

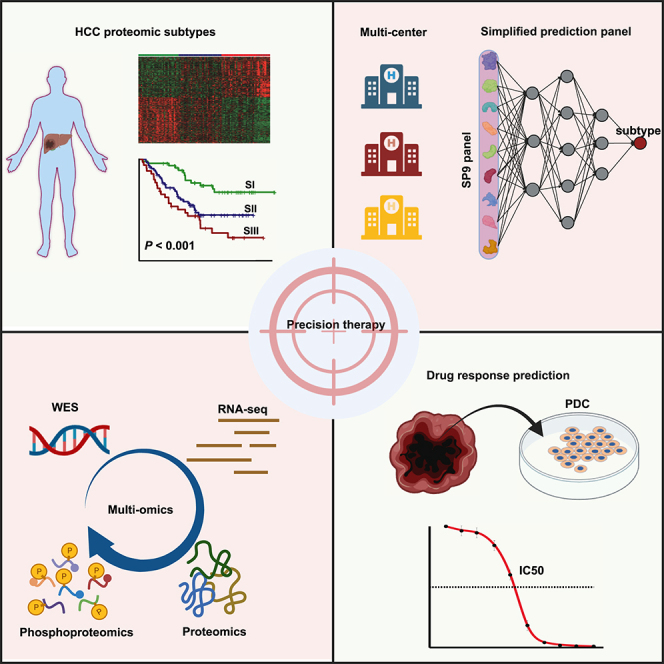
Summary
Patients with hepatocellular carcinoma (HCC) at the same clinical stage can have extremely different prognoses, and molecular subtyping provides an opportunity for individualized precision treatment. In this study, genomic, transcriptomic, proteomic, and phosphoproteomic profiling of primary tumor tissues and paired para-tumor tissues from HCC patients (N = 160) are integrated. Proteomic profiling identifies three HCC subtypes with different clinical prognosis, which are validated in three publicly available external validation sets. A simplified panel of nine proteins associated with metabolic reprogramming is further identified as a potential subtype-specific biomarker for clinical application. Multi-omics analysis further reveals that three proteomic subtypes have significant differences in genetic alterations, microenvironment dysregulation, kinase-substrate regulatory networks, and therapeutic responses. Patient-derived cell-based drug tests (N = 26) show personalized responses for sorafenib in three proteomic subtypes, which can be predicted by a machine-learning response prediction model. Overall, this study provides a valuable resource for better understanding of HCC subtypes for precision clinical therapy.
Keywords: hepatocellular carcinoma, proteomic subtypes, multi-omics, Sorafenib, machine learning, response prediction model, precision therapy
Graphical abstract
Highlights
-
•
A large-scale multi-omics study of hepatocellular carcinoma (HCC)
-
•
Robustness and simplified panel for HCC proteomic subtypes
-
•
Proteomic model to personalize prediction of sorafenib response for HCC precision therapy
-
•
Workflow from generating large-scale omics datasets to drug testing
Xing et al. develop a workflow from generating large-scale omics datasets to drug testing. The robustness and simplified panel for hepatocellular carcinoma (HCC) proteomic subtypes, as well as effective prediction of sorafenib response, demonstrate that targeting multi-omics can improve the implementation of personalized therapeutic strategies for HCC.
Introduction
Hepatocellular carcinoma (HCC) accounts for about 75%–85% of all primary liver cancer, with limited therapies and accompanied by poor prognosis.1 The majority of HCC patients are diagnosed at an advanced stage and can only receive systemic anti-tumor therapy, which is less effective and has a low response rate.2,3,4,5,6,7 In particular, the prognoses of patients with HCC at the same clinical stage have been remarkably different, making it challenging to predict the outcomes. Therefore, accurate staging and subsequent appropriate personalized treatment are the keys to improving clinical outcomes of HCC.
Extensive efforts have been devoted to identify new subtypes for HCC using genomic and transcriptomic data,8,9,10,11,12 but with limited impact on clinical decisions. Proteins ultimately perform the real biological functions and represent the real world of complex diseases; in particular, the important post-translational modifications (PTMs) of proteins cannot be informed from genomic and transcriptomic data, such as phosphorylation-related kinases that are important drug targets. Proteomics, which reflects systemic changes in functional executors, has been extensively applied for biomarker discovery and therapy selection in cancers13,14,15,16,17 and other complex diseases,18,19,20,21,22,23 and provides unlimited potential for precision medicine. Recently, Jiang et al. performed a comprehensive proteomics study and stratified early-stage HCC patients into three subtypes with different clinical outcomes (SI, SII, and SIII), and they identified a therapeutic target sterol O-acyltransferase 1 (SOAT1) for subtype SIII with the worst prognosis.24 Subsequently, Gao et al. performed the proteogenomic characterization of hepatitis B virus (HBV)-related HCC and also reported three similar HCC proteomic subtypes; meanwhile, they further identified two prognostic biomarkers involved in metabolic reprogramming.25 These two studies opened the avenue to precision typing and prognosis determination of HCC based on proteomics.
To apply HCC proteomic subtypes in clinical precision therapy, further verifying the robustness and universality of HCC proteomic subtypes and identifying signatures for HCC subtypes are crucial. In addition, occurrence and progression of HCC is an extremely complex process, and the proteomics also needs to be integrated with genomics and transcriptomics to panoramically and comprehensively understand the molecular changes of HCC from the genetic level to the protein level and even the PTM level to better predict prognosis, find personalized drug targets, and evaluate drug efficacy.26,27,28,29,30,31,32,33,34,35,36,37,38,39,40
Here, we comprehensively analyzed the genomic, transcriptomic, proteomic, and phosphoproteomic profiles of an independent HCC cohort to reveal and verify the robustness and universality of the proteomic subtypes and have developed a simplified discriminating panel of proteomic subtypes using machine learning. Integrated multi-omics analysis further revealed the functional impact of the molecular alterations for different HCC subtypes. In addition, we identified actionable drug targets and corresponding drugs for different proteomic subtypes by integrating proteomics and phosphoproteomics and further assessed the therapeutic responses in patient-derived cells (PDCs). Overall, our study highlights the importance of proteomics and phosphoproteomics in clinical decisions for HCC patients, especially in precise prognosis evaluation and personalized therapeutic strategy selection.
Results
Overview of the study
Proteomics-based precision typing of HCC leads us to the era of precision medicine. Regarding the robustness and universality of HCC proteomic subtypes, molecular characterization and a simplified panel for clinical application are the main obstacles to further advancement of proteomics to the clinic. To this end, we performed multi-omics profiling of primary tumor tissues (T) and paired para-tumor tissues (N) from 160 HCC patients (proteomics, N = 160; phosphoproteomics, N = 132 from 160; whole-exome sequencing [WES], N = 58 from 132; transcriptome sequencing [RNA_Seq], N = 57 from 58) (Figure S1A and Table S1). HCC subtypes were identified by proteomic analysis based on data-independent acquisition (DIA) (Figure S1B) and validated in three independent publicly available external validation sets to confirm its robustness and universality. Machine learning was used to develop a simplified panel for discriminating HCC proteomic subtypes for facilitating clinical applications. Furthermore, to explore the molecular characteristics and screen the potential clinically actionable drugs for different HCC proteomic subtypes, integrated multi-omics analysis was performed, especially kinase abundance and kinase-substrate regulatory network analysis. Finally, 26 PDC models were established to investigate the personalized response to sorafenib in different proteomic subtypes, and a sorafenib therapeutic effect prediction model was constructed and validated by machine learning to provide precise clinical decisions for patients who should undergo sorafenib treatment.
Proteomic characterization identified three HCC subtypes
In total, we identified 6,512 proteins (false discovery rate <0.01) across all 152 paired samples that passed quality control, accounting for 76.33% of the hybrid spectral library (Figures S1C and S1D; Table S2). The high stability of the mass spectrometry (MS) platform (median Pearson’s correlation coefficient of 0.954 in data-dependent acquisition [DDA] mode, 0.976 in DIA mode) resulted in high quantitative stability and accuracy of HCC samples (Figures S1E and S1F). As expected, the coefficient of variation values were higher in T than that in N (6.37% vs. 5.37%) (Figure S1G), and the T and N were distinctly distinguished (Figures S1H and S1I), highlighting the high inter-tumor heterogeneity of HCC, consistent with previously reported studies.24 These results strongly affirmed the high quality of our proteomic data.
Unsupervised consensus clustering identified three proteomic subtypes in 152 HCC patients, which were designated as SI (n = 49), SII (n = 47), and SIII (n = 56) (Figures 1A and S2A–S2C). Furthermore, the three proteomic subtypes significantly differed in overall survival (OS) and recurrence-free survival (RFS) (log-rank test, p < 0.0001) (Figure 1B). The three proteomic subtypes were further verified in two independent surgical specimen cohorts from Jiang et al. (N = 101)24 (Figure 1C) and Gao et al. (N = 159)25 (Figure 1D), and even in an independent biopsy specimen cohort from Ng et al. (N = 51)41 (p < 0.01) (Figure 1E), which supported the reliability of our proteomic subtypes. Consistent with the current clinical consensus, the advanced BCLC stage (p = 0.041) and TNM stage (p = 0.032, Fisher’s exact test), positive α-fetoprotein (AFP) (p < 0.001), microscopic vascular invasion (MVI+, p = 0.006), tumor low differentiation (p = 0.001), multiple tumor number (p = 0.027, Fisher’s exact test), and absence of tumor capsule (p = 0.045, Fisher’s exact test) were more prominent in SIII than in SI, and SII was intermediate (Figures 1A and S2D–S2J; Table S2). In addition, the proteomic subtypes were also authenticated as an independent prognosis indicator by multivariate analysis (hazard ratio, 2.43; 95% confidence interval, 1.22–4.82; p = 0.011) (Figure S2K).
Figure 1.

Proteomic characterization identified three HCC subtypes
(A) Consensus clustering of 152 HCC tumors. The associations of HCC proteomic subtypes with clinical characteristics are annotated in the upper panel (chi-squared test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). The heatmap depicts the relative abundance of signature proteins (log2-transformed). Each column represents a patient sample, and rows indicate proteins.
(B) Kaplan-Meier (KM) curves of OS and RFS for each proteomic subtype (log-rank test).
(C–E) KM curves of OS for each proteomic subtype in Jing et al.’s cohort (C), Gao et al.’s cohort (D), and Ng et al.’s cohort (E). The p values are calculated by log-rank test.
(F) ssGSEA reveals the pathways that are significantly enriched in the three respective proteomic subtypes. The specific enriched signaling pathways of each subtype are analyzed and summarized into four clusters. C1, upregulated in SI only; C2, upregulated in both SI and SII; C3, upregulated in SII and SIII; C4, upregulated in SIII only.
(G) Signal pathway changing trend in the four clusters (C1–C4).
See also Figures S1 and S2.
Molecular features of three HCC proteomic subtypes
To comprehensively characterize the molecular features of three HCC proteomic subtypes, the specific enriched signaling pathways of each subtype were analyzed and summarized (Figure 1F). SI was characterized by the highest enrichment of metabolism-related pathways, which accounted for the majority of all enriched pathways and was highly consistent with Jiang et al.’s and Gao et al.’s (denoted as metabolism subgroup, S-Mb) results.24,25 SII was a transitional subtype from SI to SIII with an increase in cell-growth- and immunity-related pathways but a decrease in metabolism-related pathways. The enrichment of immune-related pathways, such as antigen processing and presentation and Toll-like receptor, was consistent with the immune properties of SII in Jiang et al.’s and Gao et al.’s studies (denoted as microenvironment dysregulated subgroup, S-Me). SIII showed an increase in proliferation-, metastasis-, and immune-related pathways but almost no metabolic pathways, which validated the immune/metastatic characteristics of Jiang et al.’s SIII subtype and the proliferation characteristics of Gao et al.’s SIII subtype (denoted as proliferation subgroup, S-Pf), suggesting more aggressive characteristics in SIII than in both SI and SII (Figure 1G and Table S2).
Thus, the molecular characteristics of the three proteomic subtypes in the three independent studies were highly consistent, indicating the stability and reliability of HCC proteomic subtypes. Meanwhile, the relevant or specific pathways of HCC significantly correlated with their proteomic subtypes, further highlighting their clinical implications.
The HCC proteomic subtypes were robust and universal
To investigate the universality of HCC proteomic subtypes, we performed a cross-validation with our subtypes and the proteomic subtypes from Jiang et al.’s cohort24 and Gao et al.’s cohort,25 with patient number >100. These three cohorts belonged to different clinical centers with different inclusion/exclusion criteria and MS strategies (Figure 2A). The subtype-specific signatures (namely, signatures) contained 761 proteins in our cohort, which was significantly lower than the signatures of Jiang et al. (1,269 proteins) and Gao et al. (1,274 proteins) (Figure S3A and Table S3). However, to increase the potential for clinical application, these signatures need to be further simplified, as it is generally believed that fewer proteins are more applicable for clinical translation. For cross-validation, firstly, clustering our DIA data with Jiang et al.’s and Gao et al.’s signatures from DDA data using unsupervised consensus clustering also generated three subtypes with different OS (p = 0.00015 and p = 0.00047), and there was obvious concordance of patient allocation with our original subtypes (Jiang et al.: 85.5%; Gao et al.: 82.9%), supporting the reliable data-acquisition and subtyping procedure in this study (Figure 2B and Table S3). In turn, the consistency was up to 79.2% in Jiang et al.’s cohort (p = 0.0051) and 78.6% in Gao et al.’s cohort (p < 0.0001) when clustering their data using our signatures (Figure 2C). Furthermore, in the mutual verification of the signatures from each of Jiang et al.’s or Gao et al.’s cohort, the consistency between these subtypes and the original subtypes reached up to 88.1% in Jiang et al.’s cohort (p = 0.05) and 77.9% in Gao et al.’s cohort (p = 0.00032) (Figures 2D and S3B; Table S3). These results showed that the signatures from three proteomic studies were robust and universal.
Figure 2.

The robustness and universality of HCC proteomic subtypes and their simplified discriminating panel
(A) Workflow of cross-validation of proteomic signatures in three cohorts (Gao et al.’s cohort: N = 159; Jiang et al.’s cohort: N = 101; present cohort: N = 152).
(B) Validation of Jiang et al.’s and Gao et al.’s signatures in our cohort. The left panel shows the KM curves of OS and RFS according to the proteomic subtypes (log-rank test). The right alluvial plot shows the comparison between these subtypes and the original subtypes.
(C) Validation of our signatures in Jiang et al.’s and Gao et al.’s cohorts. The confusion matrices are provided.
(D) Validation of Jiang et al.’s and Gao et al.’s signatures in each other’s cohorts. The confusion matrices are provided.
(E) Concordance rate of three proteomic signatures in three cohorts.
(F) Workflow for developing the simplified panel for discriminating proteomic subtypes.
(G) Receiver-operating characteristic accuracy, sensitivity, and specificity of simplified panel for discriminating proteomic subtypes in the validation set.
(H) Alluvial plot shows the comparison between proteomic subtypes identified by simplified panel and the original subtypes in the validation set. The predictor represents subtypes from the SP9, while the response represents the original subtypes from the full panel.
See also Figure S3.
Generally, a strong concordance of three proteomic subtypes was validated in three independent cohorts (over 80% on average), with SIII having the highest concordance among the three subtypes in three independent cohorts, followed by SI, while SII showed the lowest consistency because of its transition properties (Figure 2E). In addition, most of the inconsistencies by different signatures in the same cohort were found in SII or SIII because of the similar molecular features in SII and SIII as mentioned above (Figures S3C–S3H).
A machine-learning-based simplified panel for distinguishing HCC proteomic subtypes
It is expressly necessary to reduce the number of signatures of HCC proteomic subtypes to facilitate potential clinical applications. For machine learning, our cohort and Jiang et al.’s cohort were used as the training set, and Gao et al.’s cohort was used as the independent external validation set, after removing the batch effect from these three cohorts (Figures 2F and S3I). The machine-learning analysis resulted in a simplified panel (SP9) comprising nine proteins (DCXR, EHHADH, ALDH4A1, ABAT, ALDH6A1, ALDH7A1, SULT2A1, SORD, and ACSM2B), which were all drastically downregulated in SIII (Figure S3J) and significant for OS to stratify the patients (Figure S3K). Mechanistically, they were involved with fatty acid metabolism (ACSM2B, EHHADH), amino acid metabolism (ALDH6A1, ALDH7A1, ALDH4A1, ABAT), glycometabolism (SORD, DCXR), and bile secretion (SULT2A1).
SP9 showed high sensitivity and specificity to discriminate SI or SIII from the total HCC cohort (Figures S3L and S3M). In the validation cohort, the sensitivity and specificity of SP9 for SI discrimination were 84.5% and 96.4%, respectively with an area under the curve (AUC) of 0.961; for SII discrimination the sensitivity and specificity were 72.7% and 89.4% with an AUC of 0.881; and the highest sensitivity (95.8%) and specificity (95.5%) with the highest AUC of 0.988 were obtained in discriminating SIII from SI and SII patients, respectively (Figure 2G). Furthermore, the subtypes using SP9 showed a similarity up to 84.3% with the original subtypes, in conformance with the similarity of the three subtypes in different cohorts (Figure 2H). These results indicated that SP9 was stable and reliable for HCC subtyping, which provided a possibility for clinical application of HCC proteomic subtypes.
Mutation and copy-number variation landscapes of three HCC proteomic subtypes
To further explore the molecular characteristics of these three proteomic subtypes, we performed WES (N = 58), RNA_Seq (N = 57), and phosphoproteomic (N = 132) analysis of paired T and N samples (Figure 3A), whose clinicopathological characteristics were not significantly different from the total samples (Figure S4A). The mutation landscape showed that TP53, TNN, OBSCN, CTNNB1, RYR2, PCLG, MUC16, RYR1, ARIDA1, and RB1 were the most frequently mutated genes in HCC (Figures 3B and S4B; Table S4). CTNNB1 mutations were the most important feature in SI and SIII and were significantly associated with OS and RFS (p < 0.05) rather than WNT pathway alterations (Figures 3C–3F and S4C–S4G). These findings were consistent with previous reports, indicating the reliability of our results.42,43
Figure 3.

Proteogenomic and immune landscape of three HCC proteomic subtypes
(A) Illustration of 152 paired HCC cases used in the individual omics experiments. The omics experiments are colored blue, and the tumor tissues and non-tumor tissues were detected in pairs.
(B) WES-based genomic landscape of the three HCC proteomic subtypes. The top panel represents the mutation profile, the middle panel represents TMB and TNB levels, and the bottom panel represents the CNV.
(C–E) Feature importance ranking by the random forest algorithm for SI (C), SII (D), and SIII (E). The feature with the highest rank of importance score indicates the highest association (either positive or negative) with the proteomic subtype.
(F) KM curves for OS of HCC patients with CTNNB1 mutation or wild type (log-rank test).
(G) Heatmap shows the immune cell populations of HCC patients belonging to different proteomic subtypes.
(H) Principal component analysis plot of immune scores of immune cell populations based on proteomic data in three proteomic subtypes.
(I) Proteome-based immune scores in three proteomic subtypes (two-tailed Wilcoxon test). Box plots show median (central line), upper and lower quartiles (box limits), and 1.5× interquartile range (whiskers).
(J) Proteomics-based immune scores of anti-tumor immunity and pro-tumor immune suppression in three proteomic subtypes (two-tailed Wilcoxon test). Box plots show median (central line), upper and lower quartiles (box limits), and 1.5× interquartile range (whiskers).
(K) Correlation between anti-tumor immunity and pro-tumor immune suppression based on proteome in three proteomic subtypes. Each point represents a patient sample, the lines represent the fitted curves of correlation in each subtype, and the shaded area represents 95% confidence interval. Pearson’s correlation coefficient (r) and p values are present in the table. The p values were calculated using Pearson’s correlation method.
See also Figure S4.
Meanwhile, the copy-number variation (CNV) landscape showed significant differences among different HCC subtypes, with different regulatory genes during hepatocarcinogenesis and progression. 1q was predominantly co-amplified in SII and SIII, suggesting that they may co-regulate gene expression (Figure 3B). Additionally, the tumor mutation burden (TMB) and tumor neoantigen burden (TNB) in individual patients revealed the heterogeneity of HCC, consistent with previous reports.44 Nevertheless, there was no significant difference in TMB and TNB among the three proteomic subtypes (Figure 3B and Table S4).
Immune landscape of three HCC proteomic subtypes
To investigate the immune landscape of three HCC proteomic subtypes, we estimated the abundance of 28 immune cell populations using the single-sample gene set enrichment analysis (ssGSEA) method based on the proteomic and transcriptomic data. The immune landscape of three HCC proteomic subtypes showed high heterogeneity, with distinctly distinguished abundance of immune cell populations (Figures 3G, 3H, S4H, and S4I). The immune infiltration score was significantly higher in SIII than that in SI and SII (p < 0.05, median ± SD: SIII [162.74 ± 258.87] vs. SII [17.72 ± 154.67] vs. SI [−186.26 ± 163.07]) (Figures 3I and S4J). In detail, SI was characterized by increasing CD56 dim natural killer cells compared to SII and SIII. Meanwhile the abundance of most immune cells, including but not limited to central memory CD4 T cells, natural killer T cells, regulatory T cells, and myeloid-derived suppressor cells, were significantly higher in SIII than in the other two subtypes (Figure S4K). The enriched ssGSEA score of immune activation cells (p < 0.05, median ± SD: SIII [1.25 ± 3.05] vs. SII [0.32 ± 2.64] vs. SI [−0.73 ± 4.05]) and immune suppression cells (p < 0.05, median ± SD: SIII [0.57 ± 3.33] vs. SII [−3.10 ± 2.37] vs. SI [−0.48 ± 2.32]) were both significantly higher in SIII than in SI and SII (Figures 3J and S4L). Meanwhile, anti-tumor immunity and pro-tumor immune suppression were significantly positively correlated in SIII, indicating a co-existence of immune activation and immunosuppression in aggressive HCC (Figures 3K and S4M).
In addition, the abundance of key molecules of the immune microenvironment were significantly different among the three proteomic subtypes. As expected, SIII displayed significant upregulation of human leukocyte antigen (HLA) molecules (including HLA class I and II molecules), immune checkpoints (such as PDCD1 [PD-1], CD86, and CD47), immune-related CT antigens (such as ADAM28, CEP55, SPAG9, and PBK), and cytokines (such as IL-16, CCL3, TGFB-3, TGFB-1, CCL21, CFS1, and IL-18) compared with the other two subtypes (Figure S4N). These results suggested that SIII HCC patients might be more suitable for immunotherapy, although this would need to be validated in other prospective clinical trials.
Phosphoproteomic and kinase profiles of three HCC proteomic subtypes
In phosphoproteomics analysis, the high inter-tumor heterogeneity in HCC that was shown in proteome was also clearly presented in phosphoproteome, and the characteristics of the phosphorylation modifications were similar to those in other reports (Figures 4A and S5A–S5G; Table S5).45 The signaling pathways enriched in three proteomic subtypes at the phosphorylation level were also very similar to that at RNA level and protein level (Figures 4B, 4C, and S5H–S5J; Table S6). Considering that protein kinases have been developed as viable drug targets for cancer therapies, we next investigated the kinase activity (reflected by the overall phosphorylation level of the substrates) in three proteomic subtypes and its correlation with the substrate phosphorylation level and kinase abundance. In total, 215 kinases were found in HCC, among which the activity of 75 kinases differed among three subtypes, and the enriched pathways also suggested the close association of kinase activity with the occurrence and development of HCC (Figures 4D and 4E, Table S5). Furthermore, the kinase-substrate correlation was significantly lower in SIII than that in SI and SII (p < 0.01, median ±SD: SIII [0.15 ± 0.22] vs. SII [0.17 ± 0.22] vs. SI [0.16 ± 0.23]) (Figures 4F and 4G). Nevertheless, there was no correlation between kinase abundance and kinase activity (Figure 4H). Moreover, the clinical value of kinase activity and kinase abundance in HCC was also significantly different. For example, the kinase activity of BCKDK, CDK16, and MAP3K2, but not the kinase abundance, was significant for the prognosis of HCC (Figures 4I, 4J, and S5K). Therefore, it is necessary to systematically analyze the abundance and activity of kinases by integrating proteome and phosphoproteome for drug screening and precise treatment.
Figure 4.

Phosphoproteomic profile and kinase-substrate regulatory network of three HCC proteomic subtypes
(A) Summary of the identification of phosphoproteins and kinases.
(B) Pathway alterations of phosphoproteins in three proteomic subtypes.
(C) Enriched functions of three proteomic subtypes by ssGSEA.
(D) Kinase activation enrichment of differentially abundant phosphosites among three proteomic subtypes. Each column represents a patient sample, and each row indicates a kinase.
(E) Kinase regulation-pathway network. Rhombus indicates signaling pathways, and roundness indicates kinases differentially expressed in phosphoproteomic data.
(F) Kinase-substrate regulation networks in three proteomic subtypes. The edges represent Pearson’s correlation coefficient between kinases and the corresponding phosphosubstrates.
(G) Distribution of Pearson’s correlation coefficients of kinase-substrate networks in (F) (two-tailed Wilcoxon test). Box plots show median (central line), upper and lower quartiles (box limits), and 1.5× interquartile range (whiskers).
(H) Correlation between kinase abundance and kinase activity. r represents Pearson’s correlation coefficient. The p values were calculated using the Pearson’s correlation method.
(I and J) KM curves of OS (I) and RFS (J) for BCKDK activity (log-rank test).
See also Figure S5.
Multi-omics landscape of three HCC proteomic subtypes
To explore the inter-omics correlation, we visualized the correlations of different omics using Pearson’s correlation coefficient. The results demonstrated that the correlation decreased along with the “central dogma,” which may be attributed to the enrichment of attenuated proteins, reflecting RNA splicing, protein synthesis, protein degradation, and even PTMs (Figures 5A–5C, S6A, and S6B). Notably, phosphorylation levels poorly correlated with CNV alterations, mRNA, and protein abundance (median Pearson’s r < 0.02), indicating the important role of the protein PTM in the development and progression of HCC (Figures 5D and 5E). The inter-omics correlations differed among the three proteomic subtypes, especially the correlation between RNA abundance and protein abundance, while the correlation between protein abundance and phosphorylation level was not significantly different among the three proteomic subtypes, mainly due to the low overall correlation between them (Figure S6C). Furthermore, we found that two-thirds of the significant mRNA-protein co-variates were positive while showing a higher number of negative regulatory interactions of protein phosphorylation, which were classified into three clusters (Figures 5F and S6D). Consistent with the characterization of proteomic subtypes, the SI-specific positive correlation cluster featured metabolism characteristics, and SIII-specific positive correlation clusters were significantly involved in proliferation and metastasis pathways (Figures 5G and S6E).
Figure 5.

Integrated multi-omics analysis of three HCC proteomic subtypes
(A) Correlation between WES, transcriptome, proteome, and phosphoproteome. The overlap patients of every two individual omics were analyzed.
(B) Spearman’s correlation of CNV to mRNA and protein. Each dot represents a transcript/protein. Attenuated proteins are represented in red using a Gaussian mixture model with two mixture components.
(C) Enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the attenuated proteins.
(D) Scatterplots depicting log2 (T/N) of protein (x axis) and phosphoprotein (y axis) abundance. The red dots indicate positive correlations, and the blue dots indicate negative correlations. The p values were calculated using the Pearson’s correlation method.
(E) Enriched KEGG pathways for negative-correlated proteins in (D).
(F) Hierarchical clustering analysis map of significantly changed phosphosite-to-protein correlations among three proteomic subtypes. Pearson’s correlation coefficients between matched pairs of phosphosite abundances versus protein abundances were calculated.
(G) Functional enrichment for significant phosphosite-to-protein correlations in each cluster.
(H) Phosphosite-to-protein co-varying MCODE complexes/subnetworks of significantly changed phosphosite-to-protein correlations among three proteomic subtypes. Top five phosphosite-to-protein co-varying MCODE complexes/subnetworks are shown in different colors.
(I–K) Top specific MCODEs in SI (I), SII (J), and SIII (K) positivity are combined with upstream kinases.
See also Figure S6.
Furthermore, MCODE complex/subnetwork analysis showed the top five key co-varying phosphosite-to-protein MCODEs (Figure 5H). We noticed that some kinases were shared within MCODEs; for example, AKT1 and CDK1, the top co-varying phosphosite-to-protein MCODEs, differed in the three proteomic subtypes. mRNA processing was the top MCODE in SI-positive cluster, and the kinase PRKCA involved in transcriptional regulation of upstream mRNA splicing was identified, consistent with previous reports (Figure 5I).46 Furthermore, nucleocytoplasmic transport was the top MCODE in the SII-positive cluster, and the experimentally verified or predicted upstream kinases (CLK1, MAPKAPK2) were also mapped (Figure 5J). In contrast, regulation of actin cytoskeleton was the top MCODE in the SIII-positive cluster, and the kinase AKT1 involved in the upstream regulation of actin cytoskeleton organization was also reported (Figure 5K).
Altogether, due to the high heterogeneity and patient specificity, the integrated analysis of multi-omics is more suitable for the comprehensive molecular characterization of HCC. In particular, the integrated analysis of proteomics and phosphoproteomics has promising prospects for drug screening and precision treatment of HCC.
Drug-response prediction for three HCC proteomic subtypes
To identify potential clinical drugs for the three HCC subtypes, we screened drug targets of solid tumor within the DrugBank database using quantitative proteomic and phosphoproteomic data. A total of 38 quantifiable drug targets (including 11 kinases) belonging to 33 clinically actionable drugs were identified, with significant difference in abundance among three proteomic subtypes (Figure 6A). We identified targets of bevacizumab (C1QC, C1QB, C1QA, FCGR3A, FCGR2C), dasatinib (RSC, HSPA8, PPAT, CSK, LYN, MAPK14, FRK), and neratinib (ORM1, ORM2), with higher abundance in SIII than in SI and SII. Notably, RAF1 and LYN, key targets of sorafenib and regorafenib, had significantly higher kinase abundance in SIII than in SI and SII. However, enzymes affecting the drug activity (UGT1A9, UGT1A1, UGT1A3) or drug metabolism (CYP2D6, CYP3A4) of sorafenib and regorafenib were identified as highly ranked targets in SI and SII but not in SIII. Furthermore, phosphorylated substrates of kinase RAF1, MAP2K1, MAP2K2, and LYN were also significantly higher in SIII, suggesting abnormal regulation of kinase activity in SIII. In addition, the inconsistency between kinase abundance and substrate phosphorylation suggested that the aberrant regulation of kinases was caused by both kinase abundance and activity (Figure 6A).
Figure 6.

Drug prediction and key drug target screening for three HCC subtypes
(A) Phosphosubstrates of kinases with clinical available drugs and fold change at proteomic and phosphoproteomic levels for kinases and substrates, respectively. The top section displays the abundance of drug-targeting proteins across the three subtypes, with each row representing a drug-targeting protein and “k” labeling representing drug-targeting kinase. The middle section shows the phosphorylation site abundance of substrate for differentially abundance kinases among the three subtypes, with each row representing the phosphorylation sites of substrate. The bottom section displays the abundance of the protein where the phosphorylation site is located, with each row representing a protein. The color gradient represents the abundance of drug-targeting proteins (kinases), phosphorylation sites, and substrate proteins, with green indicating low expression and red indicating high expression. Red labels indicate statistically significant differences in abundance among the three proteomic subtypes.
(B) Kinase activity of FDA-approved drug targets in three proteomic subtypes (two-tailed Wilcoxon test).
(C) Pathways based on the selected phosphosubstrates and kinases, with relevant drugs shown by targets.
(D) Prognostic risk scores of each target from FDA-approved HCC clinical drugs. The x axis indicates log2-based hazard ratio for each target (log-rank test); y axis indicates log2-based T/N fold change for each target (two-tailed Wilcoxon test).
(E) KM curves of OS and RFS for RAF1. The p values were calculated by log-rank test.
(F) Abundance of RAF1 among three proteomic subtypes (two-tailed Wilcoxon test).
(G) Correlation between substrate RAF1 and upstream kinase activity among three proteomic subtypes (two-tailed Wilcoxon test).
See also Figure S6.
We next analyzed the kinase activity of the drug targets and observed high heterogeneity in kinase activity among the three proteomic subtypes (Figure S6F). Specifically, the kinase activity of EGFR was significantly increased in SII compared to SI and SIII (p < 0.05); the kinase activity of ERBB2 was significantly decreased in SIII compared to SI and SII (p < 0.05); while the kinase activity of MAP2K1 was significantly increased in SIII compared to SI and SII (p < 0.05), highly consistent with the protein abundance of these kinases in the three subtypes. However, the kinase activity of RAF1 was significantly downregulated in SIII compared to SI and SII, which was completely opposite to the protein abundance (Figures 6B and S6G). Combined with the significant increase in protein abundance of BARF, MAP2K2, and MAPK1 in SIII, we speculated that the RAF-MEK-ERK signaling pathway was significantly upregulated in SIII (Figure S6G). Additionally, the decrease in kinase activity of downstream mTOR and the increase of its substrate EIF4EBP1 phosphorylation in SIII suggested that combination therapy targeting both the RAF-MEK-ERK and mTOR pathways may be a more effective approach for SIII (Figures 6C and S6H). Therefore, a more comprehensive investigation of the signaling pathway regulatory networks could provide effective evidence for expanding treatment options beyond the current Food and Drug Administration (FDA)-approved HCC therapies.
Furthermore, after stringent filtering processes, 19 out of 22 drug targets with clinically available drugs showed significant prognostic values for HCC (Figures 6D and S6I; Table S7). Among these targets, RAF1 had the highest negative correlation with OS and RFS (p < 0.01) (Figure 6E). The abundance of RAF1 was also significantly higher in SIII than in SI (p < 0.05) (Figure 6F). Additionally, the correlation between RAF1 and its upstream kinases significantly increased in SII and SIII compared to SI (p < 0.01), suggesting the important regulating role of RAF1 in SII and SIII (Figure 6G). These observations can explain the potential clinical benefits to SIII subtype HCC patients of RAF1-targeted therapies such as sorafenib, regorafenib, and dabrafenib.
SIII subtype has more specific drug sensitivity to sorafenib than SI
To further explore the potential drug response of HCC patients in three proteomic subtypes, we investigated the therapeutic effects of sorafenib on 26 PDC models (Figures 7A and 7B; Table S1). The clinicopathological features of these PDC models were not significantly different from those of the proteomic subtype discovery cohort (Figure S7A). We assigned these 26 PDCs to their corresponding proteomic subtypes by consensus clustering of their proteomic data (Figure S7B).
Figure 7.

Subtype-specific drug sensitivities and machine-learning-based efficacy prediction model for sorafenib
(A) Workflow of pharmacological tests using PDC models.
(B) Immunofluorescence of PDCs. Blue represents DAPI staining of the cell nucleus, green represents the distribution of GPC3 on the cell membrane, and red represents the localization of α-fetoprotein (AFP) and albumin (ALB) in the cytoplasm. Scale bar, 20 μm.
(C) Sorafenib sensitivity results of the PDC models in three proteomic subtypes (two-tailed Wilcoxon test). Box plots show median (central line), upper and lower quartiles (box limits), and 1.5× interquartile range (whiskers).
(D) Sorafenib sensitivity results of the PDC models under different concentrations in three proteomic subtypes (two-tailed Wilcoxon test). Box plots show median (central line), upper and lower quartiles (box limits), and 1.5× interquartile range (whiskers).
(E) Proportion of patients who achieved IC50 response in three proteomic subtypes. IC50, half-maximal inhibitory concentration.
(F) Correlations between elastic-net-predicted and observed AUC in the validation set. Correlations were calculated by Pearson’s correlation method.
(G) Regulation networks of selected protein features in sensitivity prediction model for sorafenib (Pearson’s correlation).
(H) Correlations between enriched pathways and observed AUC in PDC models.
(I) Enrichment of pathways associated with sorafenib sensitivity in three proteomic subtypes (two-tailed Wilcoxon test).
See also Figure S7.
We then measured the drug response of each PDC to sorafenib by calculating the AUC of the dose-response curve after 4 days of treatment and compared the differences in response to sorafenib among the three proteomic subtypes (Figure S7C). Overall, sorafenib effectively inhibited the proliferation of PDCs with a wide range of drug sensitivities for each subtype, indicating high heterogeneity of HCC PDCs (Figure S7D). Notably, SII and SIII tumors showed high sensitivity to sorafenib while SI was mostly resistant (p < 0.05) (Figures 7C andS7D). In particular, the inhibition of PDC growth in SII and SIII was significantly stronger than in SI at sorafenib concentration of 32 μM or even higher (Figure 7D). Moreover, the percentage of tumors achieving half-maximal inhibitory concentration (IC50) in SIII was significantly higher than that in SI (Figure 7E), suggesting that SIII tumors may have more benefit from sorafenib. These findings demonstrated the strong potential of proteomic subtypes in predicting targeted drug sensitivity of HCC patients.
Machine-learning-based efficacy prediction model for sorafenib
We developed an elastic net model to predict the responses to sorafenib using 16 tumors as the training set, while the remaining 10 tumors were regarded as the validation set. Remarkably, we observed high correlations (Pearson’s r2 = 0.753, p = 0.0011) between the predicted and observed AUC for sorafenib (Figure 7F). In total, 19 protein features were selected by elastic net in the prediction of drug response to sorafenib, with most showing negative correlations with sorafenib sensitivity (Figure 7G). Notably, most of the key proteins identified in the prediction model have not been reported to contribute to sorafenib response, such as EPHX2, ENPP1, and AKR7A3, which need further attention in future studies.
As shown in Figure 7H, metabolism-related pathways were highly enriched in the tumors resistant to sorafenib, while cell cycle and mRNA surveillance pathways were highly enriched in tumors sensitive to sorafenib (Figures 7H, 7I, and S7E). These findings were highly consistent with the molecular characteristics of SI and SIII, further demonstrating the higher sensitivity of SIII and SII tumors to sorafenib than SI. Interestingly, herpes simplex virus 1 infection, Salmonella infection, and viral life cycle HIV-1 showed high correlations with sorafenib sensitivity, suggesting that viral or bacterial infection might enhance the sorafenib sensitivity of HCC patients due to the close relationship between viral or bacterial infection and the immune microenvironment, which may also affect the efficacy of sorafenib (Figure S7E).47 Collectively, these results suggest that proteomic subtypes can serve as proxies for predicting the treatment efficacy of targeted therapies for HCC patients.
Discussion
Currently, major advances and breakthroughs in precision medicine rely entirely on genomic analysis.48,49,50 A major challenge for proteomics-driven precision medicine is how to use the comprehensive proteome to identify subtypes of patients with shared biological factors that can be targeted for treatment. In this study, we identified three HCC proteomic subtypes characterized by metabolism, proliferation & immune, and proliferation & immune & metastasis, respectively. These findings were generally consistent with the proteomic subtypes of HCC reported by Jiang et al. and Gao et al. Furthermore, the cross-validation of three independent large-scale proteomics studies resulted in a stable and universal proteomic subtype for HCC, which was not limited to multiple centers, patient characteristics (BCLC stages and HBV infection), protein quantitative strategies (labeling and label-free), and data-acquisition mode of MS. These results demonstrated the reliability of the proteomic subtype strategies for HCC reported by Jiang et al. and Gao et al. as well as the feasibility of using the proteomics for HCC classification. Moreover, we developed a multi-centric and robust simplified panel based on machine learning for distinguishing HCC proteomic subtypes, suggesting that proteomics holds great promise in identifying HCC-subtype patients associated with different prognosis who might benefit from further clinical treatment.
The integrated multi-omics analysis revealed alterations in mutation profiles, immune landscapes, and kinase-substrate regulation networks among three proteomic subtypes. SIII had stronger immune infiltration, suggesting that SIII HCC patients might derive potential benefits from immunotherapy. Therefore, a clinical trial of immunotherapy for SIII HCC patients could be attempted to test this speculation. Furthermore, the drug-targetable proteins identified through proteomic and phosphoproteomic data may provide evidence for expanding treatment selection beyond the current FDA-approved HCC therapies, which is promising for overcoming the limited availability of therapeutic drugs and their low response rates in HCC.
Sorafenib remains the first-line drug for advanced HCC.2,51 Based on the multi-omics dataset and PDC drug testing, we found that response rates to sorafenib were significantly higher in SII and SIII than in SI. We therefore hypothesize that SII and SIII patients may derive more benefit from sorafenib than SI patients. However, this hypothesis needs to be tested separately in a clinical trial. We further established a response prediction model for sorafenib based on machine learning. For patients who have lost the opportunity for surgery, we proposed a strategy that exploits the kinase-substrate regulation networks to effectively predict the benefit form sorafenib, regardless of whether tumors carry targets of sorafenib. Promisingly, the accumulation of multi-omics data combined with effective drug testing has the potential to establish a precise strategy for identifying the most appropriate drugs for HCC patients.
In summary, we validated the proteomic subtypes of HCC in multi-centric datasets and developed a simplified panel for distinguishing these subtypes. Integrated multi-omics analysis revealed the functional impact of characterized molecular alterations in three proteomic subtypes. Effective prediction of sorafenib response demonstrated that targeting multi-omics could improve the implementation of advanced personalized therapeutic strategies for HCC. These findings provided important insights into the development of precision medicine for HCC patients and offered promising strategies to improve the selection of appropriate therapies for individual patients.
Limitations of the study
A potential limitation of our study is primarily the sample size of HCC patients in the clinical cohort. Inclusion of a larger sample size could allow for more sufficient statistical analyses among the three proteomic subtypes, including prognostic and ssGSEA analyses. Especially for the construction of a sorafenib efficacy prediction model, expanding the sample size of the training set and validation set could greatly improve the reliability. Future studies in large prospective cohorts are warranted to validate the proteomic subtypes of HCC and evaluate the efficacy of the prediction model for consideration in clinical trials. Taken together, while our study has some limitations, it establishes a strong foundation for the investigation of precision medicine for HCC patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AFP Polyclonal antibody | Proteintech | Cat# 14550-1-AP; RRID: AB_2223933 |
| Albumin Polyclonal antibody | Proteintech | Cat# 16475-1-AP; RRID: AB_2242567 |
| glypican-3 (1G12) | SANTA | Cat# sc-65443; RRID: AB_831376 |
| Goat anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Invitrogen | Cat# A32723; RRID: AB_2633275 |
| Alexa Fluor® 546 goat anti-rabbit IgG (H + L) | Life Technologies | Cat# A11010; RRID: AB_2534077 |
| Alexa Fluor® 546 goat anti-mouse IgG (H + L) | Life Technologies | Cat# A11003; RRID: AB_2534071 |
| Biological samples | ||
| Tumor with paired surrounding non-tumor tissues from primary HCC patients | Mengchao Hepatobiliary Hospital of Fujian Medical University (Fujian, China) | This paper |
| Patient-derived cell (PDC) | Mengchao Hepatobiliary Hospital of Fujian Medical University (Fujian, China) | This paper |
| Chemicals, peptides, and recombinant proteins | ||
| Urea | Sigma-Aldrich | Cat# U1250 |
| β-Glycerophosphate disodium salt hydrate | Sigma-Aldrich | Cat# G9422 |
| Sodium orthovanadate | Sigma-Aldrich | Cat# S6508 |
| Sodium pyrophosphate tetrabasic decahydrate | Sigma-Aldrich | Cat# S6422 |
| Sodium dihydrogen phosphate dihydrate | Sigma-Aldrich | Cat# 04269 |
| Protease inhibitor cocktail tablets | Roche | Cat# 11697498001 |
| PhosSTOP | Roche | Cat# 04906837001 |
| Iodoacetamide | Sigma-Aldrich | Cat# I6125 |
| DL-Dithiothreitol | Sigma-Aldrich | Cat# D0632 |
| Acetonitrile, LC-MS Grade | Thermo Fisher Scientific | Cat# 51101 |
| Water, LC-MS Grade | Thermo Fisher Scientific | Cat# 51140 |
| Triethylammonium bicarbonate buffer | Sigma-Aldrich | Cat# 90360 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5111 |
| 0.1% Formic acid in water | Thermo Fisher Scientific | Cat# 85170 |
| 0.1% Formic acid in ACN | Thermo Fisher Scientific | Cat# 85174 |
| Pierce HeLa Protein Digest Standard | Thermo Fisher Scientific | Cat# 88329 |
| iRT | Biognosys | Cat# KI-3002-1 |
| Collagenase D | Roche | Cat# 11088866001 |
| Dnase I | Roche | Cat# 10104159001 |
| EBSS | Gibco | Cat# 14155063 |
| DMEM/F12 | Gibco | Cat# C11330500BT |
| GlutaMax | Gibco | Cat# 35050061 |
| HEPES | Gibco | Cat# 15630106 |
| Pen/Strep | Gibco | Cat# 15140122 |
| B27 | Gibco | Cat# 12587010 |
| N2 | Gibco | Cat# 17502048 |
| Forskolin | Sigma-Aldrich | Cat# F3917 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636 |
| N-acetyl-L-cysteine | Sigma-Aldrich | Cat# A9165 |
| recombinant human EGF | PeproTech | Cat# AF-100-15 |
| recombinant human FGF-10 | PeproTech | Cat# 100-26 |
| recombinant human HGF | PeproTech | Cat# 100-39 |
| [Leu15]-Gastrin I human | Sigma-Aldrich | Cat# G9145 |
| A83-01 | Tocris Bioscience | Cat# 2939 |
| Y-27632 | MCE | Cat# HY-10071 |
| Dexamethasone | Sigma-Aldrich | Cat# D1756 |
| Hoechst 33342 | Beyotime | Cat# C1028 |
| Sorafenib | Selleckchem | Cat# S7397 |
| Critical commercial assays | ||
| EasyⅡProtein Quantitative Kit (BCA) | TransGen Biotech | Cat# DQ111 |
| High-Select™ Fe-NTA Phosphopeptide Enrichment Kit | Thermo Fisher Scientific | Cat# A32992 |
| Pierce High pH Reversed-Phase Peptide Fractionation Kit | Thermo Fisher Scientific | Cat# 84868 |
| QIAamp DNA Mini Kit | Qiagen | Cat# 51306 |
| Agilent SureSelect Human All Exon V6 kits | Agilent Technologies | Cat# 5190-8864 |
| TransDetect Cell Counting Kit (CCK) | TransGen Biotech | Cat# FC101 |
| Deposited data | ||
| DrugBank database | NA | RRID:SCR_002700; https://go.drugbank.com/drugs |
| MSigDB database | Broad Institute | RRID: SCR_002700; https://data.broadinstitute.org/gsea-msigdb/msigdb/release/2022.1.Hs/c2.cp.kegg.v2022.1.Hs.symbols.gmt |
| NetworKIN 3.0 | Horn et al.46 | RRID: SCR_007818; http://networkin.info |
| PhosphoSitePlus | Hornbeck et al.52 | RRID:SCR_001837; https://www.phosphosite.org |
| Proteomic data of this cohort (N = 152) | This paper | iPROX: IPX0005108000; ProteinXchange: PXD046519 |
| WES data of this cohort (N = 58) | Cai et al.53 | NGDC: HRA000045 |
| RNA_Seq data of this cohort (N = 57) | Li et al.44 | NGDC: HRA000464 |
| MSigDB c2 gene sets (version 6.2) | NA | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
| Proteogenomic data of the Jiang et al. ’s cohort (N = 101) | Jiang et al.24 | PMID: 30814741 |
| Proteogenomic data of the Gao et al. ’s cohort (N = 159) | Gao et al.25 | PMID: 31585088 |
| Proteomic data of the Ng et al. ’s cohort (N = 51) | Ng et al.41 | PMID: 35508466 |
| Software and algorithms | ||
| HCC Proteomic Molecular subtypes and the SP9 | This paper | https://doi.org/10.5281/zenodo.10053446 |
| Spectronaut (version 13.2) | Biognosys Inc. | https://www.biognosys.com |
| R (version3.6.3) | R Core Team | https://www.r-project.orgs |
| NIS-Elements (version 4.40) | Nikon | RRID: SCR_014329 |
| TitanCNA (version 1.23.1) | Ha et al.54 | https://github.com/gavinha/TitanCNA |
| Gephi (version 0.9.2) | Bastian et al.55 | RRID:SCR_004293; https://gephi.org/ |
| Cytoscape (version 3.9.0) | Shannon et al.56 | RRID:SCR_003032; https://cytoscape.org/ |
| ConsensusClusterPlus R package (version 1.50.0) | Wilkerson et al.57 | RRID:SCR_016954; http://bioconductor.org/packages/release/bioc/html/ConsensusClusterPlus.html |
| GISTIC (version 2.0.23) | Mermel et al.58 | RRID:SCR_000151; https://portals.broadinstitute.org/cgi-bin/cancer/publications/view/216 |
| NMF (version 2.23.0) | Gaujoux et al.59 | https://cran.r-project.org/web/packages/NMF/index.html |
| Mutect (version 2) | Broad Institute | RRID:SCR_000151; https://portals.broadinstitute.org/cgi-bin/cancer/publications/view/216 |
| Genome Analysis Toolkit (GATK) (version 4.1.0.0) | Broad Institute | RRID:SCR_001876; https://software.broadinstitute.org/gatk/ |
| caret R package (version 1.50.0) | Github | http://topepo.github.io/caret/index.html |
| ANNOVAR (version 2017 Jul 17) | Wang et al.60 | http://annovar.openbioinformatics.org/en/latest/ |
| ggplot2 R package (version 3.3.6) | Github | https://github.com/tidyverse/ggplot2 |
| Metascape software (version 3.5) | Zhou et al.61 | https://metascape.org/gp/index.html#/main/step1 |
| Other | ||
| Centrifugal filter | Millipore | Cat# UFC5010BK |
| ACQUITY UPLC BEH C18 Column | Waters | Cat# 186002352 |
| nanoEase M/Z HSS T3 Column | Waters | Cat# 186008818 |
| Acclaim PepMap™ 100 C18 column | Thermo Fisher Scientific | Cat# 164946 |
| Cell Strainers | Biologix | Cat# 15-1070 |
| Glass bottom cell culture dish | Biosharp | Cat# BS-15-GJM |
| 96 Well cell culture plate | Corning | Cat# CLS3599 |
| SpectraMax M5e | Molecular Device | RRID: SCR_020300 |
| Waters Acquity H-class UPLC system | Waters | Cat# ACQUITY UPLC H-Class; RRID: SCR_022217 |
| EASY-nLC 1000 | Thermo Fisher Scientific | Cat# LC120; RRID: SCR_014993 |
| Q Exactive Plus mass spectrometry | Thermo Fisher Scientific | Cat# IQLAAEGAAPFALGMBDK; RRID: SCR_020556 |
| Zeiss LSM 780 Confocal Laser Scanning Microscope | Carl Zeiss | RRID: SCR_020922 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xiaolong Liu (xiaoloong.liu@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Raw data of proteome and phosphoproteome derived from HCC tissues have been deposited at iProX: IPX0005108000 and ProteomeXchange: PXD046519.62,63 Raw data of genome and transcriptome derived from HCC tissues have been deposited at Genome Sequence Achieve of National Genomics Data Center (NGDC): HRA000045, HRA000464. Accession numbers are listed in the key resources table.
-
•
All analysis code has been deposited at Zenodo: https://doi.org/10.5281/zenodo.10053446 and is publicly available as of the date of publication. DOls are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Patients and tissue samples for multi-omics analysis
A total number of 160 primary HCC patients obtained from Mengchao Hepatobiliary Hospital of Fujian Medical University in China were recruited for multi-omics analysis in this study (Figure S1A). The clinical and pathological characteristics of these HCC patients were summarized in Table S1. The included HCC patients represented all major subtypes at different pathological stages, such as age, gender, tumor number, tumor size, tumor capsule, AFP concentration, HBV-DNA, PVTT, MVI, BCLC stage, TNM stage, cirrhosis, and differentiation. The median follow-up of this cohort for overall survival was 30.5 months, and recurrence-free survival had a median of 7.2 months (Figure S1A and Table S1). This project was approved by the Institution Review Board of Mengchao Hepatobiliary Hospital of Fujian Medical University. Informed consent was obtained from each participant before the operation. The use of clinical specimens was completely in compliance with the “Declaration of Helsinki”.
Patient derived cell models of HCC
To established Patient derived cell models of HCC, a total of 26 fresh HCC tumor tissue samples from patients who underwent surgical resection were collected, and the study protocol of clinical samples was approved by the Institutional Ethics Committee of Mengchao Hepatobiliary Hospital of Fujian Medical University. Tumor tissue acquisition was approved by the ethics committees of Mengchao Hepatobiliary Hospital of Fujian Medical University (Fuzhou, China) and agreed by each patient via written informed consent, and the whole procedure was carried out according to national and institutional regulations on experimental use of human tissues. The surgical tissue samples were enzymatically dissociated using digestive lysate (2.5 mg/mL Collagenase D, 1 mg/mL DNase I, 1×EBSS), and then the tumor cells from HCC tissues were collected by centrifugation at 500g for 3 min, followed by washing with washing buffer (1×DMEM/F12, 2mM GlutaMax, 10mM HEPES, 1×Pen/Strep).64 PDCs were cultured in defined medium (1×DMEM/F12, 2mM GlutaMax, 10mM HEPES, 1×Pen/Strep, 1×B27, 1×N2, 10μM forskolin, 10mM Nicotinamide, 1.25 mM N-Ac, 50 ng/mL hEGF, 100 ng/mL FGF-10, 25 ng/mL hHGF, 10 nM Gastrin, 5 μM A83-01, 10 μM Gastrin, 10 μM Y-27632, 3 nM Dexamethasone), and seeded in 96-well plates at 50000 cells/well in quintuplicate for sorafenib (purchased from Selleckchem) treatment at 37°C in a humidified atmosphere with 5% CO2. In addition, the PDCs were inoculated in 15 mm confocal dishes (Biosharp Life sciences) with 1 × 105 cells in triplicate for immunofluorescence. The cells were cultured as described above.
Method details
Protein extraction and digestion
Total protein was extracted from tissue samples by lysis buffer containing 9 M urea, 75 mM NaCl, 10 mM Tris-Hcl (pH8.0), 10 mM IAA, 1 mM NaF, 1 mM β-glycerophosphate, 1mM Sodium orthovanatate, 10 mM Sodium pyrophosphate, 100 mM Sodium dihydrogen phosphate, 1 mM PMSF and protease inhibitors as described previously. Briefly, tissue was disrupted using a tissue homogenizer on ice for 3 min followed by centrifugation at 17000× g for 10 min at 4°C and then the supernatant was collected. The proteins were sequentially reduced with 10 mM dithiothreitol at 55°C for 30 min and alkylated with 50 mM iodoacetamide in the dark for 30 min at room temperature. Then, the protein concentration was determined using the BCA assay. Subsequently, 100 μg of total protein was loaded in 10 kDa centrifugal filter tubes (Millipore), washed twice with 400 mL 100 mM triethyl ammonium bicarbonate (TEAB), and enzymatic digestion was carried out with trypsin at a concentration of 1:50 (w/w) using filter-aided sample preparation for 18 h at 37°C. Peptides were eluted by centrifugation at 14,000g for 15 min and evaporated to dryness for LC-MS/MS analysis.
To prepare samples for spectral library construction, each 20 samples were pooled into a 1 mg protein mixture (equally to a final of 16 pools), and prepared in the same method as mentioned above (Figure S1B).
Phosphopeptide enrichment
The phosphopeptide enrichment was performed using High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Scientific) according to the kit manual. In brief, the peptide mixture from 1 mg proteins were dissolved with 200 μL binding buffer, then were added to a Fe-NTA spin column and incubated for 20 min at room temperature with end-over-end rotation. After 1 min of centrifugation at 1,000 g for 1 min, the column was washed for 4 times with wash buffer to remove nonspecific peptides. Finally, phosphopeptides were eluted with elution buffer and were immediately dried by speed-vac for MS analysis.
To prepare samples for spectral library construction, each 40 samples were pooled into a 10 mg protein mixture (equally to a final of 8 pools), and prepared in the same method as mentioned above (Figure S1B).
High pH reversed-phase separation
In order to increase the depth of protein or phosphopeptide identification for generating the spectral libraries, the pooled samples were fractionated by high-pH reverse phase liquid chromatography. Peptide mixtures were resuspended in FA/water (1/1000) for peptide fractionation. Offline basic RP UPLC fractionation was performed on a XBridge BEH C18 column (1.7 μm, 100 μm × 10 cm) using a Waters Acquity H-class UPLC system. Chromatographic separation was performed with 30 min gradient as following: 0–5 min, increase to 7% B; 5–20 min, 7–35% B; 20–25 min, 35–80% B (mobile phase A: 0.1% FA (v/v); mobile phase B: 90% ACN, 0.08% FA (v/v), flow rate 300 nL/min). A fraction was collected every 30 s; to reduce the MS running time, the 60 fractions were pooled (F01 + F13 + F25, F02 + F14 + F26 and so on) as described45 to a final of 12 samples (Figure S1B). For each sample, 2 μg were taken out and dried by vacuum centrifugation for proteome analysis.
LC-MS/MS analysis
Peptide fractions were analyzed on an EASY-nLC 1000 LC system (Thermo Fisher Scientific,Waltham, MA) coupled with an Orbitrap Exactive Plus mass spectrometry (Thermo Fisher Scientific, Waltham, MA). Briefly, the peptide was resuspended in mobile phase C (0.1% formic acid in water) and equal amounts of 11 indexed retention time (iRT) peptide standards (Biognosys) were spiked into each shotgun run in data-dependent acquisition (DDA) mode. And the peptides were separated onto the analytical column (1.8 μm, 75 μm × 25 cm) with a 120 min gradient at a constant flow rate of 300 nL/min (0–3 min, 4 to 7% of D; 3–79 min, 7 to 20% D; 79–103 min, 20 to 32% of D, 103–105 min, 32 to 90% of D and held at 90% D for 15min mobile phase D: 0.1% Formic acid in ACN). MS was operated under a data-dependent acquisition mode. The spectra of full MS scan (m/z 300–1800) were acquired by Orbitrap mass analyzer at 70,000 resolution for a maximum injection time of 60 ms with an AGC target value of 3e6. Up to 20 precursors were selected for MS2 analysis and fragmented by HCD using a normalized collision energy of 27% and analyzed using the Orbitrap at 17,500 resolution for a maximum injection time of 45 ms with AGC target value of 1e5. The dynamic exclusion was 20 s and the precursor ions with unassigned charge state as well as charge state of 1+, or superior to 8+ were excluded from fragmentation selection.
As above, equal amount of iRT peptide standards were mixed into individual sample peptide for each shotgun run at DIA mode, and nano-LC MS/MS basic parameters were as the same as described above. In DIA mode, precursor ions were acquired using an MS1 master scan (m/z range: 400–1200, max. injection time: 20 ms, AGC target: 3e6, resolution: 70K), following 32 DIA scans for MS2 with two kinds of wide isolation window as follows: m/z range 400–1000 using an isolation window width of m/z 20, and m/z range 1000–1200 using an isolation window width of m/z 100. Each isolation window overlapped by 1 m/z.
Generation of proteomic and phosphoproteomic spectral library
To generate an extensive spectral library, both DDA and DIA files were processed using Pulsar search engine inside Spectronaut with default settings. Reference FASTA files for human was downloaded from UniProt (release 2019-04, 23046 entries), combining with the fusion sequence of iRT. Enzyme specificity was set as trypsin/P. The maximum missing cleavage site was set as 2. The mass tolerances of precursor ion and fragment ion for peptides were set at 10 parts per million (ppm) and 0.02 Da, respectively. The minimal peptide length was set at 7. Carbamidomethyl (C) was set as fixed modifications. Oxidation (M) and Acetyl (Protein N-term) were set as variable modifications. For phosphoproteomic analysis, phospho (STY) was also set as a variable modification. In additional, the false discovery rate (FDR) was set to 1% at peptide spectrum matches, 1% at peptide precursor level and 1% at protein level.
DIA mode analysis to get proteomic and phosphoproteomic data
In total, 304 proteome DIA files and 264 phosphoproteome DIA files passed the quality-control were searched using Spectronaut based on the hybrid spectral library of HCC proteome or phosphoproteome, respectively. Reference FASTA files from UniProt database were combining with the fusion sequence of iRT. The samples were grouped by T (HCC tumor tissue group) and N (paratumor noncancerous tissue group). Calibration was set to non-linear iRT calibration with enabled precision iRT. Identification was performed using 5% q-value cutoff on precursor and protein level, while the maximum number of decoys was set to a fraction of 0.1 of library size. Quantity was determined on MS/MS level using area of XIC peaks with enabled cross run normalization. For phosphoproteomic analysis, minor quantified (Peptide) grouping was set by modified sequence and PTM localization was activated and the probability cutoff was set to 0, in order to summarize phosphopeptide or phosphosite later.
Quality control of the MS platform
To evaluate the performance of the MS system, the HeLa protein digest standard (Pierce) was measured every 10 samples from beginning to the end of the project as the quality-control standard. DDA analysis were performed all through the project, and DIA analysis were performed during the DIA process. The standard was analyzed using the same method and conditions as using in the HCC related tissue samples. A Pearson’s correlation coefficient was calculated for all quality-control runs in the statistical analysis environment R3.6.3.
Quality assessment of proteomic and phosphoproteomic data
The two-component mixture model implemented in the R MCLUST package v.5.2 was applied to filter the samples with insufficient peptide counts and protein identifications. In total, 152 paired samples among the 160 total involved paired samples passed the quality-control and were used for further proteomic analysis. Furthermore, 132 paired samples among the 152 paired samples passed the quality-control and were used for further phosphoproteomic analysis. All samples that passed quality control showed good consistency in the proteome quantification and phosphoproteome quantification (Figures S1F and S5F). Furthermore, PCA (Principal Component Analysis) and C.V (Coefficient of Variance) distribution of protein quantification showed that there was no batch effect in proteomic and Phosphoproteomic data (Figures S1G, S1I, S5E, and S5G).
Pre-processing of proteomic and phosphoproteomic data
In order to correct the initial loading volume for each sample, proteomic (304 samples and 6512 proteins) and phosphoproteomic (264 samples and 54789 phosphopeptides) data from Spectronaut software were first log2-transformed, and then normalized using the median centering method. Glmnet Ridge Regression (GRR) of NAguideR (https://www.omicsolution.org/)65 was used to impute missing values in tumor or paired non-tumor samples. Before missing value imputation, proteins and phosphopeptides with more than 50% missing data were taken out to make sure that each sample had enough data for imputation.
Consensus clustering for HCC in proteomic data
For proteomic subtype identification, 1128 proteins from the training set with the top 1500 of MAD (median absolute deviation) and with expression in more than 80% of the tumor samples were selected for consensus clustering using the ConseususClusterPlus R package.57 The parameters were as follows: maxK = 8, reps = 1,000 bootstraps, pItem = 0.8, pFeature = 0.8, clusterAlg = “kmdist”, distance = “euclidean”. The number of clustering was determined by the average pairwise consensus matrix within consensus clusters, the delta plot of the relative change in the area under the cumulative distribution function (CDF) curve, and the average silhouette distance for consensus clusters. To verify the reliability of the proteomic subtypes, consensus clustering was also performed in the validation set using the same parameters.
Identification of signatures for HCC subtypes
Signatures for HCC subtypes were a group of proteins that meet the following criteria: (1) the Benjamini-Hochberg-adjusted p values in each subtype should be less than 0.01 compared to the other subtypes; (2) fold change of the protein abundance between proteomic subtypes was greater than 2 for upregulation or less than 0.5 for downregulation. We performed these analyses using the “Wilcoxon test” function in the R3.6.3 environment. In total, we identified the signatures containing 761 proteins from the 3 HCC proteomic subtypes, and these signatures were then subjected to further analyses. To verify the reliability of the signatures, consensus clustering was also performed in all 152 tumor patients using both the 1128 proteins and the signatures containing 761 proteins with the same parameters. The subtypes identified using 1128 proteins were taken as the true subtypes, and the results were compared using a confusion matrix.
Correlations between proteomic subtypes and clinical features
The chi-square and Fisher-exact tests were performed to identify the correlations of proteomic subtypes with clinicopathologic feature. Survival curves were generated using the KM method, and the log rank test was applied to calculate differences between the curves. Hazard ratios (HR), 95% confidence intervals (CI) and figures were estimated by the survival and survminer or plotted by ggplot2 R packages.
The univariable and multivariable Cox analyses were further used to evaluate the prognostic power of HCC proteomic subtypes. All statistical analyses were performed in R (version 3.6.3), and a significance level of 0.05 was used.
Pathway alterations of 3 HCC proteomic subtypes
To explore the pathway alterations of 3 HCC proteomic subtypes, we performed single sample gene set enrichment analysis (ssGSEA) with 4026 proteins that were identified in more over 50% of patients using the R package GSVA,66 and the enrichment scores were scaled by R. In this study, 186 canonical pathway gene sets derived from the KEGG database were obtained from the MSigDB database (https://data.broadinstitute.org/gsea-msigdb/msigdb/release/2022.1.Hs/c2.cp.kegg.v2022.1.Hs.symbols.gmt). The clusterProfiler R package67 was used to perform pathway enrichment analysis of differentially abundant proteins or phosphoproteins. The statistical significance of pathways between 3 subtypes were analyzed by the “Wilcoxon test”, and p value less than 0.01 was considered as significant.
Cross-validation of HCC proteomic subtypes from multi-centers
To verify the robustness and universality of HCC proteomic subtypes, the protein abundance matrix, signatures, proteomic subtypes, and prognostic information were collected from Jiang et al.’s and Gao et al.’s cohorts. 1269 signatures obtained from Jiang et al.’s cohort containing 101 patients and 9252 proteins, as well as 1274 signatures were obtained from Gao et al.’s cohort containing 159 patients and 6478 proteins in total. The cross-validation was mainly focused on the two-by-two comparison of the 3 subtypes in 3 cohorts. For example, consensus clustering was performed using Gao et al.’s signatures and Jiang et al.’s signatures in our cohort, and they were further compared with our original subtypes based our signatures. When reclassifying samples in different cohort based on other signatures, the classification method in the original article related to the cohort was used. The validation results were presented in a confusion matrix, and the proportion of consistently assigned patients to the total patients was used to assess the consistency of the two subtypes. For patients with different assignments, survival analysis with two assignments by the KM method were required.
Simplified panel for discriminating proteomic subtypes
To construct the simplified panel for discriminating proteomic subtypes, a total of 412 patients (152 patients from our cohort, 101 patients from Jiang et al.’s cohort, and 159 patients from Gao et al.’s cohort) were used. Of these, our cohort and Jiang et al.’s cohort (N = 253) were used as the training set for model training and parameter tuning, and Gao et al.’s cohort was used as an external independent validation set to evaluate the model performance (N = 159). The scale function in R was used to normalize 3 datasets to eliminate the batch effect. Then, the panel for discriminating proteomic subtypes was constructed and validated by the following steps. Firstly, a random sampling method was used to screen for differentially expressed proteins in 3 subtypes, a total of 50 replicates were performed, 40 of which reached the threshold (p value < 0.01, fold change >1.5) that were initially selected as differentially expressed proteins. After removing the proteins with correlations higher than 0.9 with other proteins, a total of 1133 differentially expressed proteins were selected in the training set. Secondly, for reducing the misleading impact of random fluctuations and correlations, Boruta algorithm from the Boruta R package was used to select the signatures. Then, 59 SI specifically expressed proteins and 88 SIII specifically expressed proteins were selected in total. Thirdly, the KNN (K-Nearest Neighbors) algorithm of the caret R package was used to construct the SI discriminating panel and SIII discriminating panel, respectively. And a 5-fold cross-validation was performed to further reduce the number of signatures of the discriminating panel. The specific parameters are as follows: method = “cv”, 5, summaryFunction = twoClassSummary, and classProbs = TRUE for the trainControl function; method = “knn”, tuneLength = 30, and metric = “ROC” for the train function. Finally, 9-protein simplified panel containing 4 proteins from the SI subtype and 6 proteins from the SIII subtype (1 protein was shared) were integrated to identify HCC proteomic subtypes. In addition, the simplified panel for discriminating proteomic subtypes was validated in an external independent validation set, and the accuracy, sensitivity and specificity were calculated to evaluate the panel performance.
DNA extraction and whole-exome sequencing
Total DNA from HCC tissues and para-tumor tissues were extracted and subjected to DNA library preparation using QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. Whole-exome capture was carried out using Agilent SureSelect Human All Exon V6 kits. And sequencing was performed using the Illumina HiSeq X ten system (Annoroad Gene Tech. Co., Ltd). As described previously, qualified WES reads were aligned to hg19 human genome assembly (GRCh37) using BWA, and duplicates of all mapped reads were then marked and discarded using Picard.50
Transcriptome sequencing and mapping
RNA was extracted from HCC tumor and para-tumor tissues, and the whole transcriptome sequencing (paired-end, 150 bp) was performed on the Illumina HiSeq X10 platform by Anoda Gene Technology (Beijing) Co. All high quality reads were aligned to GRCh37 with GENCODE gene annotations using STAR as described previously.44 The abundance of all genes was quantified using transcripts per kilobase per million mapped reads (TPM).
Somatic mutation detection, CNV, TMB calculation and neoantigen prediction
Somatic mutations of HCC tumor were identified with Mutect2 in GATK (version 4.1.0.0) using paired peritumoral tissue samples as the control. Somatic mutations that meet the following criteria are retained for downstream analysis: ≥20×depth in both tumor and peritumoral samples; variant allele frequencies (VAF) ≥5% in tumor samples; VAF≤1% in peritumoral samples. CNVs were identified using TitanCNA R/Bioconductor package (version 1.23.1),54 and regions of the genome significantly amplified or deleted were identified by Gistic2.0 in “bzhanglab/gistic2”. BAM format files and the data of TMB and neoantigens were analyzed according to our previously published protocols.44,53
Random forest algorithm for ranking the importance of mutations
Random forest algorithm was applied to the WES data to identify the most important mutations associated with the proteomic subtypes. In brief, the gene mutation dataset and subtype tags were applied as input. The caret R package was used to find the best hyperparameter for the random forest model.
Immune microenvironment and immune infiltration analysis
28 immune cell types and their feature gene panels were downloaded from Charoentong et al.’s study.68 Immune cell infiltration of each cell type was estimated by ssGSEA of GSVA R package. Then, the enrichment score was scaled to represent the relative level of infiltration. The estimate R package was also used to assess the overall immune infiltration of every tumor sample. The anti-tumor immunity and pro-tumor immune suppression were calculated from the enrichment score of infiltration cell types executing anti-tumor immunity (activated CD4 T cell, activated CD8 T cell, central memory CD4 T cell, central memory CD8 T cell, effector memory CD4 T cell, effector memory CD8 T cell, type 1 T helper cell, type 17 T helper cell, activated dendritic cell, CD56 bright natural killer cell, natural killer cell and natural killer T cell) and cell types executing pro-tumor immune suppressive functions (regulatory T cell, type 2 T helper cell, CD56 dim natural killer cell, immature dendritic cell, macrophage, MDSC, neutrophil, plasmacytoid dendritic cell).69
Correlation analysis of multi-omics data
Correlations of multi-omics data were calculated for each gene/protein across the tumor samples using Pearson’s correlation coefficient, including CNV alterations and RNA abundance, CNV alterations and protein abundance, CNV alterations and phosphopeptide abundance, RNA abundance and protein abundance, RNA abundance and phosphopeptide abundance, as well as protein abundance and phosphopeptide abundance. Protein attenuation was calculated by the difference between the Pearson coefficient of the transcript correlation (correlation between CNV and transcript measurements) and the Pearson coefficient of the protein correlation (correlation between copy-number and protein measurements). Then proteins were classified according to their attenuation effect using a Gaussian mixture model with 2 mixture Components.70 Differentially expressed proteins or phosphopeptides between tumor and paired non-tumor samples were filtered using p values of less than 0.01 and fold change greater than 2.
Phosphosite-to-protein co-variation analysis
As for all proteomic and phosphoproteomic data from 132 HCC tumor samples, Pearson correlations for each protein and corresponding phosphorylation sites were also calculated in the 3 proteomic subtypes, where the upper quartile of all differences were considered as significantly co-regulated among subtypes. In total, we obtained 609 proteins corresponding to 1,185 phosphorylation sites for further analysis. Functional enrichment and interactome analysis of these proteins were performed using Metascape.61 The top 5 MCODE complexes extracted from the interactome networks were selected and visualized by Gephi software. These significantly co-regulated proteins were classified into 3 classes by the k-means algorithm. Also, for each cluster, known or predicted upstream kinases of the phosphosites in the significantly co-regulated phosphosite-to-protein correlations were given based on PhosphoSitePlus (PSP)52 or NetworKIN 3.0 (NetworKIN Score >1).46
Kinase activity prediction
To estimate the kinase activity for each patient, enrichment analysis was performed by the ssGSEA method using the GSVA R package.71 Kinase activity for each kinase was estimate using a kinase enrichment score, which was calculated based on the overall phosphorylation status of its all substrates.72,73 To deduct protein levels and abundance in the para-tumor, we used the following formula to correct each phosphorylation site abundance:
“” represented a single phosphorylation site; “” was the abundance of this phosphorylation site; “” and “” referred to the log2-transformed abundance of this phosphorylation site in tumor and non-tumor tissues, respectively; while “” and “” represented the log2-transformed abundance of the protein that corresponded to this phosphorylation site in tumor and non-tumor tissues.
The known or predicted kinase-substrate relations, were also gained from PSP or NetworKIN 3.0. The gene symbol and Uniprot ID were corrected by the Uniprot Database (https://www.uniprot.org/), and any ambiguous correspondence was excluded. In total, 239759 pairs of kinase-substrate relations were collected, which contained 381 kinases, 6906 protein substrates, and 48730 phosphorylation sites.
Clinical available drugs and target mapping
The FDA-approved drugs or candidate drugs and corresponding targets were downloaded from the DrugBank database (release version 5.1.7). Only targets with known pharmacological action as inhibitors, enzyme, carrier and transporter were selected, and 46 candidate targets were identified in this study. To get subtype-specific drug targets, we comprehensively evaluated their protein abundance, kinase activity, and correlations of kinase-substrate among the 3 proteomic subtypes.
Immunofluorescence analysis
The PDCs on confocal dishes were fixed with 4% paraformaldehyde for 15min, and then permeabilized with 0.5% Triton X-100 in PBS for 15 min at room temperature. After blocking (5% BSA in TBS containing 0.01% Triton X-100) for 1h, the PDCs were subsequently incubated overnight at 4°C with primary antibodies against AFP (1:300, proteintech), albumin (1:300, proteintech) and glypican-3 (1:300, SANTA) diluted in blocking buffer. After washing with TBS for 4 times, PDCs were incubated with secondary Alexa Fluor Plus 488 (1:1000, Invitrogen) and Alexa Fluor 546 (1:1000, Life Technologies). In addition, cell nucleus was stained with Hoechst 33342 (1:500, Beyotime). Following extensive washing, stained organoids were imaged in confocal (Zeiss LSM780, Gemany).
Drug treatment and viability assays
PDCs were treated with sorafenib in an eight-point serial dilution series from 0.25 nM to 32 μM. After 4 days of incubation, cell viability was analyzed using CCK8 assay (TransGen Biotech, Beijing, China). Viable cells were estimated using SpectraMax M5e (Molecular Device, USA). Control cells treated with dimethyl sulfoxide (DMSO) were used to calculate relative cell viability and to normalize the data. Dose-response curve fitting was performed using R (3.6.3) and was evaluated by measuring the area under curve (AUC). In brief, the plate was normalized to the mean value from the seven serial conditions compared with DMSO control. The AUC of curve was determined using R (3.6.3), ignoring regions defined by fewer than two peaks. Non-convergence or ambiguous curves were excluded in every analysis.
Drug sensitivity prediction model
To find sorafenib response associated proteomic features and build prediction models, the 26 PDC models were randomly split into two datasets. The 16 PDC models were used as the training dataset, and the remaining 10 PDC models were regarded as testing dataset. The Elastic net (EN) algorithm was used to build drug sensitivity prediction models.31,74,75 We firstly pre-selected the proteins in the training set based on their Pearson’s correlation coefficients with drug sensitivity, and then elastic net regression models were built using the 19 selected proteins. RMSE was used to select the optimal model using the smallest value. The final values used for the model were alpha = 0.770 and L1 ratio = 0.1. The model was used to predict the drug response of the 10 PDC models in the testing cohort. The Pearson’s correlation coefficients were calculated between the predicted drug sensitivity and examined ones to assess the prediction performance.
Identification of sorafenib sensitivity-related pathways
To explore the pathways that may affect sorafenib sensitivity, the ssGSEA algorithm was used to evaluate the alteration of different pathways between PDC samples. The correlation analysis of sorafenib sensitivity with pathways was performed using the Pearson test, and p values of less than 0.01 were considered as statistically significant.
Quantification and statistical analysis
Quantification and statistical analysis methods for single-omics and multi-omics analyses were mainly presented and cited in the respective STAR Methods details.
Standard statistical tests were used to analyze the clinical data, including but not limited to Wilcoxon test, Chi-square test, Fisher’s exact test, Log rank test. For categorical variables versus continuous variables, Wilcoxon test was used to test if any of the differences between the subtypes were statistically significant; for categorical variables versus categorical variables, Chi-square test and Fisher’s exact test were used; and for continuous variables versus continuous variables, Pearson correlation was used. All statistical tests were two-sided, and p value <0.05 statistical was considered statistically significant. Kaplan-Meier plots (Log rank test) were used to describe OS and RFS. Univariate Cox proportional hazards regression models were used to identify the variables associated with OS and RFS. Significant factors in the univariate analysis were further subjected to multivariate Cox regression analysis using a forward LR approach. All the analyses of clinical data were performed in R (version 3.6.3).
Acknowledgments
This work is supported by the Scientific Foundation of Fujian Provincial Health Commission (grant no. 2021ZQNZD014); the Major Science and Technology Special Project of Fujian Province (grant no. 2022YZ036012); the Natural Science Foundation of Fujian Province (grant nos. 2022J011294, 2023J011478); the Major Projects of Medicine and Health in Zhejiang Province (grant no. WKJ-ZJ-2306); the Scientific Research Project of Health and Family Planning Commission of Fujian Province (grant no. 2022GGA051); the Scientific Foundation of Fuzhou Health Department (grant no. 2022-S-009); and the Startup Fund for Scientific Research, Fujian Medical University (grant no. 2021QH1156).
Author contributions
Conceptualization, X.X. and X.L.; methodology, X.X., E.H., and J.O.; formal analysis, X.X., E.H., J.O., X.Z., and K.L.; investigation, X.X., E.H., and J.O.; resources, Y.Z., Y.W., F.W., L.C., G.C., and Z.L.; data curation, E.H. and J.O.; writing – original draft, X.X.; writing – review & editing, X.L.; visualization, X.X., E.H., and J.O.; supervision, X.L.; project administration, X.L.; funding acquisition, X.X., E.H., L.W., and X.L.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 12, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101315.
Contributor Information
Liming Wu, Email: wlm@zju.edu.cn.
Xiaolong Liu, Email: xiaoloong.liu@gmail.com.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA. Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Cheng A.L., Kang Y.K., Chen Z., Tsao C.J., Qin S., Kim J.S., Luo R., Feng J., Ye S., Yang T.S., et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 3.Kudo M., Finn R.S., Qin S., Han K.H., Ikeda K., Piscaglia F., Baron A., Park J.W., Han G., Jassem J., et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163–1173. doi: 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- 4.Llovet J., Shepard K.V., Finn R.S., Ikeda M., Sung M., Baron A.D., Kudo M., Okusaka T., Kobayashi M., Kumada H., Kaneko S., et al. 747P-A phase Ib trial of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC): Updated results. Annals of Oncology. 2019;30:v286–v287. [Google Scholar]
- 5.Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., Kudo M., Breder V., Merle P., Kaseb A.O., et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020;382:1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 6.Qin S., Bi F., Gu S., Bai Y., Chen Z., Wang Z., Ying J., Lu Y., Meng Z., Pan H., et al. Donafenib Versus Sorafenib in First-Line Treatment of Unresectable or Metastatic Hepatocellular Carcinoma: A Randomized, Open-Label, Parallel-Controlled Phase II-III Trial. J. Clin. Oncol. 2021;39:3002–3011. doi: 10.1200/JCO.21.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren Z., Xu J., Bai Y., Xu A., Cang S., Du C., Li Q., Lu Y., Chen Y., Guo Y., et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol. 2021;22:977–990. doi: 10.1016/S1470-2045(21)00252-7. [DOI] [PubMed] [Google Scholar]
- 8.Chiang D.Y., Villanueva A., Hoshida Y., Peix J., Newell P., Minguez B., LeBlanc A.C., Donovan D.J., Thung S.N., Solé M., et al. Focal gains of and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulouarn C., Factor V.M., Thorgeirsson S.S. Transforming growth factor-β gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology. 2008;47:2059–2067. doi: 10.1002/hep.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoshida Y., Nijman S.M.B., Kobayashi M., Chan J.A., Brunet J.P., Chiang D.Y., Villanueva A., Newell P., Ikeda K., Hashimoto M., et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009;69:7385–7392. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lachenmayer A., Alsinet C., Savic R., Cabellos L., Toffanin S., Hoshida Y., Villanueva A., Minguez B., Newell P., Tsai H.W., et al. Wnt-Pathway Activation in Two Molecular Classes of Hepatocellular Carcinoma and Experimental Modulation by Sorafenib. Clin. Cancer Res. 2012;18:4997–5007. doi: 10.1158/1078-0432.CCR-11-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J.S., Chu I.S., Heo J., Calvisi D.F., Sun Z., Roskams T., Durnez A., Demetris A.J., Thorgeirsson S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi: 10.1002/hep.20375. [DOI] [PubMed] [Google Scholar]
- 13.Song Q., Yang Y., Jiang D., Qin Z., Xu C., Wang H., Huang J., Chen L., Luo R., Zhang X., et al. Proteomic analysis reveals key differences between squamous cell carcinomas and adenocarcinomas across multiple tissues. Nat. Commun. 2022;13:4167. doi: 10.1038/s41467-022-31719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asleh K., Negri G.L., Spencer Miko S.E., Colborne S., Hughes C.S., Wang X.Q., Gao D., Gilks C.B., Chia S.K.L., Nielsen T.O., Morin G.B. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat. Commun. 2022;13:896. doi: 10.1038/s41467-022-28524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu W., Xie L., He Y.H., Wu Z.Y., Liu L.X., Bai X.F., Deng D.X., Xu X.E., Liao L.D., Lin W., et al. Large-scale and high-resolution mass spectrometry-based proteomics profiling defines molecular subtypes of esophageal cancer for therapeutic targeting. Nat. Commun. 2021;12:4961. doi: 10.1038/s41467-021-25202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J.Y., Zhang C., Wang X., Zhai L., Ma Y., Mao Y., Qian K., Sun C., Liu Z., Jiang S., et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell. 2020;182:245–261.e17. doi: 10.1016/j.cell.2020.05.043. [DOI] [PubMed] [Google Scholar]
- 17.Ge S., Xia X., Ding C., Zhen B., Zhou Q., Feng J., Yuan J., Chen R., Li Y., Ge Z., et al. A proteomic landscape of diffuse-type gastric cancer. Nat. Commun. 2018;9:1012. doi: 10.1038/s41467-018-03121-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niu L., Thiele M., Geyer P.E., Rasmussen D.N., Webel H.E., Santos A., Gupta R., Meier F., Strauss M., Kjaergaard M., et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat. Med. 2022;28:1277–1287. doi: 10.1038/s41591-022-01850-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson E.C.B., Carter E.K., Dammer E.B., Duong D.M., Gerasimov E.S., Liu Y., Liu J., Betarbet R., Ping L., Yin L., et al. Large-scale deep multi-layer analysis of Alzheimer's disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 2022;25:213–225. doi: 10.1038/s41593-021-00999-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wigger L., Barovic M., Brunner A.D., Marzetta F., Schöniger E., Mehl F., Kipke N., Friedland D., Burdet F., Kessler C., et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab. 2021;3:1017–1031. doi: 10.1038/s42255-021-00420-9. [DOI] [PubMed] [Google Scholar]
- 21.Virreira Winter S., Karayel O., Strauss M.T., Padmanabhan S., Surface M., Merchant K., Alcalay R.N., Mann M. Urinary proteome profiling for stratifying patients with familial Parkinson's disease. EMBO Mol. Med. 2021;13 doi: 10.15252/emmm.202013257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nie X., Qian L., Sun R., Huang B., Dong X., Xiao Q., Zhang Q., Lu T., Yue L., Chen S., et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell. 2021;184:775–791.e14. doi: 10.1016/j.cell.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L., et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Y., Sun A., Zhao Y., Ying W., Sun H., Yang X., Xing B., Sun W., Ren L., Hu B., et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567:257–261. doi: 10.1038/s41586-019-0987-8. [DOI] [PubMed] [Google Scholar]
- 25.Gao Q., Zhu H., Dong L., Shi W., Chen R., Song Z., Huang C., Li J., Dong X., Zhou Y., et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell. 2019;179:1240. doi: 10.1016/j.cell.2019.10.038. [DOI] [PubMed] [Google Scholar]
- 26.Cao L., Huang C., Cui Zhou D., Hu Y., Lih T.M., Savage S.R., Krug K., Clark D.J., Schnaubelt M., Chen L., et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell. 2021;184:5031–5052.e26. doi: 10.1016/j.cell.2021.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez H., Zenklusen J.C., Staudt L.M., Doroshow J.H., Lowy D.R. The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell. 2021;184:1661–1670. doi: 10.1016/j.cell.2021.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagaev A., Kotlov N., Nomie K., Svekolkin V., Gafurov A., Isaeva O., Osokin N., Kozlov I., Frenkel F., Gancharova O., et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:845–865.e7. doi: 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 29.Petralia F., Tignor N., Reva B., Koptyra M., Chowdhury S., Rykunov D., Krek A., Ma W., Zhu Y., Ji J., et al. Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell. 2020;183:1962–1985.e31. doi: 10.1016/j.cell.2020.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh S., Yeom J., Cho H.J., Kim J.H., Yoon S.J., Kim H., Sa J.K., Ju S., Lee H., Oh M.J., et al. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat. Commun. 2020;11:3288. doi: 10.1038/s41467-020-17139-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li C., Sun Y.D., Yu G.Y., Cui J.R., Lou Z., Zhang H., Huang Y., Bai C.G., Deng L.L., Liu P., et al. Integrated Omics of Metastatic Colorectal Cancer. Cancer Cell. 2020;38:734–747.e9. doi: 10.1016/j.ccell.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Krug K., Jaehnig E.J., Satpathy S., Blumenberg L., Karpova A., Anurag M., Miles G., Mertins P., Geffen Y., Tang L.C., et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell. 2020;183:1436–1456.e31. doi: 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillette M.A., Satpathy S., Cao S., Dhanasekaran S.M., Vasaikar S.V., Krug K., Petralia F., Li Y., Liang W.W., Reva B., et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell. 2020;182:200–225.e35. doi: 10.1016/j.cell.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dou Y., Kawaler E.A., Cui Zhou D., Gritsenko M.A., Huang C., Blumenberg L., Karpova A., Petyuk V.A., Savage S.R., Satpathy S., et al. Proteogenomic Characterization of Endometrial Carcinoma. Cell. 2020;180:729–748.e26. doi: 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart P.A., Welsh E.A., Slebos R.J.C., Fang B., Izumi V., Chambers M., Zhang G., Cen L., Pettersson F., Zhang Y., et al. Proteogenomic landscape of squamous cell lung cancer. Nat. Commun. 2019;10:3578. doi: 10.1038/s41467-019-11452-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y.J., Roumeliotis T.I., Chang Y.H., Chen C.T., Han C.L., Lin M.H., Chen H.W., Chang G.C., Chang Y.L., Wu C.T., et al. Proteogenomics of Non-smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell. 2020;182:226–244.e17. doi: 10.1016/j.cell.2020.06.012. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Q., Lou Y., Yang J., Wang J., Feng J., Zhao Y., Wang L., Huang X., Fu Q., Ye M., et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut. 2019;68:2019–2031. doi: 10.1136/gutjnl-2019-318912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasaikar S., Huang C., Wang X., Petyuk V.A., Savage S.R., Wen B., Dou Y., Zhang Y., Shi Z., Arshad O.A., et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell. 2019;177:1035–1049.e19. doi: 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark D.J., Dhanasekaran S.M., Petralia F., Pan J., Song X., Hu Y., da Veiga Leprevost F., Reva B., Lih T.S.M., Chang H.Y., et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell. 2019;179:964–983.e31. doi: 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong L., Lu D., Chen R., Lin Y., Zhu H., Zhang Z., Cai S., Cui P., Song G., Rao D., et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell. 2022;40:70–87.e15. doi: 10.1016/j.ccell.2021.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Ng C.K.Y., Dazert E., Boldanova T., Coto-Llerena M., Nuciforo S., Ercan C., Suslov A., Meier M.A., Bock T., Schmidt A., et al. Integrative proteogenomic characterization of hepatocellular carcinoma across etiologies and stages. Nat. Commun. 2022;13:2436. doi: 10.1038/s41467-022-29960-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Totoki Y., Tatsuno K., Covington K.R., Ueda H., Creighton C.J., Kato M., Tsuji S., Donehower L.A., Slagle B.L., Nakamura H., et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 43.Calderaro J., Ziol M., Paradis V., Zucman-Rossi J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019;71:616–630. doi: 10.1016/j.jhep.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Li Z., Chen G., Cai Z., Dong X., He L., Qiu L., Zeng Y., Liu X., Liu J. Profiling of hepatocellular carcinoma neoantigens reveals immune microenvironment and clonal evolution related patterns. Chin. J. Cancer Res. 2021;33:364–378. doi: 10.21147/j.issn.1000-9604.2021.03.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xing X., Yuan H., Sun Y., Ke K., Dong X., Chen H., Liu X., Zhao B., Huang A. ANXA2(Tyr23) and FLNA(Ser2152) phosphorylation associate with poor prognosis in hepatic carcinoma revealed by quantitative phosphoproteomics analysis. J. Proteonomics. 2019;200:111–122. doi: 10.1016/j.jprot.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Horn H., Schoof E.M., Kim J., Robin X., Miller M.L., Diella F., Palma A., Cesareni G., Jensen L.J., Linding R. KinomeXplorer: an integrated platform for kinome biology studies. Nat. Methods. 2014;11:603–604. doi: 10.1038/nmeth.2968. [DOI] [PubMed] [Google Scholar]
- 47.Haber P.K., Puigvehí M., Castet F., Lourdusamy V., Montal R., Tabrizian P., Buckstein M., Kim E., Villanueva A., Schwartz M., Llovet J.M. Evidence-Based Management of Hepatocellular Carcinoma: Systematic Review and Meta-analysis of Randomized Controlled Trials (2002-2020) Gastroenterology. 2021;161:879–898. doi: 10.1053/j.gastro.2021.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Francies H.E., McDermott U., Garnett M.J. Genomics-guided pre-clinical development of cancer therapies. Nat. Can. (Ott.) 2020;1:482–492. doi: 10.1038/s43018-020-0067-x. [DOI] [PubMed] [Google Scholar]
- 49.Zeng Y., Zhang W., Li Z., Zheng Y., Wang Y., Chen G., Qiu L., Ke K., Su X., Cai Z., et al. Personalized neoantigen-based immunotherapy for advanced collecting duct carcinoma: case report. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2019-000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai Z., Su X., Qiu L., Li Z., Li X., Dong X., Wei F., Zhou Y., Luo L., Chen G., et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol. Cancer. 2021;20:164. doi: 10.1186/s12943-021-01467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., de Oliveira A.C., Santoro A., Raoul J.L., Forner A., et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 52.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai Z., Chen G., Zeng Y., Dong X., Li Z., Huang Y., Xin F., Qiu L., Xu H., Zhang W., et al. Comprehensive Liquid Profiling of Circulating Tumor DNA and Protein Biomarkers in Long-Term Follow-Up Patients with Hepatocellular Carcinoma. Clin. Cancer Res. 2019;25:5284–5294. doi: 10.1158/1078-0432.CCR-18-3477. [DOI] [PubMed] [Google Scholar]
- 54.Ha G., Roth A., Khattra J., Ho J., Yap D., Prentice L.M., Melnyk N., McPherson A., Bashashati A., Laks E., et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24:1881–1893. doi: 10.1101/gr.180281.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bastian M., Heymann S., Jacomy M. Vol. 2. International AAAI Conference on Weblogs and Social Media; 2009. (Gephi: An Open Source Software for Exploring and Manipulating Networks). [Google Scholar]
- 56.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaujoux R., Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinf. 2010;11:367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F., et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47:D1211–D1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen T., Ma J., Liu Y., Chen Z., Xiao N., Lu Y., Fu Y., Yang C., Li M., Wu S., et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 2022;50:D1522–D1527. doi: 10.1093/nar/gkab1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Broutier L., Mastrogiovanni G., Verstegen M.M., Francies H.E., Gavarró L.M., Bradshaw C.R., Allen G.E., Arnes-Benito R., Sidorova O., Gaspersz M.P., et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017;23:1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S., Li W., Hu L., Cheng J., Yang H., Liu Y. NAguideR: performing and prioritizing missing value imputations for consistent bottom-up proteomic analyses. Nucleic Acids Res. 2020;48:e83. doi: 10.1093/nar/gkaa498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 69.Jia Q., Wu W., Wang Y., Alexander P.B., Sun C., Gong Z., Cheng J.N., Sun H., Guan Y., Xia X., et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat. Commun. 2018;9:5361. doi: 10.1038/s41467-018-07767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goncalves E., Fragoulis A., Garcia-Alonso L., Cramer T., Saez-Rodriguez J., Beltrao P. Widespread Post-transcriptional Attenuation of Genomic Copy-Number Variation in Cancer. Cell Syst. 2017;5:386–398.e4. doi: 10.1016/j.cels.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C., et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Casado P., Rodriguez-Prados J.C., Cosulich S.C., Guichard S., Vanhaesebroeck B., Joel S., Cutillas P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013;6:rs6. doi: 10.1126/scisignal.2003573. [DOI] [PubMed] [Google Scholar]
- 73.Wiredja D.D., Koyutürk M., Chance M.R. The KSEA App: a web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics. 2017;33:3489–3491. doi: 10.1093/bioinformatics/btx415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X., Soares J., et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw data of proteome and phosphoproteome derived from HCC tissues have been deposited at iProX: IPX0005108000 and ProteomeXchange: PXD046519.62,63 Raw data of genome and transcriptome derived from HCC tissues have been deposited at Genome Sequence Achieve of National Genomics Data Center (NGDC): HRA000045, HRA000464. Accession numbers are listed in the key resources table.
-
•
All analysis code has been deposited at Zenodo: https://doi.org/10.5281/zenodo.10053446 and is publicly available as of the date of publication. DOls are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.