

Summary
Quantitative traits are shaped by networks of pleiotropic genes [1]. To understand the mechanisms that maintain genetic variation for quantitative traits in natural populations and to predict responses to artificial and natural selection, we must evaluate pleiotropic effects of underlying quantitative trait genes and define functional allelic variation at the level of quantitative trait nucleotides (QTNs). Catecholamines up (Catsup), which encodes a negative regulator of tyrosine hydroxylase [2], the rate-limiting step in the synthesis of the neurotransmitter dopamine, is a pleiotropic quantitative trait gene in Drosophila melanogaster [2–4]. We used association mapping to determine whether the same or different QTNs at Catsup are associated with naturally occurring variation in multiple quantitative traits. We sequenced 169 Catsup alleles from a single population and detected 33 polymorphisms with little linkage disequilibrium (LD). Different molecular polymorphisms in Catsup are independently associated with variation in longevity, locomotor behavior, and sensory bristle number. Most of these polymorphisms are potentially functional variants in protein coding regions, have large effects, and are not common. Thus, Catsup is a pleiotropic quantitative trait gene, but individual QTNs do not have pleiotropic effects. Molecular population genetic analyses of Catsup sequences are consistent with balancing selection maintaining multiple functional polymorphisms.
Results and Discussion
Quantitative Genetic Analysis
Catsup is a pleiotropic quantitative trait gene affecting lifespan [4] and locomotor behavior (K.W.J. and T.F.C.M., unpublished data). Catsup mutations are resistant to starvation [3], and Catsup transcript levels are reduced in starved flies [5]. Catsup mutants also affect sensory bristle development and exhibit a suite of aberrant abdominal phenotypes [2]. We quantified phenotypic variation in longevity, locomotor behavior, sensory bristle number, and starvation resistance in a population of 169 second chromosome substitution lines (see Tables S1 and S2 in the Supplemental Data available with this article online). We observed considerable naturally occurring genetic variation for these traits, with estimates of broad-sense heritabilities of 0.20 (locomotor reactivity), 0.23 (lifespan), 0.36 (sternopleural bristle number), 0.44 (starvation resistance), and 0.47 (abdominal bristle number). In addition, we observed an abnormal abdomen-like phenotype [6] in some of the chromosome substitution lines [7], characterized by low but extremely variable numbers of abdominal bristles between adjacent abdominal sternites and between genetically identical individuals for the same abdominal sternite (“environmental plasticity”). The broad-sense heritability of environmental plasticity of abdominal bristle number was 0.60.
There were significant genetic correlations between lifespan and starvation stress resistance (rG = 0.55, p < 0.001), lifespan and sternopleural bristle number (rG = 0.33, p < 0.001), sternopleural bristle number and starvation stress resistance (rG = 0.35, p < 0.001), sternopleural and abdominal bristle number (rG = 0.18, p = 0.02), and abdominal bristle number and environmental plasticity of abdominal bristle number (CVE) (rG = −1.09, p < 0.001). The strong negative association between mean abdominal bristle number and CVE of abdominal bristle number is attributable to the lines that exhibit the abnormal abdomen phenotype [6, 7].
Molecular Variation at Catsup
We sequenced approximately 3000 bp including the Catsup transcription unit from all lines. We observed 33 polymorphisms: 30 were present more than once in the sample and included 28 single nucleotide polymorphisms (SNPs) and five insertion/deletion polymorphisms (indels) (Figure 1A, Table S3). Estimates of the population mutation rate (, where is the effective population size and is the nucleotide mutation rate), based on the number of nucleotide differences between pairs of sites ( [8]) and the number of segregating sites ( [9]), were and , which are low but within the range observed for D. melanogaster [10]. Much of this variation is localized in the second exon.
Figure 1.
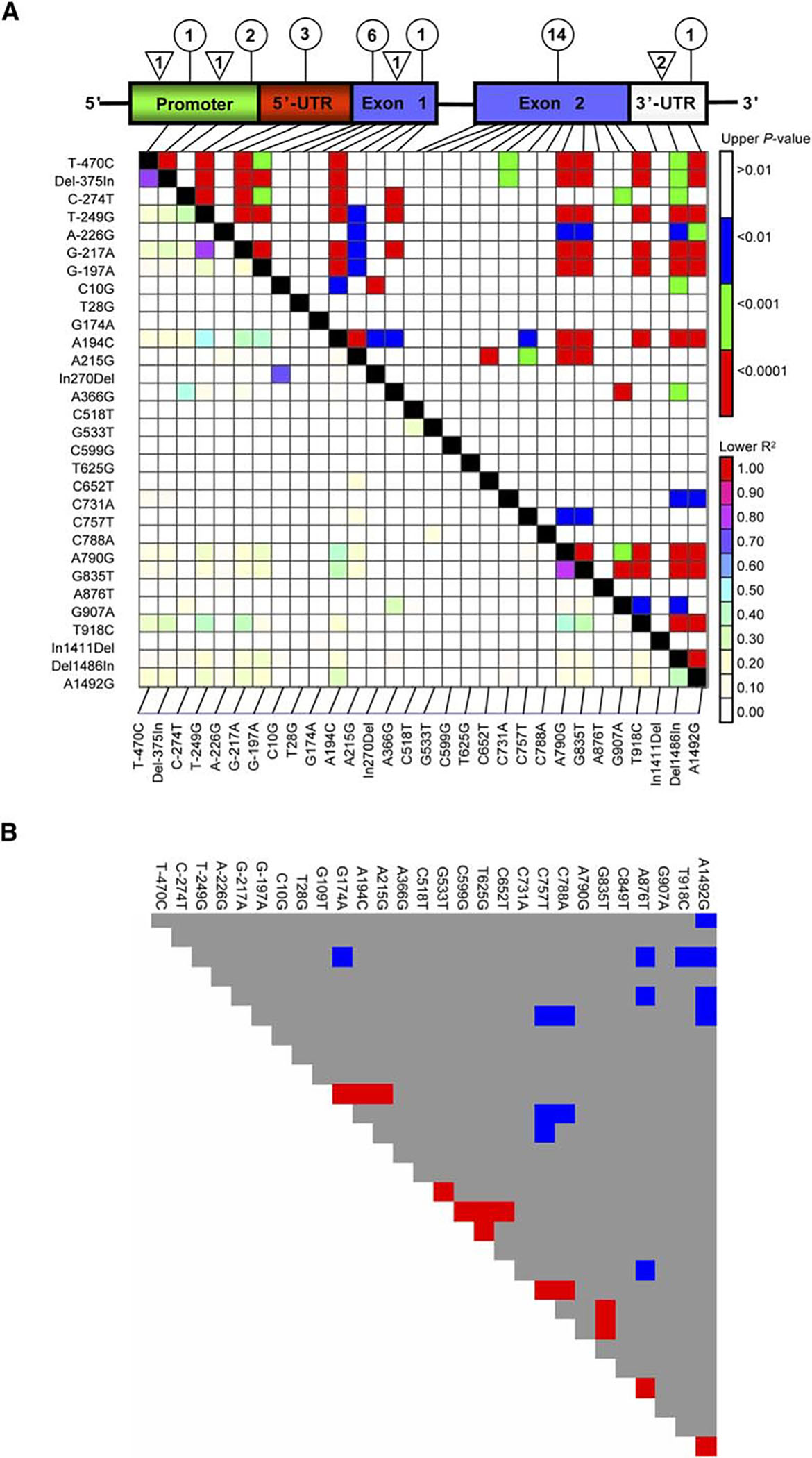
Catsup Polymorphisms (A) The Catsup gene structure is depicted with the number and distribution of SNPs (circle) and InDels (triangle) in 169 Catsup alleles sampled from the Raleigh population. The promoter region is predicted and not functionally confirmed. LD in Catsup is shown below the gene structure, with p values from Fisher’s Exact Test above the diagonal and estimates of r2 below the diagonal.
(B) Departures of LD in Catsup from the level expected given the maximum likelihood estimate of the population recombination parameter, 4Nr. Red and blue blocks depict pairwise associations that exhibit significantly less LD and significantly more LD, respectively, than expected.
We observed little significant LD except between pairs of sites at opposite ends of the Catsup region (Figure 1A). Given our large sample size and substantial number of segregating SNPs, we would expect to make a robust inference of the population recombination rate (4Nr, where r is the rate of recombination for the surveyed region [11]). Assuming a model of a single crossover [12], 4Nr = 87. Estimates of 4Nr tended to be higher than expected between pairs of sites in close proximity and lower than expected between distant pairs of sites (Figure 1B). However, because of the unusual LD pattern, the estimate of 4Nr for the entire sequenced region was not significantly different from zero according to a permutation test [12, 13].
The observed patterns of recombination and LD could be explained by localized gene conversion. We tested a model of gene conversion versus a model of single crossovers and compared their likelihoods [13]. Assuming a gene conversion model with tract lengths of 400 bp, we estimated the rate of exchange among alleles (, where is the rate of gene conversion and is the average tract length [12, 14]) as , which is significantly different from zero by the randomization procedure (p = 0.001). This suggests that gene conversion plays a large role in shaping polymorphism at this gene. Regardless of the mechanism, it is clear that recombination allows individual sites at the Catsup gene to evolve nearly independently—a highly favorable scenario for identifying Catsup QTNs causally associated with phenotypic variation.
Genotype-Phenotype Associations
We observed more Catsup polymorphisms associated with variation in all quantitative traits than expected by chance, at a 5% significance level; eight polymorphisms were associated with the traits by a Bonferroni-corrected significance level (Figures 2A and 2B). Several of these polymorphisms are possibly functional (Figure 2C). The Catsup protein has six predicted transmembrane helices, with extensive spans of histidine residues in two predicted extracellular domains (Figure 2C), the first of which may bind zinc [15]. Catsup interacts with tryosine hydroxylase (TH) and GTP cyclohydrolase (GTPCH, the rate-limiting step in the synthesis of tetrahydropterin, which is required for TH activity) via a conserved cytoplasmic loop [15]. C599G is associated with variation in longevity and results in a leucine-valine replacement in the third N-terminal transmembrane helix. In270Del is a 57 bp in-frame deletion of 19 amino acids (GHDHGHHHHGHDHDHDHDH) in the predicted zinc binding region [15], associated with variation in locomotor behavior. Polymorphisms associated with variation in locomotor behavior (A876T) and abdominal (C788A) and sternopleural (T918C) bristle number cause amino acid replacement polymorphisms in the cytoplasmic loop thought to interact with TH and GTPCH [15] (Figure 2C, Table S3). T28G is associated with both variation in mean abdominal bristle number and environmental plasticity of abdominal bristle number and results in a cysteine-glycine substitution in the cytoplasmic N-terminal domain of the protein. Four of the six amino acid polymorphisms are in exon 2. Two noncoding polymorphisms in the 5′ UTR, G-217A and A-226G, were significantly associated with variation in sternopleural bristle number and environmental plasticity of abdominal bristle number, respectively.
Figure 2.
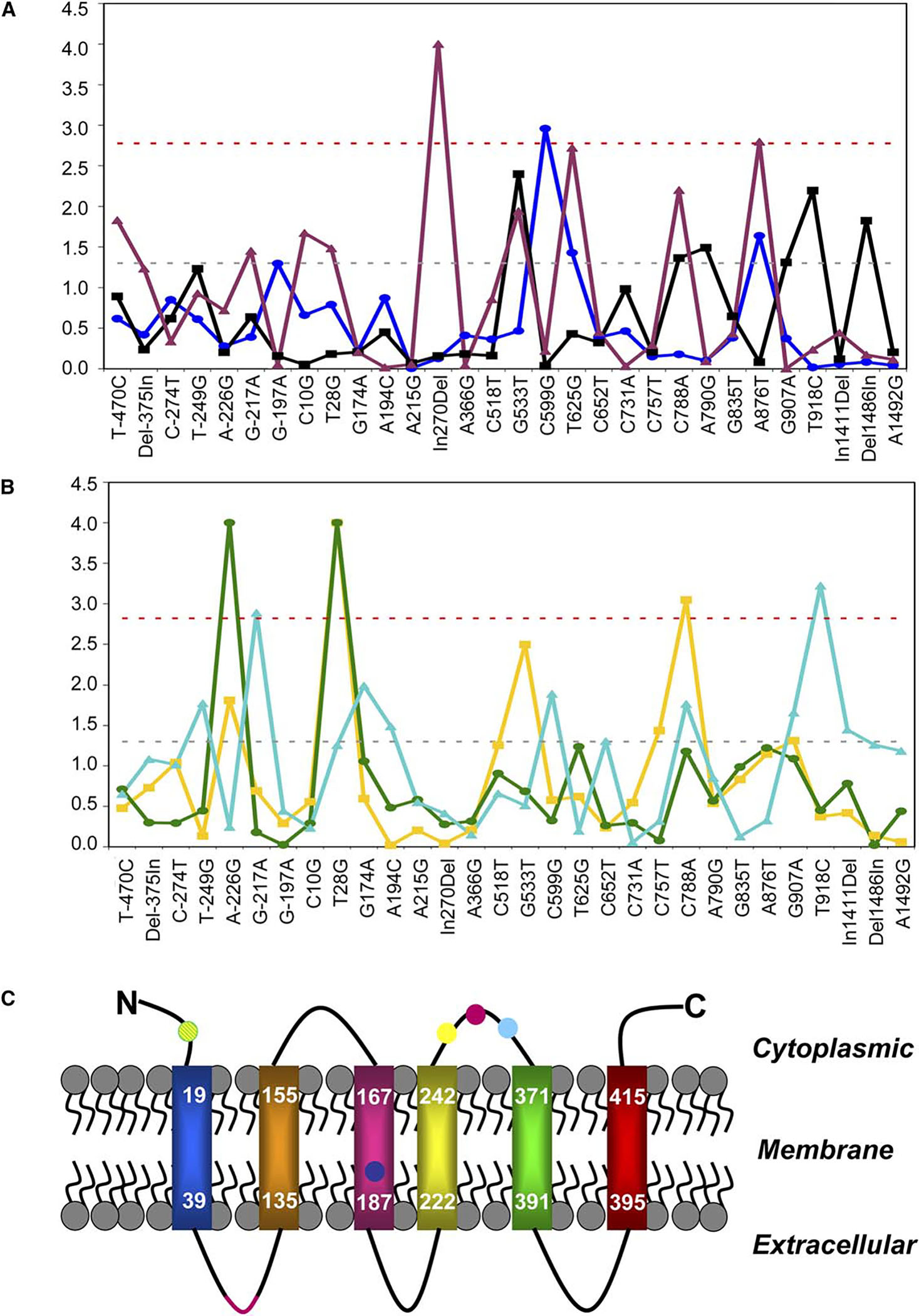
Genotype-Phenotype Associations at Catsup
(A) Plots of p values (transformed to log(1/P), y axis) from ANOVA tests of association of longevity (dark blue), locomotor behavior (purple), and starvation resistance (black) for each of the polymorphic markers at Catsup (x axis).
(B) Plots of p values (transformed to log(1/P), y axis) from ANOVA tests of association of abdominal bristle number (yellow), environmental plasticity of abdominal bristle number (green), and sternopleural bristle number (teal) for each of the polymorphic markers at Catsup (x axis). In both (A) and (B), the red horizontal dashed line indicates the experiment-wise p < 0.05 threshold given by the Bonferroni correction for multiple tests, and the black horizontal dashed line indicates the nominal p < 0.05 significance threshold.
(C) Predicted Catsup protein structure and locations of potentially functional amino acid polymorphisms associated with lifespan (dark blue), locomotor behavior (purple), abdominal bristle number (yellow), environmental plasticity in abdominal bristle number (teal), and sternopleural bristle number (light blue).
Six Catsup polymorphisms were nominally associated with variation in starvation resistance (G533T, C788A, A790G, G907A, T918C, Del1486In; Figure 2A). Del1486In is the 3′-UTR and the remaining polymorphisms are in exon 2 (Table S3). Based on a permutation test, the probability of observing six or more sites significant by chance was p = 0.022. Further, there was significant variation in resistance to starvation (p = 0.00009) among the 87 population haplotypes formed by considering all 30 Catsup polymorphisms (data not shown). This association was significant (p = 0.002) based on a permutation test. A significant association of starvation stress with population haplotypes, but not individual sites, suggests that the multiple sites with individually small effects interact to affect this trait. The two most extreme haplotypes have mean survival times of approximately 100 hr and 38 hr and differ at 12 of the 30 polymorphic sites present more than once in the sample; four of these variable sites are also nominally significantly associated with variation in starvation resistance.
Although LD is highly significant between sites at opposite ends of the sequenced region (Figure 1A, top diagonal), the absolute magnitude of the LD is very weak (Figure 1A, bottom diagonal). Further, only two of the markers that are significantly associated with the traits are in LD (G-217A and T918C, p < 0.0001, r2 = 0.4), and both are associated with sternopleural bristle number. The lack of a correlation among polymorphisms is consistent with the observed high rates of recombination, which generates independence in evolutionary histories among sites within a population. These observations are consistent with causal associations of the Catsup polymorphisms with these traits, although the mechanistic basis of the associations remains to be elucidated. Only two of the observed associations would have been detected had we implemented the common design of only genotyping the common variants. An alternative explanation is that the associated sites are not causal, but in LD with a true site outside the sequenced region. If this were true, then the true sites would have to have very large effects and be in strong (not weak) LD with the observed sites. The effect attributable to a polymorphic site in LD with the true causal site always underestimates the true effect [16].
If generally applicable to many quantitative trait genes, these observations might partly explain the notorious difficulty of replicating correlated responses to selection [17]. Although Catsup is a pleiotropic quantitative trait gene, the actual polymorphisms associated with the different traits are largely independent. Independent selection lines for any of the traits generated from sampling this population would evolve different correlated responses in the other characters, depending on the exact constellation of frequencies of marker alleles associated with the correlated traits in the initial sample.
These observations highlight the wealth and variety of naturally occurring variation and the utility of viable alleles with subtle effects in understanding the effects of candidate genes on complex traits [1]—most induced Catsup mutations are homozygous lethal. In addition, they address fundamental questions regarding the nature of standing variation for complex traits. What fraction of naturally occurring variation is attributable to polymorphisms in regulatory domains versus protein coding regions? What is the distribution of gene frequencies and effects of causal variants? Previous studies in Drosophila have shown that most (but not all) molecular polymorphisms associated with variation in quantitative traits are in noncoding regions, have moderate effects, and are at intermediate frequency (e.g., [18, 19]). However, only a few studies to date have conducted association tests with full sequence data, leaving open the possibility that the observed associations were not causal, but in LD with a true variant not assessed in the sample. Here it is likely that we have identified the actual causal variants, and find that most sites individually associated with complex trait phenotypes were nonsynonymous polymorphisms in protein coding regions, had large effects, and had minor allele frequencies less than 5% (Table S3 and S4). Indeed, we observed a significant negative correlation (r = −0.78, p < 0.05) between the effects of the significant sites in genetic standard deviation units and the frequency of the rare allele.
The use of chromosome substitution lines to assess genotype-phenotype associations greatly increased our power to detect associations with rare alleles. We restricted the genetic variation affecting the traits to genes on the second chromosome and reduced the contribution of random environmental variance by measuring multiple individuals of each line, thereby obtaining accurate measures of the mean genotypic values and high broad-sense marker heritabilities (Table S4). For comparison, we can infer the contribution of the significant sites to narrow-sense heritabilities (, where is the additive genetic variance and is the total phenotypic variance [17]) in an outbred population. Under a strictly additive model, attributable to a single biallelic locus in a random mating population is , where and are the frequencies of the common and rare allele, respectively, and a is the allelic effect (half of the difference in trait phenotype between homozygous genotypes) [17]. The estimates are 0.007 (longevity, C599G); 0.021 and 0.009 (locomotor behavior, In270Del and A876T, respectively); 0.035 and 0.009 (abdominal bristle number, T28G and C788A, respectively); 0.034 and 0.048 (environmental plasticity of abdominal bristle number, A-266G and T28G, respectively), and 0.006 and 0.007 (sternopleural bristle number, G-217A and T918C, respectively).
It will prove challenging to replicate these associations in a natural population. First, simple power calculations reveal that huge sample sizes are required to ensure sufficient individuals in the rare allele class to detect effects of the expected magnitude. The number of individuals (n) required to detect a difference between homozygous genotypes is , where is the within-genotype standard deviation; and are, respectively, the Type I and Type II significance levels set; and is the ordinate of the normal distribution corresponding to its subscript [20]. Assuming and , and evaluating the expression for observed values of (Table S4) gives the number of individuals representing the homozygous minor allele; dividing this by (the Hardy-Weinberg expected frequency of the rare homozygous genotype) gives the total sample size. For lifespan, this is more than 49,000 individuals. The best-case scenario is for the association of G-217A with sternopleural bristle number, which would “only” require 5670 individuals. The challenge is further complicated by uncontrolled environmental variation in nature, the possibility that epistasis and genotype by environment interaction causes the effects to differ between the laboratory and natural environments, and other factors [21].
Natural Selection at Catsup
Although direct observations of the effects of molecular variation at Catsup and quantitative trait phenotypes is challenging in nature, we can use molecular population genetic tests for selection to infer what evolutionary forces may be regulating quantitative and nucleotide variation at the Catsup locus.
The frequency spectrum of intraspecific mutations was slightly skewed toward an excess of per site heterozygosity given the number of segregating sites, but did not indicate a significant departure from neutrality (Tajima’s [22] D = 0.28, p > 0.05). We examined the relationship of polymorphism within species to divergence between species. The conservative McDonald-Kreitman (MK) test [23] examines departures from the neutral expectation that the ratio of nonsynonymous (DN) to synonymous (DS) divergence between species should not be significantly different from the ratio of nonsynonymous to synonymous polymorphisms within species. The observed estimates of and per site at Catsup were moderate relative to other loci [24] (Table 1). However, in contrast to the neutral expectation, the ratio of the number of nonsynonymous to synonymous polymorphisms was significantly in excess of the ratio of nonsynonymous to synonymous substitutions (Table 1).
Table 1.
Molecular Population Genetics of Catsup
| Sites |
|||
|---|---|---|---|
| All | Synonymous (S) | Nonsynonymous (N) | |
|
| |||
| Divergence (D) | 0.0421 | 0.0715 | 0.0070 |
| Polymorphism (π) | 0.0017 | 0.0019 | 0.0010 |
| No. fixed differences | 86 | 81 | 5 |
| No. segregating in | 21 | 7 | 14 |
| D. melanogaster | |||
McDonald-Kreitman test [23] comparing fixed and segregating ratios of synonymous and nonsynonymous substitutions, p = < 0.0001.
Also in contrast to the neutral expectation, there is highly significant heterogeneity in the ratio of polymorphism to divergence across Catsup. Figure 3 depicts a sliding window analysis of the ratio of nonsynonymous to synonymous variation at Catsup, which shows that exon 2 has very high ratios and low DN/DS ratios, i.e., much more amino acid polymorphism than divergence. This heterogeneity is significant based on HKA tests [25] comparing the noncoding regions versus the two exons (, p = 0.026), or comparing the noncoding regions plus exon 1 versus exon 2 (, p = 0.007). In addition, when we performed an MK test on each exon separately, we found that the second exon contributes most to the overall MK test result (Fisher’s Exact Test with Bonferroni correction; exon 2, p = 0.013).
Figure 3.
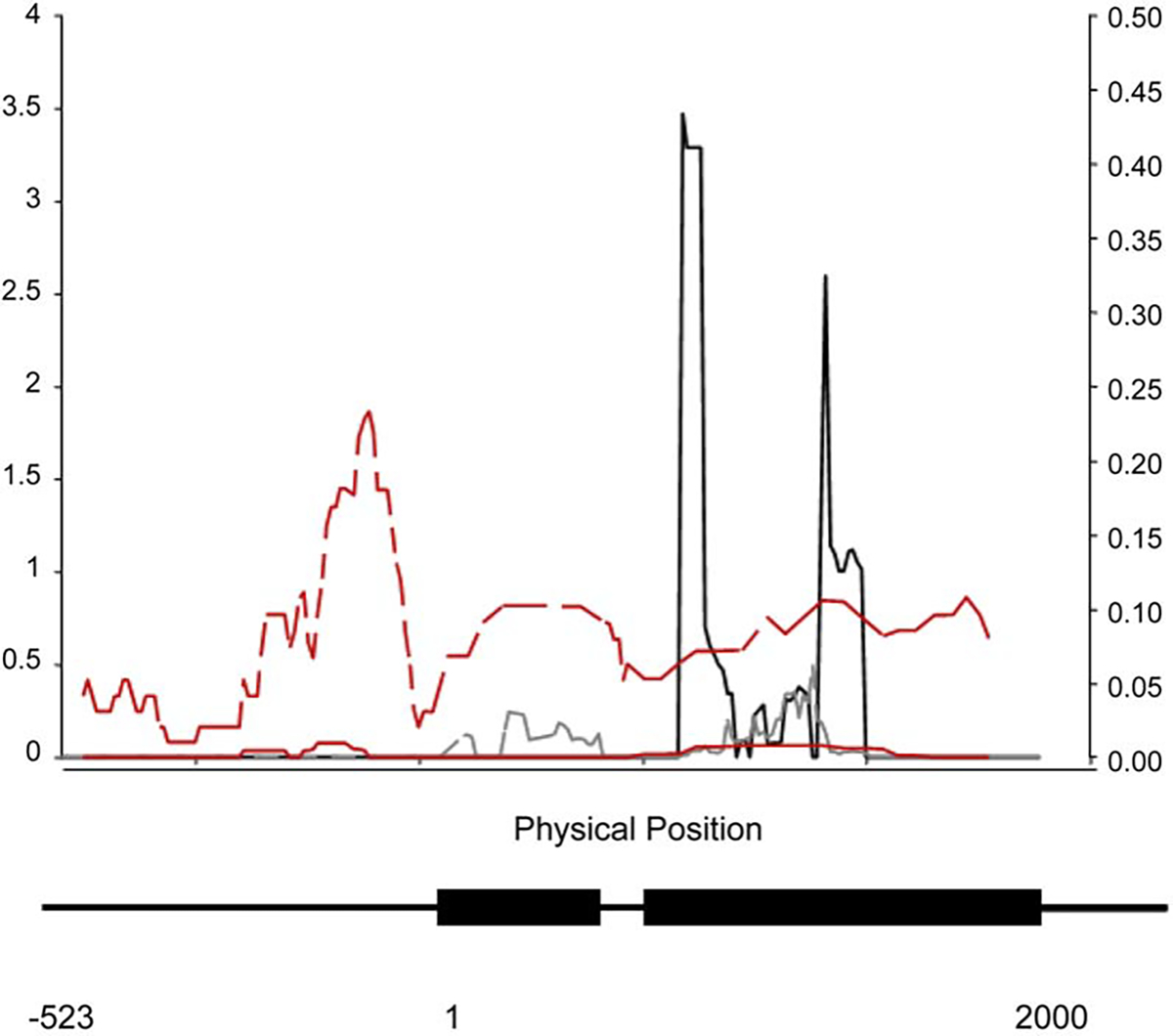
Sliding Window Analysis of the Ratio of Nonsynonymous to Synonymous Variation at Catsup
The solid black line denotes ratios and the gray line indicates DN/DS ratios (right y axis). The solid and broken red lines denote polymorphism () within D. melanogaster and divergence between D. melanogaster and D. simulans, respectively (left y axis). Windows were 100 bp with a step size of 10 bp. The gene structure of Catsup is indicated below the x axis. The scale is in bp, with 1 indicating the translation start site.
There are two contrasting explanations for the observed pattern of departure from neutrality at Catsup. can exceed DN/DS if purifying selection against deleterious nonsynonymous polymorphisms is less effective in regions of low recombination [26–29]. Alternatively, balancing selection may increase the frequency of above that expected given the amount of divergence at nonsynonymous and synonymous sites. Several lines of evidence favor the latter interpretation. (1) The frequency spectrum for nonsynonymous (or synonymous) sites at Catsup is not skewed toward rare alleles overall (Tajima’s D [22] was positive), as expected under the former interpretation. (2) Recombination is high, not low; therefore, selection should be highly effective against deleterious alleles, removing them from the population [26, 28]. (3) The excess of replacement polymorphisms is concentrated in the second exon, and many of these sites are associated with variation in quantitative trait phenotypes (Figure 2). In addition, population recombination rates are elevated in the second exon (Figure 1C). This pattern of variation in polymorphism and recombination is as expected from a local increase in the effective population size due to balancing selection [30]. (4) The effects of the rare alleles on quantitative traits were not always in the direction of reduced fitness. For example, the rare allele at the site affecting lifespan is associated with increased longevity, and the rare 57 bp deletion is associated with increased locomotor reactivity (Table S4). Since population genetic inferences about the fitness effects of alleles are often attributed to the frequency with which they segregate in the population, it appears that Catsup alleles are segregating at frequencies consistent with them being maintained in the population. Thus, overall our data are more consistent with balancing selection maintaining multiple functional polymorphisms, although the mechanism of selection is not known.
Catsup is only one component of the catecholamine biosynthetic pathway [2, 31, 32]. Previously, we found that polymorphisms at Ddc, which catalyzes the final step in the biosynthesis of dopamine and serotonin, are associated with quantitative variation in longevity [33], locomotor behavior (K.W.J. and T.F.C.M., unpublished data), and environmental plasticity of abdominal bristle number [7]. Other genes involved in dopamine synthesis, such as TH and GTPCH, may also determine variation in longevity and other quantitative traits in Drosophila and other species, including humans. Indeed, a polymorphism in TH is associated with variation in human longevity [34]. The challenge for the future will be to assess the combined effects of the network of genes that affect dopamine synthesis on natural variation in dopamine levels and on pleiotropic quantitative traits.
Experimental Procedures
Drosophila Stocks
Isofemale lines were established from wild-type gravid females collected at the Raleigh, NC, Farmer’s Market in 1999. Single second chromosomes were extracted from each of 169 isofemale lines and substituted into the highly inbred Samarkand (Sam) background by standard techniques with balancer chromosomes [33]. In addition, an ethyl methanesulfonate-induced recessive lethal null allele of Catsup (Catsup1) was substituted into the Sam genetic background. All stocks were maintained on cornmeal-agar-molasses medium at 25°C, 60%–75% relative humidity, and a 12 hr light-dark cycle.
Quantitative Trait Phenotypes
Lifespan (n = 4 replicates/sex/line, 6 same-sex individuals per replicate); resistance to starvation stress (n = 2 replicates/sex/line, 10 same-sex individuals per replicate); abdominal and sternopleural bristle number (2 replicates/line, 5 males and 5 females per replicate); and environmental plasticity of abdominal bristle number (2 replicates/line) were assessed on the homozygous chromosome 2 substitution lines as described previously [5, 7, 33].
A complementation test design was used to assess whether naturally occurring Catsup alleles contributed to quantitative variation in locomotor behavior. Each of the chromosome 2 substitution lines (C2i,) was crossed to both Sam; Catsup1 and Sam; Catsup+ stocks, and Sam; Catsup1/C2i and Sam; Catsup+/C2i F1 individuals, respectively, were collected from each cross. Locomotor behavior was assessed for 20 males and 20 females of each F1 genotype. Single 3- to 7-day-old adult flies were subjected to a gentle mechanical disturbance, and locomotor behavior was quantified as the number of seconds each fly was active in the 45 s period immediately after the disturbance. All measurements were taken in a behavioral chamber at the same time of day (8 am–12 pm) under constant temperature (25°C) and humidity (75%).
Quantitative Genetic Analysis
Mixed model analyses of variance (ANOVA) were used to partition variance in quantitative traits for the chromosome substitution lines. The model for lifespan and resistance to starvation stress was , where is the overall mean, is the random effect of line, is the fixed effect of sex, is the random effect of replicate vial, is the within-vial variance, and parentheses indicate nested effects. Similarly, the model for numbers of sensory bristles was . We partitioned the variance in of abdominal bristle number as , where here is the variance in between replicate vials. The complementation test ANOVA model for locomotor reactivity was , where is the fixed main effect of cross (Catsup1 or Catsup+). For all traits except locomotor reactivity, the total genotypic variance among lines was estimated as , where is the among-line variance component and is the variance attributable to the interaction. The total genotypic variance in locomotor reactivity was estimated as , where is the interaction variance and is the three-way interaction variance. The total phenotypic variance was estimated as for all traits, where is the environmental variance component. We estimated broad-sense heritabilities as , and genetic correlations between traits as , where is the covariance among line means for the two traits, and and are the square roots of the genotypic variance from the analyses of each trait separately. All statistical analyses were performed by SAS procedures (SAS Institute).
Catsup Sequence
We sequenced 3000 bp including the Catsup transcription unit and approximately 1000 bp upstream and 265 bp downstream of the coding region in the 169 D. melanogaster chromosome substitution lines, and one D. simulans allele. Genomic DNA was extracted with the Puregene DNA isolation kit (Gentra Systems). PCR and sequencing primers were designed by means of the published Catsup sequence (http://www.flybase.org). PCR products from each primer pair were purified with Qiaquick columns (Qiagen) and sequenced directly from both strands with the ABI PRISM Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems). Sequences were aligned and edited with VectorNTI software (InforMax). Single nucleoide polymorphisms (SNPs) and insertion/deletion variants (indels) were identified from the alignments.
Genotype-Phenotype Associations
Associations between molecular polymorphisms and all quantitative trait phenotypes except for locomotor behavior were assessed by two-way fixed effects ANOVA of line means, according to the model , where and denote the effect of marker allele (or haplotype) and sex, respectively, and E is variance within marker genotype. For locomotor behavior, we used a three-way factorial ANOVA of line means, , where is the main effect of cross.
We used permutation tests to determine empirical distributions under the null hypotheses of no association between Catsup genotypes and starvation stress resistance. To assess whether we observed more nominally significant marker-phenotype associations for resistance to starvation stress than expected by chance, we permuted the trait phenotypes among the marker genotypes 1000 times and recorded the number of significant associations at p < 0.05 for each permuted data set. To assess whether the observed association of starvation resistance with the Catsup population haplotypes was less than expected by chance, we permuted the trait phenotypes among the marker haplotypes 1000 times and recorded the lowest p value for the effect of haplotype for each permuted data set.
Molecular Population Genetic Analysis
Population haplotypes were inferred by the SNAP workbench [35]. Patterns of LD and significance tests were assessed with TASSEL (http://www.maizegenetics.net). Estimates of nucleotide diversity and tests of departure from neutrality were conducted with DnaSP [36]. LD was quantified as , where and are the allele frequencies at the first and second locus, respectively, and are the haplotype frequencies at loci and . Estimates of and recombination were calculated with LDhat [12] software. Pairwise LD estimates in a given data set are not independent; therefore, we used a permutation test to assess whether there was a significant decline of LD with physical distance [13]. This test randomizes the positions of segregating sites and for each randomization computes the correlation coefficient for physical distance between pairs of sites and LD. Estimates of 4Nr were calculated by the composite likelihood approach in LDhat, for the SNP data only. Estimates were made via a model of single crossover or gene conversion [12, 13]. The relationship between physical distance and recombination rate with gene conversion is , where is the per base rate of initiation of gene conversion and is the average gene conversion tract length (assuming an exponential distribution [37]). We fixed the average tract length and estimated the compound parameter , i.e., the population rate of recombination between two distantly linked loci caused by gene conversion.
Supplementary Material
Acknowledgments
We thank Robert Anholt, Franck Prugnolle, Janis O’Donnell, and Adam Eyre-Walker for helpful comments and Brad Toms for technical assistance. This research was supported by grants from the National Institutes of Health to T.F.C.M. This is a publication of the W.M. Keck Center for Behavioral Biology. The authors declare no competing financial interests.
Footnotes
Supplemental Data
The four supplemental tables can be found with this article online at http://www.current-biology.com/cgi/content/full/16/9/912/DC1/.
Accession Numbers
The Catsup sequences have been deposited in GenBank under accession numbers DQ375815-DQ375985 (D. melanogaster) and DQ375986 (D. simulans).
References
- 1.Mackay TFC (2001). The genetic architecture of quantitative traits. Annu. Rev. Genet. 35, 303–339. [DOI] [PubMed] [Google Scholar]
- 2.Stathakis DG, Burton DY, McIvor WE, Krishnakumar S, Wright TR, and O’Donnell JM (1999). The Catecholamines up (Catsup) protein of Drosophila melanogaster functions as a negative regulator of tyrosine hydroxylase activity. Genetics 153, 361–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Donnell JM, Wang Z, and Chaudhuri A (2004). Effects of perturbation of catecholamine regulation on resistance of Drosophila melanogaster to environmental stress. In Pterins, Folates, and Related Biogenic Amines, Blau N and Thony B, eds. (Heilbronn: SPS Publications; ), pp. 94–100. [Google Scholar]
- 4.Mackay TFC, Roshina NV, Leips JW, and Pasyukova EG (2005). Complex genetic architecture of Drosophila longevity. In Handbook of the Biology of Aging, Sixth Edition, Masaro EJ and Austad SN, eds. (Burlington: Elsevier Press; ), pp. 181–216. [Google Scholar]
- 5.Harbison ST, Chang S, Kamdar KP, and Mackay TFC (2005). Quantitative genomics of starvation stress resistance in Drosophila. Genome Biol. 6, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sobels FH (1952). Genetics and morphology of the genotype “asymmetric” with special reference to its “abnormal abdomen” character (Drosophila melanogaster). Genetica 26, 117–279. [PubMed] [Google Scholar]
- 7.Mackay TFC, and Lyman RF (2005). Drosophila bristles and the nature of quantitative genetic variation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1513–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nei M, and Tajima F (1981). DNA polymorphism detectable by restriction endonucleases. Genetics 97, 145–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watterson GA (1975). On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276. [DOI] [PubMed] [Google Scholar]
- 10.Moriyama EN, and Powell JR (1996). Intraspecific nuclear DNA variation in Drosophila. Mol. Biol. Evol. 13, 261–277. [DOI] [PubMed] [Google Scholar]
- 11.Hudson RR (2001). Two-locus sampling distributions and their application. Genetics 159, 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McVean G, Awadalla P, and Fearnhead P (2002). A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160, 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awadalla P, and Charlesworth D (1999). Recombination and selection at Brassica self-incompatibility loci. Genetics 152, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Awadalla P (2003). The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4, 50–60. [DOI] [PubMed] [Google Scholar]
- 15.O’Donnell JM, Stathakis DG, Burton D, and Chen Z (2002). Catecholamines-up, a negative regulator of tyrosine hydroxylase and GTP cyclohydrolase I in Drosophila melanogaster. In Chemistry and Biology of Pteridines and Folates, Milstein GKS, Levine R, and Shane B, eds. (Boston: Kluwer Academic Publishers; ), pp. 211–215. [Google Scholar]
- 16.Lai C, Lyman RF, Long AD, Langley CH, and Mackay TFC (1994). Naturally occurring variation in bristle number and DNA polymorphisms at the scabrous locus in Drosophila melanogaster. Science 266, 1697–1702. [DOI] [PubMed] [Google Scholar]
- 17.Falconer DS, and Mackay TFC (1996). Introduction to Quantitative Genetics, 4th edition (Harlow: Addison Wesley Longman; ). [Google Scholar]
- 18.Long AD, Lyman RF, Langley CH, and Mackay TFC (1998). Two sites in the Delta gene region contribute to naturally occurring variation in bristle number in Drosophila melanogaster. Genetics 149, 999–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robin C, Lyman RF, Long AD, Langley CH, and Mackay TFC (2002). hairy: a quantitative trait locus for Drosophila bristle number. Genetics 162, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sokal RR, and Rohlf FJ (1981). Biometry, 2nd edition (New York: W.H. Freeman and Company; ). [Google Scholar]
- 21.Macdonald SJ, and Long AD (2004). A potential regulatory polymorphism upstream of hairy is not associated with bristle number in wild-caught Drosophila. Genetics 167, 2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tajima F (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald JH, and Kreitman M (1991). Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654. [DOI] [PubMed] [Google Scholar]
- 24.Andolfatto P (2001). Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18, 279–290. [DOI] [PubMed] [Google Scholar]
- 25.Hudson RR, Kreitman M, and Aguadé M (1987). A test of neutral molecular evolution based on nucleotide data. Genetics 116, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill WG, and Robertson A (1966). The effect of linkage on limits to artificial selection. Genet. Res. 8, 269–294. [PubMed] [Google Scholar]
- 27.Nachman MW (1998). Y chromosome variation of mice and men. Mol. Biol. Evol. 15, 1744–1750. [DOI] [PubMed] [Google Scholar]
- 28.Weinreich DM, and Rand DM (2000). Contrasting patterns of nonneutral evolution in proteins encoded in nuclear and mitochondrial genomes. Genetics 156, 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Presgraves DC (2005). Recombination enhances protein adaptation in Drosophila melanogaster. Curr. Biol. 15, 1651–1656. [DOI] [PubMed] [Google Scholar]
- 30.Takahata N (1990). A simple genealogical structure of strongly balanced allelic lines and trans-species evolution of polymorphism. Proc. Natl. Acad. Sci. USA 87, 2419–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wright TRF (1987). The genetics of biogenic amine metabolism, sclerotization and melanization in Drosophila melanogaster. Adv. Genet. 24, 127–222. [PubMed] [Google Scholar]
- 32.Stathakis DG, Pentz ES, Freeman ME, Kullman J, Hankins GR, Pearlson NJ, and Wright TRF (1995). The genetic and molecular organization of the Dopa decarboxlyase gene cluster of Drosophila melanogaster. Genetics 141, 629–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Luca M, Roshina NV, Geiger-Thornsberry GL, Lyman RF, Pasyukova EG, and Mackay TFC (2003). Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat. Genet. 34, 429–433. [DOI] [PubMed] [Google Scholar]
- 34.De Luca M, Rose G, Bonafe M, Garasto S, Greco V, Weir BS, Franceschi C, and De Benedictis G (2001). Sex-specific longevity associations defined by Tyrosine Hydroxylase-Insulin-Insulin Growth Factor 2 haplotypes on the 11p15.5 chromosomal region. Exp. Gerontol. 36, 1663–1671. [DOI] [PubMed] [Google Scholar]
- 35.Price EW, and Carbone I (2005). SNAP: workbench management tool for evolutionary population genetic analysis. Bioinformatics 21, 402–404. [DOI] [PubMed] [Google Scholar]
- 36.Rozas J, and Rozas R (1999). DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15, 174–175. [DOI] [PubMed] [Google Scholar]
- 37.Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, and Di Rienzo A (2001). Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am. J. Hum. Genet. 4, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.