

Abstract
Single-cell genomic technologies are revealing the cellular composition, identities and states in tissues at unprecedented resolution. They have now scaled to the point that it is possible to query samples at the population level, across thousands of individuals. Combining single-cell information with genotype data at this scale provides opportunities to link genetic variation to the cellular processes underpinning key aspects of human biology and disease. This strategy has potential implications for disease diagnosis, risk prediction and development of therapeutic solutions. But, effectively integrating large-scale single-cell genomic data, genetic variation and additional phenotypic data will require advances in data generation and analysis methods. As single-cell genetics begins to emerge as a field in its own right, we review its current state and the challenges and opportunities ahead.
Introduction
Genome-wide association studies (GWASs) have uncovered hundreds of thousands of genetic variants associated with the risk of complex diseases and human traits. However, the majority of mechanisms linking these variants to their biological impact still need to be characterized, especially for variants found in non-protein-coding regions of the genome1. Expression quantitative trait locus (eQTL) mapping, which estimates the association between genetic variants (particularly SNPs) and RNA levels of either local or distal genes, can link variants to the putative target genes that they regulate. In addition, mapping of eQTLs, or other molecular QTLs2, can help characterize the modes of action of disease-associated genetic variation. This approach can help identify the genes – and consequently, the pathways and processes – that may be involved in disease pathogenesis3, which is a critical early step in identifying opportunities for therapeutic intervention.
For eQTL mapping to provide disease insights, changes in RNA expression levels must be assayed in the specific cell types and conditions relevant to the disease of interest, as the transcriptome and its regulatory mechanisms are dynamic and frequently context-dependent4. Seminal studies have demonstrated how eQTLs may only be detected in certain cell types5 or upon stimulation (that is, response eQTLs6,7). Additionally, recent efforts have assayed eQTLs across many human tissues; most notably, the Genotype-Tissue Expression Consortium8 has mapped eQTLs in more than 50 human tissues obtained from post-mortem donors. These traditional eQTL studies use bulk transcriptomes, which assess average expression levels across millions of cells from either whole tissues or cell-type samples. Using experimental (for example, fluorescence-activated cell sorting (FACS) and in vitro differentiation) and computational (for example, deconvolution) tools, bulk studies revealed some of the earliest insights into eQTLs specific to a cell type or transient state9-11. However, bulk studies are limited in their resolution of rare cell states or lack surface proteins with robust antibodies for FACS. Moreover, some transient or dynamic states cannot be recapitulated in vitro. These limitations reduce the utility of bulk eQTLs for understanding the biology of disease-associated variants: although tissue-level eQTLs are enriched for disease-associated genetic variants from GWASs, only 20–50% of common disease alleles colocalize with eQTLs12-14, which suggests that many variants influence biology through cell-state-specific mechanisms that cannot be identified without fundamentally new approaches.
Single-cell genomic technologies, particularly single-cell transcriptomics (that is, single-cell RNA sequencing (scRNA-seq)), offer a solution. As these approaches, which measure expression levels in individual cells, have become prevalent in recent years, they have revealed unanticipated cellular heterogeneity in many biological systems15-17. In addition, recent advances in technology, algorithms and experimental design have reduced the cost of scRNA-seq, making it more comparable to bulk RNA-seq and thus feasible to deploy across thousands of individuals18. This approach allows researchers to combine the granularity of single-cell assays with the large sample sizes required for genetic association studies, enabling a new category of ‘single-cell genetics’ studies that most prominently feature single-cell eQTL (sc-eQTL) studies.
The number of published sc-eQTL studies has more than doubled between January and December 2022 (Fig. 1), and international initiatives such as the single-cell eQTLGen Consortium (established in 2020 (ref. 19)) are attempting to harmonize efforts in this space. sc-eQTL studies have started to tackle questions that could not be asked with bulk expression data, such as finding eQTLs that vary with the cellular context or identifying the cell states in which disease-associated variants modulate gene expression. Context-specific, high-resolution maps of expression across deeply phenotyped individuals will eventually be valuable for therapeutic development.
Fig. 1∣. Overview of single-cell expression quantitative trait locus studies.

Single-cell studies published in the past 5 years. On the x axis is the date of publication, and on the y-axis is the number of unique individuals considered. The size of the dots represents the average number of cells per individual included in each study (when this number was not reported in this study, we estimated it as the total number of cells divided by the total number of individuals).
In this Review, we first briefly review single-cell genomics and human genetics, before focusing our attention on their intersection. Next, we review the first sc-eQTL studies, which demonstrate the feasibility of applying bulk analysis approaches to single-cell data. We discuss unanticipated challenges that become relevant when compared with traditional studies using bulk RNA-seq. Next, we highlight newer approaches using the single-cell resolution provided by scRNA-seq data, such as mapping eQTLs that vary along continuous trajectories. Finally, we provide an overview of key future directions for the field, including new data types and integration strategies, and translation to clinical and therapeutic applications.
A brief review of contributing fields
We define single-cell genetics as the emerging field at the intersection of single-cell genomics and human genetics. The two contributing fields each have opportunities, challenges and bottlenecks. Here, we review relevant gaps and synergies at this intersection (Fig. 2) and introduce concepts that provide the necessary context for this Review.
Fig. 2∣. Human genetics and single-cell genomics, a 20-year timeline.

Fundamental genomic resources (red), genetic studies (blue), sequencing technologies (yellow) and statistical methods and software (green) have contributed to the current state of single-cell genomics and human genetics, including expression quantitative trait locus (eQTL) mapping studies. References 42-44,49,52 and 170-176 are for landmark studies and initiatives, respectively; refs. 22,24,27,57,58,124 and 177-183 are for technological and statistical advances, respectively; and refs. 5,19,51,61,63,155,184-189 are for eQTL mapping. GWAS, genome-wide association study; HCA, Human Cell Atlas; PRS, polygenic risk score; RNA-seq, RNA sequencing; snRNA-seq, single-nucleus RNA sequencing; TWAS, transcriptome-wide association study.
Single-cell genomics
Over the past decade, single-cell genomics has rapidly demonstrated its value for studying human biology20. scRNA-seq is the most common of the single-cell modalities, and it has scaled quickly21 since its development in 2009: from only eight cells in the original publication22 to over 4 million cells in a recent study23. The most popular methods today for capturing RNA from single cells are droplet-based techniques24,25, which scale to tens of thousands of cells. Here, single cells are encapsulated inside microdroplets containing unique oligonucleotide-barcoded gel beads. When the cells are lysed, their mRNA molecules hybridize to the barcode and can be sequenced with a label corresponding to their cell of origin. Alternative methods are plate-based single-cell RNA-seq techniques (for example, Smart-seq3 (ref. 26)), in which cells are physically separated into 96-well or 384-well plates – with one cell per well – before library preparation and sequencing of full-length transcripts. Finally, in cases in which isolating viable single cells is technically challenging (for example, from frozen samples), single-nucleus RNA sequencing27 is a valuable alternative (Box 1).
Box 1. Experimental design trade-offs and considerations.
As single-cell studies expand from hundreds or thousands of individuals to even larger cohorts, experimental design will have important implications on downstream analyses.
More individuals or more cells per individual?
Assuming budget constraints limit the total number of cells that can be assayed, researchers face a trade-off between maximizing the number of cells per individual or the total number of individuals. More unique, unrelated individuals will increase the power for genetic associations, especially with rarer variants. By contrast, more cells per individual may capture rarer cell types, although it increases the chance of doublets.
Multiplexing strategies
Large-scale single-cell experiments often multiplex samples in library preparation and sequencing and computationally assign cells to individuals a posteriori. This increases throughput and reduces cost and batch effects, while improving doublet detection. Yet choosing the optimal number of individuals per pool is not trivial. Combining more samples into one pool may mitigate batch effects, but can increase doublets and decrease sequencing coverage per individual.
Single cell versus single nucleus
Single-cell transcriptomic assays measure RNA either from whole cells (single-cell RNA sequencing) or from isolated nuclei (single-nucleus RNA sequencing). The latter is preferred for frozen or hard-to-dissociate tissues, where nuclei remain intact even under stress. The transcriptomic profiles are largely concordant, but there are inherent trade-offs. Single-nucleus RNA sequencing detects intronic pre-mRNA but cannot measure transcripts outside the nucleus, for example, mitochondrial genes. Cells that are more sensitive to the stress of dissociation, such as myocytes, are under-represented in single-cell RNA sequencing.
Scaling to large data sets
Scaling experiments to thousands of individuals requires logistical considerations. First, strategies to monitor the quality of cells and consistency of output (total number of cells, cell-type composition and doublet rate) across samples can minimize compounding effects of batch as well as human error. Analyses should consider scale to optimize memory and computations for increasingly large data sets by parallelizing, using graphics processing unit and storing data in sparse matrices
In the past 10–15 years, technological improvements in single-cell data collection have produced new analytical considerations distinct from those for bulk RNA-seq data: for example, the massive number of profiles generated by a typical experiment, the sparsity of the data and a spectrum of technical artefacts. Novel methods have been developed to address these challenges. Single-cell-specific bioinformatics workflows such as Cell Ranger24 perform raw data processing tasks, for example, read-level quality control, assignment of reads to their cell barcodes and RNA molecules of origin (that is, ‘demultiplexing’), alignment to the reference genome and quantification. The data from an scRNA-seq experiment are typically represented as an integer matrix of the number of sequenced reads (or molecules, if unique molecular identifiers (UMI) were used) assigned to each gene in each cell28. For multi-individual pooled designs (particularly relevant for single-cell genetic studies), demultiplexing methods are necessary to assign cells to individuals of origin (for example, demuxlet29 and vireo30). After generating these count matrices, the next common stage in an scRNA-seq analysis workflow31-33 is pre-processing: for example, detection (and exclusion) of empty droplets, doublets and ambient RNA (which can confound associations with true single-cell expression measurements); normalization to adjust for total sequencing depth of cells (total number of reads); log transformation and correction for confounding factors including technical batch and cell cycle effects. Each of these steps is reviewed elsewhere31-33.
Subsequently, downstream analyses can be applied to the preprocessed data. To reduce the computational burden, reduce noise and facilitate visualization, it is beneficial first to reduce the dimensionality of the data set. Feature selection reduces the data to, for example, highly variable genes34,35. Then, dimensionality reduction using linear methods such as principal component analysis (PCA) and non-negative matrix factorization is typically performed to aggregate signals across genes. These reduced dimensions can be used for visualization purposes either directly or via feeding to nonlinear transformations (for example, t-distributed stochastic neighbour embedding (t-SNE)36 and uniform manifold approximation and projection (UMAP)37), which can further reduce dimensionality to two dimensions without the information loss that would occur if linear constraints were maintained.
Additionally, reduced dimensions can be used for subsequent downstream analyses. These include cell-level analyses to identify cell states and their dynamic relationships (for example, clustering, cell-type annotation or trajectory inference) and gene-level analyses to characterize the transcriptional profiles of these states (for example, differential expression or gene regulatory networks). Software to conduct these analyses is often available as part of extremely popular and comprehensive computational toolkits that create user-friendly single-cell workflows and consistent data objects. These toolkits are available in both R (for example, Seurat38 and scran39) or Python (for example, Scanpy40). Recommended methodologies and parameters for these steps are reviewed elsewhere31-33.
Impact of genetic variation on molecular phenotypes
In the two decades since the completion of the first human genome sequence41, rapid advances in sequencing technology have enabled increasingly larger genome sequencing projects and the characterization of human genetic variation across hundreds of thousands of individuals42-44. For common (population minor allele frequency >5%) and near-common (1–5%) variation, genotype arrays provide a popular solution to measure genotypes at approximately 500,000 ‘tagged’ loci systematically, and their low cost enables usage for large cohorts. DNA sequencing approaches additionally resolve rare (population minor allele frequency <1%) and structural genetic variation and can be applied to either protein-coding regions and their flanking sequences only (whole-exome sequencing) or the entire genome (whole-genome sequencing), using either cheaper short-read sequencing or more comprehensive, but substantially more expensive long-read approaches45,46.
In the setting of severe monogenic diseases, the application of DNA sequencing methods in both research and clinical settings has improved the rate of genetic diagnosis and disease gene discovery47,48. In addition, for complex traits and common diseases, GWASs have led to the identification of more than 400,000 genetic associations1 and the development of polygenic risk scores (PRSs), which combine association signals across the genome to predict the risk of disease of an individual49.
Studies of genetic variation can be combined with functional genomic assays to assess the potential biological impact of individual variants directly. The most popular approach is expression (e)QTL mapping, but similar frameworks can be used for DNA methylation, protein, histone modification, chromatin accessibility and splicing, reviewed elsewhere2. Because we expect most regulatory regions to be near their target, most QTL studies have focused on proximal (cis) mapping, for example, considering variants in and around the gene, methylation site or accessibility peak of interest. By contrast, trans-QTL mapping considers distal inter-chromosomal regulation but requires larger sample sizes50.
At present, the sample sizes of QTL studies are several orders of magnitude smaller than those of GWASs (for example, ~30,000 in the largest blood eQTL study51 versus >5 million individuals in the latest height GWASs52) owing to both cost considerations and the challenges of obtaining suitable tissue samples at the population scale. Fortunately, the magnitude of genetic effects on molecular traits is generally much larger than that on disease risk, and thus these sample sizes are sufficient to identify them. Although traditional QTL studies have considered common SNPs, approaches exist to interrogate the role of rare variants on, for example, the expression level. However, these remain largely limited to the study of rare variation in individuals with extreme phenotypes (that is, outlier analyses53,54), with few exceptions55.
Linking QTL results to GWAS results can reveal the molecular function of disease-associated genetic variants, but this task remains nontrivial56. To better understand the disease relevance of QTLs, methods have been developed to assess whether they coincide with disease loci (statistical colocalization57) or whether their effect on an intermediate molecular trait is causal for disease (two-step Mendelian randomization58), which have been reviewed elsewhere56. Transcriptome-wide association studies (TWASs) leverage eQTL information to impute gene expression for GWAS cases and controls and then perform direct association of traits and genes without directly profiling gene expression in every individual59,60.
Single-cell eQTL mapping using pseudo-bulk counts
Reduction in sequencing costs, well-established methodologies, processing pipelines, multiplexing techniques and batch-effect-removal methods enable the application of single-cell genomics (particularly transcriptomics) to large, genotyped cohorts. Furthermore, in single-cell genetics studies, using single-cell molecular profiling and genotypes from the same individuals enables the evaluation of the effects of genetic variants on molecular phenotypes at the level of a cell. Here, we focus on sc-eQTL studies, which test associations between genetic variants and changes in gene expression at single-cell resolution.
Proof-of-concept and early cell-type studies
In a 2013 study61, sc-eQTLs were first mapped, motivated by the observation that averaging expression over many cells (as is done in bulk studies) would mask certain gene expression phenotypes such as transcriptional bursting, noise and dynamic expression fluctuation. Limited to WNT pathway genes in 15 lymphoblastoid cell lines, the demonstration of the authors that SNPs are associated with transcript variance and correlation across single cells, nevertheless, served as an initial proof of concept61. It was an early example highlighting the value of single-cell-resolved gene expression in genetic studies. Within the next 5 years, a few subsequent studies demonstrated the feasibility of transcriptome-wide sc-eQTL analyses29,62. These studies leveraged single-cell advances in assaying, demultiplexing and clustering cells and focused on well-delineated immune cell types within easily accessible human peripheral blood. Despite limited sample sizes (<50 individuals), these studies found tens to hundreds of eQTLs.
These studies established a preliminary approach for sc-eQTL analyses: measure single-cell gene expression in a genotyped cohort, cluster phenotypically similar cells and associate the aggregated expression of each gene in each cluster or cell type with genotypes of individuals at nearby variants. This approach, called the ‘pseudobulk’ eQTL analysis, which we discuss further in the next section, had the advantage of building on existing bulk eQTL pipelines, making it computationally scalable to progressively larger cohorts (the current largest sc-eQTL study considers nearly 1,000 individuals63). Moreover, this approach was compatible with more sophisticated methods to organize single-cell phenotypes, such as bins along a trajectory or high-resolution cell-state clusters, allowing the approach to be extended to more heterogeneous tissues and granular cell types, including immune cells63-68 (with a particular focus on T cells65,67,68), induced pluripotent stem (iPS) cells and differentiating iPS cells69-73 (including iPS cell-derived cardiomyocytes72, dopaminergic neurons70 and retinal ganglion cells73), fibroblasts74 and brain cells75.
Methods originally devised for bulk eQTL mapping
Initial sc-eQTL studies largely used association methods originally devised for bulk eQTL mapping and other association tests between genotypes and continuous traits (Box 2). These methods assume that (1) the distribution of a phenotype across all samples is approximately Gaussian and (2) only one phenotype observation is available for each individual. These two assumptions do not necessarily hold for single-cell expression data, which in general are much sparser, and contain multiple observations of each phenotype (that is, expression level from multiple cells) per individual. To overcome this discrepancy, many studies have relied on pseudo-bulk strategies, in which gene expression levels are aggregated across multiple cells from a given individual to mimic a single bulk sample. The expression of a gene in the pseudo-bulk sample is typically either the sum of the raw counts of the gene or the mean of the normalized expression of the gene across the cells of an individual in the cell type of interest (more precisely defined using scRNA-seq data).
Box 2. Modelling considerations.
Traditional genetic association testing for quantitative traits (be it gene expression or height) uses the linear mixed model. It tests for (additive) effects of the SNP on the phenotype while accounting for covariates and population structure. The effect size coefficient (β) provides both the magnitude and the direction of the effect.
The model in the figure (part A) assumes the phenotype (y) to follow a Gaussian distribution, which is largely recapitulated when using bulk transcriptomics (see the figure, part Ba).
However, single-cell RNA sequencing data follow a distribution better described by a Poisson distribution (see the figure, part Bb). The histograms show the expression levels of the SRGAP2 gene in induced pluripotent stem cells from the same ~100 individuals78, considering bulk counts across individuals (see the figure, part Ba) and single-cell counts across cells (see the figure, part Bb).
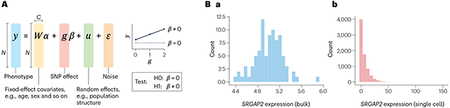
As in bulk studies, covariates may be confounded with allelic effects. Several approaches used to detect and correct for covariates affecting the expression of all (or a majority of) genes in bulk analyses can be extended to pseudo-bulk analyses. These include principal component analysis and probabilistic estimation of expression residuals (PEER), although the latter can perform suboptimally in some cases76,77. Single-cell studies have additional challenges, such as variable cell count per individual (inversely correlated with confidence in pseudo-bulk counts) or batch effects from multi-experiment study designs, which may create systematic differences in gene expression between experimental pools (Box 1). sc-eQTL models can increase power by accounting for these experimental factors with additional fixed or random effects70. There are many possible single-cell count normalization and aggregation and covariate correction strategies for pseudo-bulk sc-eQTL studies, which have been reviewed elsewhere78.
Although these studies used pseudo-bulk scRNA-seq data for eQTL mapping, contemporary studies also began to explore ways to use additional information offered by single-cell profiles. For example, in principle, these data allow one to measure the association between genetic variation and cell-to-cell gene expression variability (Fig. 3). Increased variability may reflect a lack of expression stability and increased propensity to enter extreme, pathogenic states79 or could uncover gene–environment (GxE) interactions with unmeasured environments and contexts80. Although a handful of studies have proposed methods to map such ‘variance eQTLs’ from single-cell data (borrowing from similar approaches in other settings80-82), they had limited success owing to insufficient sample sizes and the confounding correlation between the mean and variance of the expression of a gene18,71. As the size of single-cell genetic studies grows, and more sophisticated methods become available, we envision that single-cell variance eQTL studies will become more tractable. These early attempts to leverage single-cell-resolution data in genetic association models, nonetheless, have laid the foundation for new perspectives on modelling eQTLs, as well, with single-cell-resolution data.
Fig. 3∣. Types of single-cell expression quantitative trait locus.

Single-cell-resolved expression matched with genotype information allows one to consider different types of expression quantitative trait locus (eQTL) mapping strategies. When mapping cell-type-specific eQTLs, the single-cell resolution is exclusively utilized to more precisely characterize transcriptionally similar cells. Variance eQTLs test for genetic variants associated with cell-to-cell variability of gene expression (versus average expression level). Finally, to map dynamic eQTLs, single cells are ordered along a continuous trajectory, and the test consists in identifying eQTLs, the strength of which is modulated by such a trajectory.
Single-cell-resolution eQTL modelling
Cell types have historically been defined on the basis of discrete morphological and functional categories, and clustering scRNA-seq data work towards a similar ontological goal. To this end, early eQTL studies also discretized and aggregated cells of the same cell type to facilitate statistical modelling and interpretation. However, high-resolution single-cell data often reveal heterogeneity within discrete populations, which motivates modelling eQTLs at single-cell resolution. Here, we describe the second generation of sc-eQTL models, which adopt continuous frameworks to leverage granular single-cell-resolution data.
Single-cell models improve cell-state-dependent eQTL mapping
Recently, high-resolution molecular measurements (for example, transcriptomics) have been used to define and characterize single-cell phenotypes. They reveal not only discrete lineages but also continuous phenotypes and intermediate states. For example, scRNA-seq studies of human T cells have identified a continuum of cytotoxicity spanning multiple T cell sublineages83,84. During development, cells have been assayed in vitro and in vivo in intermediate differentiation states, such as the mesendoderm state preceding the determination of mesoderm or endoderm fate69. These continuous phenotypes sometimes reflect disease processes or pathogenic environmental signals, such as fibroblasts transitioning towards inflammatory states owing to NOTCH3 signalling in rheumatoid arthritis85. These examples highlight the need for more granular and continuous definitions of cell state (Box 3).
Box 3. Cell types and states.
Single-cell genomics has introduced a paradigm shift in our understanding and definitions of cellular identity, type and state. In traditional bulk assays, discrete populations of cells have been defined and sorted a priori on the basis of extracellular markers. These correspond to cell types, which may be defined as groups of cells from distinct, irreversible developmental lineages.
With single-cell transcriptomics, we can define cell populations after assaying the cells on the basis of their expression of key marker genes (see the figure). These populations are more granular than what could have been sorted on the basis of extracellular markers and reveal cell states: functionally specialized, often plastic, subpopulations of cells. These states can be discrete (for example, T helper cells) or continuous (for example, developmental states).
Single-cell resolution allows us to then define the most disease-relevant populations of cells (which might be a whole cell type or might be a transient state).
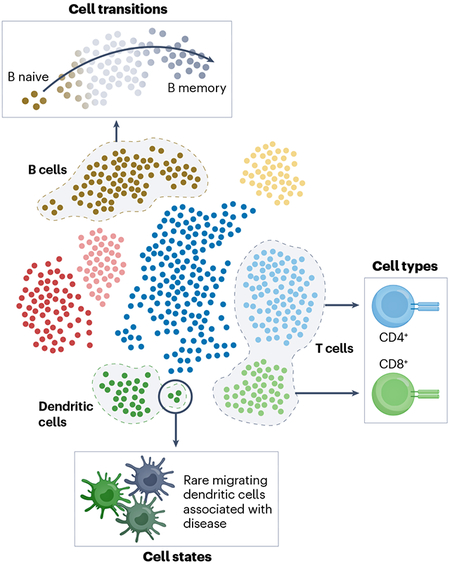
Once single-cell-resolution data are used to define these continuous states, we can model how genetic regulation varies dynamically along these trajectories. Rather than treating individuals as observations, these models treat each cell as its own observation of the expression of a gene. For example, one common model architecture is a mixed-effects interaction model, which includes random effects to account for the non-independence of cells from the same individual (which, if left unaccounted for, can inflate the false-positive rate86) and interaction terms between cell state and genotype to model state-dependent effects of genotype on expression65,87,88. These second-generation models map ‘dynamic’ eQTLs, assessing the effects of different genotype alleles on a trait that varies dynamically along a continuous axis. They have been successfully applied to continuous trajectories within differentiating iPS cells, T cells and other cell types65,87.
Other single-cell-resolution methods have adopted different approaches. For example, Gewirtz et al.89 used generative statistical (‘topic’) models to identify shared variation between genotypes and scRNA-seq profiles to identify both cis-eQTLs and trans-eQTLs across discrete cell types. As another example, Lu et al.90 used decomposition approaches to identify genetic effects on expression that are shared or specific to discrete cell types.
However, these early applications have also revealed the challenges and limitations of these models, including the non-normality of single-cell expression counts and computational tractability. We discuss these in detail in the following sections.
Sparsity and non-normality of single-cell expression data
Single-cell data are sparse (containing many 0s), owing to incomplete sampling as well as genuine biological variation in transcript presence within cells. As a result, single-cell measurements are not well described by the Gaussian distribution that linear regression-derived models assume. The large number of cells that are assayed together in bulk transcriptomes (and, to a certain extent, pseudo-bulk aggregated measurements) meant that normalized expression profiles could be approximated as Gaussian, but this does not hold for single-cell profiles91 (Box 2). Instead, discrete count distributions better describe these data. Despite their sparsity, single-cell profiles have been shown not to be zero-inflated92. Instead, a Poisson distribution offers an interpretable model of single-cell counts91 that has been used in recent studies, including the Poisson mixed-effect regression of Nathan et al.65 and the Poisson reduced-rank regression model of Fitzgerald et al.93. In some cases, more parametrized negative binomial or multinomial models may be appropriate alternatives depending on the gene expression distributions94; null testing for P-value inflation can guide those choices.
Scalability and infrastructure as sample sizes grow
Modelling each cell separately – rather than aggregating cells into pseudo-bulk measurements – requires data sets on the order of hundreds of thousands of cells, instead of hundreds of samples as in a (pseudo-)bulk study for the same number of individuals. One solution is grouping small groups of <10 phenotypically similar cells into ‘meta-cells’95,96. This type of aggregation is less disruptive than grouping thousands of cells in a cluster, or even hundreds of cells in a pseudotime bin, and is still usable for eQTL modelling87.
Moreover, because effective sample size (number of unique individuals) is also expected to grow in future studies, methods must be scalable and compatible with high-speed computing and data storage infrastructure (Box 1). This is an area where sc-eQTL methods may learn from several previous genetics tools and infrastructures built to perform efficiently at scale, such as TensorQTL97 (a graphics processing unit implementation of Matrix eQTL98,99 for QTL mapping) and Hail (a cloud-based scalable implementation of several genetic tools100). Some sc-eQTL methods with more computationally expensive frameworks have already begun leveraging graphics processing units, such as scTBLDA89, mentioned earlier. Methods may also benefit from parallelization across computing resources, cloud-based systems and algebraic and numerical approximations.
New opportunities
Current paradigms of sc-eQTL mapping offer a limited window into the overall picture of genetics and cell function. New technological advances, larger-scale studies and corresponding analytical and computational methods will be required to expand our view. In particular, we envision studies exploring more molecular traits (beyond gene expression), more types of genetic variants (beyond common SNPs) and more information about the individuals (for example, demographics, disease history and environmental exposures). Moreover, we expect data to be collected from progressively more diverse cohorts, including data from individuals of different ancestries, from individuals with diseases and from many different (disease-relevant) human tissues. As these rich data become available, new analytical and computational methods will be required to integrate information across data modalities (for example, chromatin accessibility, expression and protein level) and resolutions (from cell to tissue to individual), model context-specific and dynamic effects and predict outcomes relevant to human biology and health.
New data types
The molecular impact of DNA alleles can result in variation at the level of cells, tissues or whole organisms. A recent shift in human genetics has moved from variant discovery to exploring this multifaceted impact101. For any molecular phenotype we can measure, we can integrate genotypic information to map QTLs associated with the phenotype. With early adoption of single-cell RNA-seq and robust analysis pipelines, sc-eQTLs have been an appealing area for the first single-cell genetic association studies. However, as we are able to more efficiently measure and computationally analyse more molecular traits at single-cell resolution, we can interrogate the genetics of more cell states and molecular processes at single-cell resolution (for example, single-cell chromatin accessibility QTLs102).
Multi-omics technologies allow us to assay more than one data modality within the same cell; for example, single-cell nucleosome, methylation and transcription sequencing (scNMT-seq)103 measures chromatin accessibility, DNA methylation and expression, whereas cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)104 measures expression and surface protein level. Integrating multiple modalities provides multiple views on the phenotypes of the same cells, enabling higher-resolution definition of cell states to model dynamic sc-eQTLs and offering multiple phenotypes to model relationships with genetic variants. This integration task has been described as ‘vertical integration’105, with cells being the common link across modalities.
Simultaneously, increasing sample sizes and newer technologies are making more classes of genetic variation amenable to analysis in single-cell cohorts, such as rare variants, repeats, insertions and deletions and structural variants. These have been associated with diseases106-108, but there has been limited analysis of their effect on (whole-tissue) molecular phenotypes109-111, and none at the single-cell level. More comprehensive and systematic association studies with single-cell models and precisely defined cell states may more fully capture the impact of these variants at the molecular level.
Diverse cohorts
Ideally, to understand the mechanisms underlying biology, we need to link genetics with molecular measurements and cell states in living humans under different natural perturbations. To do so, it is necessary to assay cells across thousands of individuals with known genotypes and at least partially characterized ‘environment’, including lifestyle (for example, smoking status, diet and pollution), demographics (for example, sex, age, geography and ethnicity) and other biomedical traits (for example, medical and vaccination history, disease state and progression and medications). Incorporating these different sources of variation into single-cell genetic studies provides a clearer picture of the interactions between genetics and factors underlying changes at the cellular level. Given the demonstrated relationships between these covariates and cell-state composition, incorporating these covariates into sc-eQTL models will provide richer context for dynamic eQTLs112. Cellular-resolved, large-scale and multifaceted data sets may also enable studies of GxE interactions and their effect on molecular traits.
In addition to environmental diversity, accounting for the effect of ancestry is important. Single-cell and genetic studies more generally have failed to include ancestral diversity for many reasons, including long-standing inequities and concentration of research funding in communities with predominant European ancestries113-115. Although diversity has been a growing priority in research studies, many institutions still lack adequate infrastructure and community engagement programmes to equitably recruit participants116. Most studies continue to be conducted in European populations, and, as many have noted, genomic discoveries in Europeans are not always directly translatable to non-European individuals114. This limitation extends to sc-eQTL mapping studies, which largely consider samples of European ancestries. Yet, studying diverse populations is important, as they can have different causative alleles for diseases, different patterns of regulatory variation and different cell states and active pathways, together altering the context in which disease alleles act117-119.
The sc-eQTL analysis in ancestrally diverse cohorts can help with fine-mapping and elucidate population-specific dynamic eQTLs and their relationship with disease, improving the translation of findings of genetic studies. A few studies have already been conducted in non-European populations (in Peruvian65, Yoruban71 and African American82 populations), and large-scale cohorts from other geographical regions are being generated (for example, the Asian Immune Diversity Atlas, the African Ancestry Immune Cell Atlas and the Human Cell Map of Latin American Diversity). However, to maximize the findings that can be gleaned from these valuable data sets, it is essential to develop genetic algorithms for association testing, fine-mapping and meta-analysis that are robust to multi-ancestry data, which are currently lacking.
Studying disease tissue context
Many diseases have tissue-specific manifestations, making it critical to study the effects of genetic variation on gene regulation in disease tissue context. However, sc-eQTL studies to date have been largely limited to easy-to-access tissues (for example, skin and blood) or cell lines (for example, iPS cells), with only a minority of studies considering other tissues, such as the brain75. This limits our ability to learn about gene regulation in disease-relevant tissue (for example, colon for ulcerative colitis, or pancreas for type 1 diabetes mellitus). First, some disease-relevant cell types cannot be assessed at all in the absence of the relevant tissue. For example, neurodegenerative diseases such as Parkinson disease have proven especially difficult to study in part owing to the lack of access to data from the specific brain cells that are thought to be affected (dopaminergic neurons120). Second, even cell types that can be found, for example, in blood are found in a very different environment in tissue and thus may be subject to different context-specific genetic regulation. Finally, it is worth noting that tissues require handling, freezing and disaggregation, meaning that they are markedly more challenging to study. Moving forward, these are critical points that may be addressed by large-scale single-cell data generation projects such as the Human Cell Atlas15,121.
Although most current studies have focused on ‘healthy’ individuals, another avenue to study disease-relevant gene regulation is to obtain single-cell profiling data from genotyped individuals with diseases and other traits of interest. For example, Perez et al.64 mapped sc-eQTL in various blood cell types from patients with systemic lupus erythematosus. Additionally, the deficit of genotyped single-cell cohorts for hard-to-access tissues and people with a disease phenotype may be addressed by differentiating stem cells into cell types of interest and growing organoid models122. Recently, the concept of ‘cell villages’ has been introduced to help scale stem cell studies for larger numbers of donor lines, providing power to explore gene regulation in disease-relevant cell types and genotypes123.
Another promising avenue is to study spatial patterns of eQTLs to understand how gene regulation may interact with tissue structure to lead to disease. Spatial transcriptomics can record the in situ locations of cells along with their RNA expression profiles at near-cellular resolution124. These technologies are rapidly improving to become higher resolution, cheaper, higher fidelity and easier to implement125. In parallel, new mixture modelling strategies for spatial gene expression have already extended traditional analyses such as differential expression to spatial transcriptomics126,127, and similar refinement may be useful for eQTL models19. With further development of computational tools and spatial technologies, there could be an that vary across spatial coordinates.
Enabling disease-relevant discoveries
eQTLs provide insight into the modes of action of disease-associated genetic variation – implicating genes they regulate, the direction of effect and cell states in which they have an effect – which has several important ramifications for understanding disease processes and, down the line, helping drug development.
Single-cell genetics for identifying disease-relevant cell types
Knowing the tissues, cell types and cell states most relevant to a disease phenotype can add to clinical understanding. With the development of sc-eQTL models that can identify cell-state-specific genetic effects on gene expression, we can now integrate existing knowledge about disease alleles with their predicted regulatory targets in each cellular context. This enables inference of the contexts in which the disease alleles may be most disruptive. Methods have been developed for complex traits affected by many genetic variants to integrate bulk tissue-specific eQTL effects and to prioritize the most relevant tissue128,129. For example, Kundu et al.130 used eQTL mapping to fine-map causal disease-associated variants, finding, among other things, that the ITGA4 locus for inflammatory bowel disease is active in monocytes. Single-cell-resolved eQTL maps will provide further granularity to these types of studies by enabling subcell-type resolution. For example, two distinct studies recently combined sc-eQTLs in (iPS cell-derived) dopaminergic neurons from 215 individuals70 with GWAS results for Parkinson disease and schizophrenia, respectively, to confirm existing and identify novel genes that are likely to have a role in Parkinson disease and schizophrenia aetiology, using a Mendelian randomization approach112,131.
Other methods using single-cell data can estimate more precise cell types relevant to disease, using variant-gene expression associations and other strategies to link disease-associated variants to genes132-134 (Fig. 4). Some methods, such as single-cell disease relevance score (scDRS), estimate association of individual cells with the polygenic disease risk on the basis of their expression of genes proximal to GWAS variants135. This represents a step towards translating a PRS framework to single cells, aggregating SNP effects to predict heritable trait risk. Single-cell molecular QTL results may help construct similar predictors by further taking into account cell-type-specific regulatory effects of the genetic variants. For example, CONTENT (which stands for context-specific genetics) is an extension of transcriptome-wide association study that uses context-specific eQTLs from either single-cell or bulk analysis to identify genes with context-specific expression associated with a disease, enabling quantification of the context-specific portion of disease heritability136.
Fig. 4∣. Downstream effect of context-dependent single-cell expression quantitative trait locus.

Identification of the specific contexts in which a disease-associated genetic variant regulates gene expression may ultimately lead to new therapeutic strategies. eQTL, expression quantitative trait locus.
Moreover, these methods may benefit from more granular, single-cell data. When CONTENT was used to identify genes associated with systemic lupus erythematosus on the basis of eQTLs mapped in single-cell peripheral blood cells, it found twice as many genes when state-specific eQTLs were mapped using a single-cell-resolution decomposition method compared with pseudo-bulk meta-analysis64. This result highlights the importance of single-cell-resolution eQTL mapping approaches.
In addition to finding disease-associated genes, which may point to key pathways and drug targets, future extensions of similar methods may narrow down the cell context in which disease-associated genetics influences gene expression. This focus can also help us identify cell states to target with gene editing or other therapeutic molecules137.
Future potential in the clinic
Importantly, although recent studies have shown that drug targets with genetic evidence are twice as likely to prove clinically effective138,139, the translation of sc-eQTL results to the clinic is not a reality at present, and many critical steps are required to operationalize these data. Nonetheless, efforts using well-established data types provide hope that sc-eQTLs, too, may eventually have clinical utility.
First, complex disease heterogeneity may reflect underlying genetic and mechanistic differences. Genetic (PRSs140,141) and expression-based approaches (bulk142,143 and single cell144-148) have been used independently to stratify patients on the basis of disease risk and into disease subtypes. A recent study128 developed a method to prioritize disease-relevant tissues through Bayesian mixture modelling of the trait associations of tissue-specific bulk eQTL variants. They used this method to identify subgroups of patients with high body mass index whose genetic predisposition was most relevant to gene regulation in either brain, adipose tissue or muscle128. Using sc-eQTL studies and adapting bulk tissue methods may achieve similar results at cell-type and subcell-type resolution149, potentially allowing patients with the same clinical disease to be stratified into subgroups with different disease prognoses and optimal therapeutic strategies.
Second, incomplete functional annotation of variants limits the utility of DNA sequencing to provide accurate diagnoses for patients with monogenic diseases. Functional genomic analysis of clinical tissue samples increases diagnostic rates above those provided by DNA sequencing methods alone, with bulk RNA-seq of disease-relevant patient tissue samples in particular now well-established as substantially improving diagnosis rates by identifying disease-causing changes in gene expression or splicing150-152, leading to its incorporation into both research and clinical diagnostic workflows153,154. We can thus expect single-cell methods to increase diagnosis rates in two ways: first, by providing more accurate annotation of the genomic regions involved in the biology of specific disease-relevant cell states, leading to better in silico functional prediction for variants, and second, through direct application to patient tissue to identify variants affecting transcript structure or expression in cell types that are rare in accessible tissue.
Conclusions and perspective
This article provides an overview of the nascent field of single-cell genetics, in which single-cell resolution molecular readouts are collected from hundreds or thousands of individuals and analysed in tandem with matched genotype data. sc-eQTL mapping, in which the effects of genetic variants on RNA levels are evaluated at single-cell resolution, is one of the most technically and algorithmically advanced approaches in this area; thus, it is where many of the first single-cell genetic studies have appeared and is the focus of this article.
The field of single-cell genetics (and single-cell technologies in general) is still in its infancy, and although it holds tremendous potential, there remain areas where bulk transcriptome approaches continue to have an important role. For example, in homogeneous cell types (for example, iPS cells), a bulk eQTL study may be better powered than an sc-eQTL study in the same cell type78,155. However, as technology improves and costs decrease, this gap will progressively diminish. Emerging technologies are becoming cheaper156, require less specialized equipment157, capture longer transcripts with higher fidelity158 and may become amenable for large-scale single-cell studies in coming years.
As the second generation of eQTL mapping methods emerges, we can model regulatory differences at single-cell resolution and link them to differences in disease risk and heritability. This offers the promise of going beyond the conventional tissue and cell-type resolution that has, itself, still left the regulatory effects of many non-coding disease alleles unexplained4,13. Modelling cell-state-specific and context-specific eQTLs with single-cell data can also be used to improve inference of gene regulatory networks or haplotype-aware analyses of coordinated cis-regulatory effects on alleles159,160. However, as these single-cell data sets increase in size and algorithms seek to model heterogeneous, high-dimensional data, we face many challenges, as reviewed earlier.
Beyond these technical obstacles to implementing methods, there are additional barriers to clinical translation. Sample sizes for genetic studies are typically on the order of tens or hundreds of thousands, whereas single-cell studies have largely remained in the hundreds. Larger, more diverse cohorts of genotyped, single-cell-profiled individuals will be needed to conduct well-powered single-cell genetics studies with complex environmental or cell-state interactions. Additionally, this will enable GWAS-like studies linking genetic variants to cell-type composition and abundance estimated from scRNA-seq data (possibly adopting previous methods using FACS161,162), which are also genetically regulated and relevant to disease.
Moreover, eQTL studies often yield thousands of putative variant–gene expression associations. Although their results can be used as supporting evidence, experimental validation remains necessary to establish true causal relationships between variants and disease. This is an important open question, especially for dynamic eQTLs identified in rare or hard-to-isolate cell states. Replication in independent single-cell studies is possible, but alternative molecular validation may be challenging. For sc-eQTLs and other single-cell genetic studies to be translated to the clinic, we need parallel development of experimental techniques to test the effects of variants in specific cell states at high throughput, such as CRISPR screens163,164 or investigation in iPS cells or organoids165,166. Computational strategies that leverage the heterogeneity of other single-cell modalities measured across many individuals may also link eQTL variants to upstream regulatory elements167,168 or downstream cellular phenotypes169.
The existing and future studies described in this Review aim to provide novel insights and hypotheses into the mode of action of variants in gene regulation and disease pathogenesis. Understanding these causal pathways in a cell-state-specific manner may inform targeted therapeutic strategies.
Glossary
- Allele
One of two or more alternative DNA sequences occurring at a particular genomic locus
- Ambient RNA
Free-floating RNA captured in a single-cell RNA sequencing droplet or other reaction compartment
- Cell-type annotation
Manual or algorithmic approach to assign labels (corresponding to cell type) to unbiasedly identified cell clusters
- Cell villages
Cell lines derived from multiple donors cultured and differentiated together in a single dish. These are distinct from ‘uni-cultures’, in which each cell line is cultured independently. This makes the strategy particularly valuable for population-scale studies
- Clustering
Algorithmic approach to group cells into clusters, which are groups of similar cells based on their transcriptomes
- Colocalization
Statistical methods that aim to estimate the probability that the same genetic variant is causal for two different traits, for example, an organismal trait (for example, a disease in a genome-wide association study) and a molecular trait (for example, the expression level of a given gene in an expression quantitative trait locus study).
- Doublets
Two or more cells (also called multiplet) captured and processed in the same droplet
- Fine-mapping
The process of localizing association signals to causal variants using statistical, bioinformatic or functional methods
- Fluorescence-activated cell sorting (FACS)
Experimental technique to select cells based on physical and chemical characteristics of individual cells. Single cells from a sample are suspended in a fluid and then injected into an instrument that uses lasers to detect cell morphology and fluorescently labelled features and sort cells based on these qualities.
- Gene regulatory network (analysis)
A gene regulatory network is a set of interacting regulatory elements and genes that jointly control expression patterns that dictate a specific cell function.
- Genome-wide association studies (GWASs)
Statistical procedure to identify associations between individual genetic variants and variation in continuous traits (for example, height) or risk of disease (for example, type 2 diabetes)
- Interaction
Interplay between different sources of variation (for example, genetic variation and environmental exposure — GxE) that results in a joint effect on the trait of interest beyond the individual additive effects
- Mendelian randomization
Statistical method using measured variation in an instrumental variable (for example, a genetic variant) to test the causal effect of an exposure (for example, the expression of a gene) on an outcome (for example, a common trait or disease)
- Minor allele frequency
Population frequency for the least common (that is, minor) alleles within the population of interest
- Non-negative matrix factorization
Dimensionality reduction method to decompose a matrix of non-negative values into two matrices of vectors capturing the essential features of a data set. Unlike principal component analysis, non-negative matrix factorization components are not orthogonal
- Polygenic risk scores (PRSs)
Quantification of total risk of an individual for a given disease based on genetic contributors alone. PRSs are calculated by summing the dosage of an individual of thousands of variants weighted by the strength of their association with the trait (as estimated from a genome-wide association study for that trait).
- Principal component analysis (PCA)
Dimensionality reduction method to identify main orthogonal axes of variation in a dataset, called ‘principal components’
- Pseudotime
Approximate ordering of cells along a latent dimension based on single-cell RNA sequencing data. The ordering represents sequential changes along a transition (for example, during cell differentiation)
- Response eQTL
An association between a genetic variant and RNA level (that is, an expression quantitative trait locus) that only becomes apparent when the cells the RNA is measured in are stimulated in some way (for example, immune activation)
- Single-cell phenotypes
Cell characteristics (for example, function, gene expression and position along a transition) that can be estimated using single-cell-resolved molecular profiling (for example, single-cell RNA sequencing)
- Sparse
Containing a large number of 0s. In single-cell data, sparsity is due to the combination of inefficient sampling and true absence of expression
- Trajectory inference
Also known as trajectory mapping. A computational technique used in single-cell data to determine the form of a dynamic process experienced by cells (for example, lineage specification and differentiation) and then arrange cells based on their progression through the process, usually using a pseudotime approach
- Transcriptome-wide association studies (TWASs)
Statistical method that uses estimated associations between variants and gene expression (for example, from expression quantitative trait locus studies) to infer expression for all individuals in a genome-wide association study and to identify associations between genes and traits/diseases.
- Unique molecular identifiers (UMI)
Complex indices added to sequencing libraries before any PCR amplification steps, enabling the accurate bioinformatic identification of PCR duplicates. They are common in many single-cell RNA sequencing protocols
Footnotes
Competing interests
D.G.M. is a founder with equity in Goldfinch Bio, a paid adviser to GSK, Insitro, Third Rock Ventures and Foresite Labs and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer and Sanofi-Genzyme; none of these activities is related to the work presented here. S.R. is a founder for Mestag, Inc. and a scientific adviser for Sonoma Biotherapeutics, Pfizer, Jannsen and Sanofi. The other authors declare no competing interests.
References
- 1.Sollis E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguet F. et al. Molecular quantitative trait loci. Nat. Rev. Methods Prim 3, 4 (2023). This Primer provides a comprehensive overview of molecular QTLs, including eQTLs.
- 3.Albert FW & Kruglyak L The role of regulatory variation in complex traits and disease. Nat. Rev. Genet 16, 197–212 (2015). [DOI] [PubMed] [Google Scholar]
- 4. Umans BD, Battle A & Gilad Y Where are the disease-associated eQTLs? Trends Genet. 37, 109–124 (2021). This Review article highlights the importance of identifying the correct and dynamic cell contexts where gene regulation is active and the usefulness of single-cell data for this purpose.
- 5.Fairfax BP et al. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet 44, 502–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fairfax BP et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Jager PL et al. ImmVar project: insights and design considerations for future studies of ‘healthy’ immune variation. Semin. Immunol 27, 51–57 (2015). [DOI] [PubMed] [Google Scholar]
- 8.The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmiedel BJ et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–1715.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strober BJ et al. Dynamic genetic regulation of gene expression during cellular differentiation. Science 364, 1287–1290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westra H-J et al. Cell specific eQTL analysis without sorting cells. PLoS Genet. 11, e1005223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chun S. et al. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated Loci in three major immune-cell types. Nat. Genet 49, 600–605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connally NJ et al. The missing link between genetic association and regulatory function. eLife 11, e74970 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regev A. et al. The human cell atlas. eLife 6, e27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tabula Sapiens Consortium et al. The Tabula sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eraslan G. et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandric I. et al. Optimized design of single-cell RNA sequencing experiments for cell-type-specific eQTL analysis. Nat. Commun 11, 5504 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wijst Mvander et al. The single-cell eQTLGen Consortium. eLife 9, elife.52155 (2020). This manifesto by the single-cell eQTLGen Consortium highlights the timeliness of single-cell eQTL studies (with a focus on blood).
- 20.No authors listed. Method of the year 2013. Nat. Methods 11, 1 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Svensson V, da Veiga Beltrame E & Pachter L A curated database reveals trends in single-cell transcriptomics. Database 2020, baaa073 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Cao J. et al. A Human Cell Atlas of fetal gene expression. Science 370, eaba7721 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagemann-Jensen M et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol 38, 708–714 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Habib N. et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths JA, Scialdone A & Marioni JC Using single-cell genomics to understand developmental processes and cell fate decisions. Mol. Syst. Biol 14, e8046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94 (2018). This paper describes a method to leverage genotyping data to demultiplex single-cell data, enabling efficient experimental design to assay large cohorts.
- 30.Huang Y, McCarthy DJ & Stegle O Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference. Genome Biol. 20, 273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luecken MD & Theis FJ Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nayak R & Hasija Y A hitchhiker’s guide to single-cell transcriptomics and data analysis pipelines. Genomics 113, 606–619 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Adil A, Kumar V, Jan AT & Asger M Single-cell transcriptomics: current methods and challenges in data acquisition and analysis. Front. Neurosci 15, 591122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brennecke P. et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10, 1093–1095 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Yip SH, Sham PC & Wang J Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief. Bioinform 20, 1583–1589 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Maaten L, van der Maaten L & Hinton G Visualizing non-metric similarities in multiple maps. Mach. Learn 87, 33–55 (2012). [Google Scholar]
- 37.McInnes L, Healy J, Saul N & Großberger L UMAP: uniform manifold approximation and projection. J. Open Source Softw 3, 861 (2018). [Google Scholar]
- 38.Satija R, Farrell JA, Gennert D, Schier AF & Regev A Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarthy DJ, Campbell KR, Lun ATL & Wills QF Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 42.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lek M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaisson MJP et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T. et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baxter SM et al. Centers for Mendelian Genomics: a decade of facilitating gene discovery. Genet. Med 24, 784–797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright CF et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Li YI & Pritchard JK Trans effects on gene expression can drive omnigenic inheritance. Cell 177, 1022–1034.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Võsa U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet 53, 1300–1310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yengo L. et al. A saturated map of common genetic variants associated with human height. Nature 610, 704–712 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferraro NM et al. Transcriptomic signatures across human tissues identify functional rare genetic variation. Science 369, eaaz5900 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonder MJ et al. Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics. Nat. Genet 53, 313–321 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J, Kong N, Han B & Sul JH Rare variants regulate expression of nearby individual genes in multiple tissues. PLoS Genet. 17, e1009596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cano-Gamez E & Trynka G From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet 11, 424 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giambartolomei C. et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics 34, 2538–2545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wainberg M. et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet 51, 592–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gusev A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wills QF et al. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol 31, 748–752 (2013). The first single-cell eQTL study conducted in a cohort of 15 people and 96 genes only (not yet genome-wide).
- 62.van der Wijst MGP et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet 50, 493–497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yazar S. et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, 6589 (2022). The largest single-cell eQTL study as of 2022, with pseudobulk profiles from nearly 1,000 individuals.
- 64.Perez RK et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376, eabf1970 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nathan A. et al. Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature 606, 120–128 (2022). This study describes a method to model eQTLs in continuous cell states from single-cell data using Poisson mixed models of raw gene counts.
- 66.Oelen R. et al. Single-cell RNA-sequencing of peripheral blood mononuclear cells reveals widespread, context-specific gene expression regulation upon pathogenic exposure. Nat. Commun 13, 3267 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmiedel BJ et al. Single-cell eQTL analysis of activated T cell subsets reveals activation and cell type-dependent effects of disease-risk variants. Sci. Immunol 7, 68 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soskic B. et al. Immune disease risk variants regulate gene expression dynamics during CD4 T cell activation. Nat. Genet 54, 817–826 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuomo ASE et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun 11, 810 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jerber J. et al. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet 53, 304–312 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sarkar AK et al. Discovery and characterization of variance QTLs in human induced pluripotent stem cells. PLoS Genet. 15, e1008045 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elorbany R. et al. Single-cell sequencing reveals lineage-specific dynamic genetic regulation of gene expression during human cardiomyocyte differentiation. PLoS Genet. 18, e1009666 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daniszewski M. et al. Retinal ganglion cell-specific genetic regulation in primary open-angle glaucoma. Cell Genomics 2, 100142 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neavin D. et al. Single cell eQTL analysis identifies cell type-specific genetic control of gene expression in fibroblasts and reprogrammed induced pluripotent stem cells. Genome Biol. 22, 76 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bryois J. et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci 25, 1104–1112 (2022). This study is one of the only single-cell eQTL studies in tissue to date.
- 76.Zhou HJ, Li L, Li Y, Li W & Li JJ PCA outperforms popular hidden variable inference methods for molecular QTL mapping. Genome Biol. 23, 210 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xue A, Yazar S, Neavin D & Powell JE Pitfalls and opportunities for applying PEER factors in single-cell eQTL analyses. Genome Biol. 24, 33 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cuomo ASE et al. Optimizing expression quantitative trait locus mapping workflows for single-cell studies. Genome Biol. 22, 188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ayroles JF et al. Behavioral idiosyncrasy reveals genetic control of phenotypic variability. Proc. Natl Acad. Sci. USA 112, 6706–6711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Westerman KE et al. Variance-quantitative trait loci enable systematic discovery of gene-environment interactions for cardiometabolic serum biomarkers. Nat. Commun 13, 3993 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morgan MD et al. Quantitative genetic analysis deciphers the impact of cis and trans regulation on cell-to-cell variability in protein expression levels. PLoS Genet. 16, e1008686 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Resztak JA et al. Genetic control of the dynamic transcriptional response to immune stimuli and glucocorticoids at single cell resolution. Preprint at biorXiv 10.1101/2021.09.30.462672 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gutierrez-Arcelus M. et al. Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions. Nat. Commun 10, 687 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cano-Gamez E. et al. Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4 T cells to cytokines. Nat. Commun 11, 1801 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei K. et al. Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 582, 259–264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fonseka CY et al. Mixed-effects association of single cells identifies an expanded effector CD4 T cell subset in rheumatoid arthritis. Sci. Transl. Med 10, eaaq0305 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cuomo ASE et al. CellRegMap: a statistical framework for mapping context-specific regulatory variants using scRNA-seq. Mol. Syst. Biol 18, e10663 (2022). This paper reports a method to model eQTLs in continuous cell states from single-cell data using linear mixed models of normalized gene expression.
- 88.Kumasaka N. et al. Mapping interindividual dynamics of innate immune response at single-cell resolution. Preprint at biorXiv 10.1101/2021.09.01.457774 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gewirtz ADH, William Townes F & Engelhardt BE Expression QTLs in single-cell sequencing data. Preprint at biorXiv 10.1101/2022.08.14.503915 (2022). [DOI] [Google Scholar]
- 90.Lu A. et al. Fast and powerful statistical method for context-specific QTL mapping in multi-context genomic studies. Preprint at biorXiv 10.1101/2021.06.17.448889 (2021). [DOI] [Google Scholar]
- 91.Sarkar A & Stephens M Separating measurement and expression models clarifies confusion in single-cell RNA sequencing analysis. Nat. Genet 53, 770–777 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Svensson V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol 38, 147–150 (2020). [DOI] [PubMed] [Google Scholar]
- 93.Fitzgerald T, Jones A & Engelhardt BE A Poisson reduced-rank regression model for association mapping in sequencing data. BMC Bioinforma. 23, 529 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Townes FW, William Townes F, Hicks SC, Aryee MJ & Irizarry RA Feature selection and dimension reduction for single cell RNA-Seq based on a multinomial model. Genome Biol. 20, 295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baran Y. et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20, 206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DeTomaso D. et al. Functional interpretation of single cell similarity maps. Nat. Commun 10, 4376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taylor-Weiner A. et al. Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 20, 228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shabalin AA Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ongen H, Buil A, Brown AA, Dermitzakis ET & Delaneau O Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 32, 1479–1485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hail Team. Hail 0.2.54. https://github.com/hail-is/hail/releases/tag/0.2.54 (2020). [Google Scholar]
- 101.Lappalainen T & MacArthur DG From variant to function in human disease genetics. Science 373, 1464–1468 (2021). [DOI] [PubMed] [Google Scholar]
- 102.Benaglio P. et al. Mapping genetic effects on cell type-specific chromatin accessibility and annotating complex trait variants using single nucleus ATAC-seq. Preprint at biorXiv 10.1101/2020.12.03.387894 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun 9, 781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stoeckius M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Argelaguet R, Cuomo ASE, Stegle O & Marioni JC Computational principles and challenges in single-cell data integration. Nat. Biotechnol 39, 1202–1215 (2021). [DOI] [PubMed] [Google Scholar]
- 106.Trost B. et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 185, 4409–4427.e18 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mitra I. et al. Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mukamel RE et al. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 373, 1499–1505 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chiang C. et al. The impact of structural variation on human gene expression. Nat. Genet 49, 692–699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Scott AJ, Chiang C & Hall IM Structural variants are a major source of gene expression differences in humans and often affect multiple nearby genes. Genome Res. 31, 2249–2257 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fotsing SF et al. The impact of short tandem repeat variation on gene expression. Nat. Genet 51, 1652–1659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dang X, Zhang Z & Luo X-J Mendelian randomization study using dopaminergic neuron-specific eQTL nominates potential causal genes for Parkinson’s disease. Mov. Disord 37, 2451–2456 (2022). [DOI] [PubMed] [Google Scholar]
- 113.Petrovski S & Goldstein DB Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 17, 157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Popejoy AB & Fullerton SM Genomics is failing on diversity. Nature 538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sirugo G, Williams SM & Tishkoff SA The missing diversity in human genetic studies. Cell 177, 1080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lemke AA et al. Addressing underrepresentation in genomics research through community engagement. Am. J. Hum. Genet 109, 1563–1571 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shang L. et al. Genetic architecture of gene expression in European and African Americans: an eQTL mapping study in GENOA. Am. J. Hum. Genet 106, 496–512 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nédélec Y. et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 167, 657–669.e21 (2016). [DOI] [PubMed] [Google Scholar]
- 119. Randolph HE et al. Genetic ancestry effects on the response to viral infection are pervasive but cell type specific. Science 374, 1127–1133 (2021). This single-cell response eQTL study identifies eQTL interactions with influenza infection in a multiethnic cohort.
- 120.Stoddard-Bennett T & Pera R Stem cell therapy for Parkinson’s disease: safety and modeling. Neural Regen. Res 15, 36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rood JE, Maartens A, Hupalowska A, Teichmann SA & Regev A Impact of the human cell atlas on medicine. Nat. Med 28, 2486–2496 (2022). [DOI] [PubMed] [Google Scholar]
- 122.Kim J, Koo B-K & Knoblich JA Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol 21, 571–584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Neavin DR et al. Village in a dish: a model system for population-scale hiPSC studies. Nat. Commun (in the press; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marx V. Method of the year: spatially resolved transcriptomics. Nat. Methods 18, 9–14 (2021). [DOI] [PubMed] [Google Scholar]
- 125.Moses L & Pachter L Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022). [DOI] [PubMed] [Google Scholar]
- 126.Cable DM et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol 40, 517–526 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cable DM et al. Cell type-specific inference of differential expression in spatial transcriptomics. Nat. Methods 19, 1076–1087 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Majumdar A. et al. Leveraging eQTLs to identify individual-level tissue of interest for a complex trait. PLoS Comput. Biol 17, e1008915 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Arvanitis M, Tayeb K, Strober BJ & Battle A Redefining tissue specificity of genetic regulation of gene expression in the presence of allelic heterogeneity. Am. J. Hum. Genet 109, 223–239 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kundu K. et al. Genetic associations at regulatory phenotypes improve fine-mapping of causal variants for 12 immune-mediated diseases. Nat. Genet 54, 251–262 (2022). [DOI] [PubMed] [Google Scholar]
- 131.Dang X, Liu J, Zhang Z & Luo X-J Mendelian randomization study using dopaminergic neuron-specific eQTL identifies novel risk genes for schizophrenia. Mol. Neurobiol 10.1007/s12035-022-03160-3 (2022). [DOI] [PubMed] [Google Scholar]
- 132.Jia P, Hu R, Yan F, Dai Y & Zhao Z scGWAS: landscape of trait-cell type associations by integrating single-cell transcriptomics-wide and genome-wide association studies. Genome Biol. 23, 220 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jagadeesh KA et al. Identifying disease-critical cell types and cellular processes by integrating single-cell RNA-sequencing and human genetics. Nat. Genet 54, 1479–1492 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Corces MR et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang MJ et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nat. Genet 54, 1572–1580 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thompson M. et al. Multi-context genetic modeling of transcriptional regulation resolves novel disease loci. Nat. Commun 13, 5704 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Freimer JW et al. Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks. Nat. Genet 54, 1133–1144 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nelson MR et al. The support of human genetic evidence for approved drug indications. Nat. Genet 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 139.King EA, Davis JW & Degner JF Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15, e1008489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Benafif S. et al. The BARCODE1 Pilot: a feasibility study of using germline single nucleotide polymorphisms to target prostate cancer screening. BJU Int. 129, 325–336 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Richardson TG, O’Nunain K, Relton CL & Davey Smith G Harnessing whole genome polygenic risk scores to stratify individuals based on cardiometabolic risk factors and biomarkers at age 10 in the Lifecourse-Brief Report. Arterioscler. Thromb. Vasc. Biol 42, 362–365 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Glastonbury CA, Couto Alves A, El-Sayed Moustafa JS & Small KS Cell-type heterogeneity in adipose tissue is associated with complex traits and reveals disease-relevant cell-specific eQTLs. Am. J. Hum. Genet 104, 1013–1024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kong Y, Rastogi D, Seoighe C, Greally JM & Suzuki M Insights from deconvolution of cell subtype proportions enhance the interpretation of functional genomic data. PLoS ONE 14, e0215987 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Muus C. et al. Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med 27, 546–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reshef YA et al. Co-varying neighborhood analysis identifies cell populations associated with phenotypes of interest from single-cell transcriptomics. Nat. Biotechnol 40, 355–363 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Burkhardt DB et al. Quantifying the effect of experimental perturbations at single-cell resolution. Nat. Biotechnol 39, 619–629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nieto P. et al. A single-cell tumor immune atlas for precision oncology. Genome Res. 31, 1913–1926 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stephenson E. et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med 27, 904–916 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Davenport EE et al. Discovering in vivo cytokine-eQTL interactions from a lupus clinical trial. Genome Biol. 19, 168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cummings BB et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med 9, eaal5209 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kremer LS et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun 8, 15824 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Frésard L. et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med 25, 911–919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Montgomery SB, Bernstein JA & Wheeler MT Toward transcriptomics as a primary tool for rare disease investigation. Cold Spring Harb. Mol. Case Stud 8, a006198 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yépez VA et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 14, 38 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kilpinen H. et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546, 370–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Simmons SK et al. Mostly natural sequencing-by-synthesis for scRNA-seq using Ultima sequencing. Nat. Biotechnol 41, 204–211 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Clark IC et al. Microfluidics-free single-cell genomics with templated emulsification. Nat. Biotechnol 10.1038/s41587-023-01685-z (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Philpott M. et al. Nanopore sequencing of single-cell transcriptomes with scCOLOR-seq. Nat. Biotechnol 39, 1517–1520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jiang Y, Zhang NR & Li M SCALE: modeling allele-specific gene expression by single-cell RNA sequencing. Genome Biol. 18, 74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Qi G, Strober BJ, Popp JM, Ji H & Battle A Single-cell allele-specific expression analysis reveals dynamic and cell-type-specific regulatory effects. Preprint at biorXiv 10.1101/2022.10.06.511215 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Orrù V. et al. Genetic variants regulating immune cell levels in health and disease. Cell 155, 242–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Roederer M. et al. The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell 161, 387–403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gasperini M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 1516 (2019). [DOI] [PubMed] [Google Scholar]
- 164.Kasela S. et al. Integrative approach identifies SLC6A20 and CXCR6 as putative causal genes for the COVID-19 GWAS signal in the 3p21.31 locus. Genome Biol. 22, 242 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Warren CR et al. Induced pluripotent stem cell differentiation enables functional validation of GWAS variants in metabolic disease. Cell Stem Cell 20, 547–557.e7 (2017). [DOI] [PubMed] [Google Scholar]
- 166.Wolter JM et al. Cellular genome-wide association study identifies common genetic variation influencing lithium-induced neural progenitor proliferation. Biol. Psychiatry 93, 8–17 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sakaue S. et al. Tissue-specific enhancer-gene maps from multimodal single-cell data identify causal disease alleles. Preprint at biorXiv 10.1101/2022.10.27.22281574 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mitchell JM et al. Mapping genetic effects on cellular phenotypes with ‘cell villages’. Preprint at biorXiv 10.1101/2020.06.29.174383 (2020). [DOI] [Google Scholar]
- 170.International HapMap Consortium. A haplotype map of the human genome. Nature 437, 1299–1320 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Black JRM & Clark SJ Age-related macular degeneration: genome-wide association studies to translation. Genet. Med 18, 283–289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lango Allen H. et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.1000 Genomes Project Consortium et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Boyle EA, Li YI & Pritchard JK An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fry A. et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am. J. Epidemiol 186, 1026–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu MC et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet 89, 82–93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ramsköld D. et al. Full-length mRNA-seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol 30, 777–782 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Picelli S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
- 181.Nagano T. et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rotem A. et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stranger BE et al. Population genomics of human gene expression. Nat. Genet 39, 1217–1224 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Dixon AL et al. A genome-wide association study of global gene expression. Nat. Genet 39, 1202–1207 (2007). [DOI] [PubMed] [Google Scholar]
- 186.Montgomery SB et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464, 773–777 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pickrell JK et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464, 768–772 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lappalainen T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.GTEx Consortium. Human Genomics The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]