

Abstract
Background:
Continuously growing teeth are an important innovation in mammalian evolution, yet genetic regulation of continuous growth by stem cells remains incompletely understood. Dental stem cells are lost at the onset of tooth root formation, but this loss of continuous crown growth is difficult to study in the mouse because regulatory signaling overlaps with signals that pattern tooth size and shape. Within the voles (Cricetidae, Rodentia, Glires), species have evolved both rooted and unrooted molars that have similar size and shape. We assembled a de novo genome of Myodes glareolus, a vole with high-crowned, rooted molars, and performed genomic and transcriptomic analyses in a broad phylogenetic context of Glires (rodents and lagomorphs) to assess differential selection and evolution in tooth forming genes.
Results:
Our de novo genome recovered 91% of single-copy orthologs for Euarchontoglires and had a total length of 2.44 Gigabases, enabling genomic and transcriptomic analyses. We identified six dental genes undergoing positive selection across Glires and two genes undergoing positive selection in species with unrooted molars, Dspp and Aqp1. Transcriptomics analyses demonstrated conserved patterns of dental gene expression with species-specific variation likely related to developmental timing and morphological differences between mouse and vole molars.
Conclusions:
Our results support ongoing dental gene evolution in rodents with unrooted molars. We identify candidate genes for further functional analyses, particularly Dspp, which plays an important role in mineralizing tissues. Our expression results support conservation of dental genes between voles and model species like mice, while revealing significant effects of overall tooth morphology on gene expression.
Keywords: Evolution, selection, Glires, molar, root, dental, development, genome, rodent, tooth
BACKGROUND
Hypselodonty, or the presence of unrooted and thus ever-growing teeth, has evolved multiple times in mammals. Glires—the clade containing rodents, rabbits, and their relatives—have hypselodont incisors (1), and multiple Glires have also evolved hypselodont molars (Fig. 1). At least in rodents, molar hypselodonty evolved considerably later than hypsodont molars, which are high crowned but rooted, which in turn evolved later than hypselodont incisors. In Glires, molars appear to increase in crown height from low-crowned brachydont (low-crowned, rooted), through hypsodonty (high-crowned, rooted), toward hypselodonty (high-crowned, unrooted) (2). Mice (Mus musculus), the primary mammalian model species of dental research, have highly derived hypselodont incisors while retaining brachydont molars. Because of this, mice do not provide information about the hypsodont teeth that likely preceded hypselodonty.
Figure 1 –
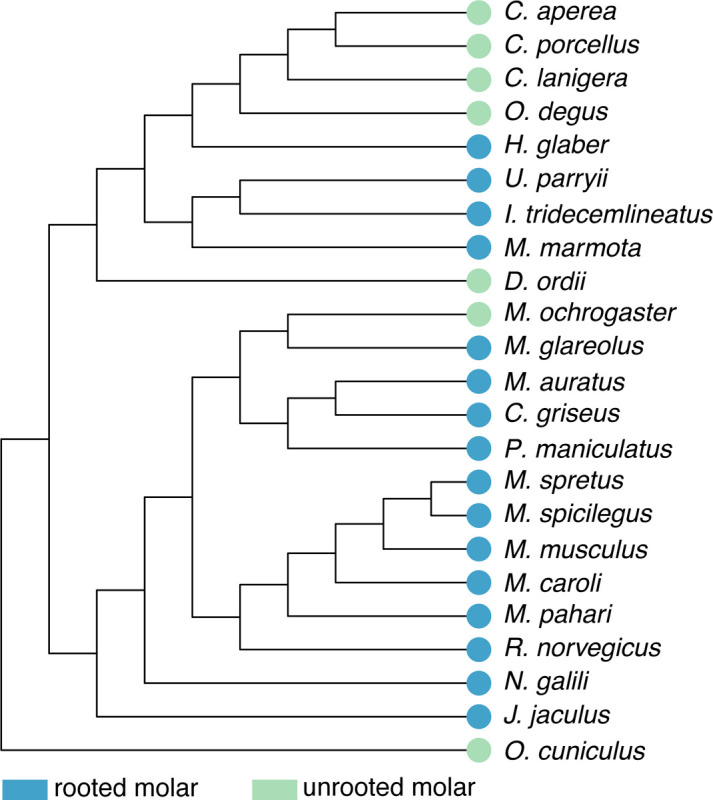
Species tree of Glires based on the Ensembl Compara species tree. Whether each species has rooted or unrooted molars is indicated at the tip of each branch. Note that unrooted, or hypselodont, molars have evolved multiple times across Glires. This topology was the basis for our orthology analysis.
Mammalian teeth sit in bony sockets, held in place by soft tissue (periodontal ligament) attached to cementum-covered tooth roots (3). Ligamentous tooth attachment may have arisen along with a reduction in the rate of tooth replacements, providing greater flexibility for repositioning the teeth as the dentary grows (4,3). Consequently, the limited replacement of mammalian teeth (two sets of teeth in most mammals and one in Glires) may have spurred the evolution of hypsodont and hypselodont teeth, both with high crowns that compensate for tooth wear from gritty or phytolith-heavy diets (5,6), and resulted in further modification of the anchoring roots. The convergent evolution of unrooted molars in Glires presents an opportunity to identify whether consistent developmental and genomic changes underlie the formation of hypselodont teeth, in turn revealing the mechanisms that must remain unchanged to produce tooth roots. Furthermore, the relatively recent evolution of molar hypselodonty, starting in the Middle Miocene (approximately 16–12 Ma) (2), should provide molecular evidence for the steps required to make a continuously growing organ.
Dental development proceeds from the tooth germ, composed of epithelium and mesenchyme, through phases known as the bud, cap, and bell (7). Multipotent enamel epithelium differentiates into the cells that form the tooth crown (8–11). As development progresses in rooted teeth, the epithelium at the tooth apex transitions first to a tissue called Hertwig’s epithelial root sheath (HERS), and eventually cementum-covered roots (9,10). Studies have identified numerous candidate genes and pathways with various roles during root development, such as Fgf10, which decreases in expression at the beginning of root formation (12–18). Although research on mouse molars has identified genetic signals of root formation, a number of the key genes studied have broad developmental roles, such as Wnt family members (14), or overlap considerably with genes also involved in patterning the size and shape of the tooth (17,19–22). This overlap between shape and root expression patterns confounds our ability to identify a clear signal initiating root formation.
Evolutionary novelties such as high-crowned hypsodont and hypselodont molars can arise from differences in gene expression and regulation (23–26). Evolutionarily conserved gene expression levels produce conserved phenotypes, and changes in gene regulatory networks have long been linked to morphological evolution (27,28). The order of genes along a chromosome (synteny) can affect gene expression and regulation, as regulatory sequences are often located near their target genes (cis-regulatory elements) (29–31). Genome rearrangements that place genes near new regulatory elements may result in changes of the expression levels and selective environment of those genes; these small-scale rearrangements of genes may be common in mammals (32–34). Genes involved in molar development are not syntenic in the mouse genome nor are genes with organ-specific expression (35), and thus the regulatory effects of co-localization need not apply to all dental genes at once. Changes in genome architecture between Glires species thus may result in different selective and expression environments for dental genes that could result in the evolution of hypselodont molars.
To establish a model rodent species with hypsodont molars for close comparison to hypselodont molars, we sequenced and annotated a highly-complete de novo genome of Myodes glareolus, the bank vole. The bank vole is increasingly used in medical and environmental research, ranging from studying zoonotic diseases (36) to immune responses (37,38), and even assessing environmental remediation efforts through heavy metals that accumulate in vole teeth (39,40), thus our efforts may be of use beyond dental research. The bank vole’s hypsodont molars bridge the gap between low-crowned mouse and hypselodont prairie vole (Microtus ochrogaster) molars. We performed a suite of genomic and transcriptomic tests of our new bank vole genome in a broad phylogenetic context to test the hypothesis that dental genes are undergoing positive selection and exhibit different expression patterns in species with unrooted, hypselodont molars. We predicted that genes without conserved syntenic relationships in these species would be more likely to have sites under positive selection or significantly different expression. Our analyses revealed positive selection among two dental genes in Glires with unrooted molars compared to those with rooted molars and demonstrated strong conservation of dental gene expression patterns between bank voles and mice, with key differences related to the timing and patterning of tooth morphology.
RESULTS
Orthology and synteny analyses
To identify which sequences in our bank vole (Myodes glareolus) genome and annotation had the same evolutionary history as dental genes identified in other Glires and assess genome rearrangements, we performed orthology and synteny analyses in a broad phylogenetic context. OrthoFinder identified 20,547 orthogroups representing 97.9% of the genes across all 24 analyzed genomes (including the human outgroup). Of the orthogroups, 6,158 had all species present. In our de novo bank vole genome, there were 27,824 annotated genes, of which 84.2% were assigned to an orthogroup. Bank vole genes were present in 16,250 orthogroups. On average, the genomes included in the OrthoFinder analysis had 19,814 genes, with 98.2% of those assigned to orthogroups.
The completeness and large scaffold N50 (4.6 Megabases) of our bank vole assembly supported its inclusion in generating a Glires synteny network. Using the infomap clustering algorithm, we produced 19,694 microsynteny clusters from this overall synteny network. We did not expect dental genes to share the same microsynteny cluster, and instead examined whether each gene was in the same microsynteny cluster in species with rooted or unrooted molars. Among the microsynteny clusters containing dental genes, 28 networks lacked synteny in at least half the species with unrooted molars or did not have a one-to-one relationship with an orthogroup (Fig. 2).
Figure 2 –
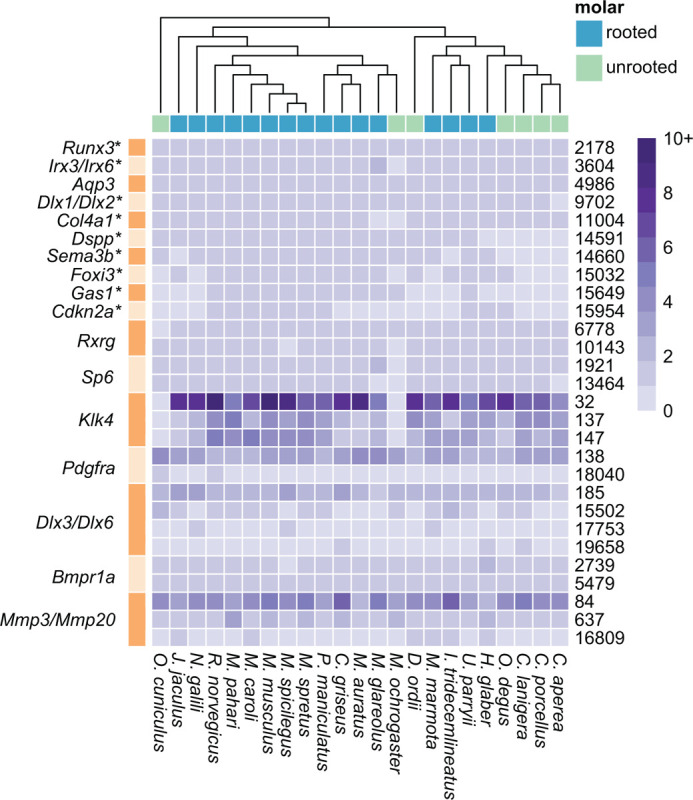
Heatmap showing the number of genes per species (inset gradient scale from 0 to 10+) in each synteny cluster. The figure shows clusters where species with unrooted molars had no representation or did not have sequences in all microsynteny clusters associated with a single gene, clusters where more than one gene mapped to the same cluster, or a single gene mapped to multiple clusters. Microsynteny cluster number is noted on the right side of the heatmap (one row per cluster), and corresponding genes are noted on the left with alternating bands showing rows to which those genes mapped. * = genes where hierarchical orthogroup did not contain genes for all 23 species. We found little evidence that non-syntenic genes in species with unrooted molars are undergoing selection for novel functions.
Positive selection analysis
We hypothesized that dental genes in species with unrooted molars are undergoing positive selection. Our positive selection analyses in PAML (phylogenetic analysis by maximum likelihood (41)) identified 6 dental gene orthogroups undergoing site-specific positive selection across Glires (Table 1). One of these genes, Col4a1, also was largely not syntenic in species with unrooted molars (Fig. 2). Another orthogroup consisted mainly of predicted sequences similar to Runx3 but only had sequences from four species; both showed site-specific positive selection. We then assessed 24 genes (those with site-specific positive selection or which lacked synteny in at least half of the species with unrooted molars) for site-specific positive selection in species with unrooted molars compared to species with rooted molars (branch-and-site specific positive selection (42)). Two genes, Dspp and Aqp1 were undergoing this branch-and-site specific positive selection. Both genes had a single highly supported site (posterior probability > 0.95) under positive selection in species with unrooted molars based on the Bayes Empirical Bayes method for identifying sites under selection implemented in PAML (43). Dspp also had multiple sites with moderate support (posterior probability > 0.75). The overall selection patterns on each gene differed. Maximum likelihood estimates of selection for Dspp showed the percentage of sites under purifying and neutral selection on all branches were nearly equal (47% and 44%, respectively). Percentages of sites under positive selection in the species with unrooted molars (foreground branches) were nearly evenly divided as well, with 5% of sites from branches where the species with rooted molars (background branches) were undergoing purifying selection and 4% of sites from branches where the species with rooted molars were under neutral selection. For Aqp1, nearly all sites were under purifying selection on all branches (91%), and few sites were under neutral selection on all branches (7%). Few sites were undergoing positive selection in the foreground branches and their distribution also was unevenly split between sites under purifying and neutral selection on background branches (0.6% and 0.04%, respectively). The complete list of dental genes with hierarchical orthogroups, microsynteny clusters, and positive selection test results are available in Additional file 1.
Table 1 –
Genes undergoing site-specific and branch-and-site-specific positive selection
| Gene | Mus transcript | Myodes transcript | Site | Branch-and-site |
|---|---|---|---|---|
| Aqpl | ENSMUST00000004774 | Mglareolus_00011822 | Yes | Yes |
| Col4a1 | ENSMUST00000033898 | Mglareolus_00032740 | Yes | No |
| Dspp | ENSMUST00000112771 | Mglareolus_00014030 | Yes | Yes |
| Fgf20 | ENSMUST00000034014 | Mglareolus_00013079 | Yes | No |
| Runx3 | EN SMU ST00000056977 | Mglareolus_00033992 | Yes | No |
| similar to Runx3 | – | – | Yes | No* |
HOG only contained four genes with one unrooted species’ sequence, could not be tested for branch-and-site specific selection.
Because genes under positive selection are often expressed at lower levels than genes under purifying selection (44–47), we also compared expression levels of Dspp and Aqp1 in postnatal first molars (M1) at postnatal days 1, 15, and 21 (P1, P15, and P21) in bank voles (rooted molars) and prairie voles (unrooted molars) using quantitative PCR. Prairie vole molars expressed Aqp1 at significantly lower levels in all three ages than bank vole molars (Fig. 3). Prairie vole P1 molars expressed significantly lower levels of Dspp than bank vole molars; at P15 and P21, their molars expressed Dspp at lower, but not statistically significantly different, levels than their bank vole equivalent. For both genes, the prairie vole had consistent expression levels across three biological replicates, while the bank vole had greater variation in expression levels across replicates.
Figure 3 –
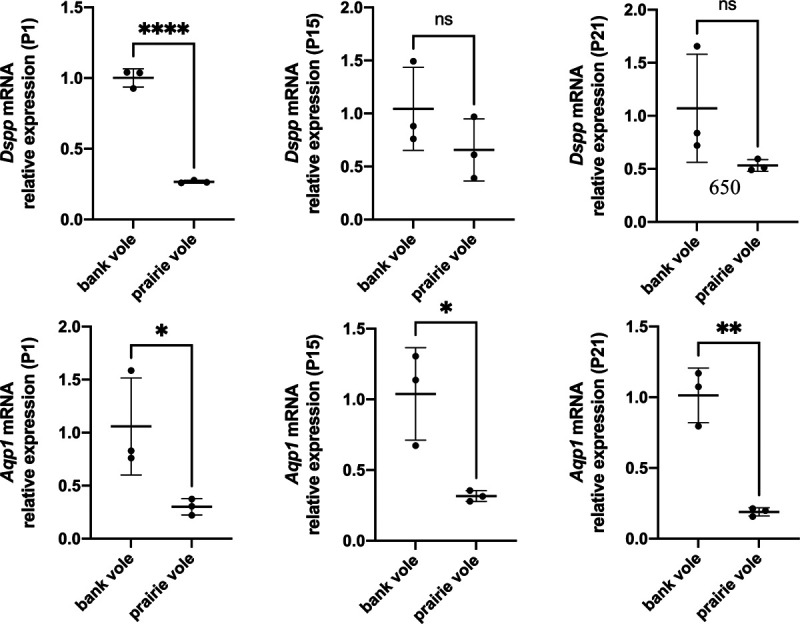
Quantitative PCR comparisons of Dspp and Aqp1 expression between bank vole and prairie vole M1 at postnatal days 1, 15, and 21 (P1, P15, P21). Expression levels for both genes are lower in the prairie vole (unrooted molars), which supports the positive selection detected for these genes in species with unrooted molars.
Sequence and secondary structure evolution
To detect whether substitutions at sites under positive selection influenced protein structure and evolution, we analyzed ancestral states and secondary structure across Glires. We first reconstructed ancestral sequences along the internal nodes of the Glires phylogeny for the genes undergoing branch-and-site specific positive selection to assess potential secondary structural changes in their protein sequences. At the best-supported site in Dspp (position 209 in the gapped alignment, Additional file 2), there were three major amino acid changes. The ancestral Glires sequence started with an asparagine (N) in this position. Two of the three species with unrooted molars represented in the Dspp dataset had amino acid substitutions at this position, with Oryctolagus cuniculus substituting a leucine (L) and Dipodomys ordii substituting an aspartic acid (D) at this position (Fig. 4A). All muroids (the clade including the voles in family Cricetidae and mice and rats in family Muridae) in our phylogeny substituted histidine (H) for the asparagine at this position. The secondary structure predicted at this position was a coil for most sequences but a helix for the D. ordii sequence (Fig. 5). Aqp1 sequences varied greatly at the position under putative positive selection in species with unrooted molars (position 294 in the gapped alignment, Additional file 3). The ancestral state reconstruction showed twelve changes of the amino acid at this position across Glires (Fig. 4B), yet these changes did not affect the predicted secondary structure of the protein near this residue, which was a coil for all sequences tested. All secondary structure predictions are available in Additional file 4.
Figure 4 –
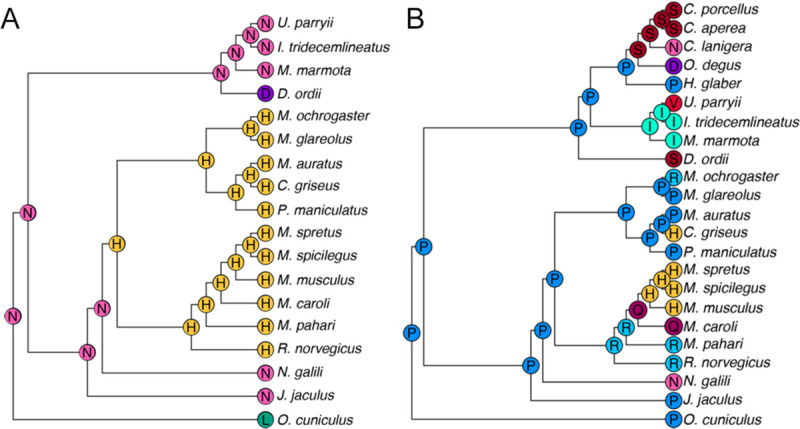
Ancestral state reconstructions of the residue under positive selection in PAML tests. Letters at tips and internal nodes represent IUPAC codes for amino acids. A Dspp; B Aqp1.
Figure 5 –
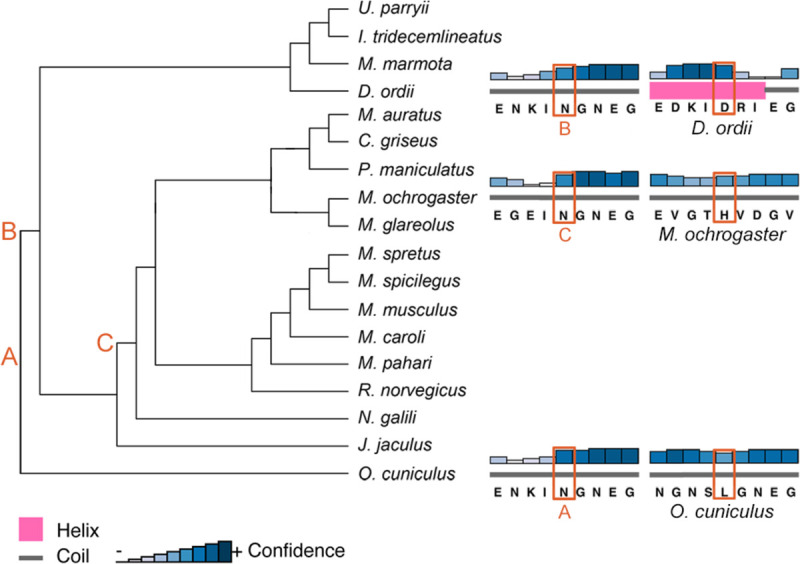
PSIPRED secondary structure predictions for the three species with unrooted molars represented in the Dspp sequences. Letters correspond to the most recent ancestor of each tip species that had a different amino acid at the position under positive selection.
Developmental gene expression
We also assessed differential gene expression between mouse and bank vole molars across early development to study the effects of morphology on expression levels of dental genes. Our gene expression analysis focused on keystone dental gene categories, which are based on the effects null mutations of each gene are reported to have during embryonic dental development (48): “shape” genes cause morphological errors, “eruption” genes prevent tooth eruption, “progression” genes stop the developmental sequence, “tissue” genes cause defects in tissues, “developmental process” genes are annotated with the “GO:0032502” gene ontology term, and “dispensable” genes, while dynamically expressed in developing teeth, have no documented effect on phenotype. The group “other” is composed of the remaining protein coding genes (48). Our bank vole genome was like the mouse and rat genomes in terms of the numbers and expression patterns of genes annotated from these keystone categories (Table 2). Ordination of gene expression results from the bank vole and mouse data at embryonic day 13, 14, and 16 (E13, E14, E16) (48) by principal components analysis showed a distinct separation between the mouse and bank vole along the first principal component (PC1) of the 500 most variable genes (Fig. 6A). PC1 explained 82.81% of the variance in these genes; there are distinct, species-specific expression patterns in these tissues. Along PC2 (7.47% of variance explained), E13 and E14 samples differ from the E16 samples, although the difference in time points is much greater in bank voles. Ordination of just the keystone dental genes showed clear separations between tissues based on species and age (Fig. 6B). Within this focused set of genes, however, PC1 and PC2 explain less variance (44.8% and 28.84% respectively), and how species and age relate to the PCs is less clear. There are two distinct, parallel trajectories for the mouse and bank vole. Although within each species there is separation by age along PC1 and PC2, mouse E16 and bank vole E13 occupy a similar position along PC1, and mouse E13 and bank vole E16 occupy a similar position along PC2.
Table 2 –
P-values of permutation tests between keystone gene categories in bank vole M1 at embryonic days 13, 14, and 16
| Tissue | Dispensable | Dev. Process | Other | ||
|---|---|---|---|---|---|
| E13 | Progression | 0.0310 | 0.0942 | 0.0436 | 0.0402 |
| Shape | 0.6431 | 0.9041 | 0.2289 | 0.0995 | |
| Double | 0.1292 | 0.1521 | 0.0716 | 0.0655 | |
| E14 | Progression | 0.0136 | 0.0383 | 0.0437 | 0.0401 |
| Shape | 0.3115 | 0.4725 | 0.0922 | 0.0454 | |
| Double | 0.1288 | 0.0945 | 0.0709 | 0.0630 | |
| E16 | Progression | 0.0140 | 0.0401 | 0.0303 | 0.0274 |
| Shape | 0.3770 | 1 | 0.1831 | 0.0662 | |
| Double | 0.1343 | 0.1099 | 0.0638 | 0.0596 |
Italicized values are statistically significant (p < 0.05)
Figure 6 –
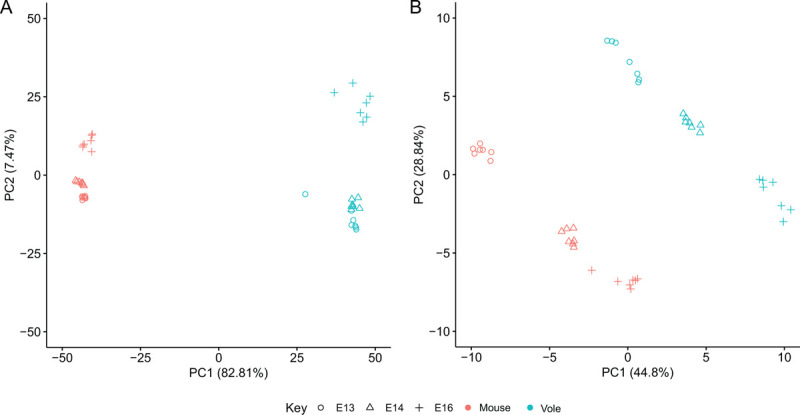
Principal component (PC) analyses of differentially expressed genes in mouse and bank vole M1. A PC1 and PC2 of the 500 most variable genes, showing a clear differentiation between species along PC1 and differentiation between age classes along PC2. B PC1 and PC2 of the keystone dental genes. Both PC1 and PC2 separate age classes within, but not between, the species, likely due to differences in developmental timing and molar morphology between mice and voles.
Examining individual genes underlying the differences between mouse and vole molars, we note several upregulated genes in our vole molars are broadly expressed in developing molars of other vole species (49,50). Relative to the mouse molars, vole molars overexpressed genes related to forming tooth cusps, including Bmp2, Shh, p21 (also known as Cdkn1a), and Msx2, a difference explained by the faster patterning and larger number of cusps in the vole molar compared to the mouse molar (50). Another gene upregulated in the patterning stage vole molar is Fgf10, which is associated with delayed root formation later in vole molar development (9).
Nevertheless, developing bank vole molars at E13, E14, and E16 expressed keystone dental genes in overall proportions like those observed at analogous stages of mouse and rat molar development (Fig. 7). Permutation tests within each bank vole sample showed that log counts for the set of genes related to the progression of dental development were significantly higher than those in the tissue, dispensable, developmental process, and “other” categories at E14 and E16. The progression gene counts in E13 molars were higher for all of these except the dispensable category. Shape category genes also were significantly higher than “other” category genes in the E14 tissue. Overall, even though we observed conserved expression patterns of dental genes at the system level, individual genes involved in cusp patterning and morphology differed between the mouse and the vole.
Figure 7 –
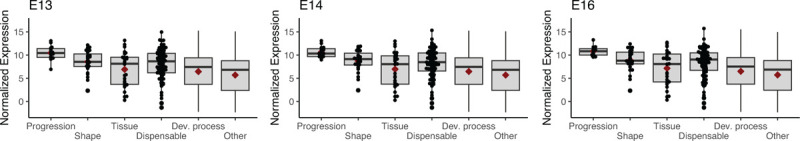
Box and whisker plots showing normalized log base 2 expression levels for each keystone gene category in bank vole M1 at embryonic days 13, 14, and 16. Gene expression profiles at these stages are comparable to mouse and rat molars at analogous developmental stages, as seen in Hallikas et al. 2021.
DISCUSSION
Our two goals in sequencing the genome of Myodes glareolus were to support the development of a comparative system for studying tooth root development and to investigate the evolution of dental genes in Glires, a clade in which ever-growing molars have evolved multiple times (1). Our new M. glareolus assembly and annotation captured nearly all of the single-copy orthologs for Euarchontoglires and provided scaffolds with sufficient length for synteny analyses. It was well represented in ortholog groups and microsynteny clusters across Glires. We tested the hypothesis that dental genes are undergoing site-specific positive selection in species with unrooted molars (branch-and-site specific positive selection (42)) and exhibit differential expression patterns. We predicted that lack of conserved syntenic relationships in species with unrooted molars could place dental genes in regulatory and selective environments that promote changes among genes relevant to tooth root formation. Our analyses revealed that most dental genes have conserved syntenic relationships across Glires, yet two dental genes, Dspp and Aqp1, were undergoing positive selection in species with unrooted molars. We also demonstrated conserved patterns of gene expression among dental keystone genes between bank voles and mice during early embryonic development, and deviations from these conserved patterns likely related to differences in molar morphology between the two species.
We identified 13 genes which were not syntenic in at least half of the species with unrooted molars, and 6 genes undergoing site-specific positive selection across all Glires. Only one gene, Col4a1, lacked synteny and had evidence of positive selection. The two genes undergoing positive selection in species with unrooted molars, Dspp and Aqp1, both maintained their synteny relationships across the Glires studied. Although we predicted loss of synteny for dental genes in Glires with unrooted molars could result in sequence evolution by placing genes in new selective contexts, our analyses did not support a relationship between non-syntenic genes and positive selection. Maximum likelihood estimates of sites under different types of selection for the genes with branch-specific positive selection did reveal different selective pressures on Dspp and Aqp1 overall; Dspp sites on background branches (i.e., branches with species that have rooted molars) were under a mix of purifying and neutral selection, while nearly all Aqp1 background branch sites were under purifying selection. These selection regimes suggest there is greater conservation for Aqp1 function across Glires than for Dspp function. Gene duplication can result in functional redundancy and evolution toward a novel function in some genes (51–54), which may explain positive selection in Aqp1, as there are other aquaporin family genes present. Dspp has no paralogs, but overlaps functionally with other SIBLING family proteins (e.g., Opn, Dmp1) (55,56).
Aqp1 and Dspp play different functional roles during dental development. Under the keystone dental development gene framework, Aqp1 is a “dispensable” gene: developing teeth express it, but tooth phenotypes do not change in its absence. Aqp1 is expressed in endothelia of microvessels in the developing tooth (57,58). Dspp may be particularly relevant for the formation of an unrooted phenotype if its expression domain or function have been modified in species with unrooted molars. Dspp is a “tissue” category keystone dental gene, meaning the main effects of a null mutation occur during the tissue differentiation stage of dental development (48). Null mutations of Dspp cause dentin defects in a condition called dentinogenesis imperfecta (59,60); in some patients, teeth form short, brittle roots (60,61). Dspp knockout mice also exhibit the shortened root phenotype, among a variety of other defects in both endochondral and intramembranous bone, due to the disruption of collagen and bone mineralization (62–64).
Our ancestral sequence reconstructions and estimated secondary protein structures allowed us to assess whether nonsynonymous substitutions at sites under positive selection resulted in structural differences, thus potentially affecting protein function. Although unrooted molars are a convergent phenotype across Glires, the sites under positive selection did not converge on the same amino acid substitution in species with unrooted molars, and Aqp1 appeared particularly labile at this residue. The non-synonymous substitutions at these sites often resulted in changes of properties of the amino acid in the sequence, for example in Dspp, polar asparagine was replaced with non-polar leucine in O. cuniculus. Only one of these substitutions changed the predicted secondary structure. Nevertheless, single amino acid substitutions do produce phenotypes for both Dspp (65) and Aqp1 (66), thus we cannot rule out functional changes in these genes in species with unrooted molars.
Although the exact relationship between gene expression and sequence divergence remains unclear (67), studies of genome evolution across small numbers of mammal species show correlations between gene sequence divergence and levels of expression (68). In particular, highly-expressed genes are more likely to experience purifying selection (44–47), while lowly-expressed genes and tissue-specific genes may experience positive selection (45). The decreased expression of Dspp and Aqp1 in prairie vole M1 compared to that of the bank vole M1 thus supports our finding of positive selection in these genes in species with unrooted molars. If all species with unrooted molars also exhibit decreased expression levels of Dspp and Aqp1, it could suggest a strong link between lower levels of the genes and the unrooted phenotype.
Without analyses of functional variation caused by positive selection at these coding sites, or spatial sampling to determine where these genes may be expressed during development, we are limited from exploring the specific effects of Dspp and Aqp1 on root formation. Nevertheless, we found evidence for evolution of these genes in Glires with unrooted molars, and Dspp especially has clinical relevance for tooth root formation. Future studies should explore the spatial distribution of Dspp expression, which could be relevant to functional changes in Glires with unrooted molars. If Dspp is relevant to the lack of root formation in hypselodont Glires incisors, the positive selection identified here may modify its expression domain or its interaction with yet-unidentified root formation co-factors, thus serially reproducing the unrooted incisor phenotype in molars.
Our RNA sequencing results supported the bank vole as a suitable system for studying dental development. Although molar morphology differs considerably across mammals, candidate-gene approaches have identified numerous conserved genes involved in tooth development and morphological patterning (69). Studies of single genes or gene families have identified shape-specifying roles common to multiple species (50,70–72), and high-throughput sequencing of mouse and rat molars demonstrate that both species express sets of dental development genes in similar proportions during early stages of tooth development (48). The similarity of our high-throughput RNA sequencing results to the mouse and rat results in previous studies suggest overall expression patterns of keystone dental development genes within each stage may be conserved in Glires. Our principal component analyses and differential expression analyses measuring changes between mouse and bank vole molars, however, showed that several dental genes’ expression levels differed significantly by species and age. Previous research has documented organ expression patterns that are conserved across species early in development and diverge over time, with some major organs displaying heterochronic shifts in some species (73). If the major source of variation in keystone dental gene expression patterns between mice and bank vole molars were solely attributable to species, we might expect to see clear separation between the species along the first or second principal component (PC1 or PC2), like that observed in PC1 of the 500 most variable genes (Fig. 6). If molar development follows the diverging expression patterns observed in other organs, we might expect just the earliest age classes to align on one, or multiple, PCs. Instead, we found two trajectories that were nearly parallel across PC1 and PC2 and multiple keystone dental genes that were significantly differentially expressed with respect to species and age. This variation between species is likely driven by the larger number of cusps in the vole molar, and corresponding upregulation of genes regulating cusp formation. The overall acceleration of patterning in vole molars likely explains the significance of the age variable in our expression results, causing a heterochronic shift in the expression patterns.
Our analyses were limited by the small number of rodent species with sufficiently annotated genomes to be included in synteny and positive selection analyses. This limitation left us with a small phylogeny for our ancestral state reconstructions, which thus did not encompass the full diversity of Glires tooth roots, and potentially weakened model-based genomic analyses. Although positive selection analyses using the Bayes Empirical Bayes criterion are robust to smaller sample sizes (43), including fossil species in ancestral state reconstructions can change estimations of ancestral characteristics (74). Innovations in paleoproteomics also offer the opportunity to compare fossil species’ dental gene sequences directly to living and estimated ancestral sequences (75,76). By incorporating data for extinct Glires in both morphological and molecular analyses, we can further elucidate links between dental gene evolution and unrooted teeth.
CONCLUSIONS
Our genomics and transcriptomics analyses, based on our newly sequenced, high-quality draft bank vole genome assembly and annotation, showed that bank vole early tooth development is comparable to other commonly used rodent models in dental development research. We identified 6 dental gene orthogroups that were undergoing site-specific positive selection across Glires and two genes, Dspp and Aqp1, that were undergoing site-specific positive selection in Glires with unrooted molars. Dspp appears particularly relevant to root formation, as loss-of-function mutations cause a dentin production defect that can result in shortened tooth roots. Future research must explore the functional role that Dspp plays in tooth root formation in Glires and other clades. The rodent dentary is an exciting system for understanding tooth development; it provides an easily manipulated set of tissues that can be produced quickly and features a lifelong population of stem cells in the incisor with genomic mechanisms that are potentially replicated across other teeth in species with unrooted molars. Our results identify candidate genes for future analyses, and our draft bank vole genome and annotation improve the utility of this species for comparative dental research that can uncover the genetic mechanisms of tooth root formation.
METHODS
Tissue collection and sequencing
To assemble the bank vole genome, we sequenced tissues from a single adult male specimen housed in a colony at the UCSF Mission Center Animal Facility. We euthanized the animal according to UCSF IACUC protocol AN189916 and harvested muscle, kidney, heart, and liver tissue, which were immediately frozen at −80°C. Tissues were sent to a third-party sequencing service, where they were combined and homogenized to achieve appropriate mass for high molecular weight DNA extraction. We targeted 60x coverage with 150 base pair (bp) reads using 10X Chromium linked-read chemistry (77,78) and sequenced on the Illumina platform. We also targeted 10x coverage with Pacific Biosciences SMRT long-read chemistry. For genome annotation and gene expression analyses, we collected seven biological replicates each of first molars at embryonic days 13–16 (E13, E14, E15, E16), second molars at E16, and jaw tissues at E14 under University of Helsinki protocols KEK16–021, KEK19–019, and KEK17–030 and stored them in RNAlater at −80°C for RNA sequencing, following a tissue harvesting protocol established for mice and rats (48). We extracted RNA from these tissues using a guanidium thiocyanate and phenol-chloroform protocol combined with an RNeasy column purification kit (Qiagen) based on the keystone dental gene protocol (48). Single-end 84 bp RNA sequencing was performed using the Illumina NextSeq 500 platform.
Genome assembly and quality control
We first assembled only the 10X Chromium linked reads using the default settings in Supernova 2.1.1. (77,78). We selected the “pseudohaplotype” (pseudohap) output format, which randomly selects between potential alleles when there are two possible contigs assembled for the same region. This option produces two assemblies, each with a single resolved length of the genome sequence (77–79). We used our lower-coverage, long-read data for gap filling and additional scaffolding. First, we estimated the genome’s length using the raw sequence data in GenomeScope (80), which predicted a length of 2.6 gigabases. We then performed error correction of the long reads using Canu (81), removing reads shorter than 500 bp and disregarding overlaps between reads of fewer than 350 bp. We kept only those reads with minimum coverage of 3x for scaffolding. Following long read error correction, we used Cobbler and RAILS (82) with a minimum alignment length of 200 bases to accept matches for gap filling and scaffolding of both pseudohap assemblies.
For quality control, we assessed both unscaffolded and long-read scaffolded pseudohap assemblies by standard assembly length statistics with QUAST (83) and presence of single-copy orthologs with BUSCO v3 (84). Both scaffolded assemblies were approximately 2.44 Gigabases long, with an N50 (the length of the shortest scaffold at 50% of the total assembly length) of 4.6 Megabases; we refer to them as Pseudohap1+LR and Pseudohap2+LR. The Pseudohap1+LR assembly had 17,528 scaffolds over 1000bp (base pairs) long, and the Pseudohap2+LR assembly had 17,518 scaffolds over 1000bp long (Table 3). BUSCO searched for universal single-copy orthologs shared by Euarchontoglires, recovering 89.4% of these genes in the scaffolded Pseudohap1+LR assembly and 92.8% of the single-copy orthologs in the scaffolded Pseudohap2+LR assembly (Fig. 8). The two assemblies were similar length and contiguity, but because the scaffolded Pseudohap2+LR assembly recovered more single-copy orthologs, we based annotation and downstream analyses on it.
Table 3 –
QUAST assembly statistics for de novo bank vole (Myodes glareolus) genome assemblies
| Pseudohap1 | Pseudohap1+LR | Pseudohap2 | Pseudohap2+LR* | |
|---|---|---|---|---|
| Largest contig | 27939478 | 32658832 | 27937749 | 32657565 |
| Total length | 2434151515 | 2441426554 | 2434099357 | 2441472313 |
| GC (%) | 41.88 | 41.89 | 41.88 | 41.89 |
| N50 | 4187179 | 4579815 | 4187179 | 4558134 |
| N75 | 1689669 | 1818134 | 1687188 | 1810460 |
| L50 | 170 | 153 | 170 | 154 |
| L75 | 388 | 357 | 388 | 358 |
| Ns per 100 kbp | 1151.99 | 1030.75 | 1151.96 | 1030.48 |
assembly used for annotation and downstream analyses in this paper.
Figure 8 –
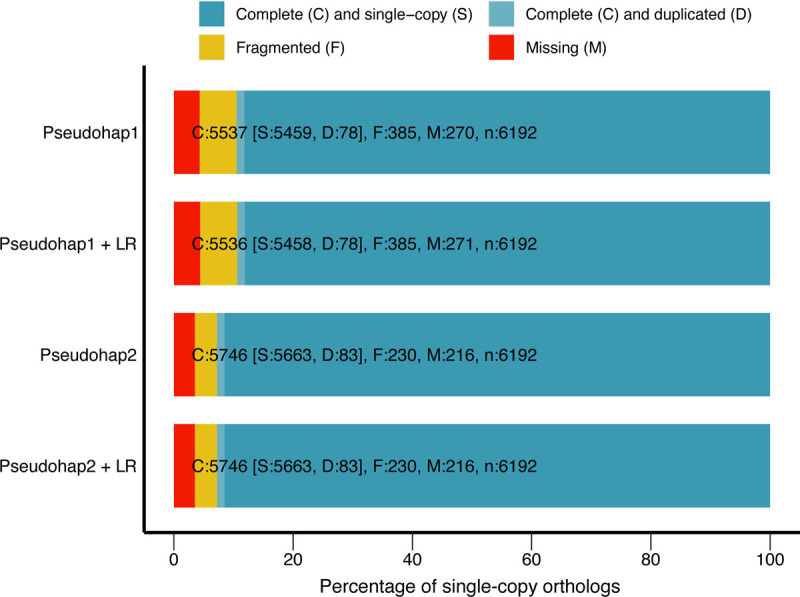
BUSCO single-copy ortholog recovery for each “pseudohaploid” version of our draft bank vole genome assembly and these version after long-read scaffolding (denoted by “+ LR”). Each bar represents the cumulative proportion of the 6,192 single-copy orthologs for Euarchontoglires identified by BUSCO represented by complete single-copy, complete-duplicated, fragmented, and missing orthologs. The Pseudohap2 and Pseudohap2 + LR assemblies had the best single-copy ortholog recovery.
Genome annotation
We annotated the genome using three rounds of the MAKER pipeline (85–87). MAKER combines multiple lines of evidence to annotate a genome. For evidence from gene transcripts, we assembled a de novo transcriptome assembly based on the single-end RNA sequencing of all molar and jaw tissues using Trinity (88). We also included cDNA sequences from the Mus musculus assembly GRCm38. We used SwissProt’s curated protein database to identify protein homology in the genome. Two libraries of repeats provided information for repeat masking: the Dfam Rodentia repeat library (89–91) and a custom library specific to the bank vole estimated based on the modified protocol of Campbell et al. (86). The custom library features miniature inverted-repeat transposable elements identified with default settings in MiteFinder (92), long terminal repeat retrotransposons extracted with the GenomeTools LTRharvest and LTRdigest functions (93) based on the eukaryotic genomic tRNA database, and de novo repeats identified with RepeatModeler (94). We combined elements identified by these programs into a single repeat library, then removed any elements that matched to a custom SwissProt curated protein database with known transposons excluded; this custom repeat library is available in Additional file 5. We trained a custom gene prediction model for MAKER as well. The first iteration of the model came from BUSCO’s implementation of augustus (95). Between each round of MAKER annotation, we updated the gene prediction model with augustus.
MAKER considered only contigs between 10,000–300,000 bp long during annotation. Our second and third iterations of MAKER used the same settings but excluded the “Est2genome” and “protein2genome” functions, as recommended in the MAKER tutorial. We included a SNAP (96) gene prediction model based on the output of the first round of annotation during the second and third iterations of MAKER annotation. Annotation quality (i.e., agreement between different lines of evidence and the MAKER annotation) was assessed visually in JBrowse after each iteration and using compare_annotations_3.2.pl (97), which calculates the number of coding and non-coding sequences in the annotation in addition to basic statistics about sequence lengths. Our MAKER annotation covered 2.41 Gb of the scaffolded Pseudohap2 assembly in 4,125 scaffolds. These scaffolds contained 27,824 coding genes (mRNA) and 15,320 non-coding RNA sequences. The average gene length was 12,705 bp. Most annotations (91.4%) had an annotation edit distance (AED) of 0.5 or better. AED is a measure of congruency between the different types of evidence for an annotation, where scores closer to zero represent betterannotated genes (98).
Orthology and synteny analyses
We analyzed orthology and synteny of the bank vole genome to understand gene and genome evolution related to dental development across Glires with rooted and unrooted molars. We obtained genomes from Ensembl for 23 Glires species and one phylogenetic outgoup, Homo sapiens (Table 4). These genomes all had an N50 over 1 Mb, which improves synteny assessment (99). We first analyzed all 24 genomes for groups of orthologous genes (orthogroups) in OrthoFinder (100), providing a tree topology based on the Ensembl reference tree (Fig. 1) to guide orthology detection. Because we would not analyze the human outgroup in downstream analyses, we implemented the OrthoFinder option that splits orthogroups at the root of Glires (hierarchical orthogroups), thus any group of orthologs studied here represents only genes with shared, orthologous evolutionary history within Glires. We selected MAFFT (101) for multiple sequence alignment and fastme (102) for phylogenetic tree searches within OrthoFinder; we retained the gene trees estimated for each orthogroup for downstream analyses.
Table 4 –
Genomes used in orthology, synteny, and positive selection analyses
| Species | Assembly | Citation |
|---|---|---|
| Myodes glareolus | CUNY_Mgla_1.0 | This paper |
| Cavia porcellus * | Cavpor3.0 | (120) |
| Cavia aperea * | CavAp1.0 | (121) |
| Marmota marmota | marMar2.1 | (122) |
| Microtus ochrogaster * | Mic0ch1.0 | (123) |
| Mus musculus | GRCm39 | (124) |
| Oryctolagus cuniculus * | OryCun2.0 | (120) |
| Dipodomys ordii * | Dord_2.0 | (120) |
| Jaculus jaculus | JacJac1.0 | (125) |
| Rattus norvegicus | Rnor_6.0 | (126) |
| Mus pahari | PAHARI_EIJ_v1.1 | (127) |
| Mus caroli | CAROLI_EIJ_v1.1 | (127) |
| Mus spretus | SPRET_EiJ_v1 | (128) |
| Mus spicilegus | MUSP714 | (129) |
| Cricetulus griseus | CHOK1GS | (130) |
| Mesocricetus auratus | MesAur1.0 | (131) |
| Peromyscus maniculatus | HU_Pman_2.1 | (132) |
| Nannospalax galili | S.galili_v1.0 | (133) |
| Octodon degus * | OctDeg1.0 | (134) |
| Heterocephalus glaber (F) | HetGla_female_1.0 | (135) |
| Chinchilla lanigera * | ChiLan1.0 | (136) |
| Urocitellus parryi | ASM342692v1 | (137) |
| Ictidomys tridecemlineatus | SpeTri2.0 | (138) |
| Homo sapiens ** | GRCh38 | (139) |
Species with unrooted molars;
Peptide annotation used as outgroup only in OrthoFinder analysis.
Although dental development genes are spread throughout the genome, we were interested in whether each gene remained in the same local arrangement across species of Glires. We prepared each genome annotation and sequence file for synteny analysis using the reformatting functions of Synima (103) to extract each peptide sequence associated with a gene coding sequence in the Ensembl annotation. Collinear synteny blocks estimated by MCScanX (104) formed the basis for microsynteny network analyses using the SynNet pipeline (105–107). We inferred networks from the top five hits for each gene, requiring any network to have a minimum of 5 collinear genes and no more than 15 genes between a collinear block, settings that perform well for analyzing mammal genomes (107). Using the infomap algorithm, we clustered the synteny blocks into microsynteny networks, from which we extracted network clusters corresponding to the list of keystone dental genes (48). For each dental gene microsynteny network, we assessed whether genes of species with unrooted molars were not syntenic with the other Glires species’ sequences.
Positive selection analysis
We aligned protein sequences for each dental gene orthogroup with clustal omega (108) using default settings. Based on universal translation tables, we obtained codon-based nucleotide alignments with pal2nal (109), removing sites in which any species had an indel (i.e., ungapped) and formatting the output for analysis in PAML (41). We pruned and unrooted the orthogroup gene trees from OrthoFinder to contain only tips representing the genes in each synteny network or orthogroup under analysis in PAML. We tested whether any of the genes were undergoing positive selection using a likelihood ratio test comparing site-specific models of “nearly neutral” and positive selection. In these models, w, the ratio of nonsynonymous to synonymous nucleotide substitutions (also known as dN/dS), can vary at each codon site. In the “nearly neutral” model, ω can take values between 0 and 1, while the positive selection model allows sites to assume ω values greater than 1 (43,110). We allowed PAML to estimate κ (the ratio of transitions to transversions) and ω from initial values of 1 and 0.5, respectively, for both tests.
Dental genes with significant site-specific positive selection or those for which over half the unrooted species’ sequences were not in the same synteny block as sequences for species with rooted molars formed the basis for our second set of positive selection tests using a branch- and-site model of positive selection. This model allows ω to vary not only among codon sites, but also between “foreground” and “background” lineages (43). We marked the species with unrooted molars as foreground lineages, then ran the model twice: once with ω unconstrained to detect sites undergoing positive selection only on foreground branches, and a second time and with ω fixed to 1, or neutral selection. A likelihood ratio test of the two models determined whether the lineage-specific positive selection model was more likely than a neutral model, and Bayes Empirical Bayes analyses (43) produced posterior probabilities to identify sites under positive selection.
Genes under positive selection also tend to have lower expression levels (45), thus we compared expression of the genes with branch-and-site specific positive selection between the prairie (unrooted molars) and the bank vole (rooted molars) to provide further support for selective differences. We collected three biological replicates of first molars from both species at three postnatal stages (P1, P15, and P21) and immediately preserved them at −80°C in lysis buffer (Buffer RLT; Qiagen) supplemented with 40 μM dithiothreitol. RNA was extracted from homogenized tissues using a RNeasy column purification kit (Qiagen). We assessed concentration and purity of extracted RNA using a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific). Using 1 μg of RNA, we synthesized cDNA using a high-capacity cDNA reverse transcription kit (ThermoFisher Scientific). We used 1 μL diluted cDNA (1:3 in ddH2O) and iTaq Universal SYBR Green Supermix (Bio-rad) in the Bio-rad CFX96 real-time PCR detection system for qPCR experiments, producing three technical replicates for each biological replicate. We normalized cycle threshold (CT) values of genes of interest to GAPDH expression levels and calculated relative expression levels as 2−ΔΔCT. A two-tailed unpaired t-test calculated in Prism 9 measured whether expression of these genes significantly differed between bank voles and prairie voles. The oligonucleotide primers for each species and gene are in Additional file 6.
Sequence and secondary structure evolution
We performed ancestral sequence reconstruction on the codon sequences of the genes that had evidence of branch-and-site specific positive selection to understand how the sequence has changed through time. The gapped clustal omega alignments were the basis for ancestral sequence reconstruction on the Glires species tree (Fig. 1) using pagan2 (111). For each gene, we plotted amino acid substitutions at the site with potential positive selection. Finally, we predicted secondary structures (i.e., helices, beta sheets, and coils) for each unrooted species’ protein sequence and the reconstructed ancestral sequence prior to the change at the site under positive selection using the PSIPRED 4.0 protein analysis workbench (112,113). Comparing these predictions across the phylogeny, we assessed how these substitutions at the site under selection may affect the structure of each protein.
Developmental gene expression
We used performed quality control and filtering of the short reads for the seven replicates of first molar tissues at E13, E14, and E16 using the nf-core/rnaseq v. 3.11.2 workflow (114) for comparability to previous mouse and rat analyses (48). RNAseq reads were evaluated and adapter sequences were filtered using FastQC v. 0.11.9 (115) and Cutadapt v. 3.4 (116), and ribosomal RNA was removed using SortMeRNA v. 4.3.4 (117). We then aligned trimmed sequences to our bank vole annotation using Salmon v. 1.10.1 (118). Counts were then normalized by gene length. We categorized gene count data into functional groups based on their established roles in tooth bud development (48) using the one-to-one orthology list between our bank vole genome and the mouse GRCm39.103 genome annotation generated from our OrthoFinder output. Using the rlog function of DESeq2 (119), we normalized gene counts within each functional group on a log2 scale. A permutation test assessed whether the mean counts of the progression, shape, and double functional groups were significantly different from genes in the tissue, dispensable, and “other” groups (which are potentially relevant later in development) based on 10,000 resampling replicates of the dataset (48).
We also assessed differential expression between the bank vole first molar and published mouse M1 data at the same three time points (GEO accession GSE142199 (48)), combining the data based on the one-to-one orthology relationships used in the functional permutation analysis. Using the mouse E13 molar as the reference level, we modeled expression as a response to species (mouse or vole), embryonic day (E13, E14, or E16), and the interaction between species and day. We considered as significant any gene with a log fold change greater than 1, log fold change standard error less than 0.5, and false discovery rate adjusted p value less than 0.05.
Supplementary Material
Additional file 1 [.xlsx] Dental gene results – Full table of orthology, synteny, and positive selection test results for all dental genes assessed.
Additional file 2 [.txt] Dspp gapped alignment – Gapped codon-based alignment for Dspp in fasta formatted sequences.
Additional file 3 [.txt] Aqp1 gapped alignment – Gapped codon-based alignment for Aqp1 in fasta formatted sequences.
Additional file 4 [.pdf] Structure predictions – PSIPRED Secondary structure predictions for each ancestral node and unrooted molar tip species for Dspp and Aqp1.
Additional file 5 [.txt] Custom repeat library – Custom repeat library of fasta formatted sequences used in annotation of the draft Myodes glareolus genome. See Methods for description of the process used to generate the library.
Additional file 6 [.pdf] Oligonucleotide primers – List of oligonucleotide primers for Dspp, Aqp1, and GAPDH used in bank vole and prairie vole qPCR experiments.
Acknowledgements:
The authors thank A. Joo, N. Ahituv, G. Amato, A. Narechania, S. Singh, A. Scott, and A. Paasch for advice on methods and access to cluster computing resources.
Funding:
This research was supported by National Science Foundation grants CNS-0958379, CNS-0855217, OAC-1126113, and OAC-2215760 through the City University of New York High Performance Computing Center at the College of Staten Island; OAC-1925590 through the MENDEL high performance computing cluster at the American Museum of Natural History; Academy of Finland to JJ; Doctoral Programme in Biomedicine, University of Helsinki to MMC; and National Institutes of Health NIDCR R01-DE027620 and R35-DE026602 to ODK.
Footnotes
Ethics approval: The University of California, San Francisco (UCSF) Institutional Animal Care and Use Program and the Finnish national animal experimentation board approved protocols for humane euthanasia and collection of tissues for animals used in this study under protocols AN189916 (UCSF) and KEK16–021, KEK19–019, and KEK17–030 (University of Helsinki).
Competing interests: The authors declare that they have no competing interests.
Availability of data and materials:
The datasets supporting the conclusions of this article are available in the GenBank repository under [GenBank reference number to be added upon acceptance] and in the article’s additional files.
REFERENCES
- 1.Renvoisé E, Michon F. An Evo-Devo perspective on ever-growing teeth in mammals and dental stem cell maintenance. Front Physiol. 2014;5 AUG(August):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tapaltsyan V, Eronen JT, Lawing AM, Sharir A, Janis C, Jernvall J, et al. Continuously growing rodent molars result from a predictable quantitative evolutionary change over 50 million years. Cell Rep. 2015;11(5):673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LeBlanc ARH, Brink KS, Whitney MR, Abdala F, Reisz RR. Dental ontogeny in extinct synapsids reveals a complex evolutionary history of the mammalian tooth attachment system. Proc R Soc B Biol Sci. 2018. Nov 7;285(1890):20181792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saffar JL, Lasfargues JJ, Cherruau M. Alveolar bone and the alveolar process: the socket that is never stable. Periodontol 2000. 1997;13(1):76–90. [DOI] [PubMed] [Google Scholar]
- 5.Davit-Béal T, Tucker AS, Sire JY. Loss of teeth and enamel in tetrapods: Fossil record, genetic data and morphological adaptations. J Anat. 2009;214(4):477–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Damuth J, Janis CM. On the relationship between hypsodonty and feeding ecology in ungulate mammals, and its utility in palaeoecology. Biol Rev. 2011;86(3):733–58. [DOI] [PubMed] [Google Scholar]
- 7.Miletich I, Sharpe PT. Normal and abnormal dental development. Hum Mol Genet. 2003. Apr 2;12(suppl_1):R69–73. [DOI] [PubMed] [Google Scholar]
- 8.Harada H, Kettunen P, Jung HS, Mustonen T, Wang YA, Thesleff I. Localization of putative stem cells in dental epithelium and their association with Notch and FGF signaling. J Cell Biol. 1999;147(1):105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tummers M, Thesleff I. Root or crown: A developmental choice orchestrated by the differential regulation of the epithelial stem cell niche in the tooth of two rodent species. Development. 2003;130(6):1049–57. [DOI] [PubMed] [Google Scholar]
- 10.Thesleff I, Tummers M. Tooth organogenesis and regeneration. In: StemBook. Cambridge, MA: Harvard Stem Cell Institute; 2008. [PubMed] [Google Scholar]
- 11.Krivanek J, Buchtova M, Fried K, Adameyko I. Plasticity of dental cell types in development, regeneration, and evolution. J Dent Res. 2023. Jun 1;102(6):589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luan X, Ito Y, Diekwisch TGH. Evolution and development of Hertwig’s epithelial root sheath. Dev Dyn. 2006;235(5):1167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumakami-Sakano M, Otsu K, Fujiwara N, Harada H. Regulatory mechanisms of Hertwig’s epithelial root sheath formation and anomaly correlated with root length. Exp Cell Res. 2014;325(2):78–82. [DOI] [PubMed] [Google Scholar]
- 14.Wen Q, Jing J, Han X, Feng J, Yuan Y, Ma Y, et al. Runx2 regulates mouse tooth root development via activation of WNT inhibitor NOTUM. J Bone Miner Res. 2020;35(11):2252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang S, Choi H, Kim TH, Jeong JK, Liu Y, Harada H, et al. Cell dynamics in Hertwig’s epithelial root sheath are regulated by β-catenin activity during tooth root development. J Cell Physiol. 2021;236(7):5387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashiro T, Tummers M, Thesleff I. Expression of bone morphogenetic proteins and Msx genes during root formation. J Dent Res. 2003;82(3):172–6. [DOI] [PubMed] [Google Scholar]
- 17.Yokohama-Tamaki T, Ohshima H, Fujiwara N, Takada Y, Ichimori Y, Wakisaka S, et al. Cessation of Fgf10 signaling, resulting in a defective dental epithelial stem cell compartment, leads to the transition from crown to root formation. Development. 2006;133(7):1359–66. [DOI] [PubMed] [Google Scholar]
- 18.Ota MS, Vivatbutsin P, Nakahara T, Eto K. Tooth root development and the cell-based regenerative therapy. J Oral Tissue Eng. 2007;4(3):137–42. [Google Scholar]
- 19.Jernvall J, Thesleff I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mech Dev. 2000;92:19–29. [DOI] [PubMed] [Google Scholar]
- 20.Harada H, Toyono T, Toyoshima K, Yamasaki M, Itoh N, Kato S, et al. FGF10 maintains stem cell compartment in developing mouse incisors. Dev Camb Engl. 2002;129(6):1533–41. [DOI] [PubMed] [Google Scholar]
- 21.Tapaltsyan V, Charles C, Hu J, Mindell D, Ahituv N, Wilson GM, et al. Identification of novel Fgf enhancers and their role in dental evolution. Evol Dev. 2016;18(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christensen MM, Hallikas O, Das Roy R, Väänänen V, Stenberg OE, Häkkinen TJ, et al. The developmental basis for scaling of mammalian tooth size. Proc Natl Acad Sci. 2023. Jun 20;120(25):e2300374120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen ZJ. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol. 2007;58(1):377–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007. Feb 9;315(5813):848–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero IG, Ruvinsky I, Gilad Y. Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet. 2012. Jul;13(7):505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Montaigu A, Giakountis A, Rubin M, Tóth R, Cremer F, Sokolova V, et al. Natural diversity in daily rhythms of gene expression contributes to phenotypic variation. Proc Natl Acad Sci. 2015. Jan 20;112(3):905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erwin DH, Davidson EH. The last common bilaterian ancestor. Development. 2002. Jul 1;129(13):3021–32. [DOI] [PubMed] [Google Scholar]
- 28.Irie N, Kuratani S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat Commun. 2011;2:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koonin EV. Evolution of genome architecture. Int J Biochem Cell Biol. 2009. Feb 1;41(2):298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007. Mar;8(3):206–16. [DOI] [PubMed] [Google Scholar]
- 31.Acemel RD, Maeso I, Gómez-Skarmeta JL. Topologically associated domains: A successful scaffold for the evolution of gene regulation in animals. WIREs Dev Biol. 2017;6(3):e265. [DOI] [PubMed] [Google Scholar]
- 32.Coghlan A, Eichler EE, Oliver SG, Paterson AH, Stein L. Chromosome evolution in eukaryotes: A multi-kingdom perspective. Trends Genet. 2005. Dec 1;21(12):673–82. [DOI] [PubMed] [Google Scholar]
- 33.Swenson KM, Blanchette M. Large-scale mammalian genome rearrangements coincide with chromatin interactions. Bioinformatics. 2019. Jul 15;35(14):i117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long HS, Greenaway S, Powell G, Mallon AM, Lindgren CM, Simon MM. Making sense of the linear genome, gene function and TADs. Epigenetics Chromatin. 2022. Jan 29;15(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das Roy R, Hallikas O, Christensen MM, Renvoisé E, Jernvall J. Chromosomal neighbourhoods allow identification of organ specific changes in gene expression. PLOS Comput Biol. 2021. Sep 10;17(9):e1008947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torelli F, Zander S, Ellerbrok H, Kochs G, Ulrich RG, Klotz C, et al. Recombinant IFN-γ from the bank vole Myodes glareolus: A novel tool for research on rodent reservoirs of zoonotic pathogens. Sci Rep. 2018;8(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloch A, Babik W, Bajer A, Siński E, Radwan J. Effects of an MHC-DRB genotype and allele number on the load of gut parasites in the bank vole Myodes glareolus. Mol Ecol. 2010;19(SUPPL. 1):255–65. [DOI] [PubMed] [Google Scholar]
- 38.Migalska M, Sebastian A, Konczal M, Kotlík P, Radwan J. De novo transcriptome assembly facilitates characterisation of fast-evolving gene families, MHC class I in the bank vole (Myodes glareolus). Heredity. 2017;118(4):348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Appleton J, Lee KM, Sawicka Kapusta K, Damek M, Cooke M. The heavy metal content of the teeth of the bank vole (Clethrionomys glareolus) as an exposure marker of environmental pollution in Poland. Environ Pollut. 2000;110:441–9. [DOI] [PubMed] [Google Scholar]
- 40.Gdula-Argasińska J, Appleton J, Sawicka-Kapusta K, Spence B. Further investigation of the heavy metal content of the teeth of the bank vole as an exposure indicator of environmental pollution in Poland. Environ Pollut. 2004;131(1):71–9. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007. Aug 1;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005. Dec;22(12):2472–9. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z, Wong WSW, Nielsen R. Bayes Empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005. Apr 1;22(4):1107–18. [DOI] [PubMed] [Google Scholar]
- 44.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci. 2005. Oct 4;102(40):14338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kosiol C, Vinař T, Fonseca RR da, Hubisz MJ, Bustamante CD, Nielsen R, et al. Patterns of positive selection in six mammalian genomes. PLOS Genet. 2008. Aug 1;4(8):e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martincorena I, Luscombe NM. Non-random mutation: The evolution of targeted hypermutation and hypomutation. BioEssays. 2013;35(2):123–30. [DOI] [PubMed] [Google Scholar]
- 47.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015. May 22;348(6237):880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hallikas O, Das Roy R, Christensen MM, Renvoisé E, Sulic AM, Jernvall J. System-level analyses of keystone genes required for mammalian tooth development. J Exp Zoolog B Mol Dev Evol. 2021;336(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keränen SVE, Åberg T, Kettunen P, Thesleff I, Jernvall J. Association of developmental regulatory genes with the development of different molar tooth shapes in two species of rodents. Dev Genes Evol. 1998;208(9):477–86. [DOI] [PubMed] [Google Scholar]
- 50.Jernvall J, Keränen SVE, Thesleff I. Evolutionary modification of development in mammalian teeth: Quantifying gene expression patterns and topography. Proc Natl Acad Sci. 2000;97(26):14444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc R Soc Lond B Biol Sci. 1997. Jan;256(1346):119–24. [DOI] [PubMed] [Google Scholar]
- 52.Wagner A. Selection and gene duplication: A view from the genome. Genome Biol. 2002. Apr 15;3(5):reviews1012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.David KT, Oaks JR, Halanych KM. Patterns of gene evolution following duplications and speciations in vertebrates. PeerJ. 2020. Mar 31;8:e8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Copley SD. Evolution of new enzymes by gene duplication and divergence. FEBS J. 2020;287(7):1262–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fisher LW. DMP1 and DSPP: Evidence for duplication and convergent evolution of two SIBLING proteins. Cells Tissues Organs. 2011. Aug;194(2–4):113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bouleftour W, Juignet L, Bouet G, Granito RN, Vanden-Bossche A, Laroche N, et al. The role of the SIBLING, bone sialoprotein in skeletal biology — Contribution of mouse experimental genetics. Matrix Biol. 2016. May 1;52–54:60–77. [DOI] [PubMed] [Google Scholar]
- 57.Felszeghy S, Módis L, Németh P, Nagy G, Zelles T, Agre P, et al. Expression of aquaporin isoforms during human and mouse tooth development. Arch Oral Biol. 2004. Apr 1;49(4):247–57. [DOI] [PubMed] [Google Scholar]
- 58.Yoshii T, Harada F, Saito I, Nozawa-Inoue K, Kawano Y, Maeda T. Immunoexpression of aquaporin-1 in the rat periodontal ligament during experimental tooth movement. Biomed Res. 2012;33(4):225–33. [DOI] [PubMed] [Google Scholar]
- 59.Zhang X, Zhao J, Li C, Gao S, Qiu C, Liu P, et al. DSPP mutation in dentinogenesis imperfecta Shields type II. Nat Genet. 2001. Feb;27(2):151–2. [DOI] [PubMed] [Google Scholar]
- 60.de La Dure-Molla M, Philippe Fournier B, Berdal A. Isolated dentinogenesis imperfecta and dentin dysplasia: Revision of the classification. Eur J Hum Genet. 2015. Apr;23(4):445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shields ED, Bixler D, El-Kafrawy AM. A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol. 1973. Apr 1;18(4):543–IN7. [DOI] [PubMed] [Google Scholar]
- 62.Sreenath T, Thyagarajan T, Hall B, Longenecker G, D’Souza R, Hong S, et al. Dentin sialophosphoprotein knockout mouse teeth display widened predentin zone and develop defective dentin mineralization similar to human dentinogenesis imperfecta type III. J Biol Chem. 2003. Jul 4;278(27):24874–80. [DOI] [PubMed] [Google Scholar]
- 63.Verdelis K, Ling Y, Sreenath T, Haruyama N, MacDougall M, van der Meulen MCH, et al. DSPP effects on in vivo bone mineralization. Bone. 2008. Dec 1;43(6):983–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Y, Zhang Y, Ramachandran A, George A. DSPP is essential for normal development of the dental-craniofacial complex. J Dent Res. 2016. Mar 1;95(3):302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.von Marschall Z, Mok S, Phillips MD, McKnight DA, Fisher LW. Rough endoplasmic reticulum trafficking errors by different classes of mutant dentin sialophosphoprotein (DSPP) cause dominant negative effects in both dentinogenesis imperfecta and dentin dysplasia by entrapping normal DSPP. J Bone Miner Res. 2012;27(6):1309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith BL, Preston GM, Spring FA, Anstee DJ, Agre P. Human red cell aquaporin CHIP. I. Molecular characterization of ABH and Colton blood group antigens. J Clin Invest. 1994. Sep 1;94(3):1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jordan IK, Mariño-Ramírez L, Koonin EV. Evolutionary significance of gene expression divergence. Gene. 2005. Jan 17;345(1):119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warnefors M, Kaessmann H. Evolution of the correlation between expression divergence and protein divergence in mammals. Genome Biol Evol. 2013;5(7):1324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jernvall J, Thesleff I. Tooth shape formation and tooth renewal: Evolving with the same signals. Development. 2012;139(19):3487–97. [DOI] [PubMed] [Google Scholar]
- 70.Mitsiadis TA. Role of Islet1 in the patterning of murine dentition. Development. 2003;130(18):4451–60. [DOI] [PubMed] [Google Scholar]
- 71.Charles C, Pantalacci S, Peterkova R, Tafforeau P, Laudet V, Viriot L. Effect of eda loss of function on upper jugal tooth morphology. Anat Rec. 2009;292(2):299–308. [DOI] [PubMed] [Google Scholar]
- 72.Zurowski C, Jamniczky H, Graf D, Theodor J. Deletion/loss of bone morphogenetic protein 7 changes tooth morphology and function in Mus musculus: Implications for dental evolution in mammals. R Soc Open Sci. 2018. Jan 3;5(1):170761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, et al. Gene expression across mammalian organ development. Nature. 2019. Jul;571(7766):505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Finarelli JA, Flynn JJ. Ancestral state reconstruction of body size in the Caniformia (Carnivora, Mammalia): The effects of incorporating data from the fossil record. Syst Biol. 2006;55(2):301–13. [DOI] [PubMed] [Google Scholar]
- 75.Welker F, Collins MJ, Thomas JA, Wadsley M, Brace S, Cappellini E, et al. Ancient proteins resolve the evolutionary history of Darwin’s South American ungulates. Nature. 2015. Jun;522(7554):81–4. [DOI] [PubMed] [Google Scholar]
- 76.Warinner C, Korzow Richter K, Collins MJ. Paleoproteomics. Chem Rev. 2022. Aug 24;122(16):13401–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol. 2016. Feb;34:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marks P, Garcia S, Martinez A, Belhocine K. Resolving the full spectrum of human genome variation using linked-reads. 2017; [DOI] [PMC free article] [PubMed]
- 79.Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. Direct determination of diploid genome sequences. Genome Res. 2017;27(5):757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, et al. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics. 2017. Jul 15;33(14):2202–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017. May 1;27(5):722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Warren RL. RAILS and Cobbler: Scaffolding and automated finishing of draft genomes using long DNA sequences. J Open Source Softw. 2016. Nov 17;1(7):116. [Google Scholar]
- 83.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics. 2013. Apr 15;29(8):1072–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015. Oct 1;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 85.Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Campbell MS, Law M, Holt C, Stein JC, Moghe GD, Hufnagel DE, et al. MAKER-P: A tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014. Feb 1;164(2):513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Campbell MS, Holt C, Moore B, Yandell M. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinforma. 2014. Dec 12;48:4.11.1–4.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011. May 15;29(7):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wheeler TJ, Clements J, Eddy SR, Hubley R, Jones TA, Jurka J, et al. Dfam: A database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 2013. Jan;41(Database issue):D70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Caballero J, Smit AFA, Hood L, Glusman G. Realistic artificial DNA sequences as negative controls for computational genomics. Nucleic Acids Res. 2014. Jul;42(12):e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016. Jan 4;44(D1):D81–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu J, Zheng Y, Shang X. MiteFinder: A fast approach to identify miniature inverted-repeat transposable elements on a genome-wide scale. In: 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2017. p. 164–8. [Google Scholar]
- 93.Gremme G, Steinbiss S, Kurtz S. GenomeTools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013. May 1;10(03):645–56. [DOI] [PubMed] [Google Scholar]
- 94.Smit A, Hubley R. RepeatModeler Open-1.0. 2008.
- 95.Keller O, Kollmar M, Stanke M, Waack S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 2011. Mar 15;27(6):757–63. [DOI] [PubMed] [Google Scholar]
- 96.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004. May 14;5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campbell MS. compare_annotations_3.2.pl [Internet]. 2015. Available from: https://github.com/mscampbell/Genome_annotation/blob/master/compare_annotations_3.2.pl
- 98.Eilbeck K, Moore B, Holt C, Yandell M. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinformatics. 2009. Feb 23;10(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu D, Hunt M, Tsai IJ. Inferring synteny between genome assemblies: A systematic evaluation. BMC Bioinformatics. 2018. Jan;19(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Emms DM, Kelly S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015. Aug 6;16(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Katoh K, Misawa K, Kuma K ichi, Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002. Jul;30(14):3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lefort V, Desper R, Gascuel O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol. 2015. Oct 1;32(10):2798–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farrer RA. Synima: A synteny imaging tool for annotated genome assemblies. BMC Bioinformatics. 2017. Nov 21;18(1):507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012. Apr;40(7):e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao T, Schranz ME. Network approaches for plant phylogenomic synteny analysis. Curr Opin Plant Biol. 2017. Apr 1;36:129–34. [DOI] [PubMed] [Google Scholar]
- 106.Zhao T, Holmer R, de Bruijn S, Angenent GC, van den Burg HA, Schranz ME. Phylogenomic synteny network analysis of MADS-Box transcription factor genes reveals lineage-specific transpositions, ancient tandem duplications, and deep positional conservation. Plant Cell. 2017. Jun 1;29(6):1278–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao T, Schranz ME. Network-based microsynteny analysis identifies major differences and genomic outliers in mammalian and angiosperm genomes. Proc Natl Acad Sci. 2019. Feb 5;116(6):2165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sievers F, Higgins DG. Clustal Omega. Curr Protoc Bioinforma. 2014;48(1):3.13.1–3.13.16. [DOI] [PubMed] [Google Scholar]
- 109.Suyama M, Torrents D, Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006. Jul 1;34(suppl_2):W609–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wong WSW, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004. Oct 1;168(2):1041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Löytynoja A, Vilella AJ, Goldman N. Accurate extension of multiple sequence alignments using a phylogeny-aware graph algorithm. Bioinformatics. 2012. Jul 1;28(13):1684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999. Sep 17;292(2):195–202. [DOI] [PubMed] [Google Scholar]
- 113.Buchan DWA, Jones DT. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019. Jul 2;47(W1):W402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020. Mar;38(3):276–8. [DOI] [PubMed] [Google Scholar]
- 115.Andrews S. FastQC: A quality control tool for high throughput sequence data. [Internet]. 2010. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 116.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011. May 2;17(1):10–2. [Google Scholar]
- 117.Kopylova E, Noé L, Touzet H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012. Dec 1;28(24):3211–7. [DOI] [PubMed] [Google Scholar]
- 118.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017. Apr;14(4):417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014. Dec 5;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011. Oct;478(7370):476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Weyrich A, Schüllermann T, Heeger F, Jeschek M, Mazzoni CJ, Chen W, et al. Whole genome sequencing and methylome analysis of the wild guinea pig. BMC Genomics. 2014. Nov 28;15(1):1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gossmann TI, Ralser M. Marmota marmota. Trends Genet. 2020. May;36(5):383–4. [DOI] [PubMed] [Google Scholar]
- 123.Di Palma F, Alföldi J, Johnson J, Berlin A, Gnerre S, Jaffe D, et al. The draft genome of Microtus ochrogaster. Broad Inst [Internet]. 2012; Available from: https://www.ncbi.nlm.nih.gov/bioproject/72443 [Google Scholar]
- 124.Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002. Dec 5;420(6915):520–62. [DOI] [PubMed] [Google Scholar]
- 125.Di Palma F, Alföldi J, Johnson J, Berlin A, Gnerre S, Jaffe D, et al. The draft genome of Jaculus jaculus. Broad Inst [Internet]. 2012; Available from: https://www.ncbi.nlm.nih.gov/bioproject/72445 [Google Scholar]
- 126.Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004. Apr;428(6982):493–521. [DOI] [PubMed] [Google Scholar]
- 127.Kolmogorov M, Armstrong J, Raney BJ, Streeter I, Dunn M, Yang F, et al. Chromosome assembly of large and complex genomes using multiple references. Genome Res. 2018. Nov 1;28(11):1720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lilue J, Doran AG, Fiddes IT, Abrudan M, Armstrong J, Bennett R, et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat Genet. 2018. Nov;50(11):1574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Couger MB, Arévalo L, Campbell P. A high quality genome for Mus spicilegus, a close relative of house mice with unique social and ecological adaptations. G3 GenesGenomesGenetics. 2018. May 24;8(7):2145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chinese hamster CHOK1GS assembly and gene annotation. Horiz Eagle [Internet]. 2017; Available from: https://www.ensembl.org/Cricetulus_griseus_chok1gshd/Info/Annotation
- 131.Di Palma F, Alföldi J, Johnson J, Berlin A, Gnerre S, Jaffe D, et al. The draft genome of Mesocricetus auratus. Broad Inst [Internet]. 2012; Available from: https://www.ncbi.nlm.nih.gov/bioproject/77669 [Google Scholar]
- 132.Lassance JM, Hopi Hoekstra. Improved assembly of the deer mouse Peromyscus maniculatus genome. Harv Univ Hughes Med Inst [Internet]. 2018; Available from: https://www.ncbi.nlm.nih.gov/bioproject/494228 [Google Scholar]
- 133.Fang X, Nevo E, Han L, Levanon EY, Zhao J, Avivi A, et al. Genome-wide adaptive complexes to underground stresses in blind mole rats Spalax. Nat Commun. 2014. Jun 3;5(1):3966. [DOI] [PubMed] [Google Scholar]
- 134.Di Palma F, Alföldi J, Johnson J, Berlin A, Gnerre S, Jaffe D, et al. The draft genome of Octodon degu. Broad Inst [Internet]. 2012; Available from: https://www.ncbi.nlm.nih.gov/bioproject/74595 [Google Scholar]
- 135.Keane M, Craig T, Alföldi J, Berlin AM, Johnson J, Seluanov A, et al. The naked mole rat genome resource: Facilitating analyses of cancer and longevity-related adaptations. Bioinforma Oxf Engl. 2014. Dec 15;30(24):3558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Di Palma F, Alföldi J, Johnson J, Berlin A, Gnerre S, Jaffe D, et al. The draft genome of Chinchilla lanigera. Broad Inst [Internet]. 2012; Available from: https://www.ncbi.nlm.nih.gov/bioproject/68239 [Google Scholar]
- 137.Federov V., Dalen L, Olsen RA, Goropashnaya AV, Barnes BM. The genome of the Arctic ground squirrel Urocitellus parryii. Inst Arct Biol [Internet]. 2018; Available from: https://www.ncbi.nlm.nih.gov/bioproject/477386 [Google Scholar]
- 138.Di Palma F, Alföldi J, Johnson J, Berlin A, Gnerre S, Jaffe D, et al. The draft genome of Ictidomys tridecemlineatus. Broad Inst [Internet]. 2012; Available from: https://www.ncbi.nlm.nih.gov/bioproject/61725 [Google Scholar]
- 139.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017. May 1;27(5):849–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 [.xlsx] Dental gene results – Full table of orthology, synteny, and positive selection test results for all dental genes assessed.
Additional file 2 [.txt] Dspp gapped alignment – Gapped codon-based alignment for Dspp in fasta formatted sequences.
Additional file 3 [.txt] Aqp1 gapped alignment – Gapped codon-based alignment for Aqp1 in fasta formatted sequences.
Additional file 4 [.pdf] Structure predictions – PSIPRED Secondary structure predictions for each ancestral node and unrooted molar tip species for Dspp and Aqp1.
Additional file 5 [.txt] Custom repeat library – Custom repeat library of fasta formatted sequences used in annotation of the draft Myodes glareolus genome. See Methods for description of the process used to generate the library.
Additional file 6 [.pdf] Oligonucleotide primers – List of oligonucleotide primers for Dspp, Aqp1, and GAPDH used in bank vole and prairie vole qPCR experiments.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the GenBank repository under [GenBank reference number to be added upon acceptance] and in the article’s additional files.