

Abstract
Sarcopenia, characterized by the insidious reduction of skeletal muscle mass and strength, detrimentally affects the quality of life in elderly cohorts. Present therapeutic strategies are confined to physiotherapeutic interventions, signaling a critical need for elucidation of the etiological underpinnings to facilitate the development of innovative pharmacotherapies. Recent scientific inquiries have associated mitochondrial dysfunction and inflammation with the etiology of sarcopenia. Mitochondria are integral to numerous fundamental cellular processes within muscle tissue, including but not limited to apoptosis, autophagy, signaling via reactive oxygen species, and the maintenance of protein equilibrium. Deviations in mitochondrial dynamics, coupled with compromised oxidative capabilities, autophagic processes, and protein equilibrium, result in disturbances to muscular architecture and functionality. Mitochondrial dysfunction is particularly detrimental as it diminishes oxidative phosphorylation, escalates apoptotic activity, and hinders calcium homeostasis within muscle cells. Additionally, deleterious feedback loops of deteriorated respiration, exacerbated oxidative injury, and diminished quality control mechanisms precipitate the acceleration of muscular senescence. Notably, mitochondria exhibiting deficient energetic metabolism are pivotal in precipitating the shift from normative muscle aging to a pathogenic state. This analytical review meticulously examines the complex interplay between mitochondrial dysfunction, persistent inflammation, and the pathogenesis of sarcopenia. It underscores the imperative to alleviate inflammation and amend mitochondrial anomalies within geriatric populations as a strategy to forestall and manage sarcopenia. An initial overview provides a succinct exposition of sarcopenia and its clinical repercussions. The discourse then progresses to an examination of the direct correlation between mitochondrial dysfunction and the genesis of sarcopenia. Concomitantly, it accentuates potential synergistic effects between inflammatory responses and mitochondrial insufficiencies during the aging of skeletal muscle, thereby casting light upon emergent therapeutic objectives. In culmination, this review distills the prevailing comprehension of the mitochondrial and inflammatory pathways implicated in sarcopenia and delineates extant lacunae in knowledge to orient subsequent scientific inquiry.
Keywords: Sarcopenia, inflammaging, mitochondrial dysfunction
Introduction
The prolongation of human life expectancy is associated with significant public health challenges on a global scale, exerting strain on healthcare infrastructures due to the degenerative health conditions prevalent in an aging demographic [1]. Such deterioration in health status not only compromises the well-being of the elderly but also imposes substantial demands on caregiving resources and financial systems [2]. The World Health Organization (WHO) advocates for the promotion of healthy aging, focusing on the preservation of functional abilities, postponement of diseases correlated with advanced age, and the improvement of autonomy and quality of life for the elderly [3]. Sarcopenia, a syndrome linked to aging, is characterized by the diminution of muscle mass and strength [4]. The onset of this condition is generally observed between the ages of 30 and 40, with muscle fiber atrophy occurring at a rate of 3-8% per decade, continuing up to the sixth decade of life, thereby adversely affecting an individual’s quality of life [5]. The European Working Group on Sarcopenia in Older People (EWGSOP) defines sarcopenia as a syndrome typified by progressive loss of skeletal muscle mass that can culminate in physical incapacitation, diminished quality of life, and mortality [6]. Although aging is a primary contributor, the initiation and progression of sarcopenia are also modulated by factors such as lifestyle choices, level of physical activity, dietary patterns, metabolic syndromes, and neuromuscular impairments [7]. Notably, a lifestyle characterized by physical inactivity or suboptimal exercise can precipitate what is known as secondary sarcopenia at an early stage [8,9].
At present, therapeutic approaches for sarcopenia predominantly involve physiotherapeutic measures aimed at augmenting muscular strength and enhancing locomotion proficiency. The pharmacological landscape, however, lacks specific agents approved for the amelioration of sarcopenia [10]. Nonetheless, advancing our comprehension of sarcopenia’s foundational mechanisms promises to catalyze the innovation of targeted therapeutic modalities. Contemporary scientific inquiry has concentrated on elucidating the molecular underpinnings of sarcopenia [11-13], with particular emphasis on the critical roles of mitochondrial impairment and inflammatory processes in its emergence and progression [14]. Mitochondria are implicated in a spectrum of pivotal cellular functions within skeletal muscle that are essential for sustaining normal muscular operation and protein homeostasis [15]. These functions encompass the orchestration of programmed cell death [16], self-digestion of cellular components [17], generation of reactive oxygen species (ROS) [18], and oversight of protein homeostasis [19]. The roles of mitochondria in mediating both apoptosis and autophagy are indispensable for the purging of defunct organelles and aberrant proteins. Furthermore, the production of mitochondrial ROS is instrumental in redox signaling, which governs gene expression and protein homeostasis [20]. Mitochondria also play a crucial role in upholding the proteome and ensuring proper protein folding through their chaperone and protease systems [21]. Their involvement in these essential pathways is fundamental to maintaining skeletal muscle integrity [22]. Emerging evidence posits that mitochondrial dysfunction is a significant factor contributing to the degeneration of skeletal muscle associated with primary aging and is implicated in the exacerbation of secondary aging processes [23,24]. In contrast, enhancing mitochondrial function has been postulated to mitigate the advent of secondary aging phenomena [25].
This revelation has facilitated the identification of innovative targets for therapeutic intervention and the formulation of advanced treatment strategies. Age-related modifications in mitochondrial morphology and functionality, encompassing disturbed mitochondrial dynamics, diminished oxidative capabilities, attenuated autophagy, compromised mitochondrial DNA (mtDNA), and disrupted protein homeostasis, exert deleterious effects on skeletal muscle architecture and functionality [14,24]. Mitochondrial dysfunction is instrumental in the etiology of sarcopenia, as it leads to a decrease in oxidative phosphorylation, an increase in programmed cell death, and compromised calcium ion regulation [17]. A pernicious cycle involving reduced mitochondrial respiratory function, escalated oxidative injury, and faltering mitochondrial quality control mechanisms contributes to the acceleration of skeletal muscle senescence [17,23]. Recent research indicates that mitochondria exhibiting diminished bioenergetic performance serve as a critical indicator that triggers the transition from normal muscular aging to pathogenic muscle aging [26].
This review is designed to explicate the complex interconnection between mitochondrial dysfunction and sarcopenia, alongside the effect of inflammaging on mitochondrial deficits linked to this muscle-wasting condition. The manuscript underscores the pivotal role of chronic inflammation and mitochondrial insufficiency management in elderly populations as a strategy for sarcopenia prevention and therapy. The initial segment offers a succinct yet thorough overview of sarcopenia, characterized by the progressive diminution of muscle mass, strength, and function. Subsequent to this, the nexus between mitochondrial dysfunction and sarcopenia is examined. In conclusion, the review explores the potential interactions between inflammatory processes and mitochondrial dysfunctions in the aging of skeletal muscle tissue.
Sarcopenia: loss of muscle and physical function decline
Skeletal musculature is essential for the sustenance of body posture and the facilitation of routine physical activities, such as standing, ambulation, and autonomous living [6]. Additionally, serving as the primary reservoir for glycogen, skeletal muscle is integral to the regulation of glycogen metabolism and the maintenance of systemic metabolic equilibrium [27]. With the progression of age, both physiological and pathological decline in skeletal muscle attributes occurs, culminating in a decrease in muscle mass, contractile function, and resistance to fatigue, which are collectively recognized in the clinical context as sarcopenia [10]. This diminished capability to sustain muscular strength and power increases the susceptibility of older individuals to a heightened incidence of falls, compromised postural stability, and a consequent erosion of autonomy [28]. Consequently, the age-associated regression in skeletal muscle function can markedly affect the health and life quality of the geriatric demographic. Therefore, it is imperative to preserve skeletal muscle functionality to ensure continued locomotion, metabolic integrity, and independence within the aging cohort.
Contemporary diagnostic criteria for sarcopenia have identified muscular strength as a more reliable indicator of negative health outcomes compared to muscle mass. Evidence from cross-sectional analyses indicates that peak levels of isometric and concentric muscular strength are attained between the second and third decades of life, with a plateau persisting into the fourth or fifth decades. Subsequent to this period, a decline commences approximately in the fifth decade, characterized by a decrement of 12% to 15% per decade, which further intensifies in advanced age [29,30]. Such a decline in muscular strength may result in an individual at the age of 80 possessing merely 30-50% of the strength typified by a man in his third decade [29].
Beyond the reduction in muscular strength, the aging process is correlated with a deceleration of contractile function, manifested by extended durations of muscle contraction and half-relaxation [31]. This elongation in muscle twitch response is indicative of underlying alterations in the kinetics of cross-bridge formation and a decelerated calcium ion turnover, both of which are factors that contribute to a decrease in the power and speed of muscle contractions [32]. For example, research utilizing murine subjects has demonstrated that the soleus and gastrocnemius muscles in aged mice (27-29 months) display a maximal shortening velocity that is approximately 60% slower than that observed in young adult counterparts (7-9 months) [33]. This reduction in shortening velocity is concurrent with extended twitch contraction durations observed in the aged specimens. Comparable shifts in contractile kinetics have been observed in human skeletal muscles during senescence [34]. The diminution in the ability to rapidly generate force is a significant determinant of functional impairment in the elderly. The necessity for maximal muscular power for activities such as standing from a seated position implies that even minor decrements in mobility can substantially influence the shift from an independent to a dependent state [35]. Consequently, interventions aimed at ameliorating the declines in muscle contractile velocity associated with aging may serve to alleviate the detrimental effects of slower muscle twitch responses on the physical capabilities of older adults [32,34].
Alterations in musculature with advancing age profoundly affect performance metrics, encompassing changes in isometric and dynamic strength as well as resistance to fatigue [36]. The diminution of motor units and muscle fibers stands as the principal etiological factor in the development of sarcopenia [6]. Notably, the decline in peak isometric strength is concomitant with the diminution of overall muscle mass, a process that is partially ascribed to increased deposition of intramuscular adipose tissue and connective tissue proliferation [37]. Concurrent with generalized muscle atrophy are modifications in the composition of muscle fiber types, with a reduction in the size of both type I and type II fibers in elderly populations when compared to their younger counterparts [38]. Moreover, type II fibers are subject to more pronounced atrophy, indicative of their more rapid degeneration as a function of the aging process [39].
Furthermore, the intrinsic contractile characteristics of muscle tissue are compromised with age. Diminished calcium responsiveness, alterations in actomyosin crossbridge cycling, and impairments in excitation-contraction coupling are fundamental to the observed decrement in muscle contractility associated with aging [40]. Quantitative measures such as peak force generation, rates of unloaded muscle shortening, and specific force are observed to wane as part of the aging process [41]. A critical underlying factor is the age-progressive loss of spinal motor neurons, which becomes more pronounced post the sixth decade of life, leading to a preferential denervation and subsequent atrophy of fast-twitch type II muscle fibers [42]. The disparity between the loss of muscular strength and the reduction in muscle mass suggests a deterioration in muscle quality, a characteristic feature of sarcopenia [6]. Consequently, the expedited reduction in motor unit numbers contributes to declines in muscular strength that surpass those attributable solely to muscle atrophy in the elderly [43].
To encapsulate, the integrity of skeletal muscle is essential for the preservation of posture, ambulation, and autonomous functioning. Nonetheless, the senescence process precipitates sarcopenia, which is delineated by a diminution in muscle mass, strength, power, and a diminished resistance to fatigue [42]. Such quantitative and qualitative deteriorations in skeletal muscle function engender profound ramifications, including compromised postural stability, an escalated risk of falls, and a decreased capacity to execute routine activities of daily life [44,45]. Sarcopenia significantly contributes to the erosion of autonomy and the degradation of life quality among the aged population. There is an imperative need for augmented research into the underlying mechanisms and potential therapeutic strategies to mitigate the effects of sarcopenia and to safeguard musculoskeletal vitality during the aging process. The sustenance of mobility and self-sufficiency stands as a cornerstone for the promotion of healthful aging.
Mitochondrial dysfunction in muscle aging
Impaired mitochondrial protein homeostasis
Mitochondria are critical to cellular function, orchestrating calcium signaling, the genesis of reactive oxygen species (ROS), and the induction of cell death via apoptosis among other pathways (Figure 1) [46,47]. Within skeletal muscle cells, the primary pathway for synthesizing adenosine triphosphate (ATP)-the pivotal molecule for energy transduction in muscle contraction-is mitochondrial oxidative phosphorylation, which is indispensable for the maintenance, proliferation, and proper functioning of skeletal muscle [48]. The maintenance of mitochondrial integrity and skeletal muscle health necessitates the meticulous regulation of mitochondrial protein homeostasis, entailing synchronized processes of protein biosynthesis, folding, trafficking, turnover, and degradation [17,49]. Nonetheless, research indicates that the aging process is associated with an exacerbated accumulation of damaged and improperly folded mitochondrial proteins, which in turn perturbs mitochondrial homeostasis. This disturbance leads to an amplification of oxidative stress due to heightened ROS production and consequent inflammation within the skeletal muscle tissue [23,26].
Figure 1.
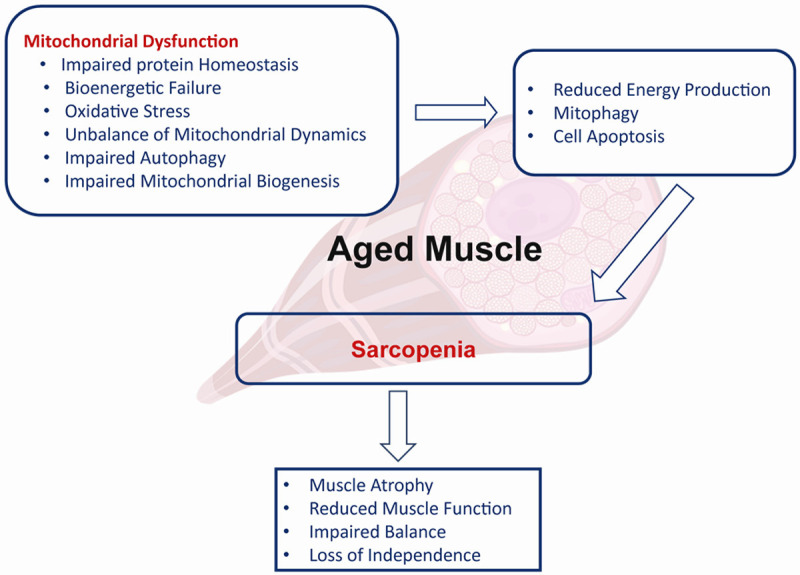
Association between mitochondrial dysfunction and sarcopenia in aged muscle.
A plethora of empirical investigations across human and various animal models have unequivocally established that the incidence of mRNA mistranslation, protein misfolding, and non-enzymatic post-translational modifications in mitochondrial proteins-specifically the formation of irreversible advanced glycation end products and oxidative alterations-are markedly elevated in the skeletal muscle of aged organisms in contrast to their youthful counterparts [50,51]. These deleterious alterations progressively amass in mitochondrial proteins and consequentially impair the reparative functions of mitochondrial protein quality control mechanisms, including but not limited to heat shock proteins, the ubiquitin-proteasome system, and mitochondrial-specific proteases [52]. This decrement in reparative proficiency precipitates a compounding cycle of disruption, further undermining the homeostasis and structural integrity of the mitochondrial proteome [17].
Moreover, mitochondrial dysfunction, precipitated by the accrual of impaired proteins, incrementally debilitates the mitochondrial energy provision by diminishing the activity of the electron transport chain and the synthesis of ATP [53]. The consequent scarcity of ATP renders the energetic requisites for concurrent normal metabolic functions and mitochondrial protein repair mechanisms increasingly onerous for muscular tissues [52]. This energetic disequilibrium prompts a metabolic shift from adaptable, ATP-intensive reactions, such as the restoration of damaged proteins, to a more inflexible, ATP-preserving basal metabolic state [17]. This shift exacerbates the disruption of mitochondrial protein equilibrium and perpetuates a progressive decrement in mitochondrial energy generation over time.
An array of interrelated elements is implicated in the perturbation of mitochondrial protein equilibrium as organisms age, encompassing primary mitochondrial malfunctions, attenuated activities in both cytoplasmic and mitochondrial protein quality control systems, modifications in mitochondrial dynamics, and the deficient autophagic elimination of defective mitochondria [52,54]. The aggregation of aberrantly folded, oxidatively modified, and incorrectly altered mitochondrial proteins compromises the operational efficacy of the electron transport chain complexes that reside within the inner mitochondrial membrane. This leads to a decrement in ATP synthesis and an overarching deterioration in mitochondrial functionality [55,56]. Such mitochondrial impairments are instrumental in exacerbating muscle atrophy, curtailing muscle protein biosynthesis, augmenting muscle protein catabolism, and they play a contributory role in the onset and advancement of sarcopenia as well as associated senescent muscle phenotypes [57,58].
For the prophylactic stabilization or active enhancement of mitochondrial function and the deceleration of skeletal muscle senescence, it is imperative to give precedence to the preservation of mitochondrial protein equilibrium as a fundamental element [23,58]. This necessitates a multifaceted strategy that encompasses the prevention of damaged mitochondrial protein accumulation by reinforcing quality control mechanisms, sustaining the reparative capabilities of mitochondrial proteins through the amplification of stress response pathways, mitigating oxidative stress and inflammation, and augmenting the total mitochondrial content alongside the exercise capacity of skeletal muscle tissue [50,58]. Such synergistic initiatives are vital to guarantee that skeletal muscle retains its capacity to execute crucial functions, including the restoration of damaged proteins, the biosynthesis of new muscle proteins at a robust pace, and the catabolism of senescent proteins at a controlled rate. Consequently, formulating strategies to preserve mitochondrial protein homeostasis is identified as an essential objective in the fight against sarcopenia and in fostering the longevity of skeletal muscle health [59].
Bioenergetic failure
Mitochondria serve as the nexus of energy metabolism within the body, fulfilling the crucial role of being the principal locus for oxidative phosphorylation and the generation of adenosine triphosphate (ATP) [60]. Nonetheless, skeletal muscle exhibits a decrement in these mitochondrial functions and bioenergetics with advancing age, a phenomenon referred to as bioenergetic failure [61]. There exists a close interrelation between such bioenergetic insufficiency in mitochondria and sarcopenia-the age-associated decline in muscle mass, strength, and functionality [62].
The onset of sarcopenia is influenced by bioenergetic insufficiency within mitochondria [63]. A reduction in ATP availability leads to a compromise in muscular contractility [64], hinders the anabolic processes of protein synthesis [65], and induces modifications in the cellular signaling cascades that are essential for muscle preservation [66]. Age-related reductions in both basal and peak oxygen (O2) consumption are observed [67], suggesting a diminution in mitochondrial efficacy or quantity with age, or possibly both [68]. The significance of the decline in mitochondrial bioenergetics on muscular aging is underscored by the established association between the rate of ATP production/O2 utilization and the preferred walking speed among elderly individuals [69].
Bioenergetic insufficiency may arise from a constellation of factors, such as diminished mitochondrial density, compromised efficacy of enzymes crucial for oxidative phosphorylation, mutations within mitochondrial DNA (mtDNA), and escalated oxidative stress [70-72]. A pivotal element leading to this bioenergetic insufficiency is the diminished oxidative capacity of mitochondria [58]. The preservation of an optimal oxidative capacity within mitochondria is essential for the assurance of proficient ATP generation and the maintenance of cellular energetic equilibrium [73]. A compromised oxidative capacity within mitochondria can precipitate a decrease in ATP output and perturbations in cellular metabolic processes [74], which in turn may adversely impact various energy-dependent cellular functions, including but not limited to, muscular contraction, protein biosynthesis, and cellular signaling pathways [67,75]. Within the context of skeletal muscle tissue, such compromised mitochondrial function assumes particular significance with respect to the pathogenesis of sarcopenia.
With the progression of age, there is a notable decrement in the capacity for oxidative phosphorylation within the mitochondria of skeletal muscles, evidenced by reduced maximal and resting oxygen consumption [64,76]. Furthermore, investigations have revealed in both aged humans and animal models a decline in the peak rate of mitochondrial ATP production, as well as a reduction in ATP generation at baseline, particularly pronounced within type I muscle fibers [77]. Additionally, aging skeletal muscles exhibit a decline in mitochondrial density and a compromised capacity for mitochondrial biogenesis [15], collectively indicating a reduction in mitochondrial oxidative capacity [78]. The efficiency of mitochondria in executing oxidative phosphorylation wanes, culminating in diminished ATP synthesis and consequent bioenergetic insufficiency [58]. This bioenergetic failure, in turn, exacerbates the decline in mitochondrial oxidative function, thereby perpetuating a deleterious feedback loop [50].
Interventional approaches aiming to ameliorate bioenergetics within aging muscular tissue-such as habitual physical activity [79], dietary modifications [80], and targeted measures to bolster mitochondrial functionality [81]-have demonstrated efficacy in attenuating the progression of sarcopenia [82]. These strategies may encompass the administration of antioxidants [72], therapies directed at mitochondria [83], or targeting specific metabolic routes to augment ATP production [66]. Advanced research is imperative to elucidate the molecular underpinnings of bioenergetic failure in mitochondria and its interaction with the sarcopenic process [50]. The identification of novel therapeutic targets to mitigate these age-associated alterations holds the potential for developing interventions aimed at preserving muscular health and functionality in the geriatric population [75].
Oxidative stress
Oxidative disequilibrium arises from a disproportionate ratio of reactive oxygen species (ROS) generation to the biological system’s capacity for their neutralization via endogenous antioxidants [84,85]. Within skeletal muscle, mitochondria constitute the principal source of ROS, significantly influencing a myriad of physiological processes [86]. At physiological concentrations, ROS are known to activate mitogen-activated protein kinase (MAPK) pathways, thereby facilitating redox signaling and supporting cellular homeostasis [87]. Conversely, persistent oxidative stress may induce pathogenic consequences, including the induction of muscle atrophy [88]. While moderate oxidative stress may potentiate skeletal muscle strength [89], an excessive oxidative burden undermines muscular strength and precipitates fatigue [81].
The mitochondrial free radical theory of aging posits that oxidative impairment of mitochondrial DNA (mtDNA) sets in motion a deleterious feedback mechanism. This mechanism involves compromised production of electron transport chain (ETC) components, attenuated adenosine triphosphate (ATP) synthesis, and escalated ROS accumulation [71,90].
The degradation of mtDNA within senescent skeletal muscle is intricately associated with ROS overproduction. Elevated ROS levels precipitate mtDNA lesions, mutagenesis, and functional impairments [71]. Given its proximity to the ETC, mtDNA is particularly susceptible to oxidative assaults [91]. MtDNA’s vulnerability is exacerbated by its location adjacent to the site of oxidant generation, absence of protective histones and introns, and an inherently limited repair capacity relative to nuclear DNA [92]. This susceptibility predisposes mtDNA to mutations induced by ROS, undermining its structural integrity and replication efficacy [93]. Such impairment in mtDNA replication contributes to mitochondrial dysfunction, which in turn perpetuates ROS overproduction and diminishes the capacity for mitochondrial biogenesis in aging skeletal muscle [94].
The age-associated augmentation in ROS within skeletal muscle tissue can be attributed to a confluence of factors, notably a diminution in the activity of critical antioxidant enzymes, including superoxide dismutase, catalase, and glutathione peroxidase [95]. Research utilizing murine models has demonstrated a progressive decline in these enzymes’ activities within skeletal muscle across the lifespan [96]. Furthermore, mitochondria extracted from aged skeletal muscle have been shown to emit more ROS per unit of ADP than those from younger counterparts, signifying qualitative mitochondrial functional discrepancies [50]. The concomitant decrease in enzymatic antioxidant activity and increased mitochondrial ROS production culminates in an accumulation of ROS due to inadequate neutralization mechanisms.
Empirical data from both preclinical and clinical research underscore the central role that the mitochondrial free radical vicious cycle plays in the aging of skeletal muscle. Studies have consistently reported that aging is associated with increased ROS emissions, heightened oxidative stress, and mtDNA damage within skeletal muscle, leading to mitochondrial functional decline [50,64,71]. Furthermore, ROS generated within mitochondria of aged skeletal muscle have been implicated in the inhibition of ATP synthase, an essential component of the ETC that facilitates ATP production [97]. This inhibition by ROS further compromises mitochondrial efficiency, perpetuating a self-sustaining cycle of dysfunction. In aggregate, aging in skeletal muscle is typified by an increase in mitochondrial ROS output, oxidative injury, and a decline in bioenergetic function [17].
In conclusion, the overproduction of ROS exacerbates mitochondrial DNA damage and dysfunction, engendering a self-reinforcing loop that results in a downturn of mitochondrial protein biosynthesis. Thus, interventions designed to preserve mtDNA integrity or amplify mtDNA abundance may offer viable therapeutic avenues to mitigate age-related mitochondrial deficits and sustain skeletal muscle function. Future research endeavors should investigate modalities such as caloric restriction, exercise regimens, and specific antioxidants for their potential to safeguard mtDNA and enhance mitochondrial protein synthesis in aged skeletal muscle tissue.
Unbalance of mitochondrial dynamics
The form and configuration of mitochondria are pivotal in defining their functionality. Notably, within myocytes, mitochondria exhibit intricate structural complexity [98]. Alterations in these structural parameters may precipitate a spectrum of mitochondrial pathologies [99]. Mitochondria are characterized by their dynamism, perpetually engaging in fusion and fission processes to preserve their form and ensure physiological function [54,100]. Fusion facilitates the intermingling of mitochondrial contents and augments the mitochondrial network. In contrast, fission is indispensable for segmenting this network into discrete units, a process essential for the selective catabolism and recycling of impaired mitochondria via the mitophagy-lysosome pathway [54]. Key proteins governing mitochondrial fusion in myocytes are mitofusin 1 and 2 (Mfn1/2), which serve to tether adjacent outer mitochondrial membranes (OMs), and optic atrophy proteins 1 and 2 (Opa1/2), which are involved in the fusion of the inner mitochondrial membranes (IM) [101]. Analogous to Mfn2’s role, dynamin-related protein 1 (Drp1) localizes to the OM and collaborates with mitochondrial fission factor (Mff) and fission protein 1 (Fis1) to encircle and constrict mitochondria, facilitating organelle separation [102].
The preservation of mitochondrial structural integrity is essential for muscular health [14]. Recent empirical research has underscored the importance of mitochondrial dynamics in sustaining muscular and mitochondrial function as well as structural integrity. It has been demonstrated that mutations in MFN2 influence mtDNA replication processes, engendering modifications in mitochondrial oxidative phosphorylation [103]. Conditional ablation of Mfn1 and Mfn2 in murine skeletal muscle induces profound dysfunction, characterized by cellular proliferation, atrophy, depletion of mtDNA, and an accumulation of mutations, thereby underlining the significance of fusion mechanisms in maintaining mtDNA integrity [104]. Additionally, deletion of MfnD1 is correlated with reduced activity in the electron transport chain and diminished physical performance [105].
Fission of mitochondria is equally vital for muscular and mitochondrial well-being. In sedentary individuals-as opposed to those who are active-an age-related decrease in OPA1 correlates with sarcopenia [106]. The immediate loss of Opa1 and Drp1 in adult musculature precipitates aberrant mitochondrial aggregation, endoplasmic reticulum (ER) stress, and disrupts autophagic and mitophagic pathways, culminating in muscle degradation and compromised force production [107]. Moreover, several studies have illuminated the physiological relevance of mitochondrial fission in the upkeep of skeletal muscle condition. Overexpression of muscle-specific Dro1 has been implicated in the impairment of muscular development in murine models [108].
In essence, the equilibrium between mitochondrial fusion and fission is imperative for maintaining mitochondrial efficacy and overall skeletal muscle fitness. The aging process in skeletal muscle is associated with disruptions in these dynamic processes, potentially leading to muscle atrophy and weakness. It is posited that a harmonious balance between these two processes, rather than the dominance of one over the other, is critical for the preservation of skeletal muscle health.
Decreased mitochondrial autophagy
Mitochondrial autophagy, termed mitophagy, constitutes a specialized autophagic process that orchestrates the catabolic removal of compromised or non-functional mitochondria. The precise modulation of this process is critical for the preservation of cellular viability and the maintenance of homeostasis [109]. Within the milieu of healthy skeletal muscle, mitophagy selectively targets and excises damaged and depolarized mitochondria [110], thus maintaining mitochondrial integrity by precluding the accrual of non-functional organelles. A correlation has been established between the disruption of mitophagy and the pathogenesis of myopathies as well as muscle wasting [111], signifying the essential role of mitophagic processes in the maintenance of skeletal muscle health.
The PINK1/Parkin axis is the most extensively elucidated pathway in mitophagy. This cascade facilitates the polyubiquitination of proteins within compromised mitochondria, thereby marking them for autophagic engulfment [112-114]. Under normative conditions, PINK1 is subject to proteolytic cleavage and subsequent degradation by PARL when translocated into intact mitochondria. Conversely, mitochondrial damage precipitates the stabilization of PINK1 on the outer mitochondrial membrane, which in turn activates Parkin’s E3 ubiquitin ligase function through phosphorylation. Parkin’s activity leads to the ubiquitination of outer mitochondrial membrane proteins, earmarking the impaired mitochondria for sequestration by autophagosomes [115]. These ubiquitinated mitochondria are then recognized by autophagy receptors that link to LC3 on the autophagosomal membrane [116]. Modulating the PINK1/Parkin pathway has been shown to enhance mitochondrial functionality and mitigate muscle degradation in atrophic conditions [117], suggesting that the activation of Parkin-mediated mitophagy may offer a therapeutic avenue to combat sarcopenia.
Mitophagic flux within skeletal muscle is regulated by a cohort of autophagy-related proteins such as LC3, ATG7, P62, Beclin 1, and Bnip3 [118,119]. Age-related changes in the expression of these proteins have been linked to diminished mitophagic capacity [120]. In mice, muscle-specific deletion of Atg7 results in pronounced atrophy, neuromuscular deficits, and a shortened lifespan [120]. Mutations in the human ATG7 gene have been associated with neurodevelopmental anomalies accompanied by neuromuscular impairments [121]. An accumulation of P62/SQSTM1 has been detected in aged skeletal muscle, suggesting a decline in autophagic processes contributes to the etiology of sarcopenia [122]. Therefore, maintaining the expression and functionality of mitophagy-related factors may present a strategy to alleviate muscle degeneration associated with aging.
Lysosomal dysfunction is a contributing factor to the reduced efficacy of mitochondrial autophagy observed in aging skeletal muscle. In aged rats, an accumulation of lipids within lysosomal structures has been associated with decreased activity of lysosome-associated membrane protein 2 (Lamp2) and a corresponding reduction in mitophagic capability [120]. A deficiency in Lamp2 within murine models leads to an aberrant build-up of mitochondria and muscular weakness [120], implicating suboptimal lysosomal degradation in mitochondrial autophagy impairments. Thus, ensuring robust lysosomal degradative capacity and Lamp2 expression could serve as a protective measure against sarcopenia.
A diminution in mitophagic capacity has been implicated in the age-associated decline in skeletal muscle function and mass. Investigations have revealed a suppressed mitophagic flux in the skeletal muscle of aged rodents [120], with a noted decrease in the expression of mitophagic regulators such as Parkin in elderly mice and rats [58,123]. These observations indicate that reduced mitophagy signaling may play a role in mitochondrial dysfunction and the consequent loss of muscular mass observed with advancing age. The impaired elimination of dysfunctional mitochondria may disrupt mitochondrial quality control and turnover, which are normally sustained through a balance with mitochondrial biogenesis. Further investigative efforts are necessary to elucidate the interplay between altered mitochondrial dynamics and compromised mitophagy in the development of sarcopenia.
Changes in mitochondrial biogenesis
Mitochondrial biogenesis represents a complex intracellular process involving the generation and integration of nascent mitochondria within the cellular matrix. This process is essential for the maintenance of mitochondrial efficacy, adenosine triphosphate (ATP) generation, and overall cellular equilibrium [83]. In the context of senescent skeletal musculature, perturbations in mitochondrial biogenesis are implicated in the onset of mitochondrial impairments, which in turn are contributory to the pathogenesis of sarcopenia [50,58].
The orchestration of mitochondrial biogenesis is subject to regulation by a cadre of transcriptional modulators. Among these, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) stands out as a pivotal regulator, which initiates and augments the transcription of genes fundamental to mitochondrial biosynthetic pathways [124]. Additionally, PGC-1α contributes to an increase in mitochondrial volume by mitigating reactive oxygen species and facilitating mitochondrial autophagy mediated by FoxO1 [125]. Research involving PGC-1α deficient murine models has demonstrated an upsurge in the accumulation of dysfunctional organelles and the expression of genes associated with muscular atrophy [126]. Moreover, mitochondrial dysfunction has been linked to age-associated remodelling of the neuromuscular junction (NMJ), resulting in a selective decrement of motor units within type II fibers, culminating in atrophy [127]. Conversely, the overexpression of PGC-1α has been observed to preserve NMJ structure with advancing age, whereas oxidative damage at the NMJ has been associated with a decline in skeletal muscle proteostasis [128]. A decline in both the expression and functionality of PGC-1α, alongside other mitochondrial biogenesis mediators, is characteristic of aging, leading to a concomitant reduction in mitochondrial biosynthesis [129].
Inter-mitochondrial communication is integral to the preservation of mitochondrial biogenesis. In senescent musculature, disturbances within this communicative network compromise the coordination necessary for effective biogenesis, engendering dysfunction [130]. The accrual of impaired mitochondria that circumvent quality control mechanisms also contributes to a decrease in mitochondrial turnover and biogenesis [131]. Moreover, aging is accompanied by elevated oxidative stress and persistent inflammation, which negatively influence both mitochondrial functionality and biogenesis signaling pathways [132].
The alteration of mitochondrial biogenesis yields multiple detrimental outcomes in relation to mitochondrial dysfunction and sarcopenia, including a diminution in mitochondrial quantity and density, a decline in ATP production with subsequent energy deficits [24], compromised oxidative phosphorylation coupled with suboptimal ATP synthesis [133], and perturbations in substrate handling and metabolic processes [134]. The elucidation of the underlying mechanisms responsible for changes in mitochondrial biogenesis associated with dysfunction and sarcopenia is vital for the development of therapeutic interventions. Future research endeavors should aim to disentangle the intricate interplay between mitochondrial biogenesis, dysfunction, and sarcopenia. The insights gleaned from such studies could pave the way for innovative approaches to foster healthy senescence and preserve muscular functionality in the aging population.
Interaction between inflammaging and age-related mitochondrial dysfunction
The senescence process is frequently concomitant with persistent, subdued inflammatory states, colloquially termed “inflammaging” [45,46]. Such a state is contributory to the deterioration of tissue integrity and the emergence of age-correlated pathologies, including type 2 diabetes mellitus, osteoarthritis, and sarcopenia [47]. Inflammaging exerts deleterious effects on glucose homeostasis [48], potentiates insulin resistance [49], and exacerbates oxidative stress [44], further enhancing the secretion of pro-inflammatory cytokines. The principal molecular entities implicated in inflammaging encompass TNF-α, IL-6, IL-1, and various chemokines, which facilitate the recruitment of inflammatory cells that exacerbate muscular degradation via the NF-κB pathway [135].
The intersection of chronic inflammation with mitochondrial dysfunction is implicated in the pathogenesis of age-related maladies [136,137]. This discourse delineates the interrelated mechanisms of inflammation and mitochondrial dysfunction, with a focus on their impact on sarcopenia, cachexia, and other disorders related to aging.
Nitric oxide (NO) signaling emerges as a critical intermediary in the interplay between inflammatory processes and mitochondrial functionality within skeletal muscle. NO challenges oxygen for binding at complex IV of the electron transport chain (ETC), thus influencing electron flow. The repercussions of this competition are contingent upon the isoforms of nitric oxide synthase (NOS) present and the concentration of NO [138]. Endothelial and neuronal NOS (eNOS/nNOS) are known to refine oxygen distribution among subsarcolemmal and intermyofibrillar mitochondria [139], whereas inducible NOS (iNOS), when activated by pro-inflammatory cytokines such as TNF-α, may lead to excessive inhibition of the ETC, an upsurge in oxidants, and increased permeability of the mitochondrial outer membrane [140].
TNF-α not only impedes the ETC but also instigates apoptotic signaling cascades. It triggers the caspase-8-mediated extrinsic apoptotic pathway and enhances the intrinsic pathway through the cleavage of Bid, a process that results in Bid’s translocation to the mitochondria and subsequent permeabilization of the outer membrane, culminating in the release of pro-apoptotic factors [141]. An increase in Bid presence and caspase activity has been documented in senescent musculature, contributing to muscular atrophy [142].
In addition, inflammatory processes are known to inhibit mitochondrial biogenesis. TNF-α mediates a reduction in PGC-1α and other regulatory proteins through the NF-κB pathway, leading to diminished mitochondrial density as observed in conditions such as cachexia associated with chronic obstructive pulmonary disease [143,144]. This results in hindered regenerative capacity and further aggravation of mitochondrial dysfunction.
In summary, inflammation perpetuates mitochondrial dysfunction through modulations in NO signaling, apoptotic induction, and the suppression of mitochondrial biogenesis (Figure 2). Unraveling these intricate mechanisms affords a deeper understanding of sarcopenia, cachexia, and other age-related ailments, thereby identifying potential targets for interventions aimed at preserving muscular health during aging. Prospective studies should investigate these complex interrelations and their tissue-specific manifestations to devise comprehensive therapeutic approaches.
Figure 2.
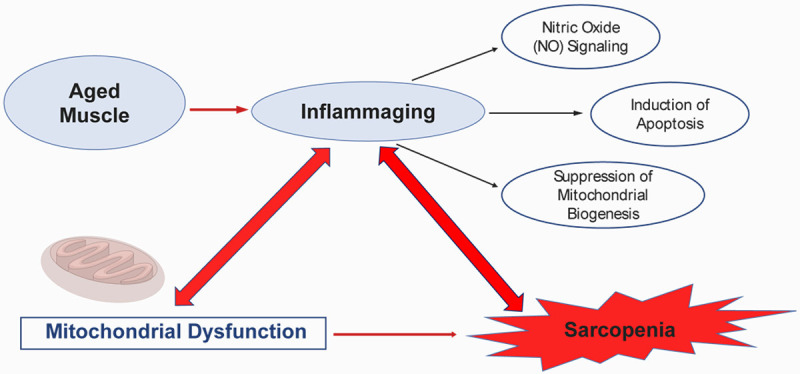
Interaction between inflammaging and age-related mitochondrial dysfunction.
Conclusions
Sarcopenia, alongside the concomitant decline in muscular mass attributable to aging, exerts a pronounced influence on the physical capabilities and overall life quality of the geriatric population. Unraveling the fundamental processes responsible for these conditions is crucial for the innovation of effective therapeutic interventions. Mitochondrial dysfunction has been identified as a contributing factor to muscular debilitation. A spectrum of mitochondrial dysfunctions associated with aging-including compromised proteostasis, augmented oxidative stress, disturbed mitochondrial dynamics, attenuated mitophagy, and diminished biogenesis-collectively impairs muscular functionality. Furthermore, it is postulated that inflammation may act as an intermediary in the pathogenesis of mitochondrial dysfunction and sarcopenia, with intensified inflammatory responses aggravating these impairments. The modulation of inflammatory processes may present a viable strategy to alleviate muscle atrophy. Intensive investigation into the inflammatory pathways that correlate with functional decline is imperative to devise interventions that preserve muscular function in the aging population. A detailed examination of the interplay between mitochondrial function and inflammation in the context of sarcopenia is essential to develop precise therapeutic approaches.
Acknowledgements
This study was supported by the program of “Chenguang Program” supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (21CGB02) and Geriatric Rehabilitation-Nursing Innovation Center Project of Shanghai Sanda University (No. 2023-14).
Disclosure of conflict of interest
None.
References
- 1.Crimmins EM. Lifespan and healthspan: past, present, and promise. Gerontologist. 2015;55:901–911. doi: 10.1093/geront/gnv130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown GC. Living too long: the current focus of medical research on increasing the quantity, rather than the quality, of life is damaging our health and harming the economy. EMBO Rep. 2015;16:137–141. doi: 10.15252/embr.201439518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beard JR, Officer A, de Carvalho IA, Sadana R, Pot AM, Michel JP, Lloyd-Sherlock P, Epping-Jordan JE, Peeters GMEEG, Mahanani WR, Thiyagarajan JA, Chatterji S. The World report on ageing and health: a policy framework for healthy ageing. Lancet. 2016;387:2145–2154. doi: 10.1016/S0140-6736(15)00516-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsson L, Degens H, Li M, Salviati L, Lee Yi, Thompson W, Kirkland JL, Sandri M. Sarcopenia: aging-related loss of muscle mass and function. Physiol Rev. 2019;99:427–511. doi: 10.1152/physrev.00061.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller K, Engelhardt M. Strength and muscle mass loss with aging process. Age and strength loss. Muscles Ligaments Tendons J. 2014;3:346–350. [PMC free article] [PubMed] [Google Scholar]
- 6.Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, Cooper C, Landi F, Rolland Y, Sayer AA, Schneider SM, Sieber CC, Topinkova E, Vandewoude M, Visser M, Zamboni M Writing Group for the European Working Group on Sarcopenia in Older People 2 (EWGSOP2), and the Extended Group for EWGSOP2. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing. 2019;48:601. doi: 10.1093/ageing/afz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kizilarslanoglu MC, Kuyumcu ME, Yesil Y, Halil M. Sarcopenia in critically ill patients. J Anesth. 2016;30:884–890. doi: 10.1007/s00540-016-2211-4. [DOI] [PubMed] [Google Scholar]
- 8.Mijnarends DM, Koster A, Schols JM, Meijers JM, Halfens RJ, Gudnason V, Eiriksdottir G, Siggeirsdottir K, Sigurdsson S, Jónsson PV, Meirelles O, Harris T. Physical activity and incidence of sarcopenia: the population-based AGES-Reykjavik Study. Age Ageing. 2016;45:614–620. doi: 10.1093/ageing/afw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du Y, Xu T, Yin Z, Espinoza S, Xie Y, Gentry C, Tian Q, Zhao LJ, Shen H, Luo Z, Deng HW. Associations of physical activity with sarcopenia and sarcopenic obesity in middle-aged and older adults: the Louisiana osteoporosis study. BMC Public Health. 2022;22:896. doi: 10.1186/s12889-022-13288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhillon RJ, Hasni S. Pathogenesis and management of sarcopenia. Clin Geriatr Med. 2017;33:17–26. doi: 10.1016/j.cger.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Constantin-Teodosiu D, Constantin D. Molecular mechanisms of muscle fatigue. Int J Mol Sci. 2021;22:11587. doi: 10.3390/ijms222111587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jimenez-Gutierrez GE, Martínez-Gómez LE, Martínez-Armenta C, Pineda C, Martínez-Nava GA, Lopez-Reyes A. Molecular mechanisms of inflammation in sarcopenia: diagnosis and therapeutic update. Cells. 2022;11:2359. doi: 10.3390/cells11152359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiedmer P, Jung T, Castro JP, Pomatto LCD, Sun PY, Davies KJA, Grune T. Sarcopenia - molecular mechanisms and open questions. Ageing Res Rev. 2021;65:101200. doi: 10.1016/j.arr.2020.101200. [DOI] [PubMed] [Google Scholar]
- 14.Leduc-Gaudet JP, Hussain SNA, Barreiro E, Gouspillou G. Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int J Mol Sci. 2021;22:8179. doi: 10.3390/ijms22158179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hood DA, Memme JM, Oliveira AN, Triolo M. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol. 2019;81:19–41. doi: 10.1146/annurev-physiol-020518-114310. [DOI] [PubMed] [Google Scholar]
- 16.Marzetti E, Calvani R, Bernabei R, Leeuwenburgh C. Apoptosis in skeletal myocytes: a potential target for interventions against sarcopenia and physical frailty - a mini-review. Gerontology. 2012;58:99–106. doi: 10.1159/000330064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romanello V, Sandri M. Mitochondrial quality control and muscle mass maintenance. Front Physiol. 2016;6:422. doi: 10.3389/fphys.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, Kavazis AN, Smuder AJ. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit Care Med. 2011;39:1749–1759. doi: 10.1097/CCM.0b013e3182190b62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cilenti L, Mahar R, Di Gregorio J, Ambivero CT, Merritt ME, Zervos AS. Regulation of metabolism by mitochondrial MUL1 E3 ubiquitin ligase. Front Cell Dev Biol. 2022;10:904728. doi: 10.3389/fcell.2022.904728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Moal E, Pialoux V, Juban G, Groussard C, Zouhal H, Chazaud B, Mounier R. Redox control of skeletal muscle regeneration. Antioxid Redox Signal. 2017;27:276–310. doi: 10.1089/ars.2016.6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123:2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 23.Hepple RT. Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci. 2014;6:211. doi: 10.3389/fnagi.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25:67–75. doi: 10.1016/j.mito.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, Bernabei R, Marzetti E. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci. 2017;18:933. doi: 10.3390/ijms18050933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen J, Rustad PI, Kolnes AJ, Lai YC. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front Physiol. 2011;2:112. doi: 10.3389/fphys.2011.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott D, de Courten B, Ebeling PR. Sarcopenia: a potential cause and consequence of type 2 diabetes in Australia’s ageing population? Med J Aust. 2016;205:329–333. doi: 10.5694/mja16.00446. [DOI] [PubMed] [Google Scholar]
- 29.Daley MJ, Spinks WL. Exercise, mobility and aging. Sports Med. 2000;29:1–12. doi: 10.2165/00007256-200029010-00001. [DOI] [PubMed] [Google Scholar]
- 30.Ikezoe T. Age-related change in muscle characteristics and resistance training for older adults. Phys Ther Res. 2020;23:99–105. doi: 10.1298/ptr.R0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung LH, Callahan DM, Kent-Braun JA. Age-related resistance to skeletal muscle fatigue is preserved during ischemia. J Appl Physiol (1985) 2007;103:1628–1635. doi: 10.1152/japplphysiol.00320.2007. [DOI] [PubMed] [Google Scholar]
- 32.Reid KF, Fielding RA. Skeletal muscle power: a critical determinant of physical functioning in older adults. Exerc Sport Sci Rev. 2012;40:4–12. doi: 10.1097/JES.0b013e31823b5f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballak SB, Degens H, de Haan A, Jaspers RT. Aging related changes in determinants of muscle force generating capacity: a comparison of muscle aging in men and male rodents. Ageing Res Rev. 2014;14:43–55. doi: 10.1016/j.arr.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Harridge SD, Lazarus NR. Physical activity, aging, and physiological function. Physiology (Bethesda) 2017;32:152–161. doi: 10.1152/physiol.00029.2016. [DOI] [PubMed] [Google Scholar]
- 35.Fiatarone MA, O’Neill EF, Ryan ND, Clements KM, Solares GR, Nelson ME, Roberts SB, Kehayias JJ, Lipsitz LA, Evans WJ. Exercise training and nutritional supplementation for physical frailty in very elderly people. N Engl J Med. 1994;330:1769–1775. doi: 10.1056/NEJM199406233302501. [DOI] [PubMed] [Google Scholar]
- 36.Clark BC, Manini TM. Functional consequences of sarcopenia and dynapenia in the elderly. Curr Opin Clin Nutr Metab Care. 2010;13:271–276. doi: 10.1097/MCO.0b013e328337819e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodpaster BH, Carlson CL, Visser M, Kelley DE, Scherzinger A, Harris TB, Stamm E, Newman AB. Attenuation of skeletal muscle and strength in the elderly: the Health ABC Study. J Appl Physiol (1985) 2001;90:2157–2165. doi: 10.1152/jappl.2001.90.6.2157. [DOI] [PubMed] [Google Scholar]
- 38.Nilwik R, Snijders T, Leenders M, Groen BB, van Kranenburg J, Verdijk LB, van Loon LJ. The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp Gerontol. 2013;48:492–498. doi: 10.1016/j.exger.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 39.Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995;50:11–16. doi: 10.1093/gerona/50a.special_issue.11. [DOI] [PubMed] [Google Scholar]
- 40.González E, Messi ML, Zheng Z, Delbono O. Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J Physiol. 2003;552:833–844. doi: 10.1113/jphysiol.2003.048165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Canepari M, Saltin B, Bottinelli R. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J Physiol. 2003;552:499–511. doi: 10.1113/jphysiol.2003.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hepple RT, Rice CL. Innervation and neuromuscular control in ageing skeletal muscle. J Physiol. 2016;594:1965–1978. doi: 10.1113/JP270561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reid KF, Pasha E, Doros G, Clark DJ, Patten C, Phillips EM, Frontera WR, Fielding RA. Longitudinal decline of lower extremity muscle power in healthy and mobility-limited older adults: influence of muscle mass, strength, composition, neuromuscular activation and single fiber contractile properties. Eur J Appl Physiol. 2014;114:29–39. doi: 10.1007/s00421-013-2728-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fielding RA, Vellas B, Evans WJ, Bhasin S, Morley JE, Newman AB, Abellan van Kan G, Andrieu S, Bauer J, Breuille D, Cederholm T, Chandler J, De Meynard C, Donini L, Harris T, Kannt A, Keime Guibert F, Onder G, Papanicolaou D, Rolland Y, Rooks D, Sieber C, Souhami E, Verlaan S, Zamboni M. Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J Am Med Dir Assoc. 2011;12:249–256. doi: 10.1016/j.jamda.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landi F, Calvani R, Cesari M, Tosato M, Martone AM, Bernabei R, Onder G, Marzetti E. Sarcopenia as the biological substrate of physical frailty. Clin Geriatr Med. 2015;31:367–374. doi: 10.1016/j.cger.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 47.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793–2809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A, Hood DA. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell. 2008;7:2–12. doi: 10.1111/j.1474-9726.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 51.Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Krüger P, Fielder E, Victorelli S, Ruswhandi RA, Giorgadze N, Pirtskhalava T, Podgorni O, Enikolopov G, Johnson KO, Xu M, Inman C, Palmer AK, Schafer M, Weigl M, Ikeno Y, Burns TC, Passos JF, von Zglinicki T, Kirkland JL, Jurk D. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 2019;29:1061–1077. e1068. doi: 10.1016/j.cmet.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quirós PM, Langer T, López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16:345–359. doi: 10.1038/nrm3984. [DOI] [PubMed] [Google Scholar]
- 53.Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yin F, Sancheti H, Cadenas E. Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim Biophys Acta. 2012;1817:401–409. doi: 10.1016/j.bbabio.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fazakerley DJ, Chaudhuri R, Yang P, Maghzal GJ, Thomas KC, Krycer JR, Humphrey SJ, Parker BL, Fisher-Wellman KH, Meoli CC, Hoffman NJ, Diskin C, Burchfield JG, Cowley MJ, Kaplan W, Modrusan Z, Kolumam G, Yang JY, Chen DL, Samocha-Bonet D, Greenfield JR, Hoehn KL, Stocker R, James DE. Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife. 2018;7:e32111. doi: 10.7554/eLife.32111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Picca A, Pesce V, Fracasso F, Joseph AM, Leeuwenburgh C, Lezza AM. Aging and calorie restriction oppositely affect mitochondrial biogenesis through TFAM binding at both origins of mitochondrial DNA replication in rat liver. PLoS One. 2013;8:e74644. doi: 10.1371/journal.pone.0074644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joseph AM, Adhihetty PJ, Buford TW, Wohlgemuth SE, Lees HA, Nguyen LM, Aranda JM, Sandesara BD, Pahor M, Manini TM, Marzetti E, Leeuwenburgh C. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell. 2012;11:801–809. doi: 10.1111/j.1474-9726.2012.00844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Konopka AR, Suer MK, Wolff CA, Harber MP. Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J Gerontol A Biol Sci Med Sci. 2014;69:371–378. doi: 10.1093/gerona/glt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kühlbrandt W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015;13:89. doi: 10.1186/s12915-015-0201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Mitochondrial bioenergetics decay in aging: beneficial effect of melatonin. Cell Mol Life Sci. 2017;74:3897–3911. doi: 10.1007/s00018-017-2619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harper C, Gopalan V, Goh J. Exercise rescues mitochondrial coupling in aged skeletal muscle: a comparison of different modalities in preventing sarcopenia. J Transl Med. 2021;19:71. doi: 10.1186/s12967-021-02737-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marzetti E, Lees HA, Wohlgemuth SE, Leeuwenburgh C. Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. Biofactors. 2009;35:28–35. doi: 10.1002/biof.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P, Wackerhage H, Taylor PM, Rennie MJ. Anabolic signaling deficits underlie amino acid resistance of wasting, aging muscle. FASEB J. 2005;19:422–4. doi: 10.1096/fj.04-2640fje. [DOI] [PubMed] [Google Scholar]
- 66.Joseph AM, Hood DA. Relationships between exercise, mitochondrial biogenesis and type 2 diabetes. Med Sport Sci. 2014;60:48–61. doi: 10.1159/000357335. [DOI] [PubMed] [Google Scholar]
- 67.Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci U S A. 2007;104:1057–1062. doi: 10.1073/pnas.0610131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Menshikova EV, Ritov VB, Ferrell RE, Azuma K, Goodpaster BH, Kelley DE. Characteristics of skeletal muscle mitochondrial biogenesis induced by moderate-intensity exercise and weight loss in obesity. J Appl Physiol (1985) 2007;103:21–27. doi: 10.1152/japplphysiol.01228.2006. [DOI] [PubMed] [Google Scholar]
- 69.Coen PM, Jubrias SA, Distefano G, Amati F, Mackey DC, Glynn NW, Manini TM, Wohlgemuth SE, Leeuwenburgh C, Cummings SR, Newman AB, Ferrucci L, Toledo FG, Shankland E, Conley KE, Goodpaster BH. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci. 2013;68:447–455. doi: 10.1093/gerona/gls196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sparks LM, Redman LM, Conley KE, Harper ME, Hodges A, Eroshkin A, Costford SR, Gabriel ME, Yi F, Shook C, Cornnell HH, Ravussin E, Smith SR. Differences in mitochondrial coupling reveal a novel signature of mitohormesis in muscle of healthy individuals. J Clin Endocrinol Metab. 2016;101:4994–5003. doi: 10.1210/jc.2016-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ryan MJ, Jackson JR, Hao Y, Leonard SS, Alway SE. Inhibition of xanthine oxidase reduces oxidative stress and improves skeletal muscle function in response to electrically stimulated isometric contractions in aged mice. Free Radic Biol Med. 2011;51:38–52. doi: 10.1016/j.freeradbiomed.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hernández-Aguilera A, Rull A, Rodríguez-Gallego E, Riera-Borrull M, Luciano-Mateo F, Camps J, Menéndez JA, Joven J. Mitochondrial dysfunction: a basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013;2013:135698. doi: 10.1155/2013/135698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kolesar JE, Safdar A, Abadi A, MacNeil LG, Crane JD, Tarnopolsky MA, Kaufman BA. Defects in mitochondrial DNA replication and oxidative damage in muscle of mtDNA mutator mice. Free Radic Biol Med. 2014;75:241–251. doi: 10.1016/j.freeradbiomed.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 75.Conley KE, Jubrias SA, Cress ME, Esselman P. Exercise efficiency is reduced by mitochondrial uncoupling in the elderly. Exp Physiol. 2013;98:768–777. doi: 10.1113/expphysiol.2012.067314. [DOI] [PubMed] [Google Scholar]
- 76.Coletti C, Acosta GF, Keslacy S, Coletti D. Exercise-mediated reinnervation of skeletal muscle in elderly people: an update. Eur J Transl Myol. 2022;32:10416. doi: 10.4081/ejtm.2022.10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dreyer HC, Blanco CE, Sattler FR, Schroeder ET, Wiswell RA. Satellite cell numbers in young and older men 24 hours after eccentric exercise. Muscle Nerve. 2006;33:242–253. doi: 10.1002/mus.20461. [DOI] [PubMed] [Google Scholar]
- 78.Ringholm S, Gudiksen A, Frey Halling J, Qoqaj A, Meizner Rasmussen P, Prats C, Plomgaard P, Pilegaard H. Impact of aging and lifelong exercise training on mitochondrial function and network connectivity in human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2023;78:373–383. doi: 10.1093/gerona/glac164. [DOI] [PubMed] [Google Scholar]
- 79.Buford TW, Anton SD, Judge AR, Marzetti E, Wohlgemuth SE, Carter CS, Leeuwenburgh C, Pahor M, Manini TM. Models of accelerated sarcopenia: critical pieces for solving the puzzle of age-related muscle atrophy. Ageing Res Rev. 2010;9:369–383. doi: 10.1016/j.arr.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McLeod M, Breen L, Hamilton DL, Philp A. Live strong and prosper: the importance of skeletal muscle strength for healthy ageing. Biogerontology. 2016;17:497–510. doi: 10.1007/s10522-015-9631-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Joseph AM, Adhihetty PJ, Leeuwenburgh C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J Physiol. 2016;594:5105–5123. doi: 10.1113/JP270659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sakuma K, Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr Aging Sci. 2010;3:90–101. doi: 10.2174/1874609811003020090. [DOI] [PubMed] [Google Scholar]
- 83.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poljsak B, Šuput D, Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792. doi: 10.1155/2013/956792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Milaeva ER. Metal-based antioxidants--potential therapeutic candidates for prevention the oxidative stress-related carcinogenesis: mini-review. Curr Top Med Chem. 2011;11:2703–2713. doi: 10.2174/156802611798040741. [DOI] [PubMed] [Google Scholar]
- 86.Akhigbe R, Ajayi A. The impact of reactive oxygen species in the development of cardiometabolic disorders: a review. Lipids Health Dis. 2021;20:23. doi: 10.1186/s12944-021-01435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Powers SK, Talbert EE, Adhihetty PJ. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J Physiol. 2011;589:2129–2138. doi: 10.1113/jphysiol.2010.201327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McClung JM, Judge AR, Powers SK, Yan Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol. 2010;298:C542–C549. doi: 10.1152/ajpcell.00192.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Henríquez-Olguín C, Díaz-Vegas A, Utreras-Mendoza Y, Campos C, Arias-Calderón M, Llanos P, Contreras-Ferrat A, Espinosa A, Altamirano F, Jaimovich E, Valladares DM. NOX2 inhibition impairs early muscle gene expression induced by a single exercise bout. Front Physiol. 2016;7:282. doi: 10.3389/fphys.2016.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 92.Kauppila TES, Kauppila JHK, Larsson NG. Mammalian mitochondria and aging: an update. Cell Metab. 2017;25:57–71. doi: 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 93.Bogenhagen DF. Mitochondrial DNA nucleoid structure. Biochim Biophys Acta. 2012;1819:914–920. doi: 10.1016/j.bbagrm.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 94.Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, Leeuwenburgh C. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol. 2013;45:2288–2301. doi: 10.1016/j.biocel.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kozakowska M, Pietraszek-Gremplewicz K, Jozkowicz A, Dulak J. The role of oxidative stress in skeletal muscle injury and regeneration: focus on antioxidant enzymes. J Muscle Res Cell Motil. 2015;36:377–393. doi: 10.1007/s10974-015-9438-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vasilaki A, Mansouri A, Van Remmen H, Van Der Meulen JH, Larkin L, Richardson AG, McArdle A, Faulkner JA, Jackson MJ. Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell. 2006;5:109–117. doi: 10.1111/j.1474-9726.2006.00198.x. [DOI] [PubMed] [Google Scholar]
- 97.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 98.Ogata T, Yamasaki Y. Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat Rec. 1997;248:214–223. doi: 10.1002/(SICI)1097-0185(199706)248:2<214::AID-AR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 99.Giulivi C, Zhang K, Arakawa H. Recent advances and new perspectives in mitochondrial dysfunction. Sci Rep. 2023;13:7977. doi: 10.1038/s41598-023-34624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 101.Romanello V, Sandri M. Implications of mitochondrial fusion and fission in skeletal muscle mass and health. Semin Cell Dev Biol. 2023;143:46–53. doi: 10.1016/j.semcdb.2022.02.011. [DOI] [PubMed] [Google Scholar]
- 102.Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vielhaber S, Debska-Vielhaber G, Peeva V, Schoeler S, Kudin AP, Minin I, Schreiber S, Dengler R, Kollewe K, Zuschratter W, Kornblum C, Zsurka G, Kunz WS. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013;125:245–256. doi: 10.1007/s00401-012-1036-y. [DOI] [PubMed] [Google Scholar]
- 104.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bell MB, Bush Z, McGinnis GR, Rowe GC. Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J Appl Physiol (1985) 2019;126:341–353. doi: 10.1152/japplphysiol.00719.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C, Loefler S, Kern H, Franceschi C, Salvioli S, Conte M, Blaauw B, Zampieri S, Salviati L, Scorrano L, Sandri M. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 2017;25:1374–1389. e1376. doi: 10.1016/j.cmet.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Romanello V, Scalabrin M, Albiero M, Blaauw B, Scorrano L, Sandri M. Inhibition of the fission machinery mitigates OPA1 impairment in adult skeletal muscles. Cells. 2019;8:597. doi: 10.3390/cells8060597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Touvier T, De Palma C, Rigamonti E, Scagliola A, Incerti E, Mazelin L, Thomas JL, D’Antonio M, Politi L, Schaeffer L, Clementi E, Brunelli S. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 2015;6:e1663. doi: 10.1038/cddis.2014.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS, Gottlieb RA. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy. 2016;12:369–380. doi: 10.1080/15548627.2015.1115172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121–2129. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016;17:300–316. doi: 10.15252/embr.201541486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. 2018;19:93–108. doi: 10.1038/nrm.2017.129. [DOI] [PubMed] [Google Scholar]
- 115.Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308:C710–C719. doi: 10.1152/ajpcell.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pansters NA, Schols AM, Verhees KJ, de Theije CC, Snepvangers FJ, Kelders MC, Ubags ND, Haegens A, Langen RC. Muscle-specific GSK-3β ablation accelerates regeneration of disuse-atrophied skeletal muscle. Biochim Biophys Acta. 2015;1852:490–506. doi: 10.1016/j.bbadis.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 119.Kim YA, Kim YS, Oh SL, Kim HJ, Song W. Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem. 2013;69:697–705. doi: 10.1007/s13105-013-0246-7. [DOI] [PubMed] [Google Scholar]
- 120.Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014;8:1509–1521. doi: 10.1016/j.celrep.2014.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Collier JJ, Suomi F, Oláhová M, McWilliams TG, Taylor RW. Emerging roles of ATG7 in human health and disease. EMBO Mol Med. 2021;13:e14824. doi: 10.15252/emmm.202114824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab. 2013;304:E1042–E1052. doi: 10.1152/ajpendo.00410.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mosole S, Carraro U, Kern H, Loefler S, Fruhmann H, Vogelauer M, Burggraf S, Mayr W, Krenn M, Paternostro-Sluga T, Hamar D, Cvecka J, Sedliak M, Tirpakova V, Sarabon N, Musarò A, Sandri M, Protasi F, Nori A, Pond A, Zampieri S. Long-term high-level exercise promotes muscle reinnervation with age. J Neuropathol Exp Neurol. 2014;73:284–294. doi: 10.1097/NEN.0000000000000032. [DOI] [PubMed] [Google Scholar]
- 124.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 125.Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rudolf R, Khan MM, Labeit S, Deschenes MR. Degeneration of neuromuscular junction in age and dystrophy. Front Aging Neurosci. 2014;6:99. doi: 10.3389/fnagi.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gill JF, Delezie J, Santos G, McGuirk S, Schnyder S, Frank S, Rausch M, St-Pierre J, Handschin C. Peroxisome proliferator-activated receptor γ coactivator 1α regulates mitochondrial calcium homeostasis, sarcoplasmic reticulum stress, and cell death to mitigate skeletal muscle aging. Aging Cell. 2019;18:e12993. doi: 10.1111/acel.12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Picard M, White K, Turnbull DM. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: a quantitative three-dimensional electron microscopy study. J Appl Physiol (1985) 2013;114:161–171. doi: 10.1152/japplphysiol.01096.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz-Bonilla V, Sala D, Planet E, Berenguer-Llergo A, Muñoz JP, Sánchez-Feutrie M, Plana N, Hernández-Álvarez MI, Serrano AL, Palacín M, Zorzano A. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016;35:1677–1693. doi: 10.15252/embj.201593084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Picard M, Ritchie D, Thomas MM, Wright KJ, Hepple RT. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell. 2011;10:1047–1055. doi: 10.1111/j.1474-9726.2011.00745.x. [DOI] [PubMed] [Google Scholar]
- 133.Porter C, Reidy PT, Bhattarai N, Sidossis LS, Rasmussen BB. Resistance exercise training alters mitochondrial function in human skeletal muscle. Med Sci Sports Exerc. 2015;47:1922–1931. doi: 10.1249/MSS.0000000000000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Joseph AM, Joanisse DR, Baillot RG, Hood DA. Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Exp Diabetes Res. 2012;2012:642038. doi: 10.1155/2012/642038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 136.Sakellariou GK, Pearson T, Lightfoot AP, Nye GA, Wells N, Giakoumaki II, Vasilaki A, Griffiths RD, Jackson MJ, McArdle A. Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci Rep. 2016;6:33944. doi: 10.1038/srep33944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Picca A, Guerra F, Calvani R, Bucci C, Lo Monaco MR, Bentivoglio AR, Coelho-Júnior HJ, Landi F, Bernabei R, Marzetti E. Mitochondrial dysfunction and aging: insights from the analysis of extracellular vesicles. Int J Mol Sci. 2019;20:805. doi: 10.3390/ijms20040805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun QA, Wang B, Miyagi M, Hess DT, Stamler JS. Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor/Ca2+ release channel (RyR1): sites and nature of oxidative modification. J Biol Chem. 2013;288:22961–22971. doi: 10.1074/jbc.M113.480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Clerc P, Rigoulet M, Leverve X, Fontaine E. Nitric oxide increases oxidative phosphorylation efficiency. J Bioenerg Biomembr. 2007;39:158–166. doi: 10.1007/s10863-007-9074-1. [DOI] [PubMed] [Google Scholar]
- 140.Sakellariou GK, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, Jackson MJ. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal. 2013;18:603–621. doi: 10.1089/ars.2012.4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.O’Brien ME, Londino J, McGinnis M, Weathington N, Adair J, Suber T, Kagan V, Chen K, Zou C, Chen B, Bon J, Mallampalli RK. Tumor necrosis factor alpha regulates skeletal myogenesis by inhibiting SP1 interaction with cis-acting regulatory elements within the Fbxl2 gene promoter. Mol Cell Biol. 2020;40:e00040-20. doi: 10.1128/MCB.00040-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rice KM, Blough ER. Sarcopenia-related apoptosis is regulated differently in fast- and slow-twitch muscles of the aging F344/N×BN rat model. Mech Ageing Dev. 2006;127:670–679. doi: 10.1016/j.mad.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 143.Remels AH, Gosker HR, Bakker J, Guttridge DC, Schols AM, Langen RC. Regulation of skeletal muscle oxidative phenotype by classical NF-κB signalling. Biochim Biophys Acta. 2013;1832:1313–1325. doi: 10.1016/j.bbadis.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 144.Fermoselle C, Rabinovich R, Ausín P, Puig-Vilanova E, Coronell C, Sanchez F, Roca J, Gea J, Barreiro E. Does oxidative stress modulate limb muscle atrophy in severe COPD patients? Eur Respir J. 2012;40:851–862. doi: 10.1183/09031936.00137211. [DOI] [PubMed] [Google Scholar]