

Summary
Despite tremendous progress in detecting DNA variants associated with human disease, interpreting their functional impact in a high-throughput and single-base resolution manner remains challenging. Here, we develop a pooled prime editing screen method, PRIME, which can be applied to characterize thousands of coding and non-coding variants in a single experiment with high reproducibility. To showcase its applications, we first identified essential nucleotides for a 716 bp MYC enhancer via PRIME-mediated single-base resolution analysis. Next, we applied PRIME to functionally characterize 1,304 GWAS-identified non-coding variants associated with breast cancer and 3,699 variants from ClinVar. We discovered that 103 non-coding variants and 156 variants of uncertain significance are functional via affecting cell fitness. Collectively, we demonstrate PRIME is capable of characterizing genetic variants at single-base resolution and scale, advancing accurate genome annotation for disease risk prediction, diagnosis, and therapeutic target identification.
Keywords: High-throughput screens, single-base resolution, prime editing, enhancer, disease variants
Graphical Abstract
eTOC Blurb
Ren et al., present PRIME, a genome-scale method to characterize genome sequences by prime editing at the base-pair resolution. PRIME enables the analysis of functional DNA elements at the nucleotide resolution by introducing all possible nucleotide substitutions. Additionally, PRIME can be leveraged to characterize disease-associated genetic variations at scale.
Introduction
Advances in genome sequencing have led to the identification of hundreds of millions of genetic variants in the human population, with a fraction conferring risk for common illnesses such as diabetes, neurological disorders, and cancers.1 A major barrier to understanding the genetic underpinnings of these complex diseases is the paucity of functional annotation for disease-associated variants, especially because such variants are predominantly located within non-coding regions. Growing evidence suggests that non-coding risk variants may contribute to disease pathogenesis by disrupting gene regulation.2 Even protein-coding variants discovered from individuals with disease are frequently classified as Variants of Uncertain Significance (VUS). Therefore, more precise and higher throughput functional characterization methods for elucidating disease-associated variant function at single-base resolution, and multiplexed across genomic loci, are necessary to realize the potential of personalized medicine.
The development of genome editing technologies has enabled us to perturb and assess DNA sequences in desired regions at a large scale. However, there are still fundamental barriers to utilizing these methods for precision genome annotation. For example, CRISPR-mediated genetic screens have been applied for characterizing both disease-associated genes and cis-regulatory regions,3–5 but CRISPR screens, including CRISPRi, CRISPRa, CRISPR deletion, and CRISPR indel, failed to directly pinpoint causal variants for diseases at the single-base resolution. Other methods of characterizing DNA variants by knock-in via CRISPR-mediated homologous recombination are inefficient6 and low throughput.7 Base editors also have limitations, as each editor introduces one specific mutation (C→T, A→G, T→C, or G→A) with varied target efficiencies.8 Thus, there is still a significant deficit in methods for effectively characterizing all possible disease-associated variants in human health and diseases. Robust high-throughput methods making desired edits at single-base resolution are urgently needed to achieve a better understanding of the genetic underpinnings of disease.
Design
Prime editing (PE), a versatile and precise genetic engineering method, has been developed to introduce any type of edit, including point mutation, insertion, and deletion.9 PE employs the Streptococcus pyogenes Cas9 (SpCas9) H840A nickase and Moloney murine leukemia virus (M-MLV) reverse transcriptase (RT). The spacer in the prime editing guide RNA (pegRNA) directs the Cas9 nickase and M-MLV complex to the target site, while the RT template sequence provides the desired editing information. Thus, both targeting and editing information can be easily programmed in the same pegRNA to perform single nucleotide substitution, insertion or deletion. PE3 can further increase editing efficiency by promoting the replacement of non-edited strands using an additional single-guide (sgRNA) for nicking.10 Prime editors’ capacity for precision genome editing suggests the possibility of comprehensive and high-throughput variant-level genome manipulation. Recently, PE screens were used to identify VUS at the NPC1 locus based on a lysosome functional assay by transfection of pegRNAs and targeted sequencing of this region.11 Although transient transfection of PE machinery followed by targeted sequencing of the edited locus enables the identification of editing events, its scope is limited to just that locus, and thus, scaling up for massively parallel assessment of multiple loci is not feasible. Besides increased throughput, improved control of transgene copy number, stable expression of PE machinery, and direct loci comparison are also desired.
Here, we enable high-throughput pooled screens of thousands of DNA variants in the human genome by lentiviral delivery of PE, namely PRIME. We demonstrate the utility of PRIME for three different applications, including the single-base resolution analysis of a 716 bp enhancer, the functional characterization of 1,304 breast cancer-associated variants, and the evaluation of 3,699 clinical variants’ impact on cell fitness of MCF7 cells. Our results establish the generalizability of PRIME for precisely characterizing genetic variants in the human genome.
Results
Optimization of PE in mammalian cells via lentiviral delivery
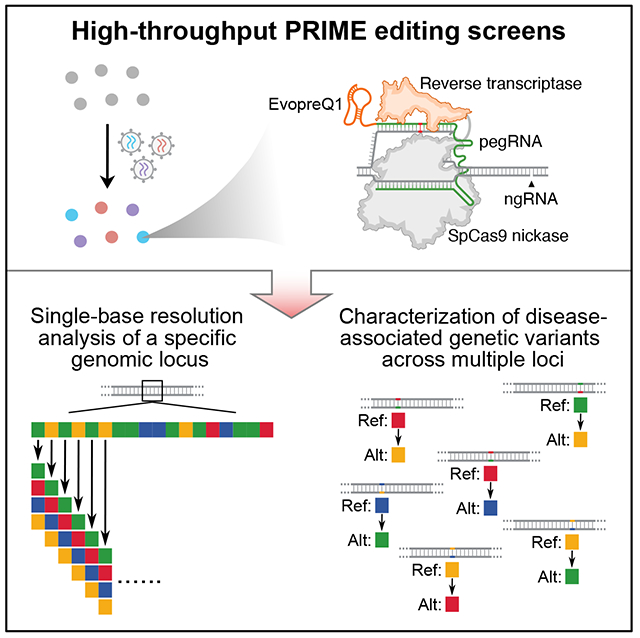
To enable PE screens with lentiviral delivery, we assessed the PE efficiency using two previously tested loci (EMX1 +1G>C, FANCF +5G>T).10 We initially installed PE3 by infecting MCF7 cells using three different viruses: 1) virus expressing SpCas9 (H840A) nickase (nCas9) and M-MLV RT; 2) virus expressing pegRNA; 3) virus expressing nick sgRNA (ngRNA). Unfortunately, this strategy yielded less than 1% PE efficiency with a relatively high indel rate due to the low efficiency of coinfecting three different viruses in the same cell (Figures 1A and S1A).
Figure 1. Optimizing PE efficiency in mammalian cells using lentiviral delivery.
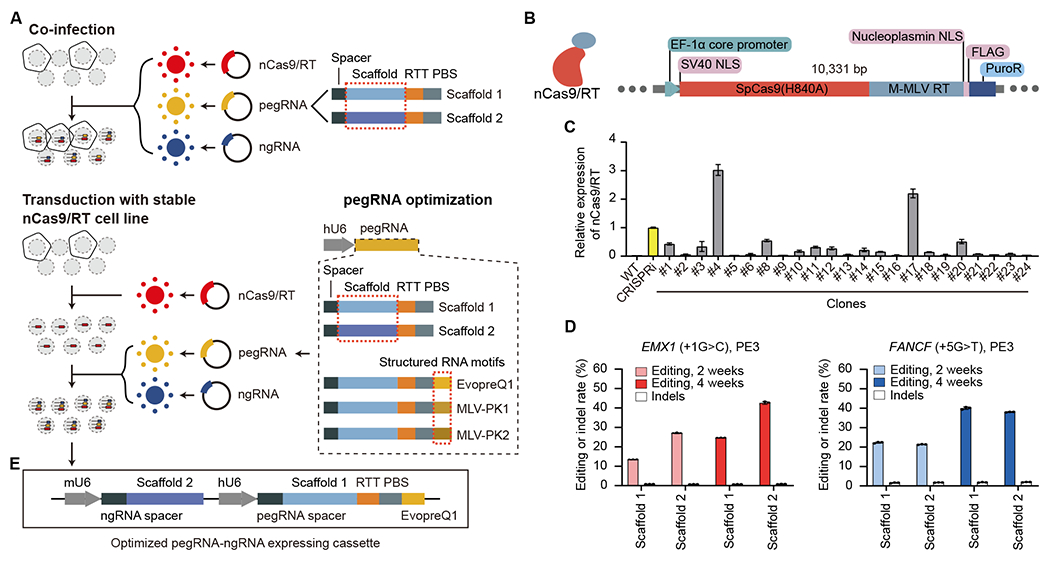
(A) Optimizing PE efficiency in MCF7 cell lines. Top: co-infecting three different viruses to deliver PE machinery. Bottom: pegRNA and ngRNA viral infection of clonal MCF7 line stably expressing nCas9 and M-MLV RT. (B) Lentiviral construct for generating nCas9/RT expressing MCF7 clones. PuroR, Puromycin resistance gene. (C) RT-qPCR analysis showing the relative expression of nCas9/RT in different clones, normalized to the dCas9 expression of an established CRISPRi iPSC line (Yellow). Error bars represent the s.e.m. (D) The editing efficiency and indel rate for EMX1 and FANCF loci at 2-week and 4-week after PE installation using two different RNA scaffolds. Error bars represent the s.d. (E) Improved vector for expression of pegRNA and ngRNA for PRIME. RTT: reverse transcription template. PBS: primer binding site. See also Figure S1.
To increase PE efficiency and facilitate a pooled screening approach with a lentiviral library, we infected MCF7 cells with lentivirus containing an nCas9 and M-MLV RT (nCas9/RT) stable expression cassette (Figure 1B). After puromycin selection, we isolated multiple clones and selected one with the highest nCas9 expression (Figure 1C, clone #4, Figure S1B) for subsequent experiments. The stable expression of nCas9/RT allows for high efficiency pegRNA/ngRNA packaging and lentiviral delivery, with greater editing efficiency than the co-infection method (Figures 1A and 1D). To further improve PE efficiency, we assessed editing efficiency using three different structured RNA motifs (EvopreQ1, MLV-PK1, and MLV-PK2) at the 3’ terminus of the pegRNA.12–14 Cells treated with pegRNAs containing structured RNA motifs exhibited consistently higher editing efficiencies at both the EMX1 and FANCF locus compared to using pegRNAs without structured RNA motifs (Figures 1A and S1C), therefore we incorporated evopreQ1 into the pegRNA design due to its shorter length compared to the other two. Scaffold 110 and 215 had no significant effects on PE efficiency, suggesting the feasibility of dual pegRNA and ngRNA delivery from the same viral particle (Figure 1D). All PE experiments in clonal MCF7 cells (MCF7-nCas9/RT) exhibited relatively low indel rates (0.7% to 1.95%). Thus, we used MCF7-nCas9/RT cells and lentiviral delivery of both the pegRNA with scaffold 1 and ngRNA with scaffold 2 in the same construct for PRIME screens (Figure 1E).
PRIME enables single-base resolution analyses of enhancer function
Enhancers can modulate cell type-specific gene expression and are highly enriched with disease-associated variants. Knowledge of the endogenous function for each nucleotide in enhancers should reveal crucial transcription factors that govern enhancer activation and facilitate the development of better models for gene regulatory networks and the prediction of disease-associated non-coding variant regulatory effects. To test whether PRIME can quantify the impact of each base in an enhancer, we focused on an MCF7-specific MYC enhancer,16 405 kb downstream of MYC, displaying enhancer signatures, including open chromatin, H3K27ac, and H3K4me1 signals, and a chromatin loop with the MYC promoter (Figure 2A). Deletion of this enhancer caused an 85% downregulation of MYC expression confirming its enhancer activity for MYC (Figure S2A). Since MYC downregulation is correlated with MCF7 cell survival,17 we performed a PRIME-enabled high-throughput single-base resolution analysis screen of this enhancer in MCF7 cells using the cell survival phenotype (Figure 2B).
Figure 2. Functional characterization of a MYC enhancer by single-base resolution analysis using PRIME.
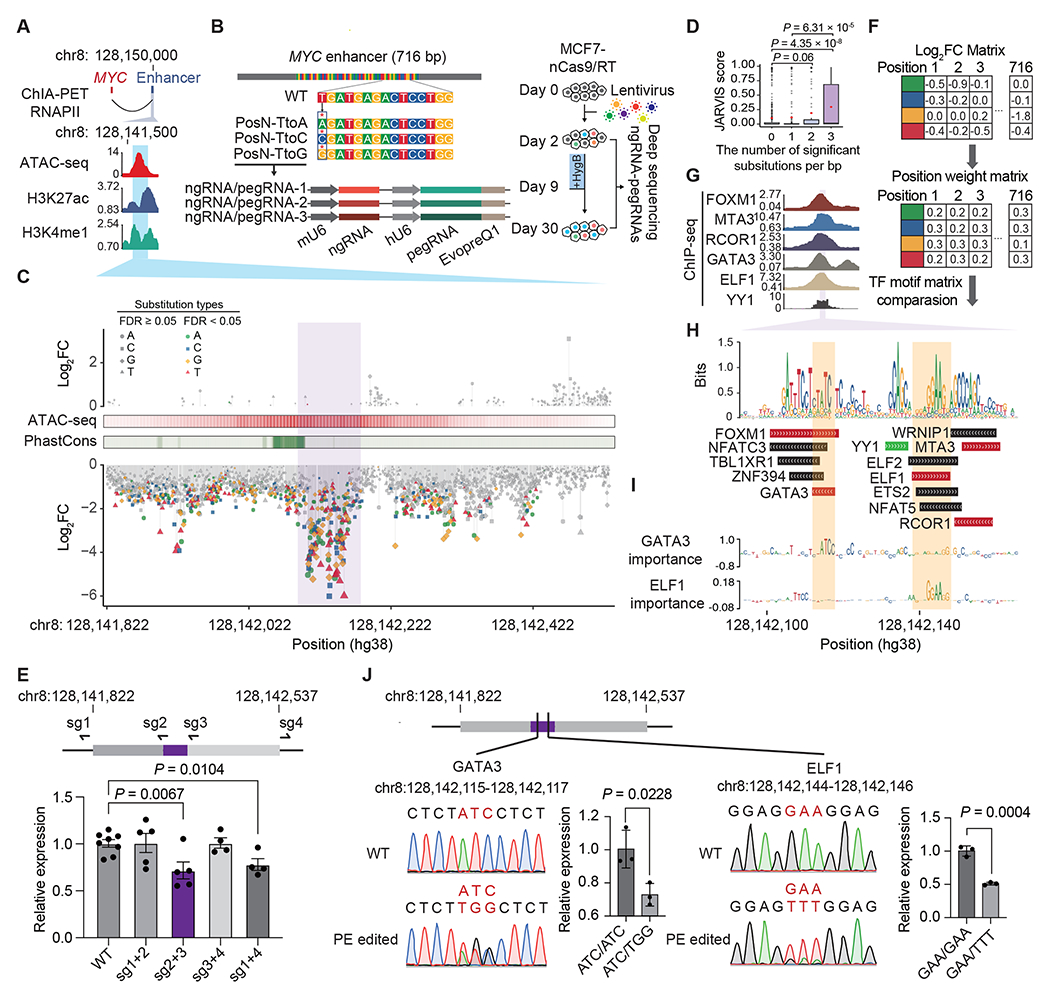
(A) The target enhancer is downstream of MYC. The blue area indicates the region selected for PRIME. (B) Diagram showing the design of single-base resolution analysis screening at the 716 bp enhancer. Each nucleotide was subjected to substitution with three nucleotides by PE. Each substitution event was covered by three uniquely designed pegRNA/ngRNA pairs. (C) Log2(fold change) of each substitution at each base pair ordered by their genomic locations. Mutations with a significant effect on cell fitness are colored. ATAC-seq signals and conservation scores calculated by PhastCons are shown. The purple area indicates the core enhancer region. (D) JARVIS scores for base pairs with different numbers of significant substitutions. Box plots indicate median, IQR, Q1 – 1.5 × IQR, and Q3 + 1.5 × IQR. Outliers are shown as gray dots. Mean values are shown as red dots. (E) Design of sgRNAs for deleting distinct regions of the MYC enhancer (Top) and MYC expression levels in different regional deletion clones (Bottom). (F) The creation of a functional PWM for identifying potential TF binding sites. (G) ChIP-seq signals of 6 TFs in MCF7. The purple region indicates the core enhancer region. (H) The sequence logo plot for the core enhancer region generated by the functional PWM and the matched TF binding sites. The TF binding supported by ChIP-seq data in G are labeled in red. The YY1 (green) binding is predicted by Avocado. (I) Dense tracks showing BPNet model-derived nucleotide importance scores for GATA3 and ELF1 binding sites. (J) The impact of mutations in GATA3 and ELF1 motifs measured by MYC expression. For E and J, dots show individual replicate values and error bars represent s.e.m. P values in D, E and J were calculated by two-tailed two-sample t-test. See also Figure S2 and Table S1.
We designed a library of 6,252 pairs of pegRNA/ngRNA to generate 2,127 single nucleotide substitutions within the 716 bp MYC enhancer region (Table S1). Specifically, we changed the original base into three other nucleotides, and each event was independently evaluated three times in the same screen (Figure 2B). We also included 94 positive control pegRNA/ngRNA pairs, which introduced stop codons (iSTOPs) in MYC, and 398 negative control pegRNA/ngRNA pairs. 245 of the negative controls were non-human genome targeting, and 153 targeted the AAVS1 safe harbor locus (Table S1). We then infected MCF7-nCas9/RT cells with lentiviral libraries expressing these pegRNA/ngRNA pairs (Figure S2B). Two days after infection, virus-transduced cells were hygromycin selected for one week and expanded in regular media for another 3 weeks. We collected cells at 2 and 30 days post-infection, amplified the integrated pegRNA/ngRNA pairs, and determined the relative depletion or enrichment of each pegRNA/ngRNA between these two time points by deep sequencing (Figure 2B). We performed this screen 3 times (Figure S2C) and used negative controls, including non-human targeting and AAVS1 targeting paired pegRNA/ngRNAs for data normalization. Fold changes (FC) for each pegRNA/ngRNA pair between day 2 and day 30 samples were calculated using the MAGeCK pipeline18 (Table S1). As expected, 78% (73/94) of iSTOPs were depleted (log2FC < 0) 30 days post-infection. iSTOP depletion rates were negatively correlated with their distance from the transcription start site (TSS) of MYC, consistent with the observation that gene knockout is more efficient when perturbations are introduced at the 5’ terminus19 (Figure S2D). In addition, two iSTOPs (amino acid position 350 and 355) targeting the region between the nuclear localization signal (NLS) and the carboxy-terminal domain (CTD) domain were also significantly depleted (Figure S2D). The N-terminus of MYC contains its core transcription transactivation domain which binds multiple partners.20 It is possible that those two iSTOPs created a truncated MYC still capable of binding to cofactors, but unable to bind to MYC DNA targets, interfering with the functions of wild type MYC and its cofactors.
To investigate the effects of each nucleotide on enhancer function, we defined sensitive base pairs (SBP) as nucleotides that affect cell fitness when substituted at least once (FDR < 0.05, |log2FC| > 1). 334 of the 716 (46.6%) tested base pairs were SBP with log2FC < −1 (Table S1), indicating that mutations at those locations reduce enhancer activity and cell fitness. 23.1% (77/334) of SBPs were depleted at day 30 with all three substitutions (FDR < 0.05, log2FC < −1). Additionally, none of the tested sequences were significantly enriched at day 30 with increased cell growth phenotype, indicating that perturbation of these sequences exclusively attenuated enhancer activity (Figure 2C). Encouragingly, SBPs with two or more significant substitutions (n = 172) were predicted to be more deleterious than SBPs with only one significant substitution (n = 162) or non-SBPs (n = 382) by JARVIS21 (Figure 2D). We further established a continuous bin density analysis, detecting variation in SBP density along the enhancer (Figures S2E and S2F) and identified the core enhancer region with a minimal slope cut-off of 0.43 (Z score-derived P < 0.05) of the cumulative curve of SBPs with three significant substitutions, as a larger slope value indicates a higher density of SBPs in the region. The core enhancer region (chr8:128,142,093-128,142,181, hg38) contains SBPs with the most extensive fold changes when mutated, indicating its strong effect on enhancer activity (Figure 2C). Notably, the enhancer’s core sequence, while colocalized with an open chromatin summit, located next to a highly conserved region (Figure 2C). This is not surprising because enhancers undergo rapid evolutionary changes compared to protein-coding sequences.22 Deletions of either the core enhancer region or the entire enhancer resulted in MYC downregulation at similar levels. Conversely, deleting other regions in the enhancer did not affect MYC expression (Figures 2E and S2G), confirming the functional significance of the core enhancer region.
Our functional data provide a unique opportunity to calculate and construct a position weight matrix (PWM). Using fold changes for each nucleotide, we generated a functional PWM (Figure 2F). Comparing our functional PWM with curated transcription factors (TFs) motifs from the JASPAR, HOCOMOCO, and SwissRegulon databases,23–25 we identified 13 TFs with matched motif PWMs (Figures 2G and 2H; Table S1). Five predicted TFs (GATA3, ELF1, FOXM1, MTA3 and RCOR1) have already been shown to bind to the MYC enhancer based on ENCODE ChIP-seq datasets,26 and YY1 is predicted to bind to this enhancer in MCF7 by Avocado through the ENCODE project27 (Figure 2G). Essential nucleotides for the GATA3 and ELF1 binding motifs identified by our screens were consistent with those imputed by BPNet28 (Figure 2I). Furthermore, we altered the GATA3 binding site by substituting the motif sequences of ATC with TGG, and the ELF1 binding site by replacing GAA with TTT (Figure 2J), respectively, as these bases represented strongest effects on enhancer activity for these two motifs based on PRIME results. Heterozygous clones for each of these alterations exhibited a significant reduction in MYC expression compared to the wild type counterparts (Figure 2J). These results confirm the pivotal functional role of these specific TF binding sites in enhancer activity. Combined, we demonstrate that PRIME is effective in annotating functional nucleotides in cis-regulatory elements.
Characterization of breast cancer-associated variants
Next, we tested the feasibility of characterizing a large number of disease-associated DNA variants across various genomic loci, including non-coding variants from GWAS and variants detected from clinical samples. For GWAS-identified variants, we focused on breast cancer, the most common cancer in women in the U.S.29 We used the summary statistics from the largest GWAS to date, including samples of mostly European ancestry.30 Candidate genes from a comprehensive fine mapping effort for this GWAS31 overlapping with growth phenotype genes prioritized by CRISPR screens32,33 were selected. These include: CCND1, PSMD6, MYC, UBA52, DYNC1I2, ESR1, MRPS18C, NOL7, EWSR1, BRCA2, and GRHL2, which were negatively selected in a CRISPR knockout screen, and CUX1, CASP8, and TNFSF10, which are tumor suppressor genes and positively selected in a CRISPR knockout screen (Figure S3A). We then selected 1,304 single nucleotide polymorphisms (SNPs) (Figure S3B; Table S2) within 500 kb upstream and downstream of these genes that were previously associated with breast cancer30 and had been implicated as possibly acting through these genes.31 We also selected 3,699 variants from the ClinVar database (Figure S3C), 2,840 of which were identified from patients who were tested for hereditary breast cancer.34 To systematically assess variants’ impact on cell fitness, we designed two libraries: one to introduce reference alleles (Ref library) and another to introduce alternative alleles (Alt library) targeting the selected variants (Figure 3A; Table S2), each with 250 non-targeting pegRNA/ngRNA pairs added as negative controls. For the Alt library, 115 pegRNA/ngRNA pairs introducing stop codons (iSTOPs) in 23 MCF7 growth-related genes were included as positive controls, while pegRNA/ngRNA pairs introducing reference sequences were used for those loci in the Ref library. The cloned plasmids were packaged into lentiviral libraries and transduced into MCF7-nCas9/RT cells. Cells were collected 2 and 32 days post infection, and pegRNA/ngRNA pairs were amplified and deep sequenced (Figure 3B). PRIME replicates using either Ref or Alt library (n = 4) were reproducible at the read count level (Figure S3D).
Figure 3. PRIME reveals functional SNPs associated with breast cancer.
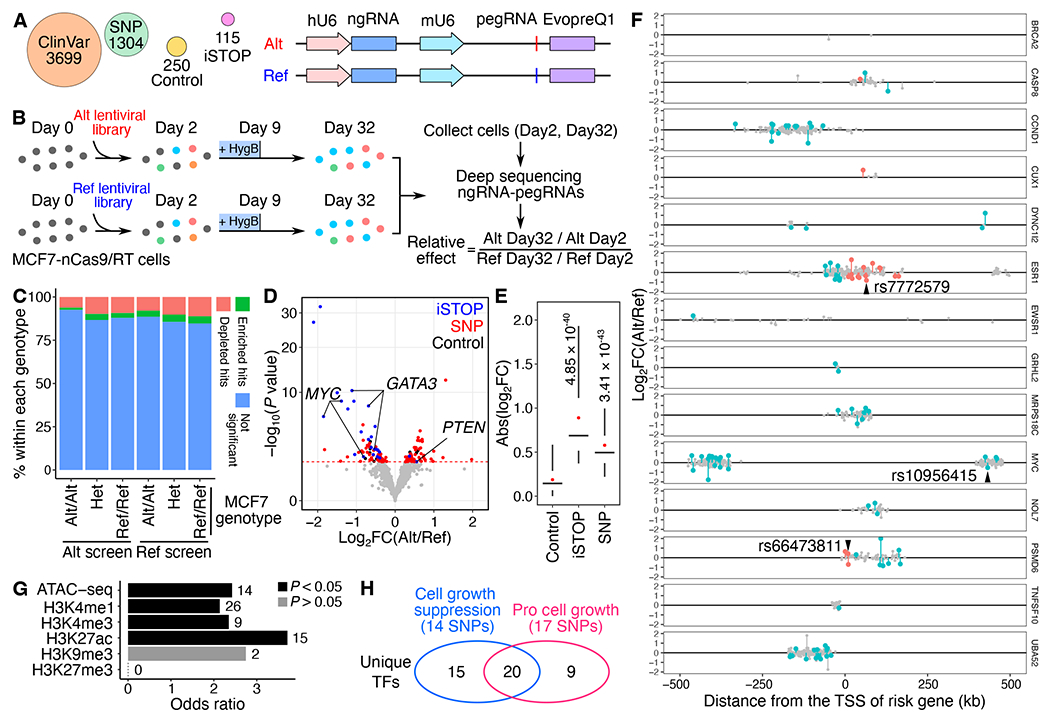
(A) Alt and Ref library design overview. For each variant, pegRNA/ngRNA pairs introducing either the Alt or Ref allele were designed. (B) Workflow of PRIME with Alt and Ref libraries. MCF7-nCas9/RT cells were infected with either lentiviral library. The relative effect of each variant was determined based on its relative impact on cell growth between Alt versus Ref alleles. (C) The percentage of significant hits (FDR < 0.05) identified from Alt and Ref screens for Alt/Alt, Het, and Ref/Ref genotypes in MCF7. (D) The functional SNPs (red) with either a positive or a negative impact on cell growth were determined by their relative effect in the Alt versus Ref screens. Blue dots represent significant iSTOPs, and black dots represent controls. The red dashed line indicates 0.05 FDR. (E) Absolute effects of identified functional iSTOPs and SNPs are higher than the effects of negative controls (P values were calculated by two-tailed two-sample t-test). Box plots indicate the median, IQR, Q1 – 1.5 × IQR, and Q3 + 1.5 × IQR. Red dots indicate the mean. (F) The genomic distance of SNPs tested at each risk locus relative to each gene’s TSS. Red dots are functional SNPs within gene bodies, blue dots are functional SNPs in distal regions, and gray dots are SNPs with non-significant effects. Three selected SNPs for validation were labeled. (G) Relative enrichment of genomic features for identified functional SNPs (P values were calculated by two-tailed Fisher’s exact test). The numbers of SNPs overlapping each genomic feature are labeled next to each bar. (H) Venn diagram showing the numbers of unique transcription factors (TFs) with differential binding sites centered on functional SNPs. The numbers of SNPs that alter TF binding sites are shown in the parentheses. See also Figure S3, Tables S2 and S3.
From Alt library screens, 33.04% (38/115) of iSTOPs showed a significant cell fitness effect (FDR < 0.05), which is comparable to the 31.8% positivity rate of iSTOPs for common essential genes reported from the base editing screen in MCF7 cells.35 Furthermore, the fold changes for iSTOPs were highly correlated with those for sgRNAs from MCF7 CRISPR knockout screens of the same genes32 (Figure S3E). More pegRNA/ngRNA pairs were depleted (FDR < 0.05, Alt screen n = 322 and Ref screen n = 337) than enriched (FDR < 0.05, Alt screen n = 148 and Ref screen n = 209) (binomial test, P = 4.78×10−8 for Alt screen and P = 6.85×10−16 for Ref screen) for both Alt and Ref screens on day 32 compared to day 2 (Figures S3F and S3G; Table S2). Theoretically, when a designed peg/ngRNA pair matches the wild type MCF7 genotypes, they should have no effect on cell growth. Notably, however, certain pegRNAs matching the wild type MCF7 genotype, exhibited significant effects on cell growth beyond what was predicted, while the proportion of significant hits for each genotype group were independent of initial MCF7 genotypes (Chi-square test P = 0.9998 on the Ref library and P = 0.999 on the Alt library, Cochran-Mantel-Haenszel test P = 0.9665 for the Ref library and Alt library together). For example, in the Ref library, 11.2% (59 out of 528) of pegRNAs at sites with a Ref/Ref MCF7 genotype exhibited significant depletion, similar to the 10.2% (55 out of 540) at heterozygous sites and 7.9% (18 out of 227) at Alt/Alt genotype sites (Figure 3C). These changes at sites where alleles were not expected to change suggests the presence of undesired consequences of constitutive nCas9 expression, similar to CRISPR inhibition (CRISPRi) once editing machinery is recruited to target sites.36 To further test for potential CRISPRi activity of nCas9 in PE, we compared the results between iSTOPs in the Alt library and the corresponding pegRNA/ngRNA pairs in the Ref library. While pegRNAs in the Ref library exhibited smaller effects on Day 32 compared to iSTOPs targeting the same loci, they were still depleted on Day 32, confirming unintended consequences due to nCas9 occupancy at target genomic loci (Figure S3H). Combined, we found that prolonged PE expression exhibits undesired activity similar to CRISPRi, a crucial factor for consideration when analyzing lentivirus-mediated PE screens.
To correct for this undesired CRISPRi effect, we compared the ratio of FC for each pegRNA/ngRNA pair from Alt and Ref screens by DESeq2.37 We determined functional SNPs based on their relative impact on cell growth between Alt and Ref PEs. In total, 56 SNPs with Ref alleles and 47 SNPs with Alt alleles were identified to promote cell growth (P < 0.05, empirical significance threshold to control type-I error at 5%, Figures 3D and S3I; Table S2). As expected, identified functional SNPs had smaller effect sizes than stop codons and significantly larger effect sizes than negative control PEs (Figure 3E). Additionally, iSTOPs for genes promoting cell growth, such as MYC and GATA3, were depleted, while the iSTOP for the cell growth suppressor PTEN was enriched, validating our analysis approach (Figure 3D).
Since risk variants can either be the Ref or Alt allele, we further annotated functional SNPs based on genetic annotation of breast cancer risk variants. Since most GWAS SNPs are likely not causal, we expected that only a fraction of the 1,304 tested SNPs would exhibit a biological effect. We calculated the mean likelihood of a variant being causal using CAVIAR and found that the mean expectation for a variant being causal was ~8.9% when we made the assumption of only one causal variant in each linkage disequilibrium (LD) clump. If we allowed for more than one causal variant in each LD clump the mean probability of being causal for the variants was ~13.0%. Compared to the reference allele, 50 risk SNPs’ alternative alleles were pro-growth, and 53 risk SNPs’ alternative alleles reduced cell growth (Figure 3F). 18.45% (19/103) of the functionally validated risk SNPs were located within the risk gene’s body. The rest were located in distal regions with an average distance of 185.8 kb from the risk gene’s TSS (Figure 3F). All tested loci contained at least one SNP with a significant effect on cell growth, except for the BRCA2 locus, in which only 2 SNPs were tested. Finally, identified functional SNPs were significantly enriched for active chromatin marks (two-tailed Fisher’s exact test, P < 0.05), including ATAC-seq, H3K27ac, H3K4me1, and H3K4me3 signals, relative to their corresponding genomic background (1 Mbp surrounding selected cell growth genes) (Figure 3G).
To explore potential mechanisms for functional SNPs’ regulation of cell fitness changes, we searched candidate TF binding motifs against the human motif database HOCOMOCO24 using 40 bp regions centered on 103 identified functional SNPs. We retrieved 281 and 391 motifs (FDR < 0.05 and TF expression > 1 FPKM) containing Alt and Ref alleles, respectively. After removing redundant motifs for each SNP locus, we identified 90 TF binding sites for 35 unique TFs associated with the cell growth suppression phenotype (log2FC(Alt/Ref) < 0) and 55 sites for 29 unique TFs associated with the pro cell growth phenotype (log2FC(Alt/Ref) > 0) (Figure 3H; Table S3).
To validate our PRIME results and explore the molecular mechanisms of those identified functional SNPs, we selected three non-coding SNPs (rs10956415, rs7772579, rs66473811) that exhibited moderate effects on cell growth (Figures 3F, 4A, 4D, and 4G). MCF7 cells are homozygous for the alternative allele (A) at rs10956415, the alternative allele (C) at rs7772579, and the reference allele (T) at rs66473811. rs10956415 is located in a candidate enhancer 432 kb downstream of the MYC TSS and 25 kb downstream of the 716 bp MYC enhancer we analyzed (Figures 4B and S4A). rs7772579 is in the ESR1 intron (Figures 4E and S4A), and rs66473811 is in the PSMD6 intron (Figures 4H and S4A). Using PE, we generated heterozygous clones for these three SNPs (Figures S4B, S4C, and S4D). Compared to control clones, PE edited clones showed approximately a 40% increase in MYC (Figures 4B and 4C) and ESR1 expression, respectively (Figures 4E and 4F). MYC and ESR1 promote MCF7 proliferation,38,39 which aligns with the cell growth inhibitory PRIME results of rs10956415 (Log2FC(Alt/Ref) = −0.55) and rs7772579 (Log2FC(Alt/Ref) = −0.82).
Figure 4. Functional validation of PRIME identified functional SNPs.
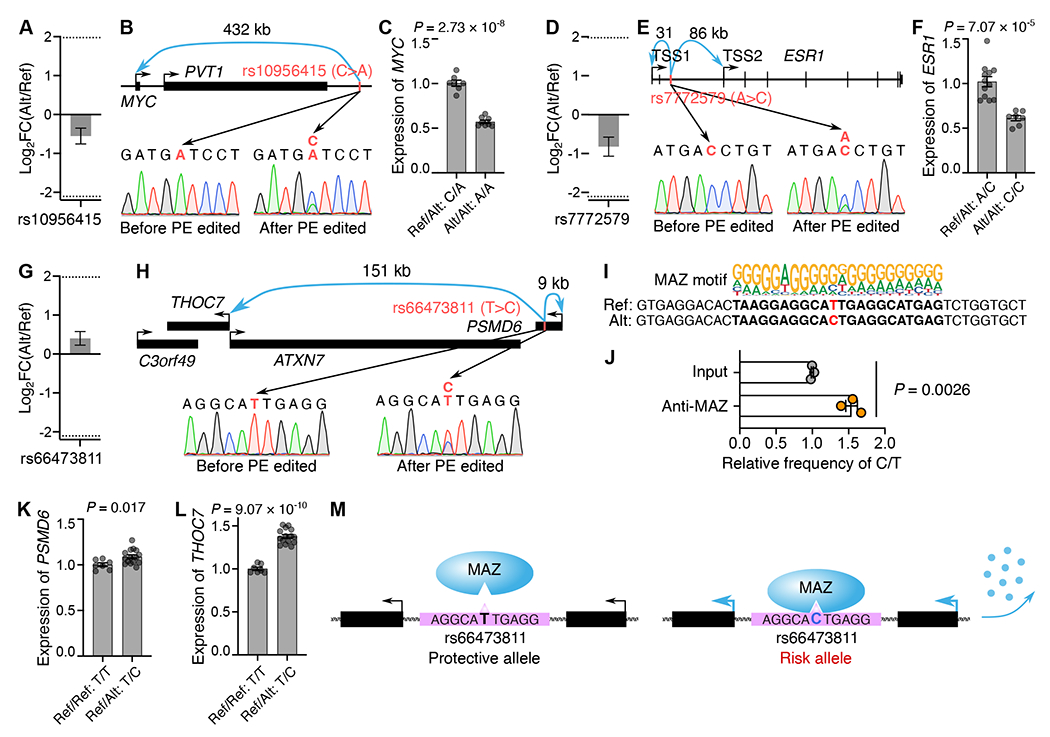
(A, D, G) The relative effect (Alt/Ref) of rs10956415, rs7772579, rs66473811 on MCF7 cell growth from PRIME. Error bars indicate s.e. (B, E, H) The genomic landscapes and sequences before and after PE for rs10956415, rs7772579 and rs66473811. (C) The relative expression of MYC in PE edited clones (Ref/Alt: C/A, n=7) and control clones (Alt/Alt: A/A, n=8). (F) The relative expression of ESR1 in PE edited clones (Ref/Alt: A/C, n=12) and control clones (Alt/Alt: C/C, n=7). (I) The MAZ binding motif at rs66473811 locus. (J) Relative enrichment of MAZ binding at Alt (C) and Ref (T) alleles by ChIP and targeted sequencing (n=3 clones). (K, L) Relative expression of PSMD6 and THOC7 in control clones (Ref/Ref: T/T, n=7) and PE edited clones (Ref/Alt: T/C, n=15). (M) An illustration of T>C substitution increasing MAZ binding at the rs66473811 locus, upregulating PSMD6 and THOC7 expression, and promoting MCF7 growth. For C, F, and J-L, data are displayed in mean with s.e.m., P values were calculated by two-tailed two-sample t-test, and dots show individual replicate values. See also Figure S4.
Regarding rs66473811, the alternative allele (C) better matched with the MAZ binding motif (Figure 4I). The binding of MAZ at rs66473811 locus was confirmed by ChIP-qPCR (Figure S4E). Further quantification of the relative binding frequency between the rs66473811 reference and alternative alleles in PE edited heterozygous clones demonstrated a higher binding affinity of MAZ at the alternative allele (C) (Figure 4J). In addition, PE edited heterozygous clones (Ref/Alt: T/C) exhibited higher expression levels of PSMD6 (8.7%) and THOC7 (37.6%) compared to control clones (Ref/Ref: T/T) (Figures 4K and 4L), suggesting that the higher binding affinity of MAZ, due to a single base T>C change at the rs66473811 locus, contributed to the elevated PSMD6 and THOC7 expression (Figure 4M). Together, our validation results at three independent SNP loci support the use of PRIME in determining functional GWAS-identified variants.
PRIME can characterize clinical variants of uncertain significance
Genetic variants detected in clinical samples provide a valuable resource for understanding the etiologies of human diseases. However, many clinically discovered variants are annotated as Variants of Uncertain Significance (VUS) due to unpredictable functional consequences, even in well-characterized protein-coding genes. To assess the capacity of PRIME to functionally annotate VUS using MCF7 growth phenotypes, we designed pegRNA/ngRNA pairs for 2,532 VUS, 745 pathogenic variants, and 422 benign variants for 17 genes (Figure S3C; Table S2). 76.78% of the variants tested were from breast cancer patients (Table S2). By comparing the relative effect sizes of each Alt and Ref allele pair, we identified 236 functional clinical variants affecting cell growth in 15 genes, including 49 pathogenic variants, 156 VUS, and 31 benign variants (Figure 5A; Table S2). The average effect sizes for pathogenic variants, VUS, and benign variants were between that of negative controls and iSTOPs (Figure 5B).
Figure 5. Functional clinical variants identified using PRIME.
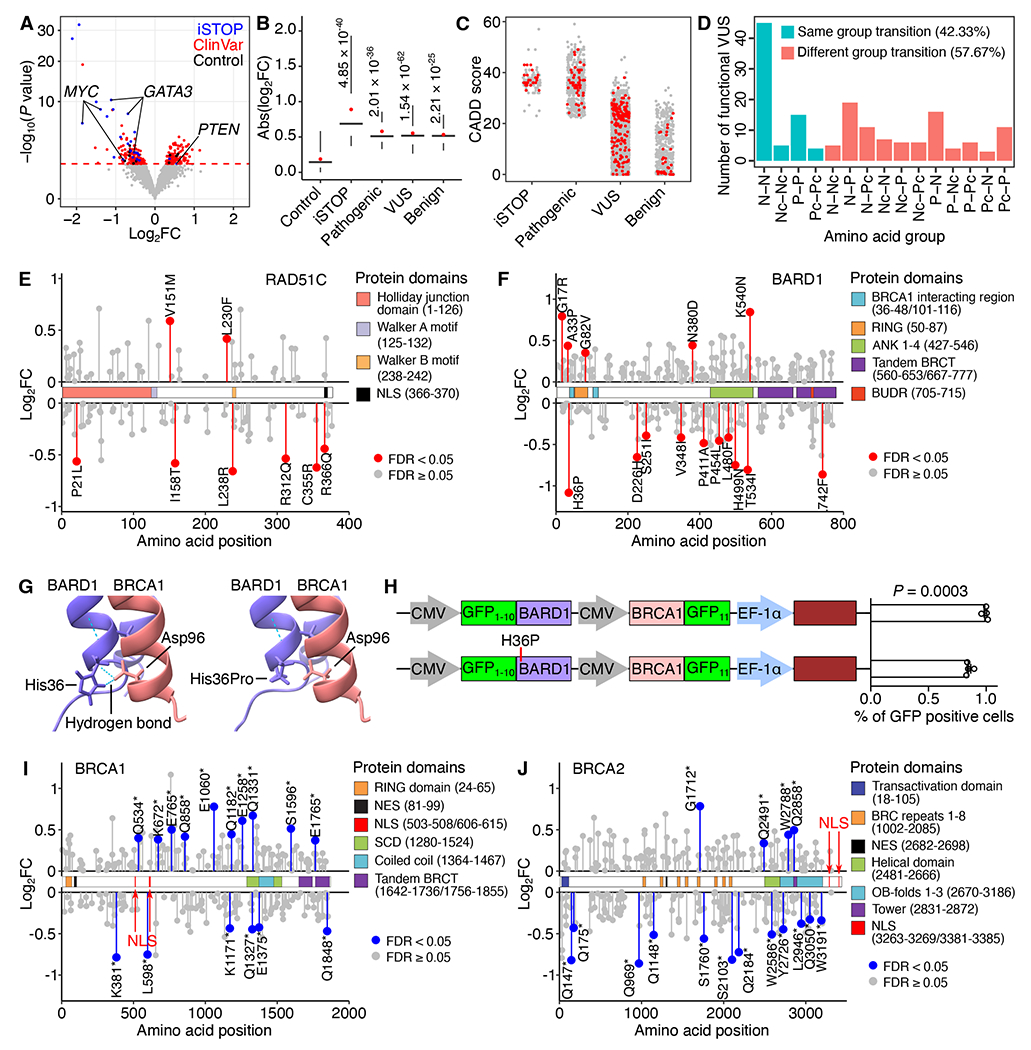
(A) Functional clinical variants (red) were determined by relative effects on cell fitness between Alt and Ref alleles. Blue dots represent significant iSTOPs, and black dots represent negative controls. The red dashed line indicates 5% FDR. (B) Effect sizes of identified functional iSTOPs and clinical variants are larger than that of negative controls (P values were calculated by two-tailed two-sample t-test). Box plots indicate the median, IQR, Q1 – 1.5 × IQR, and Q3 + 1.5 × IQR. Red dots indicate the mean. (C) CADD scores for iSTOPs and clinical variants. (D) Number of identified functional VUS causing each amino acid group transition. (N, Nonpolar; P, Polar; Pc, Positively charged; Nc, Negatively charged). (E, F) Lollipop plots of VUS in RAD51C and BARD1 mapped to their canonical isoforms. The identified functional VUSs are labeled in red. (G) The AlphaFold predicted protein structure of the BARD1 and BRCA1 complex. Two hydrogen bonds were identified between His36 in BARD1 and Asp96 in BRCA1, but lost following the BARD1 His36Pro mutation. (H) The percentage of GFP positive cells representing BARD1 and BRCA1 interactions by the split GFP system. The mCherry reporter was used to normalize the transfection rate. Data are displayed in mean with s.e.m. P values were calculated by two-tailed two-sample t-test. Dots show individual replicate values. (I, J) Lollipop plots of the nonsense variants in BRCA1 and BRCA2 mapped to their canonical isoforms. The identified significant hits are labeled in blue. See also Figure S5 and Table S2.
Several computational metrics have been used to assess the deleteriousness of variants.40,41 One such method is CADD, which integrates diverse genome annotations into a single, quantitative score estimating the relative pathogenicity of human genetic variants.40 iSTOPs and pathogenic variants have similarly high CADD scores relative to other categories (Figure 5C). The CADD scores for the VUS and benign variants exhibit a broad distribution with median scores much lower than those of iSTOPs and pathogenic variants. Interestingly, the CADD scores for identified functional variants within the VUS or benign variant groups did not have higher CADD scores as expected, indicating the limitation of solely relying on computational prediction for variants annotation and underscoring the importance of validating clinical variants with functional assays, even for those located in well-studied protein-coding genes. For example, one benign variant in BARD1 (Arg378Ser) with a low CADD score (CADD = 4.317) would not be classified as functional. However, our PRIME results revealed a significant cell growth suppression effect in MCF7 (Log2FC(Alt/Ref) = −0.81). BARD1 Arg378Ser mutant impairs the nuclear localization of the BARD1.42,43 While the BARD1 Arg378Ser mutation didn’t suppress tumorigenesis of MCF10A in mouse models,42 it appears that the Arg378Ser mutation affects cell fitness in MCF7 cells in our study. This is possibly due to the usage of different cell lines and experimental approaches. Nevertheless, our results in MCF7 cells align with the observation that the cytoplasm localization of BARD1 is associated with increased cell apoptosis.44 Furthermore, most of the identified functional VUS were missense variants, and about half of the functional VUS from our screens changed amino acid type within the same group based on polarity (Figure 5D), complicating the determination of their molecular consequences. Our results offer novel insights into the potential roles of clinical variants in disease pathogenesis through their modulation of cell fitness, and provide annotations for VUS and benign variants previously uncharacterized.
Functional and structural domains are integral contributors to protein function. 60% of the functional VUS identified are located within an annotated protein domain in the UniProt database,45 supporting their pathogenicity. For example, we identified 8 VUS in RAD51C (Figure 5E), a cancer susceptibility gene and an essential gene for MCF7 survival. Two variants, one (Pro21Leu) in the RAD51C functional domain (amino acid: 1-126) for Holliday junction processing and the other (Arg366Gln) in the NLS region (amino acid: 366-370), were associated with reduced cell growth by our screens (Figure 5E). We also identified functional variants that were not located in any annotated domain, including a functional RAD51C VUS (Arg312Gln) associated with a phenotype of reduced MCF7 growth (Figure 5E). Since Arg312Trp in RAD51C results in homologous recombination deficiency and reduced colony formation phenotypes in MCF10A cells, and abolishes RAD51C-RAD51D interaction,46 Arg312Gln may produce a similar pathogenic consequence on protein function. When comparing the RAD51C sequence with other RAD51 family proteins, we observed functional VUS were located in both conserved and non-conserved amino acids (Figure S5A), underscoring the challenge of predicting variant function based solely on protein sequence conservation.
Protein-protein interaction (PPI) is another essential functional activity in many biological processes. In this study, we also identified functional VUS located in protein binding regions with the potential to affect PPI. For example, BARD1 interacts with BRCA1 through RING domains, and BRCA1-BARD1’s ubiquitin ligase activity is indispensable for DNA double-strand break repair.47,48 We identified a functional VUS (His36Pro) in the BARD1 RING domain (Figure 5F), suggesting the structural consequences of this clinical variant affecting BARD1-BRCA1 heterodimer formation (Figure S5B). Consistent with these findings, AlphaFold predicts that the His36Pro variant disrupts hydrogen bond formation between His36 in BARD1 and Asp96 in BRCA1 (Figure 5G). Indeed, using the split GFP system,49 we confirmed a notable impact of the BARD1 His36Pro on the BARD1-BRCA1 interaction. Specifically, the BARD1 His36Pro variant (GFP1-10-BARD1H36P + BRCA1-GFP11) resulted in fewer GFP positive cells compared to wild type BARD1 (GFP1-10-BARD1 + BRCA1-GFP11) (Figures 5H, S5C, and S5D).
Nonsense mutations can generate new stop codons and truncated proteins. Although most are annotated as pathogenic variants in ClinVar, the functional consequences of many remain uncharacterized.34 In our screens, 563 nonsense clinical variants were tested in 13 breast cancer risk genes with 38 variants identified as positive hits in 7 genes. Remarkably, 39.47% (15/38) exhibited unexpected phenotypes compared to the knockout phenotypes of cell death of these genes. Specifically, a similar number of functional nonsense variants in BRCA1 (n = 15) and BRCA2 (n = 16) were identified (Figures 5I and 5J); however, 60% (9/15) in BRCA1 could promote MCF7 cell growth compared to 25% (4/16) in BRCA2. After locating variants within BRCA1 and BRCA2, we noticed that truncated proteins resulting from all gain-of-function nonsense variants in BRCA1 still retained their NLS. These results were confirmed by a different nonsense mutation at Q858, located downstream of the NLS in BRCA1, which resulted in truncated BRCA1 with NLS and increased cell growth of MCF7.35 However, for all of the functional variants identified in BRCA2, their NLSs were located at the c-terminus50 and were thus removed from the truncated proteins, leading to the loss of BRCA2 nuclear localization. Collectively, these results demonstrate the capability of PRIME to functionally characterize some nonsense mutations.
Discussion
In this study, we describe a new genomic screening method, PRIME, to interrogate DNA function at single-base resolution by adopting and optimizing prime editing.10,14 We demonstrate the success of PRIME to identify essential nucleotides in a MYC enhancer via single-base resolution analysis screen, characterize 1,304 breast cancer-associated risk SNPs and 3,699 clinical variants. Our study offers a novel strategy to elucidate genome function at an unprecedented precision and scale. The broad applications demonstrated in this work suggest that PRIME can significantly augment the functional characterization toolbox and advance our ability to elucidate the roles of disease-associated variants in the human genome.
Our analyses show that lentiviral installation of PE can result in unwanted sequence-specific repression similar to CRISPRi due to long lasting expression of the PE machinery. This bias must be corrected to produce accurate single-base resolution annotations. When assessing the functional impact of a variant, pegRNA controls should be included to introduce other alleles at the same locus. Our study normalized sequence-specific repression bias by comparing the differential effects on cell survival of all base pair substitutions at each locus in the MYC enhancer, and between Alt and Ref alleles for disease variants. Additional improvement could be achieved through controlled nCas9 expression duration. For example, a doxycycline-inducible nCas9 could be selectively expressed when editing is needed and reversibly turned off afterwards. In addition to establishing and optimizing PRIME, we defined sensitive base pairs (SBPs) and core sequences for a MYC enhancer’s function. We generated a functional PWM for this enhancer by leveraging effect sizes for all possible substitutions at each base from the screens. The functional PWM enabled us to accurately predict TF binding sites within the enhancer, providing critical annotations for delineating MYC activation in MCF7 cells.
Interpreting the effect of inherited genetic variations will dramatically advance our ability to predict an individual’s disease risk. However, utilizing GWAS data for risk prediction is still limited without substantial functional annotation. In this study, 7.9% of the 1,304 tested GWAS-identified breast cancer variants, and 6.2% of the 2,532 tested VUS were identified as significant hits with functions linked to MCF7 growth phenotypes. Our results demonstrate the feasibility of PRIME for functionally characterizing individual variants. The impact of variants was context-specific and our findings were limited to assessing variants with growth phenotype related functions in MCF7 cells. Other ClinVar did not show changes in our functional assay likely have functional consequences for breast cancer susceptibility genes in a different cell type or other biological processes.
Future work employing different phenotypic screening readouts across multiple cell lines will provide new insights into variant function. For example, screens that identify variants associated with differential drug treatment responses will help construct better predictive models for an individual’s unique benefits and risks from therapeutics. Screens of variants with readouts directly linked to physiological functions e.g. endolysosomal activities in microglia or synaptic activities in neurons using iPSC models will uncover functional variants associated with neuropsychiatric diseases. In summary, our study provides a roadmap to advance functional genomics toward the actionable disease prediction, prevention, and treatment necessary to realize personalized medicine.
Limitations of the study
In this study, we introduce PRIME, a high-throughput PE-mediated pooled screen platform for comprehensive characterizing genome function at single-base resolution. Similar to pooled CRISPR screening strategies, PRIME determines functional nucleotides based on the relative enrichment of pegRNA/ngRNA pairs in a cell population following phenotype selection. While we observed high prime editing efficiencies at EMX1 and FANCF loci, it is worth noting that PE efficiency could vary depending on the quality of pegRNA/ngRNA pairs and chromatin contexts of the targeting loci. Although we attempt to address this problem by employing multiple pegRNA/ngRNA pairs for each desired nucleotide substitute, loci with low editing efficiencies may still lead to false negative results, potentially impacting screening sensitivity. PRIME can be further optimized by adopting any current and future improved PE systems, such as introducing same-sense mutations in pegRNA51 and inhibiting DNA mismatch repair (MMR).52 The significance of PRIME will be further enhanced by incorporating more sensitive and biologically relevant readouts that go beyond cell fitness and survival. The functional variants of breast cancer identified by PRIME elucidate their roles in cell growth/finesse in our current study. Conducting additional experiments will bring further insights into the casual roles of functional variants in breast cancer.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Please direct requests for resources and reagents to lead contact: Yin Shen (yin.shen@ucsf.edu).
Materials availability
Plasmids generated in this study are available from Addgene. Additional details are provided in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-FLAG | Sigma-Aldrich | Cat# F7425, RRID:AB_439687 |
| Rabbit polyclonal anti-MAZ | Novus | Cat# NB100-86984; RRID:AB_2266238 |
| Guinea Pig anti-Rabbit Immunoglobulin G (IgG) antibody | Antibodies-Online | Cat# ABIN101961; RRID:AB_10775589 |
| Donkey anti-Rabbit IgG, Alexa Fluor 568 | Invitrogen | Cat# A10042; RRID:AB_2534017 |
| Bacterial and virus strains | ||
| Endura ElectroCompetent cells | Biosearch Technologies | Cat# 60242 |
| Stellar Competent Cells | TaKaRa | Cat# 636763 |
| Chemicals, peptides, and recombinant proteins | ||
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Hygromycin B | Gibco | Cat# 10687010 |
| PolyJet | SignaGen Laboratories | Cat# SL100688 |
| Polybrene | Millipore | Cat# TR-1003-G |
| Penicillin-Streptomycin | Thermo Fisher | Cat# 15140122 |
| 1M Tris-HCI, pH 8.0 | Invitrogen | Cat# 15568025 |
| 0.5M EDTA | Invitrogen | Cat# 15575020 |
| 10% SDS | Invitrogen | Cat# 15553027 |
| Phenol:Chloroform | Thermo Fisher Scientific | Cat# 17908 |
| Isopropanol | Thermo Fisher Scientific | Cat# BP2618500 |
| Glycine | Invitrogen | Cat# 15527013 |
| IGEPAL CA-630 | Sigma-Aldrich | Cat# I8896-50ML |
| Lithium Chloride | Sigma-Aldrich | Cat# L9650-100G |
| NaCl | Sigma-Aldrich | Cat# S9888-25G |
| Sodium Deoxycholate | Sigma-Aldrich | Cat# D6750-100G |
| Formaldehyde | Fisher Scientific | Cat# F79-500 |
| Span 80 | Sigma-Aldrich | Cat# S6760-250ML |
| Tween 80 | Sigma-Aldrich | Cat# P4780-100ML |
| Triton X-100 | Sigma-Aldrich | Cat# T9284-100ML |
| Mineral oil | Sigma-Aldrich | Cat# M5904-500ML |
| EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693159001 |
| RNase | NEB | Cat# T3018L |
| BsmBI | NEB | Cat# R0739S |
| T4 ligase | NEB | Cat# M0202M |
| Exonuclease I | NEB | Cat# M0568L |
| NEBNext High-Fidelity 2× PCR Master Mix | NEB | Cat# M0541S |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | Cat# E2621L |
| Luminaris HiGreen qPCR Master Mix | Thermo Scientific | Cat# K0992 |
| Proteinase K | NEB | Cat# P8107S |
| Trypsin-EDTA | Gibco | Cat# 25200056 |
| Dulbecco’s phosphate-buffered saline (DPBS) | Gibco | Cat# 14190144 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Gibco | Cat# 10569010 |
| Fetal Bovine Serum | HyClone | Cat# SH30396.03 |
| Cas9-NLS purified protein | QB3 MacroLab at the University of California, Berkeley | Cas9-NLS purified protein |
| Critical commercial assays | ||
| Qubit dsDNA HS Assay kit | Thermo Fisher | Cat# Q32851 |
| Precision gRNA Synthesis Kit | Invitrogen | Cat# A29377 |
| Gel and PCR Clean-Up System | Promega | Cat# A9282 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28104 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708890 |
| Plasmid DNA Mini Kit | Omega Bio-tek | Cat# D6943-02 |
| Plasmid Plus Maxi Kit | Qiagen | Cat# 12963 |
| EndoFree Plasmid Mega Kit | Qiagen | Cat# 12381 |
| LightCycler 96 | Roche | Cat# 05815916001 |
| P3 Primary Cell 4D-Nucleofector X Kit L | Lonza | Cat# V4XP-3024 |
| Wizard genomic DNA purification kit | Promega | Cat# A1120 |
| AMPure XP beads | Beckman Coulter | Cat# A63881 |
| Dynabeads Protein A beads | Invitrogen | Cat# 10001D |
| Millex-HV 0.45-μm polyvinylidene difluoride filter | Millipore | Cat# SLHV033RS |
| Ultra-15 centrifugal filter units | Amicon | Cat# UFC910008 |
| microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm | Covaris | Cat# 520045 |
| Covaris S220 Focused-ultrasonicator | Covaris | Cat# 500217 |
| MycoAlert Mycoplasma Detection Kit | Lonza | Cat# LT07-218 |
| Deposited data | ||
| Raw sequencing data | This study | NCBI SRA: BioProject PRJNA909251 |
| Imaging data | This study | Mendeley data: https://data.mendeley.com/datasets/27jrjsp527 |
| Custom code | This study | Zenodo: https://doi.org/10.5281/zenodo.10139699 |
| Experimental models: Cell lines | ||
| MCF7 cell line | ATCC | Cat# HTB-22 |
| MCF7-nCas9/RT | This study | N/A |
| Oligonucleotides | ||
| Custom library Oligos for PRIME screen: MYC enhancer | This study | Table S1 |
| Custom library Oligos for PRIME screen: Variants | This study | Table S2 |
| Custom PCR primers and DNA sequences | This study | Table S4 |
| Recombinant DNA | ||
| LentiV2-EF1α-nCas9/RT | This study | Addgene plasmid# 210188 |
| LentiV2-hU6-evopreQ1 | This study | Addgene plasmid# 210189 |
| LentiV2-mU6-evopreQ1 | This study | Addgene plasmid# 210190 |
| pCMV-GFP1-10-BARD1 | This study | N/A |
| pCMV-GFP1-10-BARD1H36P | This study | N/A |
| pCMV-GFP11-BRCA1 | This study | N/A |
| pCMV-BARD1-GFP11 | This study | N/A |
| pCMV-GFP1-10-BARD1-CMV-BARD1-GFP11-EF-1α-mCherry | This study | N/A |
| pCMV-GFP1-10-BARD1H36P-CMV-BARD1-GFP11-EF-1α-mCherry | This study | N/A |
| lentiCRISPR v2 | Addgene | Addgene plasmid# 52961 |
| pCMV-PE2 | Addgene | Addgene plasmid# 132775 |
| PsPAX2 | Addgene | Addgene plasmid# 12260 |
| pMD2.G | Addgene | Addgene plasmid# 12259 |
| Software and algorithms | ||
| CRISPResso2 | Clement et al., 201953 | https://github.com/pinellolab/CRISPResso2 |
| PrimeDesign | Hsu et al., 202154 | https://github.com/pinellolab/PrimeDesign |
| pegIT | Anderson et al., 202155 | https://pegit.giehmlab.dk/ |
| MAGeCK (0.5.9) | Li et al., 201418 | https://sourceforge.net/projects/mageck/ |
| DESeq2 (1.38.0) | Love et al., 201437 | https://github.com/thelovelab/DESeq2 |
| AlphaFold (v2.2.4) | Mirdita et al., 202256 | https://github.com/google-deepmind/alphafold |
| Fastp (v0.23.2) | Chen et al., 201857 | https://github.com/OpenGene/fastp |
| FIMO (v5.5.0) | Grant et al., 201158 | https://meme-suite.org/meme/tools/fimo |
| EditR | Kluesner et al., 201859 | http://baseeditr.com/ |
| BWA (v.0.7.17) | Heng Li | https://github.com/lh3/bwa |
| GATK (v.4.2.5.0) | Broad Institute | https://github.com/broadinstitute/gatk |
| CalcMatch (v.1.1.2) | Yun Li | https://genome.sph.umich.edu/wiki/CalcMatch |
| DeepPrime | Yu et al., 202360 | http://deepcrispr.info/DeepPrime/ |
Data and code availability
All original data are available on NCBI Sequence Read Archive database and Mendeley data. All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
All original code used for the design and analysis of pegRNA/ngRNA pairs is publicly available at Zenodo. The DOI of the code is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Study Participant Details
Mammalian cell culture
MCF7 cells and HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, 10569010) supplemented with 10% fetal bovine serum (FBS) (HyClone, SH30396.03), and were passaged with trypsin-EDTA (Gibco, 25200072). The MCF7-nCas9/RT cell line was generated by lentiviral transduction of cells with a cassette expressing the nickase Cas9 (nCas9) Moloney murine leukemia virus reverse transcriptase (M-MLV RT) fusion protein. The infected MCF7 cell pool was treated with puromycin (2.5 μg/ml; Sigma-Aldrich, P8833) for two weeks. Then, single cells were sorted into 96-well plates with one cell per well by fluorescence-activated cell sorting (FACS) to generate a clonal MCF7-nCas9/RT cell line. nCas9/RT expression levels were quantified in each clone via RT-qPCR, and normalized to the dCas9 expression level in a WTC11 doxycycline-inducible dCas9-KRAB iPSC line.61,62 All cells were cultured with 5% CO2 at 37°C and verified to be free of mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza, LT07-218).
METHOD DETAILS
Functional characterization of a MYC enhancer by CRISPR deletion
Two sgRNAs were designed to knock out a MCF7 enhancer (chr8:128,141,747-128,142,627, hg38) (sg1: GAAGTTGTAAGTATAGCGAG, sg2: AGTGCCTGGCACAAGGCAGA). sgRNAs were synthesized in vitro using the Precision gRNA Synthesis Kit (Invitrogen, A29377) according to the manufacturer protocol and concentrations were quantified with Nanodrop. To deliver genome editing machinery, 100 pmol of Cas9-NLS protein (sourced from QB3 MacroLab in University of California, Berkeley) and 120 pmol of in vitro synthesized gRNA were electroporated into 250,000 MCF7 cells with the P3 primary nucleofection solution (Lonza, V4XP-3024), using the DN-100 Lonza 4D-Nucleofector program. Cells were then plated into 6-well plates and cultured for 2 days, followed by plating into 96-well plates to pick single clones. Successful knockout clones were identified by genotyping PCR (primers are listed in Table S4). RT-qPCR was used to quantify the MYC expression with normalization to GAPDH (primers are listed in Table S4).
To target distinct segments of the MCF7 enhancer, we designed four unique guide RNAs (sg1: TCCATCACCAAACTCCCTTG; sg2: GCCAAAGGTCACAGTGTTCT; sg3: CAAAGAAAAATTTGCCCTCC; sg4: AACTTTCTAGAACCAGCATG). In vitro synthesis of sgRNAs was carried out using the Precision gRNA Synthesis Kit (Invitrogen, A29377) following the manufacturer’s protocol, with subsequent quantification of concentrations using Nanodrop. To introduce desired deletions, we electroporated 100 pmol of Cas9-NLS protein (sourced from QB3 MacroLab at the University of California, Berkeley) along with 120 pmol of sgRNAs into 250,000 MCF7 cells employing the P3 primary nucleofection solution (Lonza, V4XP-3024) and the DN-100 Lonza 4D-Nucleofector program. Subsequently, the cells were seeded into 6-well plates and cultured for 2 days, followed by transfer to 96-well plates for the isolation of single clones. Approximately two weeks later, we performed genotyping PCR followed by Sanger sequencing to identify clones with desired deletion (primers are listed in Table S4). qPCR was then performed to quantify the copy number of deleted alleles in each clone (primers are listed in Table S4). In order to ensure comparability among the resulting clones, clones that retained only two copies of the wild type enhancer sequences were used for RT-qPCR analysis to quantify MYC expression after normalizing with GAPDH (primers are listed in Table S4).
Cloning of prime editing plasmids
To construct the lentiV2-EF1α-nCas9/RT plasmid, we first excised the U6-sgRNA cassette from the lentiCRISPR v2 plasmid (Addgene, 52961) by dual KpnI and EcoRI digestion followed by blunt end ligation. We further replaced the Cas9 cassette with an nCas9/M-MLV-RT cassette from the pCMV-PE2 plasmid (Addgene, 132775). The lentiV2-pegRNA and lentiV2-ngRNA plasmids were constructed by replacing the Cas9 and Puromycin sequences in the lentiCRISPR v2 plasmid (Addgene, 52961), with hygromycin B and EGFP sequences. RNA motifs and sgRNA scaffolds were further integrated by Gibson assembly (NEB, E2621L).
Testing prime editing efficiency
To assess prime editing efficiencies at the EMX1 and FANCF loci, we cloned paired pegRNAs/ngRNAs into individual vectors. For lentivirus co-infection testing, we first infected MCF7 cells with EF1α-nCas9/RT lentivirus followed by treatment with puromycin (2.5 μg/ml; Sigma-Aldrich, P8833) for 2 weeks to eliminate uninfected cells. Then, EF1α-nCas9/RT-infected cells were seeded in 24-well plates at 12,500 cells per well for pegRNA and ngRNA co-infection. The infected cells were treated with hygromycin B (200 μg/ml; Gibco, 10687010) 48 hours after infection, and were collected one week after infection for editing efficiency assessment. For testing in the MCF7-nCas9/RT clonal line, we seeded cells in 24-well plates at 12,500 cells per well, followed by lentiviral infection (pegRNA-mCherry and ngRNA-EGFP). Two days after infection, mCherry and EGFP double-positive cells were isolated by FACS and cultured. Cultured cells were then collected at 2-week and 4-week post-infection for editing efficiency assessment. Genomic DNA was then extracted from each sample using the Wizard genomic DNA purification kit (Promega, A1120). Genomic sites of interest were amplified from purified genomic DNA and amplicons were sequenced on the Illumina NovaSeq 6000 platform. Briefly, sequencing libraries were prepared using DNA primers amplifying target genomic loci of interest for the first round of PCR (PCR1). Then, DNA primers containing index adapters were used for the second round of PCR (PCR2) to add these adapters to PCR1 amplicons. Finally, dual indexing primers were used for the third round PCR (PCR3) to add Illumina indexes to each PCR2 amplicon. Alignment of amplicons to reference sequences was performed using CRISPResso2.53 For all prime editing efficiency quantification, wild type and edited amplicon frequencies were quantified using a 21 bp window centered on either the 1 bp wild type or edited sequence. The remaining amplicons were classified as indels.
SNP prioritization
We selected 14 MCF7 growth-related genes overlapping with GWAS identified breast cancer susceptibility genes.31 For each gene, we selected SNPs using the GWAS results from the Breast Cancer Association Consortium.30 We identified genome-wide significant SNPs with GWAS P < 1×10−5, minor allele frequency > 0.02, and odds ratios < 0.9 or > 1.2 (representing approximately the top and bottom quartiles of the odds ratio distribution for SNPs meeting the location, P value, and MAF thresholds) for association with breast cancer within the locus +/− 500 kb of each transcription start site. We also separately selected SNPs with GWAS P < 1×10−5 in the ESR1 locus using GWAS results from a Latina population.63 We determined linkage disequilibrium (LD) clumps among the selected SNPs using the LD Link R package64 with an LD threshold of R2 > 0.1. We then prioritized the most likely causal variants using CAVIAR,65 as those with a causal posterior probability (> 0.1), the highest posterior probability (≤ 0.1), or most extreme odds ratio in each haplotype block. We ran CAVIAR twice for each locus, once assuming only one causal variant per LD clump, and again allowing for more than one causal variant in each LD clump.
Clinical variant prioritization
We retrieved clinical variants from the ClinVar database (accessed 2021-12-25), and all single nucleotide variants (SNVs) were kept for the PRIME design (Figure S3C). We first selected only the SNVs whose genes overlapped with breast cancer risk and MCF7 growth-related genes. Next, we only retained SNVs in the benign, pathogenic and uncertain significance categories. Further, for SNVs associated with BARD1, BRCA1, BRCA2, RAD51C, RAD51D, and PTEN, we only retained the SNVs with more than three submitters, as there are thousands of identified variants for these genes. Finally, our selection criteria yielded 5,310 SNVs, of which we successfully designed pegRNA/ngRNA pairs for 3,699 SNVs.
Design and construction of prime-editing libraries
For single-base resolution analyses of MYC enhancer function, paired pegRNAs/ngRNAs targeting a 716 bp enhancer region (chr8:128,141,822-128,142,537, hg38) were first designed using PrimeDesign’s PooledDesign-Saturation mutagenesis tool.54 We optimized pegRNA/ngRNA pairs based on ngRNA pegRNA proximity (more than 50 bp) and primer binding site (PBS) length (near 14 nt), redesigning the sequence containing the BsmBI cutting sites (GAGACG, CGTCTC) or TTTTT. Next, we used GuideScan2 to assess the specificity and efficiency of each pegRNA and ngRNA spacer sequence. Spacer sequences with low specificity were redesigned to improve the specificity. Finally, three different pegRNA/ngRNA pairs were designed to target the same base pair for 99.0% (709/716) of the substitutions. Each replicate pegRNA/ngRNA pair shared the same pegRNA and sgRNA spacer sequences, and only the substitution alleles differed in the pegRNA extension sequence. To design positive control guides, we used pegIT52 to generate pegRNA/ngRNA pairs which alter a single base pair to introduce a stop codon within the MYC coding region. We selected the best pegRNA/ngRNA pair for each position suggested by pegIT.55 The AAVS1 locus was selected as the targeting pegRNA/ngRNA pair negative control region based on previous work,66 and guides were designed as described above using PrimeDesign.54 For non-targeting pegRNA/ngRNA pairs, pegRNA and ngRNA spacer sequences and pegRNA extension sequences were selected from the ENCODE non-targeting sgRNA reference data set (https://www.encodeproject.org/files/ENCFF058BPG/). A guanine nucleotide was added to the 5’ end of all pegRNAs/ngRNAs with leading nucleotides other than G, to increase transcription efficiency from the U6 promoter. We used the following template to link these component sequences: 5’-CTTGGAGAAAAGCCTTGTTT[ngRNA-spacer]GTTTAGAGACG[5nt-random-sequence]CGTCTCACACC[pegRNA-spacer]GTTTTAGAGCTAGAAATAGCAAGTTAAAAAGTGGCACCGAGTCGGTGC[pegRNA extension]CCTAACACCGCGGTTC-3’.
Library oligos for the MYC enhancer screen were synthesized by Twist Bioscience and amplified using the NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S), forward primer: GTGTTTTGAGACTATAAATATCCCTTGGAGAAAAGCCTTGTTT and reverse primer CTAGTTGGTTTAACGCGTAACTAGATAGAACCGCGGTGTTAGG. To amplify paired pegRNA/ngRNA library oligos for enhancer single-base resolution analysis, we employed emulsion PCR (ePCR) to reduce recombination of similar amplicons during PCR. Briefly, ninety-six 20μL ePCR reactions were performed using 0.01 fmol of pooled oligos with NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S). Each 20μL PCR mix was combined with 40μL of oil-surfactant mixture (containing 4.5 % Span 80 (v/v), 0.4 % Tween 80 (v/v) and 0.05 % Triton X-100 (v/v) in mineral oil)67. This mixture was vortexed at maximum speed for 5 min, briefly centrifuged, and placed into the PCR machine for amplification. Thermocycler settings were: 98 °C for 30 s, then 26 cycles (98 °C 10 s, 60 °C 20 s, 72 °C 30 s), then 72 °C for 5 min, and finally a 4 °C hold. The ramp rate for each step was 2°C/s. After PCR, individual reactions were combined and purified using the QIAQuick PCR Purification Kit (QIAGEN, 28104) following previously established guidelines.68 Purified PCR products were then treated with Exonuclease I (NEB, M0568L) and purified using 1× AMPure XP beads (Beckman Coulter, A63881). The isolated ePCR products were then inserted into a BsmBI-digested lentiV2-mU6-evopreQ1 vector via Gibson assembly (NEB, E2621L). The assembled products were electroporated into Endura electrocompetent Escherichia coli cells (Biosearch Technologies, 60242) and approximately 4,000 independent bacterial colonies were cultured for each oligo. The resulting plasmid DNA was linearized by BsmBI digestion, gel-purified, and ligated using T4 ligase (NEB, M0202M) to a DNA fragment containing an sgRNA scaffold and the human U6 promoter. The resulting library was electroporated into Endura electrocompetent Escherichia coli cells (Biosearch Technologies, 60242) and cultured as described above. The final plasmid library was extracted using the Qiagen EndoFree Plasmid Mega Kit (QIAGEN, 12381).
For the SNP and clinical variant PRIME screen Alt library, pegRNA/ngRNA pairs were designed using PrimeDesign.54 The sequences 200 bp upstream and downstream of each variant or iSTOP were used as inputs for PrimeDesign. We generated initial pegRNA/ngRNA pairs using the following parameters: number of pegRNAs per edit: 10, length of homology downstream: 10 nt, PBS length: 13 nt, maximum reverse transcription template (RTT) length: 50 nt, number of ngRNAs per pegRNA: 10, ngRNA to pegRNA nicking distance: 50 and 75 bp. Next, a guanine nucleotide was added to the 5’ end of all pegRNAs/ngRNAs with leading nucleotides other than G to increase transcription efficiency from the U6 promoter. pegRNA/ngRNA pairs containing BsmBI sites (GAGACG, CGTCTC) or a TTTTT sequence in the pegRNA spacer, ngRNA spacer or pegRNA extension were eliminated. pegRNA/ngRNA pairs were further selected to maximize specificity, efficiency, and ngRNA to pegRNA distance while minimizing pegRNA to edit distance when multiple pairs were available for the same locus. For non-targeting pegRNA/ngRNA pairs, pegRNA spacer, ngRNA spacer and pegRNA extension sequences were selected from the ENCODE non-targeting sgRNA reference data set (https://www.encodeproject.org/files/ENCFF058BPG/). To design the Ref library, we used the same pegRNA/ngRNA pairs as the Alt library, but replaced the alternative alleles in the pegRNA extension sequences with the reference allele sequences. The final oligos adhered to the following template architecture: 5’-CTTGTGGAAAGGACGAAACACC[ngRNA-spacer]GTTTCGAGACG[6nt-random-sequence]CGTCTCTTGTTT[pegRNA-spacer]gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc[pegRNA extension]TTGACGCGGTTCTATCTAGTTAC-3’.
The Alt and Ref library oligos were synthesized by Twist Bioscience. The Alt and Ref plasmid libraries were cloned separately using two-step cloning. First, the oligo pool for each library was amplified with NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S) and the following primers: Forward primer: TCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACAC, Reverse primer: ATTTCTAGTTGGTTTAACGCGTAACTAGATAGAACCGCGTCAA. PCR products were purified via gel excision and column purification (Promega, A9282), followed by insertion into the BsmBI-digested lentiV2-hU6-evopreQ1 vector by Gibson assembly (NEB, E2621L). The assembled products were electroporated into Endura electrocompetent Escherichia coli cells (Biosearch Technologies, 60242). About 25 million bacterial colonies were cultured for each library, followed by purification with the QIAGEN Plasmid Maxi Kit (QIAGEN, 12963). For the second step, the resulting plasmid libraries from the first cloning step were linearized by BsmBI digestion, gel-purified, and ligated using T4 ligase (NEB, M0202M) to a DNA fragment containing an sgRNA scaffold and the mouse U6 promoter. The ligated products were electroporated into Endura electrocompetent Escherichia coli cells (Biosearch Technologies, 60242), and about 40 million bacterial colonies were cultured for each library. The final plasmid libraries were extracted with the Qiagen EndoFree Plasmid Mega Kit (QIAGEN, 12381).
Lentivirus production and titration
To produce the lentiviral library, we used our previously described method.62 Briefly, 5 μg of plasmid library, with 3 μg of psPAX (Addgene, 12260) and 1 μg of pMD2.G (Addgene, 12259) packaging plasmids were cotransfected into 8 million HEK293T cells in a 10-cm dish supplemented with 36μL PolyJet (SignaGen Laboratories, SL100688). The medium was replaced 12 hours after transfection and harvested every 24 hours thereafter for a total of three harvests. Harvested viral media was filtered through a Millex-HV 0.45-μm polyvinylidene difluoride filter (Millipore, SLHV033RS) and further concentrated via centrifugation using 100,000 NMWL (nominal molecular weight limit) Ultra-15 centrifugal filter units (Amicon, UFC910008).
The lentiviral titer was determined by transducing 400,000 cells with increasing volumes (0, 1, 2, 5, 10, 20, and 40μL) of concentrated virus and polybrene (6 μg/ml; Millipore, TR-1003-G). 48 hours after the transduction, cells were dissociated with Trypsin-EDTA (0.25%; Gibco, 25200056) and seeded as two separate replicates; one treated with hygromycin B (200 μg/ml; Gibco, 10687010) for four days, and another that was not. Finally, hygromycin-resistant and control cells were counted to calculate the infected cell ratios and viral titers.
Prime-editing screens
We performed MYC enhancer screens in triplicate. We transfected MCF7-nCas9/RT cells with lentivirus libraries at a multiplicity of infection (MOI) of 0.3 with a coverage of 1,000 transduced cells per pegRNA/ngRNA pair. 48 hours later, approximately 10 million cells were harvested as controls (Day 2) and the remaining cells were treated with hygromycin B (200 μg/ml; Gibco, 10687010) for 7 days. After antibiotic selection, the cells were maintained in DMEM supplemented with 10% FBS for 30 days post infection (Day 30), and 10 million cells were collected from the final cell population.
We performed Alt and Ref library screens in quadruplicate. We separately infected about 24 million MCF7-nCas9/RT cells with the lentivirus library for each replicate of the Alt and Ref screens at a MOI of 0.5, with a cell coverage of 2,000 infected cells per pegRNA/ngRNA pair. 48 hours post infection, one-third of the infected cells were collected from each cell pool as control samples (Day 2). The remaining cells were treated with hygromycin B (200 μg/ml; Gibco, 10687010) for 7 days and cultured until 32 days post infection (Day 32).
Generation of Illumina sequencing libraries
Genomic DNA was extracted from each sample via cell lysis and digestion (100 mM tris-HCl pH 8.0, 5 mM EDTA, 200 mM NaCl, 0.2% SDS, and proteinase K 100 μg/ml), phenol:chloroform (Thermo Fisher Scientific, 17908) extraction, and isopropanol (Thermo Fisher Scientific, BP2618500) precipitation. For the MYC enhancer screen, we applied ePCR during library preparation to amplify the pegRNA/ngRNA pair sequences from each sample and reduce recombination between similar sequences. Briefly, thirty 20μL ePCRs were performed using 400 ng of DNA for each reaction and NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S) with the following primers: Enh-lib-Forward: TCCCTACACGACGCTCTTCCGATCTNNNNNCCTTGGAGAAAAGCCTTGTTT, Enh-lib-Reverse: GGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNGAACCGCGGTGTTAGG. ePCR was performed as described previously to amplify pegRNA/ngRNA pairs from genomic DNA. Thermocycler settings were 98 °C for 30 s, then 25 cycles (98 °C 10 s, 60 °C 20 s, 72 °C 1 min), then 72 °C 5 min, and finally a 4 °C hold. The ramp rate for each step was 2°C/s. After PCR, individual reactions were combined and purified using the QIAQuick PCR Purification Kit (QIAGEN, 28104) following previously established guidelines.68 Purified PCR products were then treated with Exonuclease I (NEB, M0568L) and purified using 1× AMPure XP beads (Beckman Coulter, A63881). Round one PCR amplicons were used in the 2nd round of PCR to add Illumina adapter and index sequences. For the 2nd round PCR, we performed 6 ePCR reactions containing 0.023 ng of purified DNA each, using NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S). The 2nd round PCR mixture was prepared and purified similarly to the 1st. Thermocycler settings were 98 °C for 30 s, then 12 cycles (98 °C 10 s, 60 °C 20 s, 72 °C 1 min), then 72 °C 5 min, and finally a 4 °C hold. The ramp rate for each step was 2°C/s. For Alt and Ref screens, we amplified pegRNA/ngRNA pair sequences from each sample using NEBNext High-Fidelity 2× PCR Master Mix (NEB, M0541S) and the following primers: Alt-Ref-lib-Forward: TCCCTACACGACGCTCTTCCGATCTNNNNNCTTGTGGAAAGGACGAAACACC, Alt-Ref-lib-Reverse: GGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNCGTAACTAGATAGAACCGCGTCAA. Twenty-four50μL PCR reactions, each containing 600 ng genomic DNA, were performed for each sample. Individual reactions were combined for each sample and column purified (Promega, A9282). The purified products were then amplified by indexing PCR to add Illumina TruSeq adaptors and sample index sequences with the following primers: Index-Forward: aatgatacggcgaccaccgagatctacac[8 bp index]acactctttccctacacgacgctcttccgatct, Index-Reverse: caagcagaagacggcatacgagat[8 bp index]gtgactggagttcagacgtgtgctcttccgatct. The final libraries were gel purified and sequenced with 150 bp paired-ends on the Illumina NovaSeq 6000 platform.
Data processing and analysis of prime-editing data
Sequencing libraries were first trimmed with 5 bp random sequences from read1 and read2, and low quality reads were filtered out with the fastp tool (v0.23.2) before formal mapping. To calculate the read counts, each pegRNA/ngRNA pair was included if it met the following criteria: (1) Read 1 exactly matched the sequence containing a 20-21 nt ngRNA spacer and 5 bp flanking sequences; (2) Read 2 exactly matched the reverse complementary sequence containing the full pegRNA extension and 5 bp flanking sequences.
For PRIME of MYC enhancer, the original raw counts for each pegRNA were first normalized by the total read counts. Subsequently, classical multidimensional scaling was employed to calculate and visualize the distances between different samples in two-dimensional plots. K-means clustering was then applied to partition the data points into k classes, with a final outcome of 2 groups, which were used in the multidimensional scaling plot for the Figure S2C. The MAGeCK (0.5.9) pipeline18 was used to estimate the statistical significance and fold change for each pegRNA/ngRNA pair at the guide RNA level, and for each substitution at the gene level in the cell population relative to controls. The non-targeting and AAVS1 targeting pegRNA/ngRNA pairs were used as negative controls for normalization. To identify the core enhancer region for the MYC enhancer based on the screening results, we first identified base pairs with three significant substitutions (FDR < 0.05), and calculated the slopes for each continuous bin (moving step = 1 bp, bin size = 30 bp, × axis: the position of each base pair, y axis: the accumulation number of SBPs with three significant substitutions) (Figure S2E). The slopes were then transformed into Z score-derived P values accordingly. The core enhancer region was identified by merging overlapping significant bins (P value < 0.05).
For Alt and Ref library screens, oligos with zero reads for any sample were removed before the following analysis. Oligo counts from all samples were passed into DESeq2 (1.38.0)37 and a median-of-ratios method was used to normalize samples for varying sequencing depths. Normalized read counts for each oligo were then modeled by DESeq2 as a negative binomial distribution. We then used DESeq2 to check the fold changes for each oligo in Alt and Ref libraries by comparing Day 32 to Day 2 data (design= ~ Replicate + Condition). We further estimated relative effects between the reference and alternate alleles by adding an interaction term (design= ~ Replicate + Condition + Allele + Condition:Allele). Condition refers to the collection timepoint (i.e. Day 32 or Day 2), and Allele refers to the allele category (i.e. Alt or Ref). Finally, a Wald test was performed via DESeq2 to calculate the P value. To minimize false positive hits and achieve an empirical FDR less than 5%, we then selected a P value cutoff corresponding to the fifth percentile of P values from non-targeting control oligos.
Motif matrix comparison analysis
To identify potential transcription factor (TF) binding sites within the target MYC enhancer, we established a new method based on motif comparison69 to directly compare known TF motifs with our single-base resolution functional data. We first calculated the log2(fold change) for each substitution at each base pair with MAGeCK (0.5.9).18 The log2(fold changes) of the wild type alleles were set to 0. We then transformed the log2(fold change) of each substitution into the corresponding fold change value. We further constructed the position weight matrix by normalizing the fold change of each allele per base pair to the sum of all unique alleles’ fold change per base pair. We further partitioned the enhancer sequence into multiple bins with lengths of 5 and 10 base pairs. We only retained bins with an information content over 3 and an ‘N’ content less than 10%. We then collected all TF motifs from JASPAR, HOCOMOCO, and SwissRegulon databases with high expression in MCF7 cells (TPM > 10, GSE175204). Next, we compared the filtered TF motif matrices with the enhancer bin matrix using Tomtom (P value < 0.05) to identify the potential TF binding sites at the enhancer. Finally, we only retained positive TF motif hits overlapping at least 95% of the input sequences’ essential base pairs (positions with maximum probabilities > 0.5). Details about the best matching motifs are summarized in Table S1.
Predicting base pair contribution to enhancer activity with BPNet
We trained a convolutional neural network using BPNet consistent with the published approach28 to explain the GATA3, ELF1, FOXM1, MTA3, and RCOR1 ChIP-seq data from ENCODE projects. Briefly, the model inputs were 1kb sequences across each ChIP-seq peak locus, and corresponding ChIP-seq control peaks were used as the bias track for training. The region from chromosome 2 was used as the tuning set, and chromosomes 5, 6, 7, 10, and 14 were used as the test set. The X and Y chromosomes were excluded. The remaining regions from other chromosomes were used to train the model with default parameters. Once models were acquired for each TF’s ChIP-seq data, DeepLIFT was used to calculate each input sequence base pair’s contribution to enhancer activity. TF-MoDISco contribution scores were finally used to cluster and determine consolidated TF motifs and map these to input peak regions.
Prime editing of GATA3 and ELF1 motifs in MYC enhancer
To alter GATA3 and ELF1 binding motifs sequences in the MYC enhancer, we designed pegRNA/ngRNA pairs (http://deepcrispr.info/DeepPrime/)60 and cloned them into lentiV2-mU6-evopreQ1 vector (see Table S4). After verifying cloned sequence using Primordium whole-plasmid sequencing, 5 μg of the PE plasmid was transfected into 1 million MCF7-nCas9/RT cells using the P3 primary nucleofection solution (Lonza, V4XP-3024) and the DN-100 Lonza 4D-Nucleofector program. The cells were then cultured in 6-well plates for a period of 2 days before being transferred to 96-well plates for the isolation of single clones. Verification of the clones with the desired edits was conducted through PCR followed by Sanger sequencing (primers are listed in Table S4).
MCF7 genotyping analysis
Sequence Read Archive (SRA) files for SRR7707725 and SRR7707726 (paired-end, two reads per loci) were retrieved from BioProject PRJNA486532. We used bwa-mem (v.0.7.17) to align sequenced reads to the human reference genome hg38 for each run separately. The Picard tools, SortSam, MarkDuplicates, AddOrReplaceReadGroups were then used to process the BAM files. Finally, GATK (v.4.2.5.0) was used to call SNPs and indels via local haplotype re-assembly (HaplotypeCaller) followed by joint genotyping on a single-sample GVCF from HaplotypeCaller (GenotypeGVCFs). Finally, CalcMatch (v.1.1.2) was used to verify genotype consistency between two runs.
Motif scan and TF identification for alleles with functional breast cancer SNPs
The sequences 20 bp upstream and downstream of each SNP (Alt and Ref alleles) were used as input sequences for TF motif analysis. FIMO software (version 5.5.0)58 was used to identify matching motifs centered on the SNP regions against the human TF motif database HOCOMOCO (v11 FULL).24 All FIMO motif scans were performed using default settings. Finally, TFs (FPKM >1) with binding motifs overlapping target SNP loci were selected (FDR < 0.05, P value < 0.0001).
Functional validation of SNPs using prime editing and RT-qPCR
To validate the function of PRIME identified functional SNPs in MCF7 cells, we converted the alternative allele to the reference allele at rs10956415 (Ref: C, Alt: A) and rs7772579 (Ref: A, Alt: C) loci, converted the reference allele to the alternative allele at rs66473811 (Ref: T, Alt: C) locus using PE. To clone the pegRNA/ngRNA expression plasmid, we amplified the fragment containing the ngRNA-mU6-pegRNA for these edits from the screening plasmid library, and inserted the fragment into the BsmBI-digested lentiV2-hU6-evopreQ1 vector using Gibson assembly (NEB, E2621L) (see Table S4). We verified the cloned pegRNA/ngRNA plasmid sequence using Primordium whole-plasmid sequencing.
To perform PE, we transfected two million MCF7-nCas9/RT cells with 2,000 ng of pegRNA/ngRNA plasmid containing an EGFP marker using PolyJet (SignaGen Laboratories, SL100688). Five days after transfection, we sorted the cells with the highest EGFP expression level (top 2%) into 96-well plates with 100 cells per well using FACS. Approximately two weeks later, we extracted genomic DNA from half of the cells in each well and maintained the other half by seeding them in a 24-well plate. We estimated the PE efficiency for each well by performing genotyping PCR followed by Sanger sequencing. We then expanded the cells in the wells with the highest editing efficiency to isolate clonal PE edited cell lines. We sorted the cell pool into 96-well plates with one cell per well using FACS. Approximately two weeks later, we performed genotyping PCR followed by Sanger sequencing to identify successfully edited clones (primers are listed in Table S4). Deep sequencing was then performed to quantify the copy number of edited alleles.
To assess the effect of SNPs on genes expression, we used multiple PE edited clones and control clones without intended editing for each SNP. About two million cells from each sample were used to extract total RNA with the RNeasy Plus Mini Kit (QIAGEN, 74134), and 1 μg of RNA was used to generate cDNA with the iScript cDNA Synthesis Kit (Bio-Rad, 1708890). The gene expression was analyzed on a Roche LightCycler 96 instrument using Luminaris HiGreen qPCR Master Mix (Thermo Scientific, K0992) (primers are listed in Table S4). Data were normalized to GAPDH.
Protein structure prediction with AlphaFold
To explore the impact of the BARD1 His36Pro mutation on BARD1/BRCA1 complex structure, we predicted the wild type BRAD1/BRCA1 and BARD1(His36Pro)/BRCA1 complex structures with AlphaFold. We used the same amino acid chain which is used in the BARD1/BRCA1 complex structure determined by NMR spectroscopy47 (BARD1, residues 26-122; BRCA1, residues 1-103) as input for complex structure predictions. The amino acid chains of BARD1 and BRCA1 were imported into the Google Colab Version of AlphaFold V2.2.4,56,70 powered by Python 3 Google Compute Engine. AlphaFold applied a multimer model in response to the duo-sequence imputation, then searched the genetic database to determine the best suited multiple sequence alignment for the imported sequence and initiated structural prediction. To avoid stereochemical violations, all structures are relaxed with AMBER model (Assisted Model Building with Energy Refinement) using GPU acceleration. The resulting PDB files were imported into UCSF Chimera X71,72 for structure visualization. Protein chains were assigned different colors to distinguish individual chains, and selected amino acid atomic structures and hydrogen bonds were illustrated for interaction analysis. Finally, the real-time rendered complex structures were exported using the snapshot function in Chimera X at the optimal visualization angle.
Checking BARD1-BRCA1 interaction using Split GFP
To check the interaction between BARD1 and BRCA1, we tagged BARD1 with GFP1-10, BRCA1 with GFP11. Specifically, we used the same fragment used for AlphaFold prediction, and cloned CMV promoter controlled GFP1-10-BARD1, GFP1-10-BARD1His36Pro, GFP11-BARD1, and BARD1-GFP11. We transfected 0.45 million MCF7 cells with the cloned BARD1 and BRCA1 plasmids (1,000 ng) using PolyJet (SignaGen Laboratories, SL100688). By checking the GFP signals with flow cytometry two days after transfection, we found the combination of GFP1-10-BARD1 + BARD1-GFP11, GFP1-10-BARD1His36Pro+ BARD1-GFP11 showed strong GFP signals. This observation is consistent with spatial distance between BARD1 and BRCA1 termini and proves the sensitivity of the split GFP method. To quantify the effect of His36Pro mutation on the interaction between BARD1 and BRCA1, we cloned GFP1-10-BARD1 or GFP1-10-BARD1His36Pro and BARD1-GFP11 into same vector. Meanwhile, we added EF-1α-mCherry into the same vector as internal control. We transfected 0.45 million MCF7 cells with 500 ng plasmids using PolyJet (SignaGen Laboratories, SL100688), and checked the transfected cells with flow cytometry 48 hours after transfection. To minimize the impact of plasmid copy number on the test, we took the cells with relatively low expression level of mCherry for analysis.
ChIP-qPCR
Chromatin immunoprecipitation (ChIP) was performed as described62 with minor modifications. MCF7 cells were detached with 0.25% Trypsin-EDTA, collected with DMEM media supplemented with 10% FBS, and centrifuged at 200g for 10 min at room temperature. The collected MCF7 cells were cross-linked in 1% formaldehyde for 10 min at room temperature followed by quenching with 125 mM glycine for 5 min. Cross-linked cells were washed twice with ice-cold DPBS and centrifuged at 1000g for 10 min at 4°C. The resulting cross-linked cells were incubated with lysis buffer (20mM Tris-HCl pH 8.0, 0.5% SDS, 2mM EDTA, 150mM NaCl, 1% Triton X-100, 1× Protease inhibitor) for 20 min on ice. For each sample, 6 million lysed cells (3 million lysed cells in 130μL lysis buffer for one microTUBE AFA Fiber Pre-Slit Snap-Cap 6x16mm tube) were sonicated using Covaris S220 focused-ultrasonicator (Duty factor, 5%; Peak incident power, 105 W; Cycles per burst, 200) for 10 min 30 sec to shear chromatin into 200 - 500 bp fragments. The sonicated lysate was centrifuged at 12,000g for 10 min at 4°C and 20μL of the sheared chromatin was saved as input. The remaining sheared chromatin was evenly split for incubating either with anti-IgG (Antibodies-Online, ABIN101961) or anti-MAZ (Novus Biologicals, NB100-86984). After precleared with 30μL Dynabeads Protein A beads (Invitrogen, 10001D) and 760μL dilution buffer (10mM Tris-HCl pH 8.0, 159mM NaCl, 1.14mM EDTA, 1.14% Triton X-100, 0.1% SDS, 0.114% Sodium Deoxycholate, 1× Protease inhibitor) for 1 h at 4°C on a rotator, the precleared chromatin was further incubated with 5 μg anti-IgG or anti-MAZ at 4°C overnight on a rotator. Meanwhile, the Protein A beads that will be used in the chromatin immunoprecipitation were blocked with BSA buffer (10mM Tris-HCl pH 8.0, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% SDS, 0.1% Sodium Deoxycholate, 5mg/mL BSA) at 4°C overnight on a rotator. Next day, the antibody-bound chromatin was incubated with BSA blocked beads at 4°C for 4 h on a rotator. All samples were then washed three times with low salt wash buffer (10mM Tris-HCl pH 8.0, 140mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% SDS, 0.10% Sodium Deoxycholate, 1× Protease Inhibitor Cocktail), two times with high salt wash buffer (10mM Tris-HCl pH 8.0, 300mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% SDS, 0.10% Sodium Deoxycholate), one time with LiCl buffer (10mM Tris-HCl pH 8.0, 150mM Lithium Chloride, 1mM EDTA, 0.5% IGEPAL CA-630, 0.1% Sodium Deoxycholate), two times with LTE buffer (10mM Tris-HCl pH 8.0, 0.1mM EDTA). Subsequently, 200μL elution buffer (10mM Tris-HCl pH 8.0, 1mM EDTA, 1% SDS) was added to all the samples and inputs and incubated overnight at 65°C. All the samples were further treated with 4 μg RNase (NEB, T3018L) for 30 min at 37°C, and 8 μg Proteinase K (NEB, P8107S) for 1 h at 55°C. Reverse crosslinked samples were purified using AMPure XP beads (Beckman Coulter, A63881) and eluted with 30μL LTE buffer and analyzed on a Roche LightCycler 96 instrument using Luminaris HiGreen qPCR Master Mix (Thermo Scientific, K0992) and primers targeting the MAZ binding site (MAZ-qF: TGGGATTCAAGCATACTTTGGC, MAZ-qR: CCTTAGACTGGGTTATTGCCCT). All samples were run in triplicates and normalized to the input. The allele frequency of rs66473811 in input and IP samples from PE edited clones were checked with Sanger sequencing after qPCR and quantified with EditR.59
Supplementary Material
Table S1. pegRNA and ngRNA oligo sequences and PRIME results for MYC enhancer, related to Figures 2 and S2.
Table S2. pegRNA and ngRNA oligo sequences and PRIME results for GWAS SNPs and clinical variants, related to Figures 3, S3 and 5.
Table S3. TF motif analysis for alleles with functional GWAS SNPs for breast cancer, related to Figure 3.
Table S4. The primers and DNA sequences, related to STAR Methods.
Highlights.
PRIME is a prime editing-mediated high-throughput genetic screen method
PRIME enables single-base resolution characterization of genome sequences
PRIME identifies essential nucleotides critical for enhancer function
PRIME can be used to annotate functional variants associated with diseases
Acknowledgements
We thank Howard Y. Chang (Stanford University) for sharing wild type MCF7 cells. We thank Bo Huang (University of California, San Francisco) for sharing the plasmids for the split GFP system. This work was supported by the Laboratory for Genomic Research Innovation Award LGRFU1019 and the George and Judy Marcus Innovation Fund - Precision Medicine Innovation SBI2019 (to Yin Shen and E.Z.), the National Institutes of Health (NIH) grants R01AG057497, R01EY027789, UM1HG009402, and U01DA052713 (to Yin Shen), U01HG011720 (to Y.L.). Sequencing was performed at the UCSF CAT, supported by UCSF PBBR, RRP IMIA, and NIH 1S10OD028511-01 grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
X.R., H.Y., and Yin Shen have filed a patent application related to pooled prime editing screens. The other authors declare no competing interests.
Inclusion and diversity statement
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. (2021). Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299. 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.French JD, and Edwards SL (2020). The Role of Noncoding Variants in Heritable Disease. Trends Genet 36, 880–891. 10.1016/j.tig.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Wunnemann F, Fotsing Tadjo T, Beaudoin M, Lalonde S, Lo KS, Kleinstiver BP, and Lettre G (2023). Multimodal CRISPR perturbations of GWAS loci associated with coronary artery disease in vascular endothelial cells. PLoS Genet 19, e1010680. 10.1371/journal.pgen.1010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197. 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16, 299–311. 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Findlay GM, Boyle EA, Hause RJ, Klein JC, and Shendure J (2014). Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123. 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. (2015). FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373, 895–907. 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844. 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 9.Chen PJ, and Liu DR (2022). Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet. 10.1038/s41576-022-00541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, and Liu DR (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erwood S, Bily TMI, Lequyer J, Yan J, Gulati N, Brewer RA, Zhou L, Pelletier L, Ivakine EA, and Cohn RD (2022). Saturation variant interpretation using CRISPR prime editing. Nat Biotechnol 40, 885–895. 10.1038/s41587-021-01201-1. [DOI] [PubMed] [Google Scholar]
- 12.Anzalone AV, Lin AJ, Zairis S, Rabadan R, and Cornish VW (2016). Reprogramming eukaryotic translation with ligand-responsive synthetic RNA switches. Nat Methods 13, 453–458. 10.1038/nmeth.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houck-Loomis B, Durney MA, Salguero C, Shankar N, Nagle JM, Goff SP, and D’Souza VM (2011). An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature 480, 561–564. 10.1038/nature10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, An M, Newby GA, Chen JC, Hsu A, and Liu DR (2022). Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol 40, 402–410. 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang Y, Jia G, Choi J, Ma H, Anaya E, Ye C, Shankar P, and Wu H (2015). Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol 16, 280. 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen PB, Fiaux PC, Zhang K, Li B, Kubo N, Jiang S, Hu R, Rooholfada E, Wu S, Wang M, et al. (2022). Systematic discovery and functional dissection of enhancers needed for cancer cell fitness and proliferation. Cell Rep 41, 111630. 10.1016/j.celrep.2022.111630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, et al. (2018). Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412 e1322. 10.1016/j.cell.2018.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baluapuri A, Wolf E, and Eilers M (2020). Target gene-independent functions of MYC oncoproteins. Nat Rev Mol Cell Biol 21, 255–267. 10.1038/s41580-020-0215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitsios D, Dhindsa RS, Middleton L, Gussow AB, and Petrovski S (2021). Prioritizing non-coding regions based on human genomic constraint and sequence context with deep learning. Nat Commun 12, 1504. 10.1038/s41467-021-21790-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, et al. (2015). Enhancer evolution across 20 mammalian species. Cell 160, 554–566. 10.1016/j.cell.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, Medvedeva YA, Magana-Mora A, Bajic VB, Papatsenko DA, et al. (2018). HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res 46, D252–D259. 10.1093/nar/gkx1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pachkov M, Balwierz PJ, Arnold P, Ozonov E, and van Nimwegen E (2013). SwissRegulon, a database of genome-wide annotations of regulatory sites: recent updates. Nucleic Acids Res 41, D214–220. 10.1093/nar/gks1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium, E.P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreiber J, Durham T, Bilmes J, and Noble WS (2020). Avocado: a multi-scale deep tensor factorization method learns a latent representation of the human epigenome. Genome Biol 21, 81. 10.1186/s13059-020-01977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Avsec Z, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, and Zeitlinger J (2021). Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat Genet 53, 354–366. 10.1038/s41588-021-00782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cronin KA, Scott S, Firth AU, Sung H, Henley SJ, Sherman RL, Siegel RL, Anderson RN, Kohler BA, Benard VB, et al. (2022). Annual report to the nation on the status of cancer, part 1: National cancer statistics. Cancer 128, 4251–4284. 10.1002/cncr.34479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michailidou K, Lindstrom S, Dennis J, Beesley J, Hui S, Kar S, Lemacon A, Soucy P, Glubb D, Rostamianfar A, et al. (2017). Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94. 10.1038/nature24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fachal L, Aschard H, Beesley J, Barnes DR, Allen J, Kar S, Pooley KA, Dennis J, Michailidou K, Turman C, et al. (2020). Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet 52, 56–73. 10.1038/s41588-019-0537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516. 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 33.Hanna RE, Hegde M, Fagre CR, DeWeirdt PC, Sangree AK, Szegletes Z, Griffith A, Feeley MN, Sanson KR, Baidi Y, et al. (2021). Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080 e1020. 10.1016/j.cell.2021.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, Hoffman D, Jang W, Kaur K, Liu C, et al. (2020). ClinVar: improvements to accessing data. Nucleic Acids Res 48, D835–D844. 10.1093/nar/gkz972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuella-Martin R, Hayward SB, Fan X, Chen X, Huang JW, Taglialatela A, Leuzzi G, Zhao J, Rabadan R, Lu C, et al. (2021). Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097 e1019. 10.1016/j.cell.2021.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang YH, Liu S, Zhang G, Zhou CQ, Zhu HX, Zhou XB, Quan LP, Bai JF, and Xu NZ (2005). Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumor cells growth in vitro and in vivo. Breast Cancer Res 7, R220–228. 10.1186/bcr975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao XH, Lu DL, Wang N, Liu LY, Wang Y, Li YQ, Yan TB, Sun XG, Hu P, and Zhang TC (2014). Estrogen receptor alpha mediates proliferation of breast cancer MCF-7 cells via a p21/PCNA/E2F1-dependent pathway. FEBS J 281, 927–942. 10.1111/febs.12658. [DOI] [PubMed] [Google Scholar]
- 40.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315. 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121. 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W, Gu X, Liu C, Shi Y, Wang P, Zhang N, Wu R, Leng L, Xie B, Song C, and Li M (2021). A synergetic effect of BARD1 mutations on tumorigenesis. Nat Commun 12, 1243. 10.1038/s41467-021-21519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuchner S, Tembe V, Rodriguez JA, and Henderson BR (2005). Nuclear targeting and cell cycle regulatory function of human BARD1. J Biol Chem 280, 8855–8861. 10.1074/jbc.M413741200. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez JA, Schuchner S, Au WW, Fabbro M, and Henderson BR (2004). Nuclear-cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene 23, 1809–1820. 10.1038/sj.onc.1207302. [DOI] [PubMed] [Google Scholar]
- 45.UniProt C. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prakash R, Rawal Y, Sullivan MR, Grundy MK, Bret H, Mihalevic MJ, Rein HL, Baird JM, Darrah K, Zhang F, et al. (2022). Homologous recombination-deficient mutation cluster in tumor suppressor RAD51C identified by comprehensive analysis of cancer variants. Proc Natl Acad Sci U S A 119, e2202727119. 10.1073/pnas.2202727119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brzovic PS, Rajagopal P, Hoyt DW, King MC, and Klevit RE (2001). Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol 8, 833–837. 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 48.Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M, Fletcher A, Blair-Reid S, Beesley J, Johal B, et al. (2016). Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol 23, 647–655. 10.1038/nsmb.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, and Huang B (2016). Versatile protein tagging in cells with split fluorescent protein. Nat Commun 7, 11046. 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spain BH, Larson CJ, Shihabuddin LS, Gage FH, and Verma IM (1999). Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc Natl Acad Sci U S A 96, 13920–13925. 10.1073/pnas.96.24.13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Zhou L, Gao BQ, Li G, Wang X, Wang Y, Wei J, Han W, Wang Z, Li J, et al. (2022). Highly efficient prime editing by introducing same-sense mutations in pegRNA or stabilizing its structure. Nat Commun 13, 1669. 10.1038/s41467-022-29339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen PF, Chen C, Nelson JW, Newby GA, Sahin M, et al. (2021). Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652 e5629. 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37, 224–226. 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hsu JY, Grunewald J, Szalay R, Shih J, Anzalone AV, Lam KC, Shen MW, Petri K, Liu DR, Joung JK, and Pinello L (2021). PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat Commun 12, 1034. 10.1038/s41467-021-21337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anderson MV, Haldrup J, Thomsen EA, Wolff JH, and Mikkelsen JG (2021). pegIT - a web-based design tool for prime editing. Nucleic Acids Res 49, W505–W509. 10.1093/nar/gkab427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirdita M, Schutze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M (2022). ColabFold: making protein folding accessible to all. Nat Methods 19, 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kluesner MG, Nedveck DA, Lahr WS, Garbe JR, Abrahante JE, Webber BR, and Moriarity BS (2018). EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J 1, 239–250. 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G, Kim HK, Park J, Kwak H, Cheong Y, Kim D, Kim J, Kim J, and Kim HH (2023). Prediction of efficiencies for diverse prime editing systems in multiple cell types. Cell 186, 2256–2272 e2223. 10.1016/jcell.2023.03.034. [DOI] [PubMed] [Google Scholar]
- 61.Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18, 541–553. 10.1016/j.stem.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ren X, Wang M, Li B, Jamieson K, Zheng L, Jones IR, Li B, Takagi MA, Lee J , Maliskova L, et al. (2021). Parallel characterization of cis-regulatory elements for multiple genes using CRISPRpath. Sci Adv 7, eabi4360. 10.1126/sciadv.abi4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fejerman L, Ahmadiyeh N, Hu D, Huntsman S, Beckman KB, Caswell JL, Tsung K , John EM, Torres-Mejia G, Carvajal-Carmona L, et al. (2014). Genome-wide association study of breast cancer in Latinas identifies novel protective variants on 6q25. Nat Commun 5, 5260. 10.1038/ncomms6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Machiela MJ, and Chanock SJ (2015). LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557. 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, and Eskin E (2014). Identifying causal variants at loci with multiple signals of association. Genetics 198, 497–508. 10.1534/genetics.114.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CH, Xiao T, Xu H, Jiang P, Meyer CA, Li W, Brown M, and Liu XS (2018). Improved design and analysis of CRISPR knockout screens. Bioinformatics 34, 4095–4101. 10.1093/bioinformatics/bty450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, and Griffiths AD (2006). Amplification of complex gene libraries by emulsion PCR. Nat Methods 3, 545–550. 10.1038/nmeth896. [DOI] [PubMed] [Google Scholar]
- 68.Verma V, Gupta A, and Chaudhary VK (2020). Emulsion PCR made easy. Biotechniques 69, 421–426. 10.2144/btn-2019-0161. [DOI] [PubMed] [Google Scholar]
- 69.Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS (2007). Quantifying similarity between motifs. Genome Biol 8, R24. 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. pegRNA and ngRNA oligo sequences and PRIME results for MYC enhancer, related to Figures 2 and S2.
Table S2. pegRNA and ngRNA oligo sequences and PRIME results for GWAS SNPs and clinical variants, related to Figures 3, S3 and 5.
Table S3. TF motif analysis for alleles with functional GWAS SNPs for breast cancer, related to Figure 3.
Table S4. The primers and DNA sequences, related to STAR Methods.
Data Availability Statement
All original data are available on NCBI Sequence Read Archive database and Mendeley data. All data are publicly available as of the date of publication. Accession numbers and DOI are listed in the key resources table.
All original code used for the design and analysis of pegRNA/ngRNA pairs is publicly available at Zenodo. The DOI of the code is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.