

Abstract
Recent advancements in single-cell technologies have enabled expression quantitative trait locus (eQTL) analysis across many individuals at single-cell resolution. Compared with bulk RNA sequencing, which averages gene expression across cell types and cell states, single-cell assays capture the transcriptional states of individual cells, including fine-grained, transient, and difficult-to-isolate populations at unprecedented scale and resolution. Single-cell eQTL (sc-eQTL) mapping can identify context-dependent eQTLs that vary with cell states, including some that colocalize with disease variants identified in genome-wide association studies. By uncovering the precise contexts in which these eQTLs act, single-cell approaches can unveil previously hidden regulatory effects and pinpoint important cell states underlying molecular mechanisms of disease. Here, we present an overview of recently deployed experimental designs in sc-eQTL studies. In the process, we consider the influence of study design choices such as cohort, cell states, and ex vivo perturbations. We then discuss current methodologies, modeling approaches, and technical challenges as well as future opportunities and applications.
Keywords: eQTL, sc-eQTL, gene regulation, cell state, noncoding variants, single-cell sequencing
1. INTRODUCTION
Genome-wide association studies (GWASs) have identified thousands of genetic loci associated with complex traits and diseases (20). A pressing challenge is to define the molecular mechanisms by which disease-associated variants contribute to pathogenesis. Since more than 90% of GWAS hits lie within noncoding genomic regions, many disease alleles are assumed to impact gene regulation (97). One approach is to find colocalization between disease variants and expression quantitative trait loci (eQTLs)—regulatory variants that affect the expression of an associated gene (referred to as an eGene) either nearby (cis; typically within 1 Mb) or far away (trans; > 1 Mb away or on a different chromosome). In addition to improving our basic understanding of gene regulation, mapping eQTLs is a potentially powerful strategy to link disease variants to candidate target genes and pinpoint causal cell types and states (146).
However, in practice, linking eQTLs to GWAS variants has had limited success—for autoimmune diseases such as rheumatoid arthritis, for example, only ~10–30% of GWAS hits have been found to colocalize with eQTLs (25, 48, 64). Large-scale eQTL studies typically use bulk RNA sequencing (RNA-seq) to profile tissue samples from many individuals and then associate genetic variants with gene expression. For example, the latest phase of the Genotype-Tissue Expression (GTEx) project profiled more than 15,000 samples from 49 tissues of more than 800 postmortem donors to identify tissue cis-eQTLs (54). Despite the large sample size, studies have estimated that GTEx eQTLs explain only ~11% of heritability for complex traits (161).
How do we explain this missing link between existing gene regulation and disease association studies (27)? There are many possible explanations (27, 102, 146, 161); for example, a substantial portion of noncoding disease variants may affect other aspects of gene regulation beyond expression, such as splicing (119) or chromatin states that do not have observable gene expression effects (82). One commonly proposed explanation is that disease-associated eQTLs are context dependent—that is, the strength of a regulatory effect depends on the cell’s specific biological state and environmental context—and thus are poorly captured by bulk, single-condition datasets (56, 64, 103, 146).
There are several ways in which study design limits the ability to resolve context-dependent eQTLs. First, eQTLs could be specific to relatively rare but distinct cell types in a given tissue, such as microglia in the brain (166), and obscured in bulk analyses that aggregate gene expression from diverse cell types. Second, eQTLs can be stronger within finer-grained cell states within a given cell type (i.e., state dependent), as cell types themselves often comprise heterogeneous subpopulations. These eQTLs may be (a) state specific, appearing only in certain cell subpopulations, such as regulatory T cells (Tregs) (15), or (b) dynamic, where the eQTL follows a continuous gradient, such as cytotoxicity in T cells (106) or differentiation over time (137). Bulk studies have sought to address these issues by isolating cell populations through techniques such as flow sorting (23, 45, 72, 105, 113) or estimating cell-type proportions with computational deconvolution methods (42, 75, 153); however, these approaches may fail to distinguish closely related cell states or be biased toward cell states for which we have well-validated marker genes or reference datasets. Third, it may be that an eQTL exerts its effect only when a cell transitions into a state in response to an environmental factor; this state may not be one that is commonly observed in unperturbed physiological conditions. An example is response eQTLs (reQTLs), which may vary in strength depending on exposure to a given environmental stimulus, such as activation (13, 76, 85), hypoxic stress (152), or drug treatment (79). Identifying these reQTLs requires assaying tissues from disease or ex vivo perturbations, for example, by stimulating immune cells with pathogens.
Single-cell transcriptomic assays, such as single-cell RNA-seq (scRNA-seq), offer an immense opportunity to both expand eQTL discovery and map transcriptional regulation at a much finer resolution than was previously possible with bulk RNA-seq, including previously uncharacterized cell states. They provide whole-transcriptome measurements from heterogeneous single cells collected from their tissue or experimental contexts, capturing variation in expression not only among individuals but also within different cell populations. This preserves the cell-state granularity required to discover more context-dependent eQTLs, which may capture cell-specific transcriptional machinery, such as transcription factors (TFs) binding to state-specific regulatory elements. With single-cell data, one can flexibly define discrete cell states at multiple resolutions in silico rather than being limited to a priori flow sorting. When studying continuous processes such as differentiation, single-cell data offer granular information on where a cell falls along a dynamic trajectory (30, 31). When studying reQTLs, it is challenging to tease apart eQTL effects from cell-type-specific differential expression and changes in cell-type abundance—hence, bulk reQTL studies have focused mostly on one cell type at a time (13, 44, 76, 85). With single-cell data, it becomes feasible to separate these effects in mixtures of cell types (37, 110, 121), which may be more representative of in vivo contexts.
In 2013, a proof-of-concept study by Wills et al. (155) used single-cell measurements to associate single-nucleotide polymorphisms (SNPs) with gene expression phenotypes. By assaying 1,440 cells from 15 individuals and measuring 92 genes by microarray, the authors showed that many genetic associations with expression-based traits are masked when expression is averaged over many cells. Since then, advancements in the throughput of single-cell technologies and sample multiplexing (60, 70, 136) have enabled the sequencing of larger cohorts amenable to eQTL analyses. In 2018, an early single-cell eQTL (sc-eQTL) study by van der Wijst et al. (147) explored cell-type-specific eQTLs from ~25,000 peripheral blood mononuclear cells (PBMCs) from 45 individuals. Recent sc-eQTL studies have grown to hundreds or thousands of individuals (106, 115, 121, 163) with diverse tissues and cell-state contexts (18, 31, 43, 129, 133) (Figure 1). The field of sc-eQTLs is still in its infancy but holds great potential to improve our understanding of both the fundamental principles of gene regulation and their roles in complex traits and diseases. Here, we review current experimental designs and methodological approaches used to identify eQTLs in single-cell data, highlighting insights from recent sc-eQTL studies, remaining challenges, and future directions.
Figure 1.
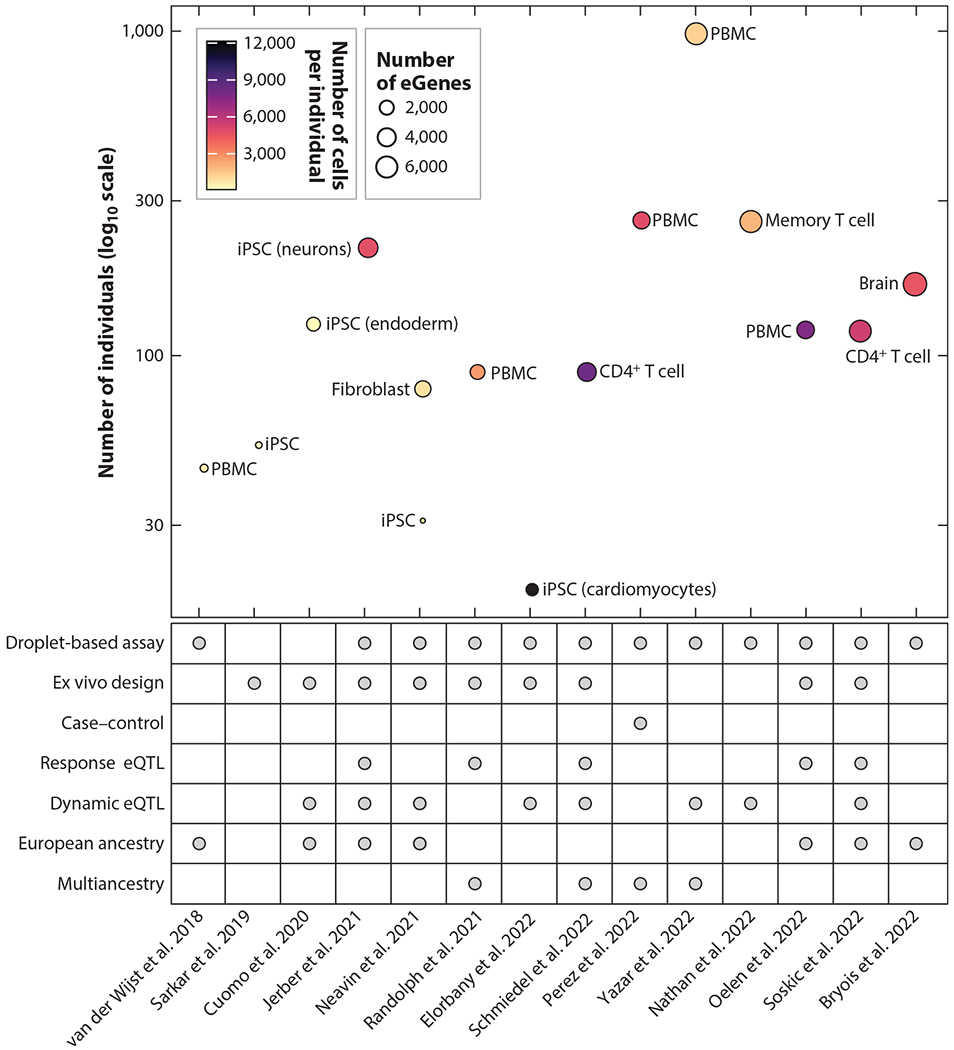
Timeline of sc-eQTL studies. The graph shows the number of individuals (y axis) included in key publications for sc-eQTL studies (x axis); the dot size indicates the number of cell-type-specific genes with at least one eQTL (eGenes), and the color corresponds to the number of cells per individual. The table shows relevant selected features (rows) for each study (columns), where a gray dot indicates that the study included the feature. [Since eGenes were not reported in the study by Sarkar et al. (127), the number of eGenes indicated for that study corresponds to the number of eQTLs instead.] Abbreviations: eQTL, expression quantitative trait locus; iPSC, induced pluripotent stem cell; PBMC, peripheral blood mononuclear cell; sc-eQTL, single-cell expression quantitative trait locus.
2. STUDY DESIGN
In this section, we discuss essential considerations for designing sc-eQTL studies (Figure 2a), including building an appropriate cohort and collecting cell types and states of interest.
Figure 2.
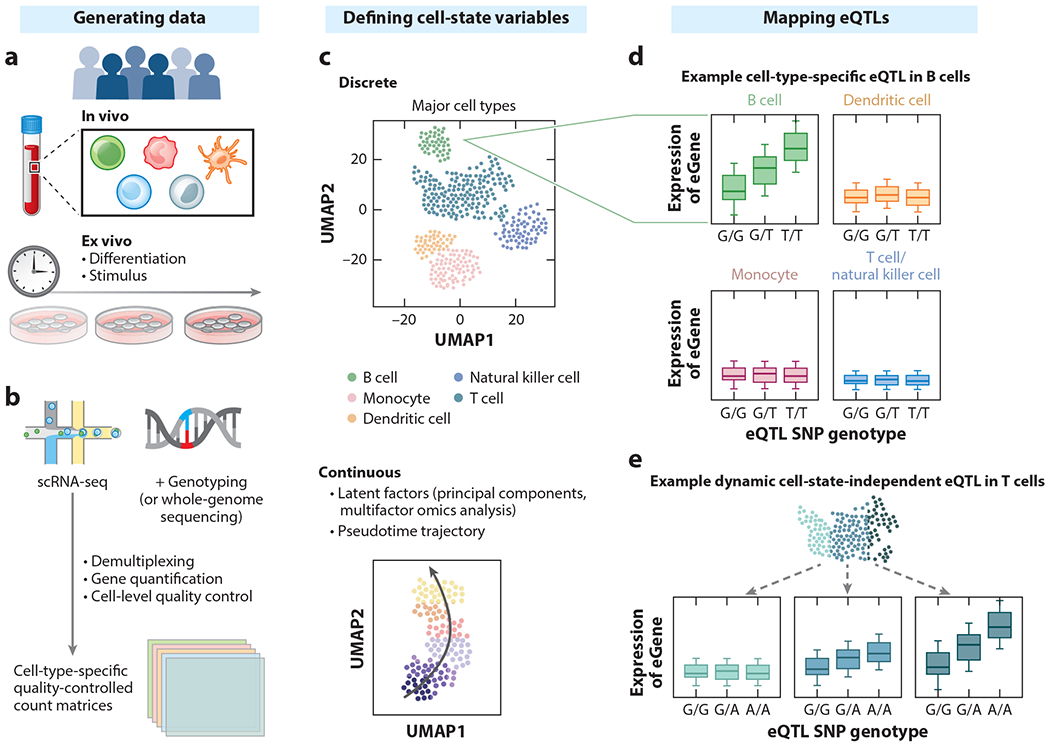
Overview of sc-eQTL study design and context-dependent eQTLs. (a) For a given cohort, cell states of interest are assayed directly from their native tissue contexts or elicited ex vivo by experimental perturbations. (b) Single-cell whole-transcriptome and genomic data are generated and serve as input to sc-eQTL analysis. (c) Single-cell data can be used to define cell types and states. Shown here is an example UMAP plot that illustrates the relationship among cells, colored by major cell type. (d) In this example of a cell-type-specific eQTL in B cells, a higher dosage of the T allele increases eGene expression. (e) In this example of a dynamic, state-dependent eQTL in T cells, the effect of the eQTL becomes stronger along a continuous cell state (increasing slope with darker teal colors). Abbreviations: eQTL, expression quantitative trait locus; sc-eQTL, single-cell expression quantitative trait locus; scRNA-seq, single-cell RNA sequencing; SNP, single-nucleotide polymorphism; UMAP, uniform manifold approximation and projection. Figure adapted from images created with BioRender.com.
2.1. Cohort Selection
Attributes of the study cohort, including disease status and genetic ancestry, can affect the ability to detect sc-eQTLs. Several bulk studies have reported that some eQTLs may be specific to disease (100, 165), arising from either disease-specific cell-state composition or disease-specific shifts in gene regulation [e.g., interferon signature in systemic lupus erythematosus (SLE) (10)]. Studies of sc-eQTLs that incorporate clinically relevant samples from people with disease can be used to compare (a) people with and without disease, (b) disease-affected and unaffected tissue from the same individuals, or (c) the same tissue across diseases. Additional information can be added by including pre- and posttreatment patients or by aggregating multiple individuals affected by similar diseases. However, heterogeneity in disease severity, clinical presentation, and treatment are important confounders that may affect expression in disease cohorts. Thus far, the scarcity and difficulty in retrieving patient tissues have partially limited case–control comparisons in sc-eQTL studies. In one of the few examples, Perez et al. (115) profiled 1.2 million PBMCs in a disease cohort of 162 individuals with SLE and 99 healthy controls.
Studying ancestrally diverse cohorts also offers unique advantages. Genetic ancestry refers to the complex patterns within an individual’s genomic DNA that reflect their ancestors’ genomic DNA (68). When fine mapping genetic signals, linkage disequilibrium can make it difficult to identify the causal variant among multiple tightly linked variants. Diverse ancestries can help disentangle variants in high linkage disequilibrium and improve fine mapping (under the assumption that causal variants are shared among ancestries) (156). They can also boost power to detect regulatory alleles that exist at low frequencies in Europeans but exist at higher frequencies in or are specific to other populations (101, 106, 121, 163) and increase sensitivity for gene-by-environment interactions due to diversity in selection pressures among ancestrally distinct groups (46).
Multiancestry eQTL studies have been used to interrogate biologically motivated questions, such as how much of observed interpopulation differences in the immune response are due to genetics versus unmeasured environmental factors correlated with genetic ancestry. For example, two pioneering bulk studies demonstrated that ancestry can be used as a proxy for the genetic contribution to the difference in the immune response to several stimuli (109, 120). A few years later, using similar experimental design, the same research groups used scRNA-seq to profile PBMCs from ~100 individuals of European and African ancestry (111, 121). Randolph et al. (121) found thousands of cell-type-specific differentially expressed genes after stimulation with influenza A virus, and increased European ancestry correlated with stronger type I and II interferon responses to influenza A virus in multiple cell types. By mapping cis-eQTLs, they demonstrated a considerable genetic component driving these differences—individual genotype and cis-eQTLs effect size alone explained more than 50% of the variance of the genetic ancestry effect on expression. As these results demonstrate, studying multiancestry cohorts at the single-cell level can provide critical references from which to understand the biological mechanisms of diseases and differences in disease susceptibility and outcomes.
2.2. Cell Type and State Selection
For sc-eQTL studies, it is important to carefully consider which cell types and states to include in the study, as this will determine whether state-specific regulatory variants will be detectable. Cell types can be narrowed by sorting cell populations, selecting appropriate tissue segments, or applying quality control after data have been collected. While the concept of cell types is nuanced (168), they can be thought of as biologically functional units playing distinct roles—for example, the pancreas contains hormone-producing types, like alpha and beta cells, as well as supporting cell types, like endothelial cells and fibroblasts. Cells can traverse a landscape of states in development and disease (145); for example, beta cells can enter an endoplasmic reticulum stress state that has been implicated in diabetes (164). eQTLs specific to endoplasmic reticulum–stressed beta cells would be missed if that particular cell state was not assayed.
After collecting samples of interest, one can either assay all cells present in an unbiased manner or enrich for one or more cell types known to be important for specific traits (e.g., disease), using techniques like flow sorting. Optionally, one can experimentally perturb the cells ex vivo to elicit cell states that may not be present in the collected sample.
The immune system lends itself well to this discussion because it comprises diverse cell types that undergo dynamic transcriptional changes when responding to pathogens and insults (139). Many existing sc-eQTL studies have focused on immune cell types from peripheral blood, including B, T, natural killer, monocyte, and dendritic cells (106, 115, 147, 163). The largest such study to date mapped eQTLs in 1.27 million PBMCs collected from 982 individuals (163). The authors identified more than 26,500 conditionally independent cis-eQTLs (for 16,597 eGenes across 14 cell types) and showed that 3,060 out of the 6,469 unique eGenes were specific to a single cell type and likely due to cell-type-specific regulatory mechanisms. Rather than analyzing all PBMCs together, it can sometimes be valuable to enrich for one or more specific cell types, especially those that display immense functional heterogeneity, like T cells. This improves the power to study finer-grained cell states. For example, two recent studies (129, 133) focused on CD4+ T cells, which have been implicated in immune-mediated diseases (91).
An important future direction is to map sc-eQTLs for immune cells not only in blood but also in solid tissues and lymphoid organs (139), which are often the site of disease activity. For example, key cell states in rheumatoid arthritis, such as T peripheral helper cells and IL1B+ proinflammatory infiltrating monocytes (122, 169, 170), are missing or occur in very low abundance in PBMCs. Future studies comparing cell types within different tissues (e.g., Tregs in blood versus kidney) will elucidate which regulatory effects are shared and which are unique to particular tissue contexts (47).
With improved solid tissue disaggregation procedures, mapping sc-eQTLs in primary tissues beyond blood is becoming possible. For example, a recent study by Bryois et al. (18) was the first to map sc-eQTLs in the brain. The authors used single-nucleus RNA-seq (snRNA-seq) to define eight cell types in postmortem samples from 192 individuals. Single-nucleus technologies may be better suited for tissues where disaggregation is challenging or causes substantial cell loss, with comparable performance to scRNA-seq in expression quantification and downstream analyses such as cell-type annotation (see Section 3.1). Bryois et al. (18) found significantly larger effect sizes for cell-type-specific cis-eQTLs—in terms of mean absolute effect sizes both within and across cell types—compared with those obtained from aggregating all the snRNA-seq data into a tissue-like profile (7,607 versus 3,058 cell-type-specific and tissue-like cis-eQTLs, respectively). In both neurons and glia, these cell-type-specific cis-eQTLs affected more evolutionarily constrained genes than the cis-eQTLs detected at a tissue level, suggesting higher disease relevance. The increasing availability of single-cell data from diverse tissues and disease conditions (18, 115, 130, 132, 169) will enable investigators to construct comprehensive maps of cell states where disease variants act.
2.3. Ex Vivo Stimulation
Cells can be perturbed ex vivo to elicit cell states absent in unperturbed tissues. While ex vivo conditions may not perfectly recapitulate complex cell dynamics in vivo, they are useful model systems.
A common approach involves exposing immune cells to stimuli known to induce activation, such as pathogens or cytokines (110, 121, 129, 133). By comparing different conditions, one can identify reQTLs that change in strength between stimulated versus unstimulated states. Bulk RNA-seq studies have identified such reQTLs and shown that they are enriched in disease-risk loci (13, 37, 44, 85). For example, reQTLs in dendritic cells stimulated with Mycobacterium tuberculosis were more likely to be enriched in the pulmonary tuberculosis GWAS signal (13). Recent single-cell studies have investigated reQTLs in multiple cell types simultaneously, as single-cell data can help tease apart changes in cell-state abundance and cell-state-specific gene regulation (110, 121). For example, Oelen et al. (110) profiled 1.3 million PBMCs collected from 120 healthy individuals and exposed in vitro to M. tuberculosis, Candida albicans, and Pseudomonas aeruginosa. They mapped eQTLs for each stimulation condition across six major cell types at multiple time points, testing a predefined set of lead eQTLs from a previous high-powered bulk meta-analysis of more than 30,000 individuals (151). Interestingly, they found that reQTLs were typically more cell-type specific than pathogen specific. In particular, myeloid cells exhibited the strongest response to stimulation and the most reQTLs, supporting their role in early infection.
Using a similar experimental setup, Randolph et al. (121) mapped cell-type-specific eQTLs in PBMCs stimulated in vitro with influenza A virus and control conditions using 235,161 cells from 90 male donors. For eQTL analysis, they divided cells into five major types and found cell-type-specific reQTLs—for example, rs10774671 is an eQTL for the OAS1 gene in CD4+ T cells only after influenza A virus stimulation. Strikingly, Kumasaka et al. (81) also found that the same OAS1 locus was a reQTL (same direction of effect) in primary dermal fibroblasts stimulated to induce an antiviral response.
2.4. Ex Vivo Differentiation
Because some tissues and developmental stages are difficult to access in vivo, several groups have differentiated induced pluripotent stem cells (iPSCs) into cell types of interest, such as neurons (67), endoderm (31), and cardiomyocytes (43). The ability to reprogram iPSCs into otherwise inaccessible cell states enables the study of dynamic differentiation and developmental processes at the single-cell level (31, 43, 67, 108).
For example, Cuomo et al. (31) generated scRNA-seq data from iPSCs of 125 individuals recruited by the Human Induced Pluripotent Stem Cell Initiative (HipSci) at four time points of differentiation toward endoderm fate. They found eQTLs unique to specific differentiation stages or modulated by specific gene programs, such as metabolism (31). In a more complex differentiation scenario, Jerber et al. (67) assayed more than 1 million cells differentiating from iPSCs into midbrain neuronal cells from 215 individuals. Generating scRNA-seq data across three different time points and one stimulated condition mimicking oxidative stress during Parkinson’s disease, they discovered more eGenes in later and stimulated stages for all cell types. The effects of many of the mapped sc-eQTLs were shared with those from GTEx brain tissues; however, 2,366 sc-eQTLs could not be detected in GTEx brain tissues at all. Furthermore, Elorbany et al. (43) mapped sc-eQTLs in 19 iPSC cell lines along seven time points in a bifurcating differentiation path toward cardiomyocytes and cardiac fibroblasts. They used cells grouped into pseudotime bins to infer dynamic eQTLs specific to each of the two lineages. Neavin et al. (108), by contrast, performed cell-type reprogramming in the reverse differentiation direction (from dermal fibroblasts into iPSCs). Most eQTLs discovered in fibroblasts disappeared during reprogramming, highlighting the importance of cell-type-specific regulatory mechanisms.
Although widely used, iPSC-derived cells present some limitations, including high costs and labor needed to develop high-quality cell lines. Moreover, in vitro models of differentiation may miss aspects of the in vivo context. For example, iPSC-derived cardiomyocytes may not experience the same cues as cardiomyocytes in native heart tissue. It will be important to compare the results from the ex vivo experiments with eQTLs identified in large in vivo tissue studies such as GTEx (43).
2.5. Power Simulations
A major consideration for sc-eQTL studies is ensuring enough statistical power to detect regulatory relationships for variants with a low (e.g., <10%) minor allele frequency. Appropriate power analysis is critical in the regime of high multiple testing burden and high cost of single-cell data generation. Given a fixed budget, researchers can modulate several experimental design parameters to increase power. These parameters include the size and genetic heterogeneity of the cohort, target number of cells per sample (though the final distribution of cells per sample may vary depending on yields from cell sorting or tissue disaggregation), sequencing coverage, and sample multiplexing.
Several tools have been developed for sc-eQTL power and sample size calculations. For example, the powerEQTL tool can be used to calculate one of power, sample size, minimum detectable slope, or minimum minor allele frequency, given values for the other three (41). Another useful approach is simulating multisample single-cell datasets to assess the effects of different study design parameters (9, 28, 94, 99, 128, 149, 167). Recently, such simulation frameworks have been able to incorporate genetic effects, making them amenable to sc-eQTL power analysis (9, 94, 128). An early study showed that, on a fixed budget, one can achieve greater power for cell-type-specific eQTL analysis by increasing sample size and cells per sample while maintaining coverage of at least 10,000 reads per cell (94). In line with this, another group developed the scPower framework (128), which models the relationship between sample size, number of cells per sample, and sequencing coverage, taking into account priors for expression level and effect size. This group also noted that shallow sequencing of more cells generally leads to higher overall power than deep sequencing of fewer cells (128). One can further decrease costs by multiplexing multiple samples on the same lane (see Section 3.1).
Certain trade-offs also depend on the goal of the study. For example, including more individuals or samples but fewer cells per sample will prioritize power to detect allelic effects, whereas increasing the number of cells per sample may improve the definition of cell states to model their interactions with a regulatory effect (163). Importantly, power calculations are tightly linked to the underlying eQTL detection model and statistical testing procedure, so when performing simulations, it is best to select the model and assumptions most in line with the planned study.
3. DATA COLLECTION AND PREPROCESSING
Performing sc-eQTL analysis requires single-cell whole-transcriptome and genotype data from the same individuals (Figure 2b). This section discusses the collection and preprocessing of each data type.
3.1. RNA Measurements
Broadly, scRNA-seq comprises plate-based and droplet-based technologies. Plate-based methods [e.g., Smart-seq2 and Smart-seq3 (58, 116)] are lower in throughput but typically provide deeper sequencing of each cell and coverage across the full length of transcripts, thereby allowing more complex libraries and clues about isoform usage. Droplet-based technologies (e.g., 10x Genomics Chromium) have much higher cell throughput—scaling to thousands of cells per library—but at the cost of lower coverage (171). Current droplet-based technologies assay only the 5′ or 3′ end of the transcript and hence offer a limited view of isoform usage. The full extent of available single-cell technologies and best practices for data preprocessing have been reviewed extensively (22, 73, 84, 92).
Sequencing solid tissue samples can be more challenging than sequencing sorted cells from blood, often requiring specialized single-cell dissociation protocols tailored to each tissue to maximize cell yield or enrich for cell types of interest (38, 148). An alternative to scRNA-seq is snRNA-seq, which sequences isolated nuclei instead of cells (40). snRNA-seq can be especially useful for tissues that are difficult to dissociate due to cell size or fragility, such as brain tissue or frozen tumor samples (11, 18, 131). Notably, transcripts measured in the nucleus will be different from those in the cytoplasm, with a greater proportion of unspliced introns but fewer mitochondrial genes. A recent study comparing snRNA-seq and scRNA-seq on fibrotic kidney samples found that snRNA-seq offered comparable gene detection and reduced biases in cell-type dissociation and dissociation-induced transcriptional stress responses (158).
For droplet-based assays, pooling multiple samples together in a single run, referred to as multiplexing, is a cost-saving approach. Multiplexing samples may also make it easier to separate interindividual sample differences from technical effects since cells from multiple individuals are in a single experimental batch. Algorithms like demuxlet (70) and souporcell (60) can use genetic variation among samples for demultiplexing (assigning cells to individuals) and identifying cross-sample doublets. Another demultiplexing approach uses sample-specific barcodes, known as cell hashing (136).
Several pipelines quantify single-cell expression by aligning reads to a reference genome or transcriptome, including CellRanger (10x Genomics), STARsolo (69), and kallisto | bustools (98). These tools can handle single-cell-specific considerations such as read-to-cell assignment, correction of cell barcodes and unique molecular identifiers (UMIs), and UMI collapsing, producing a genes-by-cells count matrix (92).
3.2. Data to Define Cell-Type and Cell-State Variables
When designing studies to investigate state-dependent regulation, it is important to collect the data types required to quantitatively represent the cell states as variables (Figure 2c). For many sc-eQTL studies, the single-cell transcriptomic data themselves are used not only as the expression phenotype but also to define cell states. For example, one can use a dimensionality reduction pipeline like principal component analysis (PCA) to define expression components, and then define cell states by clustering (121, 163). Some states can also be represented as binary variables, such as the presence or absence of a key marker gene (e.g., CD4). Alternatively, cell states can be defined continuously, such as score along a low-dimensional axis [e.g., principal component (PC)], position along a nonlinear factor [e.g., pseudotime (163)], or even the expression of another gene or gene set [e.g., interferon response (115) or cytotoxicity (57)]. These discrete or continuous variables can then be used to model cell-state-specific eQTL effects (see Section 4). For example, drawing on prior evidence for increased interferon signaling in SLE patients, Perez et al. (115) used a gene signature for type I interferon response as a cell-state variable to discover interferon-interacting eQTLs, highlighting the utility of assaying disease cell states when mapping regulatory effects.
Multimodal assays can help to define cell states by measuring additional markers beyond RNA, such as surface protein markers from the same cells with cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) (135), chromatin accessibility profiles using multimodal snRNA and assay for transposase-accessible chromatin (ATAC) sequencing (24), or even emerging trimodal measurements (140). Moreover, given the complex combination of functions that even a single cell may carry out, its state may be most precisely represented by multiple state variables (e.g., T cell, CD4+ T cell, amount of Th1-like character, or score along PC1 of transcriptional space). For example, Nathan et al. (106) recently mapped sc-eQTLs in memory T cells from a cohort of 259 Peruvian individuals assayed using CITE-seq (135). A major advantage of the study was using multiple continuously defined cell states derived from multimodal data. For example, a continuous state correlated with cytotoxicity detected more state-interacting eQTLs than discrete CD4+ versus CD8+ categories (106), often used as proxies for low- and high-cytotoxicity cells.
3.3. Removing Technical Artifacts
For quality control and downstream analysis, technical parameters commonly used to filter low-quality cells include library size (number of UMIs), number of genes detected per cell, and percentage of mitochondrial reads (65, 112). These parameters can also be controlled for in the downstream eQTL models as cell-level covariates. An important step in the processing of single-cell data is removing suspected doublets or multiplets, which can be identified as droplets with mixed sample genotypes (60, 70) or by simulating mixed transcriptional profiles (157). Several reviews have discussed quality control for single-cell data in greater depth (62, 92).
An important challenge in single-cell data is the presence of batch effects, which are technical differences that can arise from many sources, including sample processing, reagent batches, sequencing runs, sequencing technology (e.g., different chemistries of 10x Genomics technology), or even season of blood draw. Batch effects can introduce unwanted variation and make it challenging to properly define cell states. Removing batch effects using one of the many available batch correction methods (19, 80, 89, 144) can help improve downstream results and is essential to ensure that state variables are consistently defined across samples.
3.4. Genotype Data
As in bulk eQTL analysis and GWASs, genotype information in sc-eQTL studies can come from genotyping arrays (e.g., the Infinium Global Screening Array from Illumina) or whole-genome sequencing (121). Data quality control is usually performed using standard procedures (4) before haplotype phasing (16) and imputation of nonassayed variants using a reference panel of densely genotyped haplotypes, which increases the number of SNPs tested for associations and improves fine mapping of signals (35, 95).
Some groups have developed approaches for identifying eQTLs in single-cell data without requiring separate genomic data, for example, by calling SNPs from raw scRNA-seq reads (14, 93) or correlating the fraction of an allele at expressed biallelic loci to gene expression directly from scRNA-seq (87). This can be necessary in some cases, such as mapping eQTLs in species that do not have standard genotyping arrays. For example, Ben-David et al. (14) sought to map whole-organism sc-eQTLs in genetically distinct organisms of the model nematode Caenorhabditis elegans. They were able to infer genotypes from scRNA-seq reads and use the inferred variants to test for sc-eQTLs by cell type. However, because genotyping arrays are relatively cost-effective for humans, we generally recommend DNA-based genotyping when possible to ensure good coverage of variants genome-wide, rather than limiting the tested variants to those in expressed transcripts.
4. MAPPING SINGLE-CELL EXPRESSION QUANTITATIVE TRAIT LOCI
In bulk eQTL analysis, each sample’s expression of each gene is measured as a single measurement, and variants are tested for association with expression, for example, by using a regression framework. Typically, each individual in the study has a single sample. By contrast, in sc-eQTL analysis, hundreds to thousands of cells are measured from each individual, which increases the complexity of the study but simultaneously creates opportunities for new modeling approaches. In this section, we describe current strategies for identifying context-dependent eQTLs in single-cell data.
4.1. Challenges When Modeling Single-Cell Data
There are several challenges for mapping sc-eQTLs compared with bulk eQTLs. First, single-cell data inherently present a large number of transcriptional profiles, incurring challenges in data storage and computational efficiency, especially for large-scale datasets with hundreds of thousands to millions of cells. Improving computational scalability is an issue facing the single-cell field at large and is not unique to sc-eQTL analysis (83). Second, unwanted batch effects and technical variations in single-cell data, such as doublets and uneven per-cell coverage, may introduce additional noise and affect results if not carefully accounted for. Third, single-cell measurements are typically very sparse, especially for droplet-based technologies—that is, a high proportion of observations in the expression matrix have a count of zero. This can be attributed to both biological factors (e.g., truly absent expression) and technical limitations (e.g., imperfect amplification or stochastic sampling variation) (83).
One way to handle sparsity is to model it directly, which requires modeling expression–genotype associations at single-cell resolution. However, traditional linear models used in bulk eQTL mapping do not accurately reflect the distribution of single-cell expression (126, 143). Hence, there exists a trade-off between cell-state resolution and modeling strategy. At two ends of the spectrum, one can either aggregate the cells within each sample until linear assumptions hold (potentially at the loss of cell-state resolution) or preserve single-cell resolution by modeling the cells individually (requiring effective cell-state definitions across cells and models beyond linear regression, which can come with computational challenges). These two strategies describe pseudobulk models (which aggregate cells) and single-cell models (which model individual cells), respectively.
4.2. Pseudobulk Models
As the name suggests, pseudobulk analysis is similar to bulk analysis: Single-cell expression is aggregated at the sample level such that each sample has a bulk-like expression profile. The major advantages of this approach are that cell aggregation can mitigate sparsity and that one can easily apply methods developed for bulk eQTL studies to pseudobulk data.
For each sample, one can either (a) classify cells into multiple discrete groups or bins (e.g., cell-state clusters or pseudotime bins) and aggregate within each bin or (b) aggregate all the cells together into a single bin. To be effective, the bins must be large enough to mitigate sparsity for all samples; that is, individual samples in which the bin has too few cells can challenge linear model assumptions. Each genes-by-cells count matrix (one per sample–bin pair) is aggregated into a genes-by-samples matrix (Figure 3a). One can either (a) first normalize gene expression in each cell for library size and then take the mean across cells from each sample or (b) first sum expression across all cells per sample and then normalize within-sample counts (giving more weight to cells with higher coverage) (29).
Figure 3.
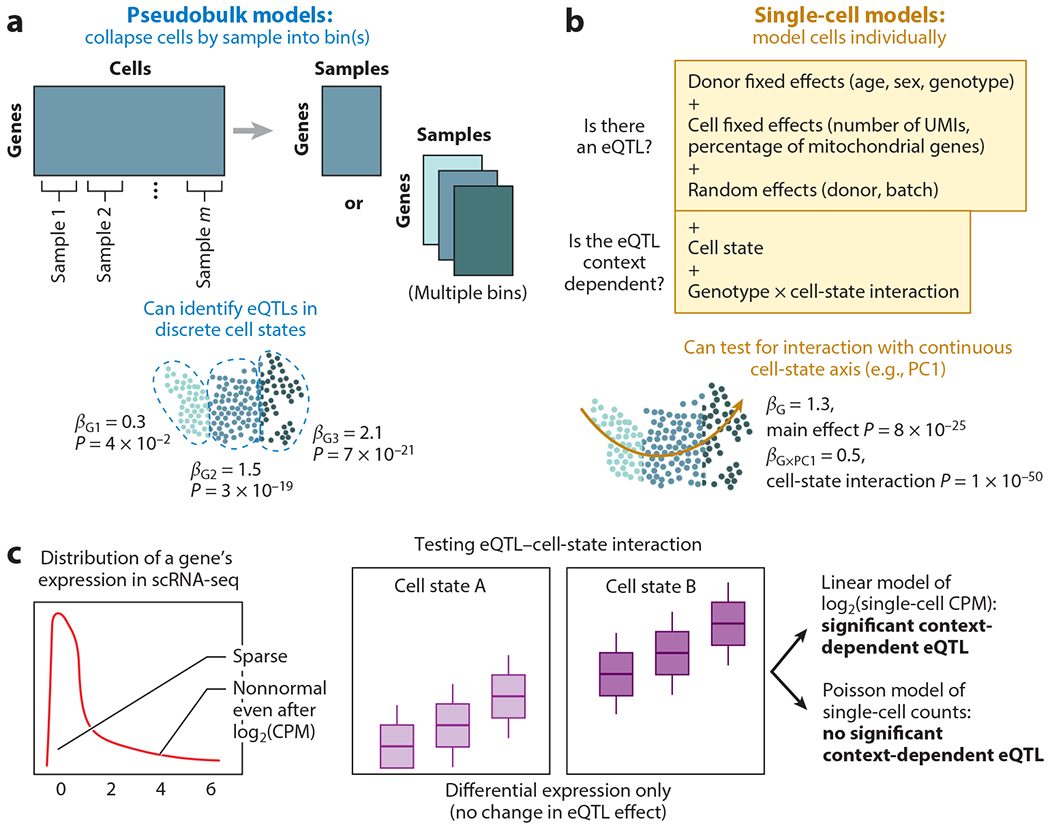
Modeling approaches for sc-eQTLs. (a) Pseudobulk models collapse cells (into potentially multiple discrete bins) and aggregate the cell profiles from each sample and bin. They can identify eQTLs by adapting methods for bulk analyses. (b) In sc-eQTL models, the expression of each cell is modeled individually; they are commonly implemented as mixed-effects models, with random effects for repeated sampling of cells from the same donor. They can test for eQTL interactions with both discrete and continuously defined cell states. (c) sc-eQTL modeling approaches must mitigate sparsity in single-cell measurements. In the presence of differential expression and sparsity, a linear mixed-effects model may identify a spurious interaction between eQTL and cell state, whereas a Poisson mixed-effects model remains robust. Abbreviations: CPM, counts per million; eQTL, expression quantitative trait locus; PC1, principal component 1; sc-eQTL, single-cell expression quantitative trait locus; scRNA-seq, single-cell RNA sequencing; UMI, unique molecular identifier. Figure adapted from images created with BioRender.com.
Each gene–SNP pair can then be tested for an association by using simple correlation or fitting a linear model:
| 1. |
where PEER stands for probabilistic estimation of expression residuals. Consistent with standard bulk eQTL analysis, expression can be modeled per sample as a function of dosage for each SNP, controlling for population stratification (e.g., using genotype PCs), sample-level covariates (e.g., age and sex), batch(es) (if applicable), and unmeasured sources of technical and biological variation as captured in expression PCs or PEER factors to increase power (134, 160). For more than two batches, one can use multiple indicator fixed effects or a random effect for the batch term. To test for the genotype main effect in each bin, the model can be run on each bin separately using linear regression (Equation 1).
To determine whether the effect of a SNP on a gene’s expression depends on cell state, one can test for heterogeneity across bins. This is modeled as interaction terms between cell-state bins and genotype,
| 2. |
and then compared with a null model without interactions. Because bins from the same donor are not independent, mixed-effects models are typically used: In Equation 2, a random effect for the donor term is included to account for the sampling of cell bins from the same donor, and one or more additional terms (the bin terms in Equation 2) are included to model potential differential expression (the bin’s effect on expression separate from its interaction with genotype). These kinds of eQTL interaction tests that include genotype interaction terms are common in bulk analyses (17, 36, 114).
Importantly, the resolution at which the bins are defined determines the cell-state variable that can be used to test for eQTL interactions using a pseudobulk model. The optimal resolution often depends on the question at hand. For example, to identify cell-type-dependent eQTLs at the level of major cell types (e.g., T cells versus monocytes; Figure 2d), one can cluster the cells at low resolution and define cell types using marker genes. For cell-state-dependent eQTLs, one can partition cells at increasing granularity, given enough cells per donor within each bin. Different approaches and challenges for unsupervised clustering have been reviewed (62, 77). Because de novo clustering and annotation can be subjective, an alternative is to perform label transfer from an external well-annotated reference dataset, using automated cell-type classification (3, 6, 26, 78, 141, 159, 173) or reference mapping (71, 90, 138), which offers speed and reproducibility advantages. For example, Yazar et al. (163) assigned 1.27 million immune cells to one of 14 different immune subtypes for pseudobulk analysis with semisupervised classification using PBMCs sorted by fluorescence-activated cell sorting as a reference (3). In the same study, the authors defined pseudobulk bins in a different way for a B cell–focused analysis: First, they defined a pseudotime trajectory from naive to memory B cells, and then they divided cells into six quantiles along the trajectory. By testing for an interaction between genotype and quantile rank using both linear and quadratic models, they found that ~17% of the 1,988 tested eQTLs showed dynamic effects during B cell maturation.
Two recent studies effectively used pseudobulk models to study sc-eQTLs in activated T cells. From a cohort of 89 donors, Schmiedel et al. (129) profiled more than 1 million CD4+ T cells activated in vitro with anti-CD3/CD28 beads. They defined 19 fine-grained subsets, which spanned from more abundant subtypes like naive Tregs and Th1 cells to rarer subtypes like CD4+ cytotoxic T lymphocytes. Pseudobulk analysis within each subset identified more than 4,000 eGenes in total, with the most prominent regulatory effects occurring in specific subsets. In a similar study, Soskic et al. (133) profiled 655,349 CD4+ T cells from 119 healthy individuals across a time course of stimulation (unstimulated and three activation time points). They defined 38 clusters, including fleeting states present only at certain activation time points, and found 6,407 eQTLs, of which approximately one-third were dynamically regulated during activation in an interaction analysis.
It is challenging to determine the appropriate resolution to model discrete cell states. Some cell types comprise a continuum of states rather than transcriptionally distinct states. Should one collapse all T cells into a single pseudobulk profile, separate profiles for CD4+ and CD8+ T cells, or profiles for even finer resolution states (e.g., Tregs)? Aggregating the cells into discrete groups may oversimplify potentially smooth and continuous gradients. Moreover, for very-high-resolution bins such as rare cell types (e.g., rare dendritic cells or innate lymphoid cell subsets), many individuals may not have enough cells of the cell type assayed, leading to reduced statistical power and inadequate mitigation of sparsity.
4.3. Single-Cell Models
Whereas pseudobulk approaches model the expression of a sample, single-cell models treat the expression for each cell as an observation (Figure 3b). Because they are not constrained by grouping cells a priori, single-cell models offer a principled strategy for unbiased identification of cell-state-specific regulatory effects. This is especially relevant when dynamic effects may be present in more granular cell states or along a continuous cell-state transition. Another advantage of single-cell models is in scenarios where the numbers of cells across individuals differ widely, which might affect variance properties when aggregating cells into pseudobulk. However, single-cell models are unable to use the established protocols for bulk eQTL mapping, and they often require more time-consuming computation to model cells individually. Single-cell models described to date include the Poisson mixed-effects (PME) model (106), the CellRegMap linear mixed model (30), and the GASPACHO (Gaussian Processes for Association Mapping Leveraging Cell Heterogeneity) Gaussian-process latent-variable model (81).
Nathan et al. (106) recently proposed the PME model to map sc-eQTLs. By modeling the raw gene expression counts in individual cells as a Poisson distribution (Equation 3; Figure 3b), this approach can handle sparsity in single-cell measurements. The model includes (a) fixed effects for donor-related covariates, including genotype, age, sex, and genotype PCs; (b) fixed effects for cell-level covariates, including expression PCs, cell quality (percentage of mitochondrial UMIs), and library size; and (c) random effects for repeated sampling of cells from donors (and batches, if applicable). To test for interactions with cell state, one can add cell-state variables and interaction terms for genotype with cell state, assessing significance by comparing with a null model without the interaction terms:
While cell states can be defined as discrete clusters, as in the pseudobulk framework, the single-cell framework also enables continuous cell-state definitions. For example, Nathan et al. (106, 107) assayed ~500,000 memory T cells from 259 individuals with prior resolved tuberculosis infection using CITE-seq (135). By integrating the RNA and protein information using canonical correlation analysis, the authors defined multiple axes of T cell state variation (canonical variates), where each cell has a score along each canonical variate. By modeling the way that each canonical variate modulated the regulatory effect (using interaction terms, e.g., genotype × cytotoxicity) and considering each cell’s unique combination of canonical variate scores, they calculated each cell’s own eQTL effect size by aggregating the contributions of all canonical variates together. Such interaction effects can be conceptualized as changes in the slope of the eQTL depending on a cell’s continuously defined cell state (Figure 2e). The memory T cell analysis revealed not only that approximately one-third of 6,511 pseudobulk lead eQTLs had significant cell-state interactions, but also that approximately two-thirds of secondary eQTLs (conditioned on the lead SNP) had dynamic effects, and some eGenes had multiple dynamic effects.
Cuomo et al. (30) recently proposed another sc-eQTL model called CellRegMap. It is conceptually similar to the PME model, except it is implemented as a linear mixed-effects (LME) model (83). Instead of modeling raw counts, CellRegMap models each cell’s log-transformed expression for a gene as a function of a persistent genotype effect, the cell’s state, and the cell-state interaction with genotype to test for dynamic effects. Like the PME model, it includes random effects to account for the repeated sampling of cells from the same donor. In an initial application, Cuomo et al. (30) reanalyzed eQTLs found in the Smart-seq2 dataset of ~30,000 iPSCs differentiating into endoderm (31). Their analysis revealed that 322 of the 4,470 tested eQTLs demonstrated significant interaction with continuous cell states defined using multiomics factor analysis (7), which infers latent factors explaining expression variation.
Importantly, when modeling very sparse measurements in large numbers of cells (e.g., droplet-based data), LME models may become less effective, especially when testing for state interactions. Nathan et al. (106) provided an example: rs2214911 is an eQTL for THAP5 in both CD4+ and CD8+ T cells and, importantly, does not interact with CD4+ versus CD8+ cell state (genotype × CD4 interaction P > 0.05). When the authors randomly downsampled THAP5 counts in CD4+ but not CD8+ cells, the LME model erroneously identified a significant eQTL interaction with CD4+ status where there is none (because the counts are downsampled randomly). Hence, with increased sparsity, LME models struggle with distinguishing differential expression from an eQTL interaction with cell state, whereas PME models remain robust (Figure 3c). For the CellRegMap iPSC example from Cuomo et al. (30, 31), this was less of an issue because of the higher library complexity in plate-based data. In a second application, Cuomo et al. (30) applied CellRegMap to ~148,000 cells from a droplet-based dopaminergic differentiation dataset. As a creative solution to handle the increased sparsity of 10x Genomics data relative to Smart-seq2, they aggregated individual cells into pseudocells (~17 cells per pseudocell) (12, 37).
Kumasaka et al. (81) introduced a third approach, GASPACHO. The underlying statistical framework uses a Gaussian-process latent-variable model—a flexible Bayesian nonparametric dimensionality reduction approach that can capture nonlinear trends, unlike PCA. While other studies have used such models to deconvolute single-cell dynamics (2, 124), GASPACHO introduced additional random-effect terms to account for donor variation or batches. This framework enables eQTL mapping by adding a term modeling the eQTL effect size as an additional Gaussian process that takes gene–environment interactions into account. Kumasaka et al. (81) applied this framework to full-length scRNA-seq of dermal fibroblasts from 68 donors stimulated to induce an antiviral response, demonstrating higher sensitivity and specificity when compared with pseudobulk eQTLs from the same data.
A major limitation of current single-cell models is the computational cost. Fitting a model to one SNP–gene pair using either the PME model or CellRegMap on a large single-cell dataset (>100,000 cells, >100 individuals, ~10 cell-state variables) takes on the order of minutes on a standard CPU, making it infeasible to test all SNPs in the cis-window for every gene. Because of this, in the PME memory T cell analysis (106) and CellRegMap iPSC analysis (30), single-cell models were used to test known eQTLs for cell-state interaction rather than for genome-wide discovery of eQTLs. The authors of these studies first nominated eQTLs with robust main effects using standard pseudobulk analyses, then used the single-cell model as a second pass to test for cell-state interactions. While effective, this approach may potentially miss hidden eQTLs that appear in specific cell states but are not detectable using pseudobulk models. In the GASPACHO fibroblast example (81), it was computationally tractable to test all variants in the cis-window for each gene because of the small dataset size (~20,000 cells). However, the authors noted that scaling to hundreds of thousands of cells will be prohibitive without more efficient inference techniques or GPUs. Apart from faster models, a potential way to improve efficiency is to reduce the effective number of cells by collapsing small neighborhoods into pseudocells (12, 30, 37), as was explored in CellRegMap.
As the field gains more experience with sc-eQTL models, it will be useful to benchmark the various strategies and ascertain the best use cases for each approach. For example, the modality of data (plate-based versus droplet-based) will likely affect model choice because some methods may handle sparsity better than others. The LME model in CellRegMap may be less appropriate for genes with lower expression levels (increased sparsity) but may offer faster model fitting for massive datasets.
5. LINKING EXPRESSION QUANTITATIVE TRAIT LOCI TO DISEASE LOCI
Unbiased eQTL discovery links variants to target genes, but identified eQTLs may not necessarily be important for disease. In fact, GTEx estimated that ~95% of all protein-coding genes have at least one eQTL (54), so an important downstream task is integrating eQTL maps with GWASs. For a given trait, linking eQTLs to associated variants can be achieved through colocalization analysis methods, such as coloc and eCAVIAR (eQTL and GWAS Causal Variants Identification in Associated Regions) (51, 52, 63). Colocalization is used to determine whether a causal variant is responsible for both the GWAS and eQTL signals in a given locus, providing stronger evidence that the regulatory effect contributes to disease (20). Designed for bulk RNA-seq, colocalization methods can be readily applied to pseudobulk sc-eQTLs (test eQTLs from each bin for colocalization), though it is less clear how to best perform colocalization with state interaction models.
In a previously mentioned study of stimulated CD4+ T cells, Soskic et al. (133) found that 127 eQTLs colocalized with GWAS variants for immune-mediated diseases, including eQTLs for CTLA4 and TYK2, which are clinically promising drug targets (8, 32). Importantly, only 40% of colocalizations could be detected in resting cells, highlighting the importance of assaying activation states. In the PBMC study by Yazar et al. (163), colocalization analysis linked 19% of the ~26,600 cis-eQTLs with GWAS risk variants. Technically, colocalization alone does not imply causality since colocalizing signals could also reflect that the GWAS hit and eQTL are two independent loci in tight linkage disequilibrium or that the variant affects both expression and the trait independently (pleiotropy) (20). Hence, the authors aimed to infer the direction of causality using Mendelian randomization, concluding that 305 loci showed evidence of modulating disease risk via affecting expression levels (163). In the study with SLE patients by Perez et al. (115), out of 43 SLE GWAS loci, 5 colocalized with cell-type-shared eQTLs, and 7 colocalized with cell-type-specific eQTLs. In a notable example, eQTLs colocalized with risk variants near ORMDL3, which was also found to harbor dynamic eQTLs by both Nathan et al. (106) and Yazar et al. (163) in T and B cells, respectively.
6. ALTERNATIVE APPROACHES AND OTHER EXPRESSION-RELATED MOLECULAR PHENOTYPES
Another way to find regulatory variation is through allele-specific expression—measuring the imbalance between the expression of alleles for a given gene in heterozygotes. Identifying allele-specific expression relies on RNA-seq reads overlapping heterozygous SNPs (21). Therefore, single-cell protocols with full-length transcript information are better for allele-specific expression. For example, Heinen et al. (61) used Smart-seq2 data from differentiating iPSCs (31) to quantify haplotype-resolved gene expression. For certain genes, the balance of expression between alleles shifted based on cell states, suggesting state-dependent activity of cis-regulatory programs. Although current full-length assays are typically plate-based and therefore limited in throughput, emerging higher-throughput full-length assays (58, 59) may enable the wider application of single-cell allele-specific expression.
Single-cell data can capture other molecular phenotypes beyond eQTLs that could not be measured in bulk RNA-seq. For example, the cell-to-cell variability of a gene’s expression may be genetically regulated. A recent study aimed to identify so-called variance QTLs using single-cell data from 5,447 iPSCs from 53 Yoruba individuals; however, they found very few variance QTLs and estimated that cohorts ofmore than 4,000 individuals are needed (127), though another group concluded otherwise (94).
Co-eQTLs are variants affecting the strength of coexpression between two genes across cells within an individual (86, 110, 147). Co-eQTLs cannot be calculated in bulk RNA-seq (with only one measurement per sample) but can quantify how genetic variation affects gene regulatory network relationships. They can also be thought of as a type of state-dependent eQTL, where one gene is the eGene for the eQTL and the other gene is a proxy capturing cell state.
Thus far, our discussion has focused on cis-eQTLs (affecting nearby genes), but bulk studies have shown that trans-eQTLs (affecting distant genes or those on different chromosomes) capture important trait-relevant regulation (49, 154). cis-eQTL effects can mediate trans-eQTLs; for example, an eQTL affecting a TF’s expression can affect downstream genes regulated by that TF (117, 154). A recent model estimated that heritability explained by trans-acting variants is at least 70% (88), though another study argued that whole-blood trans-eQTLs have limited influence on complex diseases (162). Because of the huge number of SNP-gene tests required to find trans-eQTLs genome-wide, the multiple testing burden is extremely high; historically, it has been hard to detect and reproduce trans-eQTLs in bulk without thousands of samples (117, 154). Furthermore, it is uncertain whether trans-eQTLs reflect true regulatory effects or capture cell-type abundance QTLs (151). Single-cell trans-eQTLs could be a fruitful future direction since single-cell data can deconvolute cell-type abundance from expression, and no consensus has been reached on the best modeling strategies.
7. VALIDATING SINGLE-CELL EXPRESSION QUANTITATIVE TRAIT LOCI
A key question is how to validate sc-eQTLs, especially those with predicted disease relevance. Comparing the concordance of the main effects with the results of bulk studies is an important first step. A second step would be to reproduce the signals in independent datasets with cells obtained in independent samples and ideally different expression assay platforms.
We anticipate that future studies will seek to validate eQTLs in the laboratory—for example, by using electrophoretic mobility shift assays to measure TF binding and massively parallel reporter assays to assess the effect of variants on reporter gene expression (reviewed in 123). While useful, these assays do not account for chromatin context. Another, more accurate approach is to use CRISPR-Cas systems (55, 123) to edit regulatory variants and observe their effects on expression and cellular phenotypes. However, validating context specificity in the relevant cell states where eQTLs were identified is not straightforward. For example, to prove that an eQTL depends on T cell cytotoxicity, one may sort cells by a cytotoxicity marker and show that the variant has a stronger effect in more cytotoxic populations.
Methods measuring chromatin accessibility at the single-cell level, such as single-cell ATAC sequencing (ATAC-seq) and multimodal snRNA/ATAC-seq (24), will provide an orthogonal lens by which to map functional elements to target genes and help validate sc-eQTLs. For example, in an analysis of single-nucleus ATAC-seq from eight brain cell types (18), many glial-specific peaks were enriched for eQTLs specifically discovered in glia-like cells, whereas eQTLs discovered in neurons were less specific to neuron-specific regulatory regions. It may become possible to pinpoint cell-state-specific TFs mediating regulatory effects by identifying TF binding motifs. Furthermore, mapping chromatin accessibility QTLs can help corroborate eQTLs or identify variants affecting chromatin structure in contexts without a detectable eQTL.
8. TECHNICAL CHALLENGES AND FUTURE DIRECTIONS
As the sc-eQTL field is still nascent, several methodological challenges remain. Batch effects remain a key technical issue in single-cell data. This may be especially relevant for large, population-scale data generation efforts taking place over many weeks or months and potentially involving personnel changes. Furthermore, cell types that are rare or difficult to assay due to lower RNA content or higher levels of RNases [e.g., neutrophils (53)] remain challenging to measure.
Similarly, some genes are harder to quantify accurately using standard pipelines. For example, the highly disease-relevant human leukocyte antigen (HLA) genes are very polymorphic across individuals (96, 125), and when short reads are mapped to the reference genome, this creates a bias toward individuals with alleles more similar to those in the reference. Hence, accurate mapping of eQTLs for HLA genes in single-cell data will require specialized gene quantification pipelines (34), as have been developed for bulk RNA-seq (1, 55). In fact, a recent study concluded that many genes may be inaccurately quantified in 3′ scRNA-seq datasets due to read-mapping challenges from poor 3′ untranslated-region annotation, intronic reads from unannotated exons, or reads mapping to multiple genes, which are usually discarded (118). Full-length data, 5′ data, or new assays coupling droplet-based methods with long-read sequencing (142) may yield different results since they capture different isoforms. Similarly, high-throughput single-cell full-length assays would help to map variants affecting transcript splicing (splicing QTLs), which contribute to disease risk but thus far have been systematically examined only using bulk RNA-seq (50, 75, 104, 119, 172).
As mentioned, an important direction on the modeling front is improving the efficiency of single-cell models. Applying single-cell models genome-wide in large cohorts has the potential to uncover previously hidden dynamic regulatory effects and take full advantage of single-cell resolution data. More efficient inference techniques, more powerful machines (e.g., GPUs), or approaches to reduce the effective size of a dataset with minimal information loss [e.g., pseudocells (12, 30)] may offer a way forward. Another opportunity is improving continuous cell-state definitions. Although they are highly flexible, low-dimensional cell embeddings derived from standard approaches like PCA, canonical correlation analysis, and multiomics factor analysis are not always clearly interpretable and may not encapsulate all cell states that may interact with eQTLs, including nonlinear effects. Finding the optimal interaction PCs—that is, the latent components that best maximize the eQTL effect sizes—is an active area of investigation (150).
Just as the eQTL Catalogue (74) was able to aggregate and meta-analyze data from multiple bulk studies, so we anticipate that future large-scale meta-analyses will be conducted for sc-eQTLs. How to best meta-analyze single-cell datasets—for example, defining shared cell-state variables across datasets generated from different groups—is an open question. One approach is to merge cell annotations between datasets (e.g., combining all myeloid cells from different studies), but this will lose information if some studies provide lower-resolution labels. Different groups may also use different criteria and naming schemes to define cell types. Another approach is to use single-cell reference mapping (71, 90, 138) to project cells from query datasets into continuous cell states defined using existing reference datasets. For example, Nathan et al. (106) used Symphony (71) to map memory T cells onto an ulcerative colitis colon T cell reference, using the reference’s low-dimensional embedding (PCs) to test for eQTL interactions with disease-related cell states.
Future efforts will construct cohorts comprising diverse ancestries, which will help define disease mechanisms occurring in specific populations, aid in fine mapping, and uncover evolutionary sources of phenotypic variation. Indeed, in the last three decades, technical advances have enabled investigators to perform high-coverage whole-genome sequencing on the remains of our closest known evolutionary relatives, the Neanderthals and the Denisovans, dating back more than 50,000 years ago. This sequencing revealed ~1–5% archaic introgression in the genomes of present-day Eurasians; these genomic regions are biologically important for human physiology (33) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pathogenesis (174). For example, potentially advantageous introgressed variants were found to be response-specific bulk eQTLs in monocytes (120) and subsequently experimentally validated to be responsible for potential immune responses (66). Interestingly, the previously mentioned OAS1 gene is a response eGene (81, 121) where the ancestral version of its eQTL (rs10774671-G) is derived from an introgression event from Neanderthal into Europeans. These findings motivate future efforts to unveil the ancestral origins of specific physiological and disease responses and their biological contexts at the single-cell level using similar approaches to those recently introduced by Aquino et al. (5).
As approaches for sc-eQTL studies mature, we anticipate that sc-eQTLs will deliver on their promise to help elucidate the molecular mechanisms underlying genetic associations with complex diseases. We anticipate that improved solid tissue disaggregation protocols and data quality will enable the single-cell equivalent of GTEx, effectively defining a comprehensive catalog of eQTLs in human tissue-specific single-cell states. We anticipate that at least some of the missing link between genetic association and regulatory variation will be uncovered by assaying a diverse array of precise cell types and cell-state contexts in large-scale cohorts (27, 146).
Future sc-eQTL studies will revolutionize our understanding of gene regulation in specific cellular contexts and help to illuminate fundamental biological processes, from the genetic control of differential expression to the landscape of active enhancers in different cell states. When eQTLs colocalize with disease variants, identifying the target genes and characterizing their context-dependent effects can help prioritize therapeutic targets and pathways driving pathogenesis. If disease-associated regulation varies with cell states, there may be opportunities to selectively target the most relevant states. The future of sc-eQTL analysis is exciting and will provide a high-resolution lens to understand genetically driven interindividual differences.
ACKNOWLEDGMENTS
J.B.K. is supported in part by the National Institute of General Medical Sciences (grant T32GM007753) and the National Institute of Allergy and Infectious Diseases (grant F30AI172238). N.S. is supported by the Wellcome Trust (grant 206194); the Cambridge British Heart Foundation (BHF) Centre of Research Excellence (grant RE/18/1/34212); the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre (BRC) Biomedical Resources Grant, University of Cambridge, Cardiovascular Theme (grant RG64226); and the Chan Zuckerberg Foundation. S.R. is supported in part by the National Institutes of Health (grants R01AR063759, U01HG012009, and UC2AR081023).
Glossary
- Genome-wide association study (GWAS)
a study that tests the association between common genome-wide variants and a trait (e.g., disease)
- Colocalization
analysis determining whether two genetically associated traits (e.g., a disease locus and eQTL) share a causal variant in a given locus
- Expression quantitative trait locus (eQTL)
a regulatory genetic variant that is associated with the expression of a gene nearby (cis-eQTL) or far away (trans-eQTL)
- Cell state
the potentially transient phenotype of a cell given internal and extrinsic factors, which can be defined in many ways (e.g., transcriptional state)
- Context-dependent eQTL
an eQTL that is altered by a cell’s context, such as cell state, stimulus, or environment; examples include dynamic, response, and state-dependent eQTLs
- State-dependent eQTL
a type of context-dependent eQTL that changes across finer-grained cell states within a given broader cell type
- Dynamic eQTL
a type of context-dependent eQTL that implies continuous or time-varying cell types or states
- Response eQTL (reQTL)
a type of context-dependent eQTL that changes in response to an external stimulus, such as immune cell activation
- Fine mapping
the process of narrowing the possible set of causal variants in a locus, which can be done statistically or using experimental strategies
- Linkage disequilibrium
the nonrandom assortment of alleles where certain variants tend to be inherited together, leading to a correlation between linked variants
- Cell-state variable
a variable used to define cell state quantitatively, which can be categorical (e.g., cluster) or continuous (e.g., pseudotime)
- Pseudobulk model
sc-eQTL modeling approach that aggregates gene counts for cells by donors and discrete groups (e.g., cell type or cluster)
- Single-cell model
sc-eQTL modeling approach that models each cell individually (e.g., by using mixed-effects models)
Footnotes
DISCLOSURE STATEMENT
S.R. is a scientific advisor to Pfizer, Janssen, Rheos Medicines, and Sonoma Biotherapeutics; a founder of Mestag Therapeutics; and a consultant for AbbVie and Sanofi.
LITERATURE CITED
- 1.Aguiar VRC, César J, Delaneau O, Dermitzakis ET,Meyer D. 2019. Expression estimation and eQTL mapping for HLA genes with a personalized pipeline. PLOS Genet. 15:e1008091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed S, Rattray M, Boukouvalas A. 2019. GrandPrix: scaling up the Bayesian GPLVM for single-cell data. Bioinformatics 35:47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alquicira-Hernandez J, Sathe A, Ji HP, Nguyen Q, Powell JE. 2019. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 20:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. 2010. Data quality control in genetic case-control association studies. Nat. Protoc 5:1564–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aquino Y, Bisiaux A, Li Z, O’Neill M, Mendoza-Revilla J, et al. 2022. Environmental and genetic drivers of population differences in SARS-CoV-2 immune responses. bioRxiv 2022.11.22.517073. 10.1101/2022.11.22.517073 [DOI] [Google Scholar]
- 6.Aran D, Looney AP, Liu L, Wu E, Fong V, et al. 2019. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20:163–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, et al. 2018. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol 14:e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armstrong AW, Gooderham M, Warren RB, Papp KA, Strober B, et al. 2023. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J. Am. Acad. Dermatol 88:29–39 [DOI] [PubMed] [Google Scholar]
- 9.Azodi CB, Zappia L, Oshlack A, McCarthy DJ. 2021. splatPop: simulating population scale single-cell RNA sequencing data. Genome Biol. 22:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, et al. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. PNAS 100:2610–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, et al. 2018. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLOS ONE 13:e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baran Y, Bercovich A, Sebe-Pedros A, Lubling Y, Giladi A, et al. 2019. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barreiro LB, Tailleux L, Pai AA, Gicquel B, Marioni JC, Gilad Y. 2012. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. PNAS 109:1204–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ben-David E, Boocock J, Guo L, Zdraljevic S, Bloom JS, Kruglyak L. 2021. Whole-organism eQTL mapping at cellular resolution with single-cell sequencing. eLife 10:e65857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bossini-Castillo L, Glinos DA, Kunowska N, Golda G, Lamikanra AA, et al. 2022. Immune disease variants modulate gene expression in regulatory CD4+ T cells. Cell Genom. 2:100117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Browning SR, Browning BL. 2011. Haplotype phasing: existing methods and new developments. Nat. Rev. Genet 12:703–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryois J, Buil A, Ferreira PG, Panousis NI, Brown AA, et al. 2017. Time-dependent genetic effects on gene expression implicate aging processes. Genome Res. 27:545–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryois J, Calini D, Macnair W, Foo L, Urich E, et al. 2022. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci 25:1104–12 [DOI] [PubMed] [Google Scholar]
- 19.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36:411–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cano-Gamez E, Trynka G. 2020. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet 11:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castel SE, Aguet F, Mohammadi P, GTEx Consort., Ardlie KG, Lappalainen T. 2020. A vast resource of allelic expression data spanning human tissues. Genome Biol. 21:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G,Ning B, Shi T. 2019. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet 10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Ge B, Casale FP, Vasquez L, Kwan T, et al. 2016. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167:1398–414.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Lake BB, Zhang K. 2019. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol 37:1452–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chun S, Casparino A, Patsopoulos NA, Croteau-Chonka DC, Raby BA, et al. 2017. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat. Genet 49:600–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clarke ZA, Andrews TS, Atif J, Pouyabahar D, Innes BT, et al. 2021. Tutorial: guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nat. Protoc 16:2749–64 [DOI] [PubMed] [Google Scholar]
- 27.Connally NJ, Nazeen S, Lee D, Shi H, Stamatoyannopoulos J, et al. 2022. The missing link between genetic association and regulatory function. eLife 11:e74970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crowell HL, Soneson C, Germain P-L, Calini D, Collin L, et al. 2020. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun 11:6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuomo ASE, Alvari G, Azodi CB, Single-Cell eQTLGen Consort., McCarthy DJ, Bonder MJ. 2021. Optimizing expression quantitative trait locus mapping workflows for single-cell studies. Genome Biol. 22:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuomo ASE, Heinen T, Vagiaki D, Horta D, Marioni JC, Stegle O. 2022. CellRegMap: a statistical framework for mapping context-specific regulatory variants using scRNA-seq. Mol. Syst. Biol 18:e10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuomo ASE, Seaton DD, McCarthy DJ, Martinez I, Bonder MJ, et al. 2020. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun 11:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cutolo M, Sulli A, Paolino S, Pizzorni C. 2016. CTLA-4 blockade in the treatment of rheumatoid arthritis: an update. Expert Rev. Clin. Immunol 12:417–25 [DOI] [PubMed] [Google Scholar]
- 33.Dannemann M, Prüfer K, Kelso J. 2017. Functional implications of Neandertal introgression in modern humans. Genome Biol. 18:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darby CA, Stubbington MJT, Marks PJ, Martínez Barrio Á, Fiddes IT. 2020. scHLAcount: allele-specific HLA expression from single-cell gene expression data. Bioinformatics 36:3905–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das S, Forer L, Schönherr S, Sidore C, Locke AE, et al. 2016. Next-generation genotype imputation service and methods. Nat. Genet 48:1284–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davenport EE, Amariuta T, Gutierrez-Arcelus M, Slowikowski K, Westra H-J, et al. 2018. Discovering in vivo cytokine-eQTL interactions from a lupus clinical trial. Genome Biol. 19:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Vries DH, Matzaraki V, Bakker OB, Brugge H, Westra H-J, et al. 2020. Integrating GWAS with bulk and single-cell RNA-sequencing reveals a role for LY86 in the anti-Candida host response. PLOS Pathog. 16:e1008408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denisenko E, Guo BB, Jones M, Hou R, de Kock L, et al. 2020. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeTomaso D, Jones MG, Subramaniam M, Ashuach T, Ye CJ, Yosef N. 2019. Functional interpretation of single cell similarity maps. Nat. Commun 10:4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, et al. 2020. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol 38:737–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong X, Li X, Chang T-W, Scherzer CR, Weiss ST, Qiu W. 2021. powerEQTL: an R package and shiny application for sample size and power calculation of bulk tissue and single-cell eQTL analysis. Bioinformatics 37:4269–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donovan MKR, D’Antonio-Chronowska A, D’Antonio M, Frazer KA. 2020. Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants. Nat. Commun 11:955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elorbany R, Popp JM, Rhodes K, Strober BJ, Barr K, et al. 2022. Single-cell sequencing reveals lineage-specific dynamic genetic regulation of gene expression during human cardiomyocyte differentiation. PLOS Genet. 18:e1009666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, et al. 2014. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343:1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fairfax BP, Makino S, Radhakrishnan J, Plant K, Leslie S, et al. 2012. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet 44:502–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan S, Hansen MEB, Lo Y, Tishkoff SA. 2016. Going global by adapting local: a review of recent human adaptation. Science 354:54–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farber DL. 2021. Tissues, not blood, are where immune cells function. Nature 593:506–9 [DOI] [PubMed] [Google Scholar]
- 48.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, et al. 2015. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518:337–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fehrmann RSN, Jansen RC, Veldink JH, Westra H-J, Arends D, et al. 2011. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLOS Genet. 7:e1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garrido-Martín D, Borsari B, Calvo M, Reverter F, Guigó R. 2021. Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun 12:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giambartolomei C, Liu JZ, Zhang W, Hauberg M, Shi H, et al. 2018. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 34:2538–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, et al. 2014. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLOS Genet. 10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grieshaber-Bouyer R, Radtke FA, Cunin P, Stifano G, Levescot A, et al. 2021. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat. Commun 12:2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.GTEx Consort. 2020. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369:1318–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gutierrez-Arcelus M, Baglaenko Y, Arora J, Hannes S, Luo Y, et al. 2020. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat. Genet 52:247–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutierrez-Arcelus M, Rich SS, Raychaudhuri S. 2016. Autoimmune diseases—connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet 17:160–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gutierrez-Arcelus M, Teslovich N, Mola AR, Polidoro RB, Nathan A, et al. 2019. Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions. Nat. Commun 10:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hagemann-Jensen M, Ziegenhain C, Sandberg R. 2022. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol 40:1452–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hahaut V, Pavlinic D, Carbone W, Schuierer S, Balmer P, et al. 2022. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat. Biotechnol 40:1447–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heaton H, Talman AM, Knights A, Imaz M, Gaffney DJ, et al. 2020. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods 17:615–20 [DOI] [PubMed] [Google Scholar]
- 61.Heinen T, Secchia S, Reddington JP, Zhao B, Furlong EEM, Stegle O. 2022. scDALI: modeling allelic heterogeneity in single cells reveals context-specific genetic regulation. Genome Biol. 23:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hie B, Peters J, Nyquist SK, Shalek AK, Berger B, Bryson BD. 2020. Computational methods for single-cell RNA sequencing. Annu. Rev. Biomed. Data Sci 3:339–64 [Google Scholar]
- 63.Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, et al. 2016. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet 99:1245–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu X, Kim H, Raj T, Brennan PJ, Trynka G, et al. 2014. Regulation of gene expression in autoimmune disease loci and the genetic basis of proliferation in CD4+ effector memory T cells. PLOS Genet. 10:e1004404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, et al. 2016. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jagoda E, Xue JR, Reilly SK, Dannemann M, Racimo F, et al. 2022. Detection ofNeanderthal adaptively introgressed genetic variants that modulate reporter gene expression in human immune cells. Mol. Biol. Evol 39:msab304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jerber J, Seaton DD, Cuomo ASE, Kumasaka N, Haldane J, et al. 2021. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet 53:304–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamariza M, Crawford L, Jones D, Finucane H. 2021. Misuse of the term “trans-ethnic” in genomics research. Nat. Genet 53:1520–21 [DOI] [PubMed] [Google Scholar]
- 69.Kaminow B, Yunusov D, Dobin A. 2021. STARsolo: accurate, fast and versatile mapping/quantification of single-cell and single-nucleus RNA-seq data. bioRxiv 2021.05.05.442755. 10.1101/2021.05.05.442755 [DOI] [Google Scholar]
- 70.Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, et al. 2018. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36:89–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang JB, Nathan A, Weinand K, Zhang F, Millard N, et al. 2021. Efficient and precise single-cell reference atlas mapping with Symphony. Nat. Commun 12:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kasela S, Kisand K, Tserel L, Kaleviste E, Remm A, et al. 2017. Pathogenic implications for autoimmune mechanisms derived by comparative eQTL analysis of CD4+ versus CD8+ T cells. PLOS Genet. 13:e1006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. 2020. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med 52:1419–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kerimov N, Hayhurst JD, Peikova K, Manning JR, Walter P, et al. 2021. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet 53:1290–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim-Hellmuth S, Aguet F, Oliva M, Muñoz-Aguirre M, Kasela S, et al. 2020. Cell type-specific genetic regulation of gene expression across human tissues. Science 369:eaaz8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim-Hellmuth S, Bechheim M, Pütz B, Mohammadi P, Nédélec Y, et al. 2017. Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat. Commun 8:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kiselev VY, Andrews TS, Hemberg M. 2019. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet 20:273–82 [DOI] [PubMed] [Google Scholar]
- 78.Kiselev VY, Yiu A, Hemberg M. 2018. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15:359–62 [DOI] [PubMed] [Google Scholar]
- 79.Knowles DA, Burrows CK, Blischak JD, Patterson KM, Serie DJ, et al. 2018. Determining the genetic basis of anthracycline-cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes. eLife 7:33480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, et al. 2019. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16:1289–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumasaka N, Rostom R, Huang N, Polanski K, Meyer KB, et al. 2021. Mapping interindividual dynamics of innate immune response at single-cell resolution. bioRxiv 2021.09.01.457774. 10.1101/2021.09.01.457774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kundu K, Tardaguila M, Mann AL, Watt S, Ponstingl H, et al. 2022. Genetic associations at regulatory phenotypes improve fine-mapping of causal variants for 12 immune-mediated diseases. Nat. Genet 54:251–62 [DOI] [PubMed] [Google Scholar]
- 83.Lähnemann D, Köster J, Szczurek E, McCarthy DJ, Hicks SC, et al. 2020. Eleven grand challenges in single-cell data science. Genome Biol. 21:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee J, Hyeon DY, Hwang D. 2020. Single-cell multiomics: technologies and data analysis methods. Exp. Mol. Med 52:1428–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee MN, Ye C, Villani A-C, Raj T, Li W, et al. 2014. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343:1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li S, Schmid KT, de Vries D, Korshevniuk M, Oelen R, et al. 2022. Identification of genetic variants that impact gene co-expression relationships using large-scale single-cell data. bioRxiv 2022.04.20.488925. 10.1101/2022.04.20.488925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu H, Prashant NM, Spurr LF, Bousounis P, Alomran N, et al. 2021. scReQTL: an approach to correlate SNVs to gene expression from individual scRNA-seq datasets. BMC Genom. 22:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu X, Li YI, Pritchard JK. 2019. Trans effects on gene expression can drive omnigenic inheritance. Cell 177:1022–34.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. 2018. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15:1053–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lotfollahi M, Naghipourfar M, Luecken MD, Khajavi M, Büttner M, et al. 2022. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol 40:121–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luckheeram RV, Zhou R, Verma AD, Xia B. 2012. CD4+ T cells: differentiation and functions. Clin. Dev. Immunol 2012:925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luecken MD, Theis FJ. 2019. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15:e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ma T, Li H, Zhang X. 2022. Discovering single-cell eQTLs from scRNA-seq data only. Gene 829:146520. [DOI] [PubMed] [Google Scholar]
- 94.Mandric I, Schwarz T, Majumdar A, Hou K, Briscoe L, et al. 2020. Optimized design of single-cell RNA sequencing experiments for cell-type-specific eQTL analysis. Nat. Commun 11:5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marchini J, Howie B. 2010. Genotype imputation for genome-wide association studies. Nat. Rev. Genet 11:499–511 [DOI] [PubMed] [Google Scholar]
- 96.Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. 2017. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 18:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, et al. 2021. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol 39:813–18 [DOI] [PubMed] [Google Scholar]
- 99.Millard N, Korsunsky I, Weinand K, Fonseka CY, Nathan A, et al. 2021. Maximizing statistical power to detect differentially abundant cell states with scPOST. Cell Rep. Methods 1:100120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mo A, Marigorta UM, Arafat D, Chan LHK, Ponder L, et al. 2018. Disease-specific regulation of gene expression in a comparative analysis of juvenile idiopathic arthritis and inflammatory bowel disease. Genome Med. 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moltke I, Grarup N, Jørgensen ME, Bjerregaard P, Treebak JT, et al. 2014. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512:190–93 [DOI] [PubMed] [Google Scholar]
- 102.Mostafavi H, Spence JP, Naqvi S, Pritchard JK. 2022. Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. bioRxiv 2022.05.07.491045. 10.1101/2022.05.07.491045 [DOI] [Google Scholar]
- 103.Moyerbrailean GA, Richards AL, Kurtz D, Kalita CA, Davis GO, et al. 2016. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 26:1627–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mu Z, Wei W, Fair B, Miao J, Zhu P, Li YI. 2021. The impact of cell type and context-dependent regulatory variants on human immune traits. Genome Biol. 22:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Naranbhai V, Fairfax BP, Makino S, Humburg P, Wong D, et al. 2015. Genomic modulators of gene expression in human neutrophils. Nat. Commun 6:7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nathan A, Asgari S, Ishigaki K, Valencia C, Amariuta T, et al. 2022. Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature 606:120–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nathan A, Beynor JI, Baglaenko Y, Suliman S, Ishigaki K, et al. 2021. Multimodally profiling memory T cells from a tuberculosis cohort identifies cell state associations with demographics, environment and disease. Nat. Immunol 22:781–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Neavin D, Nguyen Q, Daniszewski MS, Liang HH, Chiu HS, et al. 2021. Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells. Genome Biol. 22:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nédélec Y, Sanz J, Baharian G, Szpiech ZA, Pacis A, et al. 2016. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167:657–69.e21 [DOI] [PubMed] [Google Scholar]
- 110.Oelen R, de Vries DH, Brugge H, Gordon MG, Vochteloo M, et al. 2022. Single-cell RNA-sequencing of peripheral blood mononuclear cells reveals widespread, context-specific gene expression regulation upon pathogenic exposure. Nat. Commun 13:3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.O’Neill MB, Quach H, Pothlichet J, Aquino Y, Bisiaux A, et al. 2021. Single-cell and bulk RNA-sequencing reveal differences in monocyte susceptibility to influenza A virus infection between Africans and Europeans. Front. Immunol 12:768189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Osorio D, Cai JJ. 2021. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics 37:963–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ota M, Nagafuchi Y, Hatano H, Ishigaki K, Terao C, et al. 2021. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 184:3006–21.e17 [DOI] [PubMed] [Google Scholar]
- 114.Patel D, Zhang X, Farrell JJ, Chung J, Stein TD, et al. 2021. Cell-type-specific expression quantitative trait loci associated with Alzheimer disease in blood and brain tissue. Transl. Psychiatry 11:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Perez RK, Gordon MG, Subramaniam M, Kim MC, Hartoularos GC, et al. 2022. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376:eabf1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. 2014. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9:171–81 [DOI] [PubMed] [Google Scholar]
- 117.Pierce BL, Tong L, Chen LS, Rahaman R, Argos M, et al. 2014. Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLOS Genet. 10:e1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pool A-H, Poldsam H, Chen S, Thomson M, Oka Y. 2022. Enhanced recovery of single-cell RNA-sequencing reads for missing gene expression data. bioRxiv 2022.04.26.489449. 10.1101/2022.04.26.489449 [DOI] [Google Scholar]
- 119.Qi T, Wu Y, Fang H, Zhang F, Liu S, et al. 2022. Genetic control of RNA splicing and its distinct role in complex trait variation. Nat. Genet 54:1355–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quach H, Rotival M, Pothlichet J, Loh Y-HE, Dannemann M, et al. 2016. Genetic adaptation and Neandertal admixture shaped the immune system of human populations. Cell 167:643–56.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Randolph HE, Fiege JK, Thielen BK, Mickelson CK, Shiratori M, et al. 2021. Genetic ancestry effects on the response to viral infection are pervasive but cell type specific. Science 374:1127–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, et al. 2017. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542:110–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rao S, Yao Y, Bauer DE. 2021. Editing GWAS: experimental approaches to dissect and exploit disease-associated genetic variation. Genome Med. 13:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reid JE, Wernisch L. 2016. Pseudotime estimation: deconfounding single cell time series. Bioinformatics 32:2973–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sakaue S, Gurajala S, Curtis M, Luo Y, Choi W, et al. 2022. A statistical genetics guide to identifying HLA alleles driving complex disease. bioRxiv 2022.08.24.504550. 10.1101/2022.08.24.504550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sarkar AK, Stephens M. 2021. Separating measurement and expression models clarifies confusion in single-cell RNA sequencing analysis. Nat. Genet 53:770–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sarkar AK, Tung P-Y, Blischak JD, Burnett JE, Li YI, et al. 2019. Discovery and characterization of variance QTLs in human induced pluripotent stem cells. PLOS Genet. 15:e1008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schmid KT, Höllbacher B, Cruceanu C, Böttcher A, Lickert H, et al. 2021. scPower accelerates and optimizes the design of multi-sample single cell transcriptomic studies. Nat. Commun 12:6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schmiedel BJ, Gonzalez-Colin C, Fajardo V, Rocha J, Madrigal A, et al. 2022. Single-cell eQTL analysis of activated T cell subsets reveals activation and cell type-dependent effects of disease-risk variants. Sci. Immunol 7:eabm2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sheng X, Guan Y, Ma Z, Wu J, Liu H, et al. 2021. Mapping the genetic architecture of human traits to cell types in the kidney identifies mechanisms of disease and potential treatments. Nat. Genet 53:1322–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, et al. 2020. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26:792–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, et al. 2019. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178:714–30.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Soskic B, Cano-Gamez E, Smyth DJ, Ambridge K, Ke Z, et al. 2022. Immune disease risk variants regulate gene expression dynamics during CD4+ T cell activation. Nat. Genet 54:817–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stegle O, Parts L, Piipari M, Winn J, Durbin R. 2012. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc 7:500–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, et al. 2017. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14:865–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, et al. 2018. Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strober BJ, Elorbany R, Rhodes K, Krishnan N, Tayeb K, et al. 2019. Dynamic genetic regulation of gene expression during cellular differentiation. Science 364:1287–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, et al. 2019. Comprehensive integration of single-cell data. Cell 177:1888–902.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stubbington MJT, Rozenblatt-Rosen O, Regev A, Teichmann SA. 2017. Single-cell transcriptomics to explore the immune system in health and disease. Science 358:58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Swanson E, Lord C, Reading J, Heubeck AT, Genge PC, et al. 2021. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. eLife 10:63632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tan Y, Cahan P. 2019. SingleCellNet: a computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst. 9:207–13.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tian L, Jabbari JS, Thijssen R, Gouil Q, Amarasinghe SL, et al. 2021. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol. 22:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Townes FW, Hicks SC, Aryee MJ, Irizarry RA. 2019. Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model. Genome Biol. 20:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, et al. 2020. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Trapnell C. 2015. Defining cell types and states with single-cell genomics. Genome Res. 25:1491–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Umans BD, Battle A, Gilad Y. 2021. Where are the disease-associated eQTLs? Trends Genet. 37:109–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.van der Wijst MGP, Brugge H, de Vries DH, Deelen P, Swertz MA, et al. 2018. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet 50:493–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vieira Braga FA, Miragaia RJ. 2019. Tissue handling and dissociation for single-cell RNA-Seq. In Single Cell Methods, ed. Proserpio V, pp. 9–21. New York: Humana; [DOI] [PubMed] [Google Scholar]
- 149.Vieth B, Ziegenhain C, Parekh S, Enard W, Hellmann I. 2017. powsimR: power analysis for bulk and single cell RNA-seq experiments. Bioinformatics 33:3486–88 [DOI] [PubMed] [Google Scholar]
- 150.Vochteloo M, Deelen P, Vink B, BIOS Consort.,Tsai EA, et al. 2022. Unbiased identification of unknown cellular and environmental factors that mediate eQTLs using principal interaction component analysis. bioRxiv 2022.07.28.501849. 10.1101/2022.07.28.501849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, et al. 2021. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet 53:1300–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ward MC, Banovich NE, Sarkar A, Stephens M, Gilad Y.2021. Dynamic effects of genetic variation on gene expression revealed following hypoxic stress in cardiomyocytes. eLife 10:e57345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Westra H-J, Arends D, Esko T, Peters MJ, Schurmann C,et al. 2015. Cell specific eQTL analysis without sorting cells. PLOS Genet. 11:e1005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Westra H-J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, et al. 2013. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet 45:1238–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, et al. 2013. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol 31:748–52 [DOI] [PubMed] [Google Scholar]
- 156.Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, et al. 2019. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570:514–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wolock SL, Lopez R, Klein AM. 2019. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8:281–91.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wu H, Kirita Y, Donnelly EL, Humphreys BD. 2019. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol 30:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu C, Lopez R, Mehlman E, Regier J, Jordan MI, Yosef N. 2021. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Mol. Syst. Biol 17:e9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xue A, Yazar S, Neavin D, Powell JE. 2022. Pitfalls and opportunities for applying PEER factors in single-cell eQTL analyses. bioRxiv 2022.08.02.502566. 10.1101/2022.08.02.502566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yao DW, O’Connor LJ, Price AL, Gusev A. 2020. Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet 52:626–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yap CX, Lloyd-Jones L, Holloway A, Smartt P, Wray NR, et al. 2018. Trans-eQTLs identified in whole blood have limited influence on complex disease biology. Eur. J. Hum. Genet 26:1361–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yazar S, Alquicira-Hernandez J, Wing K, Senabouth A, Gordon MG, et al. 2022. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376:eabf3041. [DOI] [PubMed] [Google Scholar]
- 164.Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. 2021. Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat. Rev. Endocrinol 17:455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yoo T, Joo SK, Kim HJ, Kim HY, Sim H, et al. 2021. Disease-specific eQTL screening reveals an anti-fibrotic effect of AGXT2 in non-alcoholic fatty liver disease. J. Hepatol 75:514–23 [DOI] [PubMed] [Google Scholar]
- 166.Young AMH, Kumasaka N, Calvert F, Hammond TR, Knights A, et al. 2021. A map of transcriptional heterogeneity and regulatory variation in human microglia. Nat. Genet 53:861–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zappia L, Phipson B, Oshlack A. 2017. Splatter: simulation of single-cell RNA sequencing data. Genome Biol. 18:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zeng H. 2022. What is a cell type and how to define it? Cell 185:2739–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang F, Jonsson AH, Nathan A, Wei K, Millard N, et al. 2022. Cellular deconstruction of inflamed synovium defines diverse inflammatory phenotypes in rheumatoid arthritis. bioRxiv 2022.02.25.481990. 10.1101/2022.02.25.481990 [DOI] [Google Scholar]
- 170.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, et al. 2019. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol 20:928–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang X, Li T, Liu F, Chen Y, Yao J, et al. 2019. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell 73:130–42.e5 [DOI] [PubMed] [Google Scholar]
- 172.Zhang Y, Yang HT, Kadash-Edmondson K, Pan Y, Pan Z, et al. 2020. Regional variation of splicing QTLs in human brain. Am. J. Hum. Genet 107:196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang Z, Luo D, Zhong X, Choi JH, Ma Y, et al. 2019. SCINA: a semi-supervised subtyping algorithm of single cells and bulk samples. Genes 10:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou S, Butler-Laporte G, Nakanishi T, Morrison DR, Afilalo J, et al. 2021. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med 27:659–67 [DOI] [PubMed] [Google Scholar]