

Transcriptional regulation of IRF4-MYC feed-forward super-enhancer is the key to IMiD antimyeloma activity, rational therapy combinations, and mechanisms of resistance.
Abstract
Multiple myeloma (MM) is a malignancy that is often driven by MYC and that is sustained by IRF4, which are upregulated by super-enhancers. IKZF1 and IKZF3 bind to super-enhancers and can be degraded using immunomodulatory imide drugs (IMiD). Successful IMiD responses downregulate MYC and IRF4; however, this fails in IMiD-resistant cells. MYC and IRF4 downregulation can also be achieved in IMiD-resistant tumors using inhibitors of BET and EP300 transcriptional coactivator proteins; however, in vivo these drugs have a narrow therapeutic window. By combining IMiDs with EP300 inhibition, we demonstrate greater downregulation of MYC and IRF4, synergistic killing of myeloma in vitro and in vivo, and an increased therapeutic window. Interestingly, this potent combination failed where MYC and IRF4 expression was maintained by high levels of the AP-1 factor BATF. Our results identify an effective drug combination and a previously unrecognized mechanism of IMiD resistance.
Significance:
These results highlight the dependence of MM on IKZF1-bound super-enhancers, which can be effectively targeted by a potent therapeutic combination pairing IMiD-mediated degradation of IKZF1 and IKZF3 with EP300 inhibition. They also identify AP-1 factors as an unrecognized mechanism of IMiD resistance in MM.
See related article by Neri, Barwick, et al., p. 56.
See related commentary by Yun and Cleveland, p. 5.
This article is featured in Selected Articles from This Issue, p. 4
INTRODUCTION
Multiple myeloma (MM) is an incurable plasma cell malignancy that is characterized by copy-number alterations and complex genomic rearrangements. Newly diagnosed patients typically harbor one of two initiating events: primary translocations affecting the Immunoglobulin Heavy Chain locus (IGH) or select trisomies termed hyperdiploidy (1, 2). Notably, both groups display somatic rearrangements that place oncogenes near large plasma cell enhancers, termed super-enhancers. More specifically, in IGH-translocated patients, the IGH super-enhancer drives the ectopic expression of various oncogenes, such as Cyclin D (CCND1-3), NSD2, or MAF (MAF, MAFB; refs. 3–7). In hyperdiploid patients, secondary rearrangements commonly place MYC under the regulation of a plasma cell-specific super-enhancer, such as FAM46C, PRDM1, the immunoglobulin light chains (IGL, IGK), or the IGH super-enhancer loci (8–10). Moreover, all tumors arise from plasma cells and thus remain dependent on lineage factors, such as IRF4, whose expression is driven by an endogenous DUSP22 super-enhancer (11–13). In summary, all MM tumors depend on oncogenes and/or lineage factors, whose expression is driven by super-enhancers. Therefore, pharmacologically targeting both translocated and endogenous super-enhancers represents an attractive therapy for all MM genetic subtypes that has not been fully investigated.
It is generally agreed that super-enhancers consist of large H3K4-monomethylated and/or H3K27-acetylated cis-regulatory stretches of DNA occupied by a multitude of trans-factors, coactivators, and basic transcriptional machinery. These super-enhancers interact via three-dimensional loops with core promoters, resulting in extremely high levels of gene transcription (14–17). Although super-enhancer-driven oncogene dysregulation in MM is well known, there remains a great deal we do not understand about these complex loci. Case in point, it has been shown that super-enhancers are enriched for a short list of shared trans-factors such as RNAPII, MED1, BRD4, and EP300; however, super-enhancers also contain binding sites for hundreds of alternative transcription factors. Furthermore, it is unclear whether these various trans-factors that localize to super-enhancers might be functionally interchangeable or if redundancy exists within homologous transcription factor families that could offer potential mechanisms of drug resistance.
Recently, compounds that target non–DNA-binding cofactors that are enriched at super-enhancers have been developed. For example, inhibitors of the bromo- and extraterminal domains (BET inhibitors, of which JQ1 is the prototype) target the acetyl-binding bromodomain of coactivator BRD4, resulting in downregulation of oncogene MYC (18–20). Similarly, inhibition of the paralogous histone acetyl transferase proteins EP300 and CBP disrupts the DUSP22 super-enhancer, which results in the downregulation of the plasma cell lineage factor IRF4 (21–26). Both BRD4 and EP300 coactivators preferentially localize with high density to myeloma super-enhancers, and myeloma cell lines display a unique sensitivity to the inhibition of these two coactivators compared with other cancer cell lines (27). However, the coactivator proteins BRD4 and EP300 are ubiquitously expressed across all cell types, narrowing the therapeutic window in vivo (28). Indeed, recent phase I/II clinical trials using BET inhibitors have been hindered by toxicity, suggesting limits to single-agent BET inhibition in patients with myeloma (29).
In addition to cofactor targeting, super-enhancers can also be disrupted by targeting transcription factors that bind to these loci. In myeloma, the zinc-finger transcription factors IKZF1 and IKZF3 are of particular interest because they are targeted by immunomodulatory imide drugs (IMiD) with high specificity. Mechanistically, IMiDs bridge the E3 ubiquitin-ligase protein Cereblon (CRBN) to IKZF1 and IKZF3, resulting in their ubiquitination and subsequent proteasomal degradation (30–33). IKZF1 and IKZF3 are lineage factors necessary for the development of normal lymphocytes and plasma cells, respectively (34, 35). More importantly, both IKZF1 and IKZF3 can regulate hundreds of genes by binding various super-enhancers that are critical for both normal B-cell function and tumor suppression (9, 32, 36). Cellular loss of IKZF1 and IKZF3 has been shown to affect the transcription of super-enhancer-regulated genes, such as IL2 in T cells as well as MYC and IRF4 in various lymphoid tumors; however, the molecular mechanisms by which IKZF1 and IKZF3 regulate these super-enhancer regions remain unclear (37, 38).
Despite showing promise, our knowledge of how these current super-enhancer-targeting therapies function alone or in combination remains incomplete. For example, IMiD-induced degradation of the super-enhancer–binding factors IKZF1 and IKZF3 is highly specific in targeting only lymphocytes, but this does not guarantee a therapeutic response, and patients successfully treated with IMiDs ultimately relapse (39, 40). These findings necessitate better drug combinations for patients with myeloma that effectively target dysregulated super-enhancer + oncogene programs, such as IGH + MYC, as well as endogenous super-enhancer + lineage-defining programs, such as DUSP22 + IRF4, while minimizing adverse effects.
Despite being ideal therapeutic targets for myeloma, MYC and IRF4 remain “undruggable” by direct inhibition. Moreover, we do not fully understand which trans-factors maintain and activate the super-enhancers that drive MYC and IRF4 expression. Here, we show that pharmacologic targeting of coactivator proteins, in parallel with transcription factor-targeting IMiDs, disrupts MYC and IRF4 expression better than either drug alone, and with significantly reduced toxicity. Additionally, we identified an IRF4 binding partner, basic leucine zipper ATF-like factor (BATF), as a previously unidentified transcription factor that can maintain both IRF4 and MYC levels in the absence of IKZF1 and IKZF3, thus contributing to drug resistance and relapse/refractory disease. Importantly, this work identifies contributors to drug resistance in myeloma and also demonstrates a directed approach of super-enhancer disruption in order to reduce MYC and IRF4, synergistically kill myeloma cells, and improve the therapeutic windows of super-enhancer-targeting therapies in vivo.
RESULTS
Successful IMiD Response Requires the Downregulation of MYC and IRF4
We treated 48 human myeloma cell lines (HMCL) for three days with 200 nmol/L pomalidomide (POM) and then measured the effects on growth in order to capture the spectrum of sensitivity and resistance (Fig. 1A). The POM-treated growth index scores correlated well with sensitivity to lenalidomide, which is the most commonly used IMiD clinically (Supplementary Fig. S1A). Even at a low dose of 200 nmol/L, POM was highly effective at reducing known targets IKZF1 and IKZF3 in nearly all HMCLs tested (Fig. 1B and C, top two; Supplementary Fig. S1B, left). Despite significant reductions in both IKZF1 and IKZF3, 92% of the POM-treated HMCLs still maintained growth index scores above 0.5, with most IMiD-resistant cell lines retaining the highest levels of MYC and IRF4 (Fig. 1B, bottom). Consistent with this, MYC and IRF4 levels were better correlated with growth index scores than IKZF1 and IKZF3 (Fig. 1B, bottom; Supplementary Fig. S1B, left). These findings support DepMap data demonstrating that HMCLs are less dependent on IKZF1 and IKZF3 and more dependent on MYC and IRF4 (Supplementary Fig. S1C and ref. 41).
Figure 1.

IMiDs require MYC and IRF4 downregulation to be effective. A, Ranked bar plot of growth index scores for 48 HMCLs treated for 3 days with 200 nmol/L POM. B, Plots of ≥40 HMCLs (each dot is a cell line) showing changes in protein levels (y axis) with corresponding growth index scores (x axis) 3 days after treatment with 200 nmol/L POM. Changes in protein levels were determined by first normalizing to total protein using REVERT, then calculating differences relative to parental DMSO-treated control as determined by western blot. C, Representative western blots for 3 HMCLs treated with either 200 nmol/L POM (IKZF1/3 degrader), 250 nmol/L JQ1 (BRD4 inhibitor), 40 nmol/L GNE-781 (G781; EP300 inhibitor), or DMSO control. A and B, Vertical dotted red lines indicate a 50% growth index score. B, Horizontal dotted red lines indicate 50% reduction in protein levels compared with DMSO control. Shaded box indicates conditions where protein levels dropped below 50%, whereas growth index scores remained higher than 50%. Pearson correlation (R) and significance of correlation (P) as determined by linear regression are listed. A–C, Cell lines KMS11, KMS12BM, and RPMI8226 are highlighted in color as representative examples.
To target super-enhancer–driven MYC and IRF4 using an alternative non-IMiD approach, we treated HMCLs with the cofactor-targeting drugs JQ1 or GNE-781 (G781), which target cofactors BRD4 and EP300, respectively (18, 21). Although JQ1 effectively downregulated MYC, it had modest effects on IRF4, whereas G781 downregulated IRF4 and MYC more consistently and to similar degrees (Fig. 1C; Supplementary Fig. S1B). Not surprisingly, both JQ1 and G781 were less effective than POM in downregulating IKZF1 and IKZF3 (Fig. 1C; Supplementary Fig. S1B). Overall, these data support the notion that POM-induced degradation of transcription factors IKZF1 and IKZF3 is insufficient to reduce growth. Furthermore, reduced growth was better correlated with MYC and IRF4 downregulation, which was best achieved using the cofactor-targeting drugs JQ1 or G781.
JQ1 and GNE-781 Downregulate MYC and IRF4, But Are Toxic In Vivo
To further examine the effects of JQ1 and G781, we compared the IC50 values for the two compounds, which were only modestly correlated, with some cell lines being sensitive to one drug and not the other (Supplementary Fig. S2A). Increased titrations of JQ1 resulted in sharp decreases in MYC starting at 250 nmol/L, but little change in IRF4 levels (Supplementary Fig. S2B and S2C). In contrast, cells treated for 24 hours with G781 displayed a gradual decrease in MYC starting at 25 nmol/L and a decrease in IRF4 at almost every dose tested (Supplementary Fig. S2B). Both JQ1 and G781 had little-to-no effect on steady-state levels of their respective targets, BRD4 and EP300 (Supplementary Fig. S2D). Similar to POM treatment, MYC levels correlated best with growth (Supplementary Fig. S2E).
Given that MYC is driven by the IGH super-enhancer in several of the above cells, we performed chromatin immunoprecipitation (ChIP)-qPCR to examine the effects of JQ1 and G781 on the 3′ IGH super-enhancer. Interestingly, G781 reduced BRD4 occupancy as effectively as JQ1, and at doses lower than previously reported necessary for “off-target” bromodomain inhibition (ref. 23; Supplementary Fig. S3A). Although both JQ1 and G781 reduced the “active enhancer” mark H3K27Ac in the sensitive cell line KMS11, only G781 was capable of reducing H3K27Ac in the IMiD-resistant line RPMI8226 (Supplementary Fig. S3B). Subsequently, the ability of G781 to deplete H3K27Ac at the IGH 3′ super-enhancer correlated with sensitivity to G781, whereas BRD4 depletion did not (Supplementary Fig. S3C and S3D), which supports existing evidence that H3K27Ac is mechanistically required for super-enhancer function (27). In addition to reducing MYC and IRF4 levels, JQ1 and G781 treatment also reduced FGFR3 and MAF levels, which are regulated by translocations of the IGH and IGL enhancers, respectively (Supplementary Fig. S3E and S3F).
To examine the effects of coactivator targeting in vivo, we utilized our Vk*MYC mouse model of MM (42, 43). Dose titration of single-agent G781 in de novo Vk*MYC mice (aged mice that spontaneously develop plasma cell tumors) indicated that half of the 15 mpk-treated mice and one 3.75 mpk-treated mouse reached the predetermined response cutoff of <50% M-spike reduction after two weeks of treatment (Fig. 2A; Supplementary Fig. S4A). However, blood cell counts on day 14 showed progressively lower lymphocytes, monocytes, neutrophils, and platelets following increased G781 dosage, suggesting dose toxicity (Fig. 2B). To determine whether targeting BRD4 in combination with EP300 might work better, we engrafted B6 wild-type (WT) mice with myeloma tumor line Vk29790, and treated mice twice daily with either 30 mpk of the BET-inhibitor iBET-762 (molibresib; similar to JQ1; ref. 44), or with 3.75 or 15 mpk of the EP300 inhibitor G781 alone, or with the two-drug combination (Supplementary Fig. S4B). Both iBET-762 and G781 reduced the tumor burden in vivo (Fig. 2C); however, neither drug, alone or in combination, significantly extended survival (Fig. 2D). Moreover, pairing iBET-762 with G781 resulted in a significant reduction in body weight and mortality during the initial treatment, indicating combination-induced toxicity (Fig. 2D and E). Interestingly, however, the dual-treated mice that survived the initial toxicity finished with the lowest M-spikes and lived until the completion of the study (Fig. 2D and E).
Figure 2.

JQ1 and G781 are toxic in vivo. A, Changes in serum M-spike levels (a marker of tumor burden) relative to day 0 in 71–82 week-old de novo Vk*MYC mice treated twice daily for 2 weeks with increasing doses of the EP300 inhibitor G781 (shaded area). B, Day 14 blood cell and platelet counts for the same mice treated in A. C, Day 14 serum M-spike values relative to day 0 for WT mice transplanted with Vk29790 tumor line and treated twice daily for 2 weeks with vehicle, BET inhibitor (iBET-762), EP300 inhibitor (G781), or the combination. D, Survival curves for the same WT mice transplanted and treated in C. E, Day 14 decreases in % body weight relative to day 0 for same mice treated in C and D. For all panels, each dot represents an individual mouse. Survival curve statistics in D were derived from the Mantel–Cox log-ranked χ2 test. All other P values were determined by an unpaired t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. ns, not significant. Error bars display SD.
EP300 Inhibitors Synergize with IMiDs In Vitro and In Vivo
Having established that IMiD efficacy corresponds with the downregulation of MYC and IRF4, and that MYC and IRF4 can be targeted by JQ1 and G781, we reasoned that pairing an IMiD together with JQ1 or G781 would be complementary, thus limiting toxicity and increasing the therapeutic window. First, we treated multiple HMCLs with increasing doses of POM paired with increasing doses of G781, JQ1, or the standard-of-care agent, dexamethasone (DEX), to compare two-drug synergies (Fig. 3A; Supplementary Fig. S5A). In the majority of cell lines tested, the POM + G781 combination treatment produced more synergy than POM + JQ1 or the current clinical standard-of-care combination POM + DEX (Fig. 3A). RPMI8226, one of the most IMiD-resistant cell lines, showed downregulation of IRF4 and MYC as well as profound growth reduction following POM + G781 treatment (Supplementary Fig. S5B and S5C). Importantly, the efficacy of pairing POM with G781 was nearly identical to that of pairing POM with a similar EP300 inhibitor, CCS1477, which is currently in phase II clinical trials and has shown early success in treating relapsed/refractory myeloma (Supplementary Fig. S5D; refs. 45–47). Flow-cytometric analysis of drug-treated HMCLs revealed that the POM + G781 combination significantly reduced myeloma cell numbers and increased apoptosis and cell death in vitro compared with POM or G781 individually (Fig. 3B and C). Moreover, although POM treatment alone was highly effective in lowering IKZF1 and IKZF3 levels, the addition of G781 enhanced this depletion (Supplementary Fig. S5E).
Figure 3.

POM + G781 effectively target MYC and IRF4 in vitro, and synergize in vivo.A, Heat maps showing Loewe synergy scores from 6 HMCLs (listed on top) treated for 72 hours with increasing doses of POM (top; x axis) paired with increasing doses of G781 (top row; y axis), JQ1 (middle row; y axis), or DEX (bottom row; y axis). Red and yellow shading indicates drug synergy whereas blue and purple indicates drug antagonism. B, Growth curves for JJN3 (top) and KMS11 (bottom) cell lines treated with DMSO control or low doses of POM, G781, or the combination. Y axis shows the fold expansion of cells normalized to day 0. The x axis shows days posttreatment. C, Bar graphs of cell lines JJN3 (top) and KMS11 (bottom) showing the percent-positive cells (y axis) as measured by flow cytometry. Cells were treated with 200 nmol/L POM, 40 nmol/L G781, or the combination across three different time points (x axis). For each condition, the fraction of cells positive for Annexin V–only is displayed in red, added to the remaining fraction of cells double-positive for both Annexin V and Live/Dead is shown in grayscale. D, Gene set enrichment analysis (GSEA) of RNA sequencing for 200 nmol/L POM and 40 nmol/L G781, 48-hour treated myeloma cells. Enrichment scores (ES) are shown for significant (FDR ≤0.01) Hallmark gene sets enriched in both JJN3 (blue) and KMS11 (orange) ranked from most significant (top) to least significant (bottom). E, RNA expression of MYC (left) and IRF4 (right) for JJN3 (top) and KMS11 (bottom) for treatment conditions listed. F, Western blots showing changes in MYC, IRF4, IKZF1, and IKZF3 protein levels in JJN3 (top) or KMS11 (bottom) following 72-hour treatment with 200 nmol/L POM, 40 nmol/L G781, or the combination. G, Dot plots of HMCLs showing a change in MYC (left) and IRF4 protein levels 72 hours following treatment with 200 nmol/L POM, 40 nmol/L G781, or combination as detected by western blot relative to DMSO controls. JJN3 (blue) and KMS11 (orange) are denoted in blue and orange, respectively. H, Viability scores of HMCLs shown in G following 96 hours of treatment with 200 nmol/L POM, 40 nmol/L G781, or the combination. Scores were generated using CellTiter-Glo assay with values normalized to DMSO. I, Day 14 M-spike values obtained in WT mice transplanted with Vk29790 tumor line and treated for 2 weeks, twice daily, with 50 mpk of POM, 3.75 mpk G781, 15 mpk G781, or a POM + G781 combination. Values are normalized to day 0 M-spikes. J, hCRBN+ mice were treated with increasing doses of G781, low-dose POM, or low-dose POM + low-dose G781 (x axis) for 21 days, after which cell counts for white blood cells (WBC) lymphoid cells (LYM), mononuclear cells (MON), and neutrophils (NEU) were calculated using a Heska Hematology Analyzer. Day 0 cell counts prior to treatment are shown in blue. K, Survival curve of WT mice transplanted with Vk29790 tumor line and treated for 2 weeks (yellow shading), twice daily, with Vehicle, 50 mpk POM + 10 mpk DEX,15 mpk G781, or combination 50 mpk POM + 15 or 3.75 (ld) mpk G781. P values in K derived from the Mantel–Cox log-ranked χ2 test. For all other panels, P values were determined by unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Error bars display SD. Each dot in G and H represents a unique cell line. Each dot in I–J represents an individual mouse.
Next, we examined RNA sequencing (RNA-seq) data from the cell lines KMS11 and JJN3 to determine the effects of POM + G781 on transcription. Plotting the changes in gene expression showed that POM + G781 treated cells had 928 and 2,231 differentially expressed genes in JJN3 and KMS11 cells, respectively (Supplementary Fig. S5F; Supplementary Data S1). Compared with the individual treatments, combining POM + G781 resulted in at least three times more differentially expressed genes. A heat map of differentially expressed genes showed POM + G781 treated cells had similar changes in gene expression between biological replicates (Supplementary Fig. S5G). Furthermore, annotating gene-expression changes with CRISPR dropout screens from DepMap revealed that the downregulated genes were enriched for cell-specific dependencies (Supplementary Fig. S5G, right; refs. 41, 48). Notably, this downregulation of dependency genes occurred to a lesser extent in cells treated with POM or G781 alone (Supplementary Fig. S5F and S5G). Gene set enrichment analysis (GSEA) of expression changes induced by POM + G781 treatment revealed that downregulated pathways were more common, including several related to cell-cycle and MYC signaling (Fig. 3D; Supplementary Data S2). Indeed, POM + G781 significantly reduced MYC and IRF4 mRNA and protein levels (Fig. 3E and F). An extended examination of 17 myeloma cell lines revealed that the combination of POM + G781 consistently reduced MYC and IRF4 protein levels better than either drug alone (Fig. 3G). Most importantly, many cell lines resistant to single-agent POM and/or G781 were sensitive to this combination (Fig. 3H).
To determine the effects of POM + G781 in vivo, we used our recently described Vk*MYChCRBN mice, from which we obtained IMiD-responsive transplantable tumors (Vk29790; ref. 49). To isolate the effects of POM treatment on tumor cells, we engrafted Vk29790 cells in POM-resistant WT C57Bl/6 mice, and treated recipient mice twice daily with single-agent POM, low-dose G781 (3.75 mpk), high-dose G781 (15 mpk), or combined POM + G781 (Supplementary Fig. S6A). Following treatment, POM alone had a minimal effect on M-spikes (tumor burden), whereas high-dose G781 was more effective than low-dose G781 in reducing M-spikes (Fig. 3I). In agreement with our cell line data, the combination of POM + G781 was the most effective in lowering M-spikes in vivo. Importantly, POM + G781 was active, regardless of whether POM was paired with high-dose or low-dose G781 (Fig. 3I). To evaluate potential toxicities of the POM plus low-dose G781 combination in an IMiD-sensitive host, we exposed hCRBN+ mice to continual treatment for 21 days and performed complete blood count analysis, which confirmed lack of the hematologic toxicities seen with high-dose G781 (Fig. 3J). Remarkably, all tumor-bearing mice treated for only 2 weeks with POM + G781, but not POM + DEX, were cured (Fig. 3K). Examination of additional, more aggressive, and genetically distinct Vk*MYC tumor lines (bioRxiv 2023.07.25.550482) confirmed synergy and improved survival generated by pairing POM with G781 (Supplementary Fig. S6B–S6H). Overall, these data indicate that the combination of POM + G781 synergistically reduces MYC and IRF4 levels, increases tumor killing, minimizes toxicity, and extends survival in vivo.
POM + G781 Results in the Loss of Chromatin Accessibility at Translocated and Lineage-Defining Super-Enhancers
Given that POM and G781 target transcription factors and coactivators, respectively, we performed assay for transposase accessible chromatin sequencing (ATAC-seq) analysis of the same cell lines analyzed by RNA-seq to determine how POM + G781 affects chromatin accessibility. Compared with individual POM or G781 treatments, the POM + G781 combination resulted in far more significant changes in chromatin accessibility (Supplementary Fig. S7A; Supplementary Data S3). Although IKZF1 and IKZF3 can both activate or repress transcription, EP300 is predominantly recognized as a transcriptional activator, leading us to reason that the mechanism of synergy stems from enhancer decommissioning at loci harboring both EP300 and IKZF1. Indeed, regions that lost chromatin accessibility following POM + G781 treatment were enriched for both EP300- and IKZF1-binding sites (Fig. 4A, top). In contrast, the regions that gained accessibility following POM + G781 treatment were not enriched for EP300 or IKZF1 binding (Fig. 4A, bottom). Notably, loss of chromatin accessibility was observed at the IGH 3′ A1 and A2 enhancers, which drive the expression of MYC and other translocated oncogenes (Fig. 4B, top). Similar losses in chromatin accessibility were observed at other super-enhancers of lineage-defining survival genes, such as IRF4 and POU2AF1 (Fig. 4B, middle, bottom). Importantly, the aforementioned regions with diminished accessibility correspondingly had reduced expression of their associated genes, such as MYC and IRF4 (Fig. 3E), as well as POU2AF1 (Fig. 4C), which was recently identified as a regulator of B-lineage super-enhancers, a myeloma-dependency gene, and potential new therapeutic target (50).
Figure 4.

POM + G781 result in chromatin accessibility loss at MM translocated and lineage enhancers. A, Odds ratio of overlap for EP300 and IKZF1 ChIP-seq peaks (MM1S cells) with regions that lost (top) or gained (bottom) chromatin accessibility after POM (200 nmol/L) and G781 (40 nmol/L; POM + G781) treatment in JJN3 (blue) and KMS11 (orange). Confidence intervals (95%) are shown. B, Genome plot of the IGH 3′ enhancers (top), the DUSP22/IRF4 locus (middle), and POU2AF1 (bottom). Differentially accessible regions (DAR) are denoted by black marks. ATAC-seq for JJN3 (blue) and KMS11 (orange) is shown for control cells (lighter shades) along with changes induced by POM + G781 treatment (darker shades). Negative values indicate a loss in chromatin accessibility. IKZF1 and EP300 ChIP-seq are shown below for MM1S myeloma cells. Transcription-factor (TF) binding motifs enriched in regions that lose chromatin accessibility are shown (bottom, each plot). C, RNA expression of POU2AF1 as determined by RNA-seq in KMS11 (left) and JJN3 (right) for DMSO, POM (200 nmol/L), G781 (40 nmol/L), and combination (combo) treated cells. D, Odds ratio of overlap of transcription-factor binding motifs with regions that gain (top) or lose (bottom) chromatin accessibility in JJN3 (blue) and KMS11 (orange). Only TF families enriched (FDR ≤0.01) in both JJN3 and KMS11 are shown with the highest-ranking TF from each family, labelled. TF are from HOMER software with 95% confidence intervals shown. ATAC-seq data (B) represent one replicate and RNA-seq (C) represent two replicates per cell line per condition.
To further explore the determinants of POM + G781 chromatin accessibility changes, we examined the differentially accessible regions to identify transcription-factor binding motifs. Motifs most notably enriched in regions that gained chromatin accessibility with POM + G781 included RUNX and ETS factor family motifs (Fig. 4D, top). Regions that lost chromatin accessibility following POM + G781 treatment were enriched for BATF, IRF4, and FOX,bHLH (Forkhead box, basic helix–loop–helix) motifs (Fig. 4D, bottom). These motif families include BATF sites composed of adjacent basic leucine zipper (bZIP) and interferon regulator factor (IRF) motifs known as AP-1:IRF composite elements (AICE; ref. 51). The Foxa2 motif is a composite of the FOX and basic helix–loop–helix (bHLH) DNA-binding domains; bHLH domains include E-box motifs, which are known to be bound by MYC.
In total, the ATAC-seq data indicated that POM + G781 induced the loss of chromatin accessibility at myeloma-specific super-enhancers (both translocated and lineage-specific nontranslocated). Moreover, these regions of lost accessibility were associated with overlapping IKZF1 and EP300 binding sites and were enriched for BATF, IRF4, and potentially MYC motifs.
IMiD Resistance and Poor Outcome Correspond with Transcription-Factor Expression Plasticity
To complement our previous motif analysis, we examined the transcriptomes of 66 HMCLs (52) to indirectly ascertain the presence or absence of implicated transcription factors. Transcription factors were organized into defined subsets: key MM myeloma factors (IRF4 and MYC), cofactors, IKZF zinc finger, ETS, and AP-1 family members. Within each subset, cell lines were ranked by gene-expression levels (Fig. 5A; Supplementary Fig. S7B). This analysis provided several important insights relevant to myeloma. First, we identified a small number of viable HMCLs that expressed little-to-no MYC, whereas IRF4 expression appeared to be indispensable. Second, like MYC and IRF4, cofactor POU2AF1 was highly expressed in most HMCLs, is a recently identified preferential dependency for MM (41), and was downregulated by POM + G781 combination therapy. Third, an examination of the ETS and AP-1 families revealed a tendency for HMCLs to express modest-to-high levels of at least one of these factors. This pattern was most striking when viewing the mutually exclusive expression of ETS factors SPI1 (PU.1) and SPIB, or that of the BATF/FOS family of proteins, the latter of which homodimerizes with ubiquitously expressed JUN proteins to form AP-1 factors. Coincidentally, both ETS and AP-1 factors have the ability to bind DNA cooperatively with IRF4, increasing the DNA-binding affinity of IRF4 several fold (51, 53, 54). Strikingly, we noticed that many IMiD-resistant cell lines expressed high levels of at least one ETS or BATF/FOS protein (labeled in red), whereas many IMiD-sensitive cell lines expressed little-to-no ETS or BATF proteins (labeled in green; Fig. 5A). Cumulatively, these data suggest that HMCLs have unique and complex transcriptional dependencies, and that the expression of ETS and/or AP-1 transcription-factor families are implicated in IMiD resistance.
Figure 5.

Myeloma tumors have unique trans-factor expression plasticity that contributes to poor outcomes in patients. A, Gene-expression data for 66 HMCLs displayed as a heat map using log2 RPKM values (key on left). HMCLs were grouped functionally: MM (multiple myeloma factors, IRF4 and MYC); cofactors; IKZF (zinc finger proteins); ETS factors; and AP-1 factors). Once grouped, the cells were then ranked in seven different ways based on trans-factors of interest (listed diagonally above). Each black box has a unique rank order based on the gene-expression levels for the factors listed. A cell line can only occupy one row per black box. Select IMiD-resistant cell lines are denoted in red, and IMiD-sensitive cell lines in green. B, Expression of BATF, BATF2, and BATF3 in newly diagnosed (NDMM) CoMMpass samples with matching RNA-seq and whole-genome sequencing (N = 586). Primary IGH translocations, hyperdiploidy (HRD), and MYC translocations are annotated (right). C, Box plot of BATF expression in NDMM samples from the CoMMpass study categorized by gene-expression subtype for samples with RNA-seq (N = 764). The number of samples (N) in each subtype is denoted (left); subtypes are from Zhan et al. (55). CD-1, Cyclin D1; CD-2, Cyclin D1 and CD20; HP, hyperdiploid; LB, low bone disease; MF, MAF; MS, MMSET; PR, proliferation. D, Progression-free survival (PFS; left) and overall survival (OS; right) Kaplan–Meier curves of CoMMpass NDMM patients with RNA-seq and outcome data and treated with first-line IMiD-containing therapies (N = 484) stratified by median BATF expression. P values were determined by a Wald test of a Cox-proportional hazard regression treating BATF expression as a quantitative response. E, Volcano plots of gene expression correlated with BATF. The Pearson correlation coefficient of each gene with BATF is shown on the x axis, and the significance −log10(P value) is shown on the y axis. Genes significantly (FDR ≤0.01) correlated with BATF are shown in color (red, positive; blue, negative) with the number of significant genes listed (N). F, GSEA of gene expression correlated with BATF in a cross-sectional analysis of CoMMpass NDMM samples. The enrichment score is shown (top) with overlap with GSEA hallmark gene sets shown on the bottom. G,BATF expression in paired NDMM and relapse refractory (RRMM) samples from 35 CoMMpass patients with matching samples treated with IMiD-containing first-line therapies. P value was determined with a linear regression using a covariate for the patient.
Next, we examined RNA-seq data from 764 newly diagnosed multiple myeloma (NDMM) samples from the CoMMpass trial (NCT01454297) to better understand the potential impact of BATF transcription factors in primary patient samples. Similar to the cell lines, patient samples sorted by BATF family member expression showed a similar exclusivity expression pattern for BATF, BATF2, or BATF3 (Fig. 5B). Annotation of IGH translocations, hyperdiploidy (HRD), and MYC translocations using CoMMpass whole-genome sequencing did not reveal any striking correlations, except for BATF3 expression cooccurring with t(11;14) translocations (Fig. 5B, right). Further inspection, categorizing samples by gene-expression subtype based on work from Zhan and colleagues (55), indicated that BATF was expressed at higher levels in the MAF (MF), MMSET (MS), and Proliferation (PR) subtypes (Fig. 5C); BATF2 expression was low but not substantially different between gene-expression subtypes, and BATF3 was expressed at higher levels specifically in the CCND1 (CD-1) subtype, which is one of two gene-expression subtypes that harbor t(11;14) CCND1;IGH translocations (Supplementary Fig. S8A). Inspection of CoMMpass patients treated with first-line IMiD-containing therapies with RNA-seq and outcome data (N = 484) indicated that BATF and BATF3 expression was associated with worse progression-free survival (PFS) and overall survival (OS; Fig. 5D: Supplementary Fig. S8B). The expression of BATF factors was also compared with the clinical stage, which revealed that BATF3 was correlated with the increasing stage (Supplementary Fig. S8C). Importantly, BATF expression remained a significant predictor of PFS in multivariate analysis comparing age, stage, and high-risk cytogenetics (Supplementary Table S1).
The transcriptional program associated with BATF was further investigated by determining coexpressed genes, which identified thousands of genes both positively and negatively correlated with BATF expression (Fig. 5E; Supplementary Data S4). These results were annotated using GSEA, which indicated several pathways associated with BATF expression, including E2F and MYC targets (Fig. 5F; Supplementary Data S5). Finally, inspection of paired NDMM and relapse/refractory multiple myeloma (RRMM) samples from 35 patients treated with IMiD-containing regimens showed that, although BATF2 and BATF3 levels did not change upon progression, BATF was significantly upregulated upon relapse (Fig. 5G; Supplementary Fig. S8D). We investigated potential mechanisms of BATF upregulation and found that BATF copy number was increased in subset RRMM samples, but this was not significant (Supplementary Fig. S8E). We also identified chromatin accessibility regions associated with BATF expression using a cross-sectional analysis of 22 HMCLs (Supplementary Fig. S8F–S8G; ref. 56). These chromatin accessible regions contained motifs for 108 transcription factors including some that were significantly upregulated with BATF in RRMM samples (Supplementary Fig. S8H), suggesting that BATF may be upregulated by transcriptional mechanisms. Cumulatively, these data associate BATF expression with high-risk subtypes, MYC signaling, poor outcomes, and relapsed disease in MM.
BATF Proteins Promote Myeloma Tumor Survival and Contribute to IMiD and G781 Drug Resistance
Given the heterogeneous and mutually exclusive expression patterns of the BATF family proteins in both patients and cell lines, we examined DepMap data on HMCLs for BATF, BATF2, and BATF3 gene dependency. For all three BATF genes, there was a significant correlation between expression and dependency, suggesting that elevated levels of BATF, BATF2, or BATF3 promote tumor survival (Fig. 6A).
Figure 6.

BATF proteins promote myeloma viability and drug resistance. A, Scatter plot of expression (x axis) by dependency (Chronos, y axis) for BATF (left), BATF2 (middle), and BATF3 in multiple myeloma cell lines in the DepMap project. Expression data from the Cancer Cell Line Encyclopedia. Pearson correlation (R) and significance of correlation (P) as determined by linear regression are listed. Select cell lines are denoted in color (key, right). B, Diagram of the BATF2 translocation in L363 cells identified by mate-pair sequencing (top), CRISPR-Cas9 targeting sites (vertical dotted lines), and the PCR primers (red and blue arrows) used to distinguish the endogenous translocated region from the successfully deleted BATF2/enhancer region (bottom). C, PCR analysis of genomic DNA from L363 cells electroporated with CRISPR-Cas9 and sgRNAs targeting the BATF2/Enhancer locus (B). The top gel shows the detection of the translocated BATF2/enhancer across all days tested. Bottom panel shows successful deletion of BATF2/enhancer that is lost following 7 days of coculture expansion. D,BATF expression in a panel of 66 HMCLs determined by RNA-seq. Expression is shown in fragments per kilobase per million reads (FPKM) with select cell lines labeled and shown in color. E, Western blot of BATF and β-ACTIN loading control in shRNA knockdown of BATF (shBATF) or a negative control (shCtrl) of BATF in KMS12BM cells. F, Representative flow cytometry plots of KMS12BM cells infected with shRNA empty vector (top) or an shRNA targeting BATF (bottom). Cells were plated at equal densities on day 0, then treated for 3 days with DMSO control, 200 nmol/L POM, 40 nmol/L G781, or the combination. Annexin V is shown on the y axis, Live/Dead viability dye is on the x axis. Equal volumes were analyzed for all conditions and biological triplicates were measured. The percent population of each gate is listed. G, Growth curve of panel F KMS12BM cells infected with shRNA empty vector (black line) or an shRNA targeting BATF (red line) and then treated with combination 200 nmol/L POM plus 40 nmol/L G781. Fold expansion is shown on the y axis (log10 scale). Days posttreatment are shown on the x axis. H, Growth curves of 4 different HMCLs expressing negative (neg) ctrl eGFP (black lines) or exogenous BATF (blue lines) and treated with combination 200 nmol/L POM plus 40 nmol/L G781. Fold expansion is shown on the y axis (log10 scale). Cell line and translocated enhancer driving MYC expression are shown on top. Days posttreatment are shown on the x axis. P values determined by an unpaired t test.
Upon close examination of the BATF/FOS family RNA-seq data, we noticed uniquely high “all-or-none” expression for BATF2 compared with BATF and BATF3, which was limited to only three of the 66 HMCLs (MOLP8, MMM1, and L363; Fig. 5A). Significantly elevated BATF2 expression in CoMMpass patient data was likewise limited to a single patient, as compared with elevated BATF or BATF3 spread across a larger fraction of patients (Fig. 5B). Subsequent mate-pair sequencing revealed that all three BATF2-expressing HMCLs harbored previously unidentified translocations that placed BATF2 near super-enhancers, including IGH (Supplementary Fig. S9A). Similarly, whole-genome analysis of a single patient with high BATF2 expression identified an IGH translocation 1.6Mb upstream of BATF2. Unlike BATF2, however, we did not identify any translocation events near BATF or BATF3 loci in the HMCLs. These data suggest that high BATF2 expression may be selected for by translocation events and that the unique expression patterns of BATF and BATF3 may be driven by cis-elements including those identified in Supplementary Fig. S8F.
Using these cell lines, we examined L363 to determine whether elevated BATF2 expression is essential for tumor survival. L363 was chosen because of its BATF2;IGH translocation and because it possessed the lowest BATF2 Chronos score (i.e., most predicted dependency; Fig. 6A and B; Supplementary Fig. S9A). Using CRISPR/Cas9 in L363, we deleted a 37 kb region encompassing BATF2 along with part of the translocated IGH enhancer locus in a heterogeneous population of cells. Successful deletion was confirmed by PCR (Fig. 6B). Upon expansion, PCR analysis revealed a progressive loss of BATF2-deleted cells, which were undetectable after day 7 (Fig. 6C). Additionally, despite considerable effort, we were unable to generate L363BATF2–/– complete knockout clones and could only achieve minor knockdown of BATF2 in L363 cells using traditional shRNA approaches. These data support the notion that BATF2 is necessary for L363 cell survival.
Next, we used KMS12BM cells to determine whether BATF contributes to drug resistance. KMS12BM is highly IMiD-resistant, and out of 66 HMCLs, it expresses the second-highest levels of BATF (Fig. 6D). First, we performed single-guide CRISPR-Cas9 mutagenesis to knock out BATF alleles. Western blot screening of multiple KMS12BM clones revealed a ∼50% reduction of BATF protein in select clones, suggesting monoallelic knockout, but also implying that KMS12BM cells might not tolerate complete loss of BATF (Supplementary Fig. S9B). Nevertheless, even a 50% reduction in BATF was sufficient to sensitize KMS12BM clones CrC1 and CrC39 to low-dose G781 and POM + G781 combination treatment, as demonstrated by the reduced cell counts and increased Annexin V+ cells (a marker for apoptotic cells; Supplementary Fig. S9C and S9D). Using an alternative shRNA knockdown approach, we achieved 65% knockdown of BATF in KMS12BM cells (Fig. 6E). Although this level of BATF reduction did not significantly affect cell viability (Fig. 6F; DMSO) or the expansion rate of untreated KMS12BM cells (Supplementary Fig. S9E), it was sufficient to sensitize KMS12BM cells to low-dose POM, G781, and POM + G781, as shown by increased cell death (Fig. 6F). Importantly, the ability of KMS12BM cells to expand under low-dose POM + G781 was prevented following this modest knockdown of BATF (Fig. 6G).
Given that BATF2 is essential for L363 survival and that reductions of BATF sensitized KMS12BM cells to POM and G781 treatment, we next investigated whether ectopic expression of BATF proteins could confer drug resistance to cell lines normally sensitive to IMiDs. To test this, we used a doxycycline-inducible (Dox) lentiviral vector to ectopically express BATF2 in KMS11 cells—a POM-sensitive cell line that expresses little-to-no endogenous BATF, BATF2, or BATF3 (Fig. 5A, right). Increased Dox treatment resulted in a proportional increase in BATF2 expression in KMS11 cells (Supplementary Fig. S10A and S10B). We chose Dox doses of 300 ng/mL and 1,500 ng/mL to mimic the endogenous levels of BATF2 in IMiD-resistant cell lines L363 and MMM1, respectively (Supplementary Fig. S10A). At both doses, Dox-induced expression of BATF2 restored MYC levels and rescued KMS11 cells from POM-mediated cell death (Supplementary Fig. S10B–S10C). Next, we confirmed the BATF2-mediated POM resistance using a separate constitutively active pCDH-EF1α-BATF2 lentiviral vector (Supplementary Fig. S10D). Remarkably, the expression of exogenous BATF prevented low-dose POM + G781–mediated cell death in every HMCL tested (Fig. 6H). Moreover, resistance to single agents POM, G781, and to the POM + G781 combination was conferred on KMS11, KMS26, and JJN3 cells regardless of whether BATF, BATF2, or BATF3 was expressed (Fig. 7A; Supplementary Fig. S10D–S10H). Nevertheless, BATF expression was not sufficient to significantly alter the sensitivity of MM.1S and KMS11 cells to other traditional therapies, such as bortezomib, melphalan, and dexamethasone (Supplementary Fig. S10I–S10J). Taken together, these data indicate that BATF transcription factors are sufficient to induce resistance against low, but normally lethal, doses of POM, G781, and their combination; they also suggest that BATF proteins may compensate for drug-induced loss of IKZF1, IKZF3, and/or EP300.
Figure 7.

BATF maintains IRF4 and MYC expression by regulating myeloma super-enhancers. A, Growth curves showing JJN3 cells expressing eGFP, BATF, BATF2, or BATF3 constructs listed and treated with 200 nmol/L POM plus 40 nmol/L G781. The y axis shows fold expansion relative to day 0 (log10). The x axis shows days posttreatment. B, Growth curves showing JJN3 and KMS34 cells expressing various constructs listed, and treated with 200 nmol/L POM plus 40 nmol/L G781. The y axis shows fold expansion relative to day 0 (log10). The x axis shows days posttreatment. C,IRF4 expression in JJN3 cells treated with 200 nmol/L POM, 40 nmol/L G781, or both (Combo) relative to DMSO controls in cells transduced with a control (eGFP) or BATF-overexpressing construct. D, Day 3 Western blot analysis showing MYC, IRF4, IKZF1, and BATF2 levels in JJN3 cells expressing various constructs (listed at the top), and treated with 200 nmol/L POM, 40 nmol/L G781, or the combination. Actin is shown as a loading control. E, Bar plot of the number of differentially expressed genes (FDR ≤0.01) in JJN3 cells treated with 200 nmol/L POM, 40 nmol/L G781, or both (Combo) relative to DMSO controls in cells transduced with a control (eGFP) or BATF-overexpressing construct. F, GSEA for BATF overexpression (+BATF, left) as compared with control (+eGFP, right) in JJN3 (blue) and KMS11 (orange) cells treated with POM and G781. The positive enrichment score (top) denotes enrichment of the GSEA Hallmark MYC_TARGETS_V1 genes in the BATF versus control. Genes for each cell are ranked and overlap with MYC_TARGETS_V1 genes are shown in color (bottom). G, H3K27Ac and IRF4 ChIP-seq in KMS12BM empty vector (EV) cells as well as IRF4 and BATF2 ChIP-seq in KMS12BM cells overexpressing BATF2 (+BATF2) at the IGH 3′ enhancers (left) and the DUSP22/IRF4 enhancer (right). Isotype control is shown, and BATF-IRF4 composite, IRF4, and FOX-EBOX motifs are shown at the bottom. H, Motif analysis of IRF4-bound regions for KMS12BM “+BATF2” and “EV” cells where the union of the top 5 enriched motifs for each condition was combined, and the odds ratio of each motif is plotted relative to shuffled background regions. Motif logos are shown (right). I, Model of MYC and IRF4 regulation by IKZF1/3, EP300, and BATF in the context of IMiDs and EP300 inhibitors (EP300i). IKZF1/3 and EP300 are found at enhancers of IRF4 and MYC (top row). IMIDs and P300i result in IKZF1/3 depletion and inhibition of P300, respectively, resulting in repression of IRF4 and MYC (rows 2-3) that can be overcome through ectopic expression of BATF, which also binds IRF4 and MYC enhancers. Expression level of IRF4 and MYC is denoted by color (red, high; white, low).
BATF Heterodimerization with IRF4, and Maintained Expression of IRF4 Confer IMiD Resistance
Because our data suggest that IMiDs work primarily through the downregulation of MYC and IRF4, we asked whether maintained expression of either IRF4 or MYC could confer IMiD resistance. First, we overexpressed IRF4 in two IMiD-sensitive cell lines, KMS11 and MM.1S, which normally downregulate IRF4 as a result of IMiD treatment (Fig. 3F; Supplementary Fig. S11A). Indeed, vector-mediated IRF4 expression made both cell lines ∼30 × more resistant to lenalidomide (Supplementary Fig. S11B). Mechanistically, exogenous IRF4 prevented IMiD-induced cell death, as evidenced by the reduction in cleaved caspase 8 (Supplementary Fig. S11C), which is in line with previous reports demonstrating the antiapoptotic properties of IRF4 in myeloma (57). Next, given the similar drug-resistant phenotypes produced by either IRF4 or BATF expression, we wanted to examine whether these two factors might work together to confer POM + G781 drug resistance. To test this hypothesis, we generated BATFH55Q. The H55Q point mutation interferes with the AP-1 heterotrimeric interaction consisting of BATF(+JUNB):IRF4, thus greatly reducing AP-1:IRF4 complex binding at the AICE composite sites (51, 58, 59). We transduced two POM + G781-sensitive cell lines with empty vector, BATF, BATFH55Q, IRF4, or MYC-expressing vectors and treated them with POM + G781 to monitor cell growth over time. Similar to our previous results, both BATF- and IRF4-expressing cells expanded in the presence of POM + G781 (Fig. 7B). Importantly, cells expressing BATFH55Q, which blocks the ability of BATF to cooperatively interact with IRF4, did not expand (Fig. 7B). Additionally, MYC expression failed to rescue either cell line, suggesting that POM + G781 resistance was mediated less by MYC and more by IRF4 (Fig. 7B; Supplementary Fig. S11B).
Further examination of the BATF/IRF4 relationship revealed that BATF overexpression prevented POM + G781–induced downregulation of endogenous IRF4 at both the transcript and protein levels (Fig. 7C and D). The ability of BATF to maintain IRF4 levels also explains the BATF-induced repression of the proapoptotic genes BMF and BIM, which are typically induced by POM + G781 treatment (Supplementary Fig. S11D). These two genes were recently identified by Fedele and colleagues as being directly repressed by IRF4, and their downregulation likely contributes to drug resistance and antiapoptotic properties in both IRF4- and BATF-expressing cells (57).
To examine BATF/IRF4-induced drug resistance, we examined changes in protein levels by western blotting and confirmed several key findings in response to POM and G781 drug treatments: (i) POM effectively degraded IKZF1, which was not required for cell survival when IRF4 or BATF was overexpressed; (ii) MYC expression failed to rescue IRF4 levels; (iii) IRF4 expression was sufficient to rescue MYC levels; and (iv) BATF2 expression rescued both IRF4 and MYC (Fig. 7D). Notably, despite exogenous expression of MYC via “+MYC” lentivirus, POM + G781 still resulted in 35% downregulation of MYC protein, and this provided zero rescue as measured by growth curves (Fig. 7B and D). Conversely, “+IRF4” cells expanded robustly under POM + G781 treatment, despite similar 33% MYC downregulation, indicating that increased viability can be attributed to the maintained levels of BATF2 and/or IRF4 (Fig. 7D). Moreover, at the transcriptional level, BATF expression abrogated the substantial changes in gene-expression induced by POM + G781 treatment (Fig. 7E). Furthermore, BATF-transduced cells maintained their expression of MYC targets throughout POM + G781 treatment, as compared with the eGFP-transduced control cells (Fig 7F).
Previously, IRF4 was shown to enhance both its own gene expression and MYC expression through a promoter/enhancer-binding feed-forward loop (12). To examine this, we performed ChIP-seq for IRF4 with and without BATF2 overexpression in KMS12BM myeloma cells. Inspection of the endogenous DUSP22/IRF4 and translocated IGH/MYC loci showed BATF2 and IRF4 colocalization at H3K27Ac-marked super-enhancer regions, at composite AICE (BATF:IRF) motifs within the IGH 3′ A1 and A2 enhancers, as well as the DUSP22/IRF4 super-enhancer (Fig. 7G). Although IRF4 localization appeared similar with and without BATF2 overexpression at these loci, analysis of the top binding motifs indicated that bZIP/AP-1 and bZIP/AP-1:IRF composite elements (AICE) were significantly more enriched at IRF4-bound regions in BATF2 overexpressing cells (Fig. 7H). Cumulatively, data in this manuscript indicate that EP300 inhibitors potentiate IMiDs by further downregulating IRF4 and MYC, but that BATF factors can compensate for the therapeutically mediated loss of IKZF1, IKZF3, and EP300 inhibition by maintaining super-enhancer–driven IGH oncogenes as well as lineage-defining IRF4 expression (Fig. 7I), thereby promoting myeloma cell survival.
DISCUSSION
IMiDs are a backbone therapy for MM, where they have modest single-agent activity and are almost always used in combination with dexamethasone. In line with this, when screening a large panel of MM cell lines, we consistently found that single-agent IMiD activity is primarily cytostatic; moreover, when IMiDs are used in our highly predictive Vk*MYChCRBN mouse model of MM, tumor reduction is only achieved by the addition of secondary agents, such as dexamethasone (49). Here, we show that the addition of an EP300 inhibitor dramatically synergizes with IMiDs, inducing apoptosis in vitro and is curative in a subset of Vk*MYChCRBN mice in vivo, thus outperforming dexamethasone. Although IMiD effects rely on the degradation of IKZF1 and IKZF3 by itself, this is not sufficient to induce a response. Our data indicate that a better determinant of a successful IMiD response, alone or in combination with EP300 inhibitors, is the subsequent downregulation of IRF4 and MYC. This finding is consistent with previous reports (40, 60–62). Although IRF4 and MYC are essential for myeloma survival and are known to directly and positively regulate each other (12), we found that ectopic expression of MYC could not restore IRF4 expression and ultimately failed to rescue myeloma cells from a lethal dose of IMiDs and EP300 inhibition. This suggests that MYC regulation of IRF4 is dependent on IKZF1, IKZF3, and/or EP300. In contrast, forced IRF4 expression helped maintain MYC protein levels and myeloma cell growth under the same therapeutic conditions. This is consistent with MYC dysregulation primarily occurring through genomic structural alterations that result in the gene falling under the control of plasma cell super-enhancers.
Despite the initial responses, patients with myeloma eventually acquire resistance to IMiD therapy. Although analyses of relapsed myeloma samples have implicated acquired mutations and splice variants in CRBN (which may affect IMiD-mediated ubiquitination and subsequent proteolytic degradation of IKZF1 and IKZF3), this resistance mechanism is limited to a minority of cases (39, 63). Therefore, other mechanisms of IMiD resistance must exist that are independent of CRBN-mediated IKZF1 and IKZF3 degradation (64, 65). One potential mechanism recently investigated is the coexistence of multiple transcriptional states that maintain oncogene expression (66). Building on this, our data identified distinct transcription factors that have the ability to maintain MYC and IRF4 expression despite IMiD-induced loss of IKZF1 and IKZF3. Specifically, ectopic expression of any BATF family member confers resistance to IMiDs and/or EP300 inhibition and maintains IRF4 and MYC expression. Mechanistically, the heterodimeric partners BATF2 and IRF4 directly bind to the DUSP22-IRF4 and IGH super-enhancers at the same cis-elements, and BATF2 binding was detected at the IRF4 core promoter. Indeed, it is well established in B lymphocytes that BATF AP-1 factors bind with IRF4 at AP-1:IRF composite elements (AICE), resulting in increased binding affinity and transcriptional activation (67). Further supporting this mechanism of drug resistance, when BATF is expressed endogenously, myeloma cells tend to be dependent upon it. Thus, it is not surprising that IMiDs and EP300 inhibitors, which cause synergistic myeloma cell death, also result in a loss of chromatin accessibility at the same AICE motifs (otherwise known as bZIP:IRF). These AICE motifs occur at a high density in both IGH 3′ enhancers that often drive oncogene expression in myeloma, and our ChIP-seq analysis confirmed that these regions were bound by both BATF2 and IRF4.
Whether BATF transcription factors confer drug resistance through transcriptional activation of IRF4 and/or enhanced function of residual IRF4 through higher-affinity cooperative binding at AICE elements remains to be fully determined. Data herein suggest it is a combination of both mechanisms as ectopic expression of BATF maintained IRF4 transcript and protein levels; meanwhile, BATFH55Q mutant expression (which has reduced IRF4 dimerization potential) failed to fully rescue myeloma cells from IMiDs and EP300 inhibition. Cumulatively, these data demonstrate that interchangeable paralogous transcription factors (e.g., BATF, BATF2, and BATF3) can mediate myeloma cell survival even in the absence of IKZF1 and IKZF3, but also raise the question of whether other transcription-factor families may be able to maintain super-enhancer function and oncogene expression despite therapeutic modalities.
Given the transcriptional heterogeneity of MM subtypes, the possibility that distinct and redundant transcription factors can elicit the same drug resistance phenotypes presents a daunting challenge for successful IMiD therapy. One strategy that may be partially generalizable is to target cofactors that cooperate with a plethora of transcription factors. Preclinical studies on BET and EP300 inhibitors have highlighted the tremendous promise of these compounds in downregulating MYC and IRF4 in vitro and in vivo (18, 25, 27, 29). Unfortunately, the results of early-phase I/II clinical trials examining BET inhibitors have been discouraging because of common adverse events, such as thrombocytopenia, anemia, and neutropenia (68). Less is known about EP300 inhibitors, which have only entered phase II testing (47). However, early data from patients suggest that POM + EP300 inhibition may be a promising new drug combination (45, 46).
Our data support previous findings that inhibition of cofactors BRD4 and EP300 is highly effective at reducing MYC and IRF4 levels, respectively (18, 26); however, both drugs alone were ineffective at improving OS and were toxic in combination in our highly predictive murine model of myeloma. It will be important to further understand the mechanisms by which EP300 inhibitors affect BRD4 chromatin binding and function, as well as IKZF1 and IKZF3 binding and function, especially given recent reports that EP300 inhibitors do not block chromatin binding conceivably resulting in a “dominant negative”-like function. Regardless of the mechanism, EP300 inhibition showed a marked synergy with IMiDs and reduced toxicity. JQ1, however, did not pair as effectively with IMiDs. This may be due in part to JQ1 preferentially targeting MYC, whereas EP300 inhibition targeted both MYC and IRF4, the latter of which appears to be the more essential transcriptional target based on its ability to rescue myeloma cells from lethal doses of therapy.
Interestingly, data in an accompanying manuscript from Neri and Barwick and colleagues (69) show a striking overlap of EP300 and IKZF1 genomic targets, with more than 80% of EP300-bound enhancers also bound by IKZF1. This suggests that other transcription factors may compensate for the IMiD-mediated loss of IKZF1 and IKZF3, but these transcription factors require EP300. Nevertheless, our BATF data outline a cautionary tale that unique combinations of transcriptional plasticity have the potential to overcome potent therapeutic combinations. Consistent with this, we show that BATF2 and IRF4 bind the same enhancer elements as EP300 and IKZF1 at the IGH and IRF4 super-enhancers, and we propose that myeloma tumors rely on the heterogeneity and plasticity of their enhancer-regulating trans-factor programs to maintain IRF4 and MYC expression in the face of various drug treatments. As new drugs that target transcription factors and coactivators become available, studies delineating their efficacy in the context of underlying myeloma genetics, as well as the transcriptional milieu, are needed to tease apart their mechanisms of action and to identify rational combinations.
METHODS
Cell Lines and Drugs
Except as noted below, the human MM cell lines were obtained from the original investigators, with repository availability listed in parentheses. ALMC2, KP6—Diane Jelinek; AMO1—Shiro Shimizu (DSMZ); AMU-MM1—Ichiro Hanamura; ARP1, ARP1C (CAG), ARP1D (ARD)—Joshua Epstein; DELTA47—Ichiro Kubonishi (JCRB); EJM, JJN3—Ian Franklin (DSMZ); JIM3—Ian Franklin; FR4—Shinichi Tagawa; NCI-H1112—Adi Gazdar; INA6, JK6L—Martin Gramatzki; KHM1B—Hiroyuki Hata (JCRB); KMM1, KMS11, KMS12BM, KMS12PE, KMS20, KMS26, KMS28BM, KMS28PE, KMS34—Masayoshi Namba (JCRB); L363, LP1 (DSMZ), MC1286—P. Leif Bergsagel; MM-M1—Takayuki Takahashi; MM.1S—Steve Rosen (ATCC); MOLP8 (DSMZ), OCI-MY1, OCI-MY5—Hans Messner; OPM1—Shuich Katagiri care of Brad Thompson; OPM2—Shuich Katagiri care of Brad Thompson (DSMZ); RPMI-8226 (ATCC), SACHI—Masafumi Taniwaki; SKMM1—Alan Houghton; U266 (ATCC), UTMC2—Alan Solomon; XG6, XG7—Riccardo Dalla-Favera. MC1286PE1, PE2, PE3, PE5, and PE7 are five newly established cell lines from recurrent pleural effusions over the course of three years in patient MC1286. Human MM cell lines were maintained in RPMI-1640 supplemented with 5% FBS and 1× GlutaMAX (Gibco), without antibiotics (70). All cell lines were tested for Mycoplasma contamination twice per year using the MycoAlert kit (Promega) and were identity-validated quarterly using copy-number polymorphism by PCR. Cell lines expressing exogenous BATF, BATF2, BATF3, BATF(H55Q), MYC, or IRF4 were generated by lentiviral transduction using human cDNA subcloned into pCDH lentiviral vector containing an EF1α promoter with IRES-EGFP markers used for FACS purification or flow identification (Supplementary Table S2). Pure populations of EGFP+ transduced cell lines were sorted on a five-laser Fortessa (Beckton Dickinson). Purity of >90% was confirmed prior to each experiment using a Cytoflex (Beckman Coulter). Pomalidomide (Pom) >98% purity was purchased from Pharmablock USA. GNE-781 (cat. #S8665), (+)-JQ1(cat. #S7110), and Molibresib (iBET-762; cat. #S7189) were purchased online from Selleckchem; all were solubilized in DMSO for in vitro use, or in 0.5% carboxymethyl-cellulose + 0.25% Tween-80 in water for oral gavage at indicated doses and frequencies. Dexamethasone was obtained from the clinical pharmacy and solubilized in saline for i.p. injection at 10 mg/kg on days 1–5 and 8–12.
Growth and Viability Assays
HMCLs were plated at 50,000 to 100,000 cells/mL to ensure a log-growth phase during the 3-day assay. Cells were plated in 6-well plates and then treated with DMSO or drug. Growth index scores were calculated as follows: (final cell number of drug treated – starting cell number)/(final cell number of DMSO treated – starting cell number). Cell counts for growth index scores were performed in triplicate using a hemocytometer. IC50 values and viability scores were calculated using Promega CellTiter-Glo assays per kit instructions with each cell line normalized to vehicle-only–treated controls. For multiday growth curves, fold expansion was determined using normalized cell counts generated by expanded cells in triplicate in 96-well plates, treating with drug (or DMSO ctrl), then using a CytoFlex LX flow cytometer to mix and collect equal 35 μL volumes from each well then gating on viable cells (Annexin V–, Live/Dead−, excluding debris). Lenalidomide area under the curve sensitivity was determined previously (71).
Flow Cytometry and FACS
Single-cell suspensions of HMCLs or ACK-lysed tissue preparations from necropsy of mice were stained using Annexin V–PE at 1:50 (BD Pharmingen 556422) and Live/Dead Violet at 1:100 (Thermo Fisher L34955). Samples were analyzed using a five-laser CytoFlex LX (Beckman Coulter) or sorted to >97% purity using a BD Biosciences FACSAria III.
Mice
All experiments were performed under the approval of Mayo Foundation Institutional Animal Care and Use Committee (IACUC) approval and conformed to all the regulatory Environmental Safety standards. All mice were maintained on a C57Bl/6J background. The generation and initial characterization of the Vk*MYC mice (Tg(Igkv3-5*-MYC)#Plbe) and derived transplantable lines have been reported elsewhere (42, 43, 70). The original Vk*MYC mice (RRID:MMRC_68098_MU, MGI ID: 7430707 and 6856586) and their derivative lacking LoxP sites, Vk*MYCDLox (RRID: MMRRC_068099-MU, MGI ID: 6856591) have been deposited to the MMRRC repository, Human CRBN transgenic mice were generated at the Mayo Clinic by microinjection into C57BL/6J pronuclei two independent bacterial artificial chromosomes (BAC): CTD-2335G16 spanning the entire hCRBN gene and RP11-1042H15 extending to include additional regulatory regions. Two founder lines were obtained: hC123 (Tg(CRBN)123Plbe, MGI ID: 7429138), from the former BAC and hC343 (Tg(CRBN)343Plbe, MGI ID: 7429136) from the latter, both capable of germline transmission, detectable by PCR on tail DNA using primers: TAAAGGTGCAGCATGCCAAAC and AGAGCCATTCTGTGTGCATCA. Both founder lines were independently bred into murine CRBN-null background, Crbntm1.2Jjh−/− MGI_ID: 5302192 (72) and to Vk*MYC or Vk*MYCDLox mice to generate Vk*MYC-hCRBN mice. Transgenic (de novo) mice were aged and monitored via weekly tail-bleeds for the appearance of a monoclonal protein spike (“M-Spike”—a correlate for tumor burden) by serum protein electrophoresis. M-spikes were quantified by calculating the ratio of densitometric values of the M-spike and albumin bands (G/A), assuming albumin at a standard value of 27 g/L. Transplantation details are provided in Supplementary figures. Transplanted mice were enrolled in drug studies when their M-spike was detected with a G/A >0.1 depending on the aggressiveness of the line. Mice were randomized to different treatment arms, stratified by the size of their M-spikes. Unless specified, treatment duration was 12 days. Blood cell counts in Fig. 3J were calculated using a Heska Hematology Analyzer.
Western Blots
All antibodies for western blots were purchased online. BATF (D7C5, Cell Signaling 8638, used at 1:1,000), BATF2 (PCRP-BATF2-2B9, Developmental Studies Hybridoma Bank, University of Iowa; used at 1:40), BATF3 (3H1, Abnova, H00055509-M04, used at 1:1,000), c-MYC (Y69, Abcam, ab32072, used at 1:1,000), IKZF1 (H-100, Santa Cruz, sc-13039, used at 1:1,000), IKZF3 (D1C1E, Cell Signaling Technology #15103, used at 1:1,000), IRF4 (Cell Signaling Technology, #4964 used at 1:1,000), Cyclin D1 (A-12, Santa Cruz, sc-8396, used at 1:1,000), FGFR3 (C51F2, Cell Signaling Technology, #4574, used at 1:1,000), cMAF (M-153, Santa Cruz, sc7866, used at 1:1,000), b-Actin (AC-15, Abcam, ab6276, used at 1:2,000). Cell extracts were prepared by harvesting cells, washing 2× with cold PBS, adding cold RIPA buffer to the cell pellet at a 3:1 ratio, and resting on ice for 40 minutes with a 1-minute vortex every 10 minutes. Cell mixtures were then spun down at 14,000 rpm at 4 degrees and the supernatant was transferred to a clean tube. Protein concentrations of the supernatants were determined using Pierce BAC protein assay (Thermo Fisher; cat. #23225). For each western, 20 μg of total protein was loaded per lane. Western blots were run using Invitrogen Life Technologies Mini Gel systems (cat. #A25977) using 4%–12% or 8% Bis-Tris 1.0–1.5 mm mini gels at 120 V for 1–2 hours. Gels were then transferred to nitrocellulose membranes with Bio-Rad Mini-Trans Blot systems using 100 V for 1 hour at 4 degrees. To quantify the total protein signal for each gel lane, membranes were immediately stained and imaged following transfer using REVERT total protein quantification stain per kit instructions (Li-Cor #926-11010). REVERT signal was obtained using an Odyssey DLx imaging system. Following total protein imaging, REVERT was immediately removed and the membrane was washed using kit reagents. Next, membranes were blocked in 5% milk for 2 hours at RT, rinsed in TBST, and stained overnight with primary antibodies in 5% milk. The following day, membranes were rinsed and stained with appropriate secondary antibodies at 1:10,000 for 1 hour at RT, then imaged using and Odyssey DLx. All gels were analyzed using Image Studio software. Band intensity changes were calculated by first normalizing total protein (REVERT signal) across all lanes, followed by a second normalization to DMSO control (band-of-interest signal), per Li-Cor protocol. In the majority of figures, Beta-Actin is included as a visual confirmation of normalized loading.
RNA-Seq
KMS11 and JJN3 cells were treated with 200 nmol/L POM and/or 40 nmol/L GNE781 and viable cells (Annexin V– Live/Dead Dye−) were sorted to >97% purity using a BD Biosciences FACSAria III after 48 hours of treatment for RNA-seq analysis performed by GeneWiz. RNA-seq data for 66 MM cell lines were downloaded as a matrix text file from http://www.keatslab.org/data-repository containing the gene-expression levels (FPKM). These mRNA-seq data were generated by Dr. Jonathan J. Keats, which were aligned with Tophat2 and calculated expression estimates by Cufflinks2 using Ensembl64 gene models (52).
RNA-Seq Analysis
RNA-seq analysis was performed similar to previously described (73). Briefly, FASTQ files were quality trimmed with TrimGalore! (v0.6.4) and CutAdapt (v2.5), and mapped to the GRCh38 reference genome (GRCh38.d1.vd1.fa) with the GENCODE v22 transcription database using STAR aligner (v2.5.3a; ref. 74). Putative PCR duplicate reads were marked in BAM files using Samtools (v1.10; ref. 75). R (v4.1.2; ref. 76) was used to determine read counts using the “summarizeOverlaps” function of the GenomicAlignments package (v1.30.0) to identify exonic read counts. Gene-expression data were normalized for sequencing depth using fragments per kilobase per million autosomal reads (FPKM). Differential expressed genes were determined using edgeR (v3.36.0; ref. 77) imposing an FDR ≤0.01 and fold-change ≥2. Only genes with ≥1 FPKM were included in the differential analysis. GSEA; v4.2.0; ref. 78) used the Hallmark gene set and a preranked list where rank was determined as follows:
Heat maps of differentially expressed genes were produced with bespoke code available upon request. Overlap of differentially expressed genes and myeloma dependencies used DepMap (21Q2) data and significance was assessed by the Fisher exact test.
ATAC-Seq
KMS11 and JJN3 cells were treated for 48 hours with 200 nmol/L POM, 40 nmol/L GNE781, or the combination, after which Annexin V–, Live/Dead Dye– viable cells were sorted to >97% purity using a BD Biosciences FACSAria III. Nuclei isolation, transposition, and DNA sequencing library preparations were performed using the protocol from Buenrostro and colleagues (79). Briefly, nuclei were tagmented with the Nextera DNA Library Prep kit (Illumina REF15028212) followed by PCR amplification and barcode addition using IDT for Illumina Nextera DNA UD index Plate A, followed by SPRI bead purification. Sequencing was performed by Novogene (Novogene Co. Inc.), where they were checked for quality assurance, and then sequenced using 150M paired-end reads using an Illumina Hi-Seq platform.
ATAC-Seq Analysis
ATAC-seq FASTQ files were quality and adapter trimmed using Trim Galore! (v0.6.4) and CutAdapt (v2.5) and mapped to the GRCh38 reference genome using bowtie2 (v2.3.5.1; ref. 80). Aligned significance analysis of microarray (SAM) files were converted to BAM files and putative PCR duplicates were marked with Samtools (v1.10; ref. 75). Regions of chromatin accessibility were identified in each sample using MACS2 (v2.1.1.20160309; ref. 81). The union of all accessible regions was determined and reads mapping to these regions for each sample was assessed using the “summarizeOverlaps” function of the GenomicAlignments package (v1.30.0) in R (v4.1.2). Regions that overlapped ENCODE blacklisted regions were removed (82). As a quality control metric, the number of reads in peaks was determined and used to calculate a normalized accessibility score as reads per peak million (RPPM) as previously described (83) and according to the following formula:
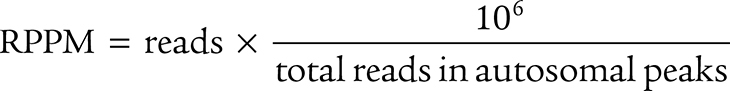 |
Differential chromatin accessible regions were determined using edgeR (v3.36.0; ref. 77) and imposing an FDR ≤0.05 and a fold change ≥1.5. Overlap of differential chromatin accessible regions with transcription-factor binding motifs used curated motifs from HOMER software (84). Here, motifs were determined genome-wide using HOMER, and the overlap of motifs with differentially accessible regions was determined using the Fisher exact test and FDR correction.
ChIP-Seq Analysis
ChIP-seq FASTQ files were processed as described for ATAC-seq (above) and factor-bound (“peaks”) regions were determined using MACS2 (v2.1.1.20160309; ref. 81) relative to IgG control. Motifs enriched in ChIP-seq peaks were determined using HOMER motifs, similar to ATAC-seq above, with enrichment determined relative to shuffled regions throughout the genome of the same number and size. Odds ratio and significance were determined by the Fisher exact test. Comparison between IRF4 in KMS12BM empty vector and BATF2 expressing cells used the same number of peaks to make odds ratios and P values comparable.
CoMMpass RNA-Seq Analysis
CoMMpass RNA-seq counts were from Genomic Data Commons where HTseq normalized read counts (FPKM) were downloaded and combined. Structural variants including translocations and copy-number alterations for the same samples were derived from whole-genome sequencing data as previously described (9). Outcome data, including PFS and OS, clinical correlates including stage (ISS), age, and beta-2-microglobulin levels, and patient self-identified race, were obtained as part of the Multiple Myeloma Research Foundation CoMMpass data package (IA17). Further details on the generation of CoMMpass data can be found at MedRxiv 2021.08.02.21261211.
CoMMpass Gene-Expression Subtypes
CoMMpass gene-expression subtypes were mapped to those defined by Zhan and colleagues (55). Here, the 50 top upregulated and 50 top downregulated genes in each subtype were obtained from the original publication (55) and the SAM score for each gene was used to weight CoMMpass RNA values, which were log transformed [log2(FPKM + 1)] and z-score normalized so as to weight each gene evenly. The product sum of SAM scores and CoMMpass RNA values was used to create an overall score for each gene-expression subtype and each sample was assigned to the gene-expression subtype with the highest score. Importantly, these gene-expression subtypes correlated with genomic structural events that partially define those gene-expression subtypes.
CoMMpass Gene Expression and Outcome
The association of gene expression and PFS and OS was determined using Cox proportional hazards regression using the “coxph” function of the survival package (v3.5.0) in R (v4.1.2). Gene expression was log transformed [log2(FPKM + 1)] and treated as a quantitative variable with respect to OS and PFS. P values were calculated using Wald's test. Multivariate analysis was also performed considering patients’ age, stage (ISS), and the presence of high-risk genetic alterations [t(4;14), t(14;16), del(17p)] as determined by whole-genome sequencing. Kaplan–Meier survival plots were also created in R using the “survfit” function of the survival package with patients separated by the median expression for visualization.
CoMMpass Gene-Expression Correlates
Gene expression associated with BATF expression was determined using a generalized linear model in edgeR where BATF expression was treated as a quantitative variable (v3.36.0; ref. 77). Gene expression was filtered for expressed genes (FPKM ≥1 in 5% of samples analyzed), BATF was removed, and only NDMM bone marrow samples were analyzed. Results were FDR corrected. GSEA used a preranked list and MSigDb Hallmark gene sets to annotate genes correlated with BATF expression.
Data Access Statement
Genomic data generated as part of this study are deposited in the Gene-Expression Omnibus under the super-series accession number GSE244003 with the individual data sets provisioned as follows: RNA-seq: GSE243990, ATAC-seq: GSE243978, and ChIP-seq: GSE244002. Genomic data from the CoMMpass study (MedRxiv 2021.08.02.21261211) were obtained from the Genomic Data Commons (GDC; https://portal.gdc.cancer.gov/projects/MMRF-COMMPASS) through NIH dbGaP project phs000748.v7.p4. Access to CoMMpass was granted by the NIH dbGaP data use committee for general research use under project #14898: “Genetic and epigenetic analysis of myelomagenesis.” All CoMMpass Genomic data deposited in GDC as of October 2020 were utilized. RNA-seq expression levels (FPKM) on 66 MM cell lines are available at http://www.keatslab.org/data-repository (52).
Supplementary Material
Multi-variate analysis of BATF, BATF2, and BATF3 expression
Primers
Supplementary Figures 1-11, which support data and conclusions provided in the main figures
Differentially expressed genes between POM (200 nM), GNE781 (40nM) or both (Combo) in JJN3 and KMS11 control (eGFP) and BATF overexpressing cells
GSEA Hallmark Gene Sets
Differential Accessible Regions in JJN3 and KMS11
Gene expression associated with BATF in NDMM bone marrow CoMMpass samples
GSEA of genes associated with BATF expression in CoMMpass
Acknowledgments
We wish to dedicate this work to W. Michael Kuehl, who conceived and inspired this work as well as so many seminal discoveries over his lifetime and in the process helped train generations of multiple myeloma researchers. B.G. Barwick was supported by an ASH Scholar Award and NCI award 1K22CA266739. B.G. Barwick and L.H. Boise were supported by NCI award 3 R01 CA192844-04S1. B.G. Barwick, L.H. Boise, M. Chesi, and P.L. Bergsagel were supported by the Paula and Rodger Riney Foundation. B.G. Barwick, P. Neri, N.J. Bahlis, and L.H. Boise were supported by an MMRF Answer Fund Award. S.J. Welsh, M. Chesi, and P.L. Bergsagel were supported by a Cancer Moonshot U54 CA224018 and SPORE grant P50 CA186781.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Blood Cancer Discovery Online (https://bloodcancerdiscov.aacrjournals.org/).
Authors’ Disclosures
S.J. Welsh reports grants from NCI during the conduct of the study. B.G. Barwick reports grants from the American Society of Hematology, NIH/NCI, and Paula and Rodger Riney Foundation during the conduct of the study and personal fees from Multiple Myeloma Research Foundation outside the submitted work. N. Meurice reports grants from NIH/NCI during the conduct of the study and personal fees from AbbVie outside the submitted work. S. Lonial reports personal fees from Janssen, Celgene, BMS, Novartis, AbbVie, Genentech, GSK, Regeneron, and Pfizer outside the submitted work; and is a member of the Board of Directors with stock for TG Therapeutics (no oncology-related agents). L.H. Boise reports personal fees from AstraZeneca and AbbVie outside the submitted work. M. Chesi reports grants from NCI during the conduct of the study; grants from Pfizer and nonfinancial support from BMS outside the submitted work; in addition, M. Chesi has a patent for hCRBN transgenic mice licensed to Novartis and a patent for VkMYC cell line licensed to Pfizer. P.L. Bergsagel reports personal fees from CellCentric, grants from NCI and Paula and Rodger Riney Foundation during the conduct of the study; personal fees from Pfizer, AbbVie, Salarius, Oncopeptides, and GlaxoSmithKline outside the submitted work; in addition, P.L. Bergsagel has a patent for hCRBN transgenic mice licensed to Novartis and a patent for VkMYC cell line licensed to Pfizer. No disclosures were reported by the other authors.
Authors’ Contributions
S.J. Welsh: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. B.G. Barwick: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. E.W. Meermeier: Methodology, writing–review and editing. D.L. Riggs: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, project administration. C.-X. Shi: Data curation, investigation, methodology. Y.X. Zhu: Data curation, investigation, methodology. M.E. Sharik: Data curation, investigation, methodology. M.T. Du: Data curation, investigation, methodology. L.D. Abrego Rocha: Investigation. V.M. Garbitt: Data curation, investigation, methodology. C.K. Stein: Data curation, formal analysis, investigation, methodology. J.L. Petit: Data curation, formal analysis, investigation, methodology. N. Meurice: Data curation, formal analysis, investigation, methodology. Y. Tafoya Alvarado: Investigation. R. Fonseca: Investigation. K.T. Todd: Investigation. S. Brown: Investigation. Z.J. Hammond: Investigation. N.H. Cuc: Investigation. C. Wittenberg: Investigation. C. Herzog: Investigation. A.V. Roschke: Investigation. Y.N. Demchenko: Investigation. W.-d.D. Chen: Investigation. P. Li: Data curation, investigation, methodology. W. Liao: Data curation, investigation, methodology. W.J. Leonard: Data curation, investigation, methodology. S. Lonial: Data curation, supervision, writing–review and editing. N.J. Bahlis: Conceptualization, investigation. P. Neri: Conceptualization, investigation. L.H. Boise: Formal analysis, supervision, investigation, methodology, writing–review and editing. M. Chesi: Conceptualization, resources, formal analysis, supervision, funding acquisition, validation, investigation, methodology, project administration, writing–review and editing. P.L. Bergsagel: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res 2004;64:1546–58. [DOI] [PubMed] [Google Scholar]
- 2. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest 2012;122:3456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, et al. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood 1998;91:4457–63. [PubMed] [Google Scholar]
- 4. Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet 1997;16:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci U S A 1996;93:13931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood 1996;88:674–81. [PubMed] [Google Scholar]
- 7. Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer 2002;2:175–87. [DOI] [PubMed] [Google Scholar]
- 8. Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia 2014;28:1725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun 2019;10:1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020;34:322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol 2006;7:773–82. [DOI] [PubMed] [Google Scholar]
- 12. Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature 2008;454:226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood 2014;124:1873–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155:934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X, Cairns MJ, Yan J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res 2019;47:11481–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 2015;521:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pott S, Lieb JD. What are super-enhancers? Nat Genet 2015;47:8–12. [DOI] [PubMed] [Google Scholar]
- 18. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conery AR, Centore RC, Neiss A, Keller PJ, Joshi S, Spillane KL, et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 2016;5:e10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romero FA, Murray J, Lai KW, Tsui V, Albrecht BK, An L, et al. GNE-781, A highly advanced potent and selective bromodomain inhibitor of cyclic adenosine monophosphate response element binding protein, binding protein (CBP). J Med Chem 2017;60:9162–83. [DOI] [PubMed] [Google Scholar]
- 24. Breen ME, Mapp AK. Modulating the masters: chemical tools to dissect CBP and p300 function. Curr Opin Chem Biol 2018;45:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vannam R, Sayilgan J, Ojeda S, Karakyriakou B, Hu E, Kreuzer J, et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem Biol 2021;28:503–14. [DOI] [PubMed] [Google Scholar]
- 26. Hogg SJ, Motorna O, Cluse LA, Johanson TM, Coughlan HD, Raviram R, et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell 2021;81:2183–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep 2018;24:1722–9. [DOI] [PubMed] [Google Scholar]
- 28. Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol 2016;3:e196–204. [DOI] [PubMed] [Google Scholar]
- 29. Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer 2021;124:1478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014;343:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014;343:305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ding Y, Zhang B, Payne JL, Song C, Ge Z, Gowda C, et al. Ikaros tumor suppressor function includes induction of active enhancers and super-enhancers along with pioneering activity. Leukemia 2019;33:2720–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu YX, Shi CX, Bruins LA, Wang X, Riggs DL, Porter B, et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J 2019;9:19. 10.1038/s41408-019-0173-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cortés M, Georgopoulos K. Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med 2004;199:209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoshida T, Georgopoulos K. Ikaros fingers on lymphocyte differentiation. Int J Hematol 2014;100:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu Y, Zhang Z, Kashiwagi M, Yoshida T, Joshi I, Jena N, et al. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev 2016;30:1971–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4CRBN. Br J Haematol 2014;164:811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gopalakrishnan R, Matta H, Tolani B, Triche T, Chaudhary PM. Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. Oncogene 2016;35:1797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dimopoulos K, Fibiger Munch-Petersen H, Winther Eskelund C, Dissing Sjö L, Ralfkiaer E, Gimsing P, et al. Expression of CRBN, IKZF1, and IKZF3 does not predict lenalidomide sensitivity and mutations in the cereblon pathway are infrequent in multiple myeloma. Leuk Lymphoma 2019;60:180–8. [DOI] [PubMed] [Google Scholar]
- 40. Bird S, Pawlyn C. IMiD resistance in multiple myeloma: current understanding of the underpinning biology and clinical impact. Blood 2023;142:131–40. [DOI] [PubMed] [Google Scholar]
- 41. De Matos Simoes R, Shirasaki R, Downey-Kopyscinski SL, Matthews GM, Barwick BG, Gupta VA, et al. Genome-scale functional genomics identify genes preferentially essential for multiple myeloma cells compared to other neoplasias. Nat Cancer 2023;4:754–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008;13:167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012;120:376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leal AS, Williams CR, Royce DB, Pioli PA, Sporn MB, Liby KT. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett 2017;394:76–87. [DOI] [PubMed] [Google Scholar]
- 45. Nicosia L, Spencer GJ, Brooks N, Amaral FMR, Basma NJ, Chadwick JA, et al. Therapeutic targeting of EP300/CBP by bromodomain inhibition in hematologic malignancies. Cancer Cell 2023;23:S1535–6108. [DOI] [PubMed] [Google Scholar]
- 46. Brooks N, Raja M, Young BW, Spencer GJ, Somervaille TC, Pegg NA. CCS1477: a novel small molecule inhibitor of p300/CBP bromodomain for the treatment of acute myeloid leukaemia and multiple myeloma. Blood 2019;134:2560. [Google Scholar]
- 47. Welti J, Sharp A, Brooks N, Yuan W, McNair C, Chand SN, et al. Targeting the p300/CBP axis in lethal prostate cancer. Cancer Discov 2021;11:1118–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell 2017;170:564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meermeier EW, Welsh SJ, Sharik ME, Du MT, Garbitt VM, Riggs DL, et al. Tumor burden limits bispecific antibody efficacy through T cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discov 2021;2:354–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doane AS, Chu C-S, Di Giammartino DC, Rivas MA, Hellmuth JC, Jiang Y, et al. OCT2 pre-positioning facilitates cell fate transition and chromatin architecture changes in humoral immunity. Nat Immunol 2021;22:1327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 2012;490:543–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yellapantula VD, Allen K, Jelinek DF, Bergsagel L, Keats JJ. The comprehensive genomic characterization of all commercially and non-commercially available multiple myeloma cell lines. Blood 2013;122:1914.23900238 [Google Scholar]
- 53. Hsing M, Wang Y, Rennie PS, Cox ME, Cherkasov A. ETS transcription factors as emerging drug targets in cancer. Med Res Rev 2020;40:413–30. [DOI] [PubMed] [Google Scholar]
- 54. Fan F, Podar K. The role of AP-1 transcription factors in plasma cell biology and multiple myeloma pathophysiology. Cancers (Basel) 2021;13:2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood 2006;108:2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gupta VA, Barwick BG, Matulis SM, Shirasaki R, Jaye DL, Keats JJ, et al. Venetoclax sensitivity in multiple myeloma is associated with B-cell gene expression. Blood 2021;137:3604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fedele PL, Liao Y, Gong J-N, Yao Y, van Delft MF, Low MSY, et al. The transcription factor IRF4 represses proapoptotic BMF and BIM to licence multiple myeloma survival. Leukemia 2021;35:2114–8. [DOI] [PubMed] [Google Scholar]
- 58. Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Lugt BV, et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1 - IRF complexes. Science 2012;338:975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, Kc W, Albring JC, et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 2012;490:502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lopez-Girona A, Heintel D, Zhang L-H, Mendy D, Gaidarova S, Brady H, et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br J Haematol 2011;154:325–36. [DOI] [PubMed] [Google Scholar]
- 61. Cippitelli M, Stabile H, Kosta A, Petillo S, Gismondi A, Santoni A, et al. Role of Aiolos and Ikaros in the antitumor and immunomodulatory activity of IMiDs in multiple myeloma: better to lose than to find them. Int J Mol Sci 2021;22:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bjorklund CC, Lu L, Kang J, Hagner PR, Havens CG, Amatangelo M, et al. Rate of CRL4CRBN substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J 2015;5:e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jones JR, Barber A, Le Bihan Y-V, Weinhold N, Ashby C, Walker BA, et al. Mutations in CRBN and other cereblon pathway genes are infrequently associated with acquired resistance to immunomodulatory drugs. Leukemia 2021;35:3017–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sievers QL, Gasser JA, Cowley GS, Fischer ES, Ebert BL. Genome-wide screen identifies cullin-RING ligase machinery required for lenalidomide-dependent CRL4CRBN activity. Blood 2018;132:1293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shirasaki R, Matthews GM, Gandolfi S, de Matos Simoes R, Buckley DL, Raja Vora J, et al. Functional genomics identify distinct and overlapping genes mediating resistance to different classes of heterobifunctional degraders of oncoproteins. Cell Rep 2021;34:108532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Frede J, Anand P, Sotudeh N, Pinto RA, Nair MS, Stuart H, et al. Dynamic transcriptional reprogramming leads to immunotherapeutic vulnerabilities in myeloma. Nat Cell Biol 2021;23:1199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF–IRF interactions control immune-regulatory networks. Nat Rev Immunol. 2013;13:499–509. [DOI] [PubMed] [Google Scholar]
- 68. Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: a systematic study of clinical trials. Front Pharmacol 2020;11:621093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Neri P, Barwick BG, Jung D, Patton JC, Maity R, Tagoug I, et al. ETV4-dependent transcriptional plasticity maintains MYC expression and results in IMiD resistance in multiple myeloma. Blood Cancer Discov 2024;5:56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chesi M, Mirza NN, Garbitt VM, Sharik ME, Dueck AC, Asmann YW, et al. IAP antagonistssss induce anti-tumor immunity in multiple myeloma. Nat Med 2016;22:1411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bonolo de Campos C, Meurice N, Petit JL, Polito AN, Zhu YX, Wang P, et al. Direct to Drug” screening as a precision medicine tool in multiple myeloma. Blood Cancer J 2020;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rajadhyaksha AM, Ra S, Kishinevsky S, Lee AS, Romanienko P, DuBoff M, et al. Behavioral characterization of cereblon forebrain-specific conditional null mice: a model for human non-syndromic intellectual disability. Behav Brain Res 2012;226:428–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Barwick BG, Gupta VA, Matulis SM, Patton JC, Powell DR, Gu Y, et al. Chromatin accessibility identifies regulatory elements predictive of gene expression and disease outcome in multiple myeloma. Clin Cancer Res 2021;27:3178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc 2012;7:1728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Amemiya HM, Kundaje A, Boyle AP. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep 2019;9:9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR, et al. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun 2018;9:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multi-variate analysis of BATF, BATF2, and BATF3 expression
Primers
Supplementary Figures 1-11, which support data and conclusions provided in the main figures
Differentially expressed genes between POM (200 nM), GNE781 (40nM) or both (Combo) in JJN3 and KMS11 control (eGFP) and BATF overexpressing cells
GSEA Hallmark Gene Sets
Differential Accessible Regions in JJN3 and KMS11
Gene expression associated with BATF in NDMM bone marrow CoMMpass samples
GSEA of genes associated with BATF expression in CoMMpass