

Abstract
An enantioselective copper-catalyzed alkynylation of unstabilized cyclic iminium ions has been developed. Whereas such alkynylations typically utilize pyridinium, quinolinium and isoquinolinium intermediates, this method enables use of cyclic iminium ions unstabilized by resonance. With the use of a Lewis acid and copper catalyst, these iminium ions are generated in situ from readily available hemiaminal methyl ethers and transformed into highly enantioenriched α-alkynylated cyclic amines. A variety of terminal alkynes can be incorporated in high yields and enantiomeric excesses.
Keywords: enantioselective catalysis, copper catalysis, alkynylation, nitrogen heterocycles, Hammett correlation
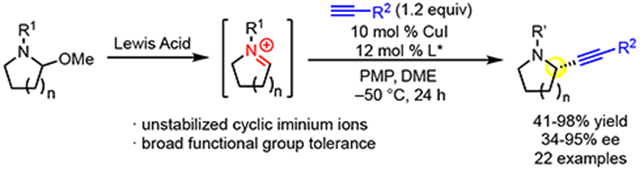
Graphical Abstract
Saturated nitrogen heterocycles are important motifs in drug discovery.1 In particular, cyclic amines bearing α-stereocenters are present in many pharmaceuticals, natural products, and bioactive molecules.2 An attractive approach to these products is nucleophilic addition to a prochiral cyclic iminium ion, and we and others have developed enantioselective alkynylations to deliver α-chiral amine heterocycles functionalized with versatile alkynyl substituents.3,4 Despite impressive advances in these alkynylations, the scope of iminium ions has been restricted to those that are stabilized by resonance, specifically isoquinolinium,5 quinolinium,6 and pyridinium7 ions (Scheme 1A). When we carried out this work, no examples of enantioselective alkynylation to unstabilized cyclic iminium ions had been reported. These iminium ions lack aromatic or other resonance stabilization, making them more difficult to form and prone to undesirable decomposition via E1 elimination under the basic conditions of a copper-catalyzed alkynylation. Although Knochel has developed an impressive enantioselective alkynylation of acyclic N-alkyl iminium ions,8 the only stereoselective alkynylation of a cyclic iminium ion, done by Royer, relied upon a chiral auxiliary (sulfiniminium ion) approach and required stoichiometric aluminum acetylide.9
Scheme 1.

Cyclic Iminium Ions in Stereoselective Alkynylation Reactions
Because of the importance of saturated nitrogen heterocycles, we embarked on a quest to develop a high-yielding and highly enantioselective alkynylation of unstabilized iminium ions. Herein, we report a copper/pyridine-(bis)oxazoline catalyst system to accomplish enantioselective alkynylation of racemic hemiaminal ether substrates 1a (Scheme 1B). In addition to identification of the optimal ligand for high enantioselectivity, the successful development of this reaction included careful balancing of the Lewis acid and base to avoid undesired decomposition. As we prepared this manuscript, the Wasa group reported alkynylation of N-aryl iminium ion intermediates, formed in situ via C─H activation, with trimethylsilylpropriolates, including enantioselective examples with cyclic systems.10 This elegant method complements our current report; in contrast to Wasa’s method, we use racemic hemiaminal ethers as substrates, employ terminal aryl acetylenes, and deliver products with readily removed carbamate protecting groups.
We selected the reaction of hemiaminal ether 1a and phenylacetylene for optimization. Aminal 1a can be readily prepared in two steps from commercially available N-(benzyloxycarbonyl)piperidone.11 Because of our previous success in alkynylations of stabilized oxocarbenium and iminium ions using copper(I)/L1 catalysts and hindered bases,5e, 12 we examined similar conditions for this alkynylation. The use of CuI along with trimethylsilyl triflate (TMSOTf) as Lewis acid led to a promising 53% yield and 47% ee, along with ~15% enamine from undesired elimination (Table 1, entry 1). A significant effect of the copper counterion was observed; other counterions, both halides and hexafluorophosphate, led to lower yields and ee’s (entries 1-4). Other commercially available pyridine(bis)oxazoline (PyBox) ligands also failed to improve the yield and ee (entries 5–6). Low yields and ee were also observed when bidentate (bis)oxazoline ligands L7 and L8 were used, despite their success in enantioselective alkynylations of stabilized cations (entries 7–8).13 Decreasing the reaction temperature improved the enantioselectivity somewhat without loss in yield; this lower temperature required that dioxane was replaced with 2-Me-THF to prevent the solvent from freezing (entry 9). Although seeing this increase in enantioselectivity was encouraging, it was also clear that we needed to rethink our catalyst design to enable the dramatic increases in enantioselectivity that we needed.
Table 1.
Optimization of Alkynylation.a

| |||||
|---|---|---|---|---|---|
| entry[a] | [Cu] | L*[b] | temp (°C)[c] |
yield (%) |
ee (%) |
| 1 | CuI | L1 | r.t. | 53 | 47 |
| 2 | CuCl | L1 | r.t. | 14 | 1 |
| 3 | CuBr | L1 | r.t. | 10 | 7 |
| 4 | Cu(MeCN)4PF6 | L1 | r.t. | 4 | 0 |
| 5 | CuI | L2 | r.t. | 31 | 37 |
| 6 | CuI | L3 | r.t. | 27 | 30 |
| 7 | CuI | L7 | r.t. | 17 | 0 |
| 8 | CuI | L8 | r.t. | 0 | ndf |
| 9b | CuI | L1 | −30 | 54 | 52 |
| 10b | CuI | L4 | −30 | 80 | 82 |
| 11b | CuI | L5 | −30 | 80 | 86 |
| 12b | CuI | L6 | −30 | 74 | 55 |
| 13b,c | CuI | L5 | −30 | 73 | 85 |
| 14d | CuI | L5 | −30 | 50 | 91 |
| 15d,e | CuI | L5 | −30 | 85 | 90 |
| 16d,e | CuI | L5 | −50 | 86 | 92 |
Conditions: aminal 1a (0.1 mmol), [Cu] (10 mol %), ligand (12 mol %), phenylacetylene (1.2 equiv), PMP (1.5 equiv), TMSOTf (1.1 equiv), dioxane (0.05 M), 24 h, unless otherwise noted. Yields determined by 1H NMR with 1,3,5-trimethoxybenzene as internal standard. Ee’s determined by HPLC using a chiral stationary phase.
2-Me-THF as solvent.
iPr2NEt as base.
Dimethoxyethane as solvent.
BF3·OEt2 as Lewis acid.
nd = not determined.
![[f]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/5101/10769448/794deb52bead/nihms-1939480-f0007.jpg)
In considering the differences between the unstabilized iminium ion formed from aminal 1a and the stabilized quinolinium and isoquinolinium ions that have been used previously, we hypothesized that the smaller size of iminium 2 might require a tighter chiral pocket in the catalyst. Because t-Bu-substituted L2 was worse than L1, we also hypothesized that aryl substituents were necessary. We thus investigated Ph-PyBox derivatives with substitution at R2, which could compress the R1 substituents about the chiral pocket.14,15 Indeed, the enantiomeric excess increased to 82% ee with methyl-substituted L4, and to 86% ee with ethyl-substituted L5 (entries 10–11). Using L5 as ligand, further increase in enantiomeric excess was realized by using dimethoxyethane (DME) as solvent, albeit in lower yield (entry 14). High yield could then be restored by using boron trifluoride diethyl etherate as the Lewis acid (entry 15). Finally, by lowering the reaction temperature to −50 °C, 86% yield and 92% ee was observed (entry 16). Alternative leaving groups on the hemiaminal ether were less effective (see Supporting Information).16
Under the optimized conditions (Table 1, entry 16), various aminal substrates were examined (Scheme 2). The model reaction can be run on 1.0-mmol scale to deliver alkyne 3 without diminished yield or ee. In addition to the benzyloxycarbonyl (Cbz) protecting group, tert-butoxycarbonyl (Boc) protected amine 4 can be formed in 68% yield and 91% ee. The absolute configuration of 4 was determined by comparison of its optical rotation to the literature value.17 The absolute configuration of other products was assigned by analogy. High yield and ee is observed in the reaction of 6-membered cyclic iminium ions, as demonstrated by formation of piperidine 5. Azepane 6 can also be delivered via this method, albeit with lower ee. Conformational analysis shows that both π-faces of the 7-membered iminium ion are more sterically encumbered than the π-faces of the 5- and 6-membered systems. Given this difference, it is not surprising that the same catalyst would not provide high ee for the 7-membered iminium ion intermediate. Substitutions on the ring are also well tolerated (7, 8, 9). With the use of (S,S)-L5, excellent yield and a single diastereomer were observed in the formation of 9 from an enantiopure substrate. However, using (R,R)-L5 afforded the same diastereomer with diminished yield. These results show that the stereoselectivity is due to substrate control, but the difference in yield highlights the influence of the chiral catalyst in the mismatched case. This strong substrate control has been observed in similar pyrrolidine systems.18
Scheme 2. Scope in Aminal.a.

a Conditions: aminal 1 (0.3 mmol), CuI (10 mol %), L5 (12 mol %), phenylacetylene (1.2 equiv), PMP (1.5 equiv), BF3·OEt2 (1.1 equiv), DME (0.05 M), −50 °C, 24 h. Average isolated yields (±5%) and ee’s (±2%) of duplicate experiments, unless noted otherwise. b 0.1 mmol scale with (R,R)-L5. Yield determined by 1H NMR with 1,3,5-trimethoxybenzene as internal standard.
The scope of terminal alkynes was then explored. A variety of aryl acetylenes are well tolerated (Scheme 3). The additional steric bulk of o-tolylacetylene is accommodated with high yield and ee observed (10). Functional groups, such as p-bromo, m-chloro, and p-Bpin, can be incorporated effectively (11, 12, 14), enabling downstream cross-couplings of the alkynylated products. High enantioselectivities were observed for other aryl acetylenes with electron-withdrawing substituents such as ether (13), nitrile (15), trifluoromethyl (16), and ester (17). Heteroaryls, such as thiophene (18) can be incorporated as well. However, aryl acetylenes with electron-donating substituents, such as p-methoxy and p-dimethylamino substituents, led to diminished enantioselectivity (19, 20). Additionally, the use of alkenyl and alkyl acetylenes afforded desired products. albeit with diminished ee and yield (21, 22, 24). Silyl acetylenes were also examined, among which triphenylsilyl acetylene produced the best result of 70% yield and 43% ee (23).19
Scheme 3. Scope in terminal alkynesa.

a Conditions: 1a (0.3 mmol), CuI (10 mol %), L5 (12 mol %), alkyne (1.2 equiv), PMP (1.5 equiv), BF3·OEt2 (1.1 equiv), DME (0.05 M), −50 °C, 24 h. Average isolated yields (±5%) and ee’s (±2%) of duplicate experiments, unless noted otherwise. b Yields were ±10%. c Single experiment.
With respect to mechanism, we hypothesize that a chiral copper acetylide is formed in situ, as is the iminium ion. Attack of the copper acetylide onto the iminium ion then provides the desired product. We note a Hammett correlation between the aryl acetylene substitution and the enantiomeric ratio (Fig. 1).20 The higher enantioselectivity observed with less electron-rich alkynes is consistent with C─C bond formation as the enantiodetermining step. Less nucleophilic acetylenes should have later transition states in the C─C bond formation, resulting in closer proximity of the iminium ion to the chiral pocket of the chiral copper acetylide, ultimately giving higher enantioselectivity. Ongoing studies are focused on more deeply understanding the nature of the active catalyst and developing a detailed model for enantioinduction.
Figure 1.

Hammett correlation of substitution on aryl acetylene with enantiomeric ratio.
In summary, a copper-catalyzed enantioselective alkynylation of unstabilized cyclic iminium ions has been described. The iminium ions are formed in situ from readily available hemiaminal ethers. This method delivers 5- and 6-membered nitrogen heterocycles with α-stereocenters in good yields and enantiomeric excess under mild reaction conditions. The reaction features excellent functional group tolerance, and a variety of aminals and alkynes can be used.
Supplementary Material
ACKNOWLEDGMENT
We thank NSF (CHE1664981). Data were acquired at UD on instruments obtained with assistance of NSF and NIH funding (NSF CHE0421224, CHE1229234, CHE0840401, and CHE1048367; NIH P20 GM104316, P20 GM103541, and S10 OD016267).
REFERENCES
- 1.(a) Taylor RD; MacCoss M; Lawson AD, Rings in Drugs. J Med Chem 2014, 57 (14), 5845–59. 10.1021/jm4017625; [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles Among U.S. FDA Approved Pharmaceuticals. J Med Chem 2014, 57 (24), 10257–74. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.(a) Mailyan AK; Eickhoff JA; Minakova AS; Gu Z; Lu P; Zakarian A, Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C-N Bonds in Total Synthesis. Chem Rev 2016, 116 (7), 4441–557. 10.1021/acs.chemrev.5b00712; [DOI] [PubMed] [Google Scholar]; (b) Grind M; Nilsson M-I; Nilsson L; Oxenstierna G; Sedvall G; Wahlén A, Remoxipride — A New Potential Antipsychotic Compound. Psychopharmacology 1989, 98 (3), 304–309. 10.1007/BF00451679; [DOI] [PubMed] [Google Scholar]; (c) Ahrén B; Landin-Olsson M; Jansson P-A; Svensson M; Holmes D; Schweizer A, Inhibition of Dipeptidyl Peptidase-4 Reduces Glycemia, Sustains Insulin Levels, and Reduces Glucagon Levels in Type 2 Diabetes. J. Clin. Endocrin. Metabol 2004, 89 (5), 2078–2084. 10.1210/jc.2003-031907. [DOI] [PubMed] [Google Scholar]
- 3.Liu J; Dasgupta S; Watson MP, Enantioselective Additions of Copper Acetylides to Cyclic Iminium and Oxocarbenium Ions. Beilstein J. Org. Chem 2015, 11, 2696–2706. 10.3762/bjoc.11.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For alternative approaches, see: (a) Olier C; Kaafarani M; Gastaldi S; Bertrand MP, Synthesis of Tetrahydropyrans and Related Heterocycles via Prins Cyclization; Extension to Aza-Prins Cyclization. Tetrahedron 2010, 66 (2), 413–445. 10.1016/j.tet.2009.10.069; [DOI] [Google Scholar]; (b) Perry MA; Hill RR; Leong JJ; Rychnovsky SD, Stereochemical Outcomes in Reductive Cyclizations To Form Spirocyclic Heterocycles. Org. Lett 2015, 17 (13), 3268–3271. 10.1021/acs.orglett.5b01422; [DOI] [PubMed] [Google Scholar]; (c) Perry MA; Morin MD; Slafer BW; Rychnovsky SD, Total Synthesis of Lepadiformine Alkaloids using N-Boc α-Amino Nitriles as Trianion Synthons. J. Org. Chem 2012, 77 (7), 3390–3400. 10.1021/jo300161x; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Perry MA; Morin MD; Slafer BW; Wolckenhauer SA; Rychnovsky SD, Fully Substituted Carbon Centers by Diastereoselective Spirocyclization: Stereoselective Synthesis of (+)-Lepadiformine C. J. Am. Chem. Soc 2010, 132 (28), 9591–9593. 10.1021/ja104250b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Li Z; MacLeod PD; Li C-J, Studies on Cu-catalyzed Asymmetric Alkynylation of Tetrahydroisoquinoline Derivatives. Tetrahedron: Asymmetry 2006, 17 (4), 590–597. 10.1016/j.tetasy.2006.02.007; [DOI] [Google Scholar]; (b) Li Z; Li C-J, Catalytic Enantioselective Alkynylation of Prochiral sp3 C─H Bonds Adjacent to a Nitrogen Atom. Org. Lett 2004, 6 (26), 4997–4999. 10.1021/ol047814v; [DOI] [PubMed] [Google Scholar]; (c) Sun S; Li C; Floreancig PE; Lou H; Liu L, Highly Enantioselective Catalytic Cross-Dehydrogenative Coupling of N-Carbamoyl Tetrahydroisoquinolines and Terminal Alkynes. Org. Lett 2015, 17 (7), 1684–1687. 10.1021/acs.orglett.5b00447; [DOI] [PubMed] [Google Scholar]; (d) Huang T; Liu X; Lang J; Xu J; Lin L; Feng X, Asymmetric Aerobic Oxidative Cross-Coupling of Tetrahydroisoquinolines with Alkynes. ACS Catalysis 2017, 7 (9), 5654–5660. 10.1021/acscatal.7b01912; [DOI] [Google Scholar]; (e) Dasgupta S; Liu J; Shoffler CA; Yap GP; Watson MP, Enantioselective, Copper-Catalyzed Alkynylation of Ketimines To Deliver Isoquinolines with alpha-Diaryl Tetrasubstituted Stereocenters. Org Lett 2016, 18 (23), 6006–6009. 10.1021/acs.orglett.6b02787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pappoppula M; Cardoso FSP; Garrett BO; Aponick A, Enantioselective Copper-Catalyzed Quinoline Alkynylation. Angew. Chem. Int. Ed 2015, 54 (50), 15202–15206. 10.1002/anie.201507848. [DOI] [PubMed] [Google Scholar]
- 7.(a) Black DA; Beveridge RE; Arndtsen BA, Copper-Catalyzed Coupling of Pyridines and Quinolines with Alkynes: A One-Step, Asymmetric Route to Functionalized Heterocycles. J. Org. Chem 2008, 73 (5), 1906–1910. 10.1021/jo702293h; [DOI] [PubMed] [Google Scholar]; (b) Sun Z; Yu S; Ding Z; Ma D, Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu─Bis(oxazoline) Complexes. J. Am. Chem. Soc 2007, 129 (30), 9300–9301. 10.1021/ja0734849. [DOI] [PubMed] [Google Scholar]
- 8.(a) Koradin C; Gommermann N; Polborn K; Knochel P, Synthesis of Enantiomerically Enriched Propargylamines by Copper-Catalyzed Addition of Alkynes to Enamines. Chem. Eur. J 2003, 9 (12), 2797–2811. 10.1002/chem.200204691; [DOI] [PubMed] [Google Scholar]; (b) Koradin C; Polborn K; Knochel P, Enantioselective Synthesis of Propargylamines by Copper-Catalyzed Addition of Alkynes to Enamines. Angew. Chem. Int. Ed 2002, 41 (14), 2535–2538. . [DOI] [PubMed] [Google Scholar]
- 9.Turcaud S; Sierecki E; Martens T; Royer J, Asymmetric α-Alkynylation of Piperidine via N-Sulfinyliminium Salts. J. Org. Chem 2007, 72 (13), 4882–4885. 10.1021/jo070631c. [DOI] [PubMed] [Google Scholar]
- 10.Chan JZ; Yesilcimen A; Cao M; Zhang Y; Zhang B; Wasa M, Direct Conversion of N-Alkylamines to N-Propargylamines Through C-H Activation Promoted by Lewis Acid/Organocopper Catalysis: Application to Late-Stage Functionalization of Bioactive Molecules. J Am Chem Soc 2020, 142 (38), 16493–16505. 10.1021/jacs.0c08599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Louwrier S; Tuynman A; Hiemstra H, Synthesis of Bicyclic Guanidines from Pyrrolidin-2-one. Tetrahedron 1996, 52 (7), 2629–2646. 10.1016/0040-4020(95)01086-6. [DOI] [Google Scholar]
- 12.Dasgupta S; Rivas T; Watson MP, Enantioselective Copper(I)-Catalyzed Alkynylation of Oxocarbenium Ions to Set Diaryl Tetrasubstituted Stereocenters. Angew. Chem. Int. Ed 2015, 54 (47), 14154–14158. 10.1002/anie.201507373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Srinivas HD; Maity P; Yap GPA; Watson MP, Enantioselective Copper-Catalyzed Alkynylation of Benzopyranyl Oxocarbenium Ions. J. Org. Chem 2015, 80 (8), 4003–4016. 10.1021/acs.joc.5b00364; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maity P; Srinivas HD; Watson MP, Copper-Catalyzed Enantioselective Additions to Oxocarbenium Ions: Alkynylation of Isochroman Acetals. J. Am. Chem. Soc 2011, 133 (43), 17142–17145. 10.1021/ja207585p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desimoni G; Faita G; Quadrelli P, Pyridine-2,6-bis(oxazolines), Helpful Ligands for Asymmetric Catalysts. Chem. Rev 2003, 103 (8), 3119–3154. 10.1021/cr020004h. [DOI] [PubMed] [Google Scholar]
- 15.Substituted aryl substituents at R1 were ineffective in increasing the enantiomeric excess. See Supporting Information.
- 16.(a) Sun S; Liu L, Catalytic Enantioselective Alkynylation of Tetrahydroisoquinoline-Based N-Acyl Hemiaminals. Synthesis 2016, 48 (16), 2627–2636. 10.1055/s-0035-1561421; [DOI] [Google Scholar]; (b) Roche SP; Samanta SS; Gosselin MM, Autocatalytic One Pot Orchestration for the Synthesis of Alpha-arylated, Alpha-amino Esters. Chem Commun (Camb) 2014, 50 (20), 2632–4. 10.1039/c3cc48884e. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y; Wen X; Cui X; Zhang XP, Enantioselective Radical Cyclization for Construction of 5-Membered Ring Structures by Metalloradical C-H Alkylation. J Am Chem Soc 2018, 140 (14), 4792–4796. 10.1021/jacs.8b01662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Martin SF; Bur SK, Vinylogous Mannich Reactions. Stereoselective Formal Synthesis of Pumiliotoxin 251D. Tetrahedron 1999, 55 (29), 8905–8914. 10.1016/S0040-4020(99)00452-4; [DOI] [Google Scholar]; (b) Hanessian S; McNaughton-Smith G, A Versatile Synthesis of a β-Turn Peptidomimetic Scaffold: An Approach towards a Designed Model Antagonist of the Tachykinin NK-2 Receptor. Bioorg. Med. Chem. Lett 1996, 6 (13), 1567–1572. 10.1016/S0960-894X(96)00275-2. [DOI] [Google Scholar]
- 19.See Supporting Information for examples of other silyl acetylenes.
- 20.Hansch C; Leo A; Taft R, A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91 (2), 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.