, 920–930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Z., Zhang L., Chen Y., Zhang H., Zhang Q., Li R., Ma J., Li Z., Yu C., Lai Y., Lin T., Zhao X., Zhang B., Ye Z., Liu S., Wang W., Liang X., Liao R., Shi W., Cdc42 deficiency induces podocyte apoptosis by inhibiting the Nwasp/stress fibers/YAP pathway. Cell Death Dis. 7, e2142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schell C., Baumhakl L., Salou S., Conzelmann A.-C., Meyer C., Helmstädter M., Wrede C., Grahammer F., Eimer S., Kerjaschki D., Walz G., Snapper S., Huber T. B., N-WASP is required for stabilization of podocyte foot processes. J. Am. Soc. Nephrol. 24, 713–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H., Suleiman H., Kim A. H. J., Miner J. H., Dani A., Shaw A. S., Akilesh S., Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol. Cell. Biol. 33, 4755–4764 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akilesh S., Suleiman H., Yu H., Stander M. C., Lavin P., Gbadegesin R., Antignac C., Pollak M., Kopp J. B., Winn M. P., Shaw A. S., Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Invest. 121, 4127–4137 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao J., Ashraf S., Tan W., van der Ven A. T., Gee H. Y., Braun D. A., Fehér K., George S. P., Esmaeilniakooshkghazi A., Choi W.-I., Jobst-Schwan T., Schneider R., Schmidt J. M., Widmeier E., Warejko J. K., Hermle T., Schapiro D., Lovric S., Shril S., Daga A., Nayir A., Shenoy M., Tse Y., Bald M., Helmchen U., Mir S., Berdeli A., Kari J. A., El Desoky S., Soliman N. A., Bagga A., Mane S., Jairajpuri M. A., Lifton R. P., Khurana S., Martins J. C., Hildebrandt F., Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J. Clin. Invest. 127, 4257–4269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kühn S., Geyer M., Formins as effector proteins of Rho GTPases. Small GTPases. 5, e983876 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohatgi R., Ho H. H., Kirschner M. W., Mechanism of N-Wasp activation by Cdc42 and phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150, 1299–1310 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy D. A., Courtneidge S. A., The “ins” and “outs” of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413–426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridley A. J., Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutartre H., Davoust J., Gorvel J. P., Chavrier P., Cytokinesis arrest and redistribution of actin-cytoskeleton regulatory components in cells expressing the Rho GTPase CDC42Hs. J. Cell Sci. 109, 367–377 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Peng J., Wallar B. J., Flanders A., Swiatek P. J., Alberts A. S., Disruption of the diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr. Biol. 13, 534–545 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Kozma R., Ahmed S., Best A., Lim L., The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 15, 1942–1952 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hale L. J., Howden S. E., Phipson B., Lonsdale A., Er P. X., Ghobrial I., Hosawi S., Wilson S., Lawlor K. T., Khan S., Oshlack A., Quinlan C., Lennon R., Little M. H., 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat. Commun. 9, 5167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanigawa S., Islam M., Sharmin S., Naganuma H., Yoshimura Y., Haque F., Era T., Nakazato H., Nakanishi K., Sakuma T., Yamamoto T., Kurihara H., Taguchi A., Nishinakamura R., Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes. Stem Cell Rep. 11, 727–740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins J. W., Chambon A., Bishard K., Hartung A., Arndt D., Brugnano J., Er P. X., Lawlor K. T., Vanslambrouck J. M., Wilson S., Combes A. N., Howden S. E., Tan K. S., Kumar S. V., Hale L. J., Shepherd B., Pentoney S., Presnell S. C., Chen A. E., Little M. H., Bioprinted pluripotent stem cell–derived kidney organoids provide opportunities for high content screening. bioRxiv , 505396 (2018). [Google Scholar]
- 28.Anastas J. N., Biechele T. L., Robitaille M., Muster J., Allison K. H., Angers S., Moon R. T., A protein complex of SCRIB, NOS1AP and VANGL1 regulates cell polarity and migration, and is associated with breast cancer progression. Oncogene 31, 3696–3708 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrel D., Hernandez K., Kwon M., Mau C., Trivedi M. P., Brzustowicz L. M., Firestein B. L., Nitric oxide synthase 1 adaptor protein, a protein implicated in schizophrenia, controls radial migration of cortical neurons. Biol. Psychiatry 77, 969–978 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richier L., Williton K., Clattenburg L., Colwill K., O’Brien M., Tsang C., Kolar A., Zinck N., Metalnikov P., Trimble W. S., Krueger S. R., Pawson T., Fawcett J. P., NOS1AP associates with scribble and regulates dendritic spine development. J. Neurosci. 30, 4796–4805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernandez K., Swiatkowski P., Patel M. V., Liang C., Dudzinski N. R., Brzustowicz L. M., Firestein B. L., Overexpression of isoforms of nitric oxide synthase 1 adaptor protein, encoded by a risk gene for schizophrenia, alters actin dynamics and synaptic function. Front. Cell. Neurosci. 10, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaffrey S. R., Snowman A. M., Eliasson M. J. L., Cohen N. A., Snyder S. H., CAPON: A protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron 20, 115–124 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Clattenburg L., Wigerius M., Qi J., Rainey J. K., Rourke J. L., Muruganandan S., Sinal C. J., Fawcett J. P., NOS1AP functionally associates with YAP to regulate Hippo signaling. Mol. Cell. Biol. 35, 2265–2277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugiyama K., Sasano T., Kurokawa J., Takahashi K., Okamura T., Kato N., Isobe M., Furukawa T., Oxidative stress induced ventricular arrhythmia and impairment of cardiac function in Nos1ap deleted mice. Int. Heart J. 57, 341–349 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Newton-Cheh C., Eijgelsheim M., Rice K. M., de Bakker P. I. W., Yin X., Estrada K., Bis J. C., Marciante K., Rivadeneira F., Noseworthy P. A., Sotoodehnia N., Smith N. L., Rotter J. I., Kors J. A., Witteman J. C. M., Hofman A., Heckbert S. R., O’Donnell C. J., Uitterlinden A. G., Psaty B. M., Lumley T., Larson M. G., Stricker B. H. C., Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat. Genet. 41, 399–406 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfeufer A., Sanna S., Arking D. E., Müller M., Gateva V., Fuchsberger C., Ehret G. B., Orrú M., Pattaro C., Köttgen A., Perz S., Usala G., Barbalic M., Li M., Pütz B., Scuteri A., Prineas R. J., Sinner M. F., Gieger C., Najjar S. S., Kao W. H. L., Mühleisen T. W., Dei M., Happle C., Möhlenkamp S., Crisponi L., Erbel R., Jöckel K.-H., Naitza S., Steinbeck G., Marroni F., Hicks A. A., Lakatta E., Müller-Myhsok B., Pramstaller P. P., Wichmann H.-E., Schlessinger D., Boerwinkle E., Meitinger T., Uda M., Coresh J., Kääb S., Abecasis G. R., Chakravarti A., Common variants at ten loci modulate the QT interval duration in the QTSCD Study. Nat. Genet. 41, 407–414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otto E. A., Hurd T. W., Airik R., Chaki M., Zhou W., Stoetzel C., Patil S. B., Levy S., Ghosh A. K., Murga-Zamalloa C. A., van Reeuwijk J., Letteboer S. J., Sang L., Giles R. H., Liu Q., Coene K. L. M., Estrada-Cuzcano A., Collin R. W. J., McLaughlin H. M., Held S., Kasanuki J. M., Ramaswami G., Conte J., Lopez I., Washburn J., MacDonald J., Hu J., Yamashita Y., Maher E. R., Guay-Woodford L., Neumann H. P., Obermüller N., Koenekoop R. K., Bergmann C., Bei X., Lewis R. A., Katsanis N., Lopes V., Williams D. S., Lyons R. H., Dang C. V., Brito D. A., Dias M. B., Zhang X., Nürnberg G., Nürnberg P., Pierce E., Jackson P., Antignac C., Saunier S., Roepman R., Dollfus H., Khanna H., Hildebrandt F., Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840–850 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sibley C. R., Blazquez L., Ule J., Lessons from non-canonical splicing. Nat. Rev. Genet. 17, 407–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karaiskos N., Rahmatollahi M., Boltengagen A., Liu H., Hoehne M., Rinschen M., Schermer B., Benzing T., Rajewsky N., Kocks C., Kann M., Müller R.-U., A single-cell transcriptome atlas of the mouse glomerulus. J. Am. Soc. Nephrol. 29, 2060–2068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu H., Malone A. F., Donnelly E. L., Kirita Y., Uchimura K., Ramakrishnan S. M., Gaut J. P., Humphreys B. D., Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J. Am. Soc. Nephrol. 29, 2069–2080 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizutani K., Miki H., He H., Maruta H., Takenawa T., Essential role of neural Wiskott-Aldrich syndrome protein in podosome formation and degradation of extracellular matrix in src-transformed fibroblasts. Cancer Res. 62, 669–674 (2002). [PubMed] [Google Scholar]
- 42.Peterson J. R., Lebensohn A. M., Pelish H. E., Kirschner M. W., Biochemical suppression of small-molecule inhibitors: A strategy to identify inhibitor targets and signaling pathway components. Chem. Biol. 13, 443–452 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizvi S. A., Neidt E. M., Cui J., Feiger Z., Skau C. T., Gardel M. L., Kozmin S. A., Kovar D. R., Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol. 16, 1158–1168 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartzman M., Reginensi A., Wong J. S., Basgen J. M., Meliambro K., Nicholas S. B., D’Agati V., McNeill H., Campbell K. N., Podocyte-specific deletion of Yes-associated protein causes FSGS and progressive renal failure. J. Am. Soc. Nephrol. 27, 216–226 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun H., Schlondorff J. S., Brown E. J., Higgs H. N., Pollak M. R., Rho activation of mDia formins is modulated by an interaction with inverted formin 2 (INF2). Proc. Natl. Acad. Sci. 108, 2933–2938 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Combes A. N., Zappia L., Er P. X., Oshlack A., Little M. H., Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med. 11, 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phipson B., Er P. X., Combes A. N., Forbes T. A., Howden S. E., Zappia L., Yen H.-J., Lawlor K. T., Hale L. J., Sun J., Wolvetang E., Takasato M., Oshlack A., Little M. H., Evaluation of variability in human kidney organoids. Nat. Methods 16, 79–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long D. A., Kolatsi-Joannou M., Price K. L., Dessapt-Baradez C., Huang J. L., Papakrivopoulou E., Hubank M., Korstanje R., Gnudi L., Woolf A. S., Albuminuria is associated with too few glomeruli and too much testosterone. Kidney Int. 83, 1118–1129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen A., Sheu L., Ho Y., Lin Y., Chou W., Wang J., Lee W., Administration of dexamethasone induces proteinuria of glomerular origin in mice. Am. J. Kidney Dis. 31, 443–452 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Manser E., Leung T., Salihuddin H., Zhao Z., Lim L., A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 367, 40–46 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Nobes C. D., Hall A., Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Randles M. J., Woolf A. S., Huang J. L., Byron A., Humphries J. D., Price K. L., Kolatsi-Joannou M., Collinson S., Denny T., Knight D., Mironov A., Starborg T., Korstanje R., Humphries M. J., Long D. A., Lennon R., Genetic background is a key determinant of glomerular extracellular matrix composition and organization. J. Am. Soc. Nephrol. 26, 3021–3034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uchio-Yamada K., Monobe Y., Akagi K., Yamamoto Y., Ogura A., Manabe N., Tensin2-deficient mice on FVB/N background develop severe glomerular disease. J. Vet. Med. Sci. 78, 811–818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Braun D. A., Lovric S., Schapiro D., Schneider R., Marquez J., Asif M., Hussain M. S., Daga A., Widmeier E., Rao J., Ashraf S., Tan W., Lusk C. P., Kolb A., Jobst-Schwan T., Schmidt J. M., Hoogstraten C. A., Eddy K., Kitzler T. M., Shril S., Moawia A., Schrage K., Khayyat A. I. A., Lawson J. A., Gee H. Y., Warejko J. K., Hermle T., Majmundar A. J., Hugo H., Budde B., Motameny S., Altmüller J., Noegel A. A., Fathy H. M., Gale D. P., Waseem S. S., Khan A., Kerecuk L., Hashmi S., Mohebbi N., Ettenger R., Serdaroğlu E., Alhasan K. A., Hashem M., Goncalves S., Ariceta G., Ubetagoyena M., Antonin W., Baig S. M., Alkuraya F. S., Shen Q., Xu H., Antignac C., Lifton R. P., Mane S., Nürnberg P., Khokha M. K., Hildebrandt F., Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Invest. 128, 4313–4328 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armougom F., Moretti S., Poirot O., Audic S., Dumas P., Schaeli B., Keduas V., Notredame C., Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 34, W604–W608 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Sullivan O., Suhre K., Abergel C., Higgins D. G., Notredame C., 3DCoffee: Combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol. 340, 385–395 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Ochoa-Montaño B., Pandurangan A. P., Blundell T. L., Ascher D. B., SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 45, W229–W235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Isogai T., van der Kammen R., Innocenti M., SMIFH2 has effects on Formins and p53 that perturb the cell cytoskeleton. Sci. Rep. 5, 9802 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howden S. E., Thomson J. A., Little M. H., Simultaneous reprogramming and gene editing of human fibroblasts. Nat. Protoc. 13, 875–898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howden S. E., McColl B., Glaser A., Vadolas J., Petrou S., Little M. H., Elefanty A. G., Stanley E. G., A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Rep. 7, 508–517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen G., Gulbranson D. R., Hou Z., Bolin J. M., Ruotti V., Probasco M. D., Smuga-Otto K., Howden S. E., Diol N. R., Propson N. E., Wagner R., Lee G. O., Antosiewicz-Bourget J., Teng J. M. C., Thomson J. A., Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takasato M., Er P. X., Chiu H. S., Little M. H., Generation of kidney organoids from human pluripotent stem cells. Nat. Protoc. 11, 1681–1692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takasato M., Er P. X., Chiu H. S., Maier B., Baillie G. J., Ferguson C., Parton R. G., Wolvetang E. J., Roost M. S., de Sousa Lopes S. M. C., Little M. H., Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/1/eabe1386/DC1