

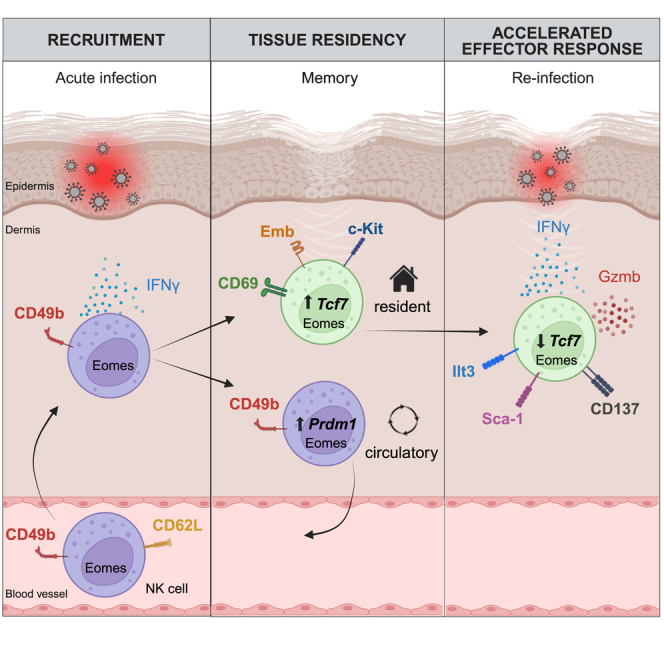
Summary
Natural killer (NK) cells are present in the circulation and can also be found residing in tissues, and these populations exhibit distinct developmental requirements and are thought to differ in terms of ontogeny. Here, we investigate whether circulating conventional NK (cNK) cells can develop into long-lived tissue-resident NK (trNK) cells following acute infections. We found that viral and bacterial infections of the skin triggered the recruitment of cNK cells and their differentiation into Tcf1hiCD69hi trNK cells that share transcriptional similarity with CD56brightTCF1hi NK cells in human tissues. Skin trNK cells arose from interferon (IFN)-γ-producing effector cells and required restricted expression of the transcriptional regulator Blimp1 to optimize Tcf1-dependent trNK cell formation. Upon secondary infection, trNK cells rapidly gained effector function and mediated an accelerated NK cell response. Thus, cNK cells redistribute and permanently position at sites of previous infection via a mechanism promoting tissue residency that is distinct from Hobit-dependent developmental paths of NK cells and ILC1 seeding tissues during ontogeny.
Keywords: natural killer cells, innate lymphoid cells, tissue-resident lymphocytes, infection, tissue immunity, innate immune memory, vaccinia virus, Staphylococcus aureus, trained immunity, vaccination
Graphical abstract
Highlights
-
•
NK cells form tissue-resident (trNK) memory-like cells after infections resolve
-
•
Tcf1hiCD69hi trNK cells share similarities with CD56bright NK cells in human skin
-
•
Hobit-independent cNK-to-trNK cell differentiation is regulated by Tcf1 and Blimp1
-
•
trNK cells mediate accelerated effector responses upon secondary infection
Conventional natural killer (cNK) cells are short-lived, circulating innate lymphocytes. Torcellan, Friedrich et al. demonstrate that cNK cells differentiate into long-lived, memory-like, tissue-resident NK (trNK) cells upon acute infections of skin. These Tcf7hiCD69hi cells share similarities with CD56bright NK cells in human skin and mediate accelerated responses to secondary infection.
Introduction
Natural killer (NK) cells are effectors of the innate immune system that play important roles in fighting infections and tumors and in regulating the immune system. NK cells are present in the circulation and within tissues, and these populations are proposed to represent developmentally distinct trajectories: conventional NK (cNK) cells develop continuously in the bone marrow and recirculate through the peripheral blood, from where they can be transiently recruited to infected or inflamed tissues.1,2 Tissue-resident NK (trNK) cells, on the other hand, settle into selected tissues early during life, in the absence of infectious or microbial triggers, to form long-lived, self-renewing local lymphocyte populations.3,4,5,6,7 These cells share similarities with innate lymphoid cells (ILCs), and as their development is independent of the transcriptional regulators Eomes and Nfil3 but fate-maps for Hobit, these cells appear to develop from ILC progenitors, independently of the cNK cell lineage.3,4,8,9
Analyses of human tissues show a heterogeneous picture in terms of local NK cell populations: although in some tissues, such as the liver, trNK cells are detectable in most individuals, there are other tissues, such as the lung, in which trNK cells are found only in a fraction of analyzed individuals.5,10,11 These observations raise the question of whether trNK cells can arise in different contexts: first, during ontogeny, as part of the development of those organs of which they are an integral part, and second, throughout life, in the context of inflammation or infection. Differences between tissues could then be explained by differences in the experience of infections or tissue inflammation. One conceivable mechanism for the establishment of trNK cells in inflamed tissues during adult life would be the local differentiation from multipotent progenitor cells, as we and others have demonstrated for ILCs.12,13,14 An alternative mechanism could be the recruitment of circulating cNK cells and their “conversion” into trNK cells, akin to the establishment of tissue-resident memory T cells at the site of a previous infection, a hallmark of adaptive immunity.
NK cells exhibit several adaptive features, including clonal expansion, epigenetic reprogramming, and differentiation into long-lived populations.1,15,16,17 Although there is strong evidence that cNK cells acquire adaptive features during infections, previous studies have either focused on NK cells in lymphoid organs and the peripheral blood1,15,16,18,19 or on NK cells in chronically infected tissues,20,21 where persisting inflammatory signals may retain or drive continuous replenishment of these cells. A major open question is whether cNK cells have the potential to establish bona fide long-lived trNK cells at the site of previous infection, long after the infection has been cleared.
To this end, we established novel experimental tools to track the differentiation of endogenous cNK cells in viral and bacterial skin infections. Our findings suggest that cNK cells are capable of forming long-lived populations of trNK cells in the skin in response to acute, resolving infections. These cells rapidly reactivated and mediated an accelerated effector response during a secondary infection. Infection history is therefore an important factor that determines the cellular composition of local pools of innate lymphocytes and their ability to respond to secondary infections. Our analyses suggest that CD56bright-like NK cells in human tissues share central features with this unique population of cNK-derived Tcf1hiCD69hi trNK cells that we identified here in murine skin. Because circulating cNK cells are the major source of cells for NK-based adoptive cell therapy, their unexpected differentiation potential into long-lived trNK cells may open new avenues for NK-based immunotherapeutic strategies.
Results
Eomes+ NK cells in the skin of naive mice are rare and predominantly intravascular cells
Eomes+ NK cells and Eomes− ILC1s can be identified in selected tissues of healthy mice, for example, in the uterus, salivary glands (SGs), and the liver (Figures 1A and 1B).3,7,8 These group 1 ILCs populate their host tissues through developmental programs that are independent of microbial challenges, as we found similar numbers in tissues of gnotobiotic and specific-pathogen-free (SPF) mice (Figures S1A–S1D).3 Phenotypic similarity to circulating cNK cells limits the ability to unambiguously identify and track cells that are being recruited into tissues during infection. Interestingly, NK cells and ILC1s were largely absent from ear skin (Figures 1A and 1B), although we readily detected Lin−NK1.1−CD127+ cells that contain ILC2 and ILC322 (Figure S1E). The rare Lin−NK1.1+ cells detected in skin digestions had the phenotype of either Eomes−CD49a+CD49b− ILC1s or of circulating Eomes+CD49a−CD49b+ cNK cells (Figures 1A and 1C), as previously reported.9 Consistent with a circulating phenotype, Eomes+ cells in skin overwhelmingly labeled with intravenously injected anti-CD45 antibody (ivCD45), suggesting their presence in the vasculature (Figures 1C and S1F). Accordingly, NKp46-expressing cells were readily detected by microscopy in tissue sections of SGs and uterus but not of the skin (Figure S1G). These analyses also indicated that the few ILC1s detected in tissue digestions by flow cytometry may actually reside in adjacent subdermal layers (Figure S1H). Together, these analyses established that the healthy skin of SPF mice is largely devoid of NK cells and that the rare cells detected represent largely intravascular cells with a circulating phenotype, providing an opportunity to track changes in the ILC1 compartment that may occur during infection.
Figure 1.

Group 1 ILCs are rare in the skin of naive mice and expand after local infection
(A) Representative FACS plots showing CD45+ live (top) and Lin−NK1.1+ gated cells (bottom) in the indicated organs.
(B) Absolute numbers of CD49a+NK1.1+ NK cells.
(C) Representative FACS plot showing Eomes+ NK cells in naive ear skin after intravascular labeling with anti-CD45 (ivCD45).
(D, E, and I) Numbers (D and I) and representative gating (E) of Lin−NK1.1+ cells in ear skin of mice infected with vaccinia virus expressing m157 (VACV) or S. aureus at the indicated times post infection.
(F and J) Confocal immunofluorescence images of skin cryosections from naive or VACV-infected mice housed in SPF (F) or wildling conditions (J). Scale bars represent 20 μm, 30 μm, or 50 μm, as indicated.
(G and H) Numbers of Eomes+ NK cells in the skin of individual ears on day 30 pi of B6 mice with WT or m157-expressing vaccinia virus (G) or of B6 versus Ly49H-deficient (Klra8−/−) mice with VACV (H).
Data in (B), (D), and (I) were pooled from n = 6–8 mice (B), n = 7–23 ears (D), or n = 4–17 ears (I) per organ or time point. Data in (A), (C), (E)–(H), and (J) are representative of three independent experiments with n = 3–5 mice (A) or n = 3–6 ears (C, E–H, and J) per group. Error bars indicate mean + SD. p values were calculated by unpaired Student’s t test. ∗∗p < 0.01 and ∗∗∗∗p < 0.0001. n.s., not significant. See also Figure S1.
Local infection shapes the skin lymphocyte niche and expands group 1 ILCs
To this end, we adopted an established model of intra-epidermal infection with a recombinant vaccinia virus (VACV) expressing the NK cell-activating ligand m157.23,24 We detected a pronounced increase of Lin−NK1.1+ cells by day 3 post infection (pi), consistent with their recruitment to the site of infection (Figures 1D and S1I). The number of skin-infiltrating cells peaked around day 7 pi and then rapidly declined, after viral clearance, leaving well-described resident CD8+ memory T cell (Trm) populations in the skin (Figures 1D, S1J, and S1K). Interestingly, Lin−NK1.1+ cells were also retained over several weeks and were strictly localized to the dermis, often in proximity to T cells and major histocompatibility complex (MHC)-II+ myeloid cells, and around the hair follicles (Figures 1D–1F). Recognition of m157 through the Ly49H receptor (encoded by Klra8) can directly activate NK cells and thereby increase their expansion and effector differentiation, but it also limits viral replication.1 Numbers of Eomes+ NK cells in the skin were independent of Ly49H expression by NK cells and were even increased when infecting with a non-recombinant wild-type (WT) VACV, raising the possibility that the increased replication potential of the WT virus, rather than triggering of the Ly49H receptor, increased the numbers of skin NK cells (Figures 1G and 1H). Similar to VACV, local infection with Staphylococcus aureus (S. aureus) induced a strong expansion of Lin−NK1.1+ cells until day 7 of infection, with the subsequent contraction and establishment of long-term persisting cells in the skin (Figure 1I). We could not detect increased numbers of Lin−NK1.1+ cells in contralateral uninfected ears, in the skin of mice that underwent systemic viral infection with murine cytomegalovirus (MCMV), or in so-called wildlings with a diverse microbial exposure (Figures 1J and S1L and data not shown). Together, these data suggest that the establishment of Lin−NK1.1+ cells in the skin is not a consequence of systemic immune activation or microbial exposure but instead requires local infection.
Infection induces NK cells with a program of tissue-resident cells
Our results indicated that the skin niche of group 1 ILCs is substantially reorganized during infection. Similar to Eomes−CD49a+ ILC1s, which are well characterized as tissue-resident cells, Eomes+ NK cells persisted long term in the skin several weeks pi (Figures 2A and 2B). These cells lacked expression of CD49a and Cxcr6, suggesting that they are phenotypically distinct from ILC1s arising during ontogeny (Figures 2A and S2A). They also lacked expression of RORγt, which was expressed by subsets of T cells in the skin (Figure S2B). mRNA-sequencing analysis confirmed that NK cells sorted from previously infected skin have a unique gene expression profile, as compared with splenic cNK and CD49a+ uterine cells (Figures 2C, 2D, S2C, and S2D). NK cells in skin and uterus expressed lower levels of genes associated with a circulating phenotype, such as Sell, S1pr5, Klf2, Zeb2, Prdm1, Cx3cr1, and Klrg1, and higher levels of Cd69, Rgs1, Rgs2, Vps37b, Tnfaip3, and Bhlhe40—a gene program that is characteristic of tissue-resident memory CD8 T cells25,26,27,28 (Figure S2D). In addition, skin-derived NK cells expressed increased levels of Emb, Tcf7, Kit, and Rora, suggesting that these cells are distinct from the uterine ILC1s.
Figure 2.

Infection induces NK cells with a program of tissue-resident cells
(A and B) Representative FACS plots (A) and numbers of Lin−NK1.1+ pregated CD49a−Eomes+ NK cells and CD49a+Eomes− ILC1s (B) in naive versus VACV-infected ears on day 35 pi.
(C and D) mRNA-sequencing analysis of Lin−NK1.1+Cxcr6− NK cells sorted from naive (uterus) or VACV-infected mice on day 35 pi (spleen and skin). (C) Euclidean distance of samples visualized by principal-component analysis. (D) Venn diagram showing numbers and examples of shared and differentially expressed genes.
(E–I) scRNA-seq analysis of Lin−NK1.1+ cells sorted on day 25 post VACV infection from indicated organs, pooled from n = 5–10 individual mice each. UMAP visualization of tissue of origin (E), identified clusters (F), selected marker genes delineating NK cells and ILC1s (G), and scores based on the top 100 DEGs between NK cells and ILC1s (I).
(H) Bubble plot representation of Z score of selected marker gene expression.
Data are representative of three (A and B) or two (E–I) independent experiments with n = 4–18 ears per group. Error bars indicate mean + SD. p values were calculated by unpaired Student’s t test. ∗∗p < 0.01 and ∗∗∗p < 0.001. See also Figure S2.
In order to resolve the heterogeneity and putative differentiation states of infection-induced skin-resident NK cells and ILC1s, we sorted Lin−NK1.1+ cells from skin, SGs, and spleen on day 25 pi, hashtagged them to mark their tissue of origin, and performed cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) single-cell mRNA sequencing (scRNA-seq) (Figures 2E and S2E–S2H). Uniform manifold approximation and projection (UMAP) dimensionality reduction revealed 14 distinct clusters (Figure 2F). ILC1s (clusters 0, 2, 11, and 12) were characterized by high expression of Il7r, Rora, Gpr183, Cxcr6, Sdc4, Tmem176a/b, and Tcrg-C4, whereas remaining clusters could be identified as Eomes and Ly49 receptor (e.g., Klra4 and Klra8) expressing NK cells, as previously suggested8,29 (Figures 2G, 2H, and S2I). Similar to ILC1s, NK cells clustered largely by their tissue of origin, consistent with tissue-specific expression patterns (Figure S2G). For example, SG NK cell clusters 1, 6, and 10 expressed Il21r, Itga1, and Osgin1, whereas splenic NK cell clusters 3, 5, and 7 were characterized by expression of Zeb2, S1pr5, and Cma1. Interestingly, NK cells isolated from the skin (clusters 4 and 8) had low expression of these genes and clustered in between SGs and splenic NK cells and in between the NK cell and ILC1 clusters (Figures 2F and 2H). Consistently, skin-derived NK cells shared expression of genes that are also expressed by ILC1s, for example, Tcf7, Rora, Emb, and Ltb. To further compare these cells, we generated scores from the top 100 differentially expressed genes (DEGs) between NK cells and ILC1s derived from a recent dataset by Lopes et al.30 NK cells from both the skin and SG had an intermediate cNK cell score, suggesting that they are distinct from splenic NK cells, whereas subsets of SG NK cells, but not of skin NK cells, appeared to have increased similarity with ILC1s (Figure 2I). Together, these data suggest that infection induces a transcriptionally unique NK cell population in the skin.
Identification of Tcf1hiCD69hi skin-resident NK cells
NK cells from the skin and SG expressed many genes associated with a program of tissue residency.27 A score calculated from these genes correlated with the surface expression of CD69, a protein that functions to prevent tissue egress by suppressing chemotactic responsiveness to sphingosin 1-phosphate gradients (Figures 3A and 3B).31 The NK cell and ILC1 clusters were predominantly composed of cells derived from either a non-lymphoid tissue (SG and skin) or the spleen, with the notable exception of cluster 9, which received input from all three tissues, suggesting that these cells may represent recirculating cells (Figure 3C). To distinguish the cells represented by these different clusters in the skin, we derived a set of marker genes and validated their expression at the protein level (Figures 3D–3H and S3A). Consistently, expression of Cxcr6 and CD49a (Itga1) distinguished Eomes− ILC1s from Eomes+ NK cells in previously infected skin (Figures 3F and S2A). As predicted by the gene expression patterns of clusters 4/8 versus 9, we identified a CD69hiCD49blo subset of NK cells that expressed high levels of Tcf1 (encoded by Tcf7), Embigin, and c-Kit and a CD49bhiCD69lo subset of cells that instead expressed increased levels of Klrg1, CD11b, perforin, as well as granzymes A (Gzma) and B (Gzmb) (Figures 3F–3H and S3A). As expression of these and other marker genes was consistent across clusters 4 and 8, the split of these clusters may be driven by differential activation or the expression of stress-related genes induced by tissue disruption. Consistent with that idea, there was a large overlap between the genes that defined separated clusters of skin ILC1s (0 vs. 2), skin NK cells (4 vs. 8), and SG NK cells (6 vs. 1), although we cannot exclude further heterogeneity among these cell types (Figures S3B and S3C; Table S1).
Figure 3.

Identification of Tcf1hiCD69hi skin-resident NK cells
(A–E) scRNA-seq analysis of Lin−NK1.1+ cells on day 25 post VACV infection as in Figure 2. (A and B) UMAP of a score based on genes associated to tissue-resident memory T cells (A) and of CD69 protein expression by CITE-seq (B). (C) Bar graph showing tissue contribution to each cluster in Figure 2F. (D) Bubble plot representation of Z score of selected marker genes in selected NK cell clusters. (E) Volcano plot showing DEGs between indicated clusters (cellular transcriptomes of skin origin only).
(F–H) Representative FACS plots of Eomes− ILC1s and Eomes+ NK cells (F), frequency (G), and geometric mean of fluorescence intensity (geoMFI) (H) of indicated protein expression within indicated cell subsets 4 weeks post infection with VACV.
(I and J) Representative FACS plots (I) and frequency (J) of intravascular (i.v. anti-CD45-labeled) NK cell subsets.
(K and L) Comparative marker expression and i.v. anti-CD45 label of ear skin NK cells from 16 pooled naive (red, 260 cells) or 1 representative VACV-WT infected (blue, 345 cells) ear skins on day 35 pi (K) and from naive or VACV-WT infected skin on day 35 and day 80 pi (L).
(M–O) Photoactivation of VACV-infected skin on day 28 pi of Dendra2 mice. Experimental design (M), representative FACS analysis (N), and frequency (O) of Dendra2Red+ label-retaining photoconverted cells within indicated subsets of cells analyzed 30 min (day 0), and 5 and 10 days post photoactivation.
(P) Representative FACS plots of skin NK cell subsets at memory time points post infection with S. aureus, heat-inactivated S. aureus, VACV-WT, or non-replicating VACV (MVA).
Data are representative of two (A–E and P) or three (F–N and O) independent experiments with n = 6 (J) or n = 8 (F–H) ears per group. FACS plots in (I) and (N) are pooled from n = 3 (I) or n = 5 (N) ears. Data in (O) are pooled from n = 2 (day 0) or n = 8 (days 5 and 10) skins from 2 independent experiments. Error bars indicate mean + SD. p values were calculated by paired one-way ANOVA (G and O) or paired Student’s t test (J). ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗∗p < 0.0001; n.s., not significant. See also Figure S3.
A large fraction of CD49bhi cells were stained with ivCD45, indicating their presence in the blood circulation. By contrast, ivCD45 failed to stain CD49bloCD69hi NK cells, suggesting that these cells located to extravascular tissue niches (Figures 3I and 3J). Although naive skins harbored some ivCD45+ NK cells, ivCD45− cells lacking CD49b and expressing CD69, Tcf1, and c-Kit were extremely rare, and could only be detected sporadically, when pooling cells from many (n = 16) ears (Figures 3K and S3D). By contrast, ivCD45− CD69hi cells persisted in the long term after viral infection (Figure 3L). To directly test whether these cells were tissue-resident, we performed non-invasive percutaneous photoswitching (Dendra2Green to Dendra2Red) of histone-Dendra2 transgenic mice, which labeled ∼80% of Lin−NK1.1+ cells (Figure S3E). Intriguingly, CD69hiEomes+ NK cells retained the photoconverted Dendra2Red signal after 5 and 10 days, and to a similar degree as ILC1s and tissue-resident CD69+CD103+CD8+ T cells (Figures 3M–3O and S3F). The CD69hi NK cell population showed no detectable loss of the frequency of Dendra2Red+ cells, nor of the signal intensity of Dendra2Red, demonstrating that these cells remained in their tissue niche as resident cells and that they were not actively dividing and diluting the photoswitched protein during the observation time. By contrast, CD49bhi NK cells had lost the Dendra2Red signal already by day 5, suggesting that these cells had been replaced by circulating cells. Together, the specialized gene expression program, extravascular localization, and label retention upon photoconversion indicated that Tcf1hiCD69hi NK cells that are generated in response to local infection are bona fide trNK cells. We found that these cells were induced in response to various infectious stimuli, including WT and m157-expressing VACV and S. aureus. CD69hi NK cells with this unique phenotype were also generated upon infection with non-replicating VACV (modified vaccinia Ankara [MVA]) or heat-inactivated S. aureus, albeit at much reduced numbers, suggesting that local pathogen replication facilitates the generation of these cells (Figures 3P and S3G).
Tcf1hiCD69hi NK cells share similarities with NK cells present in human skin
To test whether a similar population of NK cells may exist in human skin, we analyzed available scRNA-seq data of non-lesional skin.32 Similar to previously infected murine skin, digestions of non-lesional skin retrieved two distinct NK cell populations that resembled known subsets of CD56bright and CD56dim NK cells (Figures 4A and S4A–S4E).16,33 Accordingly, one of the two populations exhibited increased expression of FCGR3A (encoding CD16), B3GAT1 (encoding CD57), ZEB2, GZMB, GZMH, and PRF1, and hence it resembled CD56dim cNK cells (Figures 4B, 4C, and S4C–S4E). The other population had increased expression of genes characteristic of the CD56bright subset of NK cells, including RUNX2, ZEB1, MAML3, GZMK, and XCL1.34 Similar to the Tcf1hiCD69hi NK cells that we identified in previously infected murine skin, human skin-resident CD56bright-like NK cells had increased expression of TCF7, CD69, RORA, LTB, EMB, GPR183, and CXCR4 (Figures 4B and 4C). Importantly, a score derived from the top 100 DEGs between the human NK cell subsets further suggested conserved gene expression patterns between CD56brightTCF7hiCD69hi cells and the skin-resident trNK cells identified here (Figure 4D). Both human and murine Tcf1hiCD69hi cells shared a gene expression program related to tissue residency, with increased expression of CD69, CXCR3, RGS1, RGS2, XCL1, CRTAM, and LMNA1 and reduced expression of S1PR5, KLF2, SELL, CX3CR1, and KLRG1, which was also reflected by a high tissue-residency score (Figures 4E and S4F).5,35 By contrast, CD56dim-like cells shared similarities with murine circulating splenic NK cells, underscoring the conserved transcriptional profiles of various subsets of NK cells between mice and humans. Of note, enrichment of the Trm score in CD56bright-like NK cells was only observed in the skin, and there was a low residency score in human peripheral blood NKs, and no difference was observed between CD56bright and CD56dim subsets (Figures S4G–S4I). Together, these analyses reveal intriguing similarity with the skin-resident NK cells identified here, and TCF7hi CD56bright-like cells present in human skin.
Figure 4.

Tcf1hiCD69hi NK cells share similarities with NK cells present in human skin
Analysis of single-cell gene expression of NK cells in human skin (data generated by Alkon et al.32).
(A) UMAP showing re-clustering of NK cell clusters 0 and 3 from Figure S4A.
(B) Bubble plot representation of Z score of selected marker genes in murine (skin and spleen on day 25 pi, left) and human skin NK cells (CD56bright- and CD56dim-like subsets, right).
(C) UMAP representation of selected marker gene expression of human skin NK cells.
(D and E) Visualization of scores of gene expression in murine NK cells (D) and in human skin NK cell subsets (E). Scores were calculated based on genes significantly upregulated in indicated human skin NK cells, and then used to analyze murine NK cells (D) or based on genes associated to tissue-resident memory of T cells, as in Figure 3A, and then used to analyze human skin NK cells (E). See also Figure S4.
CD62L+ Eomes+ IFN-γ-producing effector cells give rise to Tcf1hi tissue-resident memory NK cells
Tissue-resident innate lymphocytes can develop from local or recruited progenitor cells that have multilineage potential and in principle could also generate resident NK cell populations that acquire adaptive features.4,5,12,14 An alternative possibility would be the recruitment of lineage-committed NK cells during the acute phase of infection that then differentiate into long-lived resident memory-like cells, akin to the differentiation of CD8 Trm. Consistent with this idea, NK cells started to accumulate in infected skins as early as 12 h pi (Figure S5A). To directly test whether cNK cells being recruited into infected skin have the potential to differentiate into trNK cells, we analyzed SellCre-ERT2xRosaLSL-tdTomato and EomesCre-ERT2xRosaLSL-tdTomato fate-mapping mice (Sell-FM and Eomes-FM, respectively). CD62L (encoded by Sell) is expressed in circulating cNK cells, and Eomes is induced during NK cell development and lineage commitment downstream of T-bet.36,37 We genetically marked Sell- and Eomes-expressing cells through tamoxifen (TAM) treatment before infection and then tracked their fate. In the memory phase, a significant fraction of CD69hi NK cells in the skin fate-mapped for Sell (Figures 5A–5C) and Eomes (Figures 5D–5F) expression, suggesting that circulating cNK cells can become trNK cells during infection.
Figure 5.

CD62L+ Eomes+ IFN-γ-producing effector cells give rise to Tcf1hi tissue-resident memory NK cells
(A, D, and G) Experimental design.
(B, C, E, F, and H–J) Representative FACS plots and bar graphs show fate-map (FM) labeling of indicated NK cell subsets in the skin of Sell-FM (B and C), Eomes-FM (E and F), IFN-γ-FM (H and I), or WT mice (J) at the indicated time points post infection.
(K–M) scRNA-seq analysis of spleen and skin NK cells sorted on day 8 post VACV infection, pooled from n = 5 individual mice. UMAP visualization of cell origin (K), expression of selected genes (L), and scores calculated as in Figure 4E (M).
Data in (B), (C), (F), and (H)–(J) are representative of three (B, C, F, I, and J) or two (H) independent experiments with n = 4 (H), n = 5 (F), or n = (B, C, and I) skins per group. Data in (E) were pooled from n = 5 skins and are representative of two independent experiments. Error bars indicate mean + SD. p values were calculated by paired Student’s t test. ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001; n.s., not significant. See also Figure S5.
To test whether trNK cells could arise from NK cells that participate in the effector response early during infection, we took advantage of a novel interferon (IFN)-γ-reporter/FM model (IFN-γeGFP-Cre-ERT2xRosaLSL-tdTomato). NK cells in the skin produced IFN-γ on days 3 and 4 of infection, allowing us to genetically tag IFN-γ-producing cells in this early phase of infection (Figures 5G–5I and S5B). At the peak of the NK cell infiltration, similar fractions of CD69hi and CD49bhi NK cells were FM+ (Figure 5H). Interestingly, CD69hi NK cells retained a higher level of IFN-γ fate-label in the memory phase, as compared with their CD49bhi circulating counterparts (Figure 5I). These analyses cannot distinguish whether cells may be first recruited into skin and then produce IFN-γ there or whether they start to produce IFN-γ, e.g., in the draining lymph node, and then infiltrate the skin; nevertheless, they clearly demonstrate that IFN-γ-producing cells have the potential to form long-lived populations of skin-resident NK cells. Consistent with the idea that the characteristic trNK program may be established, at least in part, already during the acute phase of infection, we detected distinct populations with heterogeneous expression of CD69 and CD49b on day 7 pi (Figure 5J). scRNA-seq indicated substantial activation and heterogeneity of cells present in the skin on day 8 pi (Figures 5K, S5C, and S5D) and revealed that many of the marker genes of the newly identified skin-resident NK cells were already upregulated in the skin in the acute phase of infection, including Cd69, Tcf7, Emb, Kit, Ltb, and Bhlhe40. By contrast, genes that are characteristically expressed by circulating and splenic NK cells were downregulated (e.g., S1pr5, Klf2, Zeb2, Itgam, and Klrg1; Figures 5L, S5E, and S5F). This was corroborated by a high score for genes associated with tissue residency in the skin, as compared with splenic NK cells (Figure 5M). Pseudotime and trajectory analysis suggested a potential trajectory originating in cluster 0 in the spleen and projecting a continuous path across clusters 8 > 3 > 1 > 4 toward cluster 2 in skin, which is marked by a high tissue-residency score (Figures 5M and S5G–S5J). Several marker genes were regulated along this trajectory, including Klf2, Cma1, Zeb2, Kit, Emb, and Bhlhe40. Although it is currently unclear when and where trNK cell commitment occurs and whether the cellular states in spleen and skin are actually connected at this day 8 pi “snapshot” of gene expression, this analysis highlighted several interesting genes regulated along this trajectory, including the following: Satb1, a chromatin organizer with a function in maintaining the naive state of CD8+ T cells before activation,38 the lysine demethylase 6b (Kdm6b) involved in effector differentiation of CD8+ T cells and NK cells,39 and the RNA-binding protein 3 (Rbm3) that has been suggested to increase β-catenin signaling, and thereby activity of Tcf/Lef transcription factors, and to restrain activation of ILC2s.40 Other interesting genes include Stat3, which regulates responsiveness to cytokines, Ifngr1, involved in ILC1 differentiation,41 and Smad7, which can antagonize TGF-β signaling and thereby may inhibit signals from concomitantly upregulated Tgfb1.42
Together, these data suggest that CD62L+Eomes+ circulating cNK cells are recruited into infected skin where they differentiate into bona fide long-lived trNK cells, revealing a remarkable conceptual similarity to the differentiation cascade of CD8 Trm.
Skin-resident NK cells require Eomes but are independent of Hobit and TGF-β
Our findings indicated that infection-induced trNK cells in skin and trNK cells that are established during ontogeny in the SG may be generated through different developmental paths. SG trNK cells develop independently of cNK cells and of their hallmark transcription factor Eomes.3,20 Indeed, conditional deletion of Eomes in Ncr1Cre Eomesfl/fl mice did not affect numbers of SG trNK cells, but it strongly reduced both CD69hiTcf7hi and CD49bhi NK cells in previously infected skin, as well as cNK cells in systemic compartments (Figures 6A, 6B, and S6A). Although CD69hiTcf7hi trNK cells in skin required Eomes for their differentiation, they lacked reporter activity and fate-mapping of Hobit—a key transcription factor that regulates programs of tissue residency and effector differentiation in SG trNK cells and ILC1s8,43,44 (Figures 6C, 6D, and S6B). Of note, skin-resident ILC1s were efficiently labeled using our Hobit fate-mapping approach, excluding the possibility that the skin environment is generally non-permissive for the induction of Hobit in ILC1s (Figure S6B). In addition, conditional deletion of Tgfbr2, which is critically required for the development of trNK cells in the SG3 (Figure S6C), did not alter the formation of trNK cells in skin during infection (Figures 6E and 6F). Together, these results confirm that different molecular programs and signals regulate the formation of trNK cells in infected skin, as opposed to trNK cells in SGs during ontogeny. Also, we did not observe the conversion of NK cells into ILC1s, as Lin−NK1.1+Eomes−FM+ cells retained Eomes expression, nor did we observe conversion of ILC1s into skin-resident NK cells (lack of Hobit fate-mapping) (Figures 6D and S6H).
Figure 6.

Differential transcription factor requirements of skin-resident and circulating NK cells
(A and B) Numbers of indicated NK cell subsets in skin on day 30 post VACV infection (A) or in SGs of naive Eomesfl/fl Ncr1cre or control mice (B).
(C and D) Representative FACS plots (C) and frequency (D) of fate-map (FM) labeling of indicated NK cell subsets in Hobit-FM mice on day 30 post VACV infection.
(E–J) Representative FACS plots (E, G, and I) and frequency and absolute numbers (F, H, and J) of indicated NK cell subsets in the skin of Tgfbr2fl/fl Ncr1cre (E and F), Tcf7fl/fl Ncr1cre (G and H), and Prdm1fl/fl Ncr1cre (I and J) and indicated control mice. Mice were analyzed at >3 weeks pi, and data are representative of three independent experiments with n = 6–8 (A, D, and E–J) skins, n = 3 (B), or n = 4 (C) mice per group. Error bars indicate mean + SD. p values were calculated by unpaired Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001; n.s., not significant. See also Figure S6.
Tcf1 and Blimp1 regulate the differentiation of resident memory versus circulating effector NK cells
Downregulation of Tcf1 by Hobit appears a critical step in the transition from central memory-like toward tissue-resident CD8+ T cells.44 By contrast, skin trNK cells, as well as CD56bright-like NK cells found in human skin, were Tcf1hi. Conditional deletion of Tcf7 strongly reduced the numbers of trNK cells in the skin (Figures 6G and 6H), suggesting a role of Tcf7 in trNK cell development. Interestingly, Tcf1-deficient cells in skin and spleen exhibited a more mature phenotype with increased expression of CD11b and Klrg1 (Figures S6D and S6E). Therefore, Tcf7, which is associated with stemness in circulating T cells, NK cells, and ILC1s,8,34,45,46 may regulate trNK cell numbers in part by preventing their terminal differentiation to effector cells. To further test this, we investigated whether trNK cell formation during infection could be promoted by inhibition of Blimp1, which drives maturation and effector differentiation of cNK cells.47 Indeed, conditional deletion of Prdm1 (the gene encoding Blimp1) in NK cells significantly increased trNK cell numbers in the skin (Figures 6I and 6J). Conversely, the circulating compartment of CD69loCD49bhi cells was significantly decreased. Lack of Blimp1 led to the accumulation of Tcf1hic-Kithi NK cells in the skin and in the spleen (Figures S6F and S6G). Interestingly, Blimp1-deficient NK cells expressed high levels of Gzma, suggesting that targeting Blimp1 could potentially be used to regulate differentiation into circulating versus resident NK cells while preserving selected effector functions.
Skin-resident NK cells rapidly reactivate and mediate accelerated effector responses during secondary infection
Our analyses revealed the establishment and long-term retention of a specialized trNK cell population at the site of previous infection, raising the question of whether these cells may contribute to accelerated or increased responses during pathogen re-encounter. Indeed, we found dramatically increased IFN-γ production when comparing NK cells isolated from skin at 12 h post primary infection versus 12 h post secondary infection (Figures 7A–7C). We found very few NK cells in the skin of naive mice challenged with VACV at this early time pi, and only few of these produced IFN-γ (Figures 7A–7C). Reinfection did not significantly increase NK cell numbers, as compared with the resting memory condition (Figure S7A). Therefore, we speculated that the strong IFN-γ production of NK cells in rechallenged skin was driven to a significant extent by the local reactivation of trNK cells that had been generated during the first pathogen encounter. To directly test this possibility, we photoconverted skin NK cells on day 30 post primary infection and waited for 5 days before rechallenge (Figure 7D). In this experimental setup, only CD69hi Tcf1hi resident NK cells retain the DendraRed photoactivation label at the time of secondary infection (Figures 3N and 3O), 12 h post rechallenge, and these produced copious amounts of IFN-γ (Figures 7E and 7F). scRNA-seq analysis showed that NK cells recruited into the skin at 12 h pi can be identified through a high cNK score characteristic of circulating and splenic NK cells (Figures 2I and 7G–7I). Such cells were only a small fraction of cells in rechallenged skin in which cells overwhelmingly exhibited an intermediate score characteristic of skin-resident NK cells (Figures 2I and 7G–7I), consistent with trNK cells being locally activated in this condition. These analyses confirmed that the few naive NK cells that were being recruited into the skin at 12 h pi produced much less IFN-γ and Gzmb, as compared with reactivated trNK cells (Figures 7J–7L, S7B, and S7C). Consistent with the scRNA-seq analysis, skin NK cells had lost expression of Tcf1 and gained expression of Gzmb by 12 h pi (Figures 7J and 7K). Together, these data suggest the rapid reconfiguration of Tcf1hi trNK cells into a Tcf1lo effector state. In addition to increased expression of effector molecules Ifng, Gzma, Gzmb, and Prf1, reactivated skin NK cells increased expression of several surface molecules (e.g., Il2ra, Tnfrsf9, Icos, Ly6a [encoding Sca1], Lilr4b [encoding Ilt3], and Cd53) that are associated with activation and effector function.48,49,50,51 Indeed, fluorescence-activated cell sorting (FACS) analysis validated the significantly higher expression of Sca1, Ilt3, CD25, CD137, and Gzmb (Figures 7M–7O). Rapid reprogramming of trNK cells was further evidenced by the differential expression of several transcription factors involved in NK cell effector function and differentiation (e.g., Irf8, Zbtb32, Hif1a, Id2, and Prdm1), the modulation of genes toward increased survival (e.g., Bax, Bcl2, Bcl10, and Bcl2l11), and genes increasing metabolic activity and fitness (e.g., Aldoa, Eno1, and Ldha) (Figure S7D).52,53,54,55,56
Figure 7.

Accelerated effector response of skin-resident NK cells during secondary infection
(A–C and G–N) (A) Experimental design for (B), (C), and (G)–(N). Representative FACS plots (B and J) and frequency and absolute numbers of skin NK cells expressing indicated proteins (C and K).
(D–F) Photoactivation of VACV-WT-infected skin in Dendra2 mice. Experimental design (D), representative FACS plots (E), and frequency of IFN-γ-producing Dendra2Red+ label-retaining skin NK cells (F).
(G–I and L) scRNA-seq analysis of sorted skin NK cells, experimental setup as in (A). (G–I) UMAP visualization of analyzed conditions and cNK cell score calculated as in Figure 2I.
(L) Bubble plot representation of Z score of selected marker gene expression.
(M–O) Representative FACS plots (M), frequency (N), and geometric mean of fluorescence intensity (geoMFI) (O) of skin NK cells expressing indicated proteins.
Data are representative of three (B, C, J, K, M, and N) or two (E and F) independent experiments with n = 6–8 ears per group. Error bars indicate mean + SD. p values were calculated by unpaired Student’s t test. ∗∗p < 0.01 and ∗∗∗∗p < 0.0001; n.s., not significant. See also Figure S7.
Together, our data reveal the differentiation of cNK cells into long-lived trNK cells during acute, resolving infections of the skin. Our work highlights mechanisms for the long-lasting reorganization of the innate lymphocyte niche at the site of previous infection, which enable an accelerated and increased NK cell response during reinfection.
Discussion
NK cells exhibit adaptive features that shape immune responses, and these properties can potentially be harnessed for immunotherapeutic approaches.1,57,58 Here, we investigate the differentiation potential of cNK cells during infection and demonstrate that these cells can give rise to long-lived trNK cells at a site of completely resolved acute infections. We used genetic fate-mapping to visualize the formation of these cells from the endogenous repertoire of CD62L+ Eomes+ cNK cells that get recruited into the infected tissue, produce IFN-γ early during infection, and can differentiate into a long-lived trNK cell population. We identified a CD69hiTcf1hic-KithiEmbhiGzmblo phenotype and associated transcriptional program and highlighted intriguing similarities to NK cells found in human skin. In addition, we mapped key transcriptional requirements for the generation of these cells, demonstrating that they differentiate independently of Hobit-expressing lineages of ILC1s and “ILC-like” NK cells and that Blimp1-driven effector maturation is detrimental to the Tcf1-dependent differentiation of these trNK cells. Finally, we could show that these Tcf1hiGzmblo cells rapidly reactivate during secondary infection and reprogram into Tcf7loIfnghiGzmbhiPrfhi effector cells. Our work therefore demonstrates the recruitment, differentiation, and long-term establishment of innate lymphocytes at the site of primary infection in a tissue niche that was not populated by such cells prior to infection. This establishment of resident NK cells likely expands the repertoire of molecules and conditions (e.g., “missing-self”) that can be sensed by the local pool of tissue-resident lymphocytes, and it enables an accelerated, early response upon reinfection, highlighting important conceptual parallels to the differentiation of resident memory cells in the adaptive immune system.
Our findings reveal mechanisms for the establishment of NK cell tissue residency during infection and provide several lines of evidence that distinguish these mechanisms from the development of known populations of ILC-like NK cells that seed tissues during ontogeny, in the absence of infection. Although future work is clearly needed, the emerging picture suggests that Eomes+ tissue-resident ILC1s can be generated at least from two different cellular sources—first, from ILC progenitors during ontogeny and second, from cNK cells during infection or tissue inflammation. This is reminiscent of the design principle of the myeloid and T cell systems, which can generate tissue-resident macrophages or innate T cells, respectively, from progenitors during early life or from a distinct cellular source, namely monocytes or naive T cells, respectively, during tissue infection and inflammation.59,60 To delineate these different paths to NK cell residency, and the different cell types emerging from it, we propose to refer to cells developing from ILC progenitors during ontogeny as ILC-like NK cells and to cells generated from cNK cells through an infection- or inflammation-triggered differentiation cascade as memory-like resident NK cells.
Our work highlights mechanisms that may have evolved to adapt the composition of local innate lymphocyte niches to prevalent environmental challenges. The skin of mice housed under both SPF and wildling conditions is largely devoid of IFN-γ-competent NK cells and ILC1s, yet is populated by interleukin-5 (IL-5)-, IL-13-, and IL-17-producing ILC2s and ILC3s.22,61,62 Local infection with viral and bacterial pathogens established resident NK cells and thereby conferred the capacity to rapidly produce IFN-γ upon subsequent infections. Shaping which cell types exist in a given niche can not only shape the local response types but also the spectrum of molecules and stress conditions that can be locally sensed, and therefore it may represent an early mechanism of anticipating future pathogen encounters. Such innate resident lymphocyte “memory” probably preceded the evolution of adaptive tissue-resident memory. Although conserved mechanisms may enable the recruitment and retention of innate and adaptive lymphocytes, it remains to be investigated whether any combination of receptors, or mechanisms of specificity or clonality, may favor the differentiation of infection-induced resident NK cells, which is a key feature of memory in the adaptive immune system. Conceptually, the “de novo” formation of skin-resident NK cells in response to infection is also different from “trained immunity,” which is described as the altered epigenetic programming of pre-existing cells in order to provide enhanced or qualitatively different responses.63 Although we anticipate that the cells that we discovered here may also differ by their epigenetic marks from circulating counterparts, a critical factor enabling their accelerated response is their acquired positioning within the tissue. Our findings also reinforce the question about what constitutes a “real-world” tissue niche of lymphocytes and emphasize the relevance of studying pathogen-experienced mice, in addition to SPF mice and wildlings.
Studies on memory NK cells have largely focused on lymphoid organs or the systemic circulation. Adaptive features of NK cells in tissues have been suggested in the context of persistent viral infections.20,21 It is therefore possible that NK cells accumulate as a consequence of persistent stimulation and inflammation in these tissues.20 This raises the possibility that such long-lived NK cells mirror the inflationary memory of CD8+ T cells characteristic of persisting herpesvirus infections, rather than that of tissue-resident memory cells that remain strategically positioned in the tissue long after infection has been cleared.64 Consistent with this idea, NK cells with certain characteristics of memory cells are detected in MCMV-infected SGs of BALB/c mice, in which latent infection persists for prolonged times, causing ongoing immune stimulation and gland pathology.20 Recruited NK cells were required to restrain pathogenic T cells that continue to be activated in the latently infected and chronically inflamed glands, as had been previously demonstrated by the same group.65 In B6 mice, in which MCMV does not cause such pathogenic persisting infections in the glands, these cells could not be detected.20
It was therefore important to address whether circulating cNK cells can differentiate into trNK cells that persist after clearance of acute infection, and if so, whether they would acquire a specific transcriptional program reflective of their newly adopted specialization. The tracking and manipulation of NK cells, and in particular the investigation of their adaptive features, have been largely limited to adoptive transfer approaches, with few exceptions.66 This approach bears the caveat of co-transferring progenitor populations and spleen-resident ILC-like NK cells,67,68 which may confound the cellular origin of the cells engrafting into tissues.20 To overcome these limitations, we established a novel experimental approach and suitable genetic tools to visualize and fate-map the progeny of “untouched” endogenous cNK cells as they differentiate in infected skin. This allowed us to demonstrate that NK cell memory extends to another hallmark of adaptive immunity, that is, the permanent deployment of lymphocytes to peripheral non-lymphoid tissues upon their differentiation into specialized memory populations.
The identification of tissue-resident memory-like NK cells raises the question about the existence of central and effector memory-like populations with distinct migration patterns. Interestingly, skin sensitization with haptens can prime Lin−NK1.1+ cells in the draining lymph nodes that then migrate to and persist in the liver or spleen.69 Several studies have confirmed the liver and spleen as sites of systemic memory cells that mediate amnestic responses to a range of haptens and viral antigens through the rapid mobilization and recruitment into challenged tissues.70,71,72,73,74,75,76 Although these cells resembled Eomes−CD127+CXCR6+ hepatic ILC1s and therefore are distinct from the Eomes+ trNK cells identified here, these observations together support the idea that memory of innate lymphocytes is organized into subsets of central, effector, and trNK cells. Important future questions are where and how memory-like NK cells are “primed,” how segregation into different subsets is regulated, and whether these cells are selected through mechanisms of specificity and functional avidity, as suggested in the context of haptens and herpesvirus infections.15,16,17,70,71,77,78
Adaptive differentiation of NK cells in the context of CMV infections is optimized by activating receptor signals,15,16,17 yet skin trNK cell differentiation to infection with VACV-m157 was equally efficient in Ly49H-deficient mice, and non-recombinant WT VACV, which has increased replication potential, generated even more skin-resident NK cells. Interestingly, poxviruses can trigger NK cells through various ligands,79 and it remains unclear whether S. aureus, which also induced skin trNK cell formation in our study, may also trigger activating receptors of NK cells. Interestingly, splenic NK cells are capable of responding to bacteria and can develop memory-like traits.80 Future studies should therefore investigate whether skin trNK cell formation requires or is optimized by signals from activating receptors, as in CD8 Trm,23 or whether it could be initiated by cytokine-mediated activation alone, which is common practice in the clinical application and adoptive transfer of NK cells.58,81
Our findings open new avenues to further investigate the mechanisms and signals that enable NK cell tissue persistence and adaptation and to identify additional molecular regulators that are relevant in this context. We found that Blimp1 drives the differentiation of circulating effector cells downstream of Tcf1hi NK cells34 and thereby limits skin trNK cell generation. Blimp1 can bind into the Tcf7 locus and downregulate Tcf7 expression,44 suggesting that Blimp1 may directly restrain Tcf7-dependent trNK cell potential. Therefore, enforcement of Tcf1-driven “stemness” and inhibition of Blimp1-regulated effector differentiation could be a strategy to improve tissue persistence of NK-based cellular products. Intriguingly, Blimp1-deficient NK cells had high levels of Gzma, suggesting that Blimp1-targeted Tcf1hi “stem-like” NK cells could retain effector potential. Tcf1 may further facilitate NK cell memory formation by protecting them from Gzmb-related toxicity and by regulating their responsiveness to homeostatic cytokines, as supported by the expansion of TCF7hi memory NK cells in HIV-infected individuals.82,83 In this context, it will be important to investigate whether NK cells in peripheral blood, which is easily accessible from patients and harbors CD56bright NK cells that resemble the TCF7hi resident NK cells found in tissues, have the intrinsic potential or could be engineered to differentiate into trNK cells.
Our discovery of the differentiation potential of cNK cells into long-lived trNK cells that can mediate immediate effector function in the tissue environment, the elucidation of the underlying molecular pathways, and the identification of their remarkable similarity to CD56bright NK cells in human tissues provide a framework for future mechanistic studies and clinical applications in humans.
Limitations of the study
Although NK cells are extremely rare in naive skins and get rapidly recruited during infection, we cannot exclude the possibility that cells present in the skin prior to infection may locally expand and contribute to the adapted lymphocyte pool. In this regard, it will be interesting to investigate ILC1s, which we also found to be expanded after infection but were not the focus of our current work. We anticipate that the strategic positioning of NK cells as “rapid responders” in the tissue will likely provide an advantage in controlling infections locally, especially in the context of pathogens that can interfere with MHC-I antigen presentation. The current lack of experimental tools to block recruitment of circulating NK cells or to specifically target the newly identified resident cells currently precludes testing this possibility. The development of novel genetic tools will be required to dissect the functions and contribution of these cells in the context of infection, tumor, and tissue inflammation and to understand in which tissue niches they can develop and under which circumstances and types of inflammation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 594 anti-mouse CD3 (clone 17A2) | BioLegend | Cat. #100240; RRID: AB_2563427 |
| Biotin anti-mouse CD3ε (clone 145-2C11) | BioLegend | Cat. #100304,; RRID: AB_312669 |
| FITC anti-mouse CD3ε (clone 145-2C11) | BioLegend | Cat. #100306; RRID: AB_312671 |
| BUV395 anti-mouse CD4 (clone GK1.5) | BD Biosciences | Cat. #563790; RRID: AB_2738426 |
| Biotin anti-mouse CD5 (clone 53-7.3) | BioLegend | Cat. #100604; RRID: AB_312733 |
| FITC anti-mouse CD5 (clone 53-7.3) | BioLegend | Cat. #100606; RRID: AB_312735 |
| Alexa Fluor 700 anti-mouse/human CD11b (clone M1/70) | BioLegend | Cat. #101222; RRID: AB_493705 |
| BUV737 Anti-mouse/human CD11b (clone M1/70) | BD Biosciences | Cat. #612800; RRID: AB_2738811 |
| InVivoMAb anti-mouse CD16/CD32 (clone 2.4G2) | BioXCell | Cat. # BE0307; RRID: AB_2736987 |
| Biotin anti-mouse CD19 (clone 6D5) | BioLegend | Cat. #115504; RRID: AB_313639 |
| FITC anti-mouse CD19 (clone 1D3) | BioLegend | Cat. #152404; RRID: AB_2629813 |
| BV421 anti-mouse CD25 (clone PC61) | BioLegend | Cat. #102043; RRID: AB_2562611 |
| BUV496 Anti-Mouse CD45 (clone 30-F11) | BD Biosciences | Cat. #749889; RRID: AB_2874129 |
| Pacific Blue anti-mouse CD45 (clone 30-F11) | BioLegend | Cat. #157212; RRID: AB_2876534 |
| Alexa Fluor 700 anti-mouse CD45.2 (clone 104) | BioLegend | Cat. #109822; RRID: AB_493731 |
| APC anti-mouse CD45.2 (clone 104) | BioLegend | Cat. #109814; RRID: AB_389211 |
| BV480 Hamster Anti-Rat/Mouse CD49a (clone H31/8) | BD Biosciences | Cat. #746244; RRID: AB_2743580 |
| BUV805 Hamster Anti-Rat/Mouse CD49a (clone H31/8) | BD Biosciences | Cat. #741976; RRID: AB_2871280 |
| APC/Fire 750 anti-mouse CD49b (clone DX5) | BioLegend | Cat. #108926; RRID: AB_2876423 |
| APC-eFluor 780 anti-mouse CD49b (clone DX5) | Thermo Fisher Scientific | Cat. #47-5971-82; RRID: AB_11218895 |
| PE anti-mouse CD49b (clone DX5) | BioLegend | Cat. #108908; RRID: AB_ 313415 |
| PE/Dazzle 594 anti-mouse CD49b (clone DX5) | BioLegend | Cat. #108924; RRID: AB_2565271 |
| BV510 anti-mouse CD62L (clone MEL-14) | BioLegend | Cat. #104441; RRID: AB_2561537 |
| BUV805 Hamster anti-mouse CD69 (clone H1.2F3) | BD Biosciences | Cat. #741927; RRID: AB_2871240 |
| PE/Cyanine7 anti-mouse CD69 (clone H1.2F3) | BioLegend | Cat. #104512; RRID: AB_493564 |
| APC/Cyanine7 nti-mouse CD117 (c-kit) (clone 2B8) | BioLegend | Cat. #105826; RRID: AB_1626278 |
| BV605 anti-mouse CD117 (c-kit) (clone 2B8) | BioLegend | Cat. #105847; RRID: AB_2783047 |
| PE/Dazzle 594 anti-mouse CD117 (c-kit) (clone 2B8) | BioLegend | Cat. #105834; RRID: AB_2564055 |
| PerCP-eFluor 710 anti-mouse CD137 (4-1BB) | eBioscience | Cat. #46-1371-82; RRID: AB_2573713 |
| BV421 anti-mouse CD186 (Cxcr6) (clone SA051D1) | BioLegend | Cat. #151109; RRID: AB_ 2802207 |
| BV711 anti-mouse CD186 (Cxcr6) (clone SA051D1) | BioLegend | Cat. #151111; RRID: AB_2721558 |
| PE anti-mouse CD186 (Cxcr6) (clone SA051D1) | BioLegend | Cat. #151108; RRID: AB_2572145 |
| PE/Dazzle 594 anti-mouse CD186 (Cxcr6) (clone SA051D1) | BioLegend | Cat. #151116; RRID: AB_2721699 |
| PE-Cyanine5 anti-mouse EOMES (clone Dan11mag) | Thermo Fisher Scientific | Cat. #15-4875-82; RRID: AB_11218895 |
| PerCP-eFluor 710 anti-mouse EOMES (clone Dan11mag) | Thermo Fisher Scientific | Cat. #46-4875-82; RRID: AB_10597455 |
| Biotin anti-mouse F4/80 (clone BM8) | BioLegend | Cat. #123106; RRID: AB_893501 |
| FITC anti-mouse F4/80 (clone BM8) | BioLegend | Cat. #123108; RRID: AB_893502 |
| Biotin anti-mouse FcεRIα (clone MAR-1) | BioLegend | Cat. #134304; RRID: AB_1626106 |
| eFluor 450 anti-mouse Granzyme A (clone GzA-3G8.5) | eBioscience | Cat. #48-5831-80; RRID: AB_2574078 |
| PerCP-eFluor 710 anti-mouse Granzyme A (clone GzA-3G8.5) | eBioscience | Cat. #46-5831-82; RRID: AB_2573775 |
| Alexa Fluor 700 anti-human Granzyme B (clone GB11) | BD Biosciences | Cat. #560213; RRID: AB_1645453 |
| Pacific Blue anti-human/mouse Granzyme B (clone GB11) | BioLegend | Cat. #515408; RRID: AB_2562196 |
| BV421 anti-mouse IFN-γ (clone XMG1.2) | BioLegend | Cat. #505829; RRID: AB_2563105 |
| APC anti-mouse IFN-γ (clone XMG1.2) | BioLegend | Cat. #505810; RRID: AB_315404 |
| PE-Cyanine7 CD218a (IL-18Ra) (clone P3TUNYA) | Thermo Fisher Scientific | Cat. #25-5183-82; RRID: AB_2762705 |
| Alexa Fluor 647 anti-mouse CD85k (Ilt3, gp49 Receptor) | BioLegend | Cat. #144906; RRID: AB_2562044 |
| Purified anti-mouse/human Keratin 14 (clone Poly9060) | BioLegend | Cat. #906004; RRID: AB_2616962 |
| BUV661 anti-mouse KLRG1 (clone 2F1) | BD Biosciences | Cat. #741586; RRID: AB_2870999 |
| BV711 anti-mouse KLRG1 (clone 2F1) | BD Biosciences | Cat. #564014; RRID: AB_2738542 |
| BUV615 Anti-Mouse Ly-49D (clone 4E5) | BD Biosciences | Cat. #751640; RRID: AB_2875633 |
| Biotin anti-mouse Ly-6G (clone 1A8) | BioLegend | Cat. #127604; RRID: AB_1186108 |
| FITC anti-mouse Ly-6G (clone 1A8) | BioLegend | Cat. # 127606; RRID: AB_12364946 |
| Alexa Fluor 700 anti-mouse I-A/I-E (MHCII) (clone M5/114.15.2) | BioLegend | Cat. #107621; RRID: AB_493727 |
| APC anti-mouse NK-1.1 (clone PK136) | BioLegend | Cat. #108710; RRID: AB_313397 |
| BV650 anti-mouse NK-1.1 (clone PK136) | BioLegend | Cat. #108736; RRID: AB_2563159 |
| PE/Cyanine7 anti-mouse NK-1.1 (clone PK136) | BioLegend | Cat. #108714; RRID: AB_389364 |
| PerCP-eFluor 710 anti-mouse CD335 (NKp46) (clone 29A1.4) | Thermo Fisher Scientific | Cat. #46-3351-82; RRID: AB_1834441 |
| Unconjugated anti-Mouse NKp46/NCR1 (polyclonal) | R&D Systems | Cat. #AF2225; RRID: AB_355192 |
| PE/Dazzle 594 anti-mouse Perforin (clone S16009A) | BioLegend | Cat. #154316; RRID: AB_2922482 |
| PE-CF594 Mouse Anti-Mouse RORγt (cone Q31-378) | BD Biosciences | Cat. #562684; RRID: AB_2651150 |
| BV605 anti-mouse Ly-6A/E (Sca-1) | BioLegend | Cat. #108134; RRID: AB_2650926 |
| PE Mouse Anti-Mouse TCF-7/TCF-1 (clone S33-966) | BD Biosciences | Cat. #564217; RRID: AB_2687845 |
| TCF1/TCF7 (C63D9) Rabbit mAb (Alexa Fluor® 488 Conjugate) | Cell Signaling Technology | Cat. #6444S; RRID: AB_2797627 |
| Biotin anti-mouse TCR β chain (clone H57-597) | BioLegend | Cat. #109204; RRID: AB_313427 |
| FITC anti-mouse TCR β chain (clone H57-597) | BioLegend | Cat. #109206; RRID: AB_313429 |
| Biotin anti-mouse TCR gamma/delta (clone eBioGL3) | Thermo Fisher Scientific | Cat. # 13-5711-85; RRID: AB_466669 |
| FITC anti-mouse TCR γ/δ (clone GL3) | BioLegend | Cat. #118106; RRID: AB_313830 |
| Biotin anti-mouse TER-119/Erythroid Cells (clone TER-119) | BioLegend | Cat. #116204; RRID: AB_313705 |
| DyLight™ 405 AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L) (polyclonal) | Jackson Immuno Research | Cat. # 703-475-155; RRID: AB_2340373 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 568 | Thermo Fisher Scientific | Cat. #A-11057; RRID: AB_2534104 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 555 | Thermo Fisher Scientific | Cat. #A32816; RRID: AB_2762839 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 647 | Thermo Fisher Scientific | Cat. #A32849; RRID: AB_2762840 |
| TotalSeq™-A0197 anti-mouse CD69 | BioLegend | Cat. #104546; RRID: AB_2750539 |
| TotalSeq™-A0302 anti-mouse Hashtag 2 | BioLegend | Cat. #155803; RRID: AB_2750033 |
| TotalSeq™-A0303 anti-mouse Hashtag 3 | BioLegend | Cat. #155805; RRID: AB_2750034 |
| TotalSeq™-A0305 anti-mouse Hashtag 5 | BioLegend | Cat. #155809; RRID: AB_2750036 |
| Bacterial and virus strains | ||
| Staphylococcus aureus strain HG001 | 77 | N/A |
| Vaccinia virus | 24 | N/A |
| Vaccinia virus m157 | 24 | N/A |
| Non-replicating Vaccinia Virus MVA (Modified Vacciniavirus Ankara) | 78 | This lab. |
| Chemicals, peptides, and recombinant proteins | ||
| Brefeldin A | Sigma Aldrich | Cat. #B7651-5MG |
| CaCl2 | Sigma Aldrich | Cat. #C7902 |
| Collagenase D | Roche | Cat. #11088882001 |
| Corn oil | Sigma Aldrich | Cat. #C8267 |
| Dispase | Corning | Cat. #354235 |
| DMEM, high glucose | Thermo Fisher Scientific | Cat. #41965062 |
| DNase I | Sigma Aldrich | Cat. #DN25 |
| DPBS | Sigma Aldrich | Cat. #D8537 |
| EDTA | Carl Roth | Cat. #8043.2 |
| Fetal Bovine Serum | Sigma Aldrich | Cat. #F7524 |
| Fixable Viability Dye eFluor® 780 | Thermo Fisher Scientific | Cat. #65-0865-18 |
| Fixable Viability Kit Zombie NIR™ | BioLegend | Cat. #423106 |
| Fluoromount-G | Thermo Fisher Scientific | Cat. #00-4958-02 |
| Gelatin from cold water fish skin (GCWFS) | Sigma Aldrich | Cat. #G7041 |
| Hanks’ Balanced Salt solution (HBSS) | Sigma Aldrich | Cat. #H8264 |
| HEPES (pH 7.2–7.5) | Gibco | Cat. #15630056 |
| Horse Serum | Sigma Aldrich | Cat. #H0146 |
| Ketamine | Pfizer | N/A |
| Liberase TL | Roche | Cat. #05401020001 |
| L-Lysine (pH 7.4) | Sigma Aldrich | Cat. #L5501 |
| NaIO4 | Sigma Aldrich | Cat. #769517 |
| Normal mouse serum | Thermo Fisher Scientific | Cat. #10410 |
| OCT freezing media | Sakura Finetek | Cat. #12351753 |
| Paraformaldehyde | Carl Roth | Cat. #0335.3 |
| Percoll | Sigma Aldrich | Cat. #P1644 |
| Streptavidin, BV605™ | BioLegend | Cat. #405229 |
| Streptavidin, V500 | BD Biosciences | Cat. #561419; RRID: AB_ 10611863 |
| Sucrose | Sigma Aldrich | Cat. #1.07687 |
| Superfrost Plus object slides | VWR | Cat. #631-0108 |
| Tamoxifen | Sigma Aldrich | Cat. #T5648 |
| TRIS Hydrochloride | Carl Roth | Cat. #9090.2 |
| Triton-X 100 | Carl Roth | Cat. #3051.2 |
| Xylazine | CP Pharma | N/A |
| Critical commercial assays | ||
| Cytofix / Cytoperm Kit | BD Biosciences | Cat. #554714 |
| Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat. #00-5523-00 |
| High Output Kit v2.5 | Illumina | N/A |
| High Sensitivity DNA kit | Agilent | N/A |
| Lysis buffer and RNAse inhibitors | Takara | N/A |
| Single cell 3’ reagent kit v3 | 10x Genomics | N/A |
| SMART-Seq® v4 Ultra® Low Input RNA Kit | Takara | N/A |
| Deposited data | ||
| Bulk RNA sequencing Raw and analyzed data | This paper | GEO:GSE227801 |
| Single cell RNA sequencing Raw and analyzed data (memory) | This paper | GEO:GSE226756 |
| Single cell RNA sequencing Raw and analyzed data (rechallenge) | This paper | GEO:GSE242313 |
| Single cell RNA sequencing Raw data | Alkon et al.32 | GEO:GSE180885 |
| Single cell RNA sequencing Raw data | Rückert et al.16 | GEO:GSE197037 |
| Single cell RNA sequencing Raw data | Lopes et al.30 | GEO:GSE189807 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson laboratories | RRID:IMSR_JAX:000664 |
| Mouse: Ly49HKO (B6.BXD8-Klra8Cmv1-del/WumJ) |
Fang et al.79 | RRID:IMSR_JAX:008633 |
| Mouse: RosaLSLtdTomato B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J |
Camarasa et al.80 | RRID:IMSR_JAX:007914 |
| Mouse: Tcf7 flox (B6(Cg)-Tcf7tm1Hhx/J) |
Gang et al.81 | RRID:IMSR_JAX:030909 |
| Mouse: EomesCreERT2 (Eomestm1.1(cre/ERT2)Sjar) |
Jeevan-Raj et al.82 | N/A |
| Mouse: FlpO | Wang et al.83 | N/A |
| Mouse: HobitCre (Zfp683-tdTomato-P2A-cre-P2A-DTR) |
Zhu et al.84 | N/A |
| Mouse: IFN-γeGFP-Cre-ERT2 | Generated and provided by R. Flavell | N/A |
| Mouse: SellCreERT2 (Sell CreERT2) |
Madisen et al.85 | N/A |
| Mouse: Vav-H2B-Dendra2 (VHD) | Yang et al.86 | N/A |
| Mouse: Ncr1iCre (B6.Cg-Ncr1tm1.1(icre)Viv/Orl) |
Kranz et al.87 | MGI:5308410 |
| Mouse: Prdm1 flox (C57BL/6-Prdm1tm1Nutt/J) |
Pimeisl et al.88 | N/A |
| Mouse: Tgfbr2 flox (B6;129-Tgfbr2tm1Karl/J) |
Behr et al.89 | RRID:IMSR_JAX:012603 |
| Mouse: Eomes flox (B6.129S1(Cg)-Eomestm1.1Bflu/J) |
Merkenschlager et al.90 | RRID:IMSR_JAX:017293 |
| Software and algorithms | ||
| Attune NxT Software version v3.1.2 | Thermo Fisher Scientific | RRID: SCR_019590 |
| bcl2fastq2 v2.20.0.422 | Illumina | N/A |
| BD FACSDiva Software | BD Biosciences | RRID: SCR_001456 |
| Cell Ranger software v3.0.2 | 10x Genomics | https://doi.org/10.1038/ncomms14049 |
| DESeq2 version 1.36.0 | Bioconductor | https://doi.org/10.1186/s13059-014-0550-8 |
| Flowjo v10 | BD Biosciences | RRID: SCR_008520 |
| GraphPad Prism v9 | GraphPad | RRID: SCR_002798 |
| Imaris v8.3 | Bitplane | RRID: SCR_007370 |
| Leica Las X | Leica | RRID: SCR_013673 |
| pHeatmap package | CRAN | https://CRAN.R-project.org/package=pheatmap |
| Seurat v4 | Stuart et al.99 | RRID: SCR_007322 |
| Skewer v.0.2.2 | https://doi.org/10.1186/1471-2105-15-182 | |
| SpectroFlo version 3.0.3 | Cytek | https://cytekbio.com/pages/spectro-flo |
| STAR v.2.7 | Github | https://doi.org/10.1093/bioinformatics/bts635 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Georg Gasteiger (georg.gasteiger@uni-wuerzburg.de).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Bulk and single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental models
Animals
B6 mice (C57BL/6J), Eomesfl/fl mice,84 RosaLSLtdTomato mice85 and Tcf7fl/fl mice86 were originally purchased from The Jackson Laboratory. Tcf7fl/fl mice were crossed with mice that express the Flp recombinase in the germ line87 to delete the neomycin cassette. Fatemap mouse lines were generated by crossing RosatdTomato mice with EomesCre-ERT2 mice,88 HobitCre mice,89 SellCre-ERT2 mice,90 IFN-γeGFP-Cre-ERT2 mice (generated and kindly provided by R.A. Flavell). Ncr1Cre mice91 were generously provided by E. Vivier and crossed to Eomesfl/fl mice, Prdm1fl/fl mice,92 generously provided by S. Nutt, Tgfbr2fl/fl mice,93 or Tcf7fl/fl mice. Dendra2-VHD mice,94 here referred to as Dendra2 mice, were used for photoconversion. Klra8-/- mice95 were generously provided by S. Vidal. All mice were bred and housed under specific-pathogen-free or germ-free conditions at the animal facility of the University Würzburg (Institute of Systems Immunology, and ZEMM). Germ-free mice were bred and maintained in flexible-film isolators, and germ-free status was routinely monitored. C57BL/6J wildlings were created through inverse germ-free rederivation as described.96The wildling mouse colony was housed and animals were bred at the Medical Center – University of Freiburg, Germany. Mice were housed under a 12:12 light:dark cycle in microisolator cages with autoclaved rodent chow, autoclaved tap water ad libitum as well as autoclaved nesting and bedding material. Experiments were performed in accordance with institutional and national guidelines. For all experiments, male and female mice at 7–21 weeks of age were used.
Bacterial and viral strains
Staphylococcus aureus (S.aureus) HG001, Vaccinia Virus (VACV-WT), m157-expressing VACV (VACV) and Modified Vaccinia Ankara (MVA) were previously described.24,97,98 For heat inactivation, HG001 was incubated for 60 min at 65°C and inactivation was validated by absence of colony formation when culturing on blood-agar-plates.
Method details
Skin infections
Intraepidermal infection of ear pinna with VACV (2x106 PFU in PBS), S.aureus (1x108 CFU in PBS) or MVA (1x107 PFU in PBS) was performed as previously described.23 For rechallenges, VACV-immune or naïve ears were infected intradermally with VACV-WT (2x106 PFU in PBS) and mice were sacrificed and analyzed after 12 hours.
Tissue lymphocyte isolation
Mice were euthanized, ears collected and separated into 2 halves. Each half was digested in DMEM with 10 mM Hepes, 20 μg/ml DNase I, 0.245 mg/ml Liberase TL at 37 °C and for 90 min. Salivary glands were collected, cut into small pieces, and digested in DMEM with 10 mM Hepes, 2% FCS, 5 mM CaCl2, 20 μg/ml DNase I, and 1 mg/ml Collagenase D at 37 °C and 100 r.p.m. for 40 min. Suspensions were passed through 100 μm filters and lymphocytes enriched by 40%/80% Percoll gradient (860g, 20 min, 21 °C, no brakes). The interface was washed with PBS. Spleens were collected, cut into small pieces, and digested in DMEM with 10 mM Hepes, 20 μg/ml DNase I, and 0.5 mg/ml Collagenase D at 37 °C and for 30 min and red blood cells were lysed. Uteri were collected, cut into small pieces, and underwent three rounds of digestions in HBSS with 2% FCS, 66.6 μg/ml DNase I, 0.5 mg/ml Collagenase D and 0.5 U/ml Dispase at 37 °C and 150 r.p.m. for 20 min each round. All single cell suspensions were passed through 100 μm filters and used for flow cytometric analysis or sorting.
Flow cytometry and cell sorting
Dead cells were excluded by fixable viability dye eF780 or Zombie NIR™ and nonspecific binding blocked by anti-CD16/CD32 blocking antibodies. Fluorochrome-conjugated monoclonal antibodies were purchased from commercial vendors. For cell sorting of spleen, skin and uterine Group 1 ILCs, single-cell suspensions were stained for CD45, NK1.1, NKp46, CXCR6 and CD49a, and biotinylated CD3ε, CD5, CD19, F4/80, TCRβ, TCRγδ and Ter119 lineage markers, and subsequently with streptavidin. For bulk RNA-sequencing, skin and spleen NK were sorted from VACV-infected mice (d35 pi) as live CD45+Lin−NK1.1+NKp46+CD49a−CXCR6− cells, and uterine trNK were sorted from naïve mice as live CD45+Lin−NK1.1+CD49a+CXCR6− cells. For sc RNA-sequencing, group 1 ILCs from skin, spleen and SG were sorted from VACV-infected mice (d25 pi) as live CD45+Lin−NK1.1+NKp46+. Cell sorting was performed on a BD FACSAria III.
For flow cytometry analysis, fixable viability dye eF780- or Zombie NIR-positive dead cells were excluded, and extracellular staining was performed for 20-40 min at 4°C. Lineage-positive cells were stained with a cocktail of biotin-labeled antibodies against CD3ε, CD5, CD19, F4/80, FcεR1α, Ly6G, TCRβ, TCRγδ, and Ter119, followed by staining with streptavidin. Intracellular staining was performed using the FoxP3/Transcription Factor Staining Buffer set. For IFN-γ staining, tissues were digested with the addition of Brefeldin A (1 μg/ml). Cells were acquired on a Thermo Fisher Attune™ NxT cytometer using Attune™ NxT software or Cytek Aurora cytometer using SpectroFlo software. ILC1s were gated as live CD45+Lin−CD3−NK1.1+Eomes−CD49a+ or CD45+Lin−CD3−NK1.1+Eomes−CXR6+. NK cells were gated as live CD45+Lin−CD3−NK1.1+Eomes+.
In vivo labeling of vascular leukocytes for flow cytometry
1.5 - 2 μg of Alexa Fluor 700- or APC-labeled anti-CD45.2 was injected in a total volume 200 μL of sterile PBS intravenously via the tail vein 5 min prior to sacrificing mice.
Inducible fate map labeling by tamoxifen
For inducible fate mapping, mice were treated intraperitoneally with 80 mg/kg (bodyweight) tamoxifen in corn oil two or three times and analyzed at indicated times post infection.
Skin photoconversion
Dendra2-VHD mice were anaesthetized and ear skin was illuminated for 1 min per side at a low intensity light from a BlueWave 75 light curing system equipped with a 390/40 band pass filter with an intensity of 120 mW/cm2. The output light was not collimated. The distance between the tip of the light source and the tissue was 1 cm and the illumination area at the target tissue was a circle with a diameter of approximately 1 cm. During illumination, tissue surrounding the ear was covered with a thick tissue to avoid unwanted photoactivation.
Confocal microscopy
Skin, SG and FRT were collected and fixed in PLP buffer (0.05 M phosphate buffer containing 0.1 M L-lysine, 2 mg/ml NaIO4 and 10 mg/ml paraformaldehyde) for 8 h or overnight at 4°C. Samples were dehydrated with 30% sucrose at 37°C for 8 h or overnight 4°C. Organs were embedded in OCT freezing media and stored at −80°C. Serial 14 μm (FRT), 20 μm (skin), 30 μm (SG) sections were cut on a Leica CM3050S cryostat and adhered to Superfrost Plus gold object slides. After rehydration with PBS for 5−10 min at 21°C, sections were permeabilized and blocked with 0.1 M Tris containing 1% FCS, 1% GCWFS, 0.3% Triton-X 100, and 4% normal mouse serum, 5% horse serum and anti-CD16/32 for 40 min at 21°C. Staining was performed in blocking buffer (without anti-CD16/32) overnight (primary antibodies) or for 4 h (secondary antibodies), both at 4°C. Sections were washed with PBS and mounted with Fluoromount-G. Acquisition was performed on a Leica SP8 confocal microscope with LasX software.
scRNA-seq, cell hashing and CITE-seq
ILC1s and NK cells were sorted as individual, hashtagged populations before pooling into one reaction. Before sorting, cells were labeled with oligonucleotide-tagged antibodies for cellular indexing of transcriptomes and epitopes by sequencing (CITE-Seq). Immediately after sorting, Library was performed according to the manufacturer’s instructions. Briefly, cells were resuspended in the master mix and loaded together with partitioning oil and gel beads into the chip to generate the gel bead-in-emulsion (GEM). The poly-A RNA from the cell lysate contained in every single GEM was retrotranscribed to cDNA, which contains an Ilumina R1 primer sequence, Unique Molecular Identifier (UMI) and the 10x Barcode. The pooled barcoded cDNA was then cleaned up with Silane DynaBeads, amplified by PCR and the appropriated sized fragments were selected with SPRIselect reagent for subsequent library construction. During the library construction Ilumina R2 primer sequence, paired-end constructs with P5 and P7 sequences and a sample index were added. It was then sequenced on a NovaSeq 6000 S2 flow cell with 100 cycles (28-10-10-90).
scRNA-seq data analysis
Data for the cDNA library fraction were demultiplexed using Cell Ranger software and aligned to mouse mm10 reference genome. The resulting gene expression matrix were analyzed using Seurat standard workflow. After log transformation with the command LogNormalize and scaling with ScaleData, basic QC has been performed. Viable cells have been selected by filtering for cells with UMI count superior at 1,000 and a percentage of UMI mapped to mitochondrial genes inferior to 8. Cells were then assigned to appropriate samples by demultiplexing the hashtags antibodies. To demultiplex hashtags, for each cell HTODemux function in Seurat package was used as described. Cross-sample doublet cell detection was performed based on hashtag signal. Only cells that were classified as ‘singlet’ were retained and used for downstream analysis. Variable genes were identified by the function VariableFeatures with an n value of 2,000. Dimensions of the data were reduced first using a principal component analysis (PCA) on variable genes, and then a uniform manifold approximation and projection (UMAP) on the 15 first PC dimensions. Cells were also assigned to clusters with the functions FindNeighbors on the same 15 PC dimensions and then FindClusters with a resolution of 0.5. Cluster specific gene signatures have been analyzed with the function FindAllMarkers from Seurat with arguments: only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25.
Analysis of human scRNA-seq data
Data from non-lesional skin was generated by Alkon et al. (GSE180885) and human CMV negative blood NK were generated by Rückert et al. (GSE197037), were downloaded and analyzed using Seurat standard workflow (see above).
Score calculation with Seurat
Scores were calculated using the Seurat function AddModuleScore with default settings. Unbiased core signatures of tissue resident memory T cells and circulating T cells were derived from the published data of Milner et al.27 (see gene list in Table S2). NK and ILC1 scores were calculated based on gene list generated by comparing the first 100 DEG based on log2 fold change between liver NK and liver ILC1 from published data (GSE189807, 30) (see gene list in Table S2). Scores of human CD56bright-like and CD56dim-like NK cells were generated by comparing the two skin NK cell populations (GSE180885, 32), filtering out mitochondrial and ribosomal genes and selecting the first 100 DEG based on log2 fold change (see Table S2). Glycolysis score was calculated based on the matching hallmark gene lists from GSEA (from https://www.gsea-msigdb.org).
Bulk mRNA sequencing
300 cells per well were sorted in 96-well plates, and then lysis buffer and RNAse inhibitors were added. cDNA libraries suitable for sequencing were prepared with SMART-Seq® v4 Ultra® Low Input RNA Kit according to manufacturer’s instructions (1/4 volume) and 22 PCR cycles. Libraries were quantified by QubitTM 3.0 Fluometer (ThermoFisher) and quality was checked using 2100 Bioanalyzer with High Sensitivity DNA kit. 0.4 ng of each library was subjected to a tagmentation-based protocol (Nextera XT, Illumina) using a quarter of the recommended reagent volumes. Libraries were quantified again by QubitTM 3.0 Fluometer and quality was checked using 2100 Bioanalyzer with High Sensitivity DNA kit before pooling. Sequencing of pooled libraries, spiked with 1% PhiX control library, was performed at 15 million reads/sample in single-end mode with 75 nt read length on the NextSeq 500 platform (Illumina) with 1 High Output Kit v2.5. Demultiplexed FASTQ files were generated with bcl2fastq2 v2.20.0.422 (Illumina).
Analysis of bulk mRNA sequencing data
The obtained FASTQ files were controlled for quality with FastQC, trimmed for adaptor sequences with Skewer and all files were aligned using STAR to the GRCm38.98 reference genome using standard settings. The aligned data were counted using the featureCounts function of the Rsubread package. Differential expression analysis based on the raw count-matrix was performed using DESeq2. Significant DEG were defined as having an adjusted P value <0.01 and log2FoldChange >1.5 or log2 FoldChange <−1.5. All significant DEG were visualized using the pHeatmap package in R.
Quantification and statistical analysis
FACS data were analyzed with FlowJo software. Confocal images were analyzed with Imaris software. Statistical analysis was performed using Prism software. The statistical significance of experimental data was determined using paired or unpaired t-test or one-way ANOVA with Tukey’s correction. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, n.s. not significant. Symbols in bar graphs represent analysis of inficated tissues of individual mice, except for the analysis of skin, where symbols represent individual ears. Error bars display means + SD.
Acknowledgments
We would like to thank C. Romagnani, T. Rückert, and C. Talavera Lopez for critical discussions and S. Klingler, P. Arampatzi, the Single Cell Center Würzburg, and the Core Units SysMed and IZKF FACS sorting for expert technical assistance and services. We thank K.P.J.M.v. Gisbergen, S.L. Nutt, J. Merkenschlager, M. Nussenzweig, S. Vidal, and E. Vivier for providing mice. This work was supported by grants through the European Research Council to G.G. (759176-TissueLymphoContexts); grants by the DFG German Research Foundation to G.G., W.K., and M.G.d.A (SFB 1525, SFB1583, TRR338, and SPP1937); grants to S.P.R. (Emmy-Noether-Program RO6247/1-1, SFB1160 RO6247/1-1, and TRR359 RO6247/1-1); and grants to S.J.A. (Heisenberg-Program AR732/3-1, CRC1140-B07, CRC992-A08, and CIBSS–EXC-2189). G.G. and W.K. are supported by the Max Planck Society. This work was also supported by the Bavarian Ministry of Economic Affairs, Regional Development, and Energy within the project “Single cell analysis in personalized medicine” at the Helmholtz-Institute for RNA-based Infection Research.
Author contributions
G.G. conceptualized the study and wrote the first draft. T.T. and C.F. planned and performed experiments, analyzed data, and wrote the manuscript. R.D.-L. analyzed RNA sequencing data. T.O., V.V.S., F.I., and S.R. performed experiments. M.U., S.P.R., S.J.A., N.G., R.A.F., and S.B. provided critical reagents. G.G., T.T., and C.F. supervised experiments. G.G. and W.K. provided intellectual input and conceptual advice. G.G., M.G.d.A., and W.K. provided research funds. All authors edited and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2023.11.018.
Supplemental information
References
- 1.Mujal A.M., Delconte R.B., Sun J.C. Natural killer cells: from innate to adaptive features. Annu. Rev. Immunol. 2021;39:417–447. doi: 10.1146/annurev-immunol-101819-074948. [DOI] [PubMed] [Google Scholar]
- 2.Zimmer C.L., Cornillet M., Solà-Riera C., Cheung K.W., Ivarsson M.A., Lim M.Q., Marquardt N., Leo Y.S., Lye D.C., Klingström J., et al. NK cells are activated and primed for skin-homing during acute dengue virus infection in humans. Nat. Commun. 2019;10:3897. doi: 10.1038/s41467-019-11878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortez V.S., Cervantes-Barragan L., Robinette M.L., Bando J.K., Wang Y., Geiger T.L., Gilfillan S., Fuchs A., Vivier E., Sun J.C., et al. Transforming growth factor-β signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity. 2016;44:1127–1139. doi: 10.1016/j.immuni.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nixon B.G., Chou C., Krishna C., Dadi S., Michel A.O., Cornish A.E., Kansler E.R., Do M.H., Wang X., Capistrano K.J., et al. Cytotoxic granzyme C-expressing ILC1s contribute to antitumor immunity and neonatal autoimmunity. Sci. Immunol. 2022;7:eabi8642. doi: 10.1126/sciimmunol.abi8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dogra P., Rancan C., Ma W., Toth M., Senda T., Carpenter D.J., Kubota M., Matsumoto R., Thapa P., Szabo P.A., et al. Tissue determinants of human NK cell development, function, and residence. Cell. 2020;180:749–763.e13. doi: 10.1016/j.cell.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray J.I., Farber D.L. Tissue-resident immune cells in humans. Annu. Rev. Immunol. 2022;40:195–220. doi: 10.1146/annurev-immunol-093019-112809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sojka D.K., Yang L., Yokoyama W.M. Uterine natural killer cells. Front. Immunol. 2019;10:960. doi: 10.3389/fimmu.2019.00960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich C., Taggenbrock R.L.R.E., Doucet-Ladevèze R., Golda G., Moenius R., Arampatzi P., Kragten N.A.M., Kreymborg K., Gomez de Agüero M., Kastenmüller W., et al. Effector differentiation downstream of lineage commitment in ILC1s is driven by Hobit across tissues. Nat. Immunol. 2021;22:1256–1267. doi: 10.1038/s41590-021-01013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sojka D.K., Plougastel-Douglas B., Yang L., Pak-Wittel M.A., Artyomov M.N., Ivanova Y., Zhong C., Chase J.M., Rothman P.B., Yu J., et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife. 2014;3 doi: 10.7554/eLife.01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marquardt N., Kekäläinen E., Chen P., Lourda M., Wilson J.N., Scharenberg M., Bergman P., Al-Ameri M., Hård J., Mold J.E., et al. Unique transcriptional and protein-expression signature in human lung tissue-resident NK cells. Nat. Commun. 2019;10:3841. doi: 10.1038/s41467-019-11632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Björkström N.K., Strunz B., Ljunggren H.G. Natural killer cells in antiviral immunity. Nat. Rev. Immunol. 2022;22:112–123. doi: 10.1038/s41577-021-00558-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeis P., Lian M., Fan X., Herman J.S., Hernandez D.C., Gentek R., Elias S., Symowski C., Knöpper K., Peltokangas N., et al. In situ maturation and tissue adaptation of type 2 innate lymphoid cell progenitors. Immunity. 2020;53:775–792.e9. doi: 10.1016/j.immuni.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghaedi M., Shen Z.Y., Orangi M., Martinez-Gonzalez I., Wei L., Lu X., Das A., Heravi-Moussavi A., Marra M.A., Bhandoola A., et al. Single-cell analysis of RORα tracer mouse lung reveals ILC progenitors and effector ILC2 subsets. J. Exp. Med. 2020;217 doi: 10.1084/jem.20182293. jem.20182293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim A.I., Li Y., Lopez-Lastra S., Stadhouders R., Paul F., Casrouge A., Serafini N., Puel A., Bustamante J., Surace L., et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. 2017;168:1086–1100.e10. doi: 10.1016/j.cell.2017.02.021. [DOI] [PubMed] [Google Scholar]
- 15.Sun J.C., Beilke J.N., Lanier L.L. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rückert T., Lareau C.A., Mashreghi M.F., Ludwig L.S., Romagnani C. Clonal expansion and epigenetic inheritance of long-lasting NK cell memory. Nat. Immunol. 2022;23:1551–1563. doi: 10.1038/s41590-022-01327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammer Q., Rückert T., Borst E.M., Dunst J., Haubner A., Durek P., Heinrich F., Gasparoni G., Babic M., Tomic A., et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018;19:453–463. doi: 10.1038/s41590-018-0082-6. [DOI] [PubMed] [Google Scholar]
- 18.Lau C.M., Adams N.M., Geary C.D., Weizman O.E., Rapp M., Pritykin Y., Leslie C.S., Sun J.C. Epigenetic control of innate and adaptive immune memory. Nat. Immunol. 2018;19:963–972. doi: 10.1038/s41590-018-0176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammer Q., Rückert T., Romagnani C. Natural killer cell specificity for viral infections. Nat. Immunol. 2018;19:800–808. doi: 10.1038/s41590-018-0163-6. [DOI] [PubMed] [Google Scholar]
- 20.Schuster I.S., Sng X.Y.X., Lau C.M., Powell D.R., Weizman O.E., Fleming P., Neate G.E.G., Voigt V., Sheppard S., Maraskovsky A.I., et al. Infection induces tissue-resident memory NK cells that safeguard tissue health. Immunity. 2023;56:531–546.e6. doi: 10.1016/j.immuni.2023.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brownlie D., Scharenberg M., Mold J.E., Hård J., Kekäläinen E., Buggert M., Nguyen S., Wilson J.N., Al-Ameri M., Ljunggren H.G., et al. Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016580118. e2016580118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bielecki P., Riesenfeld S.J., Hütter J.C., Torlai Triglia E., Kowalczyk M.S., Ricardo-Gonzalez R.R., Lian M., Amezcua Vesely M.C., Kroehling L., Xu H., et al. Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature. 2021;592:128–132. doi: 10.1038/s41586-021-03188-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muschaweckh A., Buchholz V.R., Fellenzer A., Hessel C., König P.A., Tao S., Tao R., Heikenwälder M., Busch D.H., Korn T., et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J. Exp. Med. 2016;213:3075–3086. doi: 10.1084/jem.20160888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Firth M.A., Madera S., Beaulieu A.M., Gasteiger G., Castillo E.F., Schluns K.S., Kubo M., Rothman P.B., Vivier E., Sun J.C. Nfil3-independent lineage maintenance and antiviral response of natural killer cells. J. Exp. Med. 2013;210:2981–2990. doi: 10.1084/jem.20130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackay L.K., Rahimpour A., Ma J.Z., Collins N., Stock A.T., Hafon M.L., Vega-Ramos J., Lauzurica P., Mueller S.N., Stefanovic T., et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 26.Li C., Zhu B., Son Y.M., Wang Z., Jiang L., Xiang M., Ye Z., Beckermann K.E., Wu Y., Jenkins J.W., et al. The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity. 2020;52:201–202. doi: 10.1016/j.immuni.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milner J.J., Toma C., He Z., Kurd N.S., Nguyen Q.P., McDonald B., Quezada L., Widjaja C.E., Witherden D.A., Crowl J.T., et al. Heterogenous populations of tissue-resident CD8+ T cells are generated in response to infection and malignancy. Immunity. 2020;52:808–824.e7. doi: 10.1016/j.immuni.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar B.V., Ma W., Miron M., Granot T., Guyer R.S., Carpenter D.J., Senda T., Sun X., Ho S.H., Lerner H., et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017;20:2921–2934. doi: 10.1016/j.celrep.2017.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McFarland A.P., Yalin A., Wang S.Y., Cortez V.S., Landsberger T., Sudan R., Peng V., Miller H.L., Ricci B., David E., et al. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity. 2021;54:1320–1337.e4. doi: 10.1016/j.immuni.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopes N., Galluso J., Escaliere B., Carpentier S., Kerdiles Y.M., Vivier E. Tissue-specific transcriptional profiles and heterogeneity of natural killer cells and group 1 innate lymphoid cells. Cell Rep. Med. 2022;3:100812. doi: 10.1016/j.xcrm.2022.100812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiow L.R., Rosen D.B., Brdicková N., Xu Y., An J., Lanier L.L., Cyster J.G., Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 32.Alkon N., Bauer W.M., Krausgruber T., Goh I., Griss J., Nguyen V., Reininger B., Bangert C., Staud C., Brunner P.M., et al. Single-cell analysis reveals innate lymphoid cell lineage infidelity in atopic dermatitis. J. Allergy Clin. Immunol. 2022;149:624–639. doi: 10.1016/j.jaci.2021.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crinier A., Milpied P., Escaliere B., Piperoglou C., Galluso J., Balsamo A., Spinelli L., Cervera-Marzal I., Ebbo M., Girard-Madoux M., et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. 2018;49:971–986.e5. doi: 10.1016/j.immuni.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins P.L., Cella M., Porter S.I., Li S., Gurewitz G.L., Hong H.S., Johnson R.P., Oltz E.M., Colonna M. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. 2019;176:348–360.e12. doi: 10.1016/j.cell.2018.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang F., Li J., Qi L., Liu D., Bo Y., Qin S., Miao Y., Yu K., Hou W., Li J., et al. A pan-cancer single-cell panorama of human natural killer cells. Cell. 2023;186:4235–4251.e20. doi: 10.1016/j.cell.2023.07.034. [DOI] [PubMed] [Google Scholar]
- 36.Gordon S.M., Chaix J., Rupp L.J., Wu J., Madera S., Sun J.C., Lindsten T., Reiner S.L. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67. doi: 10.1016/j.immuni.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daussy C., Faure F., Mayol K., Viel S., Gasteiger G., Charrier E., Bienvenu J., Henry T., Debien E., Hasan U.A., et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 2014;211:563–577. doi: 10.1084/jem.20131560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nüssing S., Miosge L.A., Lee K., Olshansky M., Barugahare A., Roots C.M., Sontani Y., Day E.B., Koutsakos M., Kedzierska K., et al. SATB1 ensures appropriate transcriptional programs within naive CD8+ T cells. Immunol. Cell Biol. 2022;100:636–652. doi: 10.1111/imcb.12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J., Hardy K., Olshansky M., Barugahare A., Gearing L.J., Prier J.E., Sng X.Y.X., Nguyen M.L.T., Piovesan D., Russ B.E., et al. KDM6B-dependent chromatin remodeling underpins effective virus-specific CD8+ T cell differentiation. Cell Rep. 2021;34:108839. doi: 10.1016/j.celrep.2021.108839. [DOI] [PubMed] [Google Scholar]
- 40.Badrani J.H., Strohm A.N., Lacasa L., Civello B., Cavagnero K., Haung Y.A., Amadeo M., Naji L.H., Lund S.J., Leng A., et al. RNA-binding protein RBM3 intrinsically suppresses lung innate lymphoid cell activation and inflammation partially through CysLT1R. Nat. Commun. 2022;13:4435. doi: 10.1038/s41467-022-32176-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai L., Vienne M., Tang L., Kerdiles Y., Etiennot M., Escaliere B., Galluso J., Wei H., Sun R., Vivier E., et al. Liver type 1 innate lymphoid cells develop locally via an interferon-gamma-dependent loop. Science. 2021;371:eaba4177. doi: 10.1126/science.aba4177. [DOI] [PubMed] [Google Scholar]
- 42.Nakao A., Afrakhte M., Morén A., Nakayama T., Christian J.L., Heuchel R., Itoh S., Kawabata M., Heldin N.E., Heldin C.H., et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 43.Yomogida K., Bigley T.M., Trsan T., Gilfillan S., Cella M., Yokoyama W.M., Egawa T., Colonna M. Hobit confers tissue-dependent programs to type 1 innate lymphoid cells. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2117965118. e2117965118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mackay L.K., Minnich M., Kragten N.A., Liao Y., Nota B., Seillet C., Zaid A., Man K., Preston S., Freestone D., et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 45.Li Z.Y., Morman R.E., Hegermiller E., Sun M., Bartom E.T., Maienschein-Cline M., Sigvardsson M., Kee B.L. The transcriptional repressor ID2 supports natural killer cell maturation by controlling TCF1 amplitude. J. Exp. Med. 2021;218:e20202032. doi: 10.1084/jem.20202032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pais Ferreira D., Silva J.G., Wyss T., Fuertes Marraco S.A., Scarpellino L., Charmoy M., Maas R., Siddiqui I., Tang L., Joyce J.A., et al. Central memory CD8(+) T cells derive from stem-like Tcf7(hi) effector cells in the absence of cytotoxic differentiation. Immunity. 2020;53:985–1000.e1011. doi: 10.1016/j.immuni.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Kallies A., Carotta S., Huntington N.D., Bernard N.J., Tarlinton D.M., Smyth M.J., Nutt S.L. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood. 2011;117:1869–1879. doi: 10.1182/blood-2010-08-303123. [DOI] [PubMed] [Google Scholar]
- 48.Wiedemann G.M., Santosa E.K., Grassmann S., Sheppard S., Le Luduec J.B., Adams N.M., Dang C., Hsu K.C., Sun J.C., Lau C.M. Deconvoluting global cytokine signaling networks in natural killer cells. Nat. Immunol. 2021;22:627–638. doi: 10.1038/s41590-021-00909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biron C.A., Young H.A., Kasaian M.T. Interleukin 2-induced proliferation of murine natural killer cells in vivo. J. Exp. Med. 1990;171:173–188. doi: 10.1084/jem.171.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshimori M., Imadome K., Komatsu H., Wang L., Saitoh Y., Yamaoka S., Fukuda T., Kurata M., Koyama T., Shimizu N., et al. CD137 expression is induced by Epstein-Barr virus infection through LMP1 in T or NK cells and mediates survival promoting signals. PLoS One. 2014;9 doi: 10.1371/journal.pone.0112564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evrard M., Becht E., Fonseca R., Obers A., Park S.L., Ghabdan-Zanluqui N., Schroeder J., Christo S.N., Schienstock D., Lai J., et al. Single-cell protein expression profiling resolves circulating and resident memory T cell diversity across tissues and infection contexts. Immunity. 2023;56:1664–1680.e9. doi: 10.1016/j.immuni.2023.06.005. [DOI] [PubMed] [Google Scholar]
- 52.Beaulieu A.M., Zawislak C.L., Nakayama T., Sun J.C. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat. Immunol. 2014;15:546–553. doi: 10.1038/ni.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams N.M., Lau C.M., Fan X., Rapp M., Geary C.D., Weizman O.E., Diaz-Salazar C., Sun J.C. Transcription factor IRF8 orchestrates the adaptive natural killer cell response. Immunity. 2018;48:1172–1182.e6. doi: 10.1016/j.immuni.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Min-Oo G., Bezman N.A., Madera S., Sun J.C., Lanier L.L. Proapoptotic Bim regulates antigen-specific NK cell contraction and the generation of the memory NK cell pool after cytomegalovirus infection. J. Exp. Med. 2014;211:1289–1296. doi: 10.1084/jem.20132459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ni J., Wang X., Stojanovic A., Zhang Q., Wincher M., Bühler L., Arnold A., Correia M.P., Winkler M., Koch P.S., et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1alpha unleashes NK cell activity. Immunity. 2020;52:1075–1087.e8. doi: 10.1016/j.immuni.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 56.Victorino F., Bigley T.M., Park E., Yao C.H., Benoit J., Yang L.P., Piersma S.J., Lauron E.J., Davidson R.M., Patti G.J., et al. HIF1alpha is required for NK cell metabolic adaptation during virus infection. eLife. 2021;10:e68484. doi: 10.7554/eLife.68484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myers J.A., Miller J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021;18:85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laskowski T.J., Biederstädt A., Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer. 2022;22:557–575. doi: 10.1038/s41568-022-00491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ginhoux F., Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44:439–449. doi: 10.1016/j.immuni.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 60.Bedoui S., Gebhardt T., Gasteiger G., Kastenmüller W. Parallels and differences between innate and adaptive lymphocytes. Nat. Immunol. 2016;17:490–494. doi: 10.1038/ni.3432. [DOI] [PubMed] [Google Scholar]
- 61.Ricardo-Gonzalez R.R., Van Dyken S.J., Schneider C., Lee J., Nussbaum J.C., Liang H.E., Vaka D., Eckalbar W.L., Molofsky A.B., Erle D.J., et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018;19:1093–1099. doi: 10.1038/s41590-018-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ricardo-Gonzalez R.R., Kotas M.E., O’Leary C.E., Singh K., Damsky W., Liao C., Arouge E., Tenvooren I., Marquez D.M., Schroeder A.W., et al. Innate type 2 immunity controls hair follicle commensalism by demodex mites. Immunity. 2022;55:1891–1908.e12. doi: 10.1016/j.immuni.2022.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hajishengallis G., Netea M.G., Chavakis T. Innate immune memory, trained immunity and nomenclature clarification. Nat. Immunol. 2023;24:1393–1394. doi: 10.1038/s41590-023-01595-x. [DOI] [PubMed] [Google Scholar]
- 64.Steinert E.M., Schenkel J.M., Fraser K.A., Beura L.K., Manlove L.S., Igyártó B.Z., Southern P.J., Masopust D. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell. 2015;161:737–749. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schuster I.S., Wikstrom M.E., Brizard G., Coudert J.D., Estcourt M.J., Manzur M., O'Reilly L.A., Smyth M.J., Trapani J.A., Hill G.R., et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity. 2014;41:646–656. doi: 10.1016/j.immuni.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 66.Nabekura T., Lanier L.L. Tracking the fate of antigen-specific versus cytokine-activated natural killer cells after cytomegalovirus infection. J. Exp. Med. 2016;213:2745–2758. doi: 10.1084/jem.20160726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flommersfeld S., Böttcher J.P., Ersching J., Flossdorf M., Meiser P., Pachmayr L.O., Leube J., Hensel I., Jarosch S., Zhang Q., et al. Fate mapping of single NK cells identifies a type 1 innate lymphoid-like lineage that bridges innate and adaptive recognition of viral infection. Immunity. 2021;54:2288–2304.e7. doi: 10.1016/j.immuni.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gasteiger G., Fan X., Dikiy S., Lee S.Y., Rudensky A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X., Peng H., Cong J., Wang X., Lian Z., Wei H., Sun R., Tian Z. Memory formation and long-term maintenance of IL-7Rα+ ILC1s via a lymph node-liver axis. Nat. Commun. 2018;9:4854. doi: 10.1038/s41467-018-07405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O'Leary J.G., Goodarzi M., Drayton D.L., von Andrian U.H. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 2006;7:507–516. doi: 10.1038/ni1332. [DOI] [PubMed] [Google Scholar]
- 71.Paust S., Gill H.S., Wang B.Z., Flynn M.P., Moseman E.A., Senman B., Szczepanik M., Telenti A., Askenase P.W., Compans R.W., et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 2010;11:1127–1135. doi: 10.1038/ni.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nikzad R., Angelo L.S., Aviles-Padilla K., Le D.T., Singh V.K., Bimler L., Vukmanovic-Stejic M., Vendrame E., Ranganath T., Simpson L., et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aat8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reeves R.K., Li H., Jost S., Blass E., Li H., Schafer J.L., Varner V., Manickam C., Eslamizar L., Altfeld M., et al. Antigen-specific NK cell memory in rhesus macaques. Nat. Immunol. 2015;16:927–932. doi: 10.1038/ni.3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peppa D., Pedroza-Pacheco I., Pellegrino P., Williams I., Maini M.K., Borrow P. Adaptive reconfiguration of natural killer cells in HIV-1 infection. Front. Immunol. 2018;9:474. doi: 10.3389/fimmu.2018.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gillard G.O., Bivas-Benita M., Hovav A.H., Grandpre L.E., Panas M.W., Seaman M.S., Haynes B.F., Letvin N.L. Thy1+ NK [corrected] cells from vaccinia virus-primed mice confer protection against vaccinia virus challenge in the absence of adaptive lymphocytes. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stary V., Pandey R.V., Strobl J., Kleissl L., Starlinger P., Pereyra D., Weninger W., Fischer G.F., Bock C., Farlik M., et al. A discrete subset of epigenetically primed human NK cells mediates antigen-specific immune responses. Sci. Immunol. 2020;5:eaba6232. doi: 10.1126/sciimmunol.aba6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grassmann S., Pachmayr L.O., Leube J., Mihatsch L., Andrae I., Flommersfeld S., Oduro J., Cicin-Sain L., Schiemann M., Flossdorf M., et al. Distinct surface expression of activating receptor Ly49H drives differential expansion of NK cell clones upon murine cytomegalovirus infection. Immunity. 2019;50:1391–1400.e4. doi: 10.1016/j.immuni.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 78.Adams N.M., Geary C.D., Santosa E.K., Lumaquin D., Le Luduec J.B., Sottile R., van der Ploeg K., Hsu J., Whitlock B.M., Jackson B.T., et al. Cytomegalovirus infection drives avidity selection of natural killer cells. Immunity. 2019;50:1381–1390.e5. doi: 10.1016/j.immuni.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fang M., Orr M.T., Spee P., Egebjerg T., Lanier L.L., Sigal L.J. CD94 is essential for NK cell-mediated resistance to a lethal viral disease. Immunity. 2011;34:579–589. doi: 10.1016/j.immuni.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Camarasa T.M.N., Torné J., Chevalier C., Rasid O., Hamon M.A. Streptococcus pneumoniae drives specific and lasting Natural Killer cell memory. PLoS Pathog. 2023;19 doi: 10.1371/journal.ppat.1011159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gang M., Wong P., Berrien-Elliott M.M., Fehniger T.A. Memory-like natural killer cells for cancer immunotherapy. Semin. Hematol. 2020;57:185–193. doi: 10.1053/j.seminhematol.2020.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jeevan-Raj B., Gehrig J., Charmoy M., Chennupati V., Grandclément C., Angelino P., Delorenzi M., Held W. The transcription factor Tcf1 contributes to normal NK cell development and function by limiting the expression of granzymes. Cell Rep. 2017;20:613–626. doi: 10.1016/j.celrep.2017.06.071. [DOI] [PubMed] [Google Scholar]
- 83.Wang Y., Lifshitz L., Gellatly K., Vinton C.L., Busman-Sahay K., McCauley S., Vangala P., Kim K., Derr A., Jaiswal S., et al. HIV-1-induced cytokines deplete homeostatic innate lymphoid cells and expand TCF7-dependent memory NK cells. Nat. Immunol. 2020;21:274–286. doi: 10.1038/s41590-020-0593-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu Y., Ju S., Chen E., Dai S., Li C., Morel P., Liu L., Zhang X., Lu B. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J. Immunol. 2010;185:3174–3183. doi: 10.4049/jimmunol.1000749. [DOI] [PubMed] [Google Scholar]
- 85.Madisen L., Zwingman T.A., Sunkin S.M., Oh S.W., Zariwala H.A., Gu H., Ng L.L., Palmiter R.D., Hawrylycz M.J., Jones A.R., et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang Q., Li F., Harly C., Xing S., Ye L., Xia X., Wang H., Wang X., Yu S., Zhou X., et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol. 2015;16:1044–1050. doi: 10.1038/ni.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kranz A., Fu J., Duerschke K., Weidlich S., Naumann R., Stewart A.F., Anastassiadis K. An improved Flp deleter mouse in C57BL/6 based on Flpo recombinase. Genesis. 2010;48:512–520. doi: 10.1002/dvg.20641. [DOI] [PubMed] [Google Scholar]
- 88.Pimeisl I.M., Tanriver Y., Daza R.A., Vauti F., Hevner R.F., Arnold H.H., Arnold S.J. Generation and characterization of a tamoxifen-inducible Eomes(CreER) mouse line. Genesis. 2013;51:725–733. doi: 10.1002/dvg.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Behr F.M., Parga-Vidal L., Kragten N.A.M., van Dam T.J.P., Wesselink T.H., Sheridan B.S., Arens R., van Lier R.A.W., Stark R., van Gisbergen K.P.J.M. Tissue-resident memory CD8+ T cells shape local and systemic secondary T cell responses. Nat. Immunol. 2020;21:1070–1081. doi: 10.1038/s41590-020-0723-4. [DOI] [PubMed] [Google Scholar]
- 90.Merkenschlager J., Finkin S., Ramos V., Kraft J., Cipolla M., Nowosad C.R., Hartweger H., Zhang W., Olinares P.D.B., Gazumyan A., et al. Dynamic regulation of TFH selection during the germinal centre reaction. Nature. 2021;591:458–463. doi: 10.1038/s41586-021-03187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Narni-Mancinelli E., Chaix J., Fenis A., Kerdiles Y.M., Yessaad N., Reynders A., Gregoire C., Luche H., Ugolini S., Tomasello E., et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. USA. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kallies A., Xin A., Belz G.T., Nutt S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31:283–295. doi: 10.1016/j.immuni.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 93.Levéen P., Larsson J., Ehinger M., Cilio C.M., Sundler M., Sjöstrand L.J., Holmdahl R., Karlsson S. Induced disruption of the transforming growth factor beta type II receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–568. doi: 10.1182/blood.v100.2.560. [DOI] [PubMed] [Google Scholar]
- 94.Ugur M., Kaminski A., Pabst O. Lymph node γδ and αβ CD8+ T cells share migratory properties. Sci. Rep. 2018;8:8986. doi: 10.1038/s41598-018-27339-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fodil-Cornu N., Lee S.H., Belanger S., Makrigiannis A.P., Biron C.A., Buller R.M., Vidal S.M. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol. 2008;181:6394–6405. doi: 10.4049/jimmunol.181.9.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosshart S.P., Herz J., Vassallo B.G., Hunter A., Wall M.K., Badger J.H., McCulloch J.A., Anastasakis D.G., Sarshad A.A., Leonardi I., et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science. 2019;365:eaaw4361. doi: 10.1126/science.aaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herbert S., Ziebandt A.K., Ohlsen K., Schäfer T., Hecker M., Albrecht D., Novick R., Götz F. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 2010;78:2877–2889. doi: 10.1128/IAI.00088-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gasteiger G., Kastenmuller W., Ljapoci R., Sutter G., Drexler I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J. Virol. 2007;81:11925–11936. doi: 10.1128/JVI.00903-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk and single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.