

Highlights
-
•
A comprehensive review on the historical perspective for amyotrophic lateral sclerosis is provided.
-
•
Accumulating evidence supports a cortical origin for amyotrophic lateral sclerosis.
-
•
Motor neuron degeneration in amyotrophic lateral sclerosis could be mediated by corticomotoneuronal hyperexcitability.
Keywords: Amyotrophic lateral sclerosis, Charcot, Dying-forward, Motor neuron disease
Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder of the human motor system, first described in the 19th Century. The etiology of ALS appears to be multifactorial, with a complex interaction of genetic, epigenetic, and environmental factors underlying the onset of disease. Importantly, there are no known naturally occurring animal models, and transgenic mouse models fail to faithfully reproduce ALS as it manifests in patients. Debate as to the site of onset of ALS remain, with three competing theories proposed, including (i) the dying-forward hypothesis, whereby motor neuron degeneration is mediated by hyperexcitable corticomotoneurons via an anterograde transsynaptic excitotoxic mechanism, (ii) dying-back hypothesis, proposing the ALS begins in the peripheral nervous system with a toxic factor(s) retrogradely transported into the central nervous system and mediating upper motor neuron dysfunction, and (iii) independent hypothesis, suggesting that upper and lower motor neuron degenerated independently. Transcranial magnetic stimulation studies, along with pathological and genetic findings have supported the dying forward hypothesis theory, although the science is yet to be settled. The review provides a historical overview of ALS, discusses phenotypes and likely pathogenic mechanisms.
1. Brief historical perspective
The historical description of ALS is closely associated with the founders of neurology, including Bell, Aran, Charcot, Duchenne, Erb, Gowers, Lockhart Clarke, and Brain (Gordon, 2006, Katz et al., 2015, Mitsumoto et al., 1998, Rose, 1999, Turner et al., 2010). These icons of neurology established ALS as an independent disease, although they disagreed on nosology, the definition of the disorder, and site of pathology. Jean-Martin Charcot (1825–1893) is generally credited for first describing ALS, but there were earlier descriptions (Charcot, 1880). During his Tuesday Lectures beginning in the late 1860s (Charcot and Charcot, 1987), Charcot was able to link the disease to its pathology (Charcot, 1880, Goetz, 2000, Goetz, 2010, Turner et al., 2010). Others laid the path along with, or before Charcot. Progressive muscular weakness accompanied by muscle wasting was clearly recognized by the mid-19th century. In 1848, François Aran, published a description of a new syndrome of progressive muscle weakness correctly suspecting a neurogenic cause. He subsequently published 11 such patients and suggested they had a new syndrome, progressive muscular atrophy (PMA). These patients were also seen by Amand Duchenne (de Boulogne) for galvanic electric therapy. Duchenne had worked in closely with Charcot at the Salpêtrière (Duchenne and Poore, 1883), and subsequently Charcot referred to PMA as the Aran-Duchenne type of progressive muscular weakness (Charcot, 1895).
Duchenne considered PMA as myogenic, and this view persisted until Cruveilhier in 1853 studied the same case. Because of changes in the spinal cord, he concluded the cause to be neurogenic. There were also reports of PMA associated with bulbar dysfunction, and these were probably ALS (Mitsumoto et al., 1998). In 1860, Bernard Luys, who described the corpus Luysii, reported ventral horn cell degeneration in PMA. The same year, Duchenne described progressive bulbar palsy (PBP) and distinguished it from PMA by the degree and rapidity of bulbar involvement.
Jean Martin Charcot was the first professor of neurology at the Salpêtrière, and together with Joffroy described two cases of PMA with lesions in the posterolateral spinal cord. They did not name this new syndrome ALS but determined its essential characteristics emphasizing that pathological changes in the posterior portions of the lateral columns and in the anterior horns of the spinal cord frequently occurred together. It was in his 1874 lecture series that Charcot established ALS as a distinct syndrome (Katz et al., 2015). Goldblatt expressed best Charcot's contribution: “Charcot did not give the first description of ALS, nor did Shakespeare originate the plots of his plays; the elements of the story were known, but it remained for the master to produce a masterpiece.” His description of ALS was based on 20 patients and five autopsy studies; most of the patients were women because Salpêtrière was a women's hospital. He set out the clinical and pathological characteristics of ALS that has since been modified very little (Duyckaerts et al., 2021, Rowland, 2001).
2. Introductory background
Amyotrophic lateral sclerosis (ALS/MND) is a uniquely human neurodegenerative complex disease having a variety of phenotypes, including frontotemporal dementia (Goutman et al., 2022, Hardiman et al., 2011, Kiernan et al., 2011). ALS phenotypes (age of onset, site of initial clinical presentation, disease duration, amongst others) are predicated by genetic, environmental, lifestyle and epigenetic influences (Grad et al., 2017). There are no known naturally occurring animal models, and induced animal models whilst usefully mimicking anterior horn cell death, and to a lesser extent loss of upper motor neurons (Marques et al., 2021), cannot truly recapitulate all aspects of the disorder as seen in humans (Bonifacino et al., 2021, Turner and Transgenics, 2008). The failure of ALS animal model mimics is probably multifactorial and includes, important anatomical differences, as related to the corticomotoneuronal system. But, differences in aging and longevity, lifestyle and the long and varied environmental exposures to which humans are subjected have to play a role.
Induced pluripotent stem cells (iPSC) are derived from skin or blood cells that have been reprogrammed back into an embryonic-like pluripotent state. This enables the development of an unlimited source of any type of human cell that can then be directed to therapeutic or investigative purposes. For example, iPSC can be prodded into becoming beta islet cells to treat diabetes, blood cells to create new blood free of cancer cells for leukemia, or neurons to investigate and treat neurological disorders. ALS is now increasingly studied using patient-derived iPSCs (Chen et al., 2014, Hawrot et al., 2020, Mazzini and De Marchi, 2023, Vasques et al., 2020), in which candidate risk factors with variable penetrance can be modelled and more recently manipulated using CRISPR gene editing (Kruminis-Kaszkiel et al., 2018, Pickles and Petrucelli, 2018, Vasques et al., 2020, Yun and Ha, 2020). These methodologies also have the potential advantage of investigating early stages of ALS. Akin to other neurodegenerative disorders, ALS has a preclinical period, probably of variable length, but likely extending years and possibly decades predating the onset of its classical clinical features (Eisen et al., 2014). By the time clinical features are apparent, the cellular cascades associated with neuronal death in ALS are likely irreversible and significant numbers of neurons have already degenerated (Fig. 1).
Fig. 1.

The seeds for the development of amyotrophic lateral sclerosis (ALS) may be sown shortly after conception. Motor neurons and supporting glia are susceptible to many potential insults, such as neuroinflammation, excitotoxicity, mitochondrial dysfunction, excessive oxidative stress and environmental risk factors. Epigenetic influences may further determine individual sensitivity and susceptibility. Environmental risk factors continue to exert their influence throughout life. In combination, these factors cause protein dysfunction and aggregation. Motor neurons and surrounding astrocytes are metabolically stressed, progressively losing function (MN ‘sickness’). After years or decades, cytosolic compensatory mechanisms begin to fail and a clinically identifiable pre-symptomatic stage starts in which electrophysiological and imaging abnormalities become detectable at a macroscopic level. Finally, the motor system fails, and ALS becomes symptomatic and relentlessly progressive (Eisen et al., 2014).
If so, biomarkers may yet become evident throughout the preclinical and presymptomatic stages, thereby enabling the future development of protective or preventive therapeutics such as modifying epigenetic effects. The term ‘presymptomatic’ refers to the period when there are no clinical correlates, while investigations such as neuroimaging, electrophysiology or cognitive assessment may be abnormal. ‘Preclinical’ refers to the much longer period when presently there are no identified markers of disease in sporadic ALS.
In sporadic ALS, interactive risk factors include genetic, environmental and lifestyle (Al-Chalabi and Hardiman, 2013). Since 1993, large scale genetic studies have identified more than 60 genes that are associated with ALS (Willemse and van Es, 2023). C9ORF72, SOD1, FUS, and TARDBP are the most common causative genes, accounting for more than 50 % of familial ALS (FALS) but also approximately 7.5 % of sporadic ALS (Duan et al., 2023). However, most genes that have been associated with ALS are considered risk genes. See Table 1 in Wang et al. (2023). The number of novel variants, particularly missense variants, associated with ALS are increasing but their clinical significance is unknown (Dilliott et al., 2023). Several mutations have been found to cluster within and across other major neurodegenerative diseases (Koretsky et al., 2023), underscoring the converging genetic and mechanistic pathways shared by these disorders (Gan et al., 2018, Wainberg et al., 2023). With technological and methodological advances some of the risk genes are likely to become considered causative, further blurring a firm separation of sporadic ALS from FALS.
Identification of lifestyle, environmental and occupational risk factors for ALS is important but difficult. Numerous non-genetic risk factors for ALS have been considered over many decades, but few have met the rigors required by epidemiologists, and replicable definitive non-genetic risk factors of ALS have not been identified (Duan et al., 2023). Head trauma, physical activity, electric shock, military service, pesticides, and lead, amongst others, are considered potential risk factors for ALS onset and progression (Andrew et al., 2021, Beaudin et al., 2022, Zhu et al., 2023). But, older age and male sex so far remain the only definitive non-genetic risks for ALS (Longinetti and Fang, 2019).
Even in early clinical phases, classic Charcot ALS is readily diagnosed by ALS physician specialists. A cognitively normal, 55-year-old patient with multi-myotomal muscle weakness and wasting associated with diffuse and profuse fasciculations, including the tongue, brisk reflexes in the weak, wasted limbs, but normal sensation and intact sphincter function is almost always diagnosed with ALS. Charcot (Charcot, 1874, Charcot, 1865), describing these characteristics over 150 years ago, was unaware of the extended ALS phenotypes and the multisystem nature of the disease as now accepted. He was adamant that ALS was not hereditary and made no mention of overlap with frontotemporal dementia (FTD) (Rowland, 2001, Veltema, 1975), but his clinical description of ALS cannot be bettered.
The question is often posed, and debate continues, as to whether ALS is one disease with a common fundamental pathogenic mechanism or multiple diseases with different mechanisms, having a shared final pathway. The answer may lie somewhere in between (Grad et al., 2017, Ravits and La Spada, 2009). In different patients, there is a striking dissimilarity in the degree of involvement of the upper motor neurons (UMNs) and the lower motor neurons (LMNs), the body regions affected, the degrees of involvement of other systems, especially cognition and behavior, and the progression rates among clinical phenotypes (Fig. 2).
Fig. 2.
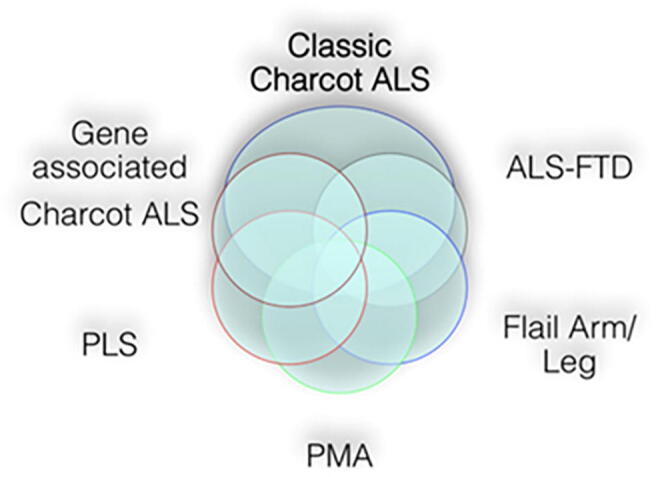
Classic Charcot ALS, characterized by mixed upper and lower motor neuron deficits can predominate as upper motor neuron (PLS) or lower motor neuron (SMA) and a small percentage of cases have a recognized causative gene (C9ORF72, SOD1, FUS, and TARDBP), but in most cases gene influences are risk factors not directly causative. Depending on the sophistication of testing up to 50 % of ALS may have cognitive impairment and some of these have definitive frontotemporal dementia. In >95 % of ALS, independent of phenotype, and 50 % of FTD have the hallmark TDP-43 inclusions.
These phenotypes sometimes appear so distinctive suggesting differing underlying biology. However, the known neuropathology, in particular extra-nuclear aggregation of TDP-43, the consistent hallmark of ALS does not clearly correlate with the different phenotypic subtypes of disease. The mechanism underlying TDP-43 cytoplasmic mislocalization and subsequent aggregation remains unclear. But TDP-43 dimerization/multimerization is impaired in the postmortem brains and spinal cords of patients with sporadic ALS and may be a critical determinant inducing TDP-43 pathology in ALS (Oiwa et al., 2023). Further, clinical phenotypes tell us nothing about lengthy pre-clinical events in ALS (Eisen et al., 2014). Our current view is that ALS is indeed one disease and the clinical phenotypes are a reflection of exposure to differing susceptibility genes, epigenetics, lifestyle and varying environmental exposures (Al-Chalabi and Hardiman, 2013).
3. Site of ALS onset – competing theories
Determining and understanding the site(s) of origin if ALS is more than just of academic interest. In common with other neurodegenerations, spread of toxic proteinaceous material, such as TDP43, appears key to disease progression. For example, mis-localized TDP-43 can induce layer V excitatory neurons in the rodent motor cortex to become hyperexcitable after 20 days of expression (Reale et al., 2023). Spread of pathogenic changes through the corticomotor system was observed and by 30 days there was a significant decrease in the number of motor neurons in the spinal cord. Cortical pathology increased excitatory inputs to the spinal cord to which local circuitry compensated with an upregulation of inhibition, indicating how TDP-43 mediated pathology spreads corticofugally in ALS (Reale et al., 2023). Pathogenic TDP-43 may spread in a prion-like propagation (Tamaki et al., 2023), but this is controversial and others suggest that ALS corticofugal propagation is likely not mediated by prion-like mechanisms, but relies on cortical hyperexcitability (Scekic-Zahirovic et al., 2021).
Charcot considered PMA as “protopathic” (meaning primary or essential), whereas ALS with both atrophic and spastic features was “deuteropathic” implying a corticofugal propagation of the lesion from white to gray matter. Sir William Gowers did not think Charcot's introduction of the term “ALS” to be very helpful and felt that he gave a new name to describe an old disease. Gowers considered PMA, PBP, and ALS were essentially one disease (Gowers, 1893, Gowers, 1896). Clinical manifestations were determined by the timing, extent, and severity of the degeneration in the upper and lower segments of the motor pathway. Brain in 1933 introduced the term “motor neuron disease” so that all these apparently different conditions could be brought together in a single general category and used the terms “motor neuron disease” and “ALS” interchangeably (Brain, 1962). These controversies lay dormant for many decades.
Specific to ALS, is dysfunction of the expanded human corticomotoneuronal system (Lemon and Griffiths, 2005). This system is the anatomical infrastructure of many early clinical features of ALS, a singularly human disorder (Eisen and Lemon, 2021). Early deficits include loss of vocalization requiring the integration of a complex respiratory system, impaired fractionation of digits and thumb opposability, responsible for manipulative agility, and difficulty with upright walking, especially the ability to navigate uneven and tricky surfaces (Fig. 3).
Fig. 3.

The CM system makes a key contribution to complex movements underlying human skilled motor repertoire, examples of which are shown. The motor deficits of ALS primarily reflect a failure of skilled movements involving muscle synergies (Eisen and Lemon, 2021).
These initial symptoms reflect dysfunction of the corticomotoneuronal system (Lemon, 2008). In addition is the association of frontotemporal dementia (FTD), causing language impairment, failing executive function, and deteriorating socialization (Snowden et al., 2013, Woolley and Strong, 2015).
These observations led Eisen et al. (Eisen et al., 1992) to develop the “the corticomotoneuronal hypothesis”, which postulates in essence that ALS is a brain disease and that demise of the spinal anterior horn cells occurs through a dying forward process (Fig. 4). There are clearly opposing views and debate continues, as it should, but a growing literature has come to support the dying forward concept (Baker, 2014, Braak et al., 2013, Eisen, 2021, Eisen et al., 2017, Ludolph et al., 2020, Vucic et al., 2021). Of interest, in 1988, Hudson and Kiernan (Hudson and Kiernan, 1988), in a rarely appreciated letter to The Lancet “Preservation of certain voluntary muscles in motorneurone disease”, proposed “that the extraocular muscle nuclei and pelvic sphincter neurons are spared in ALS because they are the only lower motor neurons that do not receive direct afferents from the cerebral cortex. Therefore, they would not be trans-synaptically affected by degeneration of the corticospinal tracts”. So, they hypothesized that the cortical neurons normally provide trophic support for the anterior horn cells and motor nuclei of the cranial nerves with which they make contact. Therefore, the primary pathology of ALS might be sought in the cortex rather than the spinal cord.
Fig. 4.

The idea of a cortical origin of ALS dates back to Charcot but lay largely dormant until 1988. Physiological studies commenced in 1992 (Eisen et al., 1992), and enhanced using threshold tracking techniques (Vucic and Kiernan, 2006, Vucic et al., 2021), strongly support a cortical origin of ALS. TDP-43 sequestered out of the cytoplasm possibly associated with cortical hyperexcitability and this is largely restricted to corticofugal fibers (Braak et al., 2017).
The identification of TAR DNA-binding protein 43 (TDP-43) positive ubiquitinated cytoplasmic inclusions in almost all patients with ALS and more than half of patients with frontotemporal dementia (FTD) has placed ALS on the so-called “ALS-FTD continuum,” highlighting the considerable clinical, pathophysiological, and neuroimaging overlap between the two neurodegenerative conditions (Neumann et al., 2006). TDP-43 proteinaceous buildup is largely restricted to corticofugal projecting neurons (“dying forward”) (Eisen et al., 2017). Evidence indicates that spinal motoneurons lose normal nuclear TDP-43 expression with subsequent formation of phosphorylated TDP-43 aggregates within their cytoplasm, but in Betz cells (and other pyramidal corticofugal neurons), by contrast, loss of nuclear TDP-43 expression is largely unassociated with the development of cytoplasmic aggregations (Braak et al., 2017). As a result, soluble and probably toxic cytoplasmic TDP-43 could then enter the axoplasm of Betz cells and other pyramidal neurons, with transmission by axonal transport to the corresponding spinal motoneurons in the lower brainstem and spinal cord, with dysregulation of normal nuclear protein. TDP-43 induced in the corticospinal neurons in a rodent model, was transported along the axons anterogradely and transferred to the oligodendrocytes along the corticospinal tract, coinciding with axon degeneration. In contrast, TDP-43 introduced in the spinal motor neurons in the same model, did not spread retrogradely to the cortical or spinal neurons (Tsuboguchi et al., 2023). Further, sophisticated MRI imaging and threshold tracking using transcranial magnetic stimulation (TMS), convincingly point to a cortical origin of ALS (Vucic and Kiernan, 2006, Vucic and Kiernan, 2013, Vucic et al., 2021, Vucic et al., 2013).
There is strong evidence for cortical hyperexcitability in ALS, derived from human electrophysiological studies as well as ALS animal models. There is a clear link between it and subsequent downstream degeneration and loss of anterior horn cells. (Kiernan and Park, 2023), Cortical hyperexcitability is a combined consequence of increased excitatory inputs to the upper motor neuron (UMN), paralleled by decreased inhibition, mediated through dysfunction of GABAergic interneurons fundamental to proper brain neural network functioning. The resulting imbalance in excitation/inhibition (E/I balance) is a feature shared by other neurodegenerative diseases (Gunes et al., 2022, Gunes et al., 2020). Threshold tracking TMS studies have established a combination of cortical disinhibition and increased facilitation, as heralded by reduction in short interval intracortical inhibition and increase of short interval intracortical facilitation respectively, in ALS and this E/I imbalance correlates with functional decline (Van den Bos et al., 2018).
In the developing nervous system, GABA is one of the earliest, and most highly evolutionary conserved neurotransmitters (Ramamoorthi and Lin, 2011). Unlike in the mature brain, during development, GABA is “excitatory” and embryonic GABA signaling is the main excitatory drive for developing cortical networks, and for corticogenesis (Kilb, 2012). The ability of embryonic GABA to depolarize primitive neurons is due to their high intracellular chloride concentration. The switch to inhibitory GABAergic neurons is completed during the first postnatal year and depends on the activity of the chloride transporters: potassium-chloride transporter member 5 (KCC2) and Na-K-Cl cotransporter 1 (NKCC1). Impaired GABA switch interferes with E/I balance which can likely have lasting effects (Kilb, 2012).
As indicated in Fig. 4, there are alternatives to dying forward. There are those that would favor a dying back mechanism and others as originally proposed by Gowers, who argue that the demise of upper and lower motor neurons occurs independently.
Further, it has been suggested that muscle degeneration is not just a passive bystander of motor neuron death but is part of an active process (Shefner et al., 2023). As recently reviewed (Shefner et al., 2023), while motor neuron degeneration clearly causes muscle weakness and atrophy, evidence also supports the hypothesis of a more active role of skeletal muscle in disease progression. Muscle fibers are precociously affected in ALS disease independently of motor neuron influence or denervation. For example, myoblasts from patients carrying ALS mutations show autonomous metabolic defects which affect muscle differentiation and muscle energy metabolism leading to decrease in muscle strength (Anakor et al., 2022, Peggion et al., 2022).
There is also evidence to support very early dysfunction of the neuromuscular junction (NMJ), in that functional deficits may appear at least 6 weeks before motor symptoms in vivo, while structural deficits occur 4 weeks later, and predominantly within NMJs (Lynch et al., 2021, McIntosh et al., 2023).
4. Split phenotypes
Early upper motor neuron deficits in ALS can be elusive and difficult to identify (Swash, 2012), although remain important for early diagnosis and admission into therapeutic drug trials (Hannaford et al., 2021). Split phenotypes are characterized by dissociated muscle weakness and wasting, where some muscles are affected whilst others having a shared peripheral nerve and spinal nerve root innervation are spared. This phenomenon which is probably unique to ALS has been recognized for several decades (Wilbourn, 2000).
The anatomical distribution of split phenotypes is reflective of corticomotoneuronal pathology (Eisen and Bede, 2021). TMS and cortical threshold tracking have been used to confirm this aspect of split phenotypes (Bae et al., 2014, Menon et al., 2014, Weber et al., 2000). F-wave persistence has been used as a surrogate for upper motor neuron dysfunction in split phenotypes (Wang et al., 2019a, Wang et al., 2019c). This is a useful measure of spinal motoneuron excitability relating to upper motor neuron dysfunction, in particular loss of cortical inhibition. But it lacks the specificity, and F-wave persistence occurs in other diseases associated with upper motor neuron dysfunction (Drory et al., 1993, Nakazumi and Watanabe, 1992).
Several split phenotypes have been reported: the split hand (Eisen and Kuwabara, 2012), the split hand plus (Menon et al., 2013), the split leg in two versions (leg and foot) (Min et al., 2020, Simon et al., 2015, Wang et al., 2019b), and the split elbow (Khalaf et al., 2019). For the split hand there is preferential thenar weakness/wasting compared to the hypothenar hand (Eisen and Kuwabara, 2012), the split hand plus also involves selective weakness/wasting of the flexor pollicis longus (Menon et al., 2013). A recently described split elbow is characterized by preferential involvement of the biceps muscle as compared to the triceps muscle (Khalaf et al., 2019, Thakore et al., 2021). In the split leg there is preferential plantar-flexion weakness/wasting compared to dorsi-flexion (Simon et al., 2015), but in contradistinction, in the split foot the extensor digitorum brevis (EDB) is preferentially involved compared to the abductor hallucis (AH) (Min et al., 2020, Wang et al., 2019b).
Ludolph et al. (Ludolph et al., 2020), have compared the strength of upper and lower limb muscle pairs in ALS. The weaker of the pair was the one with a stronger corticomotoneuronal (CM) drive, more monosynaptic connections. This too is true of the weak/wasted muscles of split phenotypes (Eisen and Bede, 2021) See Fig. 5. Preliminary data also indicate that for the upper limb it is the muscles with stronger CM connectivity that become weak before other muscles, independent of onset site (Thakore et al., 2021).
Fig. 5.

Split phenotypes appear to predominantly involve muscles that have the strongest corticomotoneuronal drive (red patches in the human figure) (Eisen and Bede, 2021). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
5. Extra motor pathology
Pathology beyond the motor system is well appreciated and is the basis of considering ALS a multisystem disorder. Extra motor pathology is not the same as non-motor symptoms, for which there can be difficult isolating as a true non-motor impairment and recently reviewed (Shojaie et al., 2023).
5.1. Frontotemporal dementia
Over the last two decades a growing appreciation that ALS is a multisystem disorder has expanded (Grossman, 2019, Silani et al., 2017). Neuropathological studies have shown the accumulation of misfolded protein aggregates in multiple cortical and subcortical areas of the CNS, including the corpus callosum (Brettschneider et al., 2013). The best recognized extra motor feature of ALS is frontotemporal dementia (Burrell et al., 2016, Burrell et al., 2011, Hudson, 1981, Kiernan, 2012, Lillo and Hodges, 2009, Snowden et al., 2013). Hexanucleotide repeat expansions in C9ORF72 gene are often associated with ALS-FTD (Renton et al., 2011), and aggregation of TDP-43 occurs in about 50 % of FTD cases (Bigio et al., 2013, Neumann et al., 2006). Neuropsychological deficits in ALS range from mild to severe and debilitating, and depending on the sophistication of testing, as many as 65 % of ALS patients exhibit some extent of cognitive or behavioral impairment (Goldstein and Abrahams, 2013). Many different neuropsychological deficits have been recorded in ALS, see Table 1 in Christidi et al. (2018).
5.2. Corpus callosum
Most axonal tracts, such as the anterior commissure, have evolved by the modified use of preexisting substrate pathways and very few entirely new axon tracts have arisen during evolution. The corpus callosum (CC), which is found only in placental mammals, is a truly new axon tract (Katz et al., 1983). Involvement of the CC as intricate to ALS has been recognized for many years (Chapman et al., 2012, Filippini et al., 2010). Mirror movements, which reflects interhemispheric disruption within the CC between the motor hemispheres during a motor task, may occur as an early manifestation of ALS (Wittstock et al., 2007).
The fibers of the corpus callosum arise from cortical pyramidal cells that are largely glutamatergic, affecting an excitatory role, although they are also known to act through GABA-mediated inhibitory neurons. In normal subjects, transcranial stimulation of one motor cortex elicits transcallosal inhibition of the other motor cortex, an approach shown to be of diagnostic value for studying callosal conduction and intracortical inhibitory mechanisms (Meyer et al., 1995). Using TMS, to measure the ipsilateral silent period, it has been shown that loss of callosal inhibition is a sensitive and early marker of disease activity in ALS (Hübers et al., 2021). CC hyperexcitability in early stages of disease might explain how TDP-43 aggregation spreads from one hemisphere to another (Hübers et al., 2021, Timmins et al., 2023). A reduction of transcallosal inhibition is associated with features indicative of cortical hyperexcitability, including reduction of short interval intracortical inhibition and increased in short interval intracortical facilitation. Reduction in transcallosal inhibition is also correlated with the rate of disease progression (Timmins et al., 2023, van den Bos et al., 2021).
5.3. Basal ganglia and parkinsonism
The ALS neuropathological staging system (Brettschneider et al., 2013), based on the distribution of TDP-43, indicates the large neurons of the thalamic nuclei that project to layer IV of the cerebral cortex develop TDP-43 aggregates in stage 2, while pTDP-43 pathology extends to caudate, putamen, and especially the ventral striatum in stage 3 of the disease. MRI studies in ALS patients show structural alterations of the basal ganglia and altered connections between the basal ganglia and frontal regions (Bede et al., 2013, Castelnovo et al., 2023). ALS is characterized by considerable degeneration in the basal ganglia, but surprisingly not associated with extrapyramidal signs (Rowland and Shneider, 2001). Basal ganglia degeneration has been shown to occur particularly in sporadic ALS-FTD-phenotype associated with cognitive decline (Masuda et al., 2016).
5.4. Hypothalamus
Atrophy of the hypothalamus is well recognized in ALS and can be visualized on MRI (Ye et al., 2021). Further, TDP-43 pathology, the pathological hallmark of ALS, has been reported in hypothalamic orexin neurons, and is associated with reduced body mass index (Cykowski et al., 2014). Gabery et al. (2021) have performed serial section microscopy to assess the degree of hypothalamic pathology (presence of TDP-43 inclusions, degree of atrophy and peptidergic neuronal loss) in ALS. Selective loss of orexin-producing neurons in the lateral hypothalamus was observed and considered to be progressive predating clinically overt disease which would result in sleep impairment.
Orexins A and B (hypocretins 1 and 2) and their two receptors (OX1R and OX2R) were discovered in 1998 (Hoyer and Jacobson, 2013). Orexin A and B are derived from the differential processing of a common precursor, the prepro-orexin peptide. The neuropeptides are expressed in a few thousand cells located in the lateral hypothalamus, but their projections and receptor distribution are widespread throughout the brain. The orexin system is involved in the regulation of sleep/wakefulness, feeding behavior, energy homeostasis, reward systems, cognition and mood (Azeez et al., 2021). Dysfunction of the orexin system has been implicated in sleep abnormalities and other issues associated with neurodegenerations (Al-Kuraishy et al., 2020). Given the substantial relationship between the glymphatic system, sleep and neurological disorders, there is likely a common key factor, and the orexin system is a good candidate (Christensen et al., 2021). Orexins are important in supressing REM sleep, whilst maintaining slow wave sleep, during which glymphatic clearance activity is highest (see section below).
6. Two lifestyle issues
Many non-genetic lifestyle factors have been considered to be associated with ALS. Examples include diet, alcohol use, stress, obesity, physical activity, and these and other environmental factors are considered elsewhere in this volume. Here we draw attention to two specific lifestyle issues that have now been shown to interact mechanistically with fundamental biological processes that could be causative in ALS and also underlie preclinical progression.
6.1. The glymphatic system, ALS, and sleep
Over the last decade there has been a growing interest in the glymphatic system, a term coined by the Danish neuroscientist Maiken Nedergaard to reflect its key components of glia cells and lymph (Hablitz and Nedergaard, 2021, Hablitz and Nedergaard, 2021, Jessen et al., 2015, Mestre et al., 2020, Ng Kee Kwong et al., 2020, Plog and Nedergaard, 2018, Rasmussen et al., 2018). Sleep is critically related to the glymphatic system which is an essential mechanism for clearance of protein aggregates, metabolic waste, and other toxic material from the brain (Anzai and Minoshima, 2021), replacing more classical cellular protein degradation pathways, autophagy and ubiquitination (Nedergaard and Goldman, 2020). Morphological and functional remodelling of astrocytes, with reduction and repositioning in aquaporin 4 (AQP4) channels in astrocyte end-feet, occurs in ALS. As a result, there is decline in the glymphatic clearance abilities, which includes TDP-43 aggregates and glutamate clearance (Verkhratsky et al., 2021), both mechanistically causative in ALS progression (see Fig. 6).
Fig. 6.

Neurodegeneration in ALS and neuronal demise involves a complex array of genetic, molecular, functional and pathological pathways. Genetic risk may exert influences across the lifespan, including ageing and lifestyle factors, particularly sleep. Dysfunction of the glymphatic system contributes to altered brain and neuronal function. Separately, glutamate excitotoxicity generates free radicals which further contribute to the process of neurodegeneration, with oxidative stress, mitochondrial dysfunction, intracellular aggregates that adversely affect transport processes, with imbalance of excitatory and inhibitory brain function promoting the emergence of hyperexcitability as a key factor linked to neurodegeneration in ALS.
Nightly sleep is critical for a wide array of brain functions (Lewis, 2021). Missing just a single night’s sleep results in memory, mood, and attentional impairments the next day and disrupted sleep across the lifespan is linked to neurodegeneration (Krause et al., 2017, Sabia et al., 2021). A variety of sleep disturbances have been described in Alzheimer’s and Parkinson’s diseases (Calderon-Garciduenas et al., 2023, Pillai and Leverenz, 2017, Stefani and Hogl, 2021). The sleep abnormalities are typically identified years or even decades prior to the diseases becoming clinically overt, and as such are potentially important early biomarkers of disease. In contrast, in ALS even though sleep abnormalities are common and have been well documented as coincident with the clinical disease (Lucia et al., 2021), impaired sleep in the preclinical setting, or as a possible risk factor for ALS, has not been reported.
6.2. What we eat – The gut microbiome
The microbiome refers to genes belonging to the myriad different microorganisms that live within and upon us, collectively known as the microbiota. Most of these microbes are found in the intestines, where they play important roles in digestion and generation of key metabolites including neurotransmitters (Boddy et al., 2021). In recent years, there has been increasing interest in the gut-brain neuroaxis and the gut microbiome, which modulates immune and metabolic health. In so doing it relates intricately with inflammation and bioenergetics, two major elements in the pathogenesis of ALS (Sun et al., 2021).
It is of interest that the diversity of the microbiome has changed significantly when compared to our hunter-gatherer ancestors (Carter et al., 2023). More than 100 gut-resident species have vanished with the advent of industrialized times, and this might be one reason why ALS, and other neurodegenerative disorders were not apparent until the mid- to late 19th century. Lifestyle aspects that have contributed to reduced diversity of the gut microbiome include consumption of highly processed foods, high rates of antibiotic administration, birth via cesarean section and use of baby formula, sanitation of the living environment, and reduced physical contact with animals and soil (Sonnenburg and Sonnenburg, 2019).
Martin et al. (2022) have reviewed data suggesting the novel roles of intestinal dysfunction and microbiota in ALS etiology and progression. ALS patients exhibit differences in their gut microbial communities compared with spouse controls, and modifying the gut microbiome, may have translational value for ALS treatment (Hertzberg et al., 2022, Kargbo, 2023).
7. Senescence and energy metabolism homeostasis
A relatively recent and rapid increase in longevity, largely determined by adequate shelter, good nutrition, medical advances, and reduced mortality in early life (Olshansky, 2022). This predicates an increasing incidence in neurodegenerations (Morris, 2013). As humans age, neocortical neurons are particularly vulnerable to the effects of senescence, which includes impaired energy metabolism homeostasis. This results in functional cellular failure and ultimately clinical disease (Kay et al., 1988).
Frontal and prefrontal cortical areas are preferentially involved early in ALS and FTD. An important component of frontal lobe development was the discrete modifications in local circuitry and interconnectivity of selected parts of the brain associated with scaling of the number and distribution of neurons. As the need for cognitive and motor skills increased, evolutionary pressures demanded greater cerebral energy metabolism of neurons with many synaptic connections and high synaptic activity that was very energy demanding (Cunnane et al., 1993, Wang et al., 2014). Intrinsic to ageing is a slowing of cerebral metabolism with disruption of neuronal homeostasis, mainly due to deficient energy metabolism (Ances et al., 2009, Villa et al., 2013). Although senescent neurons may remain metabolically active and can function within the neuronal network, their reduced metabolic efficiency will impact overall network integrity and performance.
The cascade of events that determine cellular senescence are poorly defined, but amongst other factors, a failing response to cellular energy demands ranks highly (Henderson et al., 2022). Over time there is metabolic exhaustion of energy-demanding neocortical neurons (Fig. 7). A possible mechanism for the protein aggregation lies in the energy costs of misfolded protein turnover, but other possibilities exist. Protein aggregation, the hallmark of neurodegenerations (FTP-43 in ALS and FTD), occurs as the increasing metabolic burden develops.
Fig. 7.

Neurons display high energy consumption with metabolic consumption of the brain representing 20% of the whole-body oxygen uptake. With increased evolutionary pressures as human cognitive and motor skills were fine-tunes neuronal energy demands increased. Energy deficiency induces mitochondrial dysfunction with failure of ATP production. Aging and senescence impair energy balance in homeostasis. At a cellular level this increases the tendency to protein degradation, aggregation and mis-localization with subsequent development of neurodegeneration (Henderson et al., 2022).
8. Concluding comments
It was not the intent to cover all aspects of ALS in this review. Many clinical aspects are well recognized and other important aspects such as new investigative methods and therapeutic approaches are considered elsewhere in this volume. Our intent was to draw attention to some controversial and unsettled issues. We appreciate that some of our views may be considered biased. Presently, our summary view is that ALS is a primary brain degeneration manifesting prominently as dysfunction of the CM system but that clearly the brain pathology is widespread and non-neuronal cells play an important role. Excitotoxicity is a prominent and early pathogenic mechanism, but how it and other pathogenic considerations interact continues to be an enigma. We accept this in the knowledge that the many aspects of ALS that are unresolved will continue to be modified through progressive scientific rigor.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Al-Chalabi A., Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013;9(11):617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- Al-Kuraishy H.M., Abdulhadi M.H., Hussien N.R., Al-Niemi M.S., Rasheed H.A., Al-Gareeb A.I. Involvement of orexinergic system in psychiatric and neurodegenerative disorders: A scoping review. Brain Circ. 2020;6(2):70–80. doi: 10.4103/bc.bc_42_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anakor E., Duddy W.J., Duguez S. The Cellular and Molecular Signature of ALS in Muscle. J. Pers. Med. 2022;12(11) doi: 10.3390/jpm12111868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ances B.M., Liang C.L., Leontiev O., Perthen J.E., Fleisher A.S., Lansing A.E., Buxton R.B. Effects of aging on cerebral blood flow, oxygen metabolism, and blood oxygenation level dependent responses to visual stimulation. Hum. Brain Mapp. 2009;30(4):1120–1132. doi: 10.1002/hbm.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew A.S., Bradley W.G., Peipert D., Butt T., Amoako K., Pioro E.P., et al. Risk factors for amyotrophic lateral sclerosis: A regional United States case-control study. Muscle Nerve. 2021;63(1):52–59. doi: 10.1002/mus.27085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzai Y., Minoshima S. Why We Need to Sleep: Glymphatic Pathway and Neurodegenerative Disease. Radiology. 2021;300(3):669–670. doi: 10.1148/radiol.2021211140. [DOI] [PubMed] [Google Scholar]
- Azeez I.A., Igado O.O., Olopade J.O. An overview of the orexinergic system in different animal species. Metab. Brain Dis. 2021;36(7):1419–1444. doi: 10.1007/s11011-021-00761-0. [DOI] [PubMed] [Google Scholar]
- Bae J.S., Menon P., Mioshi E., Kiernan M.C., Vucic S. Cortical hyperexcitability and the split-hand plus phenomenon: pathophysiological insights in ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014;15(3–4):250–256. doi: 10.3109/21678421.2013.872150. [DOI] [PubMed] [Google Scholar]
- Baker M.R. ALS – dying forward, backward or outward? Nat. Rev. Neurol. 2014;10(11):660. doi: 10.1038/nrneurol.2013.221-c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin M., Salachas F., Pradat P.F., Dupre N. Environmental risk factors for amyotrophic lateral sclerosis: a case-control study in Canada and France. Amyotroph. Lateral Scler. Frontotemporal Degener. 2022;23(7–8):592–600. doi: 10.1080/21678421.2022.2028167. [DOI] [PubMed] [Google Scholar]
- Bede P., Elamin M., Byrne S., McLaughlin R.L., Kenna K., Vajda A., et al. Basal ganglia involvement in amyotrophic lateral sclerosis. Neurology. 2013;81(24):2107–2115. doi: 10.1212/01.wnl.0000437313.80913.2c. [DOI] [PubMed] [Google Scholar]
- Bigio E.H., Weintraub S., Rademakers R., Baker M., Ahmadian S.S., Rademaker A., et al. Frontotemporal lobar degeneration with TDP-43 proteinopathy and chromosome 9p repeat expansion in C9ORF72: clinicopathologic correlation. Neuropathology. 2013;33(2):122–133. doi: 10.1111/j.1440-1789.2012.01332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy S.L., Giovannelli I., Sassani M., Cooper-Knock J., Snyder M.P., Segal E., et al. The gut microbiome: a key player in the complexity of amyotrophic lateral sclerosis (ALS) BMC Med. 2021;19(1):13. doi: 10.1186/s12916-020-01885-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino T., Zerbo R.A., Balbi M., Torazza C., Frumento G., Fedele E., et al. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. Int. J. Mol. Sci. 2021;22(22) doi: 10.3390/ijms222212236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Brettschneider J., Ludolph A.C., Lee V.M., Trojanowski J.Q., Tredici K.D. Amyotrophic lateral sclerosis-a model of corticofugal axonal spread. Nat. Rev. Neurol. 2013;9(12):708–714. doi: 10.1038/nrneurol.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Ludolph A.C., Neumann M., Ravits J., Del Tredici K. Pathological TDP-43 changes in Betz cells differ from those in bulbar and spinal alpha-motoneurons in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133(1):79–90. doi: 10.1007/s00401-016-1633-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain, W.R. (Ed.), 1962. Motor Neurone Disease. In: Diseases of the Nervous System. Oxford University Press, Oxford, UK.
- Brettschneider J., Del Tredici K., Toledo J.B., Robinson J.L., Irwin D.J., Grossman M., et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013;74(1):20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell J.R., Kiernan M.C., Vucic S., Hodges J.R. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134(Pt 9):2582–2594. doi: 10.1093/brain/awr195. [DOI] [PubMed] [Google Scholar]
- Burrell J.R., Halliday G.M., Kril J.J., Ittner L.M., Gotz J., Kiernan M.C., Hodges J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388(10047):919–931. doi: 10.1016/S0140-6736(16)00737-6. [DOI] [PubMed] [Google Scholar]
- Calderon-Garciduenas L., Torres-Jardon R., Greenough G.P., Kulesza R., Gonzalez-Maciel A., Reynoso-Robles R., et al. Sleep matters: Neurodegeneration spectrum heterogeneity, combustion and friction ultrafine particles, industrial nanoparticle pollution, and sleep disorders-Denial is not an option. Front. Neurol. 2023;14:1117695–1117701. doi: 10.3389/fneur.2023.1117695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter M.M., Olm M.R., Merrill B.D., Dahan D., Tripathi S., Spencer S.P., et al. Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell. 2023;186(14):3111–3124.e13. doi: 10.1016/j.cell.2023.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelnovo V., Canu E., De Mattei F., Filippi M., Agosta F. Basal ganglia alterations in amyotrophic lateral sclerosis. Front. Neurosci. 2023;17:1133758. doi: 10.3389/fnins.2023.1133758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman M.C., Jelsone-Swain L., Fling B.W., Johnson T.D., Gruis K., Welsh R.C. Corpus callosum area in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012;13(6):589–591. doi: 10.3109/17482968.2012.708935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charcot J.-M. Sclérose des cordons latéraux de la moelle épinière chez une femme hystérique atteinte de contracture permanente des quatre membres. Bull De La Societe Med. Hopit. De Paris. 1865;10:24–35. [Google Scholar]
- Charcot J.-M. Sclerose laterale amyotrophique. Oeuvres Complètes. Bureaux Du Progrès Médical. 1874;2:249–266. [Google Scholar]
- Charcot J.-M. Lecons Sur Les Maladies Du Systeme Nerveux Faites a La Salpetriere. 1880;2:227–242. [Google Scholar]
- Charcot J.M., Charcot G.CG. The Clinician: the Tuesday Lessons, Excerpts from Nine Case Presentations on General Neurology Delivered at the Salpêtrière Hospital in 1887–88 by Jean-Martin Charcot. 1987;xxx:193. [PubMed] [Google Scholar]
- Charcot, J.-B., 1895. Contribution à l'étude de l'atrophie musculaire progressive, type Duchenne-Aran. Paris: Progrès médical [etc.]. Medical Heritage Library. Online resource.
- Chen H., Qian K., Du Z., Cao J., Petersen A., Liu H., et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell. 2014;14(6):796–809. doi: 10.1016/j.stem.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J., Yamakawa G.R., Shultz S.R., Mychasiuk R. Is the glymphatic system the missing link between sleep impairments and neurological disorders? Examining the implications and uncertainties. Prog. Neurobiol. 2021;198:101917. doi: 10.1016/j.pneurobio.2020.101917. [DOI] [PubMed] [Google Scholar]
- Christidi F., Karavasilis E., Rentzos M., Kelekis N., Evdokimidis I., Bede P. Clinical and Radiological Markers of Extra-Motor Deficits in Amyotrophic Lateral Sclerosis. Front. Neurol. 2018;9:1005. doi: 10.3389/fneur.2018.01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnane S.C., Harbige L.S., Crawford M.A. The importance of energy and nutrient supply in human brain evolution. Nutr. Health. 1993;9(3):219–235. doi: 10.1177/026010609300900307. [DOI] [PubMed] [Google Scholar]
- Cykowski M.D., Takei H., Schulz P.E., Appel S.H., Powell S.Z. TDP-43 pathology in the basal forebrain and hypothalamus of patients with amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2014;2:171. doi: 10.1186/s40478-014-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilliott A.A., Kwon S., Rouleau G.A., Iqbal S., Farhan S.M.K. Characterizing proteomic and transcriptomic features of missense variants in amyotrophic lateral sclerosis genes. Brain. 2023 doi: 10.1093/brain/awad224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drory V.E., Neufeld M.Y., Korczyn A.D. F-wave characteristics following acute and chronic upper motor neuron lesions. Electromyogr. Clin. Neurophysiol. 1993;33(7):441–446. [PubMed] [Google Scholar]
- Duan Q.Q., Jiang Z., Su W.M., Gu X.J., Wang H., Cheng Y.F., et al. Risk factors of amyotrophic lateral sclerosis: a global meta-summary. Front. Neurosci. 2023;17:1177431. doi: 10.3389/fnins.2023.1177431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchenne G.B., Poore G.V. Selections from the Clinical Works by Duchenne (de Boulogne) 1883;xxii:472. [Google Scholar]
- Duyckaerts C., Maisonobe T., Hauw J.J., Seilhean D. Charcot identifies and illustrates amyotrophic lateral sclerosis. Free Neuropathol. 2021;2:2–12. doi: 10.17879/freeneuropathology-2021-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A. The Dying Forward Hypothesis of ALS: Tracing Its History. Brain Sci. 2021;11(3):300. doi: 10.3390/brainsci11030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A., Bede P. The strength of corticomotoneuronal drive underlies ALS split phenotypes and reflects early upper motor neuron dysfunction. Brain Behav. 2021;11(12):e2403. doi: 10.1002/brb3.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen A., Kuwabara S. The split hand syndrome in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 2012;83(4):399–403. doi: 10.1136/jnnp-2011-301456. [DOI] [PubMed] [Google Scholar]
- Eisen A., Braak H., Del Tredici K., Lemon R., Ludolph A.C., Kiernan M.C. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 2017;88(11):917–924. doi: 10.1136/jnnp-2017-315573. [DOI] [PubMed] [Google Scholar]
- Eisen A., Lemon R. The motor deficit of ALS reflects failure to generate muscle synergies for complex motor tasks, not just muscle strength. Neurosci. Lett. 2021;762:136171. doi: 10.1016/j.neulet.2021.136171. [DOI] [PubMed] [Google Scholar]
- Eisen A., Kim S., Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve. 1992;15(2):219–224. doi: 10.1002/mus.880150215. [DOI] [PubMed] [Google Scholar]
- Eisen A., Kiernan M., Mitsumoto H., Swash M. Amyotrophic lateral sclerosis: a long preclinical period? J. Neurol. Neurosurg. Psychiatry. 2014;85(11):1232–1238. doi: 10.1136/jnnp-2013-307135. [DOI] [PubMed] [Google Scholar]
- Filippini N., Douaud G., Mackay C.E., Knight S., Talbot K., Turner M.R. Corpus callosum involvement is a consistent feature of amyotrophic lateral sclerosis. Neurology. 2010;75(18):1645–1652. doi: 10.1212/WNL.0b013e3181fb84d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabery S., Ahmed R.M., Caga J., Kiernan M.C., Halliday G.M., Petersen A. Loss of the metabolism and sleep regulating neuronal populations expressing orexin and oxytocin in the hypothalamus in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021;47(7):979–989. doi: 10.1111/nan.12709. [DOI] [PubMed] [Google Scholar]
- Gan L., Cookson M.R., Petrucelli L., La Spada A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018;21(10):1300–1309. doi: 10.1038/s41593-018-0237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz C.G. Amyotrophic lateral sclerosis: early contributions of Jean-Martin Charcot. Muscle Nerve. 2000;23(3):336–343. doi: 10.1002/(sici)1097-4598(200003)23:3<336::aid-mus4>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Goetz C.G. Chapter 15: Jean-Martin Charcot and the anatomo-clinical method of neurology. Handb. Clin. Neurol. 2010;95:203–212. doi: 10.1016/S0072-9752(08)02115-5. [DOI] [PubMed] [Google Scholar]
- Goldstein L.H., Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol. 2013;12(4):368–380. doi: 10.1016/S1474-4422(13)70026-7. [DOI] [PubMed] [Google Scholar]
- Gordon P. In: New York. Mitsumoto H., Przedborski S., Gordon P.H., editors. Taylor & Francis; London: 2006. History of ALS. [Google Scholar]
- Goutman S.A., Hardiman O., Al-Chalabi A., Chio A., Savelieff M.G., Kiernan M.C., Feldman E.L. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21(5):465–479. doi: 10.1016/S1474-4422(21)00414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers W.R. Churchill; London: 1893. A manual of diseases of the nervous system. [Google Scholar]
- Gowers W.R. An Address on some Aspects of Diseases of the Nervous System and their Study. Br. Med. J. 1870;1896(2):1310–1312. doi: 10.1136/bmj.2.1870.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad L.I., Rouleau G.A., Ravits J., Cashman N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS) Cold Spring Harb. Perspect. Med. 2017;7(8) doi: 10.1101/cshperspect.a024117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman M. Amyotrophic lateral sclerosis - a multisystem neurodegenerative disorder. Nat. Rev. Neurol. 2019;15(1):5–6. doi: 10.1038/s41582-018-0103-y. [DOI] [PubMed] [Google Scholar]
- Gunes Z.I., Kan V.W.Y., Ye X., Liebscher S. Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 2020;14:573. doi: 10.3389/fnins.2020.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunes Z.I., Kan V.W.Y., Jiang S., Logunov E., Ye X.Q., Liebscher S. Cortical Hyperexcitability in the Driver’s Seat in ALS. Clin Transl Neurosci. 2022;5:1–19. [Google Scholar]
- Hablitz L.M., Nedergaard M. The Glymphatic System: A Novel Component of Fundamental Neurobiology. J. Neurosci. 2021;41(37):7698–7711. doi: 10.1523/JNEUROSCI.0619-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hablitz L.M., Nedergaard M. The glymphatic system. Curr. Biol. 2021;31(20):R1371–R1375. doi: 10.1016/j.cub.2021.08.026. [DOI] [PubMed] [Google Scholar]
- Hannaford A., Pavey N., van den Bos M., Geevasinga N., Menon P., Shefner J.M., et al. Diagnostic Utility of Gold Coast Criteria in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2021;89(5):979–986. doi: 10.1002/ana.26045. [DOI] [PubMed] [Google Scholar]
- Hardiman O., van den Berg L.H., Kiernan M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011;7(11):639–649. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- Hawrot J., Imhof S., Wainger B.J. Modeling cell-autonomous motor neuron phenotypes in ALS using iPSCs. Neurobiol. Dis. 2020;134 doi: 10.1016/j.nbd.2019.104680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R.D., Kepp K.P., Eisen A. ALS/FTD: Evolution, Aging, and Cellular Metabolic Exhaustion. Front. Neurol. 2022;13 doi: 10.3389/fneur.2022.890203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzberg V.S., Singh H., Fournier C.N., Moustafa A., Polak M., Kuelbs C.A., et al. Gut microbiome differences between amyotrophic lateral sclerosis patients and spouse controls. Amyotroph. Lateral Scler. Frontotemporal Degener. 2022;23(1–2):91–99. doi: 10.1080/21678421.2021.1904994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer D., Jacobson L.H. Orexin in sleep, addiction and more: is the perfect insomnia drug at hand? Neuropeptides. 2013;47(6):477–488. doi: 10.1016/j.npep.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Hübers A., Kassubek J., Müller H.P., Broc N., Dreyhaupt J., Ludolph A.C. The ipsilateral silent period: an early diagnostic marker of callosal disconnection in ALS. Ther. Adv. Chronic Dis. 2021;12 doi: 10.1177/20406223211044072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson A.J. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain. 1981;104(2):217–247. doi: 10.1093/brain/104.2.217. [DOI] [PubMed] [Google Scholar]
- Hudson A.J., Kiernan J.N. Preservation of certain voluntary muscles in motoneurone disease. Lancet. 1988;1(8586):652–653. doi: 10.1016/s0140-6736(88)91455-9. [DOI] [PubMed] [Google Scholar]
- Jessen N.A., Munk A.S., Lundgaard I., Nedergaard M. The Glymphatic System: A Beginner's Guide. Neurochem. Res. 2015;40(12):2583–2599. doi: 10.1007/s11064-015-1581-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargbo R.B. Microbiome-Gut-Brain Axis Modulation: New Approaches in Treatment of Parkinson's Disease and Amyotrophic Lateral Sclerosis. ACS Med. Chem. Lett. 2023;14(7):886–888. doi: 10.1021/acsmedchemlett.3c00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J.S., Dimachkie M.M., Barohn R.J. Amyotrophic Lateral Sclerosis: A Historical Perspective. Neurol. Clin. 2015;33(4):727–734. doi: 10.1016/j.ncl.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Katz M.J., Lasek R.J., Silver J. Ontophyletics of the nervous system: development of the corpus callosum and evolution of axon tracts. PNAS. 1983;80(19):5936–5940. doi: 10.1073/pnas.80.19.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay M.M., Bosman G., Notter M., Coleman P. Life and death of neurons: the role of senescent cell antigen. Ann. N. Y. Acad. Sci. 1988;521:155–169. doi: 10.1111/j.1749-6632.1988.tb35274.x. [DOI] [PubMed] [Google Scholar]
- Khalaf R., Martin S., Ellis C., Burman R., Sreedharan J., Shaw C., et al. Relative preservation of triceps over biceps strength in upper limb-onset ALS: the 'split elbow'. J. Neurol. Neurosurg. Psychiatry. 2019;90(7):730–733. doi: 10.1136/jnnp-2018-319894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan M.C. Amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry. 2012;83(4):355. doi: 10.1136/jnnp-2012-302357. [DOI] [PubMed] [Google Scholar]
- Kiernan M.C., Park S.B. Hyperexcitability, neurodegeneration, and disease progression in amyotrophic lateral sclerosis. Muscle Nerve. 2023 doi: 10.1002/mus.27843. [DOI] [PubMed] [Google Scholar]
- Kiernan M.C., Vucic S., Cheah B.C., Turner M.R., Eisen A., Hardiman O., et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- Kilb W. Development of the GABAergic system from birth to adolescence. Neuroscientist. 2012;18(6):613–630. doi: 10.1177/1073858411422114. [DOI] [PubMed] [Google Scholar]
- Koretsky M.J., Alvarado C., Makarious M.B., Vitale D., Levine K., Bandres-Ciga S., et al. Genetic risk factor clustering within and across neurodegenerative diseases. Brain. 2023 doi: 10.1093/brain/awad161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause A.J., Simon E.B., Mander B.A., Greer S.M., Saletin J.M., Goldstein-Piekarski A.N., Walker M.P. The sleep-deprived human brain. Nat. Rev. Neurosci. 2017;18(7):404–418. doi: 10.1038/nrn.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruminis-Kaszkiel E., Juranek J., Maksymowicz W., Wojtkiewicz J. CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS) Int. J. Mol. Sci. 2018;19(3) doi: 10.3390/ijms19030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008;31:195–218. doi: 10.1146/annurev.neuro.31.060407.125547. [DOI] [PubMed] [Google Scholar]
- Lemon R.N., Griffiths J. Comparing the function of the corticospinal system in different species: organizational differences for motor specialization? Muscle Nerve. 2005;32(3):261–279. doi: 10.1002/mus.20333. [DOI] [PubMed] [Google Scholar]
- Lewis L.D. The interconnected causes and consequences of sleep in the brain. Science. 2021;374(6567):564–568. doi: 10.1126/science.abi8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo P., Hodges J.R. Frontotemporal dementia and motor neurone disease: overlapping clinic-pathological disorders. J. Clin. Neurosci. 2009;16(9):1131–1135. doi: 10.1016/j.jocn.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Longinetti E., Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr. Opin. Neurol. 2019;32(5):771–776. doi: 10.1097/WCO.0000000000000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucia D., McCombe P.A., Henderson R.D., Ngo S.T. Disorders of sleep and wakefulness in amyotrophic lateral sclerosis (ALS): a systematic review. Amyotroph. Lateral Scler. Frontotemporal Degener. 2021;22(3–4):161–169. doi: 10.1080/21678421.2020.1844755. [DOI] [PubMed] [Google Scholar]
- Ludolph A.C., Emilian S., Dreyhaupt J., Rosenbohm A., Kraskov A., Lemon R.N., et al. Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J. Neurol. Neurosurg. Psychiatry. 2020;91(9):991–998. doi: 10.1136/jnnp-2020-323331. [DOI] [PubMed] [Google Scholar]
- Lynch E.M., Robertson S., FitzGibbons C., Reilly M., Switalski C., Eckardt A., et al. Transcriptome analysis using patient iPSC-derived skeletal myocytes: Bet1L as a new molecule possibly linked to neuromuscular junction degeneration in ALS. Exp. Neurol. 2021;345 doi: 10.1016/j.expneurol.2021.113815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques C., Burg T., Scekic-Zahirovic J., Fischer M., Rouaux C. Upper and lower motor neuron degenerations are somatotopically related and temporally ordered in the sod1 mouse model of amyotrophic lateral sclerosis. Brain Sci. 2021;11(3) doi: 10.3390/brainsci11030369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Battistini C., Sun J. A Gut Feeling in Amyotrophic Lateral Sclerosis: Microbiome of Mice and Men. Front. Cell. Infect. Microbiol. 2022;12 doi: 10.3389/fcimb.2022.839526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M., Senda J., Watanabe H., Epifanio B., Tanaka Y., Imai K., et al. Involvement of the caudate nucleus head and its networks in sporadic amyotrophic lateral sclerosis-frontotemporal dementia continuum. Amyotroph. Lateral Scler. Frontotemporal Degener. 2016;17(7–8):571–579. doi: 10.1080/21678421.2016.1211151. [DOI] [PubMed] [Google Scholar]
- Mazzini L., De Marchi F. iPSC-based research in ALS precision medicine. Cell Stem Cell. 2023;30(6):748–749. doi: 10.1016/j.stem.2023.05.008. [DOI] [PubMed] [Google Scholar]
- McIntosh J., Mekrouda I., Dashti M., Giuraniuc C.V., Banks R.W., Miles G.B., Bewick G.S. Development of abnormalities at the neuromuscular junction in the SOD1-G93A mouse model of ALS: dysfunction then disruption of postsynaptic structure precede overt motor symptoms. Front. Mol. Neurosci. 2023;16:1169075. doi: 10.3389/fnmol.2023.1169075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon P., Bae J.S., Mioshi E., Kiernan M.C., Vucic S. Split-hand plus sign in ALS: differential involvement of the flexor pollicis longus and intrinsic hand muscles. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013;14(4):315–318. doi: 10.3109/21678421.2012.734521. [DOI] [PubMed] [Google Scholar]
- Menon P., Kiernan M.C., Vucic S. Cortical excitability differences in hand muscles follow a split-hand pattern in healthy controls. Muscle Nerve. 2014;49(6):836–844. doi: 10.1002/mus.24072. [DOI] [PubMed] [Google Scholar]
- Mestre H., Mori Y., Nedergaard M. The Brain's Glymphatic System: Current Controversies. Trends Neurosci. 2020;43(7):458–466. doi: 10.1016/j.tins.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B.U., Roricht S., Grafin von Einsiedel H., Kruggel F., Weindl A. Inhibitory and excitatory interhemispheric transfers between motor cortical areas in normal humans and patients with abnormalities of the corpus callosum. Brain. 1995;118(Pt 2):429–440. doi: 10.1093/brain/118.2.429. [DOI] [PubMed] [Google Scholar]
- Min Y.G., Choi S.J., Hong Y.H., Kim S.M., Shin J.Y., Sung J.J. Dissociated leg muscle atrophy in amyotrophic lateral sclerosis/motor neuron disease: the 'split-leg' sign. Sci. Rep. 2020;10(1):15661. doi: 10.1038/s41598-020-72887-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsumoto, H., Chad, D.A., Pioro, E.P., 1998. History, Definition, and Classicification of ALS. In: Mitsumoto, H., Chad, D.A., Pioro, E.P. (Eds.), Amyotrophic Lateral Sclerosis. Contemporary Neurology Series. 49 ed. Oxford Press, New York. pp. 3–17.
- Morris J.C. Neurodegenerative disorders of aging: the down side of rising longevity. Mo. Med. 2013;110(5):393–394. [PMC free article] [PubMed] [Google Scholar]
- Nakazumi Y., Watanabe Y. F-wave elicited during voluntary contraction as a monitor of upper motor neuron disorder. Electromyogr. Clin. Neurophysiol. 1992;32(12):631–635. [PubMed] [Google Scholar]
- Nedergaard M., Goldman S.A. Glymphatic failure as a final common pathway to dementia. Science. 2020;370(6512):50–56. doi: 10.1126/science.abb8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ng Kee Kwong K.C., Mehta A.R., Nedergaard M., Chandran S. Defining novel functions for cerebrospinal fluid in ALS pathophysiology. Acta Neuropathol. Commun. 2020;8(1):140. doi: 10.1186/s40478-020-01018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oiwa K., Watanabe S., Onodera K., Iguchi Y., Kinoshita Y., Komine O., et al. Monomerization of TDP-43 is a key determinant for inducing TDP-43 pathology in amyotrophic lateral sclerosis. Sci. Adv. 2023;9(31) doi: 10.1126/sciadv.adf6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky S.J. From Life Span to Health Span: Declaring “Victory” in the Pursuit of Human Longevity. Cold Spring Harb. Perspect. Med. 2022;12(12) doi: 10.1101/cshperspect.a041480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggion C., Scalcon V., Massimino M.L., Nies K., Lopreiato R., Rigobello M.P., Bertoli A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants (Basel) 2022;11(4) doi: 10.3390/antiox11040614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S., Petrucelli L. CRISPR expands insight into the mechanisms of ALS and FTD. Nat. Rev. Neurol. 2018;14(6):321–323. doi: 10.1038/s41582-018-0005-z. [DOI] [PubMed] [Google Scholar]
- Pillai J.A., Leverenz J.B. Sleep and Neurodegeneration: A Critical Appraisal. Chest. 2017;151(6):1375–1386. doi: 10.1016/j.chest.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Plog B.A., Nedergaard M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018;13:379–394. doi: 10.1146/annurev-pathol-051217-111018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthi K., Lin Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol. Med. 2011;17(8):452–462. doi: 10.1016/j.molmed.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen M.K., Mestre H., Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17(11):1016–1024. doi: 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravits J.M., La Spada A.R. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73(10):805–811. doi: 10.1212/WNL.0b013e3181b6bbbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reale L.A., Dyer M.S., Perry S.E., Young K.M., Dickson T.C., Woodhouse A., Blizzard C.A. Pathologically mislocalised TDP-43 in upper motor neurons causes a die-forward spread of ALS-like pathogenic changes throughout the mouse corticomotor system. Prog. Neurobiol. 2023;226:102449. doi: 10.1016/j.pneurobio.2023.102449. [DOI] [PubMed] [Google Scholar]
- Renton A.E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., Gibbs J.R., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, F.C., 1999. A short history of neurology: the British contribution, 1660–1910. Butterworth-Heinemann, Oxford; Boston, ix, 282p.
- Rowland L.P. How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot. Arch. Neurol. 2001;58(3):512–515. doi: 10.1001/archneur.58.3.512. [DOI] [PubMed] [Google Scholar]
- Rowland L.P., Shneider N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001;344(22):1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Sabia S., Fayosse A., Dumurgier J., van Hees V.T., Paquet C., Sommerlad A., et al. Association of sleep duration in middle and old age with incidence of dementia. Nat. Commun. 2021;12(1):2289. doi: 10.1038/s41467-021-22354-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scekic-Zahirovic J., Fischer M., Stuart-Lopez G., Burg T., Gilet J., Dirrig-Grosch S., et al. Evidence that corticofugal propagation of ALS pathology is not mediated by prion-like mechanism. Prog. Neurobiol. 2021;200:101972. doi: 10.1016/j.pneurobio.2020.101972. [DOI] [PubMed] [Google Scholar]
- Shefner J.M., Musaro A., Ngo S.T., Lunetta C., Steyn F.J., Robitaille R., et al. Skeletal muscle in amyotrophic lateral sclerosis. Brain. 2023 doi: 10.1093/brain/awad202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaie A., Rota S., Al Khleifat A., Ray Chaudhuri K., Al-Chalabi A. Non-motor symptoms in amyotrophic lateral sclerosis: lessons from Parkinson's disease. Amyotroph. Lateral Scler. Frontotemporal Degener. 2023:1–10. doi: 10.1080/21678421.2023.2220748. [DOI] [PubMed] [Google Scholar]
- Silani V., Ludolph A., Fornai F. The emerging picture of ALS: a multisystem, not only a “motor neuron disease. Arch. Ital. Biol. 2017;155(4):99–109. doi: 10.12871/00039829201741. [DOI] [PubMed] [Google Scholar]
- Simon N.G., Lee M., Bae J.S., Mioshi E., Lin C.S., Pfluger C.M., et al. Dissociated lower limb muscle involvement in amyotrophic lateral sclerosis. J. Neurol. 2015;262(6):1424–1432. doi: 10.1007/s00415-015-7721-8. [DOI] [PubMed] [Google Scholar]
- Snowden J.S., Harris J., Richardson A., Rollinson S., Thompson J.C., Neary D., et al. Frontotemporal dementia with amyotrophic lateral sclerosis: A clinical comparison of patients with and without repeat expansions in C9orf72. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013;14:172–176. doi: 10.3109/21678421.2013.765485. [DOI] [PubMed] [Google Scholar]
- Sonnenburg J.L., Sonnenburg E.D. Vulnerability of the industrialized microbiota. Science. 2019;366:6464. doi: 10.1126/science.aaw9255. [DOI] [PubMed] [Google Scholar]
- Stefani A., Hogl B. A step forward in understanding the role of sleep and its link to neurodegeneration. Brain. 2021;144(3):700–702. doi: 10.1093/brain/awab047. [DOI] [PubMed] [Google Scholar]
- Sun J., Huang T., Debelius J.W., Fang F. Gut microbiome and amyotrophic lateral sclerosis: A systematic review of current evidence. J. Intern. Med. 2021;290(4):758–788. doi: 10.1111/joim.13336. [DOI] [PubMed] [Google Scholar]
- Swash M. Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J. Neurol. Neurosurg. Psychiatry. 2012;83(6):659–662. doi: 10.1136/jnnp-2012-302315. [DOI] [PubMed] [Google Scholar]
- Tamaki Y., Ross J.P., Alipour P., Castonguay C.E., Li B., Catoire H., et al. Spinal cord extracts of amyotrophic lateral sclerosis spread TDP-43 pathology in cerebral organoids. PLoS Genet. 2023;19(2) doi: 10.1371/journal.pgen.1010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore N.J., Drawert B.J., Lapin B.R., Pioro E.P. Progressive arm muscle weakness in ALS follows the same sequence regardless of onset site: use of TOMS, a novel analytic method to track limb strength. Amyotroph. Lateral Scler. Frontotemporal Degener. 2021:1–8. doi: 10.1080/21678421.2021.1889000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins H.C., Vucic S., Kiernan M.C. Cortical hyperexcitability in amyotrophic lateral sclerosis: from pathogenesis to diagnosis. Curr. Opin. Neurol. 2023 doi: 10.1097/WCO.0000000000001162. [DOI] [PubMed] [Google Scholar]
- Tsuboguchi S., Nakamura Y., Ishihara T., Kato T., Sato T., Koyama A., et al. TDP-43 differentially propagates to induce antero- and retrograde degeneration in the corticospinal circuits in mouse focal ALS models. Acta Neuropathol. 2023 doi: 10.1007/s00401-023-02615-8. [DOI] [PubMed] [Google Scholar]
- Turner M.R., Swash M., Ebers G.C. Lockhart Clarke's contribution to the description of amyotrophic lateral sclerosis. Brain. 2010;133(11):3470–3479. doi: 10.1093/brain/awq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B.J., Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008;85(1):94–134. doi: 10.1016/j.pneurobio.2008.01.001. https://doi.org/S0301-0082(08)00002-6 [pii] [DOI] [PubMed] [Google Scholar]
- Van den Bos M.A.J., Higashihara M., Geevasinga N., Menon P., Kiernan M.C., Vucic S. Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology. 2018;91(18):e1669–e1676. doi: 10.1212/WNL.0000000000006438. [DOI] [PubMed] [Google Scholar]
- van den Bos M.A.J., Higashihara M., Geevasinga N., Menon P., Kiernan M.C., Vucic S. Pathophysiological associations of transcallosal dysfunction in ALS. Eur. J. Neurol. 2021;28(4):1172–1180. doi: 10.1111/ene.14653. [DOI] [PubMed] [Google Scholar]
- Vasques J.F., Mendez-Otero R., Gubert F. Modeling ALS using iPSCs: is it possible to reproduce the phenotypic variations observed in patients in vitro? Regen. Med. 2020;15(7):1919–1933. doi: 10.2217/rme-2020-0067. [DOI] [PubMed] [Google Scholar]
- Veltema A.N. The case of the saltimbanque Prosper Lecomte. A contribution to the study of the history of progressive muscular atrophy (Aran-Duchenne) and amyotrophic lateral sclerosis (Charcot) Clin. Neurol. Neurosurg. 1975;78(3):204–209. doi: 10.1016/s0303-8467(75)80050-3. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A., Augusto-Oliveira M., Pivoriunas A., Popov A., Brazhe A., Semyanov A. Astroglial asthenia and loss of function, rather than reactivity, contribute to the ageing of the brain. Pflugers Arch. 2021;473(5):753–774. doi: 10.1007/s00424-020-02465-3. [DOI] [PubMed] [Google Scholar]
- Villa R.F., Gorini A., Ferrari F., Hoyer S. Energy metabolism of cerebral mitochondria during aging, ischemia and post-ischemic recovery assessed by functional proteomics of enzymes. Neurochem. Int. 2013;63(8):765–781. doi: 10.1016/j.neuint.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Vucic S., Kiernan M.C. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain. 2006;129(Pt 9):2436–2446. doi: 10.1093/brain/awl172. [DOI] [PubMed] [Google Scholar]
- Vucic S., Kiernan M.C. Utility of transcranial magnetic stimulation in delineating amyotrophic lateral sclerosis pathophysiology. Handb. Clin. Neurol. 2013;116:561–575. doi: 10.1016/B978-0-444-53497-2.00045-0. [DOI] [PubMed] [Google Scholar]
- Vucic S., Ziemann U., Eisen A., Hallett M., Kiernan M.C. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights. J. Neurol. Neurosurg. Psychiatry. 2013;84(10):1161–1170. doi: 10.1136/jnnp-2012-304019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic S., Pavey N., Haidar M., Turner B.J., Kiernan M.C. Cortical hyperexcitability: Diagnostic and pathogenic biomarker of ALS. Neurosci. Lett. 2021;759:136039. doi: 10.1016/j.neulet.2021.136039. [DOI] [PubMed] [Google Scholar]
- Wainberg M., Andrews S.J., Tripathy S.J. Shared genetic risk loci between Alzheimer's disease and related dementias, Parkinson's disease, and amyotrophic lateral sclerosis. Alzheimers Res. Ther. 2023;15(1):113. doi: 10.1186/s13195-023-01244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.L., Cui L., Liu M., Zhang K., Liu S., Ding Q. Split-Hand Syndrome in Amyotrophic Lateral Sclerosis: Differences in Dysfunction of the FDI and ADM Spinal Motoneurons. Front. Neurosci. 2019;13:371. doi: 10.3389/fnins.2019.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.L., Cui L., Liu M., Zhang K., Liu S., Ding Q., Hu Y. Reassessment of Split-Leg Signs in Amyotrophic Lateral Sclerosis: Differential Involvement of the Extensor Digitorum Brevis and Abductor Hallucis Muscles. Front. Neurol. 2019;10:565. doi: 10.3389/fneur.2019.00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Guan L., Deng M. Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Front. Neurosci. 2023;17:1170996. doi: 10.3389/fnins.2023.1170996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.L., Liu M., Ding Q., Hu Y., Cui L. Split-hand index in amyotrophic lateral sclerosis: an F-wave study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2019;20(7–8):562–567. doi: 10.1080/21678421.2019.1646770. [DOI] [PubMed] [Google Scholar]
- Wang S.P., Yang H., Wu J.W., Gauthier N., Fukao T., Mitchell G.A. Metabolism as a tool for understanding human brain evolution: lipid energy metabolism as an example. J. Hum. Evol. 2014;77:41–49. doi: 10.1016/j.jhevol.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Weber M., Eisen A., Stewart H., Hirota N. The split hand in ALS has a cortical basis. J. Neurol. Sci. 2000;180(1–2):66–70. doi: 10.1016/s0022-510x(00)00430-5. [DOI] [PubMed] [Google Scholar]
- Wilbourn A.J. The “split hand syndrome”. Muscle Nerve. 2000;23(1):138. doi: 10.1002/(sici)1097–4598(200001)23:1<138::aid-mus22>3.0.co;2–7. [DOI] [PubMed] [Google Scholar]
- Willemse S.W., van Es M.A. Susceptibility and disease modifier genes in amyotrophic lateral sclerosis: from genetic associations to therapeutic implications. Curr. Opin. Neurol. 2023 doi: 10.1097/WCO.0000000000001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittstock M., Wolters A., Benecke R. Transcallosal inhibition in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2007;118(2):301–307. doi: 10.1016/j.clinph.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Woolley S.C., Strong M.J. Frontotemporal Dysfunction and Dementia in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015;33(4):787–805. doi: 10.1016/j.ncl.2015.07.011. [DOI] [PubMed] [Google Scholar]
- Ye S., Luo Y., Jin P., Wang Y., Zhang N., Zhang G., et al. MRI Volumetric Analysis of the Thalamus and Hypothalamus in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2021;13:610332. doi: 10.3389/fnagi.2021.610332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Y., Ha Y. CRISPR/Cas9-Mediated Gene Correction to Understand ALS. Int. J. Mol. Sci. 2020;21(11) doi: 10.3390/ijms21113801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q., Zhou J., Zhang Y., Huang H., Han J., Cao B., et al. Risk factors associated with amyotrophic lateral sclerosis based on the observational study: a systematic review and meta-analysis. Front. Neurosci. 2023;17:1196722. doi: 10.3389/fnins.2023.1196722. [DOI] [PMC free article] [PubMed] [Google Scholar]