

Summary
Endurance exercise training is beneficial for skeletal muscle health, but it is unclear if this type of exercise can target or correct the molecular mechanisms of facioscapulohumeral muscular dystrophy (FSHD). Using the FLExDUX4 murine model of FSHD characterized by chronic, low levels of pathological double homeobox protein 4 (DUX4) gene expression, we show that 6 weeks of voluntary, free wheel running improves running performance, strength, mitochondrial function, and sarcolemmal repair capacity, while slowing/reversing skeletal muscle fibrosis. These improvements are associated with restored transcriptional activity of gene networks/pathways regulating actin cytoskeletal signaling, vascular remodeling, inflammation, fibrosis, and muscle mass toward wild-type (WT) levels. However, FLExDUX4 mice exhibit blunted increases in mitochondrial content with training and persistent transcriptional overactivation of hypoxia, inflammatory, angiogenic, and cytoskeletal pathways. These results identify exercise-responsive and non-responsive molecular pathways in FSHD, while providing support for the use of endurance-type exercise as a non-invasive treatment option.
Subject areas: Biological sciences, Pathophysiology, Transcriptomics;
Graphical abstract
Highlights
-
•
FLExDUX4 mice are weaker and run shorter distances per day vs. wild-type (WT) mice
-
•
FLExDUX4 mice have sarcolemmal repair deficits and elevated fibrosis vs. WT mice
-
•
Voluntary wheel running (VWR) restores FLExDUX4 gene expression toward WT levels
-
•
VWR improves FLExDUX4 membrane repair, strength, mitochondrial function, and fibrosis
Biological sciences; Pathophysiology; Transcriptomics
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is a highly prevalent (affecting 1 in 7500 worldwide) autosomal dominant muscular dystrophy, for which there is no known cure.1 FSHD typically manifests as muscle weakness in the facial and scapulohumeral muscles during the teen years and progresses distally over time, leading to difficulties with activities of daily living and a loss of ambulation in severe cases.2,3 Genetically, FSHD is subcategorized into FSHD1 and FSHD2. FSHD1 is caused by the loss of D4Z4 repeats in the sub-telomeric region of chromosome 4q35 in the presence of a permissive 4qA haplotype, leading to DNA hypomethylation, epigenetic de-repression, and stable transcription of double homeobox protein 4 (DUX4).4,5,6,7 FSHD2 is linked to mutations in the structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1), de novo methyltransferase 3B (DNMT3B), or ligand-dependent nuclear receptor-interacting factor 1 (LRIF1) genes in the presence of the 4qA haplotype, leading to hypomethylation of the D4Z4 region and DUX4 transcription.8,9,10 DUX4 is a transcription factor normally expressed in the early embryo, where it activates retroelements and germline genes that are subsequently deactivated in somatic cells.4,11 In mature skeletal muscle fibers, the aberrant expression of DUX4 has been shown to activate inflammatory genes, reduce expression of oxidative stress response genes, reduce anti-oxidant defenses, disrupt mitochondrial function, and mitigate sarcolemmal repair capacity—changes that promote apoptosis, interfere with muscle regeneration, and contribute to fibrosis and fatty infiltration.12,13,14
Significant progress has been made in identifying the genetic and epigenetic basis of FSHD, which are being used to develop novel pharmacological treatment approaches. There has been comparatively less progress in determining how lifestyle interventions, such as increasing levels of physical activity or exercise, might be used to improve muscle health despite decades of research showing that exercise targets pathways also affected by DUX4 expression. Aerobic and resistance exercise have been shown to be well tolerated in individuals with FSHD, and current exercise guidelines for FSHD recommend non-vigorous physical activity to reduce fatigue and improve fitness.15,16 However, these recommendations are based on few randomized controlled trials with small sample sizes and conflicting results, resulting in low-certainty evidence.17 Although some randomized clinical trials have reported improvements in fatigue, strength, peak oxygen consumption, endurance, muscle function, and indirect measures of mitochondrial function (e.g., citrate synthase enzyme activity) from aerobic,18,19,20,21,22,23 resistance/muscle strengthening,24,25 or combined16 exercise programs, others have reported no benefit on outcomes such as workload, 6-min walk distance, 5-time sit-to-stand time, and muscle strength.18,19,26
To better understand the potential utility of endurance exercise as a treatment for FSHD, we aimed to (1) more thoroughly define the healthy molecular response to endurance-type exercise in skeletal muscle; (2) determine if these molecular responses are the same in an FSHD-like murine model; and (3) identify atypical responses in DUX4-affected muscle and their potential regulatory mechanisms. Therefore, in this study, we utilized the FLExDUX4 murine model of FSHD characterized by low levels of leaky DUX4 expression to investigate the phenotypic and molecular adaptations to 6 weeks of voluntary wheel running in comparison to their wild-type (WT) littermates.
Results
Running characteristics are altered in FLExDUX4 vs. WT mice and are different between sexes
To study the molecular effects of an endurance-type exercise intervention on FSHD skeletal muscle, five-month-old male and female heterozygous B6(Cg)-Gt(ROSA)26Sortm1.1(DUX4∗)Plj/J (FLExDUX4) mice and their C57BL/6 wild-type (WT) littermates were randomly assigned to endurance-type voluntary, non-resisted wheel running (VWR, Exercise, Ex), or a no-wheel-running control condition (Control, Con). Mice in the training group were provided 24-h access to an 11 cm wheel for 40 days, whereas the control group remained in cages without a wheel. Using data from Schefer and Talan (1996)27 and running velocity data from Figure 1, we estimate mice in this study exercised at ∼65%–85% VO2max when running at 0.2–0.35 m/s, which corresponds to “vigorous” exercise intensity.28 Running analysis also indicated mice perform intermittent bursts of running that last 2–5 min, similar to interval aerobic exercise training.
Figure 1.

DUX4 expression, running performance, and grip strength characteristics
(A) Relative expression of DUX4 mRNA in the triceps brachii of FLExDUX4 and WT male and female mice. “∗” denotes significant difference between WT vs. FLExDUX4 (p < 0.05) (n = 3 per group, two-way ANOVA).
(B) Running distance (number of wheel turns per day) among FLExDUX4 Ex and WT Ex mice separated by sex over the 40-day training period. “∗” denotes WT > FLExDUX4 groups (p < 0.05) and FLExDUX4 F > FLExDUX4 M (n = 29, n = 7 WT M Ex, n = 6 WT F Ex, n = 8 FLExDUX4 M Ex, n = 8 FLExDUX4 F Ex, three-way repeated measures ANOVA).
(C) Percentage of the day spent running (%) over the 40-day study period. “∗” denotes female > male across WT and FLExDUX4 groups (p < 0.05) (n = 29, n = 7 WT M Ex, n = 6 WT F Ex, n = 8 FLExDUX4 M Ex, n = 8 FLExDUX4 F Ex, three-way repeated measures ANOVA).
(D) Average running velocity (meters/s) recorded over the 40-day study period. “∗” denotes female > male across WT and FLExDUX4 groups (p < 0.05) (n = 29, n = 7 WT M Ex, n = 6 WT F Ex, n = 8 FLExDUX4 M Ex, n = 8 FLExDUX4 F Ex, three-way repeated measures ANOVA).
(E) Changes in body-weight-normalized grip strength (KgF/Kg) among WT and FLExDUX4 male mice in the control and exercise groups was analyzed using three-way ANOVA (sex x genotype x training status) at each time point (n = 5 WT M Con, n = 7 WT M Ex, n = 7 FLExDUX4 M Con, n = 8 FLExDUX4 M Ex). “∗” denotes significantly different than “Baseline” within that group after adjusting for sex, training status, and time. “‡” denotes significant main effect of training status (exercise > control) in both WT and FLExDUX4 groups at 3 and 6 weeks (both p < 0.01).
(F) Changes in grip strength (KgF/Kg) among WT and FLExDUX4 female mice in the control and exercise groups was analyzed using three-way ANOVA (sex x genotype x training status) at each time point (n = 6 WT F Con, n = 6 WT F Ex, n = 9 FLExDUX4 F Con, n = 8 FLExDUX4 F Ex). “∗” denotes significant difference between time points within the given group (p < 0.05). “‡” denotes significant main effect of training status (exercise > control) in both WT and FLExDUX4 groups at 3 and 6 weeks (both p < 0.01). See main text for additional grip strength analyses. All values are mean ± SEM.
The FLExDUX4 mouse model of FSHD expresses a human DUX4-fl transgene in a C57BL/6 background under the control of the Rosa26 promoter after Cre-recombinase-mediated inversion.29 In the absence of Cre recombinase (uninduced model), these mice present with low levels of “leaky” DUX4 mRNA expression through antisense transcription consistent with the levels of DUX4 mRNA measured in human FSHD myocytes.29 We found similar evidence for leaky DUX4 mRNA in our FLExDUX4 male and female mice (Figure 1A, p < 0.05). Jones et al. (2020) recently developed a double transgenic model of FSHD by crossing the FLExDUX4 model with the skeletal-muscle-specific and tamoxifen-inducible ACTA1-MerCreMer mouse, allowing graded induction of muscle-specific DUX4 expression through increasing doses of tamoxifen.30 However, there are several limitations of this bitransgenic mouse model for exercise studies. First, although induced mice demonstrate transient declines in running performance following DUX4 induction, this phenotype is transient—with mice demonstrating recovery after only 2 weeks.30 This confounds the identification of exercise effects vs. natural recovery in the bitransgenic model. Although repeated injections of tamoxifen can be used to generate stable phenotypic changes, this is undesirable due to (1) the off-target effects of tamoxifen on molecular pathways affected by exercise, such as those involved in cell proliferation, apoptosis, fibrosis, inflammation, and mitochondrial respiration30 and (2) an increased study burden/confounding from stress of repeated injections. As we present in this paper, the 5- to 6-month-old FLExDUX4 model avoids these pitfalls while recapitulating stable, clinically relevant FSHD skeletal muscle phenotypes.
When assessing running performance, we found that mice in all exercise groups increased their running distance over the duration of training (Figure 1B, p < 0.001). FLExDUX4 mice (male and female grouped together) ran significantly shorter distances per day than their WT littermates (main effect of genotype, p = 0.03) (Figure 1B). Additionally, we identified a significant interaction between genotype and sex (p < 0.001) for running distance (Figure 1B), with FLExDUX4 male mice running significantly shorter distances than FLExDUX4 female mice (p < 0.01) and no difference between male and female WT mice (p > 0.05) (Figure 1B). Likewise, we found a significant main effect of sex (p < 0.01) for percentage of day spent running, with female mice demonstrating a greater percentage than male mice across both FLExDUX4 and WT groups (Figure 1C). Finally, we identified a main effect of sex (p < 0.05) and time (p < 0.001) for running velocity (m/s), with male mice running slower than female mice regardless of genotype (Figure 1D).
Despite similar body weight, FLExDUX4 mice are weaker than WT mice
Next, we assessed for changes in body weight and grip strength at baseline, three, and 6 weeks. At baseline, we found a significant main effect of sex after adjusting for genotype and training status on body weight (p < 0.001), with female mice weighing 3.736 g less than males on average (Figure S1). At 3 weeks, after adjusting for genotype, there were main effects of training status (p = 0.0014) and sex (p < 0.001) (Figure S1). Specifically, trained mice weighed an average of 1.84 g less than untrained mice, whereas female mice weighed an average of 3.37 g less than male mice. At 6 weeks, we found main effects of training status (p < 0.01), genotype (p < 0.05), and sex (p < 0.001). On average, trained mice were 1.81 g lighter than un-trained mice, FLExDUX4 mice were 1.53 g lighter than WT mice, and female mice were 3.74 g lighter than male mice (Figure S1A). Likewise, at 6 weeks we found a main effect of sex on muscle weight (grams) of the triceps brachii, with female triceps weighing less than male triceps overall (p < 0.001, Figure S1B).
At baseline, we identified a main effect of genotype (p < 0.01) and sex (p < 0.01) for body-weight-normalized grip strength, with FLExDUX4 mice significantly weaker than WT mice and male mice weaker than female mice (both p < 0.05). At 3 weeks and 6 weeks, there were only main effects of training status (3 weeks: p = 0.006, Ex > Con, 6 weeks: p < 0.001, Ex > Con) and sex (3 weeks: p = 0.001 female > male, 6 weeks: p < 0.001, female > male). Together, these results indicate that free wheel running increases grip strength significantly in both FLExDUX4 and WT mice and restored grip strength to WT levels in FLExDUX4 mice.
Given the effect of sex at each time point, we sought to determine the interaction between genotype, training status, and duration of training (time) in male and female mice separately. In male mice, we found main effects of time (p < 0.01), training status (p = 0.001, Ex > Con), and a significant interaction between time x training status (p < 0.01) and genotype x training status (p < 0.01) (Figure 1E). Multiple comparisons indicated FLExDUX4 Ex mice increased their grip strength at 3 (p < 0.05) and 6 (p < 0.01) weeks relative to baseline, with no difference between 3 and 6 weeks (Figure 1E). WT male Ex mice also demonstrated increased strength at 3 and 6 weeks compared with baseline, but it did not reach significance (Figure 1E). In females, we found main effects of time (p < 0.001), training status (p < 0.05, Ex > Con), and significant time x genotype (p < 0.01) and time x training status (p < 0.01) interactions. Multiple comparisons indicated female FLExDUX4 Ex mice demonstrated increased grip strength from baseline to 3 weeks (p < 0.05) and a continued increase from 3 to 6 weeks (p < 0.05, Figure 1F). WT female Ex mice also demonstrated increase in grip strength through 6 weeks (p < 0.05) (Figure 1F).
Free wheel running is associated with reduced skeletal muscle fibrosis in FLExDUX4 mice, reduced myofiber diameter, and altered fiber type composition
To identify changes in muscle morphometry from free wheel running exercise, we analyzed tissue sections from the triceps brachii of FLExDUX4 and WT mice in the control and exercise groups for the percentage of centrally nucleated fibers, percent fibrosis, and changes in myofiber diameter. The triceps brachii was chosen because it is one of the earliest and most-consistently affected muscles in patients with FSHD and because it demonstrates a high muscle activity profile during VWR in rodents.31,32 We did not find any effect of genotype, training status, or sex on the proportion of centrally nucleated fibers in the triceps. However, we did identify a significant interaction between genotype and training status (p < 0.05) for muscle fibrosis (Figure 2A). Multiple comparisons indicated that at baseline, male FLExDUX4 control mice had a significantly greater amount of fibrosis than all WT groups (p < 0.01), as well as trained FLExDUX4 male (p < 0.05) and female (p < 0.05) mice (Figure 2B). These results indicate that free wheel running exercise may prevent or delay fibrotic development in the FLExDUX4 mouse model (Figure 2B). Additionally, we identified a main effect of training status (p = 0.001) and sex (p < 0.01) on average feret diameter, with Ex mice having reduced minimal feret diameters than Con mice and female mice having smaller diameters than male mice (Figures 2C–2F). Likewise, the average myofiber cross-sectional area was significantly reduced in trained mice (p < 0.001), and was significantly lower in female vs. male mice (p < 0.05) (Figures S2A–S2C).
Figure 2.

Free wheel running reduces skeletal muscle fibrosis in FLExDUX4 male mice and alters fiber type composition
(A) Sample images of Masson’s trichrome staining in the triceps brachii from all groups. Blue staining indicates presence of fibrous connective tissue. Scale bar: 100 μm.
(B) Percentage (of total area) of triceps brachii fibrosis in each group. “∗” denotes FLExDUX4 male control > WT control male and female mice, WT male Ex mice, and FLExDUX4 male and female Ex mice (∗ p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001) (n = 44, n = 6 WT M Con, n = 6 WT M Ex, n = 5 WT F Con, n = 6 WT F Ex, n = 6 FLExDUX4 M Con, n = 5 FLExDUX4 M Ex, n = 5 FLExDUX4 F Con, n = 5 FLExDUX4 F Ex, three-way ANOVA).
(C) Sample immunofluorescence (laminin = green, nuclei = blue) images of myofibers from the triceps brachii of all groups. Scale bar: 100 μm.
(D and E) Distribution of fibers according to minimal feret diameter (um) in all male mice (D) and female mice (E) from all groups (n = 50, n = 5 WT M Con, n = 7 WT M Ex, n = 6 WT F Con, n = 6 WT F Ex, n = 8 FLExDUX4 M Con, n = 6 FLExDUX4 M Ex, n = 4 FLExDUX4 F Con, n = 7 FLExDUX4 F Ex, three-way ANOVA).
(F) Average minimal feret diameter for male and female, control and exercise groups (three-way ANOVA). “∗” denotes mean diameter is lower in trained vs. control mice across groups (main effect of training status, p < 0.01). “‡” denotes female diameter < male diameter across groups (main effect of sex, p < 0.01). Mean ± SEM are plotted for all charts.
(G) Immunofluorescence staining of skeletal muscle fiber types in the triceps brachii using antibodies targeting MyHC-I (blue), MyHC-IIa (green), MyHC-IIx (red), and MyHC-IIb (unstained). Representative images are presented for each study group. Scale bar: 50 μm.
(H) Quantification of the proportion of each fiber type in each study group (n = 32 mice, 4 per group, mean of 4396 ± 1048 fibers per section, three-way ANOVA). “∗” denotes significantly different than corresponding male untrained control. “§” denotes female mice have significantly greater proportions of type IIb fibers (p < 0.05). “#” denotes main effect of training status, with trained mice demonstrating a significant reduction in type IIb fibers vs. untrained mice (p < 0.05). “Δ” denotes training × genotype interaction (p < 0.05). WT mice show a significant decline in type IIx fibers with training, whereas FLExDUX4 mice show an increase in type IIx fibers with training (p < 0.05).
Additionally, aerobic exercise has been associated with changes in the proportion of the four muscle fiber types in mice, which can be delineated by their contractile and metabolic properties.33 They include “slow-twitch” oxidative type I (expressing Myh7), fast-twitch oxidative glycolytic type IIa (expressing Myh2), fast glycolytic type IIb (expressing Myh4), and fast glycolytic type IIx (expressing Myh1), which are smaller and more oxidative than type IIb fibers.34 Based on this previous evidence, we evaluated the triceps brachii for changes in fiber type composition, which revealed that male WT and male FLExDUX4 mice demonstrate significant increases in the proportion of type IIa myofibers with exercise (p < 0.05), trained mice have significantly reduced proportion of type IIb myofibers vs. untrained mice (main effect of training status, p < 0.05), and that overall, female mice have a significantly greater proportion of type IIb fibers than male mice (main effect of sex, p < 0.05). In response to endurance-type exercise training WT mice as a group show a significant decline in type IIx fibers, whereas FLExDUX4 mice show an increase in type IIx fibers with training (significant genotype x training status interaction, p < 0.05).
Six-month-old FLExDUX4 muscles present with multiple transcriptional abnormalities
Assessment of running performance, grip strength, and histological changes indicated that male FLExDUX4 mice present with a more severely affected phenotype that recapitulates key clinical features of FSHD than their female FLExDUX4 littermates. Therefore, we focused our follow-up molecular testing on male study groups. We performed RNA sequencing on the triceps brachii of male FLExDUX4 and WT, Ex and Con mice, thereby allowing us to relate transcriptional and histological findings in the same muscle. Our principal-component analysis (PCA) demonstrated clear separation between FLExDUX4 Con and WT Con groups along principal components 1 and 2 that was reduced with exercise (Figure 3A). The chord diagram in Figure S3A shows the proportion of unique and common genes (represented by the threaded connections) between these groups. Ingenuity pathway analysis (IPA) and gene set enrichment analysis (GSEA) were used to provide functional categorization of DEG (p < 0.05) between groups, as well as to identify core enrichment genes, upstream regulators, and regulatory networks (Tables S2, S3, S4, S5, and S6). We validated our RNA sequencing results by examining the mRNA expression of eight DEG across the main group comparisons using RT-qPCR (Figures S4A–S4E). We found statistically significant correlations between analyses (Pearson r = 0.68, p < 0.001) and strong concordance between methods (Figure S4F).
Figure 3.

Transcriptional profiling in FLExDUX4 compared with WT mice and in WT mice following voluntary wheel running
(A) Principal-component analysis (PCA) showing transcriptional separation between FLExDUX4 and WT mice that is reduced with exercise (n = 14, n = 3 WT M Con, n = 4 WT M Ex, n = 3 FLExDUX4 M Con, n = 4 FLExDUX4 M Ex).
(B and C) Dot plots depicting the top 10 significantly enriched gene ontologies (all FDR <0.25) (GOMF = molecular function, GOCC = cell component, and GOBP = biological process) among upregulated (left, red) and downregulated (right, green) DEG between FLExDUX4 Con and WT Con mice (B) and WT Ex and WT Con mice (C). NES = net enrichment score.
(D and E) Top 18 significantly enriched IPA canonical pathways among DEG between FLExDUX4 Con and WT Con mice (D) and Wt Ex and WT Con (E) (FDR p value of <0.05), denoted by the yellow line. Bars are colored according to their predicted activation Z score (green tones = downregulated pathways, red tones = upregulated pathways, gray = no predicted Z score).
(F) Volcano plot depicting IPA upstream regulators of the transcriptional differences between WT Ex and WT Con mice. Highly significant active/inhibited regulators are denoted by the red and green circles, respectively. The top 19 regulators according to FDR p value are labeled.
FLExDUX4 Con mice maintained transcriptional features consistent with elevations in actin cytoskeletal signaling, including predicted activation of integrin, integrin-linked kinase, paxillin pathways, and positive enrichment of actin contractile fiber and actin-binding gene sets (Figures 3B and 3D; Tables S2A–S2E, and S2G). These mice also presented with transcriptional evidence of (1) increased vascular remodeling, such as activation of the pro-angiogenic vascular endothelial growth factor (VEGF) signaling pathway and its downstream regulator myocardin (MYOCD) (Figures 3D and S3B); (2) inflammation, such as activation of the inflammatory cytokine/pro-catabolic interleukin-1 (IL-1) signaling cascade (Table S2E); (3) fibrosis, including enrichment of the hepatic fibrosis signaling cascade (Figure 3D); and (4) muscle wasting, including downregulation of the anti-fibrotic/anti-cachectic p21-activated kinase (PAK) signaling pathway (Figure 3D, and Table S2E),35 among other changes (Figure 3; and Table S2).
Transcriptional responses to voluntary wheel running in wild-type skeletal muscle
Compared with WT Con mice, WT Ex mice demonstrated activation of pathways related to oxidative metabolism (Figure 3C) and mitigated expression of genes involved in muscle inflammation and fibrosis, including (1) reduced activation of IL-22, IL-2, IL-15, and IL-6 cytokine signaling pathways (Table S3E) and (2) downregulation of pro-fibrotic platelet-derived growth factor (PDGF) pathway activity (Table S3E). WT Ex mice also demonstrated transcriptional changes consistent with downregulation of the calcium signaling pathway vs. WT Con mice—an adaptation that could reflect improved intracellular calcium homeostasis (Figure 3E).36 Exercise was also associated with: (1) predicted activation of angiogenesis and vascular homeostasis pathways (Table S3F); (2) activation of muscle structure/stability pathways, such as the ILK pathway (Figure 3E); (3) activation of positive regulators of satellite cell differentiation, such as pro-differentiation serine/threonine kinase 11 (STK11) (Figure 3F), (4) reduced oxidative stress response signaling (e.g., NRF2 pathway) (Figure 3E; and Table S3E); and (5) deactivation of pro-atrophic pathways, including the upstream regulator Forkhead box protein O4 (FOXO4) in WT Ex vs. WT Con mice (Table S3F).
Exercise targets dysregulated transcriptional pathways in FLExDUX4 mice
To determine if exercise restores healthy transcriptional signatures in FLExDUX4 muscle, we first compared FLExDUX4 Ex with WT Con mice. We identified transcriptional differences consistent with upregulation of oxidative metabolism and mitochondrial function, as well as upregulation of insulin receptor signaling, vascular remodeling, and oxidative stress signaling in FLExDUX4 Ex relative to WT Con samples (Figures 4A, 4B, and S3C). We found 27 signaling pathways significantly enriched in both the FLExDUX4 Con vs. WT Con analysis and the FLExDUX4 Ex vs. WT Con analysis (FDR p < 0.05). Of these 27, 6 had no predicted Z score. Within the remaining 21 pathways, three pathways (FDR p < 0.05) were downregulated in FLExDUX4 control mice and were partially or fully reversed in FLExDUX4 exercise mice (Table S4I; Figures 4C and 4D), including estrogen receptor and PAK signaling. Seven pathways were upregulated in FLExDUX4 control mice (FDR p < 0.05) and were partially or fully reversed in FLExDUX4 exercise mice compared with WT control mice (Table S4I; Figures 3C and 3D), including actin cytoskeleton, integrin-linked kinase (ILK), integrin, paxillin, and IL-1 signaling. These partially or fully reversed pathways demonstrate signaling systems likely altered by exercise in FLExDUX4 mice. Likewise, we also identified genes that were differentially expressed between FLExDUX4 Con vs. WT Con mice that were not different between FLExDUX4 Ex vs. WT Con mice (genes normalized by voluntary wheel running) (Table S4J).
Figure 4.

Transcriptional responses to exercise in FLExDUX4 mice
(A) Dot plot depicting the top 10 significantly enriched gene ontologies among upregulated (left, red) and downregulated (right, green) DEG between FLExDUX4 Ex and WT Con mice. Dots are colored according to their FDR p value and sized according to the number of genes from our dataset belonging to that gene ontology.
(B) Top 18 significantly enriched (FDR p < 0.05) IPA canonical pathways among DEG between FLExDUX4 Ex and WT Con mice. Bars are plotted according to -log10(FDR p value) and colored according to their predicted activation Z score (green = downregulated pathways, red = upregulated, gray = no predicted Z score).
(C) Comparison analysis identifies pathways (FDR p < 0.05) that were activated (positive Z score) or inhibited (negative Z score) in the [FLExDUX4 Con vs. WT Con] analysis that demonstrated a full (change in Z score from positive to negative or vice versa) or partial (change in Z score toward zero) reversal in the [FLExDUX4 Ex vs. WT Con] analysis. Pie chart depicts number of activated and inhibited pathways showing partial or full reversal and common pathways that were not reversed.
(D) Heatmap of reversed or partially reversed pathways from (C), shaded according to activation Z score (denoted in each block). Left-sided color code indicates group assignment from (C).
(E) Dot plot depicting the top 10 significantly enriched gene ontologies among upregulated (left, red) and downregulated (right, green) DEG between FLExDUX4 Ex and FLExDUX4 Con mice.
(F) Top 18 significantly enriched (FDR p < 0.05) canonical pathways between FLExDUX4 Ex and FLExDUX4 Con mice as in (D).
(G) Volcano plot depicting IPA upstream regulators of the transcriptional differences between FLExDUX4 Ex and FLExDUX4 Con mice. Highly significant active/inhibited regulators are denoted by the red and green circles, respectively. The top 19 regulators according to FDR p value are labeled.
To identify the unique effects of exercise on DUX4-affected muscle, we performed IPA and GSEA analyses on DEG between FLExDUX4 Ex vs. FLExDUX4 Con mice. Most notably, we found a strong transcriptional signature consistent with augmented mitochondrial function in FLExDUX4 Ex mice, including significant enrichment and predicted activation of the oxidative phosphorylation signaling pathway, as well as positive enrichment of gene sets related to mitochondrial proteins, mitochondrial membranes, the respiratory chain, and ATP synthesis (Figures 4E and 4F; Tables S5A, S5C, and S5E). IPA also predicted activation of pathways involved in (1) vascular function, such as upstream regulator vascular endothelial growth factor A (VEGFA) (Figure 4G; and Table S5F); (2) downregulation of inflammatory and pro-atrophic pathways such as the Janus kinase signal transducer and activator of transcription (JAK/STAT) and hepatic fibrosis pathways (Figure 4F; and Table S5E); (3) activation of pro-hypertrophic pathways, including upregulation of Akt serine/threonine kinase 1, and its downstream target p70 S6 kinase 1 (P70S6K, Table S5F); (4) reduced activation of cytokine signaling pathways, such as the IL-1 signaling pathway (Table S5E); (5) increased estrogen receptor pathway activation (Figure 4F); and (6) upregulation of pathways known to regulate the structure of the neuromuscular junction, such as ephrin B and ephrin receptor signaling pathways (Figure 4F). We did not find any change in skeletal muscle DUX4 mRNA expression in FLExDUX4 male and female mice after exercise (p > 0.05).
Transcriptional similarities and differences between FLExDUX4 and healthy muscle in response to voluntary wheel running
As confirmed by our PCA results, WT Ex and FLExDUX4 Ex mice demonstrated similar patterns of predicted activity across multiple pathways, including increased oxidative metabolism, reduced inflammatory and fibrotic signaling, downregulation of the calcium signaling, predicted activation of angiogenesis and vascular homeostasis, activation of muscle structure/stability pathways, increased activity of satellite cell differentiation pathways, reduced oxidative stress response signaling, and deactivation of pro-atrophic pathways (Figures 3C, 3E, 3F, 4E, 4F, and 4G; Tables S3 and S5).
However, despite these similarities, IPA and GSEA did detect several differences in the transcriptional response to exercise in FLExDUX4 and WT muscle. Although both FLExDUX4 and WT mice showed strong transcriptional signatures of increased oxidative metabolism compared with their untrained littermates (Figures 3C, 3E, 4A and 4B), FLExDUX4 Ex mice presented with negative enrichment of gene sets related to mitochondrial matrix, mitochondrion, and oxidoreductase activity vs. WT Ex mice (FDR p < 0.05) (Figures S3D; and Table S6A). There were also disparate transcriptional changes across pathways related to inflammation and fibrosis signaling, including higher predicted activation of the hepatic fibrosis signaling pathway (Figure S3E), the transforming growth factor β (TGFβ) signaling pathway (Table S6E), and the fibroblast growth factor (FGF) signaling pathway (Table S6E) in FLExDUX4 Ex mice. Transcriptional differences also included the following: (1) the presence of a gene network consistent with inhibition of runt-related transcription factor 3 (RUNX3) in FLExDUX4 Ex mice, which regulates proprioceptive neuron differentiation in Golgi tendon organs and muscle spindles37 (Figure S3F); (2) greater activation of pathways regulating vascular growth (e.g., of endothelin-1 pathway) (Table S6E); (3) greater activation of oxidative stress signaling (e.g., NRF2 oxidative stress response pathway) (Table S6E); (4) greater upregulation of actin cytoskeleton signaling (Table S6E); (5) greater activation of hypoxic signaling responses, including higher predicted activation of the hypoxia-inducible factor 1 alpha (HIF1α) pathway (Table S6E); (6) increased activation of muscle hypertrophic pathways (e.g., pro-hypertrophic insulin-like growth factor 1 [IGF-1], pathway) (Table S6E); (7) activation of the estrogen receptor signaling pathway (Table S6E); and (8) elevated insulin receptor signaling pathway activation (Figure S3E) in FLExDUX4 Ex vs. WT Ex mice. Additional differences are included in Table S6.
FLExDUX4 skeletal muscles present with deficits in mitochondrial function that are improved with exercise
Exercise resulted in activation of gene sets and pathways related to subunits of the electron transport chain and the mitochondrial ribosome in both FLExDUX4 and WT mice (Sankey diagram, Figure 5A). To determine if these molecular changes were sufficient to improve mitochondrial function, we performed high-resolution mitochondrial respirometry in isolated myofibers of the flexor digitorum brevis (FDB) from control and trained male WT (Figure 5B) and FLExDUX4 (Figure 5C) mice to overcome limitations associated with the use of permeabilized muscle bundles or isolated mitochondria.38 The FDB was selected due to the ease of isolating many full-length, intact (containing cytoplasm and mitochondria), undamaged myofibers small enough to fit into Seahorse XFe24 (Agilent, Santa Clara, CA, USA) microplates. WT mice had higher respiration than FLExDUX4 mice at baseline and after administration of the ATP synthase inhibitor oligomycin (main effect of genotype p < 0.01, Figure 5D). WT mice also had higher levels of uncoupled respiration (post-FCCP administration, main effect of genotype p < 0.05), which was increased in both WT and FLExDUX4 mice after training (main effect of training status p < 0.05) (Figure 5D). Likewise, WT mice had higher respiration FLExDUX4 mice after administration of the complex I and II inhibitors rotenone/antimycin A (main effect of genotype, p < 0.01) (Figure 5D).
Figure 5.

Free wheel running increases mitochondrial function in FLExDUX4 and WT mice
(A) Sankey plot of the top 28 leading edge genes in positively enriched mitochondrial gene sets (FDR p < 0.25) among DEGs between FLExDUX4 Ex and FLExDUX4 Con male mice. Plot depicts the gene, number of gene sets (size) it appears in, and the enzyme it codes for. CI = electron transport chain (ETC) complex I (NADH dehydrogenase), CIV = ETC complex IV (cytochrome c oxidase), CII = ETC complex II (succinate reductase), CIII = ETC complex 3 (cytochrome reductase), ETF = electron-transferring flavoprotein.
(B) Oxygen consumption rates (OCR) normalized to myofiber confluence level and 16S rRNA mtDNA/ncDNA ratio, measured during the baseline/basal period, and following injections of oligomycin, FCCP, and antimycin A/rotenone in WT male mice (n = 5 WT M Con, n = 7 WT M Ex).
(C) Normalized oxygen consumption rates in FLExDUX4 male mice as in B (n = 4 FLExDUX4 M Con, n = 5 FLExDUX4 M Con).
(D) Overlaid plots from (C) and (D) (n = 21, two-way ANOVA). “∗” denotes WT > FLExDUX4 OCR (main effect of genotype, p < 0.05). “‡” denotes trained > control (main effect of training status, p < 0.05).
(E) Maximal respiration rate in FLExDUX4 and WT male Con and Ex groups (n = 21, two-way ANOVA). “∗” denotes trained > control in both genotypes (main effect of training status, p < 0.05).
(F) Basal respiration measured in all four male study groups (n = 21, two-way ANOVA). “∗” denotes WT > FLExDUX4 (main effect of genotype, p < 0.05).
(G) Non-mitochondrial respiration in FLExDUX4 and WT Con and Ex groups (n = 21, two-way ANOVA). “∗” denotes WT > FLExDUX4 (main effect of genotype, p < 0.05).
(H) Spare respiratory capacity (n = 21, two-way ANOVA). “∗” denotes trained > control in both genotypes (main effect of training status, p < 0.05).
(I and J) Regulatory diagram showing the predicted activation of transcriptional co-activator PGC1α in FLExDUX4 Ex male mice (I) and WT Ex male mice (J). The prediction legend for (I) and (J) is shown to the left of (I). Arrows point to genes regulated by PGC1α. Arrow heads denote stimulation, whereas blunted ends denote inhibition. Target genes are colored according to their log2FC (green = downregulated, red = activated). There are comparatively fewer downstream targets with expression consistent with PGC1α activation in FLExDUX4 Ex than Wt Ex mice.
(K) Nd1/Hk2 mitochondrial DNA (mtDNA) copy-number analysis in FDB fibers from seahorse experiments (n = 21, n = 5 WT M Con, n = 7 WT M Ex, n = 4 FLExDUX4 M Con, n = 5 FLExDUX4 M Ex, two-way ANOVA). “∗” denotes significantly different than WT Con (all p < 0.05).
(L) FDB 16S rRNA/Hk2 mtDNA copy-number analysis (n = 21, two-way ANOVA). “∗” denotes significantly different than WT Con (all p < 0.05).
(M) Triceps brachii Nd1/Hk2 mtDNA copy-number analysis (n = 22, n = 5 WT M Con, n = 7 WT M Ex, n = 5 FLExDUX4 M Con, n = 5 FLExDUX4 M Ex, two-way ANOVA). “∗” denotes significantly different than WT Ex (all p < 0.05).
(N) Triceps brachii 16S rRNA/Hk2 mtDNA copy-number analysis (n = 22, two-way ANOVA). “∗” denotes significantly different than WT Ex (all p < 0.05). All values are mean ± SEM.
In addition to changes in oxygen consumption rate, we also assessed for changes across several measures of mitochondrial function. Exercise significantly increased maximal respiration in both WT and FLExDUX4 mice (main effect of training status, p < 0.05) (Figure 5E). WT mice also had overall higher basal respiration (main effect of genotype, p < 0.05) and non-mitochondrial respiration regardless of training status (main effect of genotype, p < 0.01) (Figures 5F and 5G). Finally, exercise increased spare capacity similarly in both WT and FLExDUX4 mice (main effect of training status, p < 0.05) (Figure 5H). We did not find any main effects of genotype or training status for proton leak, ATP-production-coupled respiration, or coupling efficiency (all p > 0.05).
Exercise increases the skeletal muscle mtDNA/ncDNA ratio in WT but not FLExDUX4 mice
IPA identified PGC1α as an activated upstream regulator in both FLExDUX4 Ex and WT Ex mice. However, the predicted activation score was lower in FLExDUX4 mice (z = 2.3 vs. z = 5.6), as was the number of downstream targets with expression consistent with PGC1α upregulation (FLExDUX4 Ex: 28/40 genes, WT Ex: 75/95 genes) (Figures 5I and 5J). Because PGC1α activity promotes mitochondrial biogenesis, we assessed for differences in triceps brachii and FDB mitochondrial content in male mice. The ratio of the mitochondrial genome copy number (mtDNA) to nuclear genome copy number (ncDNA) was calculated as the ratio of Nd1 or 16S rRNA DNA content (mitochondrial) to Hk2 (nuclear) DNA content using qPCR. We used two mtDNA genes to confirm our results.
In the FDB, we found a significant interaction between genotype and training status for both Nd1/Hk2 and 16S rRNA/Hk2 ratios (p < 0.05). Multiple comparisons indicated the following: (1) WT mice increased the Nd1/Hk2 (p < 0.05) and 16S rRNA/Hk2 (p < 0.05) ratios with exercise, whereas FLExDUX4 demonstrated no change (Figures 5K and 5L); (2) FLExDUX4 control FDB myofibers had significantly greater Nd1/Hk2 (p < 0.05) and 16S rRNA/Hk2 (p < 0.01) ratios than WT Con mice; and (3) FLExDUX4 Ex FDB myofibers had significantly greater Nd1/Hk2 (p = 0.01) and 16S rRNA/Hk2 (p < 0.05) ratios than WT Con mice (Figures 5K and 5L). In the triceps brachii, we also found a significant interaction between genotype and training status (p < 0.05) for both ratios. Multiple comparisons indicated WT Ex mice had a significantly greater Nd1/Hk2 ratio than WT control mice (p = 0.01), as well as both FLExDUX4 groups (both p < 0.05) (Figure 5M). Likewise, WT Ex mice had a higher 16S rRNA/Hk2 ratio than WT Con mice (p < 0.05), as well as both FLExDUX4 groups (both p < 0.05) (Figure 5N). There was no difference between FLExDUX4 Con and WT Con for either ratio in the triceps (Figures 5M and 5N).
Free wheel running improves sarcolemmal repair capacity in FLExDUX4 mice
We previously reported that FSHD myoblasts present with membrane repair deficits, which were improved by antioxidant treatment and DUX4 knockdown.12 We found a similar deficit in FLExDXU4 biceps myofibers.12 Given that aerobic exercise is associated with reductions in oxidative stress and improves muscle health, we wanted to determine if the exercise treatment could improve membrane repair using a laser ablation assay in male FLExDUX4 and WT mice. In this study, unused FDB myofibers isolated for our mitochondrial respiration measurements were exposed to focal laser ablation in the presence of cell-impermeant FM1-43 dye (Figure 6A). We found a significant interaction between time and genotype (p < 0.001) and time x training status (p < 0.05) for the extent of FM dye entry post-injury. Multiple comparisons identified that FLExDUX4 Con mice demonstrate significantly worse repair than their WT Con and WT Ex groups, as well as FLExDUX4 Ex mice (p < 0.05) (Figure 6B). We also found a significant interaction between genotype and training status for percentage of myofibers that failed to repair (p < 0.05). Multiple comparisons revealed FLExDUX4 mice had a greater percentage of myofibers that failed to repair compared with WT mice (p < 0.05), which was significantly reduced with exercise (p < 0.05, Figure 6C).
Figure 6.

Voluntary wheel running improves membrane repair capacity in the FLExDUX4 mouse
(A) Representative images from membrane repair assays in isolated flexor digitorum brevis (FDB) fibers from FLExDUX4 and WT mice. Arrow denotes site of focal laser ablation injury. The extent of FM1-43 dye entry into the fiber (green dye in myofiber interior) is used to determine the kinetics of membrane resealing. Scale bar: 100 μm.
(B) Mean ± SEM for change in fluorescence intensity of FM1-43 dye entry (ΔF/F) into the myofiber after laser injury in each group (n = 19, n = 5 WT M Con, n = 5 WT M Ex, n = 5 FLExDUX4 M Con, n = 4 FLExDUX4 M Ex, 12–20 fibers/mouse, three-way repeated measures ANOVA). “∗” denotes significant difference FLExDUX4 vs. WT (p < 0.05). “‡” denotes main effect of training status (p < 0.05).
(C) Percentage of myofibers that failed to repair following membrane injury in FLExDUX4 and WT mice. “∗” denotes significantly different than FLExDUX4 Con (p < 0.05).
Discussion
In this study, we used transcriptional profiling to identify abnormal signaling in skeletal muscle in response to low, chronic DUX4 expression in the FLExDUX4 model of FSHD. Using a 6-week free wheel running intervention, we aimed to determine if dysregulated pathways could be targeted and corrected by endurance-type VWR to generate phenotypic improvements in muscle health and function downstream. Our results indicate that 6-month-old FLExDUX4 mice recapitulate key skeletal muscle clinical characteristics of human FSHD including (1) significant reductions in running performance; (2) significant reductions in grip strength; (3) elevations in skeletal muscle fibrosis; (4) deficits in mitochondrial respiration; and (5) impaired sarcolemmal repair capacity—all of which improved with VWR training. Likewise, FLExDUX4 male mice demonstrate dysregulation of molecular pathways involved in skeletal muscle actin cytoskeletal signaling, vascular remodeling, inflammation, fibrosis, and muscle wasting. However, we noted that heterozygous female FLExDUX4 mice demonstrated a more mild phenotype than their male FLExDUX4 littermates, including only slight (non-significant) reductions in running distance and marginal (non-significant) increases in fibrosis vs. WT female mice. Epidemiological studies have revealed that females with FSHD are more frequently asymptomatic or minimally affected compared with males, and there is a significantly greater proportion of affected sons than daughters among offspring of asymptomatic mothers with FSHD.39,40,41 The observed differences between male and female FLExDUX4 mice further supports the validity of the FLExDUX4 mouse model to recapitulate clinical features of chronic low levels of DUX4 expression characteristic of the human disease.
Several of these pathways have been implicated in the pathophysiology of FSHD. Importantly, exercise promoted transcriptional changes across several of these pathways and reduced the dystrophic phenotype, indicating that molecular adaptations may have contributed to improved muscle health and function post-training. Overall, these results provide further support for the use of exercise to target the skeletal muscle molecular abnormalities in FSHD.
Our IPA and GSEA analysis of the transcriptional differences between FLExDUX4 and WT mice highlighted significant involvement of actin cytoskeleton signaling, which has also been reported in FSHD patient biopsies,42 as well as primary myoblasts and induced pluripotent stem cells from individuals with FSHD1 and FSHD2 with atrophic phenotypes.43,44 Upregulation of the actin cytoskeletal signaling could represent an initial compensatory mechanism to counteract declining muscle strength in the FLExDUX4 mouse, which we observed here and in our previous paper using maximal electrical stimulation of the tibialis anterior.12 Indeed, structural and chemical compensations for declining force production have been identified in myofibers from FSHD human muscle biopsies, including condensed myofilament lattice spacing, titin stiffening, and increased calcium sensitivity.45
During the training period we identified significant improvements in running distance over time across all groups, suggestive of improved exercise tolerance. This is encouraging, as individuals with FSHD often present with reduced exercise tolerance secondary to deficits in oxygen extraction and ADP-stimulated mitochondrial oxygen consumption.46,47,48 At baseline, we observed declines in mitochondrial respiration across all respiratory states in FLExDUX4 male mice vs. WT mice. With exercise, both WT and FLExDUX4 mice demonstrated significant increases in maximal respiration and spare respiratory capacity, which reflects an increased ability to respond to energy demand or stress. These results are similar to those reported by Bankole et al. (2016), who showed that 6 weeks of cycling aerobic exercise increased maximal oxygen consumption, muscle strength, and physical function in individuals with FSHD1.16 Likewise, male FLExDUX4 and WT mice showed significant increases in the proportion of fast oxidative type IIa myofibers, which could support observed increases in exercise tolerance and strength. These findings are similar to those reported in human muscle in response to sprint interval training.49,50 Unlike WT mice, male and female FLExDUX4 mice demonstrated a significant increase in fast glycolytic type IIx fibers with training. The cause of this difference in DUX4-affected muscle is unclear, but it was recently shown that type IIx fibers have higher levels of mitochondrial proteins than type IIb fibers and the highest levels of both oxidative and glycolytic enzymes, making them behave like intermediate fibers.51 DUX4-affected muscle may rely on increased intermediate fiber function than WT mice, but this hypothesis requires further investigation.
Our RNAseq analysis provides potential molecular insight into improved mitochondrial function after exercise, including upregulation of genes regulating mitochondrial structure, respirasome, ATP synthesis, coupled respiration, and the respiratory chain in both WT Ex and FLExDUX4 Ex mice. In both groups, PGC1α—a master regulator of mitochondrial biogenesis and glucose and fatty acid metabolism—was identified as a significant activated upstream regulator of the transcriptional response.52,53 Activation of the PGC1α transcriptional network may be especially important in FSHD, as Banerji et al. (2019) recently identified insufficient activation of PGC1α target genes in differentiating immortalized FSHD myoblasts and FSHD myotubes, leading to atrophic myotube formation.54
However, although we observed an increase in mtDNA content in WT male mice with exercise in the triceps brachii and FDB, we did not observe this response in FLExDUX4 male mice. This diminished change in mtDNA content could be linked to the blunted activation of the PGC1α regulatory pathway in FLExDUX4 males relative to WT males. Additionally, we measured a significantly higher mtDNA content in the FDB of FLExDUX4 control male mice compared with their WT control littermates. Given that these same myofibers present with diminished mitochondrial respiratory capacity, these findings suggest that some muscles in the FLExDUX4 mouse model might be adversely affected by the accumulation of defective mitochondria. Indeed, previous studies have found similar phenotypic changes in FSHD human biopsies, including the accumulation of large pools of intermyofibrillar and subsarcolemmal mitochondria, as well as aggregation of mitochondria near capillaries.46,47
FLExDUX4 improvements in myofiber oxygen consumption without increases in mitochondrial content could be due to exercise-induced improvements in per-mitochondria respiratory capacity secondary to changes in mitochondrial dynamics or morphology. Aerobic exercise has been shown to stimulate production of new, healthy mitochondria through mitochondrial biogenesis while also removing damaged/defective mitochondria through autophagy or mitophagy, generating a pool of high-functioning mitochondria without a net change in overall number.55,56,57 Exercise also induces metabolic changes favorable to mitochondrial respiration independent of mitochondrial number, such as increased availability of phospholipid intermediates for mitochondrial membrane integrity and mitochondrial function58 and increased levels of muscle acylcarnitines to reduce mitochondrial stress and enhance metabolic flexibility.59
We also identified molecular changes in pathways related to vascular function, including the pro-angiogenic VEGF and hypoxia-responsive HIF1α networks. Vascular abnormalities have been identified in human FSHD tissues and animal models of FSHD and may contribute to muscle dysfunction.60,61,62 For example, Banerji et al. (2015) reported evidence linking upregulated VEGF and HIF1α expression to DUX4/β-catenin-driven proteomic rewiring in human FSHD biopsies.63 Here, the VEGF signaling pathway was significantly upregulated in FLExDUX4 control relative to WT control mice, which could reflect a compensatory mechanism to stimulate vascular growth in the face of poor oxygen utilization.
Unlike VEGF, exercise has been shown to inhibit HIF1α signaling, which is typically stimulated under hypoxic conditions to promote glycolytic metabolism but suppressed by exercise training to facilitate oxidative metabolism.64 While we observed this response in WT mice after 6 weeks of wheel running, FLExDUX4 mice presented with persistent predicted HIF1α regulatory network activation, similar to human FSHD muscle tissue.63 These findings coincide with those of Lek et al. (2020), who reported that DUX4 expression triggers HIF1α activation and cell death signaling downstream.65 The persistent activation of the HIF1α pathway may leave FLExDUX4 mice increasingly vulnerable to hypoxic stress and DUX4 toxicity.65
Accumulation of skeletal muscle fibrosis, fatty deposition, and inflammatory cell infiltration has been extensively reported in FSHD and is most severe in muscles with greater deficits in oxidative metabolism.66,67,68 While FSHD shares common histological features with other muscular dystrophies, the FSHD human biopsies have been shown to present with higher levels of perivascular inflammation, limited T-cell-mediated cytotoxicity, development of fibrosis without inflammation, and asymmetry of symptoms.66,69 Fibro-fatty replacement of lean muscle is a multifactorial process linked to myofiber fragility and/or poor damage-repair capacity,12,70 as well as accumulation/activation of pro-fibrotic cell subtypes, such as fibro-adipogenic progenitors (FAPs) and myofibroblasts.67,71 Janssen et al. (2016) previously reported that both aerobic exercise training and cognitive behavioral therapy designed to increase daily physical activity effectively reduced the rate of fatty infiltration of the thigh in humans with FSHD.20 In agreement with these findings, we found that FLExDUX4 male control mice had elevated skeletal muscle fibrosis, which was mitigated by the exercise intervention. Exercise may contribute to slowed or reversed fibrotic development in FLExDUX4 mice secondary to (1) reduced activity of the hepatic fibrosis pathway—a cascade of inflammatory signals that activate myofibroblasts and stimulate excessive connective tissue deposition—and/or (2) downregulation of pro-inflammatory cytokine signaling, including IL-6 and IL-22 pathways, which activate FAPs and contribute to muscle atrophy.72
The reduction in muscle inflammatory signaling and fibrosis could also be linked to activation of the estrogen receptor signaling pathway—one of only two pathways downregulated in male FLExDUX4 mice in comparison to their WT littermates that was reversed by exercise. Estrogen receptor activity has been shown to improve membrane stability, reduce myofiber susceptibility to injury, improve myofiber regeneration, and mitigate fibrosis.73,74 Improved membrane stability could be critical to prevent skeletal muscle pathology and fibrosis in FSHD, given the evidence for membrane repair deficits in the biceps12 and FDB of the FLExDUX4 mouse. Treatments to improve sarcolemmal repair have been shown to reduce fibro-fatty muscle degeneration and restore muscle strength in other muscular dystrophies.75 Indeed, in this study, we demonstrate, for the first time, that endurance-type VWR training improves membrane repair capacity in the FLExDUX4 mouse, which could improve overall muscle health and reduce fibrosis.
Increases in mitochondrial function may have also contributed to improved membrane repair, as mitochondria have been shown to accumulate at the site of injury to clear excess calcium and promote wound closure.76,77 Likewise, Vila et al. (2017) demonstrated that excessive calcium influx from repetitive sarcolemmal disruptions in the mdx mouse model of Duchenne muscular dystrophy leads to calcium overload and mitochondrial damage/loss, which further compromises repair.77 We identified predicted downregulation of the calcium signaling pathway in FLExDUX4 Ex and WT Ex mice relative to the untrained littermates, which could reflect enhanced calcium homeostasis or calcium buffering capacity, thereby improving repair. Additionally, PGC1α overexpression was shown to increase myofiber resistance to eccentric contraction-induced injury in the mdx mouse.78 Exercise generated a transcriptional signature consistent with PGC1α activation and improved sarcolemmal repair in the FLExDUX4 Ex mice, suggesting a similar effect of PGC1α in this study.
In conclusion, we utilized the FLExDUX4 mouse model of FSHD to identify functional and molecular deficits in DUX4-affected skeletal muscle and to determine if 6 weeks of voluntary wheel running could target and improve these impairments. FLExDUX4 mice presented with elevations in skeletal muscle fibrosis, as well as deficits in running performance, grip strength, mitochondrial respiration, sarcolemmal repair, and VWR-induced increases in mtDNA content. Using transcriptional profiling, we identified dysregulated gene ontologies, signaling pathway/networks, and regulators in FLExDUX4 mice with roles in actin cytoskeletal signaling, vascular remodeling, inflammation, fibrosis, and muscle wasting. Exercise resulted in transcriptional adaptations across several of these gene networks in FLExDUX4 mice to support observed improvements in running performance, strength, fibrosis, mitochondrial function, and sarcolemmal repair (Figure 7). These pathways are also implicated in the pathophysiology of FSHD in human muscles, which provides strong mechanistic evidence for the efficacy of endurance-type exercise training as a targeted, non-pharmacologic intervention to reduce dystrophic symptoms in this population.
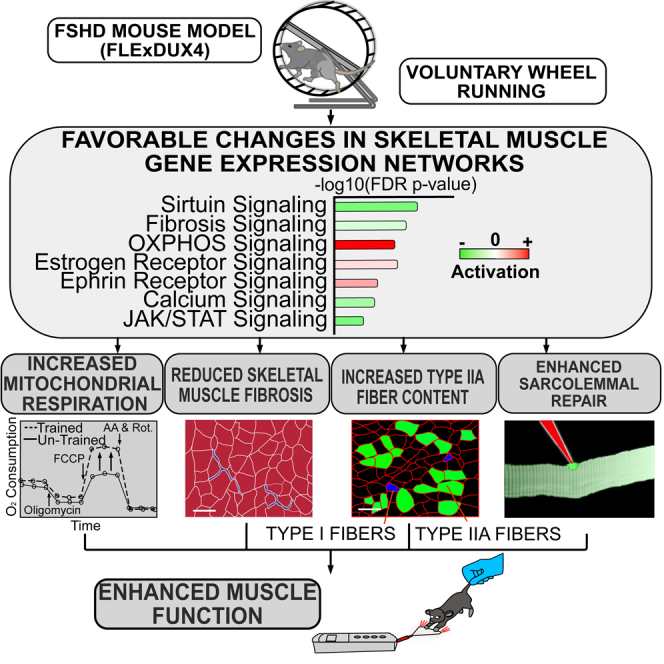
Figure 7.

Schematic conclusion
Based on the results in this study, FLExDUX4 mice skeletal muscle presents with transcriptional abnormalities compared with WT mice, including increased expression of genes in calcium, integrin, vascular remodeling (vascular endothelial growth factor, VEGF), and inflammatory (interleukin-8, IL-8) signaling pathways. These transcriptional abnormalities contribute to reduced mitochondrial respiration, dystrophic changes (e.g., fibrosis), and sarcolemmal repair deficits, which may lead to poor muscle function, including reduced grip strength and running distance in FLExDUX4 mice. Voluntary wheel running promotes favorable changes in the skeletal muscle transcriptome, including reduced expression of genes in fibrosis, calcium, and muscle wasting (Janus kinase/signal transducer and activator of transcription, JAK/STAT) signaling pathways and increased expression of genes involved in oxidative phosphorylation (OXPHOS) and estrogen receptor signaling compared with untrained FLExDUX4 mice. These transcriptional changes promote increases in mitochondrial respiration, reduced dystrophic features, increases in type IIa muscle fiber content, and improved sarcolemmal repair capacity. Together, these adaptations may contribute to measured improvements in muscle function, such as increased grip strength post-training.
Limitations of the study
In this study, we used a voluntary wheel running exercise program, as opposed to forced/stimulated exercise. We employed free wheel running because it does not rely on the use of aversive stimuli to force active movement, which may not be consistent with normal mouse behavior. Moreover, voluntary wheel running occurs within the mouse’s routine diurnal rhythm in a non-stressed laboratory environment, thereby reducing the risk of inducing confounding effects on the molecular effects of exercise. Further examination of tailored exercise interventions at different intensities and duration using stimulated exercise approaches are warranted. We estimated VWR to be similar to moderate-vigorous, interval aerobic exercise training based on running characteristics and observed molecular and phenotypic adaptations that are consistent with aerobic exercise training. However, we did not have access to cages with open circuit calorimetry to confirm the aerobic nature of VWR.
Additionally, we collected samples 6–8 h following the final exercise bout in the trained groups, which may affect the transcriptional patterns observed in trained vs. untrained mice. As identified in the study by Kuang et al. (2022)79 the expression of genes associated with mitochondrial respiration rise rapidly (within 3–9 h) post-exercise, which may explain the strong transcriptional signature associated with mitochondrial respiration observed in trained WT and FLExDUX4 mice. Due to the temporal nature of transcriptional responses to exercise across different genes, collecting samples further from the final exercise bout may allow identification of additional novel transcriptional responses across other signaling pathways.
Although these results indicate that FLExDUX4 mice recapitulate many impairments found in humans with FSHD, and mouse VWR induces adaptations similar to aerobic exercise in humans,80 our results require further validation in human FSHD skeletal muscles. Studies utilizing large and homogeneous samples will be needed to make clinical recommendations. Additional studies should also investigate the effects of exercise in induced models of FSHD with more severe phenotypes, investigate molecular responses in additional muscle groups, and include assessments of transcriptional responses to VWR in female FLExDUX4 mice, which were not performed in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal (rat IgG1 isotype) Anti-Laminin-2 (alpha-2 Chain) antibody - clone 4H8-2 | Sigma-Aldrich | Cat# L0663; RRID: AB_477153 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 488 | Invitrogen | Cat# A32766; RRID: AB_2866493 |
| Mouse Myosin heavy chain, Type IIX Monoclonal (MIgM) | Developmental Studies Hybridoma Bank | Cat# 6H1; RRID: AB_1157897 |
| Mouse Myosin heavy chain Type IIA Monoclonal (MIgG1) | Developmental Studies Hybridoma Bank | Cat# SC-71; RRID: AB_2147165 |
| Mouse Myosin heavy chain Type I Monoclonal (MIgG2b) | Developmental Studies Hybridoma Bank | Cat# BA-D5; RRID: AB_2235587 |
| Rabbit polyclonal antibody to Dystrophin (IgG) | Abcam | Cat# ab15277; RRID: AB_301813 |
| Goat anti-Mouse Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody 405 | Invitrogen | Cat# A-31553; RRID: AB_221604 |
| Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Invitrogen | Cat# A-21121; RRID: AB_2535764 |
| Goat Anti-Mouse IgM Antibody, μ chain, Cy3 conjugate | Millipore Sigma | Cat# AP128C; RRID: AB_92478 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | ThermoFisher | Cat# A-21245; RRID: AB_2535813 |
| Biological samples | ||
| Mouse Skeletal Muscles (triceps, FDB) | This Study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Hoescht 33342 dye | Abcam | Cat# ab228551 |
| Epredia™ Shandon™ Bluing Reagent | Epredia™ | Cat# 6769001 |
| Epredia™ Richard-Allan Scientific™ Eosin-Y Alcoholic | Epredia™ | Cat# 71204 |
| Epredia™ Signature Series™ Hematoxylin 7211 | Epredia™ | Cat# 7211 |
| Epredia™ Signature Series™ Clarifier™ 1 | Epredia™ | Cat# 7441 |
| Fisher Chemical™ Permount™ Mounting Medium | FisherScientific | Cat# SP15-100 |
| Xylenes (CAS 1330-20-7) | Sigma-Aldrich | Cat# 214736 |
| Epredia™ Dehydrant Alcohol 100% | Epredia™ | Cat# 6201 |
| Epredia™ Dehydrant Alcohol 100% | Epredia™ | Cat# 6301 |
| Bovine Serum Albumin (CAS 9048-46-8) | Sigma-Aldrich | Cat# A3294-500G |
| Normal Goat Serum | Vector Laboratories | Cat# S-1000-20 |
| Gibco Horse Serum, heat inactivated, New Zealand origin | ThermoFisher | Cat# 26050088 |
| M.O.M blocking reagent | Vector Laboratories | Cat# MKB-2213-1 |
| Gibco PBS, pH 7.4 | ThermoFisher | Cat# 10010023 |
| ProLong Gold Antifade Mountant | Invitrogen | Cat# P36930 |
| Collagenase, Type II, powder | ThermoFisher | Cat# 17101015 |
| Gibco Penicillin-Streptomycin (5,000 U/mL) | ThermoFisher | Cat# 15070063 |
| Gibco DMEM, high glucose, pyruvate | ThermoFisher | Cat# 11995065 |
| Gibco Sodium Pyruvate (100 mM) | ThermoFisher | Cat# 11360070 |
| Gibco Sodium Bicarbonate 7.5% solution | ThermoFisher | Cat# 25080094 |
| Matrigel Matrix | Corning Inc. | Cat# 356234 |
| Sodium Chloride (NaCl) (CAS 7647-14-5) | Sigma-Aldrich | Cat# S9888-25G |
| Ethylenediaminetetraacetic acid (EDTA) (CAS 60-00-4) | Sigma-Aldrich | Cat# E9884 |
| Sodium dodecyl sulfate (SDS) (CAS 151-21-3) | Sigma-Aldrich | Cat# 436143 |
| TRIS hydrochloride (Tris-HCl) (CAS 1185-53-1) | Sigma-Aldrich | Cat# 10812846001 |
| Proteinase K, recombinant, PCR grade | ThermoFisher | Cat# EO0491 |
| Fetal Bovine Serum, Premium | Bio-Techne | Cat# S11150 |
| HEPES (CAS 7365-45-9) | Sigma-Aldrich | Cat# 54457 |
| Hanks Balanced Salts Solution | Sigma-Aldrich | Cat# H9269 |
| FM1-43 dye ((N-(3-Triethylammoniumpropyl)-4-(4-(Dibutylamino) Styryl) Pyridinium Dibromide) | ThermoFisher | Cat# T3163 |
| Calcium Chloride (CaCl2) (CAS 10043-52-4) | Sigma-Aldrich | Cat# C4901 |
| Trichrome Stain Kit (Connective Tissue Stain) | Abcam | Cat# ab150686 |
| Critical commercial assays | ||
| Agilent Fragment Analyzer high-sensitivity assay | Agilent | Cat# DNF-472-1000 |
| QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina | Lexogen | N/A |
| Seahorse XFe24 Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | Cat# 18090010 |
| Oligo(dT)12-18 Primer | Invitrogen | Cat# 18418012 |
| SYBR Green Master Mix | Applied Biosystems | Cat# 4309155 |
| GoTaq Green Master Mix | Promega | Cat# M712 |
| 100 uM dNTPs | Invitrogen | Cat# 18427013 |
| GoTaq polymerase | Promega | Cat# M3001 |
| QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina | Lexogen | N/A |
| NextSeq 500 v2 High Output SR 75 Cycle | Illumina | N/A |
| Deposited data | ||
| RNA sequencing of mouse triceps brachii muscles | NCBI Gene Expression Omnibus (GEO) database | GSE233340 |
| Experimental models: Organisms/strains | ||
| Male and female heterozygous FLExDUX4 mice: B6(Cg)-Gt(ROSA)26Sortm1.1(DUX4∗)Plj/J | Peter & Takako Jones Laboratory (University of Nevada, Reno) | N/A |
| Male and female wild type (WT) mice: C57BL/6 | Peter & Takako Jones Laboratory (University of Nevada, Reno) | N/A |
| Oligonucleotides | ||
| Primers for RT-qPCR, see Table S7 | This Paper | N/A |
| Genotyping PCR Primers, see Table S7 | Peter & Takako Jones Laboratory (University of Nevada, Reno) | N/A |
| Software and algorithms | ||
| cutadapt v1.16 | Martin M. (2011)81 | https://doi.org/10.14806/ej.17.1.200 |
| FastQC v0.11.7 | Andrews S. (2018)82 | Babraham Institute https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR version 2.5.3a | Dobin et al. (2013)83 | https://doi.org/10.1093/bioinformatics/bts635 |
| featureCounts 1.6.2 from Subread package | Liao et al. (2013)84 | https://subread.sourceforge.net |
| DESeq2 | Love et al. (2014)85 | https://doi.org/10.1186/s13059-014-0550-8 |
| Gene set enrichment analysis (GSEA v4.1.0) | Subramanian et al. (2005)86 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Ingenuity Pathway Analysis (IPA | Qiagen | N/A |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/ |
| Cell Profiler | Laghi et al. (2022)87 | https://cellprofiler.org/ |
| VitalView Software | STARR Life Sciences | https://www.starrlifesciences.com/activity/activity-software/ |
| Parse-Running-Wheel | Uapinyoying (2018)88 | https://zenodo.org/doi/10.5281/zenodo.10210699 |
| Python | Python.org | N/A |
| Other | ||
| Borosilicate glass bottom chamber slides | ThermoFisher | Cat# 154461 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yi-Wen Chen (YChen@childrensnational.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Data: RNA-seq data have been deposited to the NCBI Gene Expression Omnibus (GEO) database, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All running performance, grip strength and body weight, histological, respirometry, mitochondrial DNA content, and sarcolemmal repair data will be shared by the lead contact upon request
-
•
Code: All original code has been deposited at Zenodo: https://zenodo.org/doi/10.5281/zenodo.10210699 88 and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
Experimental model and study participant details
Animal models
Five-month-old male and female heterozygous B6(Cg)-Gt(ROSA)26Sortm1.1(DUX4∗)Plj/J (FLExDUX4) mice and their C57BL/6 wild type (WT) littermates were used for this study. Both FLExDUX4 and WT mice were drug-naive, and FLExDUX4 mice were uninduced. Mice were originally obtained from the Jones Lab (University of Nevada, Reno, Reno, NV). Male and female, FLExDUX4 and WT littermates were housed singly in cages with sufficiently controlled temperature and humidity under a 12/12-hour light/dark cycle with access to chow and water ad libitum. All mice were fed a standard, irradiated, 18% protein formula from Teklad (Teklad Global 2918, Inotiv, Chicago, IL, USA). All mice were handled in accordance with the guidelines established by the Institutional Animal Care and Use Committee (IACUC) of the CNMC in Washington, D.C., and all procedures were carried out under the approved animal protocol.
Genotyping
Genotyping was performed according to the protocol outlines by Jones et al. 2018.29 Briefly, ∼100 ng genomic DNA was isolated from tail snips of ≤21 day-old mice was used for PCR amplification using 2x GoTaq Master Mix (Promega, Madison, WI, USA), 100 uM dNTPs (Invitrogen), dH2O, and 0.5 U GoTaq polymerase (Promega), 400 uM primer TJ76F (5’-CAATACCTTTCTGGGAGTTCTCTGCTGC), 400nM primer TJ77R (5’- CTCGTGTAGACAGAGCCTAGACAATTTGTTG), and 200nM primer TJ78R (5’- TGCAGGACAACGCCCACACACC). Reactions were incubated at 94°C for 3 min, cycled 30x (94°C for 20 sec, 62°C for 20 sec, 72°C for 35 sec), with a final 2 minute extension at 72°C.
Study groups
FLExDUX4 and WT mice were randomly assigned to voluntary, non-resisted wheel running (VWR, Exercise, Ex), or a no-wheel-running control condition (Control, Con). Mice in the training group were provided 24-hour access to an 11cm wheel for 40 days, while the control group remained in cages without a wheel. Daily running performance (n=29, n=7 WT M Ex, n=6 WT F Ex, n=8 FLExDUX4 M Ex, n=8 FLExDUX4 F Ex) was recorded for each mouse throughout the duration of exercise using VitalView Software (STARR Life Sciences, Oakmont PA). Data was analyzed for changes in running distance (wheel turns) per day, % of time running and resting, and running velocity throughout the intervention period using the custom python package parse-running-wheel.88,89 Post-training testing was completed on day 40, after which mice were sacrificed for tissue collection. We selected a 40-day training period based on previous literature demonstrating 3-5 weeks of free wheel running promotes robust adaptations in skeletal and cardiac muscle in mice,17,80 and the frequent use of 6 week training programs in human exercise studies.90,91,92,93,94
Grip strength & body weight
Forelimb grip strength (n=56, n=5 WT M Con, n=7 WT M Ex, n=6 WT F Con, n=6 WT F Ex, n=7 FLExDUX4 M Con, n=8 FLExDUX4 M Ex, n=9 FLExDUX4 F Con, n=8 FLExDUX4 F Ex) was measured at baseline before the beginning of the study, and at 3 weeks and day 40. Forelimb grip strength was determined using a force transducer (Model 1027CSM; Columbus Instrument Co., Columbus, Ohio) according to the Treat NMD guidelines.95 Grip strength (kgF) was normalized to body mass (Kg) for each mouse. Likewise, body weight (grams) was measured at baseline, 3 weeks, and 40 days using a standard tabletop scale in the same mice.
Method details
Skeletal muscle histology and immunofluorescence
Immediately after euthanasia, the triceps brachii were surgically removed, mounted on cork with tragacanth gum, flash-frozen in liquid nitrogen-chilled isopentane and stored at −80°C. 8 μm serial sections of the triceps brachii were sectioned using a Leica CM1900 cryostat (Leica Microsystems, Wetzlar, Germany). Accumulation of intrafascicular connective tissue was assessed using Masson’s trichrome staining (Trichrome Stain Kit, Abcam, Cambridge, UK) (n=44, n=6 WT M Con, n=6 WT M Ex, n=5 WT F Con, n=6 WT F Ex, n=6 FLExDUX4 M Con, n=5 FLExDUX4 M Ex, n=5 FLExDUX4 F Con, n=5 FLExDUX4 F Ex). Mean, median, minimal, maximal, and minimal feret’s diameters, as well as myofiber cross-sectional area, were quantified using immunofluorescence staining and the CellProfiler Muscle Analyzer pipeline (n=50, n=5 WT M Con, n=7 WT M Ex, n=6 WT F Con, n=6 WT F Ex, n=8 FLExDUX4 M Con, n=6 FLExDUX4 M Ex, n=4 FLExDUX4 F Con, n=7 FLExDUX4 F Ex).87 Immunofluorescence was performed as previously described96 with anti-laminin primary (1:100; Sigma-Aldrich, clone 4H8-2) and Alexa-Fluor 488 secondary antibodies (Invitrogen A32766; 1:500, Waltham, MA, USA), and counterstained with Hoescht dye (abcam ab228551, 1:2000, Cambridge, UK). Stained sections were imaged using a VS120 slide-scanning microscope (Olympus America) at ×40 magnification for use in CellProfiler. The occurrence centrally located myonuclei, as an indicator of degenerative and regenerative events, was assessed using hematoxylin-eosin staining and expressed as a percentage of total myofibers counted per muscle section (n=30, n=3 WT M Con, n=3 WT M Ex, n=3 WT F Con, n=3 WT F Ex, n=3 FLExDUX4 M Con, n=5 FLExDUX4 M Ex, n=3 FLExDUX4 F Con, n=7 FLExDUX4 F Ex).97 The relative proportion of type I, type IIa, type IIb, and type IIx fibers (percentage of total fibers) was assessed in whole triceps cross-sections according to the protocol outlined by Murach et al. (2019)98 (n=32 mice, 4 per group, mean of 4396 ± 1048 fibers per section). Briefly, slides were blocked in 1% bovine-serum albumin, 1% goat normal serum (Vector Laboratories S-1000-20, Newark, CA, USA), and 3.6% M.O.M blocking reagent (Vector Laboratories, MKB-2213-1) in PBS for 1 hour at room temperature, then incubated with primary antibodies against myosin heavy chain type 1 (BA-D5, 3.05 μg /ml), type IIa (SC-71, 0.85 μg /ml), type IIx (6H1, 10.5 μg /ml) (Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA, USA), and dystrophin (ab15277, 1:500, abcam) for 2 h at room temperature in blocking buffer, washed 3x10 min with PBS, and incubated with a secondary antibody cocktail containing Alexa Fluor 405 goat anti-mouse IgG2b (Invitrogen A-31153; 1:500), Alexa Fluor 488 goat anti-mouse IgG1 (Invitrogen A21121; 1:500), Cy3 goat anti-mouse IgM (Millipore-Sigma AP128C; 1:500, Millipore-Sigma, Burlington, MA, USA), and Alexa Fluor 647 goat anti-rabbit IgG (Invitrogen A-21245; 1:500) in blocking buffer for 1 hour at room temperature. After incubation, slides were washed 3x10 min in PBS, and mounted with ProLong Gold Antifade Mountant (Invitrogen P36930).
RNA isolation, qRT-PCR, and RNA sequencing
Total RNA was isolated from powered triceps brachii muscle as previously described (12). RNA purity was assessed using nanodrop spectrophotometry (Thermo Fisher Scientific, Waltham, MA, USA). RNA integrity was assessed using the Agilent Fragment Analyzer (Agilent, Santa Clara, CA, USA) high-sensitivity assay. For quantitative reverse transcription PCR (qRT-PCR), cDNA was reverse-transcribed from 2 μg total RNA using Superscript IV (Invitrogen, Waltham, MA, USA) and oligo dT primers. 20 ng cDNA in SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA) was used to quantify DUX4 expression normalized to the housekeeping gene GAPDH as previously described.12 Primer sequences are included in Table S7. RNA was submitted to Lexogen Gmbh (Vienna, Austria) for 3’ mRNA sequencing (n=14, n=3 WT M Con, n=4 WT M Ex, n=3 FLExDUX4 M Con, n=4 FLExDUX4 M Ex). Sequencing-ready libraries were produced using a QuantSeq 3' mRNA-Seq Library Prep Kit FWD for Illumina (015UG009V0252) using 50 ng RNA per sample. Concentration of obtained sequencing libraries was measured with a Qubit dsDNA HS assay (Thermo Fisher). A sequencing-ready pool of indexed libraries was prepared according to these quantifications. The library pool was quality controlled via Bioanalyzer (Agilent, Santa Clara, CA, USA) .Sequencing was performed on an Illumina NextSeq 500 sequencer with a v2 SR75 High Output kit at Lexogen GmbH. Reads were processed using cutadapt v1.16 92, analyzed using FastQC v0.11.7 93, and aligned to the spike-in complemented Ensembl release 90 of the Mus musculus assembly GRCh38 from the Genome Reference Consortium using the splice-aware aligner STAR version 2.5.3a94. The alignments were quantified based on the annotations of Ensembl GRCm38.90 and the spike-in specific annotations of Lexogen with the featureCounts software program version 1.6.2 of the subread analysis package.84 Adjusted p-values (q-values) were calculated using the bejamini-hochberg procedure. Principal component analysis was used to visualize sample-to-sample variance in the skeletal muscle transcriptome. Volcano plots were used to highlight the most significantly differentially expressed genes (DEG) between groups (-log10(FDR p-value) ≥ 1 and |log2(FC)| >1) (Figure S2). Heatmaps were used to observe clustering of study groups across the top 500 DEG (|Fold Change| ≥ 1.5, p<0.05) in our dataset, and within each of the five main group comparisons (Figure S3). We performed experimental validation of eight DEGs identified by DESeq2 from RNAseq analysis using qRT-PCR in the original samples (see Figure S4).85 Total RNA was isolated from the triceps and reverse transcribed to cDNA as described above. qRT-PCR was used to measure the relative expression of Acta2, Egln3, Mrpl4, Ppara, Sod2, Tab 1, Cox7a1, and Pim3 normalized to Gapdh using the 2ˆ-(△Ct) method according to the protocol outlined above. Primer sequences are included in Table S7. The log2(Fold-Change) was calculated from RT-qPCR data to directly compare to the RNA-Seq results. The Pearson correlation coefficient was calculated between RNA sequencing and qRT-PCR-derived log2(fold change) values.
Skeletal muscle mitochondrial content
Total DNA was isolated from frozen tissue sections of the triceps brachii (30 mg) and myofibers from the flexor digitorum brevis (FDB, isolated for mitochondrial respiration analysis, see “Mitochondrial Function” and “FDB Fiber Isolation”) for analysis of the mtDNA/ncDNA ratio a cording to the protocol outlined by Quiros et al. (2017) (n=21, n=5 WT M Con, n=7 WT M Ex, n=4 FLExDUX4 M Con, n=5 FLExDUX4 M Ex).99 For the triceps brachii, frozen tissue sections were homogenized under liquid nitrogen by mortar and pestle (n=22, n=5 WT M Con, n=7 WT M Ex, n=5 FLExDUX4 M Con, n=5 FLExDUX4 M Ex). The fine powder (triceps) or isolated FDB fibers was resuspended in 600 ul lysis buffer (100 mM NaCl, 10 mM EDTA, .5% SDS, and 20 mM Tris-HCl) supplemented with .2 mg/ml proteinase K (6 μl of 20 mg/ml stock), and incubated overnight at 55°C. A phenol-chloroform extraction was used to isolate DNA for use in quantitative PCR (qPCR). The DNA was amplified in triplicate in SYBR Green PCR Master Mix using 2 μl DNA template (20 ng) and 200 nM (final concentration) of forward and reverse primers in a total reaction volume of 20 μl. The thermal cycling conditions were: 50°C for 2 min, and 95°C for 10 min, followed by 40 cycles of amplification using the condition of 95°C for 15s, then 60°C for 1 min. All primer sequences are included in Table S7. The mtDNA/ncDNA ratio was calculated using the ΔΔct method: Δct = ct(Nd1 or 16s rRNA) – ct(Hk2), and ΔΔct = Δct (sample of interest) – Δct(average WT male group). Finally, expression for each sample was calculated as 2-ΔΔCt.
Flexor digitorum brevis (FDB) Fiber Isolation
Isolation of individual FDB fibers was achieved according to the protocol outlined by Keire et al. (2013).100 Briefly, the FDB was released from its origin at the calcaneus and from its insertion into the digits using sterile forceps. Visible connective tissue was removed and the muscle bundle was placed in 1 ml of digestion solution (400 U/ml collagenase type II, 10% fetal Bovine Serum, 1% pen-strep in DMEM supplemented with 4500 mg/L D-glucose, 110 mg/L sodium pyruvate, and 3700 mg/L sodium bicarbonate) in a 24-well well pre-coated in horse serum. The FDB was incubated in digestion solution for 3 hours in a 37°C, 5% CO2 incubator with constant agitation (swirling of the plate every 10 min) until individual myofibers began shedding from the edges of the muscle. To release individual myofibers, the FDB was triturated with a wide bore glass pipette in washing solution (DMEM, 10% FBS, 1% pen-strep) in 10-minute intervals. Individual myofibers were transitioned to Matrigel-coated (Corning Inc., Corning, NY, USA) Seahorse XFe24 (Agilent, Santa Clara, CA, USA) microplates for mitochondrial respiration using a small-bore glass pipette pre-coated in horse serum, or to Matrigel-coated chamber slides for use in sarcolemmal injury assays.
Mitochondrial Function
Bioenergetic analyses of intact single cultured muscle fibers were performed using an XFe24 Extracellular Flux Analyzer (Agilent) according to the protocol outlined in101 (n=21, n=5 WT M Con, n=7 WT M Ex, n=4 FLExDUX4 M Con, n=5 FLExDUX4 M Con). Briefly, myofibers were seeded in triplicate into XFe24 microplate wells coated in matrigel (diluted 1:1 in DMEM) to cover approximately 50-75% of the surface area of the well. The confluency of the myofibers was determined using the automated confluence function on the Personalized Automated Lab Assistant (PAULA, Leica Microsystems) Cell Imager. Myofibers were cultured overnight in growth media (DMEM supplemented with 20% FBS, 1% Pen-Strep). After 24 hours, growth media was exchanged for 675 uL Seahorse XF assay media. The XFe24 microplates were equilibrated for one hour in a non-CO2 incubator, during which the assay cartridge was loaded with the following substrates [Port, final concentration in well]: ATP synthase inhibitor oligomycin [Port A, 1 uM], mitochondrial uncoupler carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone FCCP [Port B, 800 nM], inhibitors of complexes III and I of the electron transport chain Antimycin and Rotenone A [Port C, 2 uM], and 50 uL assay media (Port D) as part of the XF Cell Mito Stress Test Assay (Agilent). Oxygen consumption rates (OCR, pmol/min) and extracellular acidification rates (ECAR, milli-pH/min) were recorded using 3-min mix, 2-min wait, and 3-min measure (looped 3 times) cycles under basal conditions before substrate administration, and following the titration of each substrate from Ports A-D. All respiration measures were normalized to FDB fiber confluence levels, and then the sample 16S rRNA mtDNA/ncDNA ratio to adjust for potential differences in fiber loading and mitochondrial content. The average of the three looped readings in each respiratory state were used for statistical analysis.
The Mito Stress Test Assay can be used to calculate key parameters of mitochondrial function, including: 1) Non-mitochondrial oxygen consumption = the minimum rate of OCR after Rotenone/Antimycin A injection; 2) Basal respiration = (last OCR measurement before oligomycin injection) – (non-mitochondrial OCR); 3) Maximal respiration = (maximal OCR After FCCP injection) - (non-mitochondrial OCR); 4) Proton Leak = (minimum OCR after FCCP injection) – (non-mitochondrial oxygen consumption); 5) ATP-Production Coupled Respiration = (Last rate measurement before oligomycin injection) – (minimum rate measurement after oligomycin injection); 6) Spare respiratory capacity = (Maximal Respiration) – (Basal Respiration); and 7) Coupling efficiency = (ATP-Production coupled respiration)/(Basal Respiration) x 100%.
Myofiber injury analysis
Following isolation, FDB myofibers were cultured overnight in Matrigel-coated No. 1.5 borosilicate glass bottom chamber slides in washing media to allow adherence prior to repair assessment. The next day, washing media was replaced with cell imaging media (CIM; Hanks Balanced Salts Solution with 10 mM HEPES, 2 mM Ca2+, pH 7.4) containing 1 mg/mL of the cell impermeant FM1-43 dye to the chamber. The slides were then placed in a Tokai Hit microscopy stage-top ZILCS incubator (Tokai Hit Co., Fujinomiya-shi, Japan) maintained at 37°C for laser injury. To induce sarcolemmal damage, a 1–2 mm2 area was irradiated for 10 ms with a pulsed laser (Ablate!, 3i Intelligent Imaging Innovations, Inc. Denver, CO, USA). Fibers were imaged using an inverted IX81 Olympus microscope (Olympus America, Center Valley, PA, USA) custom-equipped with a CSUX1 spinning disc confocal unit (Yokogawa Electric Corp., Tokyo, Japan). Images were acquired using Evolve 512 EMCCD (Photometrics, Tucson, AZ, USA) at 2 Hz. Image acquisition and laser injury was controlled using Slidebook 6.0 (Intelligent Imaging Innovations, Inc., Denver, CO, USA). The change in FM1-43 dye fluorescence intensity (ΔF/F0) during the course of the imaging (120 s) was averaged for each condition and plotted (n=19, n=5 WT M Con, n=5 WT M Ex, n=5 FLExDUX4 M Con, n=4 FLExDUX4 M Ex, 12-20 fibers per mouse). Successful repair was determined by the entry of FM1-43 dye into the cell interior, where a plateau in FM1-43 dye increase indicated successful repair, and failure to repair was indicated by unabated FM1-43 dye increase.
Gene Set Enrichment Analysis (GSEA)
Gene set enrichment analysis (GSEA v4.1.0) was performed on differentially expressed genes between groups to test for enrichment of specific gene ontologies (molecular function, biological process, and cell component).86 Normalized enrichment scores and false discovery adjusted q-values (q <0.25 unless otherwise stated) were used to identify significantly enriched ontologies within our gene sets. Leading edge analyses were used to identify core genes present in multiple enriched gene sets.86
Ingenuity Pathway Analysis (IPA)
Ingenuity Pathway Analysis (IPA, Qiagen GmbH, Hilden, Germany) was used identify enriched cell signaling pathways (canonical pathways) among DEG in our dataset (p value calculated using the right-tailed Fisher's Exact Test). Predicted Activation or inhibition of canonical pathways were expressed in z-scores, as calculated in Krämer et al.102 The Benjamini-Hochberg (B-H) method was used to control for multiple comparisons. IPA was also used to identify upstream regulators, regulator effects, and causal networks, which are defined in the Supplemental Methods. IPA identifies upstream regulators that are predicted to be activated or inhibited to explain the observed up-regulated and down-regulated genes in our DEG dataset. Activation z-scores, and Benjamini-Hochberg corrected p-values were used to identify significant upstream regulators. To elucidate the biological impact of these upstream regulators, we used IPA to generate regulator effects networks. Finally, we generated Causal Networks to create small hierarchical networks of regulators controlling the expression of the DEGs in our dataset– allowing an analysis of larger regulatory networks influencing our gene expression data through multiple levels (eg. regulators affecting other regulators further up the network). When assessing for changes in pathway activity between comparisons, we defined “reversed” pathways as those with a change in activation |z-score| toward zero by ≥ 0.3. We defined “reversed” pathways, as those with a change in the z-score sign from positive to negative, or vice versa.
Quantification and statistical analysis
Normality of all continuous outcomes was assessed using the Shapiro-Wilk test and confirmed visually using Q-Q plots. Three-way repeated measures ANOVAs were used to assess for the influence of genotype, training status, and sex on body weight and grip strength measures at baseline, 3 weeks, and 6 weeks. Likewise, three-way repeated measures ANOVAs were used to assess for the effect of sex, genotype, and time (baseline, 3 weeks, 6 weeks) on running outcomes (distance, percent of day running, velocity). Histological outcomes assessed at a single timepoint (triceps brachii muscle weight, central nuclei, fibrosis, fiber diameter, cross-sectional area, and fiber type composition) were analyzed using three-way ANOVAs with genotype, training status, and sex entered as variables. We used a two-way ANOVA with genotype and training status entered as between-subjects factors for measures of normalized oxygen consumption rate, as well as for measures of mitochondrial function, the mtDNA/ncDNA ratio, and percentage of myofibers that failed to repair. To measure differences in myofiber repair kinetics, we used a 3-way repeated measures ANOVA with genotype, training status, and (time) as factors. For all ANOVAs, significant interactions were explored with pairwise comparisons with the Benjamini-Hochberg procedure to control for alpha error inflation. For RNA sequencing, differential gene expression analysis was conducted using DESeq2 (version v1.18.1; https://bioconductor.org/packages/release/bioc/html/DESeq2.html).85 The analysis used the counts of unique alignments. Significance was determined at FDR-adjusted p<0.05 unless otherwise specified. Differences in expression of genes selected for validation we analyzed using t-tests between study groups. The Pearson correlation coefficient and the coefficient of determination was calculated between log2FC values from RNA sequencing and qRT-PCR to further validate RNAseq results. For IPA, we focused on highly significant upstream regulators, defined as those with activation |Z-score| >2.00 and an FDR p-value for enrichment of <0.10. Highly significant causal networks were identified as those having an activation |Z-score| >2.00 and an FDR p-value of overlap for enrichment of <0.10.
Acknowledgments
We thank Prech Uapinyoying for access to a custom python pipeline to analyze percentage of day running vs. resting. This research was supported by the National Institutes of Health T32 AR056993-06 (Partridge) and 1R21HD103993-01 (Chen), as well as the Muscular Dystrophy Association—Strength, Science, and Stories of Inspiration Fellowship (A. Bittel), FSHD Canada (Chen), an FSHD Society Grant Award (A. Bittel), and the American Physical Therapy Association New Investigator Fellowship Training Initiative (A. Bittel).
Author contributions
A.J.B. and Y.W.C. conceived and designed the study; A.J.B. and D.C.B. conducted the experiments; A.J.B., D.C.B., Y.W.C., and H.G.D. analyzed and interpreted the data; A.J.B. drafted the manuscript, which was edited by all authors; A.J.B. and Y.W.C. obtained funding for the study.
Declaration of interests
The authors declare no competing interests.
Published: December 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108632.
Contributor Information
Adam J. Bittel, Email: abittel@childrensnational.org.
Yi-Wen Chen, Email: ychen@childrensnational.org.
Supplemental information
Table contains all identified genes from RNAseq analysis, and the DEseq2 summary outputs. BaseMean = mean of normalized counts of all samples, normalizing for sequencing depth; lfcSE = standard error estimate for the log2 fold change; stat = wald statistic; p-value = unadjusted p-value; padj = B-H adjusted p-value.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Con and WT Con Groups. Table 2A. Positively enriched gene sets (GOMF = gene ontology molecular function, GOCC = gene ontology cell component, GOBP = gene ontology biological process) among DEG, including the number of genes in the study dataset present in the identified gene set, net enrichment score, nominal p-value of enrichment, and FDR p-value of enrichment. Table 2B. Negatively enriched gene sets among DEG. Table 2C. Leading Edge analysis among the positively enriched gene sets. Table 2D. Leading Edge analysis among the negatively enriched gene sets. Table 2E. Canonical pathways from IPA analysis, including the -log(B-H adjusted p-value), percentage of pathway members present in our dataset, and z-score of predicted activity. Table 2F. IPA upstream regulators of the transcriptional differences between groups, including the type of molecule, its predicted activation or inhibition, the B-H adjusted p-value of regulator overlap (based on the number of downstream targets of the identified regulator in the DEG dataset), and the genes regulated in the DEG dataset. Table 2G. IPA regulator effects, identifying how upstream regulators connect to DEG target molecules, and downstream functions or diseases. This includes the list of regulators, number of targets from the DEG dataset interacting with the regulators, the target genes in the regulator network, linked diseases or functions, and the proportion of regulator-disease/function relationships present in the IPA Knowledge Base. Table 2H. IPA Causal Networks, which constitute small hierarchical networks of regulators that control the expression of the dataset targets. This includes identified master regulators, downstream regulator targets, predicted activation of the master regulator, significance of overlap between network genes and genes in the DEG dataset, and a list of the target genes in the DEG dataset.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between WT Ex vs. WT Con Groups. Data in Tables S3A-S3H are as described in Tables S2A–SH.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Ex vs. WT Con Groups. Data in Tables S4A–S4H are as described in Tables S2A–S2H. Table 4I. Canonical pathways enriched (FDR p < 0.05) in the FLExDUX4 Con vs. WT Con and FLExDUX4 Ex vs. WT Con comparisons with a z-score. The pattern of change in the activation z-score is also indicated. Partially reversed = z-score change toward zero (by at least |z = 0.3|); Increased = upregulated in FLExDUX4 Con vs. WT Con and z-score increased in FLExDUX4 Ex vs. WT Con; Decreased = downregulated in FLExDUX4 Con vs. WT Con and z-score decreased in FLExDUX4 Ex vs. WT Con; Reversed = Z-score changed from positive in FLExDUX4 Con vs. WT Con comparison to negative in the FLExDUX4 Ex vs. WT Con comparison, or vice versa; No Change = change in z-score did not exceed 0.3 in any direction. Table 4J. List of DEG, including their log2(fold change) and p-value, between FLExDUX4 Con vs. WT Con groups (p < 0.05) that are not differentially expressed between FLExDUX4 Ex vs. WT Con groups.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Ex vs. FLExDUX4 Con Groups. Data in Tables S5A–S5H are as described in Tables S2A–S2H.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Ex vs. WT Ex Groups. Data in Tables S6A–S6H are as described in Tables S2A–S2H.
References
- 1.Hanel M.L., Sun C.-Y.J., Jones T.I., Long S.W., Zanotti S., Milner D., Jones P.L. Facioscapulohumeral muscular dystrophy (FSHD) region gene 1 (FRG1) is a dynamic nuclear and sarcomeric protein. Differentiation. 2011;81:107–118. doi: 10.1016/j.diff.2010.09.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hewitt J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy: Figure 1. Hum Mol Gen. 2015;24:R17–R23. doi: 10.1093/hmg/ddv237. [DOI] [PubMed] [Google Scholar]
- 3.Statland J.M., Tawil R. Facioscapulohumeral Muscular Dystrophy. Continuum. 2016;22:1916–1931. doi: 10.1212/CON.0000000000000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dixit M., Ansseau E., Tassin A., Winokur S., Shi R., Qian H., Sauvage S., Mattéotti C., van Acker A.M., Leo O., et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemmers R.J.L.F., de Kievit P., Sandkuijl L., Padberg G.W., van Ommen G.-J.B., Frants R.R., van der Maarel S.M. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 2002;32:235–236. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 6.Lemmers R.J.L.F., van der Vliet P.J., Klooster R., Sacconi S., Camaño P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W., et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wijmenga C., Hewitt J.E., Sandkuijl L.A., Clark L.N., Wright T.J., Dauwerse H.G., Gruter A.-M., Hofker M.H., Moerer P., Williamson R., et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 8.Lemmers R.J.L.F., Tawil R., Petek L.M., Balog J., Block G.J., Santen G.W.E., Amell A.M., van der Vliet P.J., Almomani R., Straasheijm K.R., et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Boogaard M.L., Lemmers R.J.L.F., Balog J., Wohlgemuth M., Auranen M., Mitsuhashi S., van der Vliet P.J., Straasheijm K.R., van den Akker R.F.P., Kriek M., et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016;98:1020–1029. doi: 10.1016/j.ajhg.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamanaka K., Sikrova D., Mitsuhashi S., Masuda H., Sekiguchi Y., Sugiyama A., Shibuya K., Lemmers R.J.L.F, Goossens R., Ogawa M., et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology. 2020;94:e2441–e2447. doi: 10.1212/WNL.0000000000009617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vuoristo S., Bhagat S., Hydén-Granskog C., Yoshihara M., Gawriyski L., Jouhilahti E.-M., Ranga V., Tamirat M., Huhtala M., Kirjanov I., et al. DUX4 is a multifunctional factor priming human embryonic genome activation. iScience. 2022;25 doi: 10.1016/j.isci.2022.104137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bittel A.J., Sreetama S.C., Bittel D.C., Horn A., Novak J.S., Yokota T., Zhang A., Maruyama R., Rowel Q Lim K., Jaiswal J.K., Chen Y.W. Membrane Repair Deficit in Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020;21:5575. doi: 10.3390/ijms21155575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heher P., Ganassi M., Weidinger A., Engquist E.N., Pruller J., Nguyen T.H., Tassin A., Declèves A.E., Mamchaoui K., Banerji C.R.S., et al. Interplay between mitochondrial reactive oxygen species, oxidative stress and hypoxic adaptation in facioscapulohumeral muscular dystrophy: Metabolic stress as potential therapeutic target. Redox Biol. 2022;51 doi: 10.1016/j.redox.2022.102251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen B.H., Voet N.B.M., Nabuurs C.I., Kan H.E., de Rooy J.W.J., Geurts A.C., Padberg G.W., van Engelen B.G.M., Heerschap A. Distinct disease phases in muscles of Facioscapulohumeral Dystrophy patients identified by MR detected fat infiltration. PLoS One. 2014;9 doi: 10.1371/journal.pone.0085416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersen S.P., Sveen M.-L., Hansen R.S., Madsen K.L., Hansen J.B., Madsen M., Vissing J. Creatine kinase response to high-intensity aerobic exercise in adult-onset muscular dystrophy: CK Response to Exercise in BMD. Muscle Nerve. 2013;48:897–901. doi: 10.1002/mus.23846. [DOI] [PubMed] [Google Scholar]
- 16.Bankolé L.C., Millet G.Y., Temesi J., Bachasson D., Ravelojaona M., Wuyam B., Verges S., Ponsot E., Antoine J.-C., Kadi F., Féasson L. Safety and efficacy of a 6-month home-based exercise program in patients with facioscapulohumeral muscular dystrophy: A randomized controlled trial. Medicine. 2016;95:e4497. doi: 10.1097/MD.0000000000004497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voet N.B., van der Kooi E.L., van Engelen B.G., Geurts A.C. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst. Rev. 2019;12:CD003907. doi: 10.1002/14651858.CD003907.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersen G., Prahm K.P., Dahlqvist J.R., Citirak G., Vissing J. Aerobic training and postexercise protein in facioscapulohumeral muscular dystrophy: RCT study. Neurology. 2015;85:396–403. doi: 10.1212/WNL.0000000000001808. [DOI] [PubMed] [Google Scholar]
- 19.Andersen G., Heje K., Buch A.E., Vissing J. High-intensity interval training in facioscapulohumeral muscular dystrophy type 1: A randomized clinical trial. J. Neurol. 2017;264:1099–1106. doi: 10.1007/s00415-017-8497-9. [DOI] [PubMed] [Google Scholar]
- 20.Janssen B., Voet N., Geurts A., van Engelen B., Heerschap A. Quantitative MRI reveals decelerated fatty infiltration in muscles of active FSHD patients. Neurology. 2016;86:1700–1707. doi: 10.1212/WNL.0000000000002640. [DOI] [PubMed] [Google Scholar]
- 21.Olsen D.B., Ørngreen M.C., Vissing J. Aerobic training improves exercise performance in facioscapulohumeral muscular dystrophy. Neurology. 2005;64:1064–1066. doi: 10.1212/01.WNL.0000150584.45055.27. [DOI] [PubMed] [Google Scholar]
- 22.Voet N.B.M., Bleijenberg G., Padberg G.W., van Engelen B.G.M., Geurts A.C.H. Effect of aerobic exercise training and cognitive behavioural therapy on reduction of chronic fatigue in patients with facioscapulohumeral dystrophy: Protocol of the FACTS-2-FSHD trial. BMC Neurol. 2010;10:56. doi: 10.1186/1471-2377-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voet N., Bleijenberg G., Hendriks J., De Groot I., Padberg G., Van Engelen B., Geurts A. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in FSHD: An RCT. Neurology. 2014;83:1914–1922. doi: 10.1212/WNL.0000000000001008. [DOI] [PubMed] [Google Scholar]
- 24.Bostock E.L., O’Dowd D.N., Payton C.J., Smith D., Orme P., Edwards B.T., Morse C.I. The effects of resistance exercise training on strength and functional tasks in adults with Limb-Girdle, Becker, and Facioscapulohumeral Dystrophies. Front. Neurol. 2019;10:1216. doi: 10.3389/fneur.2019.01216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colson S.S., Benchortane M., Tanant V., Faghan J.-P., Fournier-Mehouas M., Benaïm C., Desnuelle C., Sacconi S. Neuromuscular Electrical Stimulation Training: A Safe and Effective Treatment for Facioscapulohumeral Muscular Dystrophy Patients. Arch. Phys. Med. Rehabil. 2010;91:697–702. doi: 10.1016/j.apmr.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 26.van der Kooi E.L., Vogels O.J.M., van Asseldonk R.J.G.P., Lindeman E., Hendriks J.C.M., Wohlgemuth M., van der Maarel S.M., Padberg G.W. Strength training and albuterol in facioscapulohumeral muscular dystrophy. Neurology. 2004;63:702–708. doi: 10.1212/01.wnl.0000134660.30793.1f. [DOI] [PubMed] [Google Scholar]
- 27.Schefer V., Talan M.I. Oxygen consumption in adult and aged C57BL/6J mice during acute treadmill exercise of different intensity. Exp. Gerontol. 1996;31:387–392. doi: 10.1016/0531-5565(95)02032-2. [DOI] [PubMed] [Google Scholar]
- 28.American College of Sports Medicine, Liguori G., Feito Y., Fountaine C., Roy B., editors. ACSM’s guidelines for exercise testing and prescription. Eleventh edition. Wolters Kluwer; 2021. [Google Scholar]
- 29.Jones T., Jones P.L. A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS One. 2018;13 doi: 10.1371/journal.pone.0192657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones T.I., Chew G.-L., Barraza-Flores P., Schreier S., Ramirez M., Wuebbles R.D., Burkin D.J., Bradley R.K., Jones P.L. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle. 2020;10:8. doi: 10.1186/s13395-020-00227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rijken N.H.M., Van Der Kooi E.L., Hendriks J.C.M., Van Asseldonk R.J.G.P., Padberg G.W., Geurts A.C.H., Van Engelen B.G.M. Skeletal muscle imaging in facioscapulohumeral muscular dystrophy, pattern and asymmetry of individual muscle involvement. Neuromuscul. Disord. 2014;24:1087–1096. doi: 10.1016/j.nmd.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 32.Schumann N.P., Biedermann F.H.W., Kleine B.U., Stegeman D.F., Roeleveld K., Hackert R., Scholle H.C. Multi-channel EMG of the M. triceps brachii in rats during treadmill locomotion. Clin. Neurophysiol. 2002;113:1142–1151. doi: 10.1016/s1388-2457(02)00143-8. [DOI] [PubMed] [Google Scholar]
- 33.Lee S.-H., Kim B.-J., Park D.-R., Kim U.-H. Exercise induces muscle fiber type switching via transient receptor potential melastatin 2-dependent Ca 2+ signaling. J. Appl. Physiol. 2018;124:364–373. doi: 10.1152/japplphysiol.00687.2017. [DOI] [PubMed] [Google Scholar]
- 34.Zhang M.Y., Zhang W.J., Medler S. The continuum of hybrid IIX/IIB fibers in normal mouse muscles: MHC isoform proportions and spatial distribution within single fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;299:R1582–R1591. doi: 10.1152/ajpregu.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerquone Perpetuini A., Re Cecconi A.D., Chiappa M., Martinelli G.B., Fuoco C., Desiderio G., Castagnoli L., Gargioli C., Piccirillo R., Cesareni G. Group I Paks support muscle regeneration and counteract cancer-associated muscle atrophy: Group I Paks support muscle regeneration and counteract cancer-associated muscle atrophy. J. Cachexia Sarcopenia Muscle. 2018;9:727–746. doi: 10.1002/jcsm.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu R., Krüger K., Pilat C., Fan W., Xiao Y., Seimetz M., Ringseis R., Baumgart-Vogt E., Eder K., Weissmann N., Mooren F.C. Excessive accumulation of intracellular ca2+ after acute exercise potentiated impairment of t-cell function. Front. Physiol. 2021;12 doi: 10.3389/fphys.2021.728625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Wu H., Zelenin P., Fontanet P., Wanderoy S., Petitpré C., Comai G., Bellardita C., Xue-Franzén Y., Huettl R.-E., et al. Muscle-selective RUNX3 Dependence of Sensorimotor Circuit Development. Development. 2019;146 doi: 10.1242/dev.181750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agilent Technologies Measuring Mitochondrial Respiration in Intact Skeletal Muscle Fibers with an Agilent Seahorse XF24/XFe24 Analyzer. 2016. https://www.agilent.com/cs/library/applications/5991-7148EN.pdf
- 39.Sakellariou P., Kekou K., Fryssira H., Sofocleous C., Manta P., Panousopoulou A., Gounaris K., Kanavakis E. Mutation spectrum and phenotypic manifestation in FSHD Greek patients. Neuromuscul. Disord. 2012;22:339–349. doi: 10.1016/j.nmd.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Tonini M.M.O., Passos-Bueno M.R., Cerqueira A., Matioli S.R., Pavanello R., Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD) Neuromuscul. Disord. 2004;14:33–38. doi: 10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Zatz M., Marie S.K., Cerqueira A., Vainzof M., Pavanello R.C., Passos-Bueno M.R. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am. J. Med. Genet. 1998;77:155–161. [PubMed] [Google Scholar]
- 42.Rahimov F., King O.D., Leung D.G., Bibat G.M., Emerson C.P., Kunkel L.M., Wagner K.R. Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkers. Proc. Natl. Acad. Sci. USA. 2012;109:16234–16239. doi: 10.1073/pnas.1209508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laberthonnière C., Novoa-del-Toro E., Delourme M., Chevalier R., Broucqsault N., Mazaleyrat K., Streichenberger N., Manel V., Bernard R., Salort Campana E., et al. Facioscapulohumeral dystrophy weakened sarcomeric contractility is mimicked in induced pluripotent stem cells-derived innervated muscle fibres. J. Cachexia Sarcopenia Muscle. 2022;13:621–635. doi: 10.1002/jcsm.12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tassin A., Leroy B., Laoudj-Chenivesse D., Wauters A., Vanderplanck C., Le Bihan M.-C., Coppée F., Wattiez R., Belayew A. FSHD myotubes with different phenotypes exhibit distinct proteomes. PLoS One. 2012;7 doi: 10.1371/journal.pone.0051865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lassche S., Stienen G.J.M., Irving T.C., van der Maarel S.M., Voermans N.C., Padberg G.W., Granzier H., van Engelen B.G.M., Ottenheijm C.A.C. Sarcomeric dysfunction contributes to muscle weakness in facioscapulohumeral muscular dystrophy. Neurology. 2013;80:733–737. doi: 10.1212/WNL.0b013e318282513b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turki A., Hayot M., Carnac G., Pillard F., Passerieux E., Bommart S., Raynaud de Mauverger E., Hugon G., Pincemail J., Pietri S., et al. Functional muscle impairment in facioscapulohumeral muscular dystrophy is correlated with oxidative stress and mitochondrial dysfunction. Free Radic. Biol. Med. 2012;53:1068–1079. doi: 10.1016/j.freeradbiomed.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 47.Wilson V.D., Thomas C., Passerieux E., Hugon G., Pillard F., Andrade A.G., Bommart S., Picot M., Pincemail J., Mercier J., et al. Impaired oxygen demand during exercise is related to oxidative stress and muscle function in Facioscapulohumeral Muscular Dystrophy. JCSM Rapid Commun. 2018;1:1–13. [Google Scholar]
- 48.Vera K.A., Mcconville M., Glazos A., Stokes W., Kyba M., Keller-Ross M. Exercise intolerance in facioscapulohumeral muscular dystrophy. Med. Sci. Sports Exerc. 2022;54:887–895. doi: 10.1249/MSS.0000000000002882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andersen J.L., Klitgaard H., Saltin B. Myosin heavy chain isoforms in single fibres from m. vastus lateralis of sprinters: Influence of training. Acta Physiol. Scand. 1994;151:135–142. doi: 10.1111/j.1748-1716.1994.tb09730.x. [DOI] [PubMed] [Google Scholar]
- 50.Jansson E., Sjödin B., Tesch P. Changes in muscle fibre type distribution in man after physical training. A sign of fibre type transformation? Acta Physiol. Scand. 1978;104:235–237. doi: 10.1111/j.1748-1716.1978.tb06272.x. [DOI] [PubMed] [Google Scholar]
- 51.Eggers B., Schork K., Turewicz M., Barkovits K., Eisenacher M., Schröder R., Clemen C.S., Marcus K. Advanced fiber type-specific protein profiles derived from adult murine skeletal muscle. Proteomes. 2021;9:28. doi: 10.3390/proteomes9020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baar K., Wende A.R., Jones T.E., Marison M., Nolte L.A., Chen M., Kelly D.P., Holloszy J.O. Adaptations of skeletal muscle to exercise: Rapid increase in the transcriptional coactivator PGC-1. Faseb. J. 2002;16:1879–1886. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 53.Liang H., Ward W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006;30:145–151. doi: 10.1152/advan.00052.2006. [DOI] [PubMed] [Google Scholar]
- 54.Banerji C.R.S., Panamarova M., Pruller J., Figeac N., Hebaishi H., Fidanis E., Saxena A., Contet J., Sacconi S., Severini S., Zammit P.S. Dynamic transcriptomic analysis reveals suppression of PGC1α/ERRα drives perturbed myogenesis in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2019;28:1244–1259. doi: 10.1093/hmg/ddy405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laker R.C., Drake J.C., Wilson R.J., Lira V.A., Lewellen B.M., Ryall K.A., Fisher C.C., Zhang M., Saucerman J.J., Goodyear L.J., et al. AMPK phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017;8:548. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iqbal S., Ostojic O., Singh K., Joseph A.-M., Hood D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse: Mitochondrial fission and fusion in skeletal muscle. Muscle Nerve. 2013;48:963–970. doi: 10.1002/mus.23838. [DOI] [PubMed] [Google Scholar]
- 57.Nielsen J., Gejl K.D., Hey-Mogensen M., Holmberg H.-C., Suetta C., Krustrup P., Elemans C.P.H., Ørtenblad N. Plasticity in mitochondrial cristae density allows metabolic capacity modulation in human skeletal muscle: Enlarged mitochondrial cristae density in athletes. J. Physiol. 2017;595:2839–2847. doi: 10.1113/JP273040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mendham A.E., Goedecke J.H., Zeng Y., Larsen S., George C., Hauksson J., Fortuin-de Smidt M.C., Chibalin A.V., Olsson T., Chorell E. Exercise training improves mitochondrial respiration and is associated with an altered intramuscular phospholipid signature in women with obesity. Diabetologia. 2021;64:1642–1659. doi: 10.1007/s00125-021-05430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huffman K.M., Koves T.R., Hubal M.J., Abouassi H., Beri N., Bateman L.A., Stevens R.D., Ilkayeva O.R., Hoffman E.P., Muoio D.M., Kraus W.E. Metabolite signatures of exercise training in human skeletal muscle relate to mitochondrial remodelling and cardiometabolic fitness. Diabetologia. 2014;57:2282–2295. doi: 10.1007/s00125-014-3343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bosnakovski D., Shams A.S., Yuan C., da Silva M.T., Ener E.T., Baumann C.W., Lindsay A.J., Verma M., Asakura A., Lowe D.A., Kyba M. Transcriptional and cytopathological hallmarks of FSHD in chronic DUX4-expressing mice. J. Clin. Invest. 2020;130:2465–2477. doi: 10.1172/JCI133303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hibino S., Takeda A., Nishino I., Iwata N., Nakano M., Tanaka K., Yamakawa S., Nagai T., Uemura O. Severe glomerular endothelial injury associated with a short d4z4 repeat on chromosome 4q35. Intern. Med. 2017;56:1849–1853. doi: 10.2169/internalmedicine.56.7441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Statland J.M., Odrzywolski K.J., Shah B., Henderson D., Fricke A.F., van der Maarel S.M., Tapscott S.J., Tawil R. Immunohistochemical characterization of Facioscapulohumeral Muscular Dystrophy muscle biopsies. J. Neuromuscul. Dis. 2015;2:291–299. doi: 10.3233/JND-150077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banerji C.R.S., Knopp P., Moyle L.A., Severini S., Orrell R.W., Teschendorff A.E., Zammit P.S. β - catenin is central to DUX4 -driven network rewiring in facioscapulohumeral muscular dystrophy. J. R. Soc. Interface. 2015;12 doi: 10.1098/rsif.2014.0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mason S.D., Rundqvist H., Papandreou I., Duh R., McNulty W.J., Howlett R.A., Olfert I.M., Sundberg C.J., Denko N.C., Poellinger L., Johnson R.S. HIF-1α in endurance training: Suppression of oxidative metabolism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R2059–R2069. doi: 10.1152/ajpregu.00335.2007. [DOI] [PubMed] [Google Scholar]
- 65.Lek A., Zhang Y., Woodman K.G., Huang S., DeSimone A.M., Cohen J., Ho V., Conner J., Mead L., Kodani A., et al. Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aay0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arahata K., Ishihara T., Fukunaga H., Orimo S., Lee J.H., Goto K., Nonaka I. Inflammatory Response in Facioscapulohumeral Muscular Dystrophy (FSHD): Immunocytochemical and Genetic Analyses. Muscle Nerve Suppl. 1995:S56–S66. [PubMed] [Google Scholar]
- 67.Bosnakovski D., Chan S.S.K., Recht O.O., Hartweck L.M., Gustafson C.J., Athman L.L., Lowe D.A., Kyba M. Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun. 2017;8:550. doi: 10.1038/s41467-017-00730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kan H.E., Klomp D.W.J., Wohlgemuth M., van Loosbroek-Wagemans I., van Engelen B.G.M., Padberg G.W., Heerschap A. Only fat infiltrated muscles in resting lower leg of FSHD patients show disturbed energy metabolism. NMR Biomed. 2010;23:563–568. doi: 10.1002/nbm.1494. [DOI] [PubMed] [Google Scholar]
- 69.Banerji C.R.S., Zammit P.S. Pathomechanisms and biomarkers in facioscapulohumeral muscular dystrophy: Roles of DUX4 and PAX7. EMBO Mol. Med. 2021;13 doi: 10.15252/emmm.202013695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paleo B.J., McElhanon K.E., Bulgart H.R., Banford K.K., Beck E.X., Sattler K.M., Goines B.N., Ratcliff S.L., Crowe K.E., Weisleder N. Reduced sarcolemmal membrane repair exacerbates striated muscle pathology in a mouse model of Duchenne Muscular Dystrophy. Cells. 2022;11:1417. doi: 10.3390/cells11091417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klingler W., Jurkat-Rott K., Lehmann-Horn F., Schleip R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012;31:184–195. [PMC free article] [PubMed] [Google Scholar]
- 72.Madaro L., Passafaro M., Sala D., Etxaniz U., Lugarini F., Proietti D., Alfonsi M.V., Nicoletti C., Gatto S., De Bardi M., et al. Denervation-activated STAT3–IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat. Cell Biol. 2018;20:917–927. doi: 10.1038/s41556-018-0151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amelink G.J., Koot R.W., Erich W.B., Van Gijn J., Bär P.R. Sex-linked variation in creatine kinase release, and its dependence on oestradiol, can be demonstrated in an in-vitro rat skeletal muscle preparation. Acta Physiol. Scand. 1990;138:115–124. doi: 10.1111/j.1748-1716.1990.tb08823.x. [DOI] [PubMed] [Google Scholar]
- 74.McClung J.M., Davis J.M., Wilson M.A., Goldsmith E.C., Carson J.A. Estrogen status and skeletal muscle recovery from disuse atrophy. J. Appl. Physiol. 2006;100:2012–2023. doi: 10.1152/japplphysiol.01583.2005. [DOI] [PubMed] [Google Scholar]
- 75.Bittel D.C., Sreetama S.C., Chandra G., Ziegler R., Nagaraju K., Van der Meulen J.H., Jaiswal J.K. Secreted acid sphingomyelinase as a potential gene therapy for limb girdle muscular dystrophy 2B. J. Clin. Invest. 2022;132 doi: 10.1172/JCI141295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horn A., Van der Meulen J.H., Defour A., Hogarth M., Sreetama S.C., Reed A., Scheffer L., Chandel N.S., Jaiswal J.K. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aaj1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vila M.C., Rayavarapu S., Hogarth M.W., Van der Meulen J.H., Horn A., Defour A., Takeda S., Brown K.J., Hathout Y., Nagaraju K., Jaiswal J.K. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017;24:330–342. doi: 10.1038/cdd.2016.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan M.C., Rowe G.C., Raghuram S., Patten I.S., Farrell C., Arany Z. Post-natal induction of PGC-1α protects against severe muscle dystrophy independently of utrophin. Skelet. Muscle. 2014;4:2. doi: 10.1186/2044-5040-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuang J., McGinley C., Lee M.J.-C., Saner N.J., Garnham A., Bishop D.J. Interpretation of exercise-induced changes in human skeletal muscle mRNA expression depends on the timing of the post-exercise biopsies. PeerJ. 2022;10 doi: 10.7717/peerj.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manzanares G., Brito-da-Silva G., Gandra P.G. Voluntary wheel running: Patterns and physiological effects in mice. Braz. J. Med. Biol. Res. 2019;52 doi: 10.1590/1414-431X20187830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martin M. 2011. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. [Google Scholar]
- 82.Andrews S. FastQC [1/10/2018] https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 83.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao Y., Smyth G.K., Shi W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Laghi V., Ricci V., De Santa F., Torcinaro A. A user-friendly approach for routine histopathological and morphometric analysis of skeletal muscle using cellprofiler software. Diagnostics. 2022;12:561. doi: 10.3390/diagnostics12030561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Uapinyoying P. Parse-Running-Wheel. 2018. https://zenodo.org/records/10210700
- 89.Sonaimuthu P., Senkevitch E., Haskins N., Uapinyoying P., McNutt M., Morizono H., Tuchman M., Caldovic L. Gene delivery corrects N-acetylglutamate synthase deficiency and enables insights in the physiological impact of L-arginine activation of N-acetylglutamate synthase. Sci. Rep. 2021;11:3580. doi: 10.1038/s41598-021-82994-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vigelsø A., Gram M., Wiuff C., Andersen J.L., Helge J.W., Dela F. Six weeks’ aerobic retraining after two weeks’ immobilization restores leg lean mass and aerobic capacity but does not fully rehabilitate leg strength in young and older men. J. Rehabil. Med. 2015;47:552–560. doi: 10.2340/16501977-1961. [DOI] [PubMed] [Google Scholar]
- 91.Pettersson S., Edin F., Hjelte C., Scheinost D., Wagner S., Ekblom B., Jessen N., Madsen K., Andersson-Hall U. Six weeks of aerobic exercise in untrained men with overweight/obesity improved training adaptations, performance and body composition independent of oat/potato or milk based protein-carbohydrate drink supplementation. Front. Nutr. 2021;8 doi: 10.3389/fnut.2021.617344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wiklund P., Alen M., Munukka E., Cheng S.M., Yu B., Pekkala S., Cheng S. Metabolic response to 6-week aerobic exercise training and dieting in previously sedentary overweight and obese pre-menopausal women: A randomized trial. J. Sport Health Sci. 2014;3:217–224. [Google Scholar]
- 93.Van Der Stede T., Blancquaert L., Stassen F., Everaert I., Van Thienen R., Vervaet C., Gliemann L., Hellsten Y., Derave W. Histamine H 1 and H 2 receptors are essential transducers of the integrative exercise training response in humans. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abf2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perry C.G., Heigenhauser G.J., Bonen A., Spriet L.L. High-intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl. Physiol. Nutr. Metab. 2008;33:1112–1123. doi: 10.1139/H08-097. [DOI] [PubMed] [Google Scholar]
- 95.De Luca A. Treat-NMD; 2019. Use of Grip Strength Meter to Assess the Limb Strength of Mdx Mice.https://treat-nmd.org/sop/dmd_m-2-2-001/ [Google Scholar]
- 96.Novak J.S., Spathis R., Dang U.J., Fiorillo A.A., Hindupur R., Tully C.B., Mázala D.A.G., Canessa E., Brown K.J., Partridge T.A., et al. Interrogation of Dystrophin and Dystroglycan Complex Protein Turnover After Exon Skipping Therapy. J. Neuromuscul. Dis. 2021;8:S383–S402. doi: 10.3233/JND-210696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruegg M.A. Treat-NMD; 2016. Histopathology in Hematoxylin & Eosin Stained Muscle Sections.https://treat-nmd.org/sop/mdc1a_m-1-2-004/ [Google Scholar]
- 98.Murach K.A., Dungan C.M., Kosmac K., Voigt T.B., Tourville T.W., Miller M.S., Bamman M.M., Peterson C.A., Toth M.J. Fiber typing human skeletal muscle with fluorescent immunohistochemistry. J. Appl. Physiol. 2019;127:1632–1639. doi: 10.1152/japplphysiol.00624.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Quiros P.M., Goyal A., Jha P., Auwerx J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017;7:47–54. doi: 10.1002/cpmo.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Keire P., Shearer A., Shefer G., Yablonka-Reuveni Z. In: Helgason C.D., Miller C.L., editors. Vol. 946. Humana Press; 2013. Isolation and Culture of Skeletal Muscle Myofibers as a Means to Analyze Satellite Cells; pp. 431–468. (Basic Cell Culture Protocols). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schuh R.A., Jackson K.C., Khairallah R.J., Ward C.W., Spangenburg E.E. Measuring mitochondrial respiration in intact single muscle fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012;302:R712–R719. doi: 10.1152/ajpregu.00229.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table contains all identified genes from RNAseq analysis, and the DEseq2 summary outputs. BaseMean = mean of normalized counts of all samples, normalizing for sequencing depth; lfcSE = standard error estimate for the log2 fold change; stat = wald statistic; p-value = unadjusted p-value; padj = B-H adjusted p-value.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Con and WT Con Groups. Table 2A. Positively enriched gene sets (GOMF = gene ontology molecular function, GOCC = gene ontology cell component, GOBP = gene ontology biological process) among DEG, including the number of genes in the study dataset present in the identified gene set, net enrichment score, nominal p-value of enrichment, and FDR p-value of enrichment. Table 2B. Negatively enriched gene sets among DEG. Table 2C. Leading Edge analysis among the positively enriched gene sets. Table 2D. Leading Edge analysis among the negatively enriched gene sets. Table 2E. Canonical pathways from IPA analysis, including the -log(B-H adjusted p-value), percentage of pathway members present in our dataset, and z-score of predicted activity. Table 2F. IPA upstream regulators of the transcriptional differences between groups, including the type of molecule, its predicted activation or inhibition, the B-H adjusted p-value of regulator overlap (based on the number of downstream targets of the identified regulator in the DEG dataset), and the genes regulated in the DEG dataset. Table 2G. IPA regulator effects, identifying how upstream regulators connect to DEG target molecules, and downstream functions or diseases. This includes the list of regulators, number of targets from the DEG dataset interacting with the regulators, the target genes in the regulator network, linked diseases or functions, and the proportion of regulator-disease/function relationships present in the IPA Knowledge Base. Table 2H. IPA Causal Networks, which constitute small hierarchical networks of regulators that control the expression of the dataset targets. This includes identified master regulators, downstream regulator targets, predicted activation of the master regulator, significance of overlap between network genes and genes in the DEG dataset, and a list of the target genes in the DEG dataset.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between WT Ex vs. WT Con Groups. Data in Tables S3A-S3H are as described in Tables S2A–SH.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Ex vs. WT Con Groups. Data in Tables S4A–S4H are as described in Tables S2A–S2H. Table 4I. Canonical pathways enriched (FDR p < 0.05) in the FLExDUX4 Con vs. WT Con and FLExDUX4 Ex vs. WT Con comparisons with a z-score. The pattern of change in the activation z-score is also indicated. Partially reversed = z-score change toward zero (by at least |z = 0.3|); Increased = upregulated in FLExDUX4 Con vs. WT Con and z-score increased in FLExDUX4 Ex vs. WT Con; Decreased = downregulated in FLExDUX4 Con vs. WT Con and z-score decreased in FLExDUX4 Ex vs. WT Con; Reversed = Z-score changed from positive in FLExDUX4 Con vs. WT Con comparison to negative in the FLExDUX4 Ex vs. WT Con comparison, or vice versa; No Change = change in z-score did not exceed 0.3 in any direction. Table 4J. List of DEG, including their log2(fold change) and p-value, between FLExDUX4 Con vs. WT Con groups (p < 0.05) that are not differentially expressed between FLExDUX4 Ex vs. WT Con groups.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Ex vs. FLExDUX4 Con Groups. Data in Tables S5A–S5H are as described in Tables S2A–S2H.
Results from Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA) of DEG between FLExDUX4 Ex vs. WT Ex Groups. Data in Tables S6A–S6H are as described in Tables S2A–S2H.
Data Availability Statement
-
•
Data: RNA-seq data have been deposited to the NCBI Gene Expression Omnibus (GEO) database, and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All running performance, grip strength and body weight, histological, respirometry, mitochondrial DNA content, and sarcolemmal repair data will be shared by the lead contact upon request
-
•
Code: All original code has been deposited at Zenodo: https://zenodo.org/doi/10.5281/zenodo.10210699 88 and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request