

Abstract
The Human Phenotype Ontology (HPO) is a dictionary of more than 15,000 clinical phenotypic terms with defined semantic relationships, developed to standardize phenotypic analysis. Over the last decade, the HPO has been used to accelerate the implementation of precision medicine into clinical practice. In addition, recent research in representation learning, specifically in graph embedding, has led to notable progress in automated prediction via learned features. Here, we present a novel approach to phenotype representation by incorporating phenotypic frequencies based on 53 million full-text health care notes from more than 1.5 million individuals. We demonstrate the efficacy of our proposed phenotype embedding technique by comparing our work to existing phenotypic similarity-measuring methods. Using phenotype frequencies in our embedding technique, we are able to identify phenotypic similarities that surpass current computational models. Furthermore, our embedding technique exhibits a high degree of agreement with domain experts’ judgment. By transforming complex and multidimensional phenotypes from the HPO format into vectors, our proposed method enables efficient representation of these phenotypes for downstream tasks that require deep phenotyping. This is demonstrated in a patient similarity analysis and can further be applied to disease trajectory and risk prediction.
Keywords: Human phenotype ontology, Representation learning, Dimension reduction, Electronic health record, Phenotype embedding, Patient Similarity
1. Introduction
Electronic medical records (EMRs) have been implemented in the majority of US hospitals and accumulate clinical data at a massive scale [1]. While initially created for billing purposes [2], EMRs increasingly represent a major source of data in clinical research efforts to improve patient care[3]. However, automated interpretation of EMRs is challenging, particularly for diagnoses that incorporate dynamic and diverse sets of clinical features. Phenotypes, a set of observable characteristics and clinical traits, have an essential role in connecting clinical research and practice. Algorithms that use phenotypes to find similarities and differences between patients play a foundational role in EMR research [4]. However, phenotypic descriptions available in EMRs are often stored in the form of unstructured data and do not allow for direct comparisons.
The Human Phenotype Ontology (HPO) is one approach to overcome these limitations (human-phenotype-ontology.org). The HPO is a standardized representation of more than 15,000 clinical phenotypic concepts and their relationships based on expert knowledge. The clinical and scientific community has contributed to HPO terminology since 2010 [5–8]. The HPO has been widely used for harmonization of clinical features in various studies, including, but not limited to, semantic unification of common and rare diseases [9], genetic discoveries in pediatric epilepsy [10, 11], and delineation of longitudinal phenotypes [12, 13]. In addition, the HPO is commonly used for genomic studies and allows for analyses of clinical data at a scale that is required by current and future initiatives. For example, large national and international initiatives have started to systematically link biorepositories to EMR data, including up to 80,000 cases and 500,000 controls [14–16], highlighting the possibilities for novel biological insight at scale.
The HPO can be modeled as a directed acyclic graph (DAG) in a computational system, where each phenotype is presented as a node with a unique identifier and is connected to its parent phenotypes by “is a” relationships in the form of directed edges. This structure guarantees that if a disease or gene is annotated to a phenotypic term, it will also be annotated to all its ancestral terms (higher-level concepts within the larger phenotypic tree). The HPO is regularly updated to incorporate advances in phenotypic conceptualization.
Extraction of phenotypic concepts is a crucial step in any automated pipeline to exploit the scale of EMR data in clinical research. Natural Language Processing (NLP) pipelines such as cTAKES [17], ClinPhen [18], and MetaMap [19] are commonly used to derive phenotypic concepts from the EMR, effectively allowing the transition from unstructured free text to structured representations. While these NLP pipelines share a common goal, that is, extracting phenotypes from clinical free text, they have different components and employ a variety of procedures, thus, should be evaluated from different endpoints such as precision, sensitivity, ease of use, and speed [18, 20]. Furthermore, phenotype extraction typically serves as only a starting point to more complex analyses. Therefore, studies need to perform additional analyses on the extracted phenotypes and their relationships to accomplish tasks like patient comparisons and predicting patient status [21–23]. Manual analysis of phenotypes is non-scalable, resource-intensive, and virtually impossible for larger cohorts. Accordingly, reliable algorithms for computational phenotype analysis are urgently needed.
Comparing phenotypes to one another is a common building block for downstream clinical tasks. Methods for measuring phenotypic similarities, using measures such as the Resnik score [24], information coefficient [25], and graph information content [26], have shown promise for diagnoses and open doors to novel biological insights such as genetic discoveries [10]. However, these methods are not generally transferable to other tasks and require a significant amount of computation, even with minor changes to the data. While these methods perform well on data with hundreds of patients, they are computationally intensive for research questions involving thousands of patients and can become impractical for cases with millions of patients [18].
Representation learning is a group of machine learning algorithms that discover and learn representations of data, making it easier to extract information that can be used for various tasks such as classification and prediction [27]. Recent work in representation learning has demonstrated success in discovering useful representations without relying on procedural techniques, especially in domains with complex and large data [27]. Embedding algorithms, discussed in Section 2, are a branch of representation learning that model discrete objects as continuous vectors. They offer a compact representation that captures similarities between the original objects and have revolutionized data processing and analysis in many domains, including text processing, by representing words in a compact space [28, 29]. Embedding algorithms have also been extended to encode other data structures such as nodes and graphs [30, 31]. A few studies have applied representation learning and embedding techniques in the clinical domain and presented promising results on specific phenotypes for a limited number of diseases [32, 33].
Here, we map 53 million full-text healthcare notes of more than 1.5 million individuals to HPO terms and perform graph embedding to assess the possibilities and limitations of representation learning techniques. We demonstrate that phenotype embedding, in some scenarios, can exceed more computationally intense measures of phenotype similarity, providing a framework for computationally efficient analysis of large-scale phenotyping data. The source code for our computational model is available on GitHub [34].
2. Materials and Methods
2.1. Clinical data extraction
The data used in this study represents the entirety of available full-text patient notes from the Children’s Hospital of Philadelphia (CHOP). All medical documentation is performed using a unified Electronic Health Record (EHR) system, Epic (Verona, WI), for all care documentation (www.epic.com). The EHR system documents contacts with patients, including telephone call records, refills, visits for laboratory and imaging, hospital admissions, and clinic and emergency room visits. Each contact point is referred to as an encounter. Patient data from the resulting medical record is then merged into a separate reporting database provided by Epic (Clarity) as well as an internal database system, the Clinical Data Warehouse (CDW). For research purposes, a subset of this data is modeled within Arcus, an institutional informatics platform at CHOP [35]. The Arcus Data Repository (ADR) de-identifies the clinical data allowing researchers to safely access and conduct studies on the data, reducing administrative burden and lifting Institutional Review Board (IRB) oversight requirements. We refer to this de-identified clinical data as “data” throughout the paper.
Additionally, we selected 53 individuals with epilepsy and a genetic diagnosis of SCN1A from the data set. Most of these individuals have Dravet Syndrome (n=45), a childhood developmental and epileptic encephalopathy typically characterized by febrile seizures in the first year of life followed by other severe seizure and neurodevelopmental abnormalities [36]. These individuals were enrolled as part of the Epilepsy Genetic Research Program (EGRP) at Children’s Hospital of Philadelphia [11]. Since 2014, EGRP has been enrolling individuals with known or presumed genetic epilepsy and collected individuals’ clinical and genomic data [10, 11, 37]. As part of EGRP, all collected diagnoses were evaluated and verified in a clinical and research setting, and if necessary, reclassified according to the criteria of the American College of Medical Genetics and Genomics (ACMG). We refer to this subset of data from 53 individuals as the “EGRP-subset” and use it in our patient similarity analysis (see Section 2.8).
It is worth mentioning that due to the sensitive nature of the research supporting data, including patient notes and the extracted phenotypes, are not available in their original form. However, the input data to our proposed method, i.e., frequency of phenotypes, is available in the supplementary materials and can be used for replicating the experiments (Section 8 Supplemental).
2.2. Mapping to Human Phenotype Ontology terms
Many commercial NLP systems struggle with recognizing clinical text, and those trained on clinical data are usually costly, proprietary, and lack customizable features. Apache Clinical Text Analysis and Knowledge Extraction System (cTAKES), an open-source project, tries to fill this gap [17]. cTAKES performs clinical named-entity recognition (NER), a natural language processing task that searches for observations of medical concepts within the text [38, 39]. In doing so, cTAKES passes the text through a series of steps within a pipeline. These steps vary based on the selected configuration. Although cTAKES provided an NLP solution specialized to health data, the analysis of large sets of clinical notes is time- and resource-intense. Accordingly, in its default configuration, this framework is not practical for institution-size data, and many institutions use cTAKES only on a small subset of their records [40, 41].
We have developed a pipeline that is capable of running cTAKES on millions of clinical notes in parallel, allowing for the utilization of institution-size data at full capacity [42]. We employed cTAKES 4.0.0 [43] with the default configuration that splits a document into a series of sentences and then each sentence is split into individual words in a process called tokenization (see https://cwiki.apache.org/confluence/display/CTAKES/Default+Clinical+Pipeline).
We applied two modifications to the default cTAKES pipeline. First, we used a dictionary based on the Human Phenotype Ontology, HPO release version 2020-10-12, as included with the 2021AA release of the Unified Medical Language System (UMLS) [44]. This mechanism assigns a part of speech or phrase type to each token before the entity recognizer matches the tokens against a concept dictionary derived from the UMLS. Secondly, we included the NegEx negation annotator in addition to the default machine learning negation classifier available in cTAKES [45]. NegEx is a regular expression algorithm that uses a list of patterns in an attempt to better capture negated terms in encounter notes, filtering out sentences containing phrases that incorrectly appear to be negating terms. This process allowed us to accurately map each encounter in the EMR to a set of HPO terms at a large scale. Figure 1 illustrates a schematic of our distributed cTAKES pipeline.
Figure 1.
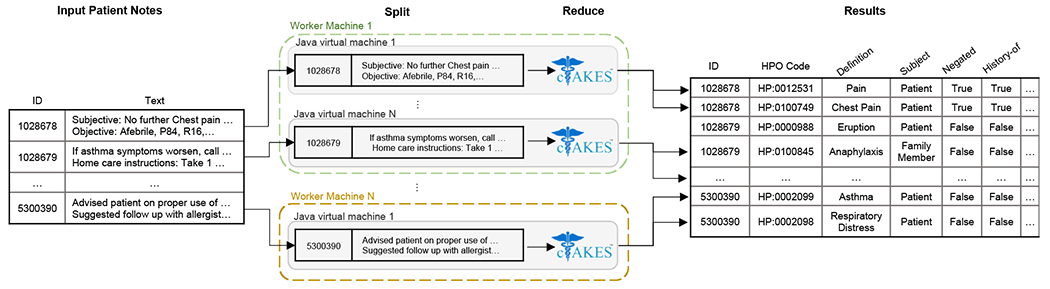
Schematic of our fault-tolerant, distributed, and scalable cTAKES pipeline using the Apache Spark programming model [42].
Although cTAKES pipeline, particularly with our changes to the default configuration, is one of the most dependable systems available, as with any NLP system, there are some shortcomings. The most prominent one is its failure to detect all phenotypes in clinical notes that are absent or have non-unique short forms. For example, medical cannabis or cannabidiol (CBD) has been recently considered as a treatment option for Alzheimer’s disease and epilepsy, especially for Dravet Syndrome [46, 47]. On the other hand, CBD is also an alias for “red-green color blindness” and deuteranomaly. Upon further examination, we realized that in individuals with epilepsy, the cTAKES pipeline was incorrectly categorizing CBD as Deuteranomaly (HP:0011520). Another area of failure for cTAKES is the manner it deals with phenotypes containing multiple phenotypic terms in their names, such as the term “myoclonic seizures” which is classified into both Myoclonic seizures (HP:0032794) and Seizure (HP:0001250). Typically, this shortcoming is inconsequential as in most methodologies, Seizure (HP:0001250) is inferred after “propagation” of Myoclonic seizures (HP:0032794) and duplicated terms are then removed. However, when Myoclonic seizures is negated, cTAKES falsely assigns negation to both Myoclonic seizures and Seizure. Nevertheless, since clinicians typically include major detected or presumed disorders separately and repeatedly in their notes, in this case Seizure, our employed query which only incorporates positive categorizations overcomes this failure as well (Figure 2).
Figure 2. Sample patient note with its extracted phenotypes using the cTAKES pipeline.
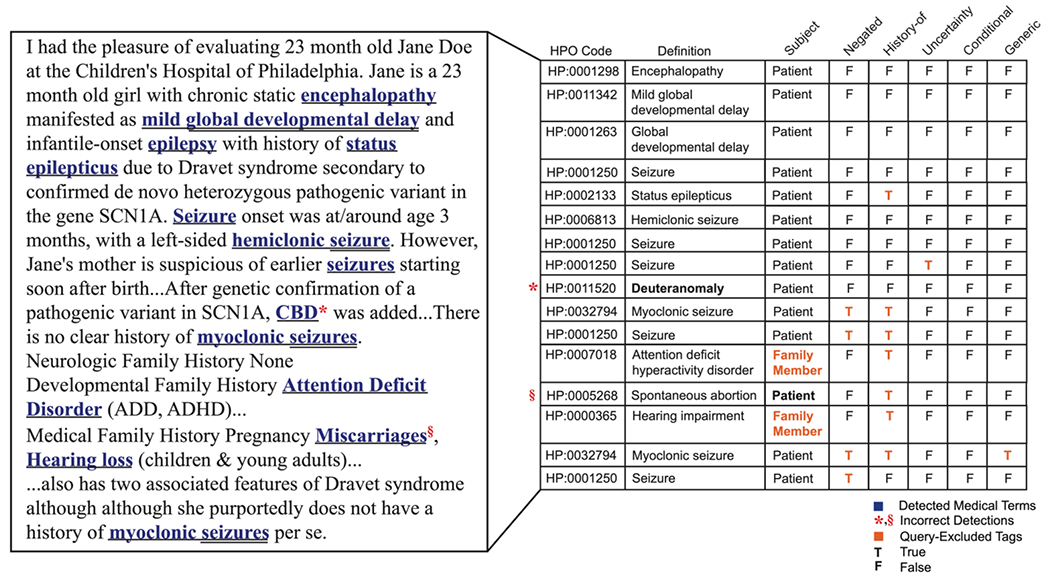
While cTAKES could correctly categorize most medical terms, it falsely detected rows marked with red signs with their mistaken tags marked in bold. In our customized query, we excluded cTAKES modifier tags that represented phenotypes identified based on family members or patients’ history, were negated, generic, conditional, or detected with uncertainty. These records are marked in orange.
Several studies with small corpora attempt to correct such errors by performing manual review on the data or by verifying the significant findings with clinical staff. However, this is not a feasible solution for large-scale hospital data such as that used in our study. Thus, we tried to reduce the number and effects of such samples by (1) Customizing data queries that incorporate additional modifier tags provided by cTAKES (Figure 1). (2) Relying on the robustness provided by algorithms that categorize large corpus samples as a whole and can generalize their findings.
Thus, in querying the data, we excluded phenotypes with tags that represented “family members” as opposed to the “patient”, had a positive “history”, were “negated”, “generic”, “conditional”, or identified with “uncertainty”. Figure 2 provides a sample patient note with its identified phenotypes obtained by running the cTAKES pipeline marked in blue. Rows marked with a red sign represent wrong categorization by cTAKES with the mistaken tag marked in bold. Furthermore, the phenotypes that were excluded based on our customized query, as discussed above, are shown with their corresponding (query-excluded) tag in orange.
2.3. Assessing HPO term frequencies corrected for frequencies of higher-level phenotypic concepts
We calculated the frequency of each phenotype by dividing the number of individuals mapped with that HPO term in at least one encounter by the total number of individuals. However, assessing the baseline frequency does not fully represent the complete frequency of each phenotypic term as higher-level terms are not typically included in encounters. Clinical descriptions tend to be annotated with the phenotypic term at the greatest applicable level of detail. Consequently, the analysis of the direct translation of clinical records into HPO terms underestimates the frequency of higher-level, conceptually broader, phenotype terms. However, it can be reasoned that these higher-level terms are often essential as they may capture large groups of individuals with phenotypically broad but clinically or biologically important similarities. Accordingly, we performed a method referred to as “propagation” that we have used extensively in past work [7, 11, 12, 48, 49]. In brief, for each individual’s set of explicitly annotated HPO terms, we add all HPO terms that can be inferred by taking the union of all those terms extracted and following all possible paths along their “is a” relationships to the root of the HPO. For example, if an individual was coded with Mild global developmental delay (HP:0011342), we can infer that they also have the parent terms Global developmental delay (HP:0001263) and Neurodevelopmental delay (HP:0012758). This technique ensures this individual is counted when calculating the frequency of the higher-level terms. The term frequency of the propagated terms provides a more accurate representation of the true term frequencies for concepts coded in the HPO [10]. We refer to the frequency derived from the propagated counts as propagated frequency. Thus, the propagated frequency of the root term in the HPO, , is always equal to 1.
2.4. Similarity analysis using the Resnik Score
Various frameworks have been introduced to automatically measure the semantic similarity between concepts [50, 51]. Among them, many focus on deriving statistical information from cohorts and combining them with lexical resources and knowledge graphs [52]. These techniques have been effective in NLP tasks such as information extraction based on WordNet [53]. In 1995, Resnik introduced information content (IC) as a measure of specificity for a concept [24]. The IC of each concept represented in an ontology can be calculated based on the occurrence frequency of that concept in a large and relevant corpus. As a result, a generic concept would be associated with a lower IC value, and a very specific and rarely encountered concept would have a higher IC. In calculating the IC of a concept, the frequency of all concepts encountered following all possible paths along the “is a” hierarchy to the main concept should be counted. Formally, the IC of a concept can be calculated as:
| (1) |
where c represents the concept, and returns the propagated frequency value of a given concept. We used logarithms to the base two in our analysis.
Having the IC, the Resnik score computes the similarity between concepts c1 and c2 based on their Most Informative Common Ancestor (MICA), the concept that subsumes c1 and c2 and has the maximum IC. Formally,
| (2) |
where is the lowest common subsumer of concepts c1 and c2, and IC(.) returns the information content of a concept.
In the HPO, represented as a DAG, phenotypes closer to the root (HP:0000001) are more general and have a lower IC value. The of two phenotypes is the least common ancestor (LCA) of those phenotypes in the HPO. As a result, the quantity of shared information between two phenotypes would be higher if the LCA is a rare term and thus has a high IC.
Among the leading methods for comparing patients and correlating phenome and genome data, phenotypic similarity measures based on information content (IC) or, more specifically, the most informative common ancestor (MICA), or Resnik method, remains the most dependable [24].
Recent studies have demonstrated the effectiveness of the Resnik method in developing novel diagnostic approaches, generating statistical evidence for disease causation, and even discovering novel genes [10, 11, 54]. Research on real-world datasets has repeatedly demonstrated Resnik to be a consistently more versatile and robust phenotype similarity measure compared to several other phenotype similarity methods, such as the Lin measure[55] , the Jiang-Conrath measure [56], the Relevance measure [57], the information coefficient measure [25], and the graph IC measure [16, 58, 59]. As such, Resnik remains one of the most popular and effective methods of measuring the similarity between phenotypes. Thus, we chose this method as a reference to evaluate the efficacy of our proposed techniques.
However, there are certain limitations associated with the application of Resnik. For instance, in some cases, the Resnik score cannot make a fine-grained distinction as many phenotypes may share the same LCA and thus receive equal similarity scores. In addition, the frequency values involved in calculating the information content may suffer from different sources of biases in the reference cohort, including but not limited to selection bias, information bias, and confounding bias [60]. Although some bias sources can be identified and their potential impact assessed, they may require extensive data cleaning and supervised analysis, which are very costly for institutional data. Many sources of bias, however, will likely remain hidden. Thus, techniques like Resnik that directly use the phenotypic frequencies in measuring similarities are inevitably susceptible to such biases.
2.5. Node embeddings using Node2Vec
Inspired by advancements in word embeddings for NLP tasks, the Node2Vec model maps nodes in the graph to a d-dimensional vector space [30]. There are several sampling strategies that are used to explore nodes in the graph [30, 61, 62]. Different sampling strategies generate different sentence-like samples that result in distinct learned representations. Some of these strategies rely on a rigid notion of a network neighborhood and are insensitive to connectivity patterns unique to networks [61, 62]. As a result, they have shortcomings in generalizing different prediction tasks and graph structures. Therefore, it is essential to employ a flexible sampling strategy to explore the graph and learn node representations with two rules in mind: (1) learning representations where nodes with similar roles in the graph receive similar embeddings, and (2) learning representations where nodes from the same substructure or community are set closer together in space. Node2Vec introduces a biased randomized procedure that samples neighborhoods for each given node where the transition probability to the next node depends on both the current and previous node. This procedure overcomes the mentioned shortcomings and follows the listed rules. The generated biased random walks from each node preserve the mutual proximity among the nodes, effectively exploring a node’s local neighborhood. By running multiple biased random walks on each node, Node2Vec generates sets of sentence-like sequences, which serve as inputs to the Skip-gram model used in word embedding [29] (Supplementary Method S1).
The Skip-gram model aims to learn a continuous representation of words by optimizing a neighborhood likelihood objective in a semi-supervised fashion. The Skip-gram objective is based on a distribution hypothesis stating that words that appear in a similar context (windows over sentences) tend to have similar meanings. Particularly, similar words tend to appear in similar word neighborhoods and should be moved closer in vector space. For example, the words “epilepsy” and “illness” will likely be much closer in vector space than “table”, as they are much more likely to appear within the same windows of text. The biased random walks introduced in Node2Vec sample the nodes’ neighborhoods and serve the same purpose as context/sentences for the Skip-gram model.
2.6. HPO2Vec and Node2Vec+ as extensions of the Node2Vec framework
Phenotype embedding for HPO data has been developed by applying Node2Vec on the phenotypic nodes with weights equal to 1, a tool referred to as HPO2Vec [32]. Despite HPO2Vec’s promising results on specific phenotypes for a limited number of diseases, the equal weighting strategy prevented connection strength from being incorporated into exploring the graph, placing phenotypes equally close to their general and rare neighbors. A possible solution to this problem is to include connection strength in the form of edge weight in the HPO (see Section 2.7). While Node2Vec was designed to work on both weighted and unweighted (equal-weight) graphs, it does not distinguish weak connections from stronger ones and thus cannot detect cases where the potential next node is weakly connected to the previous one. The Node2Vec+ model proposed an extension of Node2Vec that resolved the issue with weak connections [63]. Intuitively, a connection is called “loose” based on some threshold edge value; however, it is hard to determine a reasonable threshold value for networks without having the distribution of edge weights of all nodes. Accordingly, they set a relative threshold for each node based on its surroundings. Formally, to evaluate if a connection is loose, where and are two nodes in the graph, they compare its weight with the average edge weight from u, defined as:
| (3) |
If , the edge, , is referred to as a loose connection. For simplicity, is also considered a loose connection if . The extended bias factor is defined as:
| (4) |
where represents the current, the previous, and the next visited node in a biased random walk. Note that for an unweighted graph, Equation 4 acts as the bias factor in Node2Vec (Supplementary Method S1), and there is no difference between the two methods. Also, similar to Node2Vec, if p and q are set to 1, the biased random walk will work as a simple first-order random walk.
2.7. Frequency-based human phenotype (FQ-HP) embedding
Here we propose a frequency-based human phenotype (FQ-HP) embedding model which consists of three steps, including: (1) updating the HPO graph by incorporating the propagated frequencies, (2) creating phenotype embeddings using Node2Vec+, and (3) calculating the similarity between phenotypes. Available studies on phenotype embedding, including HPO2Vec [32] and HPO2Vec+ [33] (see Section 4), apply their techniques to equally-weighted graphs which are equivalent to unweighted graphs. This results in choosing among surrounding phenotypes evenly rather than incorporating any connection strengths and priorities. This shortcoming would ultimately create equivalent similarity values between a phenotype with its rare child and the same phenotype with its relatively general neighbor (which could be another child or its parent). We call this category of techniques Equal-weight human phenotype (E-HP) embeddings and employ them in our evaluations. See Section 3 for more details.
2.7.1. Updating the HPO graph
The HPO version used in the current project contains 15,371 nodes (phenotypes) connected with unweighted and directed edges. To apply the Node2Vec+ algorithm, we created a copy of the HPO graph, including all 15,371 nodes and their accompanying 19,523 undirected edges. Assigning undirected edges helps the biased random walks in Node2Vec+ explore each given phenotype’s lower-level and higher-level neighborhoods. Additionally, we assigned weights to the edges of our graph. Weighted edges affect the probability of choosing the next nodes to visit in the biased-random walks. More specifically, at each step in the biased-random walk, if the algorithm needs to choose amongst a group of nodes to visit next, it will most likely choose the node (phenotype) with the strongest connection (largest weight) to the current node (Equation S7). We used the frequency calculated in Section 2.3 to weight the edges. More precisely, the weight between two connected nodes is calculated by the minimum frequency value of the two nodes, formally defined as:
| (5) |
where and are two connected nodes (phenotypes), and determines the frequency of a given phenotype. Here, b is a constant bias value that prevents zero weights. In our study, we used the difference between the maximum frequency and the second maximum frequency among all phenotypes as (0.00146). The weighting mechanism introduced in Equation 5 creates stronger connections between phenotypes with higher frequency values (more general terms) compared to rare terms. Furthermore, if phenotype p has multiple neighbors, its connection(s) to its parent(s) will be stronger than to its children — that are equally rare or rarer. Conceptually, this puts rare phenotypes further away in vector space compared to common ones as we argue that they represent more unique information about a patient. We have provided the example of embedding the HPO terms Generalized-onset seizure (HP:0002197) and Motor seizure (HP:0020219), which represent children of the phenotype Seizure (HP:0001250) in Figure 3 and Supplementary Method S1.
Figure 3. An illustration of the weighting mechanism on a portion of the HPO.
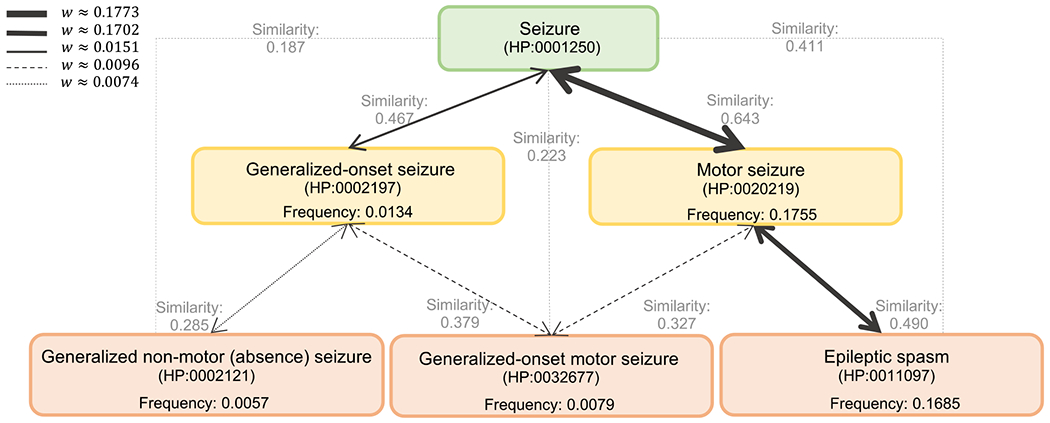
The thickness of an edge represents its weight. Stronger weights have a higher chance of being selected by the biased random walks in the sampling algorithm. The propagated frequency values are calculated as described in Section 2.3. The cosine similarity values between each phenotypic pair are shown in gray. Higher similarity values represent a higher degree of closeness in the embedding space.
2.7.2. Creating phenotype embedding
We used the Node2Vec algorithm, described in Section 2.5, with the extended bias factor introduced in Node2Vec+ (Section 2.6) to incorporate our weighting mechanism in embedding the phenotypes. We ran the embedding algorithm on all 15,371 phenotypes available in the HPO.
2.7.3. Hyper-parameters
Since our task is unsupervised and there is no label involved in the fine-tuning process, we relied on the provided hyper-parameters used in HPO2Vec+ for the sampling strategy [33]. The select hyperparameters used in the sampling strategy as well as training the Skip-gram model are provided in Table 1.
Table 1.
Select hyper-parameters used for the phenotype embedding methods.
| Hyper-parameter | Value |
|---|---|
| vector length (dimension) | 128 |
| p | 1 |
| q | 0.05 |
| number of walks | 10 |
| number of steps | 5 |
| learning rate | 0.001 |
| number of negative samples | 4 |
| batch size | 1024 |
| number of epochs | 15 |
2.7.4. Representing the embedding space
With the embedding model, each phenotype can be mapped into the embedding space with its vector representation. However, the embedding space has high dimensionality, where dimensionality is defined by the hyper-parameter vector length. Thus, visualizing how phenotypes occupy the embedding space is impossible. We apply two dimensionality-reduction techniques, namely, Principal Component Analysis (PCA) [64] and t-distributed stochastic neighbor embedding (t-SNE) [65] to the embedding vectors to visualize them in 2-D and 3-D space. Since the dimensionality reduction techniques lose important information from the original data, we use them only for visualization purposes and work with the original vectors when comparing phenotypes and calculating their similarities.
2.7.5. Calculating phenotype similarities
There are three main techniques available in the literature to calculate the similarity between two embedding vectors: Euclidean distance, Cosine similarity, and dot product. Among these techniques, the dot product is proportional to the vector length, which is a hyper-parameter of the sampling strategy. Thus, we only used the Euclidean distance and Cosine similarity to measure the similarities between the two vectors in our experiments (Table 2).
Table 2. Techniques for measuring the similarity between two vectors u and v of size d.
Similarity between the two vectors has an inverse relationship with metric value in a “decreasing” relationship, such as Euclidean distance.
| Similarity Technique | Description | Relation | Formula |
|---|---|---|---|
| Euclidean distance | Length of the line between ends of vectors | Decreasing | |
| Cosine similarity | Cosine of the angle between vectors () | Increasing | |
| Dot product | Cosine multiplied by lengths of the vectors | Increasing |
2.8. Calculating phenotypic similarity between two individuals
Phenotypic similarity between two individuals can be assessed based on the similarity between their phenotype pairs. As discussed in the previous sections, we employed three methods to calculate phenotypic similarity. These methods are Resnik, E-HP embedding, and FQ-HP embedding (see Sections 2.4, 2.6, and 2.7). We relied on available metrics for calculating individuals’ similarity when using Resnik for measuring the phenotypic similarity [11, 24].
This metric, referred to as , generates a symmetric score, measuring the similarity between two individuals P1 and P2 by summing over the maximum phenotypic pair similarity,
| (6) |
where represents a phenotype reported for individual , with a total of phenotypes, and represents a phenotype reported for individual , with a total of phenotypes. represents the similarity between phenotype of and phenotype of calculated by (see Equation 2). Note that based on the definition of , the similarity score between two individuals, with shared rare phenotypic terms is greater than that between two patients with only shared general phenotypes. This is due to the higher IC value of the MICA of two rare phenotypes.
Similarly, we propose a new metric for measuring patient similarity when using the embedding techniques, i.e., E-HP and FQ-HP embeddings, in calculating the similarity between phenotypes. In doing so, we employ a weighting mechanism to suppress the effects of common general phenotypes thereby increasing the similarity between individuals, prioritizing shared rare phenotypes between individuals. This concept is also available in by incorporating MICA (see Equation 2). Formally, we define
| (7) |
Here, is the propagated frequency of phenotype as described in Section 2.3.
We calculated the phenotypic similarity between all individuals (n = 53) in EGRP-subset resulting in 1,378 patient pairs with a median of 174 distinct phenotypic term per individual. We followed the same procedure in extracting the phenotypes as described in Section 2.2. In contrast to many studies that have relied on manual data cleansing and further clinician analysis for patient similarity [11, 66] in this study, we have analyzed the extracted phenotypes without undergoing any additional pruning. Our aim was to challenge existing and proposed similarity measuring techniques and evaluate their effectiveness on large scale data in which manual expert analysis is not feasible.
3. Results
53 million patient notes were translated into 9,477 phenotypes
We analyzed data from 1,504,582 patients with a wide range of syndromes available on 53,955,360 electronic notes in the Arcus Data Repository version 1.4.4 [35]. By applying the cTAKES algorithm on all available full-text notes from the EMR, we extracted 8,425 distinct HPO terms with more than one occurrence and 9,477 distinct phenotypes with more than one propagated occurrence. Patient encounters over time and the histogram of the propagated frequency of phenotypes are presented in Figure 4.
Figure 4. More than 53 million patient health records can be translated into phenotypes.
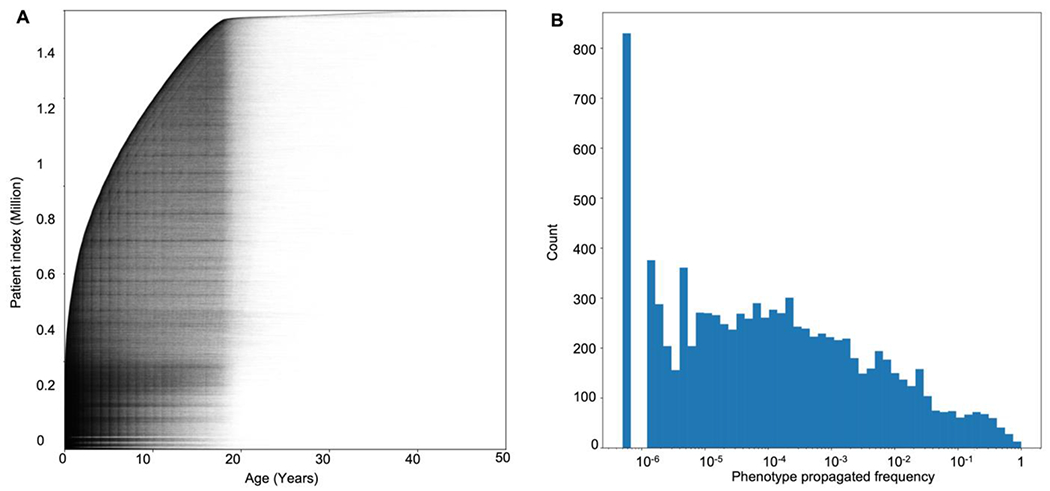
(A) Individual data points in the Arcus Data Repository used in our analysis (n = 53,955,360). The X-axis shows patients’ age at the time of the encounter, and the Y-axis represents patients’ indices stacked and sorted by their age at the earliest encounter with a total of 1,504,582 individuals. (B) Histogram of the phenotypes’ propagated frequency available in the HPO, a total of 15,371 phenotypes. Note that the X-axis has a logarithmic scale, and the phenotypes with a frequency less than 10−6 were not available in any of the encounters ().
Representing the Human Phenotype Ontology in a lower-dimensional space
We qualitatively evaluated our embedding method by visualizing the 15,371 phenotypes in the embedding space. Since we designed the embedding vectors to be of length 128, it is impossible to visualize the space directly. We used the PCA and t-SNE algorithms to reduce the embedding dimension to 2D and 3D. Figure 5 displays a 3D representation of the original (128D) embedding space using PCA, where phenotypes that are closer together are more similar.
Figure 5. The HPO can be represented in a lower-dimensional space where similarities and differences between the phenotypes are preserved.
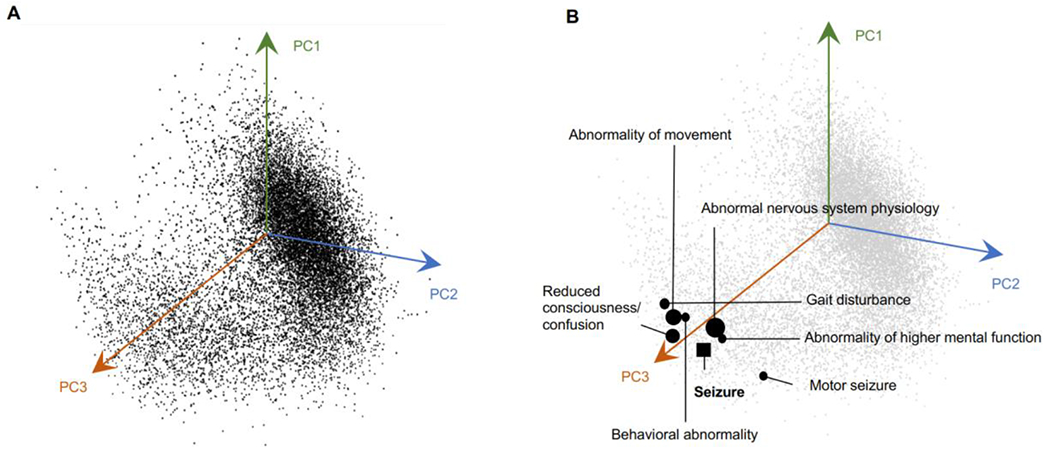
(A) A 3D representation of all phenotypes in the embedding space using the PCA algorithm. The axes are based on the first three principal component vectors (PC) of the PCA algorithm. (B) Seven closest phenotypes to Seizure (HP:0001250) are marked in the 3D space. Closer phenotypes in the original space (128D) are represented with larger circles.
Graph representation learning preserves relationships between phenotypes in the embedding space
We next assessed the Skip-gram objective to determine if proximity to similar contexts would drive the phenotypes to be closer in the embedding space. In doing so, we examined a group of phenotypes, including Seizure (HP:0001250) and its neighbors in the HPO, and measured their similarities in the embedding space. Table 3 shows the similarity values between Seizure and some of its closest neighbors in the original vector space. We calculated the similarity using the Cosine Similarity and Euclidean Distance metrics with larger values in the Cosine System representing a higher degree of similarity while smaller values in the Euclidean System representing shorter distances, thus greater similarities. Even in a lower-dimensional 3D space generated by PCA, the clos e neighbors of Seizure are evident (Figure 5). We also implemented the t-SNE algorithm to reduce the embedding space to three dimensions. Our results demonstrate that while t-SNE can preserve the similarity and difference between most phenotypes, it requires that we tune its hyper-parameters (perplexity, learning rate, and iteration number) to obtain a stable low-dimensional representation adding additional complexity that does not necessarily improve the representation (Supplementary Figure S1).
Table 3. Closest phenotypes to the node Seizure (HP:0001250) in the embedding space using the Cosine Similarity System, as shown in black, versus the Euclidean Distance System, as shown in blue.
While the two systems share most phenotypes, there are minor differences in phenotypes’ ranks.
| Cosine Rank | Euclidian Rank | HPO | Closest Phenotypes Cosine. | Cosine Similarity | Euclidian Distance | Euclidian Rank | Cosine Rank | HPO | Closest Phenotypes Euclidian. | Cosine Similarity | Euclidian Distance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | HP:0012638 | Abnormal nervous system physiology | 0.781 | 3.089 | 1 | 1 | HP:0012638 | Abnormal nervous system physiology | 0.781 | 3.089 |
| 2 | 32 | HP:0000707 | Abnormality of the nervous system | 0.705 | 4.078 | 2 | 5 | HP:0020219 | Motor seizure | 0.644 | 3.297 |
| 3 | 3 | HP:0011446 | Abnormality of higher mental function | 0.694 | 3.530 | 3 | 3 | HP:0011446 | Abnormality of higher mental function | 0.694 | 3.530 |
| 4 | 5 | HP:0002493 | Upper motor neuron dysfunction | 0.648 | 3.629 | 4 | 7 | HP:0011442 | Abnormal central motor function | 0.614 | 3.603 |
| 5 | 2 | HP:0020219 | Motor seizure | 0.644 | 3.297 | 5 | 4 | HP:0002493 | Upper motor neuron dysfunction | 0.648 | 3.629 |
| 6 | 59 | HP:0100022 | Abnormality of movement | 0.625 | 4.131 | 6 | 15 | HP:0020221 | Clonic seizure | 0.509 | 3.695 |
| 7 | 4 | HP:0011442 | Abnormal central motor function | 0.614 | 3.603 | 7 | 20 | HP:0032855 | Photosensitive myoclonic-tonic-clonic seizure | 0.495 | 3.725 |
| 8 | 12 | HP:0002527 | Falls | 0.583 | 3.906 | 8 | 14 | HP:0004372 | Reduced consciousness/confusion | 0.521 | 3.783 |
Incorporating phenotype frequencies in the HPO graph transfers likelihood distribution to embeddings
We next analyzed the effects of incorporating weights in the HPO graph. In doing so, we first calculated pair-wise cosine similarity values between all phenotype pairs. We observed changes in the similarity values when using the FQ-HP embedding (our technique) compared to the E-HP embedding (conventional equal weights). Figure 6A represents the changes in the cosine similarity values between a sample phenotype (Seizure (HP:0001250)) and all other phenotypes in the HPO. In this example, the range of cosine similarities is wider for FQ-HP embedding (our technique, y-axis) compared to the E-HP embedding (the conventional equal weights, x-axis). Considering that cosine similarity is bound by a constrained range of −1 and 1, the increased range for FQ-HP (y-axis) suggests that more phenotypes have a notable, more extreme positioning with respect to the main phenotype in the embedding space. We observed various patterns of changes in the similarity values on different phenotypes (Supplementary Figure S2).
Figure 6. Incorporating phenotypes frequencies in the HPO graph transfers likelihood distribution to embeddings.
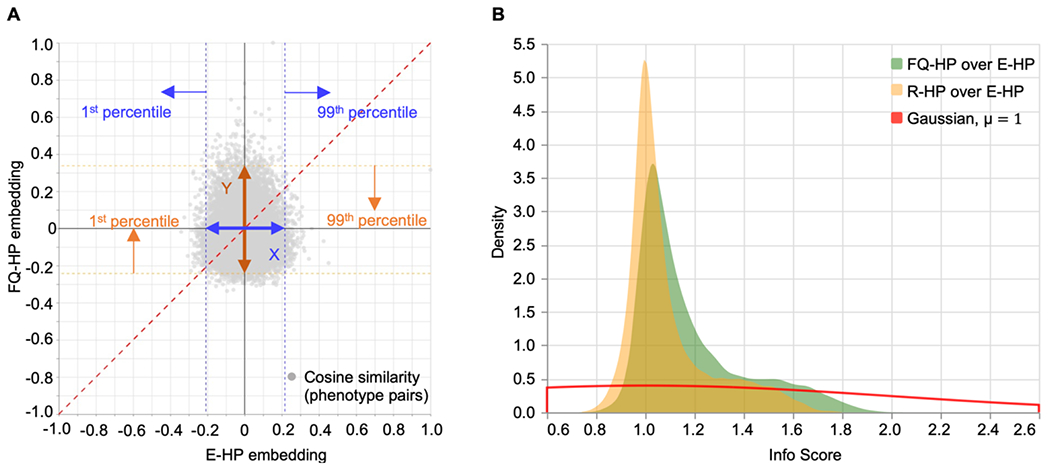
(A) Pair-wise cosine similarity changes between a sample phenotype, Seizure (HP:0001250), and all other phenotypes in the HPO using the FQ-HP and E-HP embedding methods. Info Score captures the similarity changes between these two methods by dividing the similarity range using FQ-HP by the similarity range using E-HP, i.e., Y/X. (B) A comparison between the PDF of the Info Score of FQ-HP over E-HP (shown in green) and the Info Score of Random Gaussian weights (R-HP) over E-HP (shown in yellow). The PDF of Info Score of FQ-HP/E-HP with a smaller peak around 1 and a longer tail indicates that FQ-HP could generate reliable phenotype clusters in the embedding space and shows the importance of assigning weights that represent the actual distribution of the phenotypes.
In order to assess the patterns of distributions between FQ-HP and E-HP across all phenotypes, we defined a metric that compares the ratio between the range for 99% of all cosine similarity values for FQ-HP and E-HP, the ratio of Y to X as demonstrated in Figure 6A. We referred to this metric as “Info Score” (Supplementary Method S3) and compared the distribution of Info Scores of FQ-HP over E-HP with that of embeddings with randomly assigned weights (R-HP) over E-HP. The observed Info Score distribution of FQ-HP/E-HP (Figure 6B, green) suggests stronger similarity values compared to R-HP/E-HP (Figure 6B, yellow), indicated by the “longer tail” of the FQ-HP/E-HP distribution. This difference between embeddings using true frequencies and random frequencies indicates that, on average, the spectrum of similarities is broader when using frequency-based embedding. Accordingly, when using the additional information of term frequencies, the existing proximities and distances in the phenotypic graph can be represented more efficiently.
Frequency-based phenotype embeddings improved recognition of expert-curated phenotype similarities
Next, we assessed how similarity using embeddings compared to human assessment of clinical similarities. To evaluate the utility of the frequency-based phenotype embeddings over conventional methods to assess clinical similarity, we generated an expert-curated dataset comparing a reference to two choices of phenotypes, here referred to as candidate phenotypes. The domain experts were asked to assess which of the two choices is more closely related to the reference phenotype in their opinion. They were also given the option to indicate uncertainty if they viewed the candidate phenotypes as equally related to the reference phenotype or found it impossible to choose. We reasoned that this would generate a gold standard dataset that provides a valuable framework to assess the utility of similarity-measuring algorithms. In brief, an algorithm that is more aligned with the expert-curated dataset is considered superior to an algorithm that results in less overlap.
To generate such a gold standard dataset, we provided a group of 13 domain experts in epilepsy and neurogenetics, including 12 clinicians and a researcher, with 100 neurology-related reference terms (i.e., Abnormality of the nervous system (HP:0000707) and all descendent (child) terms). These terms were balanced between common and rare phenotypes as well as phenotypes above or below in the phenotypic hierarchy compared to the candidate phenotypes (see Table 4 and Supplementary Table S1). In total, this resulted in 1,300 individual phenotype prioritizations. Across each phenotypic trio, a voting system was implemented, assigning the most frequent decision among the expert raters as the gold standard.
Table 4. Scenarios for generating expert-curated phenotypic trios.
Each scenario defines the relationship between the reference phenotype with its candidate phenotypes, represented with a three-letter acronym, where the first letter represents the reference term, and the second and third letters represent Candidate 1 and Candidate 2, respectively. C and R define common and rare phenotypic terms, respectively. Similarly, A and D define ancestor and descendant relationships with the reference term, respectively. The negation sign, i.e., ¬, represents the logic complement of a hierarchy relationship. For instance, CRDR¬D represents a common reference phenotype and two rare candidates, Candidate 1 is a descendent of the reference term and Candidate 2 is not a descendent term.
| Scenario | Abbreviation | Description | #(total:100) |
|---|---|---|---|
| S1 | CRDRD | Common1 reference with two rare candidates and both candidates as descendants | 10 |
| S2 | CRDR¬D | Common reference with two rare candidates and only one candidate as a descendant (positive control) | 5 |
| S3 | CR¬DR¬D | Common reference with two rare candidates and neither as a descendant | 5 |
| S4 | CCDCD | Common reference with one rare and one common candidate and both candidates as descendants | 5 |
| S5 | CC¬DC¬D | Common reference with two common candidates not descending from the reference | 5 |
| S6 | R¬A¬AR¬A¬AR¬A¬A | Rare reference with two rare candidates, none being an ancestor of either of the other two terms | 50 |
| S7 | RCACAA | Rare reference with two common candidates, both being ancestors and one candidate being an ancestor of the other (positive control) | 5 |
| S8 | RCAC¬A | Rare reference with two common candidates, one being its ancestor and the other not (positive control) | 5 |
| S9 | RCAR | Rare reference with one common candidate and one rare candidate with the common candidate being an ancestor of the reference (positive control) | 5 |
| S10 | RC¬A¬DR¬A¬D | Rare reference with one common candidate and one rare candidate and both unrelated | 5 |
Next, we performed a leave-one-out analysis [67] and identified a 59.85% agreement between our raters. This value provided a reference for the expected accuracy of similarity-measuring algorithms. While this reflects more than 1.5 × accuracy of random agreement (38.46%), it still indicates a wide variability in the clinical assessment by expert reviewers (Supplementary Method S4).
We then assessed the similarity-measuring algorithms in various scenarios, comparing the conventional Resnik algorithm, phenotype-embedding using equal weight (E-HP), and frequency-based phenotype-embedding (FQ-HP). For the E-HP and FQ-HP algorithms, we also included a variation with a safety threshold (ST) that would declare ties if the distances between the two candidates and the reference phenotype were close (<0.06 in the cosine system). This safety threshold was implemented to prevent false-positive prioritizations based on marginal frequency differences of the candidate phenotypes and was chosen using a trial-and-error strategy.
We found that FQ-HP, with an overall accuracy of 68%, surpasses other similarity-measuring algorithms in matching the experts’ gold standard (Figure 7A) and is even higher than the agreement level between our raters. When applying the safety threshold, we see a significant difference between the performance of FQ-HP and FQ-HP(ST) with an accuracy of 68% and 54%, respectively. These results emphasize the importance of incorporating estimated frequencies in phenotype embeddings. We also observed a wider confidence interval when using Resnik compared to the other methods, indicating its susceptibility in generalization.
Figure 7. FQ-HP embeddings improve recognition of expert-curated phenotype similarities.
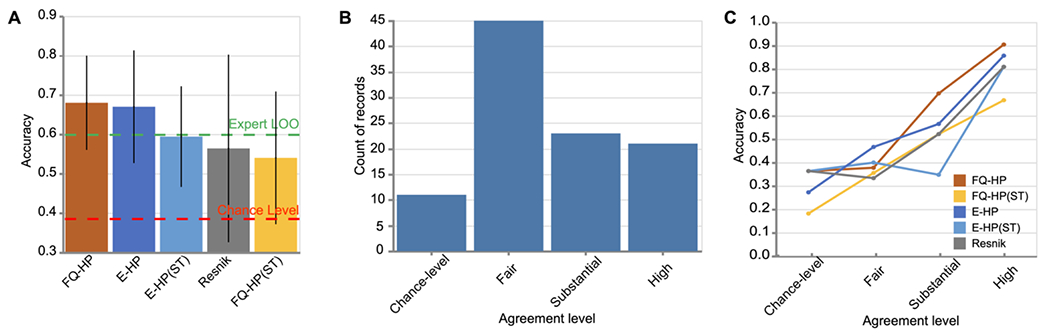
(A) A comparison on the overall accuracy of the similarity-measuring algorithms on 100 expert-curated phenotypic trios. The accuracy of our method (FQ-HP) surpasses the other similarity-measuring techniques where black bars show 95% confidence intervals, and the green dashed line represents the experts’ agreement level. (B) Histogram of experts’ agreement level for the 100 trios. Almost half of the records belong to the fair-level agreement category, indicating a wide variability in the clinical assessment of the experts and the challenging nature of such evaluations. (C) Performance of the similarity-measuring algorithms on the records of each agreement level. While all techniques have almost an increasing pattern in their performance as the agreement level improves, FQ-HP achieves better accuracy than other techniques, including Resnik, in the substantial and high agreement levels.
Next, we categorized our 13 experts’ decisions into four categories based on their agreement level (Figure 7B, Supplementary Method S4). We examined the performance of the similarity-measuring algorithms on the records of each agreement level. In short, if a similarity-measuring method performs inaccurately even when the experts have a high-level agreement, it would be considered a low-quality method. We observed a frequent increasing pattern in the performance of all similarity measuring techniques as the agreement level increased which is in line with our original hypothesis. Furthermore, we found FQ-HP to be more accurate than the other techniques, including Resnik, in the “substantial” and “high” agreement levels. These findings suggest that FQ-HP is more aligned with human experts’ decisions specially in cases with a higher-level agreement (Figure 7C).
Finally, we analyzed the efficacy of similarity-measuring algorithms in specific scenarios that were used in generating the expert-curated phenotypic trios (e.g., the combination of common and rare term frequencies as well as term hierarchies; Table 4). We sought to evaluate the effectiveness and significance of our proposed technique on scenarios that do not follow straightforward relationships based on the HPO DAG, i.e., direct hierarchical relationship between the reference terms and only one of their candidates. Figure 8 demonstrates the performance of the similarity-measuring algorithms in each scenario.
Figure 8. Comparison of scenario-based accuracy of the similarity-measuring algorithms.
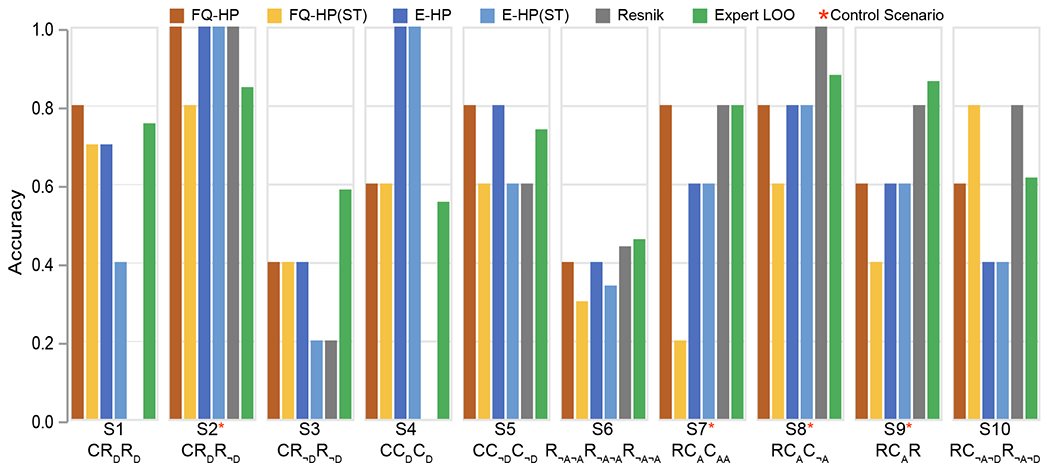
The conventional Resnik method is more aligned with experts’ assessments only in two similar scenarios: where the reference term is rare and is compared with only one hierarchically related common candidate (S8: RCAC¬A, S9: RCAR). However, Resnik fails on four other scenarios where such relationships do not exist (S1: CRDRD, S3: CR¬DR¬D, S4: CCDCD, S5: CC¬DC¬D), two of which with 0% accuracy (S1, S4). Table 4 provides details outlining each scenario.
We refer to the scenarios with straightforward relationships between the reference term and its candidate as positive controls (Table 4). We found that the conventional Resnik method performed best for positive controls (S22, S71, S8, S9), i.e., when only one candidate is in a hierarchical relationship with the reference term, providing accuracies close to expert assessments (Figure 8, marked with *). Also, among positive controls, Resnik performed better than embedding techniques in only two scenarios where the reference term was rare (S8 and S9). On the other hand, for scenarios where the reference term is not in a hierarchical relationship with the candidates (S1, S3, S4, S5), embedding methods are superior. These findings indicate that the accuracy of various strategies to assess phenotypic relatedness may depend on the relationship and frequencies of the specific terms, and Resnik may not be the preferred choice in challenging scenarios that do not follow straightforward relationships based on HPO.
Frequency-based phenotype embeddings can be used to measure patient similarity reliably
As discussed in the previous sections, our computationally efficient, interpretable, and versatile phenotype embedding technique effectively measures phenotypic similarity and aligns better with expert assessments, especially in challenging scenarios where conventional MICA-oriented techniques typically fail.
In this section we further confirm the reliability of our proposed method in a separate case study by evaluating its performance in measuring patient similarity. More specifically, we assess the phenotypic similarity of the 53 individuals with a genetic diagnosis of SCN1A-related disorders by employing metrics discussed in Section 2.8. We hypothesis that these individuals have a higher phenotypic similarity compared to a random cohort. The expected similarity scores for individuals with disease-causing variants in SCN1A (n=53) were assessed by determining their average similarity scores compared with 100 draws of random cohorts of size 53, leading to 137,800 permutations of patient pairs. Figure 9 demonstrates the distribution of patient similarity scores among random draws presented by the average similarity score in each cohort. Note that similarity scores based on the embedding techniques (FQ-HP and E-HP) has a different range than Resnik since cosine similarity used in is bound by [−1,1] while is bound by the of the rarest phenotype, which is 20.51 in our cohort. Hence, Resnik and embedding techniques cannot be directly compared based on reported similarity scores. In summary, all three methods easily identify the group of individuals with SCN1A-related disorder as significantly different than a random group of individuals. Our results demonstrate the applicability and reliability of our proposed frequency-based embedding technique, in this case on measuring patient similarity for a rare genetic childhood epilepsy.
Figure 9. Distributions of patient similarity scores, using 100 randomly chosen cohorts of size 53 of phenotypic similarities between paired individuals (137,800 paired patient permutations).
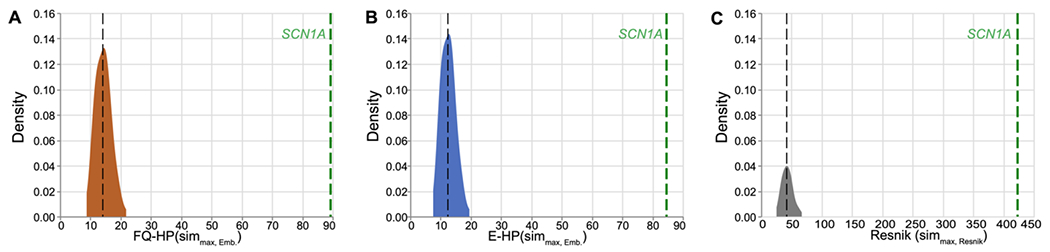
In each distribution plot, the vertical black line indicates the median similarity score of random cohorts using the specified similarity metric. The vertical green line represents the observed value for individuals with SCN1A-related disorder.
4. Discussion
This study aimed to assess the properties of phenotype embedding techniques to analyze the Human Phenotype Ontology (HPO) terms in a more computationally efficient manner while accurately reflecting their complex biological and clinical relationships. We examined representations of HPO terms using node embeddings and compared the performance of embeddings with and without including HPO term frequencies derived from more than 53 million patient notes. Finally, we assessed the degree to which phenotype embedding methods align with expert opinion and how these methods performed in comparison to conventional phenotype similarity-measuring techniques. We demonstrate that incorporating phenotype frequencies from a large patient corpus resulted in phenotype embeddings surpassing conventional techniques in calculating phenotypic similarity and aligning more closely with assessments by domain experts. In contrast to conventional methods such as Resnik, our proposed method is fast and computation-efficient and can also be used in many downstream tasks such as patient similarity without any recalculation for new patients, directly from the embedding space. In summary, our results demonstrate that incorporating phenotypes frequencies from a large patient corpus as a weighting mechanism can transfer the phenotype distributions to the embedding space and ultimately provide a superior representation that aligns more closely with domain experts.
Our study has five main findings. First, we used Human Phenotype Ontology (HPO) to analyze clinical data in our study, which has its own advantages and disadvantages. HPO contains more than 15,000 clinical phenotypic terms with defined semantic relationships, developed to standardize their representation for phenotypic analysis. In addition, HPO is modeled as a directed acyclic graph (DAG) in which each phenotype is presented as a node and is connected to its parents by “is a” relationships using directed edges. Despite the fact that this structure facilitates phenotypic analysis, HPO is limited to symptoms, and diseases and syndromes are not directly mapped to its structure. While this limitation affects the in-depth analysis of clinical features, incorporating more than 15,000 phenotypic terms in our analysis allowed us to cover a wide range of complex clinical relationships.
Second, we demonstrate that clinical information from full-text patient notes can be translated at scale through Natural Language Processing. In our study, we mapped the entirety of available full-text patient notes of a large, tertiary pediatric care network to Human Phenotype Ontology terms, generating the most comprehensive estimates for frequencies of clinical concepts in the pediatric population currently available (Supplementary Table S2). Given the increasing use of HPO for both common and rare diseases, having valid term frequencies will be essential for the development of algorithms aimed to assess phenotypic overlaps, such as the assessment of autistic-like traits [68] and rare neurodevelopmental disorders [69].
Third, we demonstrate that phenotype embedding is a useful tool to represent the >15,000 HPO concepts in a lower-dimensional space while preserving existing phenotypic relationships. Additionally, the inclusion of observed term frequencies improves the vector embedding. This was demonstrated by the embedding’s ability to generate meaningful proximities and distances between clinical terms. When compared to randomly assigned term frequencies, the inclusion of term frequencies in the embedding allows for more closely related phenotypes to be “drawn in” and more distantly related phenotypes to be “pushed out.” This exemplifies how additional information incorporated in the embedding technique such as term frequency can provide more specificity and nuance in the connections between phenotypes. Future iterations including temporal components [12, 70] or inclusion of additional data such as medication and procedure information may further improve phenotype embeddings and exceed the currently available frameworks.
Fourth, we demonstrate that the phenotype embedding methodology is largely superior to conventional similarity-measuring techniques; however, its strengths and weaknesses are dependent on the specific clinical scenario. For all comparisons, frequency-based embedding is slightly superior to equal-weight embeddings and Resnik similarity. Additionally, we only see the excellence of Resnik in scenarios where the reference terms are in a hierarchical relationship with one of the candidates. We reason that this strong effect of specific scenarios reflects the inherent mechanics of conventional methods that are designed based on the HPO graph. For example, the Resnik algorithm cannot generate meaningful similarities in situations when terms are not in a direct hierarchy, which is demonstrated by our results for Scenarios S1 and S3-S5. In contrast, frequency-based embeddings are susceptible to potential errors in frequency assessments, which may be critical when rare references are used.
Fifth, we demonstrate the efficacy of our proposed frequency-based phenotypic representation in measuring phenotypic similarity between individuals. As a case study, we show that in addition to accurately categorizing patients with certain diagnoses, in our case a genetic diagnosis of SCN1A, our proposed method is more cost-efficient. More precisely, in conventional techniques such as Resnik minor changes in patient data, e.g., recording a newly observed phenotype, require additional computation to find the least common ancestor for all new phenotypic pairs which in worst case scenarios leads to 15,371 times more than what embedding algorithms require (Supplementary Method S5).
Despite the large amount of data, our mapping algorithm and proposed similarity measuring approach have several limitations. First, we observed several phenotypes with unexpectedly large frequency values in our cohort; for example, Alveolar rhabdomyosarcoma (HP:0006779) had a propagated frequency of 27.31% while it was reported for a total of 987 children between 1975 and 2005 in the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program [71]. Given the large and diverse source of encounter notes available in our corpus and the multi-layer phenotype extraction process, we could not trace back these instances and update the extracted phenotypes in the encounters and their consequent frequencies. These frequencies could potentially shift the biased random walks, reducing their effectiveness in exploring neighborhoods; however, our proposed embedding method attempts to reduce the effect of such potential artifacts in the data by assigning the minimum propagated frequency of two phenotypes as their edge weight as well as using relative weights in graph exploration.
Furthermore, like other distance-based embedding algorithms, the biased random walks in our method are bound within a defined walk length and tend to exploit the local neighborhood more rather than explore further nodes. Consequently, denser sub-graphs of HPO are treated differently. Potential future solutions to overcome this problem may involve flexible exploring criteria, using dynamic p and q hyper-parameters depending on the sub-graph structure to navigate both sub-structures of the HPO more evenly. Unlike conventional machine learning problems, we did not have ground-truth similarity values for the phenotype pairs to tune the hyper-parameters used in our model, a limitation imposed by the structure of the data assessed in this study. These limitations shed light on the importance of incorporating domain experts’ knowledge in tuning the model parameters and potentially achieving higher accuracy.
Moreover, in evaluating our proposed method against domain experts’ judgments, we limited ourselves to neurological phenotypes. Given the expertise of our domain experts, we only validated the similarity-measuring techniques for phenotypes under “Abnormality of the nervous system (HP:0000707)” with thirteen domain experts in epilepsy and neurogenetics. The agreement level among our domain experts suggests that measuring phenotypic similarity could be a challenging task for experts in their particular field. This is particularly the case when experts are asked to compare pairs of phenotypes that are distant and hard to compare on anatomic or functional grounds. Having access to a larger and more diverse pool of domain experts could help us evaluate our model more thoroughly and obtain insights into overcoming such challenges. Additionally, based on the unsupervised nature of our embedding algorithm, we expect our proposed technique to work with approximately the same efficacy on phenotypes related to other fields of medicine.
In summary, we demonstrate that assessing clinical similarity in large EHR-derived datasets using phenotype embeddings may have significant advantages in scenarios where other similarity-measuring techniques have difficulties. We believe incorporating information from patient records, such as the intra-patient co-occurring phenotypes, will improve phenotypic similarity-measuring techniques. Incorporating additional disease-phenotype information has been explored before in the form of adding additional edges to the HPO in embedding phenotypes, a method referred to as HPO2Vec+ [33]. Although this technique was found to be more successful than embedding phenotypes purely based on the HPO, it required expert-curated data, has been evaluated on a limited set of phenotypes and diseases, and is not practical for hospital-size datasets. Our goal, however, is to provide unsupervised methods that are more robust and generalizable to large-scale data. Given the increasing availability of electronic health records and phenotype extraction techniques and the transformative results of applying representation learning in natural language processing, such as BERT [72] and GPT-3 [73], our phenotype embedding algorithm has the potential to be used in downstream tasks such as accessing similarities between clinical trajectories, identifying novel genetic etiologies. Ultimately, this will allow for providing better care to individuals by extracting useful information from unstructured medical records in a highly efficient manner. Representation learning can also be used to learn more general representations of clinical data, such as patient demographics, lab results, and imaging studies, that can be used as input to machine learning models for tasks such as risk prediction and prognosis estimation.
Supplementary Material
5. Acknowledgments and Funding
I.H. was supported by The Hartwell Foundation (Individual Biomedical Research Award), the National Institute for Neurological Disorders and Stroke (K02 NS112600, U24 NS120854, U54 NS108874), the Intellectual and Developmental Disabilities Research Center (IDDRC) at Children’s Hospital of Philadelphia and the University of Pennsylvania (U54 HD086984), and by the German Research Foundation (HE5415/3-1, HE5415/5-1, HE5415/6-1, HE5415/7-1). Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1 TR001878), by the Institute for Translational Medicine and Therapeutics’ (ITMAT) at the Perelman School of Medicine of the University of Pennsylvania, and by Children’s Hospital of Philadelphia through the Epilepsy NeuroGenetics Initiative (ENGIN). This research was funded in whole, or in part, by the Wellcome Trust [203914/Z/16/Z] supporting D.L.S.. For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Footnotes
Common indicates phenotypes with the relative propagated frequency > 20% with respect to Abnormality of the nervous system (HP:0000707).
Also, a tie between embedding techniques and Resnik.
References
- 1.Jha AK, et al. , Use of electronic health records in US hospitals. New England Journal of Medicine, 2009. 360(16): p. 1628–1638. [DOI] [PubMed] [Google Scholar]
- 2.Evans RS, Electronic health records: then, now, and in the future. Yearbook of medical informatics, 2016. 25(S 01): p. S48–S61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen T, Keravnou-Papailiou E, and Antoniou G, Medical analytics for healthcare intelligence–Recent advances and future directions. Artificial Intelligence in Medicine, 2021. 112: p. 1–5. [DOI] [PubMed] [Google Scholar]
- 4.Weng C, Shah NH, and Hripcsak G, Deep phenotyping: embracing complexity and temporality—towards scalability, portability, and interoperability. Journal of biomedical informatics, 2020. 105: p. 103433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kohler S, et al. , The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res, 2014. 42(Database issue): p. D966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kohler S, et al. , The Human Phenotype Ontology in 2017. Nucleic Acids Res, 2017. 45(D1): p. D865–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis-Smith D, et al. , Modeling seizures in the Human Phenotype Ontology according to contemporary ILAE concepts makes big phenotypic data tractable. Epilepsia, 2021. 62(6): p. 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohler S, et al. , The Human Phenotype Ontology in 2021. Nucleic Acids Res, 2021. 49(D1): p. D1207–D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groza T, et al. , The human phenotype ontology: semantic unification of common and rare disease. The American Journal of Human Genetics, 2015. 97(1): p. 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galer PD, et al. , Semantic similarity analysis reveals robust gene-disease relationships in developmental and epileptic encephalopathies. The American Journal of Human Genetics, 2020. 107(4): p. 683–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helbig I, et al. , A recurrent missense variant in AP2M1 impairs clathrin-mediated endocytosis and causes developmental and epileptic encephalopathy. The American Journal of Human Genetics, 2019. 104(6): p. 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis-Smith D, et al. , Phenotypic homogeneity in childhood epilepsies evolves in gene-specific patterns across 3251 patient-years of clinical data. European Journal of Human Genetics, 2021. 29(11): p. 1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis-Smith D, et al. , Computational analysis of neurodevelopmental phenotypes—harmonization empowers clinical discovery. Human Mutation, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewey FE, et al. , Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N Engl J Med, 2016. 374(12): p. 1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gusarova V, et al. , Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat Commun, 2018. 9(1): p. 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abul-Husn NS, et al. , A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med, 2018. 378(12): p. 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savova GK, et al. , Mayo clinical Text Analysis and Knowledge Extraction System (cTAKES): architecture, component evaluation and applications. Journal of the American Medical Informatics Association, 2010. 17(5): p. 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deisseroth CA, et al. , ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genetics in Medicine, 2019. 21(7): p. 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aronson AR and Lang F-M, An overview of MetaMap: historical perspective and recent advances. Journal of the American Medical Informatics Association, 2010. 17(3): p. 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodríguez-González A, et al. , Extracting diagnostic knowledge from MedLine Plus: a comparison between MetaMap and cTAKES Approaches. Current Bioinformatics, 2018. 13(6): p. 573–582. [Google Scholar]
- 21.Shivade C, et al. , A review of approaches to identifying patient phenotype cohorts using electronic health records. Journal of the American Medical Informatics Association, 2014. 21(2): p. 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Najafabadipour M, et al. , Reconstructing the patient’s natural history from electronic health records. Artificial Intelligence in Medicine, 2020. 105: p. 101860. [DOI] [PubMed] [Google Scholar]
- 23.Gérardin C, et al. , Multilabel classification of medical concepts for patient clinical profile identification. Artificial Intelligence in Medicine, 2022. 128: p. 102311. [DOI] [PubMed] [Google Scholar]
- 24.Resnik P, Using information content to evaluate semantic similarity in a taxonomy. arXiv preprint cmp-lg/9511007, 1995. [Google Scholar]
- 25.Li B, et al. , Effectively integrating information content and structural relationship to improve the GO-based similarity measure between proteins. arXiv preprint arXiv:1001.0958, 2010. [Google Scholar]
- 26.Pesquita C, et al. Evaluating GO-based semantic similarity measures. in Proc. 10th Annual Bio-Ontologies Meeting. 2007. Citeseer. [Google Scholar]
- 27.Bengio Y, Courville A, and Vincent P, Representation learning: A review and new perspectives. IEEE transactions on pattern analysis and machine intelligence, 2013. 35(8): p. 1798–1828. [DOI] [PubMed] [Google Scholar]
- 28.Le Q and Mikolov T. Distributed representations of sentences and documents. in International conference on machine learning. 2014. PMLR. [Google Scholar]
- 29.Mikolov T, et al. , Efficient estimation of word representations in vector space. arXiv preprint arXiv:1301.3781, 2013. [Google Scholar]
- 30.Grover A and Leskovec J. node2vec: Scalable feature learning for networks. in Proceedings of the 22nd ACM SIGKDD international conference on Knowledge discovery and data mining. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayanan A, et al. , graph2vec: Learning distributed representations of graphs. arXiv preprint arXiv:1707.05005, 2017. [Google Scholar]
- 32.Shen F, et al. Constructing node embeddings for human phenotype ontology to assist phenotypic similarity measurement. in 2018 IEEE International Conference on Healthcare Informatics Workshop (ICHI-W). 2018. IEEE. [Google Scholar]
- 33.Shen F, et al. , HPO2Vec+: Leveraging heterogeneous knowledge resources to enrich node embeddings for the Human Phenotype Ontology. Journal of biomedical informatics, 2019. 96: p. 103246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daniali M Phenotype Embedding. Source Code; 2023. [cited 2023; Available from: https://github.com/maryamdaniali/phenotype_embedding. [Google Scholar]
- 35.Arcus Data Repository Team, Deidentified Arcus Data Repository, Version 1.4.4. Extracted: 2021/07/09: Arcus at Children’s Hospital of Philadelphia. [Google Scholar]
- 36.Wheless JW, Fulton SP, and Mudigoudar BD, Dravet syndrome: a review of current management. Pediatric neurology, 2020. 107: p. 28–40. [DOI] [PubMed] [Google Scholar]
- 37.Ganesan S, et al. , A longitudinal footprint of genetic epilepsies using automated electronic medical record interpretation. Genet Med, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pagad NS and Pradeep N. Clinical named entity recognition methods: an overview. in International Conference on Innovative Computing and Communications. 2022. Springer. [Google Scholar]
- 39.Wu Y, et al. Clinical named entity recognition using deep learning models. in AMIA Annual Symposium Proceedings. 2017. American Medical Informatics Association. [PMC free article] [PubMed] [Google Scholar]
- 40.Šuster S, Tulkens S, and Daelemans W, A short review of ethical challenges in clinical natural language processing. arXiv preprint arXiv:1703.10090, 2017. [Google Scholar]
- 41.Straw I and Callison-Burch C, Artificial Intelligence in mental health and the biases of language based models. PloS one, 2020. 15(12): p. e0240376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thayer J, Miller JM, and Pennington JW. Fault-Tolerant, Distributed, and Scalable Natural Language Processing with cTAKES. in AMIA. 2019. [Google Scholar]
- 43.Masanz JJ and Finan S. CTAKES 4.0. 2021. [cited 2022; Available from: https://cwiki.apache.org/confluence/display/CTAKES/cTAKES+4.0.
- 44.Bodenreider O, The unified medical language system (UMLS): integrating biomedical terminology. Nucleic acids research, 2004. 32(suppl_1): p. D267–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman WW, et al. , A simple algorithm for identifying negated findings and diseases in discharge summaries. Journal of biomedical informatics, 2001. 34(5): p. 301–310. [DOI] [PubMed] [Google Scholar]
- 46.Tzadok M, et al. , CBD-enriched medical cannabis for intractable pediatric epilepsy: the current Israeli experience. Seizure, 2016. 35: p. 41–44. [DOI] [PubMed] [Google Scholar]
- 47.Li H, et al. , Overview of cannabidiol (CBD) and its analogues: Structures, biological activities, and neuroprotective mechanisms in epilepsy and Alzheimer’s disease. European journal of medicinal chemistry, 2020. 192: p. 112163. [DOI] [PubMed] [Google Scholar]
- 48.Xian J, et al. , Assessing the landscape of STXBP1-related disorders in 534 individuals. Brain (accepted), 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crawford K, et al. , Computational analysis of 10,860 phenotypic annotations in individuals with SCN2A-related disorders. Genet Med, 2021. 23(7): p. 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng Y, et al. , The state of the art in semantic relatedness: a framework for comparison. The Knowledge Engineering Review, 2017. 32. [Google Scholar]
- 51.Harispe S, et al. , Semantic similarity from natural language and ontology analysis. Synthesis Lectures on Human Language Technologies, 2015. 8(1): p. 1–254. [Google Scholar]
- 52.Slimani T, Description and evaluation of semantic similarity measures approaches. arXiv preprint arXiv:1310.8059, 2013. [Google Scholar]
- 53.Stevenson M and Greenwood MA. A semantic approach to IE pattern induction. in Proceedings of the 43rd Annual Meeting of the Association for Computational Linguistics (ACL’05). 2005. [Google Scholar]
- 54.Masino AJ, et al. , Clinical phenotype-based gene prioritization: an initial study using semantic similarity and the human phenotype ontology. BMC bioinformatics, 2014. 15(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin D. An information-theoretic definition of similarity. in Icml. 1998. [Google Scholar]
- 56.Jiang JJ and Conrath DW, Semantic similarity based on corpus statistics and lexical taxonomy. arXiv preprint cmp-lg/9709008, 1997. [Google Scholar]
- 57.Schlicker A, et al. , A new measure for functional similarity of gene products based on Gene Ontology. BMC bioinformatics, 2006. 7(1): p. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Q, et al. , Xrare: a machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genetics in Medicine, 2019. 21(9): p. 2126–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gong X, et al. , A new method to measure the semantic similarity from query phenotypic abnormalities to diseases based on the human phenotype ontology. BMC bioinformatics, 2018. 19(4): p. 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lambert J, Statistics in brief: how to assess bias in clinical studies? 2011, Springer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perozzi B, Al-Rfou R, and Skiena S. Deepwalk: Online learning of social representations. in Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining. 2014. [Google Scholar]
- 62.Tang J, et al. Line: Large-scale information network embedding. in Proceedings of the 24th international conference on world wide web. 2015. [Google Scholar]
- 63.Liu R, Hirn M, and Krishnan A, Accurately Modeling Biased Random Walks on Weighted Graphs Using Node2vec+. arXiv preprint arXiv:2109.08031, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pearson K, LIII. On lines and planes of closest fit to systems of points in space. The London, Edinburgh, and Dublin philosophical magazine and journal of science, 1901. 2(11): p. 559–572. [Google Scholar]
- 65.Van der Maaten L and Hinton G, Visualizing data using t-SNE. Journal of machine learning research, 2008. 9(11). [Google Scholar]
- 66.Stamberger H, et al. , Natural history study of STXBP1-developmental and epileptic encephalopathy into adulthood. Neurology, 2022. 99(3): p. e221–e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Molinaro AM, Simon R, and Pfeiffer RM, Prediction error estimation: a comparison of resampling methods. Bioinformatics, 2005. 21(15): p. 3301–3307. [DOI] [PubMed] [Google Scholar]
- 68.Ronald A, et al. , Phenotypic and genetic overlap between autistic traits at the extremes of the general population. Journal of the American Academy of Child & Adolescent Psychiatry, 2006. 45(10): p. 1206–1214. [DOI] [PubMed] [Google Scholar]
- 69.Cogliati F, Forzano F, and Russo S, Overlapping Phenotypes and Genetic Heterogeneity of Rare Neurodevelopmental Disorders. Frontiers in Neurology, 2021. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Skaf Y and Laubenbacher R, Topological data analysis in biomedicine: A review. Journal of Biomedical Informatics, 2022: p. 104082. [DOI] [PubMed] [Google Scholar]
- 71.Ognjanovic S, et al. , Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer: Interdisciplinary International Journal of the American Cancer Society, 2009. 115(18): p. 4218–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Devlin J, et al. , Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:1810.04805, 2018. [Google Scholar]
- 73.Brown T, et al. , Language models are few-shot learners. Advances in neural information processing systems, 2020. 33: p. 1877–1901. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.