

SUMMARY
Dermal adipocyte lineage cells are highly plastic and can undergo reversible differentiation and dedifferentiation in response to various stimuli. Using single-cell RNA sequencing of developing or wounded mouse skin, we classify dermal fibroblasts (dFBs) into distinct non-adipogenic and adipogenic cell states. Cell differentiation trajectory analyses identify IL-1-NF-κB and WNT-β-catenin as top signaling pathways that positively and negatively associate with adipogenesis, respectively. Upon wounding, activation of adipocyte progenitors and wound-induced adipogenesis are mediated in part by neutrophils through the IL-1R-NF-κB-CREB signaling axis. In contrast, WNT activation, by WNT ligand and/or ablation of Gsk3, inhibits the adipogenic potential of dFBs but promotes lipolysis and dedifferentiation of mature adipocytes, contributing to myofibroblast formation. Finally, sustained WNT activation and inhibition of adipogenesis is seen in human keloids. These data reveal molecular mechanisms underlying the plasticity of dermal adipocyte lineage cells, defining potential therapeutic targets for defective wound healing and scar formation.
Graphical Abstract
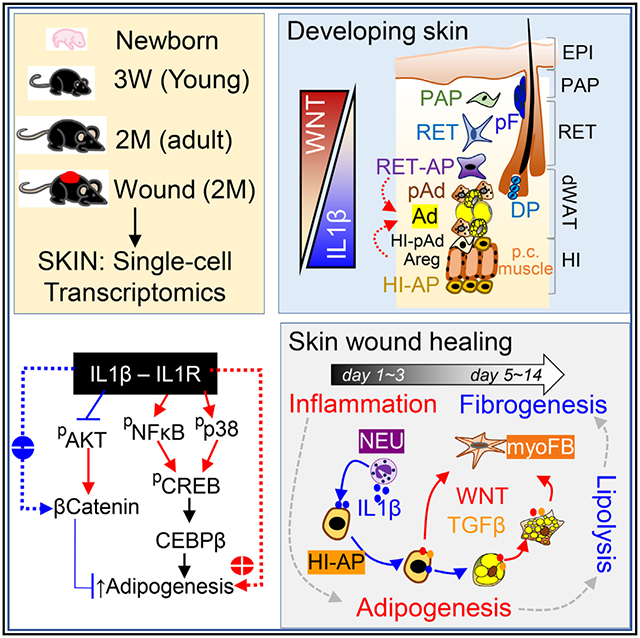
In brief
Dermal adipocyte lineage cells are highly plastic cells that expand or retract in response to stimuli. Zhang and colleagues define the heterogeneity of these cells in developing or wounded mouse skin and demonstrate that dermal adipogenesis and lipolysis are regulated by an antagonistic interplay between the IL-1-pCREB and the WNT-β catenin pathways.
INTRODUCTION
The skin, comprised of layers that include the epidermis, dermis, and dermal white adipose tissue (dWAT), is the largest organ of the human body and a critical barrier against external insults. Emerging studies show that dWAT has a high degree of plasticity and can undergo drastic expansion and/or regression during skin development, during normal hair cycling, and in response to external stimuli, such as wounding, inflammation, and/or bacterial infection.1–3 However, the cellular and molecular mechanisms underlying dWAT remodeling are poorly understood.
Dermal fibroblasts (dFBs), the most prevalent cell type in the dermis, are highly heterogeneous. In neonatal mouse skin, dFBs are committed to forming the upper dermal papillary (DPP4+LY6A−), follicle-associated dermal papilla (DP), lower dermal reticular (DPP4−DLK1+LY6A−), and hypodermal (DLK1−/+LY6A+) layers.4–6 Adipocyte progenitors (APs) and/or preadipocytes (pAds) are present in both reticular and hypodermal layers, and these adipocyte-lineage cells are poised to differentiate into adipocytes, forming the dWAT layer.4 However, the origin, cellular complexity/heterogeneity, and molecular mechanism of dermal adipocyte differentiation from their heterogeneous progenitors are only partially understood.
Skin wound healing is a highly orchestrated process; it includes an early hemostatic and inflammatory phase, then a proliferative phase, and finally a remodeling phase that can last weeks to years.7 During the inflammatory phase, myeloid immune cells are recruited, and fibroblasts migrate from the wound periphery, forming granulation tissue (GT), a temporary scaffold holding the wound.8 During the wound proliferative phase, myofibroblasts are transdifferentiated from fibroblasts to synthesize extracellular matrix (ECM) molecules; however, sustained activation of myofibroblasts leads to the development of fibrotic diseases like keloids.9
Emerging roles of adipocyte-lineage cells in skin wound healing have been reported. Following inflammation, APs proliferate, and new lipid-laden adipocytes are generated in wound GT, and inhibition of adipogenesis impairs dermal repair.10 Recently, a subset of hypodermal DPP4+LY6A+ fascia fibroblasts with remarkable reparative potential was identified, and genetic ablation or blocking of their migration into wound sites led to chronic wounds. In adipose tissue, DPP4+LY6A+ fascia fibroblasts have been identified as APs that give rise to ICAM1+DLK1+ pAds.11,12 However, whether skin DPP4+LY6A+ dFBs are adipogenic and their cell differentiation trajectory during wound repair remain largely unknown. Furthermore, the molecular cues that trigger wound-related adipogenesis responses remain undefined.
Here, we obtained skin samples from developing, homeostatic, and wounded mice and applied single-cell RNA sequencing (scRNA-seq) followed by immunostaining (immunohistochemistry [IHC]) and cell sorting to define dFB populations. scRNA-seq predicted the cellular trajectories through which dermal adipocytes differentiated from their heterogeneous APs. The behavior of these cells was examined during wound repair. Finally human keloid samples were analyzed to determine the human relevance of our findings. Our study provides insight into the molecular and cellular mechanisms underlying dermal adipogenesis and/or lipolysis during skin development and wound regeneration.
RESULTS
Characterization of heterogeneous dermal adipocyte-lineage cells in developing neonatal skin
We performed scRNA-seq of cells isolated from neonatal mouse skin, in which dFBs are actively developing from their precursors,4 allowing us to capture a continuum of cell states spanning differentiation and specification. Pdgfra+ (a pan-fibroblast marker4,13) dFBs, Krt10/14+ keratinocytes, Ptprc/Cd45+ immune cells, and Rgs5+ pericytes were identified as the major cell types present in neonatal mouse skin (Figures 1A and S1A).
Figure 1. Characterization of heterogeneous dermal adipocyte lineage cells in developing neonatal skin.
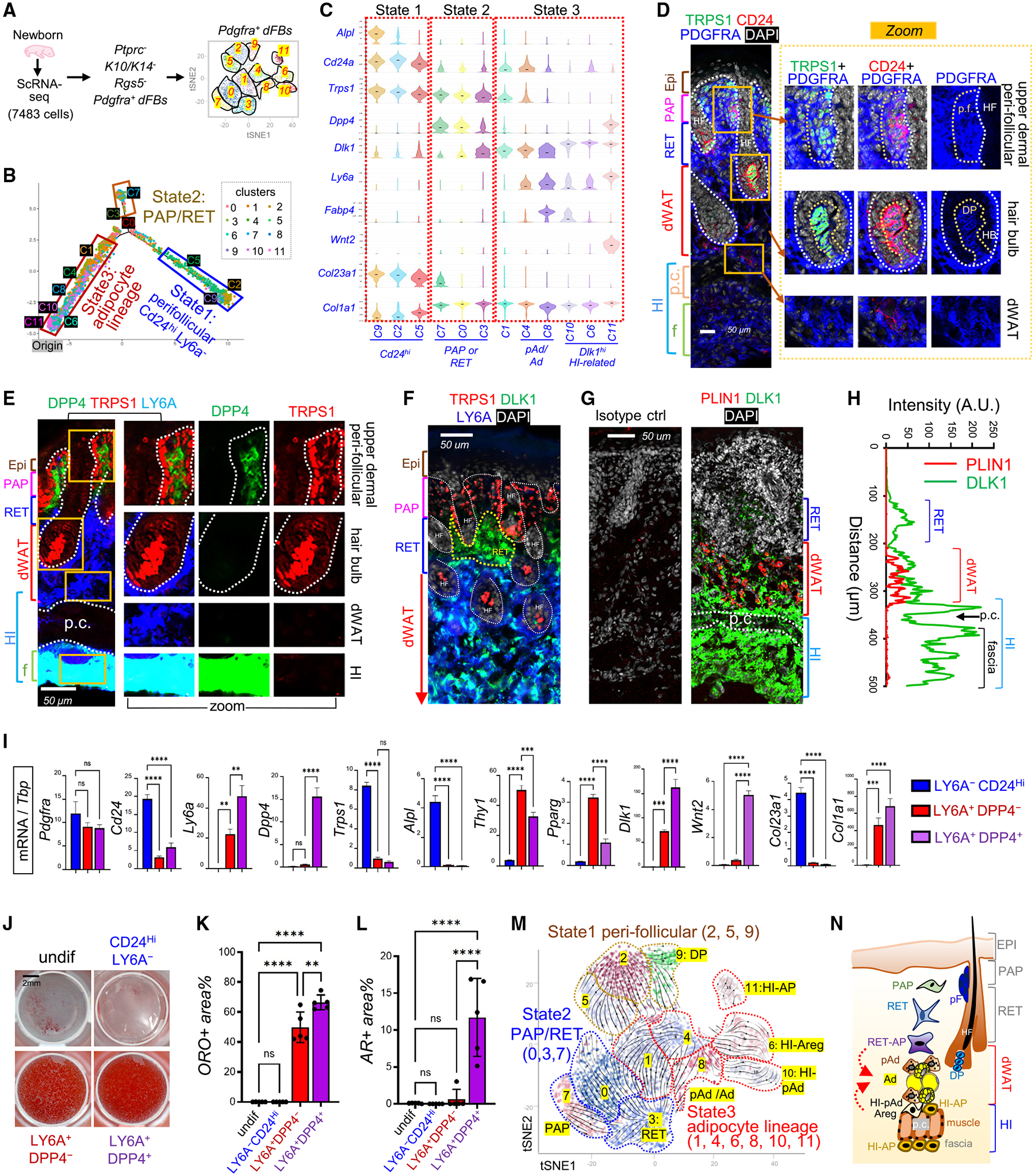
(A) scRNA-seq analysis of cells isolated from neonatal skin. Pdgfra+ dFBs were reclustered into 12 clusters.
(B) Neonatal dFBs were grouped into three cell states along differentiation trajectories.
(C) Violin plots of indicated genes.
(D–G) Neonatal mouse skin sections were immunostained with antibodies against various dFB markers as indicated, and the nuclei were counterstained with DAPI in either blue or white. Scale bars, 50 μm. Abbreviations: p.c., panniculus carnosus muscle layer; p.f., perifollicular; DP, dermal papilla; HF, hair follicle; HB, hair bulb. Brackets show specific anatomical skin layers as indicated.
(H) Quantified result of (G) showing the fluorescence intensity (arbitrary unit, A.U.) of PLIN1 (red) and DLK1 (green) across skin section from top to bottom.
(I–L) Perifollicular dFBs (CD24hiLY6A−), pAds (DPP4−LY6A+), and HI-APs (DPP4+LY6A+) were sorted and then subjected to qRT-PCR analysis (n = 3/group) of indicated genes (I) or in vitro adipocyte differentiation for adipocytes (stained with oil red O [ORO]) (J). Quantified ORO+ area for each group is shown in (K) (n = 5/group). Sorted cells were also subjected to osteocyte differentiation (stained with alizarin red [AR]; see Figure S1V), and quantified AR+ area is shown in (L) (n = 5/group). Scale bar, 2 mm. All error bars indicate mean ± SEM; **p < 0.01, ***p < 0.01, ****p < 0.0001.
(M) RNA velocity analysis overlaid on tSNE projection of developing dFBs.
(N) A model for the proposed spatial location of various dFB subpopulations in the developing skin. Abbreviations: PAP, papillary dFBs; RET, reticular dFBs; RET-AP, reticular adipocyte progenitors; pAd, preadipocytes; Ad, adipocytes; HI-pAd, hypodermal interstitial pAd; HI-AP, hypodermal interstitial AP.
Selected Pdgfra+ dFBs were reclustered into 12 clusters (C0–C11) (Figures 1A, S1B, and S1C), which were grouped into three distinct developmental cell states (Figures 1B, 1C, and S1D) and defined based on marker gene expression as shown by bubble and/or violin plots (Figures 1C and S1E). Specifically, state 1 cells (C9, C2, C5) expressed high levels of Cd24 and Trps1 but lacked Ly6a (Figures 1C and S1E). IHC analysis of neonatal skin with distinct anatomical layers (Figures S1F–S1G) showed that CD24 and TRPS1 were co-expressed at high levels in PDGFRA+ dFBs located around hair follicles at the upper dermis or in the hair bulb DP structure (Figure 1D), where DPP4, LY6A (Figure 1E), and THY1 (Figure S1H) were not expressed or expressed at low levels. ALPL, another marker enriched in state 1 cells (Figures 1C and S1E), co-expressed specifically with TRPS1 in the DP (Figure S1H). We therefore termed the state 1 cluster cells as perifollicular dFBs.
State 2 cells (C7, C0, C3) expressed markers related to papillary (PAP) and/or reticular (RET) dFBs and lacked Ly6a expression (Figure 1C). Specifically, the C7 cluster expressed established PAP dFB markers (Dpp4, Lrig, Entpd1, and Lrig1) and medium level of Trps1, whereas the C3 cluster expressed genes associated with RET dFBs (Dlk1 and Ptgfr) and a lower level of Trps1 (Figures 1C and S1E). IHC confirmed that the upper dermal PAP cells were TRPS1+DPP4+LY6A−DLK1−, whereas the lower dermal RET cells were TRPS1−DPP4−LY6A−DLK1+ (Figures 1F, S1I, and S1J). Therefore, the C7 and C3 clusters were defined as PAP and RET dFBs, respectively.
State 3 cells (C1, 4, 8, 10, 6, 11) are highly heterogeneous (Figure S1D). These cells expressed marker genes related to adipogenesis but lacked Trps1 (Figures 1C and S1E); thus these clusters were termed adipocyte lineage cells. Specifically, C4 cells (pAd) and C8 cells (pAd/Ad) expressed conventional pAd and/or adipocyte markers but lacked Dpp4. IHC analysis confirmed that the LY6A+DPP4− pAds were detected exclusively in dWAT, where BODIPY+ adipocytes were located (Figures 1E and S1G). Interestingly, we found that C4 pAd cells expressed high levels of Lgr5 (Figures S1K and S1L), a well-established marker for hair follicle stem cells.14 Furthermore, cluster C1 was termed RET-AP, because it lacked Ly6a but expressed a medium level of Dlk1 and high levels of Cebpb (Figures 1C and S1E), a central transcription factor for dermal adipogenesis.15
Among all state 3 clusters, Dlk1 was expressed the highest in clusters C6, C10, and C11 (Figures 1C and S1E). IHC (Figures 1G and 1H) showed that DLK1 was highly expressed within the interstitial space above and below the panniculus carnosus (p.c.) muscle, a space termed “hypodermal interstitium” (HI) in this study. This HI space was also enriched with LY6A+DPP4+ cells (Figure S1M). Furthermore, C10 was termed HI-pAd because it co-expressed pAd/Ad marker genes (Lpl and Icam1); C6 was termed HI-Areg (adipogenesis regulator cells), because it expressed a panel of Areg marker genes, including F3, Fmo2, and Mgp.16,17 Finally, cluster C11 was termed HI-AP, because it co-expressed AP marker genes (Dpp4, Ly6a, Cd34, and Wnt2) (Figures 1C and S1E). IHC validated that WNT2+ PDGFRA+ (Figure S1N) and WNT2+DPP4+LY6A+cells (Figure S1O) were detected primarily in the HI space.
dFBs are a major cellular source of skin ECM.9 Analysis of ECM-related genes revealed that the Cd24hi clusters expressed Col23a1 and Col13a1 but lacked Col1a1 and Col3a1 (Figures 1C and S1E). Interestingly, the HI-AP cluster was enriched with a panel of collagen genes and ECM genes related to elastic network and fibrillogenesis,18,19 such as Mfap5, Fbn1, and Fn1 (Figures 1C and S1E), indicating that HI-AP could be involved in providing structural support to the skin.
Next, we developed fluorescence-activated cell sorting (FACS) strategies, based on our and others’ previously published FACS protocol for dFBs,20–23 to isolate perifollicular dFBs (CD24hiLY6A−), pAds (LY6A+DPP4−), and HI-APs (LY6A+DPP4+) from neonatal dFBs (Figure S1P). qRT-PCR profiling analysis (Figures 1I and S1Q) showed that the sorted CD24hiLY6A− cells highly expressed genes that were enriched in C2/C9 perifollicular clusters, such as Trps1, Alpl, and Col23a1; the sorted LY6A+DPP4− dFBs expressed a panel of pAd/Ad-related genes; and the sorted LY6A+DPP4+ dFBs expressed higher levels of genes that were enriched in the C11 HI-AP cluster, such as Dlk1, Wnt2, Smpd3, and Mfap5. Using in vitro adipocyte and osteocyte differentiation assays, we found that the CD24+LY6A− cells were non-adipogenic, and both the pAds and the HI-APs were highly adipogenic (Figures 1J and 1K), whereas only HI-APs were osteogenic (Figures 1L and S1R), suggesting that DPP4+LY6A+ cells are multipotent progenitors and DPP4−LY6A+ cells are committed pAds.
In line with these functional results, trajectory analysis predicted that C11 (HI-AP) was the origin of the differentiation trajectory of adipocyte lineage cells (Figures 1B and S1S). In addition, RNA velocity analysis (Figure 1M) predicted that both HI-pAd/Areg and RET-AP dFBs actively differentiated into pAd/Ad, and C0, C1, C4, and C9 further converge into C2− perifollicular dFBs. These trajectories need to be validated by lineage tracing studies in the future.
Together, using scRNA-seq, we classified the neonatal developing dFBs into three distinct states (Table S1) and defined the heterogeneous adipocyte-lineage cells located in the RET dermis, dWAT, and HI (Figure 1N).
The antagonistic interplay between WNT-β-catenin and IL-1-immune signaling pathways in regulating dermal adipogenesis
We next sought to identify the key molecular pathways that regulate adipogenesis in the developing skin. Gene ontology (GO) analyses identified the IL-1 and WNT pathways as among the top activated or silenced pathways, respectively, during the conversion of various dFB subsets to pAd/Ad (Figures 2A and S2A). Gene expression plots (Figures 2B and S2B) showed that WNT-related genes were specifically enriched in the perifollicular and/or PAP dFB clusters, whereas a panel of immune- and inflammation-related genes was selectively enriched in the pAd/Ad-related clusters. In addition, transcription factor (TF) analysis identified Cebpb, Pparg, and several inflammatory TFs (Nfkb1 and Junb) as top TFs activated in pAd/Ad-related clusters (Figure S2C).
Figure 2. Interplay between the WNT-β-catenin and IL-1-immune signaling pathways in regulating dermal adipogenesis.
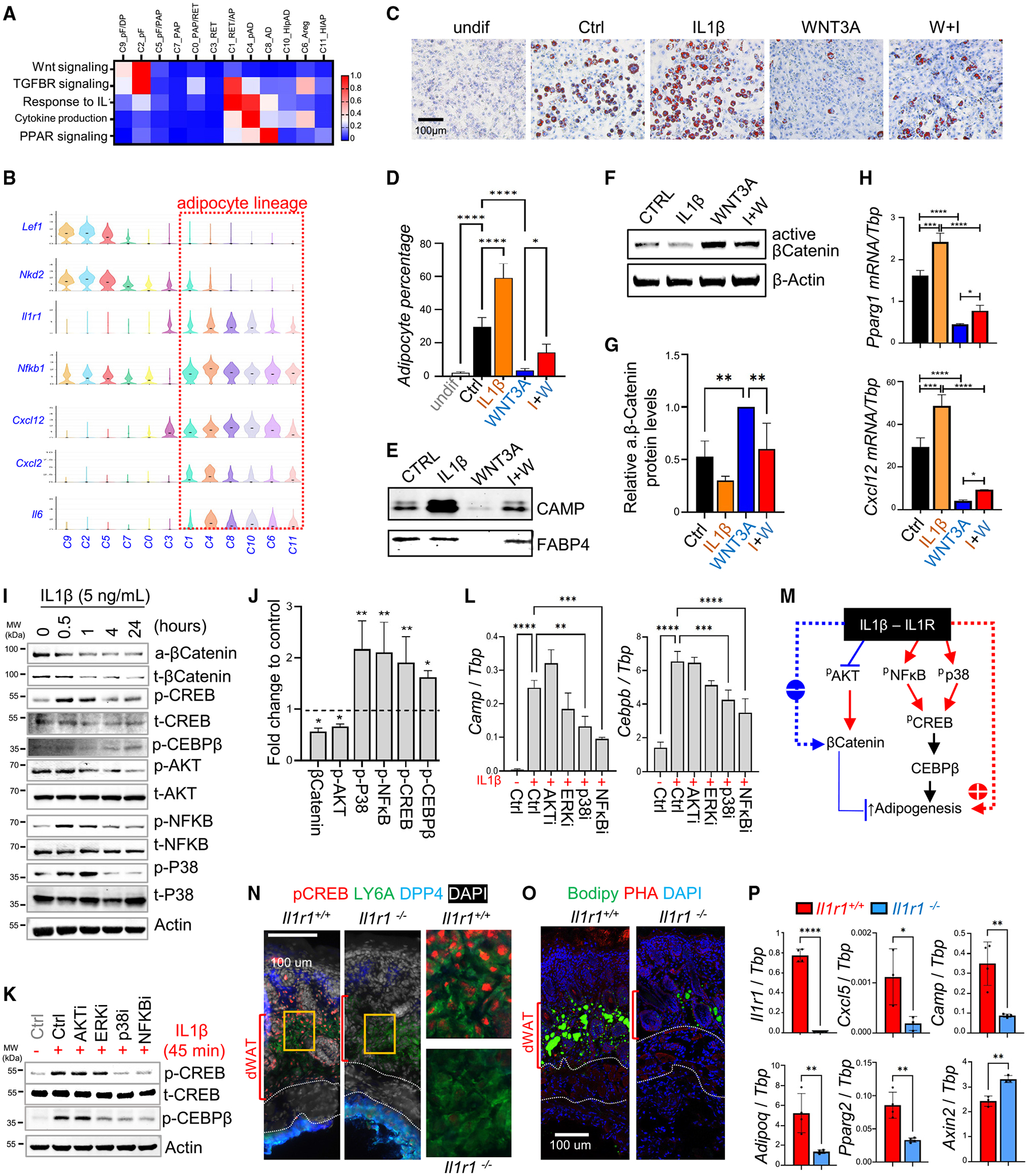
(A) Signaling enrichment analysis of neonatal dFB clusters shown by heatmap. Color scale of red to blue indicates enrichment score.
(B) Violin chart showing the expression of indicated genes.
(C–E) Neonatal dFBs were pretreated with WNT3A and/or IL-1β and then subjected to adipocyte differentiation, and differentiated cells were stained with ORO (C).The percentages of ORO+ adipocytes was quantified (D) (n = 6/group). Western blotting analysis of CAMP and FABP4 in adipocyte conditioned medium (E).
(F–H) Treated undifferentiated cells were subjected to western blotting analysis using the indicated antibodies (F; quantified results in G, n = 3/group) or qRT-PCR analysis (n = 3/group) of indicated genes (H).
(I) Neonatal dFBs were treated with IL-1β over a time course of 24 h and cell extracts were subjected to western blotting using indicated antibodies.
(J) Bar graphs showing fold changes of indicated proteins from (I) at 1 h of IL-1β treatment compared with unstimulated controls (normalized to actin or total proteins, average of n = 3/group).
(K and L) Neonatal dFBs were pretreated with specific inhibitors against the indicated signaling molecules before being stimulated with IL-1β, and cells were subjected to western blotting analysis (K) or qRT-PCR analysis (L) for the expression of indicated genes (n = 3/group).
(M) Proposed model for the antagonistic interplay between IL-1β-IL-1R and WNT-β-catenin signaling in regulating adipogenesis.
(N–P) Neonatal skin sections from WT or Il1r1 knockout mice were subjected to immunostaining analysis using indicated antibodies or dyes (N, O) or qRT-PCR analysis of indicated genes (P) (n = 3–4/group). Scale bars, 100 μm. All error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the direct effect of WNT and IL-1 on adipocyte differentiation, neonatal dFBs were treated with WNT3A, lithium, and/or IL-1β. Lithium, a GSK3β inhibitor, is commonly used to achieve broad-spectrum activation of the WNT-β-catenin pathway.24 RNA-seq results revealed that WNT3A and lithium treatment robustly induced the expression of genes associated with perifollicular dFBs (Cd24, Alpl), whereas IL-1β treatment induced the expression of a panel of pro-inflammatory genes (Cxcl12, Il1r1) and several pAd/Ad marker genes (Pparg2, Fabp4) (Figure S2D). Venn diagram analysis showed that genes upregulated by IL-1β shared a high degree of similarity with genes downregulated by WNT3A + lithium (Figure S2E). Functionally, WNT3A potently suppressed the adipogenic potential of neonatal dFBs (Figures 2C and 2D) and secretion of CAMP and FABP4, which can be partially restored by adding IL-1β (Figure 2E). Furthermore, WNT3A-mediated activation of β-catenin (Figures 2F and 2G), suppression of adipocyte- or immune-related genes, and induction of Axin2, Acta2, and Col7a1 were partially reversed by IL-1β (Figures 2H and S2F–S2I).
Time-course phosphoblotting analyses (Figures 2I and 2J) revealed that IL-1β treatment increased NF-κB, p38 and ERK phosphorylation levels but suppressed AKT phosphorylation, which is required for β-catenin stabilization.25 IL-1β treatment led to a continuous loss of protein levels of β-catenin (active or total) (Figures 2I, 2J, and S2J). In contrast, IL-1β treatment rapidly induced phosphorylation of CREB, a critical TF involved in CEBPβ activation and promoting adipogenesis,26 and IL-1β-mediated CEBPβ phosphorylation occurred in a delayed manner (Figures 2I and 2J).
We found that AKT inhibition by wortmannin promoted loss of β-catenin expression, whereas p38 inhibition by SB202190 or NF-κB inhibition by Bay117028 led to a decrease in the phosphorylation levels of both CREB and CEBPβ (Figures 2K and S2K–S2M). In line with these results, the induction of adipocyte- or immune-related genes by IL-1β was largely blocked by p38 or NF-κB inhibitors (Figure 2L). These results indicate that IL-1β promotes β-catenin degradation by inhibiting AKT and promotes activation of pro-adipogenic molecules by stimulating p38 and NF-κB (Figure 2M).
We next analyzed skin biopsies from neonatal Il1r1−/− and Il1r1+/+ littermates to determine the role of IL-1 signaling in adipogenesis. We found that Il1r1 deficiency led to a blockage of CREB phosphorylation in the neonatal mouse skin (Figure 2N). As the result, formation of BODIPY+ adipocytes (Figure 2O) and the expression of adipocyte-related genes and the inflammatory cytokine Cxcl5 were suppressed, whereas expression of WNT-related Axin2 was enhanced upon Il1r1 deletion (Figure 2P). Taken together, our results indicate that activation of the IL-1R-CREB signaling axis may play a role in promoting dermal adipogenesis during skin development.
Tracking dermal adipogenesis and fibrogenesis during skin maturation
To track dermal adipogenesis during skin maturation and aging, dorsal skin samples were collected from male mouse skin throughout the postnatal lifespan for analysis. Lipid staining and bulk RNA-seq of skin samples (Figures 3A and S3A) showed that dermal adipocytes progressed from an immature state at birth (characterized by small lipid droplets and high expression levels of Camp and Mest), to a mature state at 3 weeks (large lipid droplets), following which dermal cells and adipocytes were gradually lost during skin maturation and aging from 2 months to 1–2 years. In contrast, dermal collagen content and the expression levels of several ECM molecules increased up to 2 months of age and then progressively decreased during aging (Figures 3A and S3A).
Figure 3. Tracing dermal adipogenesis and fibrogenesis during skin maturation.
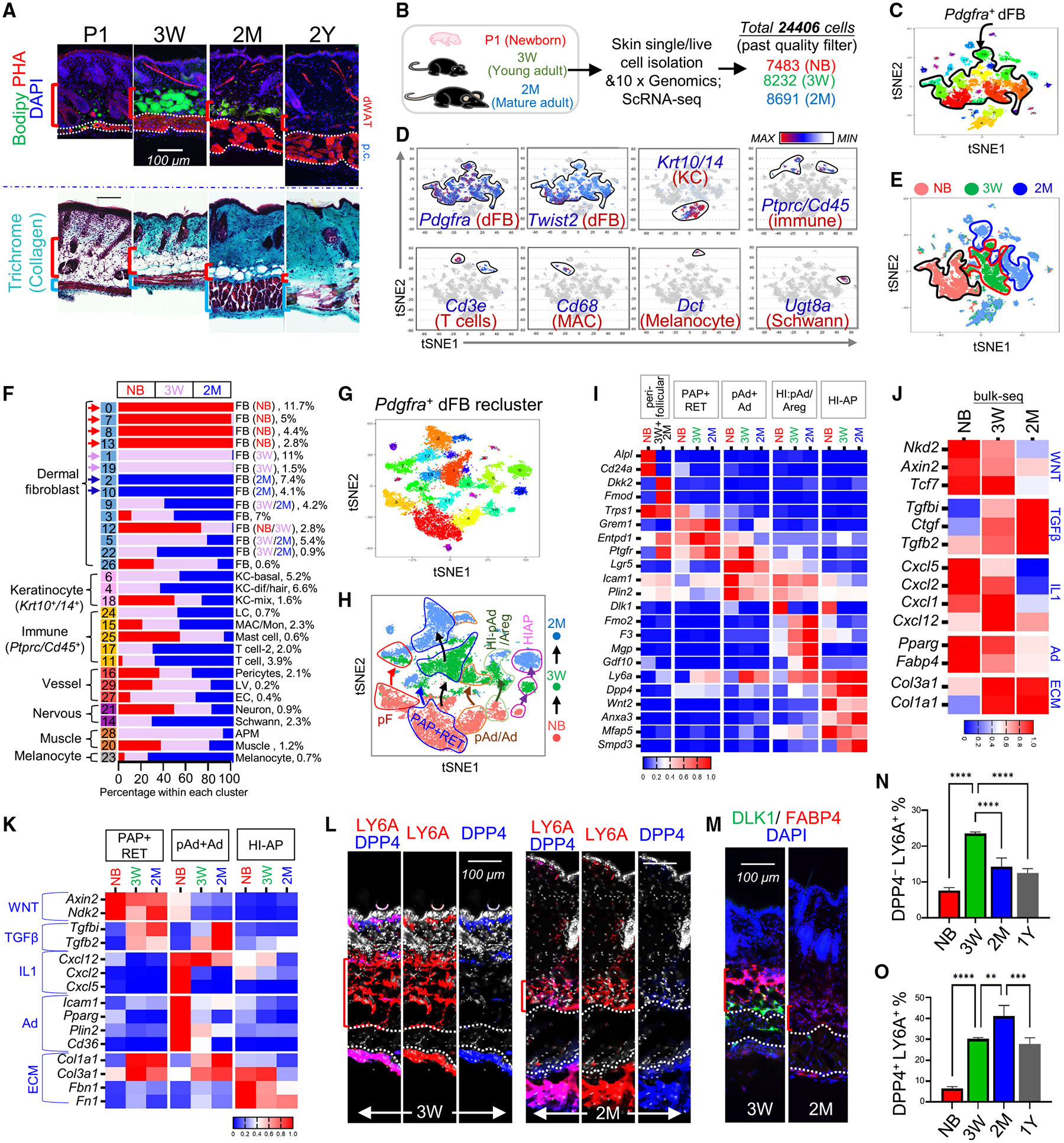
(A) BODIPY (green dye for lipid) and phalloidin (red dye for actin fiber) staining (top) and Gomori trichrome staining (bottom) of skin sections from mice at indicated ages. Scale bar, 100 μm
(B) Schematic of scRNA-seq of dorsal skin cells isolated from NB, 3-week, and 2-month mice.
(C) tSNE plots showing cell distribution by clusters indicated with unique color.
(D) tSNE plots showing the expression of indicated marker genes.
(E) tSNE plots showing cell distribution by age of NB (red), 3-week (green), and 2-month (blue) mice.
(F) Bar chart showing age-related changes in the percentage of various cell types.
(G–I) Pdgfra+ dFBs were reclustered into tSNE plots by cluster (G) or by age (H). Age-related changes in the expression of the indicated genes are shown by heatmap (I).
(J) Heatmap showing the mRNA expression (based on bulk RNA-seq) of the listed genes in mouse skin at the indicated ages.
(K) Heatmap showing age-dependent kinetics of the expression of the listed genes in the indicated dFB clusters.
(L and M) Skin sections from mice at 3 weeks or 2 months of age were immunostained with antibodies against the indicated dFB/AD markers. Scale bars, 100 μm.
(N and O) Quantified bar graphs of FACS plots in Figure S3G showing the percentage of DPP4−LY6A+ pAds (N) or DPP4+Ly6A+ HI-APs (O) in CD31−CD45−PDGFRA+ dFBs (n = 3/group). All error bars indicate mean ± SEM; **p < 0.01, ***p < 0.001, ****p < 0.0001.
To determine the cellular and molecular changes in dermal adipocyte-lineage cells during skin maturation, scRNA-seq was performed on skin cells isolated from mice at neonatal stage (P1) and 3 weeks and 2 months of age (Figure 3B). Clustering analysis identified 30 clusters, which were classified into nine cell types (Figures 3C–3F and S3B–S3D). Interestingly, we found that among all cell types, Pdgfra+ dFBs contained several age-unique clusters (Figure 3F).
To further inspect dFB heterogeneity, Pdgfra+ dFBs were reclustered (Figure 3G), and these dFB clusters from the three age groups were clearly separated on the tSNE (t-distributed stochastic neighbor embedding) plots in an age-dependent manner (Figure 3H). Based on the established neonatal dFB markers (Figure 1), we identified the corresponding adult dFB clusters (Figures 3H, 3I, and S3E). In contrast to neonatal DP cells that expressed Alpl, Sox2, and Lef1, adult DP cells lost activated WNT signals (Lef1, Nkd2) and expressed Crapb1 and the WNT antagonist Dkk2, in line with previous studies27,28 (Figures 3I and S3E). Similar to neonatal dFBs, adult PAP dFBs were enriched in Grem1 and Entpd1 (Figures 3I and S3E), and ENTPD1 (also known as CD39) has been shown to mark human adult skin PAP dFBs.29 In adult skin, Trps1 was also an exclusive marker for all Ly6a+ adipocyte-lineage cells (Figures 3I and S3E). Interestingly, we found that HI-pAd/Areg progressively lost Dlk1 but gained Areg signature gene signatures (F3, Fmo2, Mgp, and Gdf10) during maturation (Figures 3I and S3E).
Next, we examined how genes related to the IL-1, WNT, and TGF-β pathways were altered in key dFB clusters during skin maturation. First, analysis of bulk skin RNA-seq showed that the expression of WNT- and IL-1-related genes was progressively lost, whereas TGF-β-related genes were induced postnatally during skin maturation (Figure 3J). In line with these results, analysis of key dFB clusters showed that WNT- or IL-1-related genes were inhibited in pAd/Ad and HI-AP, whereas TGF-β-related genes were notably induced in PAP/RET and pAd/Ad but not HI-AP clusters age dependently (Figure 3K). As a result, ECM genes were induced and adipogenesis-related genes were inhibited in PAP/RET or pAd/Ad clusters age dependently, whereas HI-APs started to lose ECM gene expression at 2 months of age (Figure 3K). These age-dependent changes in WNT, IL-1, and TGF-β signaling activities in adipocyte-lineage cells may contribute to the adipogenic-to-fibrogenic switch of these cells during skin maturation.
IHC confirmed that, similar to neonatal skin, the fascia layer of adult skin was enriched with LY6AhiDPP4hi HI-APs, and LY6A+DPP4− pAds were detected exclusively in the dWAT layer of adult skin (Figure 3L). In addition, DLK1+ cells were detected only within the interstitial space around the p.c. muscle layer in the 3-week skin and were largely lost by 2 months (Figure 3M), which is consistent with the Dlk1 expression pattern shown by scRNA-seq (Figures 3I and S3E). scRNA-seq showed that HI-AP% increased during skin maturation (Figure S3F). FACS analysis of PDGFRA+ dFBs validated that, while pAd% increased postnatally for up to 3 weeks, HI-AP% continued to increase for up to 2 months and then declined in 1-year-old skin (Figures S3G, 3N, and 3O). In contrast, CD24+LY6A− dFBs, identified as the neonatal perifollicular dFBs, were rapidly lost postnatally (Figures S3H and S3I), which is consistent with the Cd24 scRNA-seq plots (Figures 3I and S3E). Together, these results showed that skin maturation is associated with age-dependent inactivation of WNT and IL-1 signaling, depletion of adipogenic pAd, and accumulation of inactive HI-APs.
Tracking dermal adipogenesis and fibrogenesis during skin wound healing
Next, we studied how dermal adipogenesis may be reinitiated upon wounding in adult homeostatic skin. BODIPY staining showed that numerous adipocytes were transiently detected in the wound center GT, peaking at wound day (w.d.) 3, and lipolysis was apparent at w.d. 5–7, indicated by the presence of dispersed small lipid droplets, and by w.d. 14, adipocytes were cleared from the wound bed (Figure 4A). To determine whether wound-associated adipocytes were generated de novo from adipogenesis, we induced CRE activity in Adipoq-CreERT2;mTmG mice by tamoxifen (TAM) application prior to wounding to label preexisting adipocytes with GFP (Figure S4A). Results showed that more than one-third of wound-associated COLIV+FABP4+ adipocytes were GFP− (Figures S4B and S4C), demonstrating that newly differentiated adipocytes partially contribute to wound-induced adipogenesis.
Figure 4. Tracing dermal adipogenesis and fibrogenesis during skin wound healing.
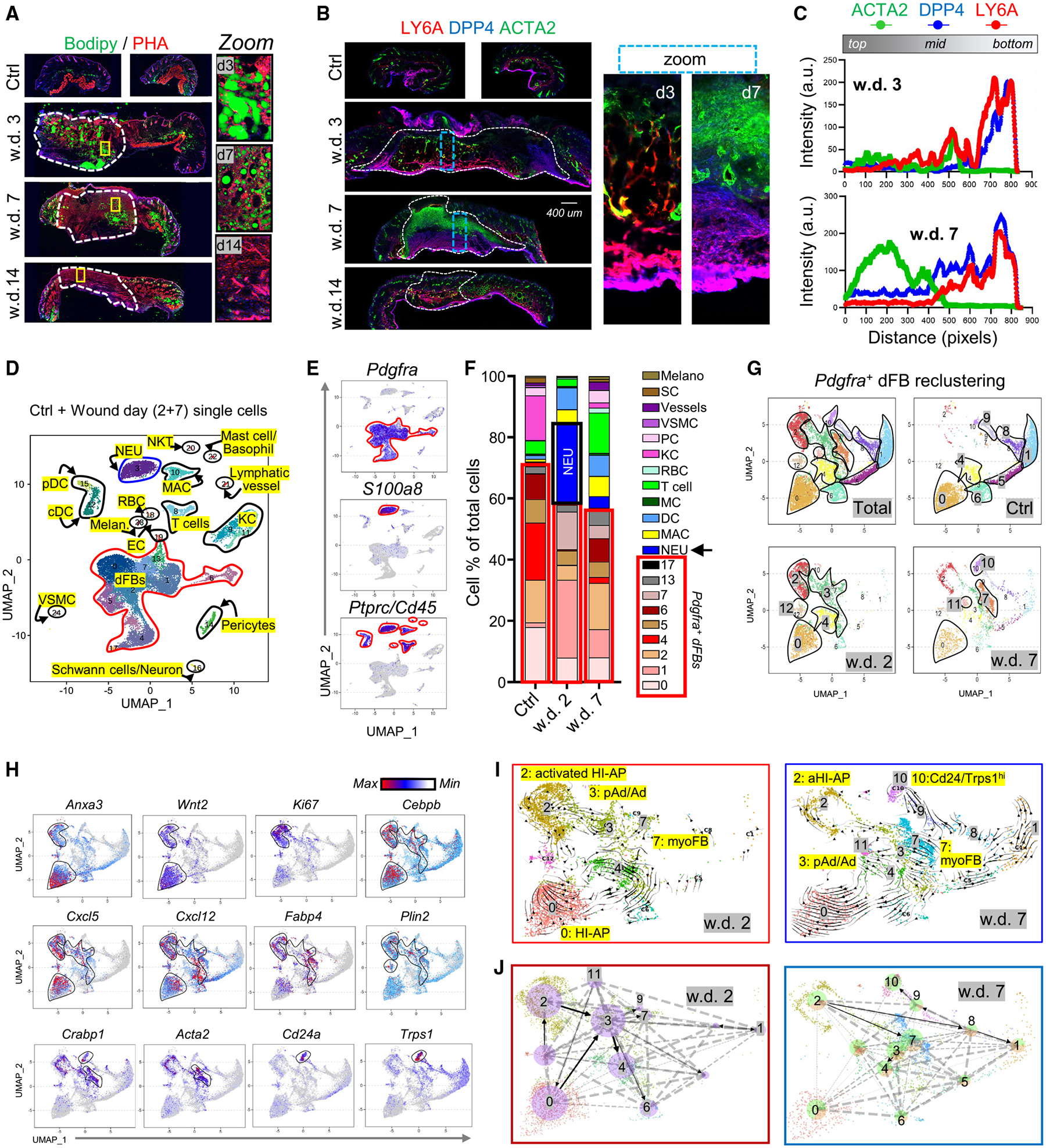
(A) Full-thickness mouse wounds were collected at indicated wound days (w.d.) for PHA (red) and BODIPY (green) staining. The dotted lines mark wound central granular tissues. Scale bar, 400 μm.
(B) LY6A (red), DPP4 (blue), and ACTA2 (green) immunostaining. Dotted lines mark wound GT. Scale bar, 400 μm.
(C) Quantified intensity profiles for (B), showing signals from all three fluorescence channels from wound top to bottom (representative of n = 3/group).
(D–F) Wound day 2 and 7 and unwounded control skin samples were subjected to scRNA-seq analysis. UMAP (uniform manifold approximation and projection) plot (D) showing cell distribution by clusters. UMAP plots (E) showing the expression of indicated marker genes for each cluster. Stacked bar graphs (F) showing the percentage of each cell cluster.
(G and H) Pdgfra+ dFBs were reclustered. UMAP plots (G) showing cell cluster distribution in different samples. UMAP plots (H) showing the expression of indicated marker genes.
(I and J) RNA velocity analyses (I) and cell rank analyses (J) were mapped onto the UMAP plot of dFB reclusters of w.d. 2 or 7 samples as indicated.
During the wound-proliferative phase, fibroblasts are activated and differentiate into ACTA2+ myofibroblasts.9 Here, IHC showed that at w.d. 3, DPP4+LY6A+ HI-APs were detected at the wound periphery and bottom, whereas DPP4−LY6A+ pAds/Ads were mainly detected in the wound mid-region (Figures 4B and 4C). By w.d. 7, pAds/Ads were largely lost, and ACTA2+DPP4−LY6A− myofibroblasts were abundantly detected in the upper wound bed (Figures 4B and 4C). By day 14, myofibroblasts were largely lost, and DPP4−LY6A+ pAds were regenerated above the HI-AP fascia layer (Figure 4B).
scRNA-seq analysis of wound skin cells was next performed to determine the cell differentiation trajectory (Figures 4D and 4E). A total of 15 different cell types, including dFBs, neutrophils, macrophages, T cells, and keratinocytes (KCs), were identified (Figures 4D, 4E, S4D, and S4E). We found that neutrophils (~30% of all cells) and dFB clusters (~50% of all cells) were the major cell types that infiltrated wound GT on w.d. 2, and a subsequent decrease in neutrophils at w.d. 7 was accompanied by an increase in dFB heterogeneity and regeneration of other cell types, such as KCs and vessel cells (Figure 4F).
To define heterogeneous dFBs, Pdgfra+ dFBs were reclustered into 13 subclusters (r0–r12; Figures 4G–4H and S4F–S4G), which were defined based on the expression of dFB marker genes identified in Figure 1 and myofibroblast markers (Acta2, Tagln). We found that adipocyte-lineage cells, including HI-AP (r0, r2, r12) and pAd/Ad (r3) cell clusters, represented >85% of dFBs in w.d. 2 GT, whereas by w.d. 7, pAd/Ad cells were largely lost, and Acta2+ myofibroblasts and Cd24hiAlpl+ dFBs represented ~1/3 of the w.d.7 dFBs (Figure S4H). Gene expression plots (Figures 4H and S4F–S4J) showed that the r2 HI-AP cluster, which was transiently generated in w.d. 2 GTs, expressed high levels of proliferation marker genes (Ki67 and Pcna) and IL-1-inducible chemokine genes (Cxcl2/5/12), suggesting that these cells were highly proliferative and inflammatory. RNA velocity analysis (Figure 4I) and cell-rank analysis (for calculation of cell-cell transition probabilities30) (Figure 4J) predicted that at w.d. 2, the activated r2 HI-APs were poised to differentiate toward the r3 pAds/Ads, which were in turn poised to differentiate into other dFB clusters, including the r7 myofibroblasts. Together, these results suggest that activated HI-APs may contribute to pAd/Ad generation during the wound-inflammatory phase, and pAd/Ad-to-myofibroblast transdifferentiation may occur during the subsequent wound-proliferative phase. Future studies using lineage tracing mouse models specific for HI-APs and pAd/Ad are needed to confirm these cell differentiation trajectories.
Wound-induced adipogenesis is regulated by neutrophils and the IL-1 signaling axis
FACS analysis confirmed that the percentage of LY6A+DPP4+ HI-AP peaked at approximately w.d. 2, and loss of HI-APs was accompanied by gain of ACTA2+LY6A+ myofibroblasts at w.d. 7 in wound GTs (Figures 5A–5C and S5A). The transition from HI-AP (Dpp4+) to pAd/Ad (Cebpbhi Fabp4+) and myofibroblasts (Acta2+) during wounding can also be visualized at the single-cell level (Figure S5B).
Figure 5. Wound-induced adipogenesis is regulated by neutrophils and the IL-1 signaling axis.
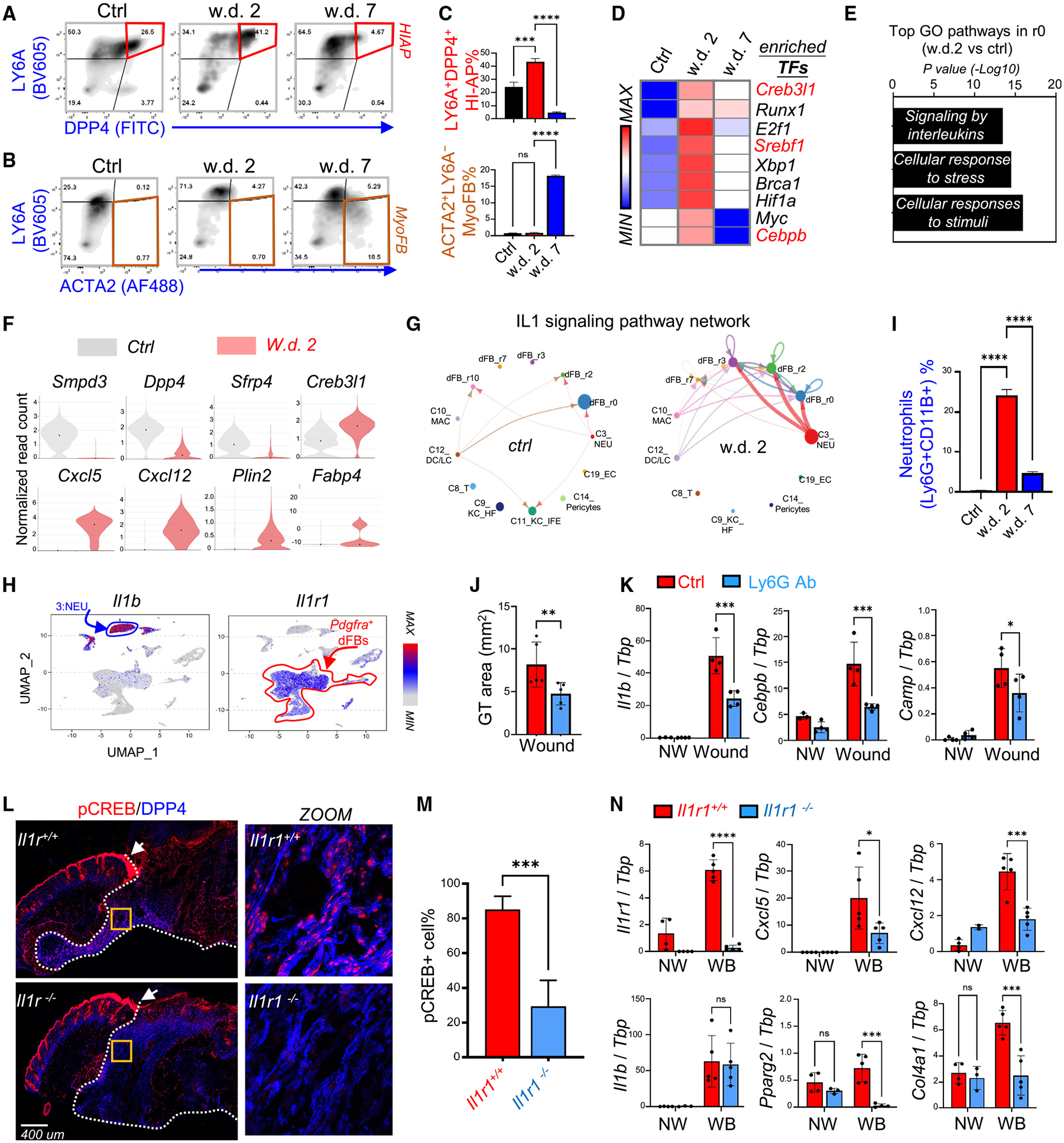
(A–C) FACS plots (A and B) and quantified bar graphs (C) showing the percentage of HI-APs (DPP4+LY6A+) and myofibroblasts (ACTA2+LY6A−) in control, w.d. 2, and w.d. 7 skin samples (n = 3/group).
(D) Heatmap showing the top enriched transcriptional factors in w.d. 2 cells compared with control and w.d. 7 cells.
(E) KEGG/GO pathway analysis comparing the r0 HI-AP cluster of control and w.d. 2 skin samples showing top enriched signaling pathways activated in HI-APs after wounding.
(F) Violin plots showing the expression of indicated genes in the control and w.d. 2 samples.
(G) Circle plot showing the inferred intercellular communication network for IL-1β signaling in control and w.d. 2 skin cells.
(H) UMAP plots showing the expression of indicated genes in total wound cell clusters.
(I) Quantified bar graphs (from FACS plots shown in Figure S5G) showing neutrophils (Ly6G+CD11B+)% (n = 3/group).
(J and K) Mice were injected intraperitoneally with isotype (Ctrl) or anti-Ly6G antibodies to deplete neutrophils and then skin wounds were collected at w.d. 3 for granular tissue (GT) area measurement (J). qRT-PCR analysis (K) of the listed genes (n = 4/group).
(L and M) Wound samples were collected from WT or Il1r1 knockout mice at w.d. 3 for immunostaining (L) of pCREB (red) and DPP4 (blue), and quantified percentage of pCREB+ cells is shown in (M) (n = 7–10/group). In (L), the white dotted line marks the granule tissue and arrow points to the unaffected pCREB signaling in the migrating epidermis. Scale bar, 400 μm.
(N) qRT-PCR analysis of the listed genes (n = 4–5/group). All error bars indicate mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001.
SCENIC TF analysis identified several key adipogenesis TFs (Cebpb, Creb, Srebf1) as the top enriched TFs driving dFB activation at w.d. 2 (Figure 5D). In addition, GO pathway analysis of the r0 HI-AP cluster identified cellular responses to stress/stimuli and IL-1 signaling as the top upregulated pathways after wounding (Figure 5E). Furthermore, violin plots showed that wounding led to a decrease in the expression of HI-AP marker genes (Smpd3, Dpp4) and an increase in the expression of IL-1- and adipogenesis-related genes in HI-APs. (Figure 5F). In addition, CellChat analysis identified IL-1 as one of the top incoming signals for HI-AP dFBs (Figure S5C), and IL-1-IL-1R signaling network analysis identified neutrophils as the most prominent source of IL-1 ligand acting on dFB clusters at w.d. 2 (Figures 5G and S5D). Expression plots confirmed that Il1b was predominantly produced by neutrophils, whereas the IL-1 receptor (Il1r1) and IL-1-inducible chemokines (Cxcl1/12/5) were expressed primarily by dFBs (Figures 5H, S5E, and S5F). Together, these results suggest that IL-1 released from neutrophils may drive the wound-induced adipogenesis response during the early wound-inflammatory phase.
FACS and immunostaining analyses (Figures S5G, S5H, and 5I) confirmed that Ly6G+ neutrophils were the dominant infiltrating immune cell type (>60% of all CD11B+ myeloid cells) during w.d. 2–3, and neutrophils were cleared from the wound by w.d. 7. Depletion of neutrophils (Figures S5I and S5J) led to a significant reduction in the DPP4+ wound GT area (Figure 5J) and impaired wound-induced adipogenesis (Figure 5K), decreased CAMP protein expression (Figure S5K), and decreased pCREB signal (Figures S5L and S5M) in the migrating DPP4+ HI-APs. In addition, depletion of neutrophils led to decreased wound-induced Il1b expression (Figure 5K), demonstrating that neutrophils are the major cellular source of IL-1β during the early stages of wound healing.
Wild-type (WT) and Il1r1−/− mice were subjected to skin wounding to validate the role of IL-1 in wound-induced adipogenesis. We found that Il1r1 deficiency led to profound inhibition of pCREB activation in DPP4+ HI-APs (Figures 5L and 5M), but not in the migrating epidermal KCs (white arrow) or other DPP4− dermal cells, indicating that this IL-1-pCREB signaling axis is specific for the activation of hypodermal cells. In line with pCREB inhibition, the wound-mediated induction of Cxcl5, Cxcl12, and adipogenesis-related genes was significantly reduced in Il1r1−/− mice compared with WT controls (Figure 5N). Furthermore, administration of an NF-κB inhibitor to skin wounds also led to decreased pCREB expression in HI-APs and pAds (Figure S5N). These results indicate that neutrophils contribute to HI-AP activation and wound-induced adipogenesis by activating the IL-1R-NF-κB-CREB pathway.
Activation of WNT and TGF-β signaling pathways in adipocyte-lineage cells during the wound-proliferative phase
We found that a peak in the expression of lipogenesis genes at w.d. 3 was followed by the induction of lipolysis genes (Atgl and Hsl) at approximately w.d. 5, prior to the induction of Acta2 and ECM genes around w.d. 5–7 (Figures 6A and S6A). In addition, loss of FABP4+ adipocytes was accompanied by transient formation of ACTA2+ myofibroblasts on w.d. 7 (Figure S6B), suggesting that adipocyte lipolysis and dedifferentiation may contribute to myofibroblast formation during the wound-proliferative phase.
Figure 6. Activation of WNT and TGF-β signaling pathways in adipocyte-lineage cells during the wound-proliferative phase.
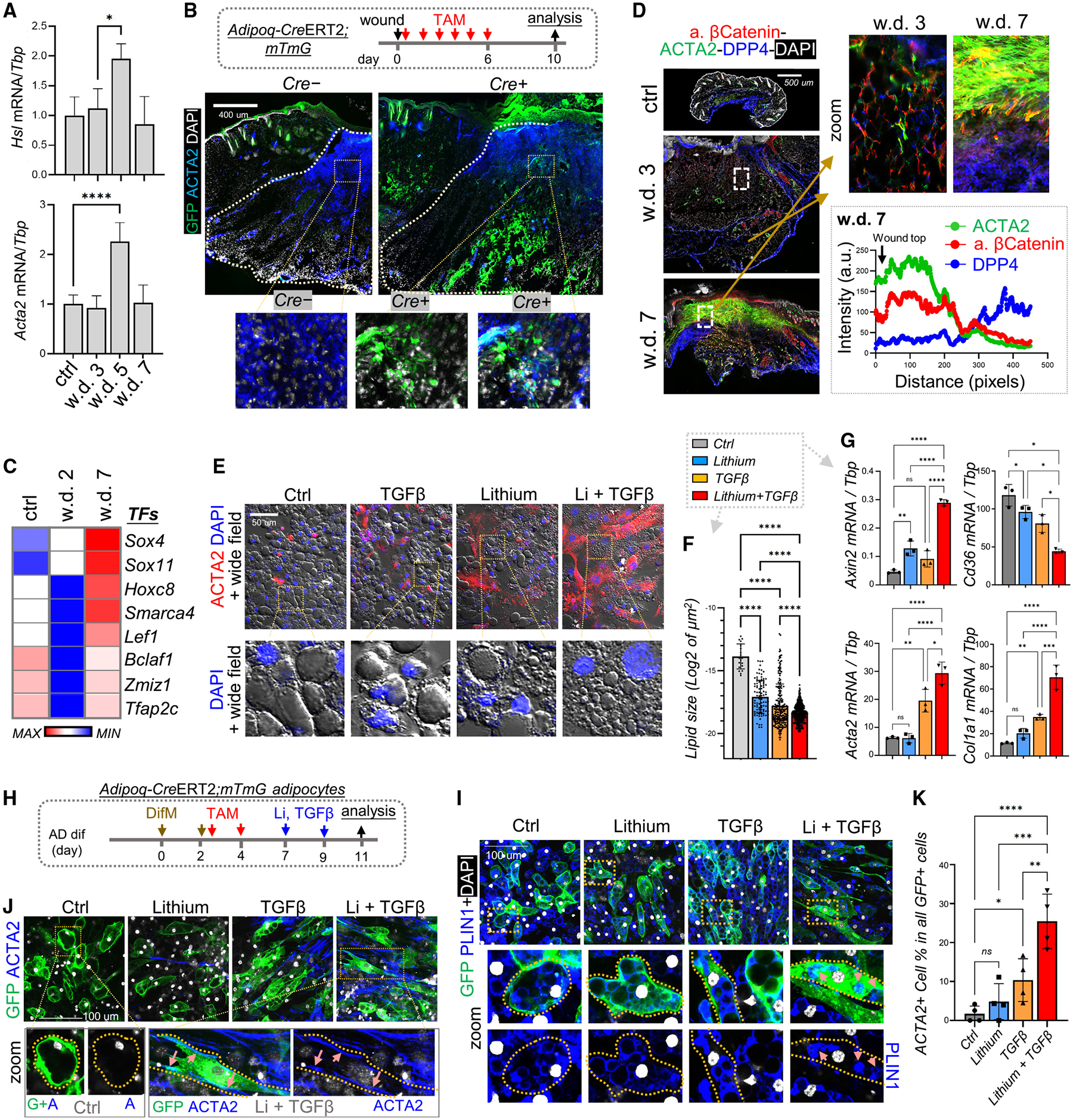
(A) qRT-PCR analysis showing the mRNA expression kinetics of listed genes during skin wound healing (n = 3 per group).
(B) Adipoq-CreERT2;mTmG mice were administered tamoxifen (TAM) from day 1 to 6 during wound healing, and wound tissues were collected at w.d. 10 for immunostaining of ACTA2 (blue) and GFP (green). Scale bar, 400 μm.
(C) Heatmap of SCENIC TF analysis showing the top enriched transcriptional factors.
(D) Wound tissues were stained with active β-catenin, ACTA2, and/or DPP4 as indicated. Scale bar, 500 μm. Quantified intensity profiles showing signals from all three fluorescence channels from wound top to bottom (the lower right box) are shown (representative of n = 3/group).
(E–G) Fully differentiated adipocytes were treated with lithium and/or TGF-β2 for 2 days. (E) Immunostaining of ACTA2 (red) and DAPI (blue). Fluorescence and wide-field images were overlaid. Scale bar, 50 μm. (F) Quantification of changes in lipid droplet (LD) size (LD quantified from n = 5 fields/group). (G) qRT-PCR analyses of the indicated genes (n = 3/group).
(H–K) Primary neonatal dFBs isolated from Adipoq-CreERT2;mTmG were differentiated into mature adipocytes in the presence of TAM and then treated with lithium and TGF-β2 (H) before being subjected to immunostaining with the indicated antibodies (I and J) or percentage quantification (K) of ACTA2-expressing myo-FBs in all GFP-positive cells (n = 4/group). Scale bars, 100 μm. All error bars indicate mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001.
To determine whether adipocytes (preexisting or newly generated) contribute to the formation of myofibroblasts, we induced CRE activity in Adipoq-CreERT2;mTmG mice by TAM application during w.d. 0–6, thereby investigating both the inflammatory and the proliferative phases of wounding (Figure 6B). Mature adipocytes labeled with GFP were detected in the upper wound bed region, where these GFP+ cells co-expressed ACTA2 (Figures 6B and S6C), indicating that myofibroblasts can be partially derived from mature adipocytes during wound healing.
SCENIC TF analysis identified several WNT-related TFs as top enriched TFs in dFBs at w.d. 7 (Figure 6C), including Lef1, Sox4, Sox11, Hoxc8, and Smarca4, all of which regulate cell development or carcinogenesis by activating the WNT-β-catenin signaling pathway.31–35 Co-staining of active β-catenin with DPP4 and/or ACTA2 revealed that at w.d. 3 the active β-catenin signal was barely detectable, but at w.d. 7 β-catenin was robustly activated in the upper wound region, where ACTA2+ myofibroblasts were generated, and by w.d. 14, active β-catenin was lost and myofibroblasts disappeared from the wound GT (Figures 6D, S6D, and S6E). Although nuclear β-catenin signal can be detected in cultured dFBs upon WNT3A stimulation (Figure S6F), it was difficult to distinguish β-catenin within nuclei from that within the cytosol in cells within whole wound tissue.
Gene plot analysis showed that both WNT and TGF-β signaling activities were prominently activated in the r7 myofibroblast cluster (Figure S6G), indicating that the crosstalk between TGFB and WNT signaling may promote adipocyte-to-myofibroblast transition during wound healing.
WNT activation in adipocytes promotes adipocyte dedifferentiation and myofibroblast formation
The TGF-β pathway plays a role in triggering lipolysis and dedifferentiation of mature adipocytes36,37; moreover, physical stress was observed to trigger adipocyte dedifferentiation via WNT-β-catenin signaling.38 Here, we found that a combination of both lithium and TGF-β robustly promoted cell elongation and the breakdown of large lipid droplets into numerous smaller lipid droplets (Figures 6E and 6F), which are characteristic of dedifferentiating adipocytes.38,39 Furthermore, co-treatment with both lithium and TGF-β2 led to the most significant increase in ACTA2+ cell percentage (Figure S6H), induction of pro-fibrotic genes, and suppression of Cd36 expression (Figures 6G and S6I).
To lineage trace adipocyte dedifferentiation in vitro, adipocytes were differentiated from Adipoq-CreERT2;mTmG dFBs and labeled by GFP in the presence of TAM, then labeled cells were treated with lithium and TGF-β to trigger lipolysis (Figures 6H, S6J, and S6K). We found that, while lithium or TGF-β alone had some effects, co-treatment profoundly changed GFP+ adipocytes from rounded to elongated morphology and promoted the internalization of GFP from membrane to cytosol and the breakdown of the PLIN1+ lipid droplets (Figures 6I and S6L). Furthermore, ACTA2 was induced along the peripheral membrane of the elongated GFP+ cells upon co-treatment (Figures 6J and 6K), supporting the idea that ACTA2+ myofibroblasts can be generated de novo from dedifferentiating adipocytes.
Gsk3 deletion in adipocytes promotes myofibroblast formation during wound healing
To examine whether WNT activation in adipocytes promotes fibrogenesis in vivo, we genetically targeted both Gsk3a and Gsk3b, key inhibitory molecules for β-catenin,40 from adipocytes by crossing Adipoq-CreERT2 mice with Gsk3a/bfl/fl mice (Gsk3AD-KO) (Figure 7A). Both GSK3 alleles must be deleted to achieve hyperactivation of the WNT-β-catenin signaling.41 We found that TAM application to Gsk3AD-KO mice during wound healing led to specific depletion of GSK3 in PLIN+ adipocytes, but not in other non-adipocyte cells (Figures 7A and 7B), indicating that targeted Gsk3 deletion in adipocytes was successful. Furthermore, targeted Gsk3 deletion in adipocytes not only increased activated β-catenin signaling in ATGL+ (a critical lipolysis enzyme42) adipocytes (Figures 7C and 7D), but also promoted myofibroblast formation (Figures 7E, 7F, and S7A–S7C) and increased CRABP1 expression (Figure S7D). However, Gsk3 deletion decreased the pool of LY6A+DPP4− pAds (Figures 7E, 7G, and S7C) and inhibited Cxcl5 and Camp expression (Figures S7E and S7F) in skin wounds. These results show that WNT activation mediated by GSK3 deletion in adipocytes dampens wound-induced inflammatory and adipogenic responses and promotes adipocyte-to-myofibroblast transition during the wound-proliferative phase.
Figure 7. WNT activation in adipocytes promotes myofibroblast formation during wound healing.
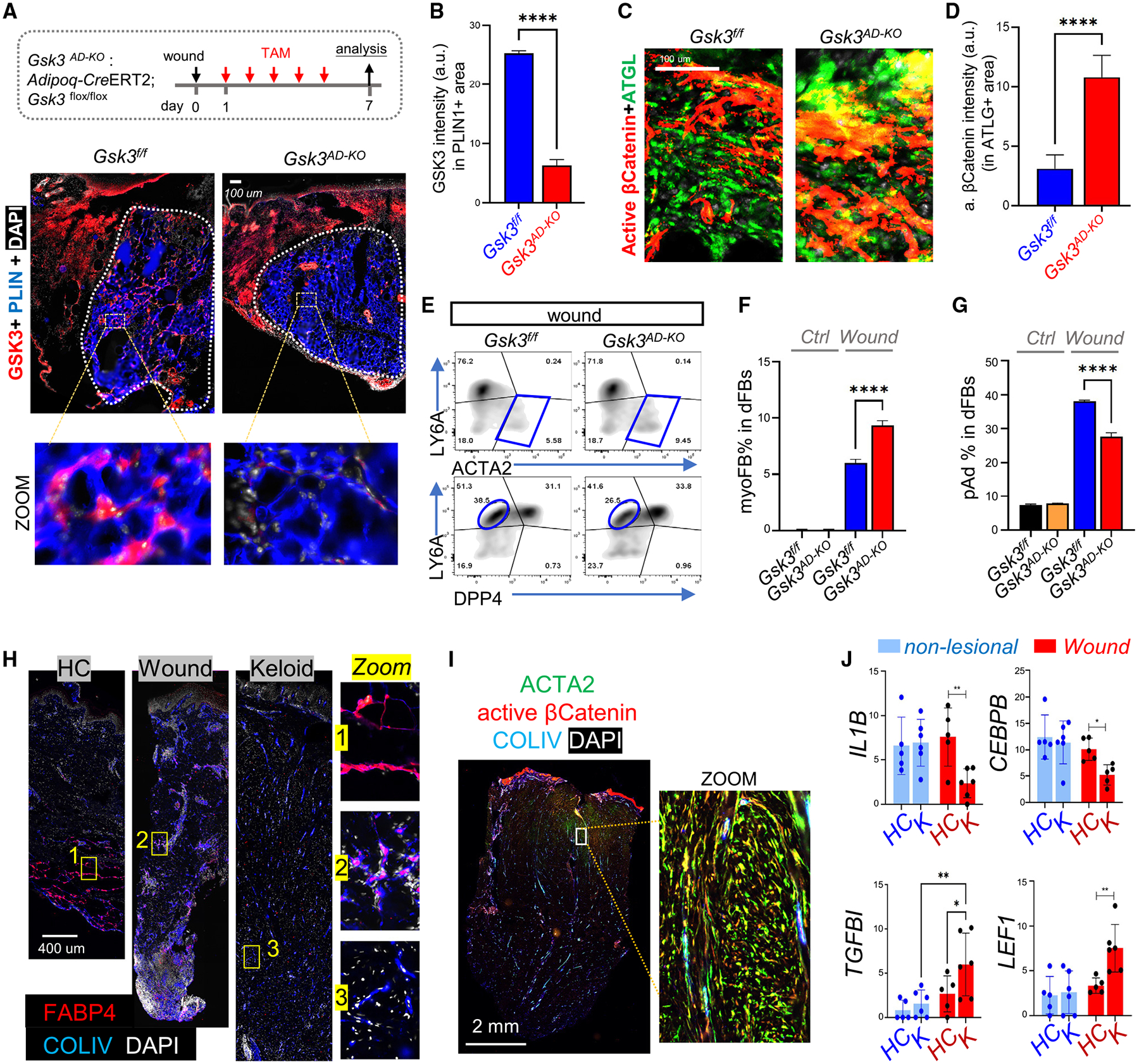
(A–G) Adipoq-CreERT2;Gsk3flox/flox mice were administered TAM from w.d. 1 to 6, and w.d. 7 tissues were collected for analysis. (A) Staining of GSK3 (red), PLIN (blue), and DAPI (white), and (B) quantified GSK3 intensity in PLIN1+ cells (n = 4/group). (C) Staining of active β-catenin (red) and ATGL (green) and (D) quantified active β-catenin intensity in ATGL+ area (n = 4/group). Scale bars, 100 μm. (E–G) FACS plots and quantified bar graphs (n = 3/group) showing the percentage of ACTA2+LY6A− myofibroblasts or LY6A+DPP4− preadipocytes in PDGFRA+ dFBs (n = 3/group).
(H) FABP4 (red), COLIV (blue), and DAPI (white) staining in healthy control (HC), wound (day 7), and keloid skin sections. Scale bar, 400 μm.
(I) Active β-catenin (red), COLIV (blue), and ACTA2 (green) staining in keloid skin sections. Scale bar, 2 mm.
(J) Bar graphs showing relative mRNA expression (based on RNA-seq FPKM [fragments per kilobase per million mapped fragments] values) of listed genes in non-lesional and wound tissues from HC or keloid (K) individuals (n = 4–5/group). All error bars indicate mean ± SEM; *p < 0.05, **p < 0.01.
Human keloid is associated with sustained WNT activation and inhibition of dermal adipogenesis
Finally, to determine whether our observations in mice are also relevant to humans, we evaluated the changes in dWAT in human skin samples from normal wounds and keloids. IHC staining showed that numerous lobular structures enriched with COLIV+FABP4+ adipocytes were present in the granular tissue of normal wounded skin, whereas COLIV+FABP4+ adipocytes were largely absent in the keloid skin dermis (Figure 7H), suggesting that adipogenesis was blocked in keloids. In addition, immunostaining revealed a strongly activated β-catenin signal in both ACTA2+ myo-FBs and COLIV+ cells in the keloid skin dermis (Figure 7I). Furthermore, reanalysis of published transcriptomic data of non-lesional skin or wounded skin from keloid-prone individuals and healthy matched subjects43 showed that compared with normal wounds, keloid wounds expressed significantly lower levels of immune- or adipogenesis-related genes (IL1B, CEBPB, PLIN3), but higher levels of WNT-and/or TGF-β-related genes and ECM genes (Figures 7J and S7G). Together, these results indicate that unbalanced activation of WNT and immune pathways during wound healing may inhibit adipogenesis and promote fibrogenic development in keloid.
DISCUSSION
dWAT has been recognized as a functional skin layer with remarkable plasticity; it expands or regresses during development, wound healing, bacterial infection, and hair cycling.3,44 To trace the cellular and molecular changes in dWAT cells, we present here a compendium of skin development, maturation, and regeneration processes by scRNA-seq. First, we grouped the developing dFBs into distinct non-adipogenic and adipogenic cell states. Second, our data identified the WNT and IL-1 pathways as the key suppressing and activating signals, respectively, for dermal adipogenesis. Third, we found that skin injury triggered a sequence of events in wound GT, including rapid infiltration/activation of HI-APs and onset of adipogenesis during the wound-inflammatory phase, followed by adipocyte lipolysis/dedifferentiation and myofibroblast generation during the wound-proliferative phase. Wound-induced adipogenesis was in part mediated by neutrophils and IL-1, and adipocyte dedifferentiation was in part mediated by activation of the WNT-β-catenin pathway. Finally, we showed that inhibition of dermal adipogenesis and sustained activation of dermal WNT signal was also associated with human keloid scar. Taken together, this study provides in-depth knowledge of the regulatory mechanisms of dWAT plasticity during skin development and wound regeneration at the single-cell level.
Defining the highly heterogeneous adipogenic and non-adipogenic dermal fibroblast subpopulations
scRNA-seq has recently been applied to understanding skin fibroblast heterogeneity.42,45–47 However, none of these studies focused on adipogenic cells, and most of these studies defined dWAT or fascia-related dFBs generally as only hypodermal or fascia dFBs, without further defining cell heterogeneity in the hypodermis. In this study, we defined several adipocyte-lineage cell subpopulations, including RET-APs, pAds, adipocytes, and three hypodermal populations (HI-pAds, Aregs, and HI-APs). In addition, we identified a distinct pool of CD24hi perifollicular dFB clusters that were non-adipogenic, enriched in developing skin, and rapidly lost during skin maturation.
This study also identified several new dFB markers at both the transcript and the protein levels. We identified Trps1 as an exclusive marker for Ly6a+ adipocyte-lineage cells. TRPS1 (both mRNA and protein) was found highly expressed in Cd24hi developing or follicle-associated dFBs, at intermediate levels in Dpp4+Ly6a− PAP dFBs, and almost undetectable in Ly6a+ pAd/APs. We also identified Lgr5, a previously known hair follicle stem cell marker,14 as a novel pAd marker in mouse skin and in vitro primary pAd culture (Figures 1 and 3). Furthermore, we found that a cluster of Fmo2+Gdf10+ Areg fibroblasts accumulated in the skin during maturation. Emerging studies have unraveled how Areg cells negatively regulate adipogenesis by secreting GDF10 and/or RSPO2,16,17,48 but whether the age-dependent increase in skin Areg contributes to the loss of the adipogenic potential of skin AP/pAds during aging needs further investigation.
Balance between the WNT-β-catenin and the IL-1-NF-κB pathways in controlling dermal adipogenesis
WNT/β-catenin signaling is known to suppress adipogenesis by inhibiting the adipogenic TFs CEBP and PPARγ, preventing the differentiation of MSCs into fat cells.49–51 During skin development, WNT-β-catenin signaling is considered the earliest signaling event that instructs the migration of dFB progenitors from the somatic mesoderm to the dorsal skin dermis and the specification of these cells to fibroblasts.52 Inhibition of WNT-β-catenin signaling promotes the conversion of dermal-lineage cells to cartilage.53 Here, we identified the WNT-β-catenin pathway as one of the most differentially suppressed pathways in adipogenic dFBs compared with other non-adipogenic dFB subclusters. Consistent with our results, active WNT gene signatures have been shown to be preferentially associated with Lrig1+ PAP dFBs, but not with Ly6a+ pAds.54 Furthermore, forced stabilization of β-catenin in Pdgfra+ dFBs leads to a reduction in dWAT, followed by the development of prominent fibrosis.55 These data suggest that, while activation of WNT-β-catenin in mesodermal stem cells is required for stem cell commitment to the dFB lineage during early development, the subsequent inhibition of WNT signaling during later skin development may be required for the commitment of dFBs to adipogenic subpopulations.
Our results suggest that IL-1 may antagonize WNT activity during dermal adipogenesis. Mechanistically, we showed that IL-1β treatment not only triggered the activation of the pro-adipogenic CREB-CEBPβ pathway, but also promoted β-catenin degradation by inhibiting AKT phosphorylation. Consistently, several immune-related and inflammatory pathways have been found to be enriched and activated upon targeted deletion of β-catenin in adipocytes.56 Furthermore, IL-1β increases the expression/activation status of CREB and its target gene CEBPβ in a chondrocyte cell line.57 IL-1B signaling also activated CEBPβ in several other cell types, such as gastric epithelial cells58 and myocytes,59 suggesting that CREB-CEBPβ activation maybe a common downstream event of IL-1 receptor signaling. In contrast, an inhibitory role for IL-1 has been observed in regulating adipogenesis in skeletal muscle tissues,50,60 suggesting that a tissue-specific microenvironment may shape the differential response of AP to IL-1.
The crosstalk between WNT and TGF-β pathways in regulating adipocyte lipolysis and dedifferentiation
Using several independent approaches, we demonstrated that WNT cooperates with TGF-β signaling to promote adipocyte lipolysis and dedifferentiation of adipocytes to myofibroblasts during the wound-proliferative phase. In line with our findings, TGF-β signaling was shown to be the top enriched pathway activated by WNT3A treatment in adipocytes.56 In addition, TGF-β and WNT are both potent pro-fibrotic signaling molecules involved in abnormal wound healing responses following skin or lung injury.54,61–63 During the pathogenesis of idiopathic pulmonary fibrosis, myofibroblast differentiation is dependent on TGF-β-WNT crosstalk.64 Further studies are required to determine how signaling events downstream of the TGF-β-WNT axis differ between dermal development and wound regeneration.
Hypodermal interstitium, a newly recognized skin structure enriched with progenitors
The interstitium, a fluid-filled interstitial space enriched with fibroblasts and ECM proteins found in numerous tissues, has been recently recognized as an important anatomical structure in edema, fibrosis, and mechanical functioning of many organs.12,65 Our study defines a multipotent subpopulation of dFBs, the WNT2+DPP4+ HI-APs, that may not only give rise to dermal adipocytes during development but also function as the origin of regenerating dFBs during wound healing. This HI-AP population is similar to the reported hypodermal fibroblasts,66 fascia fibroblasts,67 or interstitial APs,12 which have a unique ECM production profile, multipotent differentiation potential, and ability to migrate to the wound in response to injury. These converging lines of evidence suggest that HI-APs may be key progenitor cells contributing to dermal cell regeneration.
Limitations of the study
A limitation of this study is that we did not use a genetic approach to ablate or lineage trace HI-APs to confirm the function of HI-APs in dermal adipogenesis. We found that WNT2 is a highly specific marker gene for HI-APs; therefore Wnt2-Cre mice can be used to genetically ablate or lineage trace these cells to validate their roles in dermal adipogenesis during skin development or wound regeneration.
Altogether, our data defined the cellular and molecular mechanisms through which adipocyte-lineage cells are regulated during skin development and wound regeneration. Future lines of investigation examining the crosstalk between immune cells, APs, and adipocytes under conditions of abnormal wound responses, such as keloid and diabetic wounds, and other skin fibrotic diseases such as scleroderma, could lead to breakthroughs in treating human skin diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources may be directed to and will be fulfilled by the lead contact, Ling-juan Zhang (lingjuan.zhang@xmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals and animal cares
All animal experiments were approved by the Xiamen University and/or the University of California San Diego (UCSD), Institutional Animal Care and Use Committee. C57BL/6 WT mice were purchased from Xiamen University Laboratory Animal Center. Adipoq-CreERT2 mice (Stock No:025124) and ROSA-mT/mG mice (Stock No: 07676) were originally purchased from Jackson laboratory. Gsk3aflox/floxGsk3bflox/flox mice were generously provided by professor Wen-Hsien Liu.68 Adipocyte specific Gsk3 knockout mice were generated by breeding Gsk3aflox/floxGsk3bflox/flox with Adipoq-CreERT2. Il1r1−/− mice were generously provided by professor Jiahuai Han (Xiamen university, Xiamen, Fujian, China). All mice were breeded and maintained in standard pathogen free environment of the Laboratory Animal Center in Xiamen University, and all animal experiments were approved by the Animal Care and Use Committee of Xiamen University.
Human skin sample collection and analysis
Fresh adult human full thickness skin biopsies, from age and sex matched healthy, wound and keloid donors (female or male) were collected by the department of dermatology at the First Affiliated Hospital of Fujian Medical University. Keloid disorder was diagnosed based on their clinical appearance, history and treatment history. All keloid samples analyzed in this study were consistent with clinical and pathological criteria for keloid patients. Wound skin biopsies were collected from healthy individual who had no history of keloid formation and receiving wound debridement procedure. All sample acquisitions were approved and regulated by Medical Ethics Committee of the First Affiliated Hospital of Fujian Medical University (reference number No. 2020[146]). The informed consent was obtained from all subjects prior to surgical procedures. Upon collection, these samples were directly fixed with PFA then proceed for paraffin embedding for histological or immunofluorescent analyses.
Skin maturation and aging model
For skin maturation and aging mouse model shown in Figure 3, dorsal skin biopsies were collected from C57BL/6 wildtype adult male mice with various ages (3W, 2 months and 1 year old). Similar to our previous report,21 here we used male exclusively to study age-related changes because loss of dermal fat and adipogenic AP in females is relatively slower compared to male counterparts, but the overall trend to lose functional dWAT is similar between female and male.
Mouse wound healing model
7~9 weeks old C57BL/6 mice (male or female) were wounded during telogen phase of hair cycling. Mice were anesthetized using isoflurane, hair was removed by clipper and Nair, and one full-thickness biopsy (1.0 cm × 0.5 cm in size) was generated on each side of dorsal skin (two wounds per mouse). Animal were sacrificed at indicated intervals after injury and wound beds were collected for analysis. To deplete neutrophils, mice were intraperitoneally administrated with 100 ug anti-LY6G (Biolegend, 127649) or IgG2a isotype control antibody (Biolegend, 400565) one day prior to wounding and then were injected daily with 50ug antibodies from day 0 ~ day 2 post wounding, and mice were sacrificed at day 3 post wounding for sample collection and subsequent analysis.
Adipocyte lineage tracing mouse model
Adipoq-CreERT2;mTmG mice (male or female) were daily administrated intraperitoneally with tamoxifen (TAM) (Sigma,T5648) solution (20 mg/mL stock solution dissolved in corn oil (Selleck, s6701)) at 32.5mg/kg body weight for consequent 4~6 days during indicated time period of wound healing. To tracing the cell fate of mature adipocyte in vitro, primary dFBs were isolated from Adipoq-CreERT2;mTmG neonatal mice, cultured and differentiated into adipocytes in the presence of tamoxifen (20 μM) to label mature adipocyte by GFP. Measurement of PLIN1+ lipid droplet size or ACTA2-expressing in GFP+ adipocytes were performed by ImageJ software.
METHOD DETAILS
Single cell RNA library preparation, sequencing and data process
For developmental analyses, dorsal skin biopsies were collected from neonatal (P1), male young adult (3 weeks) and male mature adult (2 month) C57BL/6 mice. To minimize batch effects due to non-biological sources, samples from all age groups were processed, sequenced, and analyzed together. Skin biopsies were minced and digested with collagenase D and DNase1 to isolate single skin cells as previously described (Zhang et al., 2019; Zhang et al., 2021). Dead cells were removed using DeadCell Removal kit (Miltenyi Biotic, 130-090-101) according to manufacturer’s instruction. Live cells were counted using a hemocytometer and resuspended in 2% BSA at a concentration of 3,000 cells/uL. All samples were loaded on a 10x Genomics GemCode Single-cell instrument that generates single-cell Gel Bead-In-EMlusion (GEMs). Barcoded, full-length cDNAs were reverse-transcribed from polyadenylated mRNA. Silane magnetic beads were used to remove leftover biochemical reagents and primers from post GEM reaction mixture. Next, libraries were generated and sequenced from the cDNAs with Chromium Next GEM Single Cell 3’ Reagent Kits v2. cDNA libraries were sequenced on an Illumina Novaseq6000 platform (Illumina). The raw sequencing data was demultiplexed and aligned to the reference genome mm10–1.2.0 using Cell Ranger v3.0.2 pipiline(10x Genomics). The generated raw gene expression matrix was converted into Seurat objects using the R package Seurat v2.0. To remove doublets and poor-quality cells, we utilized the following procedures to control for cell quality: >200 genes/cell, <5000 genes/cell; >25,000 unique molecular identifiers(U-MIs); and <8% mitochondrial gene expression. Low quality cells and outliers were discarded, and only ~24,409 viable cells were used for downstream analysis. These included ~7,483 cells – NB; ~8,235 cells – young (3weeks mice); ~8,691cells-adult (2month mice).
For mouse wound samples, skin biopsies were collected from wound day 2 and day 7 granular tissues or unwounded control skin. Single cells were isolated and subjected to single cell RNAseq procedures including libraries construction and data processing as described above. After low quality cells and outliers were discarded, 10233 cells from non-lesional skin, 7807 cells from wound beds at post-wounding day 2 (PWD2), and 4925 cells from wound tissue at PWD7 were used for downstream analysis.
Unsupervised clustering and gene expression were visualized with the Seurat 2.0 on R studio, and assignment of cell clusters was based on expression of validated marker genes. To investigate the developmental path of dermal fibroblasts, pseudotime analysis was performed on selected dFB subsets by Monocle 2.10.1 (Xiaojie Qiu et al., 2017) to infer the pseudotime trajectories of these cell, followed by scEpath to identify pseudotime-dependent gene expression changes and classify into distinct differentiation states. To further determine the cell fate of dFBs, RNA velocity was performed using the package velocyto (Gioele La Manno et al., 2018) with the default parameter, and the BAM files were used as inputs. To analyze activity of transcriptional regulators in skin development or wound healing, SCENIC (Van de Sande et al., 2020) conjunction with the mm9 Rcis Target database was used and UMIs for transcriptionally defined fibroblasts as input to construct regulon networks between transcription factors and potential target genes and score the active value of regulon for each FB clusters. Single cell RNA libraries construction, sequencing and bioinformatic analysis was assisted by GENE DENOVO Inc (Guangzhou, China).
Cellrank (v1.5.0) analysis was performed to calculate cell-cell transition probabilities according to literature.30 Briefly, scVelo (v0.2.4) was first performed on selected dFB clusters (r0 - r12) to estimate RNA velocities and velocity graphs. Secondly, transition probabilities were calculated in a weighted transition matrix with 80% RNA velocity and 20% similarity. The Generalized Perron Cluster Analysis(GPCCA) was used to compute fate probabilities. The terminal states were estimated from the so-called ‘eigengap heuristic’ of the spectrum of the transition matrix. Analysis was repeated for the initial states. Probability of each cell to transit to the terminal states was computed. Finally, directed PAGA was used to aggregate the individual fate maps into a cluster-level fate map.
Bulk RNA sequencing and bioinformatic analysis
Total cellular RNA were extracted using TRIzol reagent (Sigma, T9424) and RNAExpress Total RNA Kit (NCM, M050). RNA quality was analyzed by bioanalyzer and RNA samples with RIN value >7 were used for sequencing. Next generation sequencing library preparations were constructed according to the manufacturer’s protocol (NEBNext Ultra RNA Library Prep Kit for Illumina). The poly(A) mRNA isolation was performed using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB). The mRNA fragmentation and priming were performed using NEBNext First Strand Synthesis Reaction Buffer and NEBNext Random Primers. First strand cDNA was synthesized using ProtoScript II Reverse Transcriptase and the second-strand cDNA was synthesized using Second Strand Synthesis Enzyme Mix. The purified double-stranded cDNA was then treated with End Prep Enzyme Mix to repair both ends and add a dA-tailing in one reaction, followed by a T-A ligation to add adaptors to both ends. Size selection of Adaptor-ligated DNA was then performed using AxyPrep Mag PCR Clean-up (Axygen), and fragments of ~420bp (with the approximate insert size of 300 bp) were recovered. Each sample was then amplified by PCR. The PCR products were cleaned up using AxyPrep Mag PCR Clean-up (Axygen), validated using an Agilent 2100 Bioanalyzer (Agilent Technologies), and quantified by Qubit 2.0 Fluorometer (Invitrogen). Then libraries with different indexs were multiplexed and loaded on an Illumina Navoseq instrument for sequencing using a 2×150 paired-end (PE) configuration according to manufacturer’s instructions. The sequences were processed and analyzed by GENEWIZ. Venn diagrams between gene sets were made by BioVenn. KEGG and gene ontology (GO) pathway analysis for differentially expressed genes and correlation analysis between single cell RNA-seq with bulk RNA-seq were performed by R package clusterProfile 3.12.0.
Pearson correlation analysis
Pearson correlation analysis was performed between the scRNA-seq datasets and bulk RNAseq datasets to determine the degree of correlation/similarity between dFB clusters identified by scRNA-seq with skin samples at various ages or different dFB treatment states. The scRNA-seq and bulk RNAseq transcriptomic datasets were normalized and matrixed, and Pearson correlation coefficient of the matrix was calculated, and hierarchical clustering analysis was performed to generate correlation heatmap in R Studio software.
Cell-chat signaling network analysis
R package CellChat 1.3.0 (Jin et al., 2021) was used to evaluate the potential cell-cell communication between dFB subclusters and other cell types, especially immune cells, during wound healing. The scRNA-seq data was imported to implement CellChat platform in R software. The process includes projecting the gene expression data onto protein-protein interaction (PPI) network, then assigning a probability value to infer biological cell-cell communication network, and calculating the centrality indicator of interacting network to identify the role / contribution of each cell population in distinct signaling pathways. The number and strength of identified cell-cell communication is displayed through hierarchical graphs, circle charts, heatmaps, etc. to visualize single or multiple signaling pathways.
Primary mouse dermal fibroblast isolation and in vitro adipocyte differentiation:
Primary mouse dermal fibroblasts were isolated from mouse skin as described previously (Zhang et al., 2019; Zhang et al., 2021). In brief, mouse dorsal skin was first into strips and digested with dispase overnight to remove epidermis. The dermis tissue was then cut into small pieces and digested with 2.5 mg/mL Collagenase D and 30 ng/mL DNAse1 for 2 hous at 37°C to release dermal fibroblasts. Cell mixture was then filtered through 30 μm filter and treated with red blood cell lysis buffer. Isolated dermal fibroblasts were cultured in growth medium (DMEM supplemented with 10% FBS and antibiotics/antimycotics) in a humidified incubator at 5% CO2 and 37°C under sterile conditions. Fresh medium was replenished daily to remove debris or dead cells. Primary cells were then trypsinized within 3 days and replated at 5 × 104/mL for in vitro assays, and only passage 1 cells were used for experiment. To induce adipocyte differentiation, two day post-confluent dFB were switched to adipocyte differentiation medium containing 2 μM Dexamethasone, 250 μM IBMX, 200 μM Indomethacin and 10 μg/mL recombinant human insulin. Fresh differentiation medium was changed at day 2 then medium was switched to maintenance medium (growth medium supplemented with 10 μg/mL recombinant human insulin) to promote maturation and hypertrophy of differentiated adipocytes.
Histology, collagen trichrome staining and immunohistochemistry (IHC)
To prepare paraffin sections, tissue biopsies were fixed with 4% PFA (Alfa Aesar, Shanghai, China) overnight then dehydrated and embedded in paraffin and sectioned at a thickness of 5–8 μm. For OCT embedding, fresh dorsal skin tissues were directly embedded in OCT then sectioned at a thickness of 15–20 μm, and frozen sections were fixed with 4% PFA for 15min prior to staining. Histology was assessed by Hematoxylin and Eosin (HE) staining using solutions from ZSGB-BIO (Beijing, China), and collagen was stained by the Masson’s Trichrome Stain Kit (Solarbio, Beijing, China), and elastic fiber staining was performed by elastic fiber stain kit (Saint-Bio, Shanghai, China) according to manufacturer’s instruction. Dermal cell density count shown in Figure S3C was performed manually by counting the number of nuclei per 1mm2 dermal area. Lipid staining was performed on frozen sections using BODIPY (ThermoFisher Scientific, Waltham, MA, USA) and PHA (Cytoskeleton, Denver, CO, USA) dyes. For IHC, fixed sections were permeabilized with 0.1% saponin (Sigma-Aldrich, St Louis, MO) and blocked in 5% BSA (Bioforxx, Hessen, Germany), and blocked sections were incubated with primary antibodies at 4°C overnight followed by appropriate 488-, Cy5 or Cy3-coupled secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA) in the dark for 4 hours at 4°C. Finally, sections were mounted by ProLong Gold Antifade Mountant with DAPI (Thermo Scientific Inc., Rockford, IL, US). All images were taken with Aperio VERSA Brightfield, Fluorescence Digital Pathology Scanner or Leica TCS SP8 White Light Laser Confocal Microscope, and processed by photoshop and/or Aperio ImageScope software (Leica Biosystems, Nußloch, Germany). Quantification of the fluorescence integrated intensity for each fluorophore was performed using ImageJ software.
Flow cytometry and analysis (FACS) and cell sorting
FACS procedure of primary dermal fibroblasts and immune cells was modified from previously established or reported methods.21,69 Briefly, freshly isolated mouse dFBs were first stained with zombie violet viability dye (BioLegend, 423114) to label dead cells. Cells were then blocked with anti-mouse CD16/32 (eBioscience, 14016185), followed by staining with an antibody cocktail for fibroblasts containing PECy7-CD45 (BioLegend, 147704), PerCP-Cy5.5-CD31 (BioLegend, 102522), FITC-CD26(Biolegend, 137806), AF700-CD24(Biolegend, 101836), PE-THY1 (BioLegend, 105308), APC-PDGFRa (eBioscience, 17140181) and BV605-LY6A (BioLegend, 108133), or an antibody cocktail for immune cells containing FITC-Ly6G (eBioscience, 11593182), PE-F4/80 (eBioscience, 12480182), APC-CD11C (BioLegend, 117310), AF700-MHCII (eBioscience, 56532182), PerCP-Cy5.5-Ly6C (BioLegend, 128012), PECy7-CD11B (Biolegend, 101216), APC-Cy7-CD3 (BioLegend, 100222). To stain intracellular ACTA2 in dFBs, stained cells were then fixed and permeabilized using the intracellular fixation and permeabilization buffer set (eBioscience) and stained by AF488-SMA/ACTA2 (eBioscience, 53976082). FACS analysis for protein expression of each cell marker was performed by the Thermo Attune NxT machine and analyzed by FlowJo V10 software. Dead cells stained positive with zombie violet dye were excluded from the analyses. FACS sorting for dFB subpopulations from neonatal mouse skin was performed by the BD Aria Fusion machine. After sorting, freshly isolated cells were directly lyzed in trizol for qRTPCR analysis or cultured in DMEM supplemented with FBS and antibiotics/ antimicotics for in vitro differentiation assays. Adipogenic differentiation was induced in sorted cells according to the “Primary mouse dermal fibroblast isolation and in vitro adipocyte differentiation” method section. Differentiated adipocytes were stained with Oil-red-O (ORO) to detect lipid-droplets as described previously1. To induce osteogenic differentiation, post-confluent sorted cells were treated with osteoblast differentiation medium containing 50ug/mL ascorbic acid and 10mM β-glycerophosphate, and medium was refreshed every other day for upto 20 days before being stained with Alizarin Red (AR) to visualize osteogenesis.70 The percentages of ORO- or AR-positive areas were quantified using ImageJ software.
Quantitative reverse transcription-quantitative PCR (qRT-PCR) analyses
Total cellular RNA was extracted using the RNAExpress Total RNA Kit (NM, M050) and 500 ng of RNA was reverse transcribed to cDNA using HiScript II Q RT SuperMix kit (Vazyme, R222–01). Quantitative, realtime PCR was performed on the Qtower real time system (Analytikjena, Swavesey, Cambridge, UK) using SYBR Green Mix (Bimake, Houston, Texas, USA). All of the primers used with SYBR green were designed to span at least one exon to minimize the possibility of nonspecific amplification from the genomic DNA. The expression of Tbp gene (TATA-Box Binding Protein) was used as a housekeeping gene to normalize data for the expression of mouse genes. Specific primer sequences are shown in Table S4.
Cell extract preparation and western blotting analyses
Primary dFBs were lysed in a denaturing lysis buffer containing 20 mM HEPES pH 7.4, 250mM NaCl, 2 mM EDTA, and 1% SDS supplemented with completed proteinase inhibitor cocktail (apexbio, Houston, USA) and phosphatase inhibitor cocktail (Roche, Basel, Switzerland) to maximally preserve protein post-transational modifications as described previously (Zhang et al., 2016). Lysates were boiled for 3 mins and homogenized by sonication using digital sonifier FS- 350T (Sxsonic, Shanghai, China) followed by centrifugation to remove DNA and cell debris. Protein concentrations were measured by BCA protein assay kit (Thermo Scientific Inc., Rockford, IL). For western blotting, 20ug of protein was mixed with same volume of 2x loading buffer with 20% β- mercaptoethanol (Sigma-Aldrich, St Louis, MO) and boiled for 5 mins at 95°C Then samples were separated on a 10–20% Tris-Tricine precast gel (Thermo Scientific Inc., Rockford, IL), transferred to PVDF membrane (Roche, Basel, Switzerland), followed by immunoblotting using indicated primary antibodies followed by fluorescent secondary antibodies (LI-COR, Lincoln, Nebraskal) and imaging using fluorescent Odyssey System (LI-COR). All western blot images shown in the manuscript are representative of 3 independent experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments were repeated at least 3 times with similar results and were statistically analyzed by GraphPad Prism 8 software. Quantification analyses of immunostained tissue sections showing the fluorescence integrated intensity of indicated fluorophores were performed by ImageJ (version 1.53). Quantified intensity profiles showing signals from indicated fluorescent channels across skin sections (from top to bottom) is adapted from publications.71,72 Quantification analyses of Oil-red-O or alizarin staining (Figure 1) were performed by ImageJ software according to literature.73 To quantify lipid-droplet (LD) sizes of cultured adipocytes (Figure 6), LDs were first circled based on the unique circular and bumped morphology of LDs shown by wide-field or PLIN1-staining images, then LD sizes were quantified by ImageJ software. For experiments with two groups, statistical significance was determined using Student’s unpaired two-tailed t-test. Normality was tested using the Shapiro-Wilk test, and for datasets that were not normally distributed, nonparametric tests were used to determine statistical significance. For experiments with more than two groups, one-way ANOVA multiple comparison test was performed as indicated in the legend. A p value of <0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bodipy | ThermoFisher Scientific | D3922 |
| PHA | Cytoskeleton | PHDH1-A |
| PE anti-THY1 | BioLegend | Cat# 105308; RRID: AB_313179 |
| BV605 anti-LY6A | BioLegend | Cat# 108133; RRID: AB_2562275 |
| AF700-CD24 | BioLegend | Cat# 101836; RRID: AB_2566730 |
| FITC anti-mCD26(DPP4) | Biolegend | Cat# 137806; RRID: AB_10663402 |
| APC anti-PDGFRA | eBioscience | Cat# 17140181; RRID: AB_529482 |
| PECy7 anti-CD45 | Biolegend | Cat# 147704; RRID: AB_2563536 |
| PerCP-Cy5.5 anti-CD31 | Biolegend | Cat# 102522; RRID: AB_2566761 |
| FITC anti-Ly6G | eBioscience | Cat# 11593182; RRID: AB_465314 |
| PECy7 anti-CD11B | Biolegend | Cat# 101216; RRID: AB_312799 |
| AF488 anti-SMA | eBioscience | Cat# 53976082; RRID: AB_2574461 |
| PE anti-F4/80 | eBioscience | Cat# 12480182; RRID: AB_465923 |
| PerCP-Cy5.5 anti-Ly6C | BioLegend | Cat# 128012; RRID: 1659241 |
| APC anti-CD11C | BioLegend | Cat# 117310; RRID: AB_313779 |
| AF700 anti-MHCII | eBioscience | Cat# 56532182; RRID: AB_494009 |
| APC-Cy7 anti-CD3 | BioLegend | Cat# 100222; RRID: AB_2242784 |
| Goat anti-PLIN | Abcam | Cat# AB61682; RRID: AB_944751 |
| Rat anti-CD24 | BioLegend | Cat# 101801; RRID: AB_312834 |
| Goat anti-PDGFRA | R&D system | Cat# AF1062-SP ; RRID: AB_2236897 |
| Rabbit anti-THY1 | Cell Signaling Technology | Cat# 13801S ; RRID: AB_2798316 |
| Rabbit anti-COL1A1 | abcam | Cat# ab34710; RRID: AB_731684 |
| Rat anti-LY6A | R&D system | Cat# MAB1226; RRID: AB_2243980 |
| Rat anti-Lgr5 | R&D system | Cat# mab8240; RRID: AB_2753324 |
| Goat anti-DPP4 | R&D system | Cat# AF954-SP ; RRID: AB_355739 |
| Goat anti-DLK1 | R&D system | Cat# AF8277; RRID: AB_2861313 |
| Rabbit anti-WNT2 | proteintech | Cat# 27214-1-ap ; RRID: AB_2880804 |
| Rabbit anti-TRPS1 | ABclonal | Cat# A7743; RRID: AB_2772733 |
| Goat anti-ALPL | R&D system | Cat# AF2910; RRID: AB_664062 |
| Sheep anti-ATGL | R&D system | Cat# AF5365; RRID: AB_2165678 |
| Rabbit anti-CRABP1 | Proteintech | Cat# 12588-1-AP ; RRID: AB_2292271 |
| Rabbit-anti-active β-Catenin | Cell Signaling Technology | Cat# 8814; RRID: AB_11127203 |
| Rabbit anti-β-Catenin | Cell Signaling Technology | Cat# 8480; RRID: AB_11127855 |
| Rabbit anti-GSK3β | Cell Signaling Technology | Cat# 12456; RRID: AB_2636978 |
| Rabbit anti-phospho-GSK3β(Ser9) | Cell Signaling Technology | Cat# 5558; RRID: AB_10013750 |
| Rabbit anti-CREB | Cell Signaling Technology | Cat# 9197; RRID: AB_331277 |
| Rabbit anti-Phospho-CREB(Ser133) | Cell Signaling Technology | Cat# 9198; RRID: AB_2561044 |
| Rabbit anti-AKT | Cell Signaling Technology | Cat# 4691S ; RRID: AB_915783 |
| Rabbit anti-Phospho-AKT(Ser473) | Cell Signaling Technology | Cat# 4060S ; RRID: AB_2315049 |
| Rabbit anti-ERK1/2 | Cell Signaling Technology | Cat# 9102S ; RRID: AB_330744 |
| Rabbit anti-Phospho-ERK1/2(Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370S ; RRID: AB_2315112 |
| Rabbit anti-Phospho-NF-κB p65 (Ser536) | Cell Signaling Technology | Cat# 3033; RRID: AB_331284 |
| Rabbit anti-P38 MAPK | Cell Signaling Technology | Cat# 9212S ; RRID: AB_330713 |
| Rabbit anti-Phospho-p38 MAPK (Thr180/Tyr182) | Cell Signaling Technology | Cat# 4631S ; RRID: AB_331765 |
| Mouse anti-β-Actin | Proteintech | Cat# 66009-1-Ig ; RRID: AB_2687938 |
| Goat anti-COLIV | Abcam | Cat# ab769; RRID: AB_92262 |
| Rabbit anti-FABP4 | Proteintech | Cat# 15872-1-AP ; RRID: AB_2102440 |
| Rabbit anti-CRAMP | University of California San Diego | Dr. Richard Gallo |
| Rabbit anti-SMA | Proteintech | Cat# 14395-1-ap ; RRID: AB_2223009 |
| Rabbit anti-NF-κB p65 | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| Rabbit anti-Phospho-C/EBPβ (Thr235) | Cell Signaling Technology | Cat# 3084; RRID: AB_2260359 |
| IRDye 680RD Donkey anti-Mouse | LICOR | Cat# 926-68072; RRID: AB_10953628 |
| IRDye 680RD Donkey anti-Goat IgG (H+L) | LICOR | Cat# 926-68074; RRID: AB_10956736 |
| IRDye 680RD Donkey anti-Rabbit IgG (H + L) | LICOR | Cat# 926-68073; RRID: AB_10954442 |
| IRDye 800CW Donkey anti-Goat IgG (H + L) | LICOR | Cat# 926-32214; RRID: AB_621846 |
| IRDye 800CW Donkey anti-Mouse | LICOR | Cat# 926-32212; RRID: AB_621847 |
| IRDye 800CW Donkey anti-Rabbit | LICOR | Cat# 926-32213; RRID: AB_621848 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat# 705-545-147; RRID: AB_2336933 |
| Cy3 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat# 705-165-147; RRID: AB_2307351 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-545-150; RRID: AB_2340683 |
| Cy3-AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-165-150; RRID: AB_2340666 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 711-545-152; RRID: AB_2313584 |
| Cy3 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 711-165-152; RRID: AB_2307443 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 715-545-150; RRID: AB_2340846 |
| Cy3-AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 715-165-150; RRID: AB_2340813 |
| Brilliant Violet 480-conjugated AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-685-150; RRID: AB_2651112 |
| Brilliant Violet 421-conjugated AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-675-150; RRID: AB_2651110 |
| DyLight 405 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat# 705-476-147; RRID: AB_2632563 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 711-606-152; RRID: AB_2340625 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-606-150; RRID: AB_2340695 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Goat IgG (H+L) | Jackson ImmunoResearch | Cat# 705-606-147; RRID: AB_2340438 |
| Biological samples | ||
| mouse skin samples | Xiamen University Laboratory Animal Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Eosin Staining Solution | ZSGB-BIO | ZLI-9613 |
| Hematoxylin Staining Solution | ZSGB-BIO | ZLI-9610 |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma | I5879 |
| Indomethacin | Sigma | I8280 |
| Dexamethasone | Sigma | D4902 |
| Insulin | Sigma | 91077C |
| DMEM, high glucose | ThermoFisher Scientific | C11965500BT |
| Glutamax | ThermoFisher Scientific | 35050-061 |
| Collagenase D from Clostridium histolyticum | Roche | 11088882001 |
| Antibiotic-Antimycotic | ThermoFisher Scientific | 15240062 |
| 2x SYBR Green qPCR Master Mix | Bimake | B21202 |
| red blood cell lysis buffer | TianGen | RT122-02 |
| SB431542 | Selleckchem | S1067 |
| HyClone Trypsin Protease | Hyclone | SH30042.01 |
| BD Bacto™ Dehydrated Agar | Fisher Scientific | DF0140-01-0 |
| TRI Reagent/TRIzol | Sigma | T9424-200ML |
| UltraPure™ DNase/RNase-Free Distilled Water | ThermoFisher Scientific | 10977015 |
| RNAlater™ Stabilization Solution | ThermoFisher Scientific | AM7021 |
| DnaseI | Solarbio | D8071-100mg |
| Saponin | Sigma | 47036-50G-F |
| Protease inhibitor | Sigma | 11836153001 |
| Wortamannin | Selleck | S2758 |
| U0126-EtOH | Selleck | S1102 |
| SB202190 (FHPI) | Selleck | S1077 |
| BAY 11-7082 | Selleck | S2913 |
| XAV-939 (NVP-XAV939) | Selleck | S1180 |
| 16% PFA | Alfa Aesar | 43368 |
| ProLong™ Gold Antifade Mountant with DAPI | ThermoFisher Scientific | P36931 |
| Collagenase/Dispase | Sigma | 10269638001 |
| Stabilizing Fixative 3X Concentrate | BDIS | 338036 |
| Hifair®II 1st Strand cDNA Synthesis SuperMix for qPCR | Vazyme | 11123ES60 |
| 2x M-PCR OPTI™ Mix (Dye Plus) | Bimake | B45012 |
| Recombinant mouse TGFβ2 | R&D System | 7346-B2-005 |
| recombinant mouse IL1β | R&D system | 401-ML-025 |
| recombinant mouse WNT3A | R&D system | 1324-WN-002 |
| Critical commercial assays | ||
| RNAExpress Total RNA Kit | NCM Biotech | M050 |
| Masson’s Trichrome Stain Kit | Solarbio | G1340-7 |
| Dead Cell Removal Kit | Miltenyi Biotic | 130-090-101 |
| Oil Red O stain kit | Solarbio | G1262 |
| Mouse Direct PCR Kit | Bimake | B40015 |
| Ailzarin red S (Osteoblast stain) | Sigma | A5533 |
| Intracellular Fixation & Permeabilization Buffer Set | eBioscience | 88-8824-00 |
| Deposited data | ||
| Skin developmental scRNAseq data | This study | GSE189241 |
| scRNAseq data of skin wound tissues | This study | GSE196800 |
| Experimental models: Cell lines | ||
| Primary mouse dermal fibroblasts | This study | N/A |
| Experimental models: Organisms/strains | ||
| B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | The Jackson Laboratory | JAX: 007676 |
| C57BL/6-Tg(Ad/poq-Cre/ERT2)1Soff/J | The Jackson Laboratory | JAX: 025124 |
| C57BL/6- Gsk3aflox/floxGsk3bflox/flox | From Wen-Hsien Liu | Chenfeng Liu et al.(https://doi.org/10.1126/sciadv.abg6262) |
| C57BL/6- Il1r1−/− | From Jiahuai Han | Generated by Xiamen University Laboratory Animal Center, Transgenic Animal Facility Core |
| C57BL/6 | From Xiamen University Laboratory Animial Center | N/A |
| Oligonucleotides | ||
| PCR primers, see Table S3 | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Software, Inc. | N/A |
| FlowJo V10 | FlowJo LLC | N/A |
| Adobe photoshop | Adobe software | N/A |
| Aperio ImageScope | Leica Biosystems | N/A |
| Rstudio | Rstudio | N/A |
Highlights.
Highly heterogeneous adipocyte lineage cells are defined in developing mouse skin
WNT and IL-1 are identified as key suppressing or activating signals for dermal adipogenesis
Wound-induced adipogenesis is triggered by neutrophils through IL-1R signaling
WNT activation in adipocytes promotes lipolysis and myofibroblast formation
ACKNOWLEDGMENTS
L.-j.Z. is supported by the National Key R&D Program of China (2020YFA0112901), NSFC (81971551 and K2918001), and Fujian Provincial NSF (2020J06006). L.S. is supported by NSFC (82103702) and the China Postdoctoral Science Foundation (2020M682095). R.L.G. is supported by NIH P50AR080594, R01AR074302, R37AI052453, U01AI152038, and R01AR076082. M.V.P. is supported by the LEO Foundation (AW-RAM-19-400008 and OC-20-000611), Chan Zuckerberg Initiative (AN-0000000062), W.M. Keck Foundation (5634988), NSF DMS1951144, and NIH U01-AR073159, R01-AR079470, R01-AR079150, R21-AR078939, and P30-AR075047. We thank Dr. Jiahuai Han from Xiamen University for providing the IL1r1−/− mice and Dr. Wen-Hsien Liu from Xiamen University for providing the Gsk3a/3b-floxed mice. We thank the flow cytometry and confocal microscopic core facility at Xiamen University for flow cytometry, sorting, and imaging studies.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112647.
DECLARATION OF INTERESTS
R.L.G. is a co-founder, scientific advisor, consultant for, and has equity in MatriSys Biosciences.
Data and code availability
The accession numbers for the raw data files of the scRNA-seq analyses reported in this paper are deposited in the GEO database under accession codes: GSE196800 and GSE189241.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
REFERENCES
- 1.Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, and Gallo RL (2015). Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347, 67–71. 10.1126/science.1260972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Z, Shao M, Hepler C, Zi Z, Zhao S, An YA, Zhu Y, Ghaben AL, Wang MY, Li N, et al. (2019). Dermal adipose tissue has high plasticity and undergoes reversible dedifferentiation in mice. J. Clin. Invest 129, 5327–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen SX, Zhang LJ, and Gallo RL (2019). Dermal white adipose tissue: a newly recognized layer of skin innate defense. J. Invest. Dermatol 139, 1002–1009. 10.1016/j.jid.2018.12.031. [DOI] [PubMed] [Google Scholar]
- 4.Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, Ferron SR, Herault Y, Pavlovic G, Ferguson-Smith AC, and Watt FM (2013). Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504, 277–281. 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, Driskell RR, Rosenthal N, Biernaskie J, and Horsley V (2021). Fibroblasts: origins, definitions, and functions in health and disease. Cell 184, 3852–3872. 10.1016/j.cell.2021.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joost S, Annusver K, Jacob T, Sun X, Dalessandri T, Sivan U, Sequeira I, Sandberg R, and Kasper M (2020). The molecular anatomy of mouse skin during hair growth and rest. Cell Stem Cell 26, 441–457.e7. 10.1016/j.stem.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Wen Q, Mithieux SM, and Weiss AS (2020). Elastin biomaterials in dermal repair. Trends Biotechnol. 38, 280–291. 10.1016/j.tibtech.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Alhajj M, and Goyal A (2022). Physiology, granulation tissue. In Stat-Pearls. [PubMed] [Google Scholar]
- 9.Huang J, Heng S, Zhang W, Liu Y, Xia T, Ji C, and Zhang LJ (2022). Dermal Extracellular Matrix molecules in skin development, homeostasis, wound regeneration and diseases. Semin. Cell Dev. Biol 128, 137–144. 10.1016/j.semcdb.2022.02.027. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt BA, and Horsley V (2013). Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Development 140, 1517–1527. 10.1242/dev.087593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakers A, De Siqueira MK, Seale P, and Villanueva CJ (2022). Adipose-tissue plasticity in health and disease. Cell 185, 419–446. 10.1016/j.cell.2021.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, and Seale P (2019). Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364, eaav2501. 10.1126/science.aav2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lynch MD, and Watt FM (2018). Fibroblast heterogeneity: implications for human disease. J. Clin. Invest 128, 26–35. 10.1172/JCI93555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, and Toftgård R (2008). Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet 40, 1291–1299. 10.1038/ng.239. [DOI] [PubMed] [Google Scholar]
- 15.Guo L, Li X, and Tang QQ (2015). Transcriptional regulation of adipocyte differentiation: a central role for CCAAT/enhancer-binding protein (C/EBP) beta. J. Biol. Chem 290, 755–761. 10.1074/jbc.R114.619957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrero R, Rainer P, and Deplancke B (2020). Toward a consensus view of mammalian adipocyte stem and progenitor cell heterogeneity. Trends Cell Biol. 30, 937–950. 10.1016/j.tcb.2020.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Camps J, Breuls N, Sifrim A, Giarratana N, Corvelyn M, Danti L, Grosemans H, Vanuytven S, Thiry I, Belicchi M, et al. (2020). Interstitial cell remodeling promotes aberrant adipogenesis in dystrophic muscles. Cell Rep. 31, 107597. 10.1016/j.celrep.2020.107597. [DOI] [PubMed] [Google Scholar]
- 18.Kudo A (2011). Periostin in fibrillogenesis for tissue regeneration: periostin actions inside and outside the cell. Cell. Mol. Life Sci 68, 3201–3207. 10.1007/s00018-011-0784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kielty CM, Sherratt MJ, and Shuttleworth CA (2002). Elastic fibres. J. Cell Sci 115, 2817–2828. [DOI] [PubMed] [Google Scholar]
- 20.Zhang LJ, Guerrero-Juarez CF, Chen SX, Zhang X, Yin M, Li F, Wu S, Chen J, Li M, Liu Y, et al. (2021). Diet-induced obesity promotes infection by impairment of the innate antimicrobial defense function of dermal adipocyte progenitors. Sci. Transl. Med 13, eabb5280. 10.1126/scitranslmed.abb5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang LJ, Chen SX, Guerrero-Juarez CF, Li F, Tong Y, Liang Y, Liggins M, Chen X, Chen H, Li M, et al. (2019). Age-related loss of innate immune antimicrobial function of dermal fat is mediated by transforming growth factor beta. Immunity 50, 121–136.e5. 10.1016/j.immuni.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivera-Gonzalez GC, Shook BA, Andrae J, Holtrup B, Bollag K, Betsholtz C, Rodeheffer MS, and Horsley V (2016). Skin adipocyte stem cell self-renewal is regulated by a PDGFA/AKT-Signaling Axis. Cell Stem Cell 19, 738–751. 10.1016/j.stem.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chia JJ, Zhu T, Chyou S, Dasoveanu DC, Carballo C, Tian S, Magro CM, Rodeo S, Spiera RF, Ruddle NH, et al. (2016). Dendritic cells maintain dermal adipose-derived stromal cells in skin fibrosis.J. Clin. Invest 126, 4331–4345. 10.1172/JCI85740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdul AURM, De Silva B, and Gary RK (2018). The GSK3 kinase inhibitor lithium produces unexpected hyperphosphorylation of beta-catenin, a GSK3 substrate, in human glioblastoma cells. Biol. Open 7, bio030874. 10.1242/bio.030874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JY, Kang MB, Jang SH, Qian T, Kim HJ, Kim CH, Kim Y, and Kong G (2009). Id-1 activates Akt-mediated Wnt signaling and p27(Kip1) phosphorylation through PTEN inhibition. Oncogene 28, 824–831. 10.1038/onc.2008.451. [DOI] [PubMed] [Google Scholar]
- 26.Reusch JE, Colton LA, and Klemm DJ (2000). CREB activation induces adipogenesis in 3T3-L1 cells. Mol. Cell Biol 20, 1008–1020. 10.1128/MCB.20.3.1008-1020.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harshuk-Shabso S, Dressler H, Niehrs C, Aamar E, and Enshell-Seijffers D (2020). Fgf and Wnt signaling interaction in the mesenchymal niche regulates the murine hair cycle clock. Nat. Commun 11, 5114. 10.1038/s41467-020-18643-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins CA, and Watt FM (2008). Dynamic regulation of retinoic acid-binding proteins in developing, adult and neoplastic skin reveals roles for beta-catenin and Notch signalling. Dev. Biol 324, 55–67. 10.1016/j.ydbio.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 29.Philippeos C, Telerman SB, Oulès B, Pisco AO, Shaw TJ, Elgueta R, Lombardi G, Driskell RR, Soldin M, Lynch MD, and Watt FM (2018). Spatial and single-cell transcriptional profiling identifies functionally distinct human dermal fibroblast subpopulations. J. Invest. Dermatol 138, 811–825. 10.1016/j.jid.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lange M, Bergen V, Klein M, Setty M, Reuter B, Bakhti M, Lickert H, Ansari M, Schniering J, Schiller HB, et al. (2022). CellRank for directed single-cell fate mapping. Nat. Methods 19, 159–170. 10.1038/s41592-021-01346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno CS (2020). SOX4: the unappreciated oncogene. Semin. Cancer Biol 67, 57–64. 10.1016/j.semcancer.2019.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z, Zhong Y, Chen YJ, and Chen H (2019). SOX11 regulates apoptosis and cell cycle in hepatocellular carcinoma via Wnt/beta-catenin signaling pathway. Biotechnol. Appl. Biochem 66, 240–246. 10.1002/bab.1718. [DOI] [PubMed] [Google Scholar]
- 33.Carroll LS, and Capecchi MR (2015). Hoxc8 initiates an ectopic mammary program by regulating Fgf10 and Tbx3 expression and Wnt/beta-catenin signaling. Development 142, 4056–4067. 10.1242/dev.128298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, et al. (2009). Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460, 66–72. 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eastman Q, and Grosschedl R (1999). Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol 11, 233–240. 10.1016/s0955-0674(99)80031-3. [DOI] [PubMed] [Google Scholar]
- 36.Côté JA, Lessard J, Pelletier M, Marceau S, Lescelleur O, Fradette J, and Tchernof A (2017). Role of the TGF-beta pathway in dedifferentiation of human mature adipocytes. FEBS Open Bio 7, 1092–1101. 10.1002/2211-5463.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor B, Shah A, and Bielczyk-Maczyńska E (2020). TGF-beta is insufficient to induce adipocyte state loss without concurrent PPARgamma downregulation. Sci. Rep 10, 14084. 10.1038/s41598-020-71100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Mao AS, Seo BR, Zhao X, Gupta SK, Chen M, Han YL, Shih TY, Mooney DJ, and Guo M (2020). Compression-induced dedifferentiation of adipocytes promotes tumor progression. Sci. Adv 6, eaax5611. 10.1126/sciadv.aax5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei S, Duarte MS, Zan L, Du M, Jiang Z, Guan L, Chen J, Hausman GJ, and Dodson MV (2013). Cellular and molecular implications of mature adipocyte dedifferentiation. J. Genomics 1, 5–12. 10.7150/jgen.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorzadeh S, Kohan L, Ghavami S, and Azarpira N (2021). Autophagy and the Wnt signaling pathway: a focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res 1868, 118926. 10.1016/j.bbamcr.2020.118926. [DOI] [PubMed] [Google Scholar]
- 41.Doble BW, Patel S, Wood GA, Kockeritz LK, and Woodgett JR (2007). Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 12, 957–971. 10.1016/j.devcel.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shook BA, Wasko RR, Mano O, Rutenberg-Schoenberg M, Rudolph MC, Zirak B, Rivera-Gonzalez GC, López-Giráldez F, Zarini S, Rezza A, et al. (2020). Dermal adipocyte lipolysis and myofibroblast conversion are required for efficient skin repair. Cell Stem Cell 26, 880–895.e6. 10.1016/j.stem.2020.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onoufriadis A, Hsu CK, Ainali C, Ung CY, Rashidghamat E, Yang HS, Huang HY, Niazi U, Tziotzios C, Yang JC, et al. (2018). Time series integrative analysis of RNA sequencing and MicroRNA expression data reveals key biologic wound healing pathways in keloid-prone individuals. J. Invest. Dermatol 138, 2690–2693. 10.1016/j.jid.2018.05.017. [DOI] [PubMed] [Google Scholar]
- 44.Guerrero-Juarez CF, and Plikus MV (2018). Emerging nonmetabolic functions of skin fat. Nat. Rev. Endocrinol 14, 163–173. 10.1038/nrendo.2017.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng CC, Hu YF, Zhu DH, Cheng Q, Gu JJ, Feng QL, Zhang LX, Xu YP, Wang D, Rong Z, and Yang B (2021). Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases. Nat. Commun 12, 3709. 10.1038/s41467-021-24110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solé-Boldo L, Raddatz G, Schütz S, Mallm JP, Rippe K, Lonsdorf AS, Rodríguez-Paredes M, and Lyko F (2020). Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol 3, 188. 10.1038/s42003-020-0922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Phan QM, Fine GM, Salz L, Herrera GG, Wildman B, Driskell IM, and Driskell RR (2020). Lef1 expression in fibroblasts maintains developmental potential in adult skin to regenerate wounds. Elife 9, e60066. 10.7554/eLife.60066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong H, Sun W, Shen Y, Baláz M, Balázová L, Ding L, Löffler M, Hamilton B, Klöting N, Blüher M, et al. (2022). Identification of a regulatory pathway inhibiting adipogenesis via RSPO2. Nat. Metab 4, 90–105. 10.1038/s42255-021-00509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Winter TJJ, and Nusse R (2021). Running against the Wnt: how wnt/beta-catenin suppresses adipogenesis. Front. Cell Dev. Biol 9, 627429. 10.3389/fcell.2021.627429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reggio A, Rosina M, Palma A, Cerquone Perpetuini A, Petrilli LL, Gargioli C, Fuoco C, Micarelli E, Giuliani G, Cerretani M, et al. (2020). Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/beta-catenin axis. Cell Death Differ. 27, 2921–2941. 10.1038/s41418-020-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, and MacDougald OA (2000). Inhibition of adipogenesis by Wnt signaling. Science 289, 950–953. 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 52.Morosan-Puopolo G, Balakrishnan-Renuka A, Yusuf F, Chen J, Dai F, Zoidl G, Lüdtke THW, Kispert A, Theiss C, Abdelsabour-Khalaf M, and Brand-Saberi B (2014). Wnt11 is required for oriented migration of dermogenic progenitor cells from the dorsomedial lip of the avian dermomyotome. PLoS One 9, e92679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thulabandu V, Chen D, and Atit RP (2018). Dermal fibroblast in cutaneous development and healing. Wiley Interdiscip Rev Dev Biol 7, e307. 10.1002/wdev.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rognoni E, Gomez C, Pisco AO, Rawlins EL, Simons BD, Watt FM, and Driskell RR (2016). Inhibition of beta-catenin signalling in dermal fibroblasts enhances hair follicle regeneration during wound healing. Development 143, 2522–2535. 10.1242/dev.131797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mastrogiannaki M, Lichtenberger BM, Reimer A, Collins CA, Driskell RR, and Watt FM (2016). Beta-catenin stabilization in skin fibroblasts causes fibrotic lesions by preventing adipocyte differentiation of the reticular dermis. J. Invest. Dermatol 136, 1130–1142. 10.1016/j.jid.2016.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bagchi DP, Nishii A, Li Z, DelProposto JB, Corsa CA, Mori H, Hardij J, Learman BS, Lumeng CN, and MacDougald OA (2020). Wnt/beta-catenin signaling regulates adipose tissue lipogenesis and adipocyte-specific loss is rigorously defended by neighboring stromal-vascular cells. Mol. Metabol 42, 101078. 10.1016/j.mol-met.2020.101078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petrella BL, Armstrong DA, and Vincenti MP (2011). CCAAT-enhancer-binding protein beta activation of MMP-1 gene expression in SW1353 cells: independent roles of extracellular signal-regulated and p90/ribosomal S6 kinases. J. Cell. Physiol 226, 3349–3354. 10.1002/jcp.22693. [DOI] [PubMed] [Google Scholar]
- 58.Resende C, Regalo G, Durães C, Pinto MT, Wen X, Figueiredo C, Carneiro F, and Machado JC (2016). Interleukin-1B signalling leads to increased survival of gastric carcinoma cells through a CREB-C/EBPbeta-associated mechanism. Gastric Cancer 19, 74–84. 10.1007/s10120-014-0448-x. [DOI] [PubMed] [Google Scholar]
- 59.Khanjani S, Terzidou V, Lee YS, Thornton S, Johnson MR, and Bennett PR (2011). Synergistic regulation of human oxytocin receptor promoter by CCAAT/enhancer-binding protein and RELA. Biol. Reprod 85, 1083–1088. 10.1095/biolreprod.111.092304. [DOI] [PubMed] [Google Scholar]
- 60.Vumbaca S, Giuliani G, Fiorentini V, Tortolici F, Cerquone Perpetuini A, Riccio F, Sennato S, Gargioli C, Fuoco C, Castagnoli L, and Cesareni G (2021). Characterization of the skeletal muscle secretome reveals a role for extracellular vesicles and IL1alpha/IL1beta in restricting fibro/adipogenic progenitor adipogenesis. Biomolecules 11, 1171. 10.3390/biom11081171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. (2017). Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun 8, 14532. 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, et al. (2012). Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun 3, 735. 10.1038/ncomms1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gay D, Ghinatti G, Guerrero-Juarez CF, Ferrer RA, Ferri F, Lim CH, Murakami S, Gault N, Barroca V, Rombeau I, et al. (2020). Phagocytosis of Wnt inhibitor SFRP4 by late wound macrophages drives chronic Wnt activity for fibrotic skin healing. Sci. Adv 6, eaay3704. 10.1126/sciadv.aay3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kadota T, Fujita Y, Araya J, Watanabe N, Fujimoto S, Kawamoto H, Minagawa S, Hara H, Ohtsuka T, Yamamoto Y, et al. (2021). Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-beta-WNT crosstalk. J. Extracell. Vesicles 10, e12124. 10.1002/jev2.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benias PC, Wells RG, Sackey-Aboagye B, Klavan H, Reidy J, Buonocore D, Miranda M, Kornacki S, Wayne M, Carr-Locke DL, and Theise ND (2018). Structure and distribution of an unrecognized interstitium in human tissues. Sci. Rep 8, 4947. 10.1038/s41598-018-23062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haydont V, Neiveyans V, Perez P, Busson E, Lataillade J, Asselineau D, and Fortunel NO (2020). Fibroblasts from the human skin dermo-hypodermal junction are distinct from dermal papillary and reticular fibroblasts and from mesenchymal stem cells and exhibit a specific molecular profile related to extracellular matrix organization and modeling. Cells 9, 368. 10.3390/cells9020368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Correa-Gallegos D, Jiang D, Christ S, Ramesh P, Ye H, Wannemacher J, Kalgudde Gopal S, Yu Q, Aichler M, Walch A, et al. (2019). Patch repair of deep wounds by mobilized fascia. Nature 576, 287–292. 10.1038/s41586-019-1794-y. [DOI] [PubMed] [Google Scholar]
- 68.Liu C, Ma L, Wang Y, Zhao J, Chen P, Chen X, Wang Y, Hu Y, Liu Y, Jia X, et al. (2021). Glycogen synthase kinase 3 drives thymocyte egress by suppressing beta-catenin activation of Akt. Sci. Adv 7, eabg6262. 10.1126/sciadv.abg6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rose S, Misharin A, and Perlman H (2012). A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. CytometryA. 81, 343–350. 10.1002/cyto.a.22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu R, Yallowitz A, Qin A, Wu Z, Shin DY, Kim JM, Debnath S, Ji G, Bostrom MP, Yang X, et al. (2018). Targeting skeletal endothelium to ameliorate bone loss. Nat. Med 24, 823–833. 10.1038/s41591-018-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Brien CE, Bonanno L, Zhang H, and Wyss-Coray T (2015). Beclin 1 regulates neuronal transforming growth factor-beta signaling by mediating recycling of the type I receptor ALK5. Mol. Neurodegener 10, 69. 10.1186/s13024-015-0065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Majzoub RN, Chan CL, Ewert KK, Silva BFB, Liang KS, and Safinya CR (2015). Fluorescence microscopy colocalization of lipid-nucleic acid nanoparticles with wildtype and mutant Rab5-GFP: a platform for investigating early endosomal events. Biochim. Biophys. Acta 1848, 1308–1318. 10.1016/j.bbamem.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zachara M, Rainer PY, Hashimi H, Russeil JM, Alpern D, Ferrero R, Litovchenko M, and Deplancke B (2022). Mammalian adipogenesis regulator (Areg) cells use retinoic acid signalling to be non- and anti-adipogenic in age-dependent manner. EMBO J. 41, e108206. 10.15252/embj.2021108206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the raw data files of the scRNA-seq analyses reported in this paper are deposited in the GEO database under accession codes: GSE196800 and GSE189241.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.