

Abstract
So far, the mechanisms that impede AAV transduction, especially in the human heart, are poorly understood, hampering the introduction of new, effective gene therapy strategies. Therefore, the aim of this study was to identify and overcome the main cellular barriers to successful transduction in the heart, using induced pluripotent stem cell (iPSC)-derived cardiomyocytes (iPSC-CMs), iPSC-derived cardiac fibroblasts (iPSC-CFs), and primary endothelial cells to model vector-host interactions. Through phosphoproteome analysis we established that casein kinase 2 (CK2) signaling is one of the most significantly affected pathways upon AAV exposure. Transient inhibition of CK2 activity substantially enhanced the transduction rate of AAV2, AAV6, and AAV9 in all tested cell types. In particular, CK2 inhibition improved the trafficking of AAVs through the cytoplasm, impaired DNA damage response through destabilization of MRE11, and altered the RNA processing pathways, which were also highly responsive to AAV transduction. Also, it augmented transgene expression in already transduced iPSC-CFs, which retain AAV genomes in a functional, but probably silent form. In summary, the present study provides new insights into the current understanding of the host-AAV vector interaction, identifying CK2 activity as a key barrier to efficient transduction and transgene expression, which may translate to improving the outcome of AAV-based therapies in the future.
Keywords: gene therapy, adeno-associated viral vectors, AAV, transduction efficiency, induced pluripotent stem cells
Graphical abstract
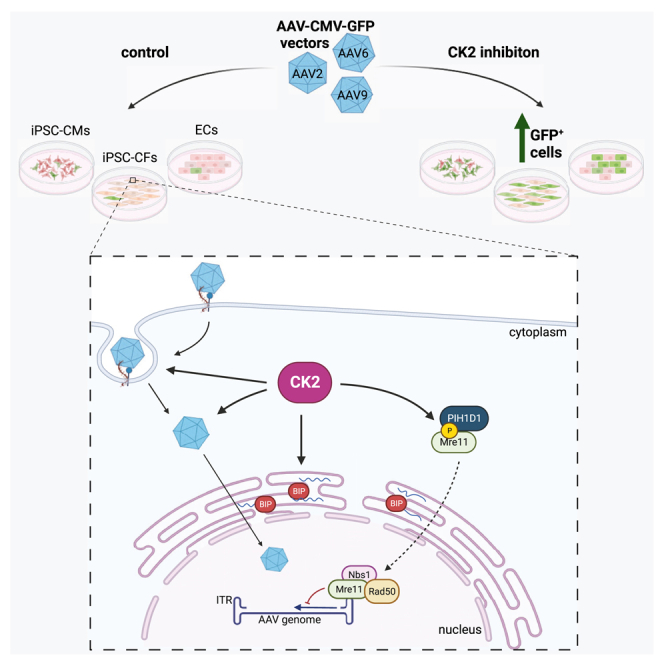
Kraszewska and colleagues, through analysis of vector-host interactions, identified casein kinase 2 (CK2) activity as a key barrier to efficient AAV vector transduction in cardiac cells. Inhibition of CK2 significantly improved transduction rate due to modulation of AAV trafficking, DNA damage response, and RNA processing pathways.
Introduction
Adeno-associated viral vectors (AAVs) are one of the most widely used transgene carriers in the clinic, as they provide transduction of numerous cell types in vivo, with limited activation of immune response. Versatility of AAV application was partially possible through usage of various serotypes exhibiting different cellular tropism. Despite successful targeting of the central nervous system, liver, or retina with AAVs,1 expanding their potential to the cardiovascular system still seems challenging.
Initially, numerous studies in murine models have proven the safety and efficiency of targeting cardiac tissue with AAV serotype 9 (AAV9) vectors (reviewed in Zhang et al.2). Considerable mitigation of cardiomyopathy symptoms in small animal models encouraged further studies using large model organisms and translation of several strategies into the clinic.3 Currently, out of the 174 active clinical trials (clinicaltrials.gov database, accessed on 17.12.22) that use AAV vectors as transgene carriers, only 4 aim at cardiomyopathy treatment. Such a limited development of therapeutic strategies in patients suggests that targeting the human heart with AAV vectors is not an easy task, and to some extent may be the aftermath of an underwhelming outcome of the CUPID (calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease) trial.4 In that study, the lack of functional improvement after AAV-SERCA2a delivery was associated with a very low efficiency of transgene delivery, with the median value of only 43 copies per 1 μg of DNA highlighting the real challenge in development of AAV-based therapeutics in the heart.
Although, to some extent, the issue of low transduction efficacy can be addressed by simply increasing the vector dose, numerous studies report significant hepatotoxicity of high AAV doses.5 While in the case of lower doses of AAVs the initial response is rather mild and can be easily controlled with administration of steroids, high doses tend to provoke severe adverse effects.6 These include thrombotic microangiopathy and, most commonly, hepatotoxicity, which led to the death of several patients undergoing treatment for X-linked myotubular myopathy with AAVs.5,6
To eventually overcome the issue of insufficient transgene delivery, it is essential to understand the mechanisms that impede AAV transduction. Prior research has shown that the main barriers to successful AAV transduction include: availability of appropriate receptors on the cell surface, phosphorylation and subsequent ubiquitination and degradation of the capsids,7,8 nuclear translocation,9 restriction of the second strand synthesis for single-stranded AAVs (ssAAV),10 and interaction of AAV genomes with DNA damage response (DDR) machinery.11 To date, several host restriction factors outside of these pathways were also identified, namely AAVR,12 GPR108,13 TM9SF2,14 and U2 snRNP.15
However, so far this topic has been studied primarily in HeLa or HEK293 cells. Although the HeLa cell line may be a convenient model in various applications, its cancer origin implies some limitations in the interpretation of the obtained data. HeLa cells are highly heterogenic, but what is important is that their genomic and transcriptomic instability differs even between batches of cells cultured for years in different laboratories.16 The sequencing of the HeLa Kyoto cell line highlighted extensive genomic rearrangements that particularly affect cell-cycle progression and DNA repair pathways.17 HEK293 cells, despite not being considered as a cancer line, exhibit significant differences in karyotype characteristics (reviewed by Stepanenko and Dmitrenko18), suggesting genome instability and selection of distinct populations during long-term culture.
More reliable in vitro modeling of vector-host interactions in the heart can be achieved using primary cells isolated directly from the tissue. Even though this strategy is feasible for cells of animal origin, human heart tissue explants are nearly impossible to obtain, especially in the amount sufficient for continuous experiments. The most convenient alternative are cells obtained from differentiation of induced pluripotent stem cells (iPSCs).19 Since the discovery of the iPSC generation method through reprogramming of somatic cells by Takahashi and co-workers,20,21 they have been used for numerous studies, serving as a source of virtually any desired cell type.
Therefore, the aim of this study was to identify the main barriers to successful AAV transduction of the human heart using iPSC-derived cardiomyocytes (iPSC-CMs) and iPSC-derived cardiac fibroblasts (iPSC-CFs) as a model. In addition, we include primary endothelial cells in the study, as they constitute up to 50% of cells in the heart22 and are particularly refractory to AAV transduction. Our data provide strong evidence that casein kinase 2 (CK2) is a crucial modulator of the AAV transduction process, as it acts on multiple levels, starting from trafficking of the vector up to DDR to AAV genomes.
Results
As a model in this study, we selected two different cardiac cell types: cardiomyocytes and cardiac fibroblasts, obtained through differentiation of human iPSCs. We confirmed the phenotype of the differentiated cells through flow cytometry analysis of two fibroblast markers—collagen type I and discoidin domain-containing receptor 2 (DDR2); and epicardial origin marker—transcription factor 21 (TCF21). Average content of triple-positive cells reached more than 95%, showing very high efficiency of the differentiation process. Furthermore, epicardial origin was confirmed by qPCR (Figure S1A). As expected, epicardial progenitor cells exhibited the highest transcript level, which later decreased in iPSC-CFs. Here, as a negative control, we used BJ human skin fibroblasts, where TCF21 expression was not detected at all. iPSC-CFs were comparable in terms of DDR2 expression with the BJ line (Figure S1B) and responded to TGF-β stimulation through upregulation of transcripts for ECM components—collagen type I (Figure S1C) and fibronectin 1 (Figure S1D). Obtained cells had a quiescent phenotype, which was confirmed by lack of α-smooth muscle actin (α-SMA)-positive cells in culture (Figure S1E), but could be easily activated after TGF-β stimulation. Such activated cells contained well-defined α-SMA fibers in the cytoplasm and produced increased amounts of collagen type I (Figure S1E). For cardiomyocytes, expression of α-actinin and its characteristic localization pattern was also confirmed using immunofluorescent staining (Figure S1F). Differentiation efficiency for cardiomyocytes ranged between 85% and 95% basing on flow cytometry staining of cardiac troponin T (Figure S1G).
CK1 and CK2 act as host restriction factors during AAV transduction
To identify the main signaling pathways involved in host-vector interaction, in the first step we analyzed the phosphoproteome of iPSC-CFs after exposure to such vectors. Since silencing of nuclear proteins results in substantial improvement in AAV transduction,11,23,24 we selected a late time point (16 h after exposure to the vectors) and enriched for nuclear fraction proteins before mass spectrometry (MS) analysis. In total, we observed that 82 phosphorylation sites (mapped to 53 unique proteins) had statistically significant ssAAV9-responsive alterations (Figure 1A). On the other hand, for self-complementary (scAAV9) we identified only 17 (mapped to 17 unique proteins) affected phosphosites (Figure 1C). Importantly, the vast majority of these motifs could be targeted by either casein kinase 1 (CK1) or CK2 (Figures 1B and 1D). Gene ontology (GO) enrichment analysis pointed to RNA processing as the main affected pathway (Figures 1E and 1F), with significant alteration detected also in ER stress response (Figure 1E).
Figure 1.

CK2 activity regulates AAV transduction efficiency
(A) Volcano plot representing detected phosphorylation sites in iPSC-CFs 16 h after ssAAV9-CMV-GFP transduction vs. control untransduced cells. Only proteins with phosphorylation fold change above 1.5 with p < 0.05 are marked in blue and were considered in any further analyses. Data normalized to full proteome analysis. (B) Differential phosphorylation motifs grouped by the putative kinases targeting particular sites. (C) Volcano plot representing detected phosphorylation sites in iPSC-CFs 16 h after scAAV9-CMV-GFP transduction vs. control untransduced cells. (D) Differential phosphorylation motifs grouped by the putative kinases targeting a particular site. (E) Dot plot showing results of gene ontology (GO) analysis of differentially regulated phosphosites by ssAAV. (F) The STRING protein-protein interaction network—graphical representation of the RNA processing cluster—confidence of interaction between proteins is marked by line thickness. Analysis of transduction efficiency: percentage of GFP+ cells and MFI of iPSC-CFs (G and H), iPSC-CMs (I and J), and HAECs (K and L) transduced with scAAV-CMV-GFP vectors in the presence of silmitasertib or SR-3029 assessed by flow cytometry 5 days after exposure to the vectors. Two-way ANOVA ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
To test the impact of CK1 on transduction efficiency, we selected SR-3029, an inhibitor of δ and ε isoforms of CK1, which, according to the Human Protein Atlas, are enriched in cardiac muscle. CK2 was targeted with silmitasertib, a potent and selective inhibitor, known also as CX-4945. Both inhibitors significantly augmented AAV transduction in iPSC-CFs (Figures 1G and 1H) for all tested serotypes. A similar effect was observed for AAV6 and AAV9 in iPSC-CMs (Figures 1I and 1J). Especially promising results were observed for silmitasertib, which led to an up to 20-fold increase in GFP+ iPSC-CFs following transduction with scAAV9 vectors (Figure 1G). Furthermore, CK1 and CK2 inhibition was also effective for primary endothelial cells (human aortic endothelial cells [HAECs]) (Figures 1K and 1L). In addition, silmitasertib treatment allowed for significant improvement of transduction efficiency in primary cells—human cardiac fibroblasts (Figure S1H) and human adventitial fibroblasts (Figure S1I).
Since SR-3029 was severely toxic, leading to death of more than half of the cells during the 5 days needed for transgene to be expressed, we focused only on silmitasertib, which did not significantly affect the viability of the cells (based on DAPI [4′,6-diamidine-2′-phenylindole dihydrochloride] staining, not shown). The specificity of silmitasertib, was verified with another CK2 inhibitor—(E/Z) GO-289 (Figures S2A–S2D). The feasibility of CK2 inhibition as a strategy to improve AAV transduction was additionally confirmed in iPSC-CMs and iPSC-CFs differentiated from alternative iPSC lines, where similar results were obtained (Figures S2E–S2H). What is important, the impact of CK2 inhibition was similar for scAAV (Figure 1G) and ssAAV (Figure S2I) vectors, showing its versatility in regulation of AAV transduction.
CK2 activity modulates AAV trafficking
Since CK2 is a broad-spectrum kinase, to unravel its role in AAV transduction we tracked all the steps of AAV processing by the host cells. Imaging of the intact AAV capsids in the cells following AAV9 transduction revealed increased amount of vectors in iPSC-CMs and iPSC-CFs upon CK2 inhibition (Figures 2A and 2B), with very limited effect on HAECs (Figure 2C). These results were confirmed by qPCR, where more AAV genome copies were present in iPSC-CFs following silmitasertib treatment (Figure 2D), but not in HAECs (not shown). Such observation may imply either improved internalization of the vectors or decreased degradation of AAV capsids in the cytoplasm. Interestingly, we noticed direct association between CK2 and AAV9 capsid 2 h after transduction (Figures 2E and 2F), and the presence of a putative CK2 phosphorylation motif in VP1 protein, based on NetPhos 3.1 database analysis (Table S5). To address the question whether CK2 may directly interact with viral proteins (VP1, VP2, and VP3) of the AAV capsid, we performed co-immunoprecipitation (coIP) using anti-CK2 antibody. After probing for VP proteins, we observed interaction of CK2 with VP1 (top band of the input), but not with the other VP proteins (Figure 2G).
Figure 2.

The impact of CK2 on AAV trafficking
Immunofluorescent staining of AAV9 intact capsid and calnexin in iPSC-CMs (A), iPSC-CFs (B), and HAECs (C) 2 and 8 h after exposure to scAAV9-CMV-GFP vectors. Scale bar, 10 μm. (D) qPCR quantification of scAAV9 genome copies in iPSC-CFs 2 h after transduction. Student’s t test vs. control ∗∗p < 0.01. (E) Proximity ligation assay for CK2 and AAV9 intact capsid in iPSC-CFs 2 h after exposure to the vectors. Scale bar, 10 μm. (F) Quantitative analysis of PLA for CK2-AAV9 as number of spots, normalized to number of nuclei. Student’s t test vs. control ∗∗p < 0.01. (G) coIP of CK2 and AAV VP proteins with anti-CK2 antibody in iPSC-CFs, 2 h after transduction with AAV9 vectors. The membrane was probed for VP1/VP2/VP3. Specific bands are marked with an asterisk. Input sample corresponds to 50% of the protein used for coIP to ensure detection of VP proteins in the starting sample. iso, isotype control; untr, untransduced control; AAV9, cells transduced with scAAV9 (2 h). (H) Western blot analysis of CAV1 level in silmitasertib-treated iPSC-CFs with densitometry analysis as ratio of CAV1 to tubulin peak intensity area. (I) Analysis of transduction efficiency of scAAV-CMV-GFP vectors in iPSC-CFs (AAV2 and AAV9) (J), iPSC-CMs (AAV9) (K), and HAECs (AAV9) (L) after silencing of CAV1 expression. Vectors were added 48 h after siRNA; percentage of GFP+ cells was assessed by flow cytometry 5 days after exposure to the vectors. Student’s t test vs. appropriate control ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (M) Immunofluorescent staining of AAV9 intact capsid and PIH1D1 in iPSC-CFs with silenced CAV1, 2 h after exposure to scAAV-CMV-GFP vectors; comparison with silmitasertib treatment. Scale bar, 10 μm. (N) qPCR quantification of scAAV9 genome copies in iPSC-CFs 2 h after transduction. Student’s t test vs. control ∗∗∗p < 0.001.
Although current studies suggest that AAVs can utilize several internalization routes,25,26,27 there is no agreement on the exact contribution of each pathway to productive transduction. In fact, CK2 affects both clathrin- and caveolin-based transport.28,29 As CK2 phosphorylates and thus stabilizes CAV1 and CAV2 proteins,29 we observed a decrease in CAV1 level after silmitasertib treatment in iPSC-CFs (Figures 2H and 2I). In line with these data, knockdown of CAV1 (confirmed at the transcript level, Figure S2J) enabled more efficient transduction of iPSC-CFs with AAV2 and AAV9 (Figures 2J and S2K), the two AAV serotypes most efficient in iPSC-CFs after silmitasertib treatment (Figure 1G). In addition, similar results were obtained for iPSC-CMs, where the percentage of GFP+ cells as well as fluorescence intensity was increased in siCAV-transfected cells (Figures 2K and S2L). For HAECs, only the percentage of GFP+ cells was elevated (Figures 2L and S2M). In accordance with elevated transduction rate, we found an increased amount of AAVs in siCAV-transfected iPSC-CFs in comparison with siMOCK cells (Figure 2N, silmitasertib treatment for comparison), which was also confirmed on qPCR (Figure 2N).
Since silmitasertib also promotes autophagy,30 we assessed the role of this process in AAV transduction. Silencing of ATG7, an essential effector of the autophagy mechanism,30 augmented transduction efficiency for AAV9 in silmitasertib-treated iPSC-CFs (Figure S2M) but had no effect in control, untreated cells (not shown). Moreover, this observation was limited only to AAV9, possibly due to differences in trafficking between AAV2 and AAV9 serotypes. Of note, this effect was specific for fibroblasts, as in iPSC-CMs or HAECs ATG7 downregulation did not affect the transduction outcome (not shown).
Processing of AAV vectors through ER
Silmitasertib treatment resulted in self-limited ER stress response, evidenced by altered BIP, CHOP, and GADD34 expression (Figures S3A–S3C) and phosphorylation of eIF2α (Figure S3D). Yet, no apparent protein aggregation could be observed (Figure S3E). While AAVs were earlier shown to promote unfolded protein response (UPR) activation,31 this effect, associated with elevated GADD34 and BIP expression and reduced level of CHOP, was rather mild and transient in iPSC-CFs transduced with AAV2 (Figures S3F–S3H), with no significant changes in other serotypes (not shown). On the contrary, iPSC-CMs did not exhibit any alterations in gene expression that may point to UPR activation following AAV exposure, regardless of the serotype used (not shown). In addition, we did not observe any correlation between genome type of AAV vectors (ssAAV or scAAV) and UPR activation in any of the tested cell types (Figures S3F–S3H). The discrepancies between iPSC-CFs and iPSC-CMs in UPR activation prompted a question, whether AAVs are actually processed through the ER in both cell types. Staining of transduced cells 6 h after exposure to AAV vectors revealed interaction of the AAV9 capsid with the BIP chaperone, which was much more apparent in iPSC-CFs (Figures 3A and 3B). BIP knockdown (silencing confirmed on transcript level in Figure S3I) induced accumulation of misfolded proteins in the ER (Figure 3C) and hampered the transduction process in iPSC-CFs (Figures 3D and 3E) but not in iPSC-CMs (Figures 3F and 3G). Here only serotypes with the highest transduction efficiency after silmitasertib treatment were used—AAV2 and AAV9 for iPSC-CFs; AAV6 and AAV9 for iPSC-CMs. A similar reduction in transduction efficiency could be observed when AAVs were added together with HA15 (Figures 3H and 3I), an inhibitor of BIP ATPase activity, in iPSC-CFs and surprisingly also in iPSC-CMs (Figures 3J and 3K). Silencing of BIP also had a visible effect on trafficking of AAVs in iPSC-CFs. While, under control conditions, 6 h after infection AAVs started to group in larger foci in the cytoplasm, and this effect was completely abrogated in siBIP-transfected cells, where the signal for AAV capsids remained dispersed in the whole cytoplasm volume (Figure 3L).
Figure 3.

ER trafficking and interaction with BIP chaperone
(A) Proximity ligation assay for AAV9 intact capsid and BIP protein in iPSC-CMs (upper panel) and iPSC-CFs (lower panel). Scale bar, 10 μm. (B) Quantitative analysis of PLA for BIP-AAV9 as number of spots normalized to number of nuclei. Student’s t test vs. control ∗p < 0.05. (C) Immunofluorescent staining of BIP and protein aggregates (PROTEOSTAT probe) in iPSC-CFs after BIP silencing. Scale bar, 10 μm. Analysis of scAAV-CMV-GFP transduction efficiency: percentage of GFP+ cells and MFI in iPSC-CFs (D and E) and iPSC-CMs (F and G) after silencing of BIP expression. Vectors were added 72 h after siRNA. Multiplicity of infection (MOI) of scAAV9 for iPSC-CFs was increased to 105 from standard 104 for better visualization of the silencing impact. Analysis of scAAV-CMV-GFP transduction efficiency: percentage of GFP+ cells and MFI in iPSC-CFs (H and I) and iPSC-CMs (J and K) in the presence of HA15 assessed by flow cytometry 5 days after exposure to the vectors. Student’s t test vs. appropriate control ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (L) Immunofluorescent staining of BIP and AAV9 intact capsid, 6 h after exposure to scAAV9-CMV-GFP vectors. Scale bar, 10 μm.
CK2 regulates transgene expression through destabilization of DDR mechanisms
Analysis of the CK2 direct interactome, using the records deposited in the PhosphoSitePlus (v.6.6.0.4) database, revealed significant enrichment in proteins responsible for DDR and chromatin organization (Figure 4A; detailed characteristics of the DDR cluster are shown in Figures S4A and S4B). Indeed, we found that transient inhibition of CK2 impaired ATM/ATR-related signaling (involved in the DNA damage sensing and downstream signal transduction from both ssDNA and dsDNA breaks), thus likely interfering with DDR execution. Phosphorylation of the ATM/ATR target motif was reduced in all analyzed cell types—iPSC-CFs (Figures 4B–4D), iPSC-CMs (Figure S5A), and HAECs (Figure S5B)—following exposure to silmitasertib. Moreover, it was associated with downregulation of MRE11 level (but did not affect its chaperone PIH1D1) and formation of MRE11/CK2 as well as ATM foci in the nuclei (Figures 4C, 4E, S5A, and S5B). The primary function of MRE11 is recognition of DNA breaks and cleavage of DNA ends to allow HR-directed repair.32 Such observations may imply disturbed resolution of DNA breaks in silmitasertib-treated cells. We also detected increasing levels of p53 in iPSC-CFs (Figures 4C and 4F) and HAECs (Figure S5B), which indicates activation of mitotic checkpoints, presumably in response to DNA damage.33 Silencing of ATM expression (knockdown confirmed on the transcript level, Figure S5C) in both iPSC-CFs and HAECs effectively reduced the signal from phosphorylated ATM substrates (not shown) and augmented AAV transduction in iPSC-CFs (Figure S5D) and HAECs (Figure S5E), with no effect on iPSC-CMs (Figure S5F). Still, this approach was far less effective than CK2 inhibition.
Figure 4.

The impact of CK2 activity inhibition on DNA damage-response machinery
(A) Analysis of GO term pathway enrichment in CK2 interactome (from PhosphoSitePlus v. 6.6.0.4 database) based on the STRING protein-protein interaction network. (B) Western blot analysis of the ATM/ATR phosphorylation motif in iPSC-CFs after 18 h treatment with silmitasertib; α-tubulin as a loading control. (C) Immunofluorescent staining of iPSC-CFs for ATM/ATR phosphorylated substrate (abbreviated as ATMpS in the figure), MRE11, PIH1D1, CK2, ATM, p-ATM, and p53 12 and 24 h after silmitasertib treatment. Scale bar, 10 μm. (D) Analysis of ATMpS signal area normalized to number of nuclei. (E) Analysis of MRE11 MFI in the nucleus area. (F) Analysis of p53 MFI in the nucleus area. Student’s t test vs. control ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Under steady-state conditions, MRE11 can be phosphorylated by CK2, which enables its stabilization by the PIH1D1 chaperone.34 PLA assay clearly confirmed the presence of such interactions in iPSC-CMs as well as iPSC-CFs (Figures 5A and 5B). Upon silmitasertib treatment, the signal for both CK2-MRE11 and Mre11-PIH1D1 was nearly abrogated in iPSC-CMs, and was significantly reduced in iPSC-CFs (Figures 5A–5D). Disruption of the MRE11-PIH1D1 interaction in iPSC-CFs was further confirmed through coIP, where silmitasertib treatment reduced the amount of detected MRE11 protein (Figures 5E and 5F). Unfortunately, we were not able to verify MRE11-CK2 interaction with the same method (not shown), suggesting that it may be more transient, or mediated by other proteins.
Figure 5.

Inhibition of CK2 disruptsMRE11-PIH1D1 interaction and activates transgene expression from AAV vectors
Proximity ligation assay for MRE11 and CK2 (upper panel) or MRE11 and PIH1D1 (lower panel) in iPSC-CMs (A) and iPSC-CFs (B) with quantification as number of spots per nucleus (C and D, respectively). Scale bar, 10 μm. Two-way ANOVA vs. control ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. coIP of PIH1D1 and MRE11 in iPSC-CFs with anti-PIH1D1 antibody—the membrane was probed for PIH1D1 (E) or MRE11 (F). Specific bands are marked with an asterisk. Input sample corresponds to 10% of the protein used for coIP. ctl, control; sil, cells treated for 24 h with 20 μM silmitasertib. (G) Immunofluorescent staining of MRE11 and NBS1 with MFI analysis (H) in iPSC-CFs 24 h after exposure to scAAV9-CMV-GFP vectors. Scale bar, 10 μm. (I) Quantification of genomes immunoprecipitated with anti-MRE11 antibody normalized to the signal obtained for control IgG. (J) qPCR quantification of AAV genomes in iPSC-CFs transduced with scAAV9-CMV-GFP. Analysis of transduction efficiency: percentage of GFP+ cells (K) and median fluorescence intensity (L) in iPSC-CFs treated with silmitasertib or (E/Z) GO289 assessed 5 days after scAAV9 transduction. Student’s t test vs. appropriate control ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Although MRE11 was suggested to mainly interfere with second-strand synthesis of the ssAAV vectors or engage in degradation of vector genomes,11 similar effectiveness of CK2 inhibition in ssAAV and scAAV transduction and direct interaction of MRE11 also with scAAV genomes (Figure 5I) do not support this. Concomitantly, hotspots of MRE11 and NBS1 colocalization were present in the cells transduced with AAVs (Figures 5G and 5H).
To challenge this view even further, we analyzed the AAV genome content in transduced iPSC-CFs over time. To differentiate between dilution of genomes during cell division and degradation of vector DNA, the cells were maintained at full confluency. We noticed that AAV genome number remains fairly stable during long-term culture, with similar counts obtained for days 1 and 7 after transduction and some decline at later time points (Figure 5J). This observation led to a hypothesis that AAV genomes, even though treated by DDR components as damaged DNA, are not resolved by this machinery. Therefore, we transduced iPSC-CFs with AAV9 and applied CK2 inhibitors only after 7 days of culture. Importantly, this resulted in efficient upregulation of transgene expression (Figures 5K and 5L), confirming that AAV genomes are somehow trapped in the cells in a functional but silent state.
CK2 inhibition affects RNA processing mechanisms, which modulate AAV transduction process
We have once again looked into the cellular phosphoproteome after transduction of iPSC-CFs upon CK2 inhibition (Figures 6A and 6C). According to our expectations, the majority of the phosphorylated motifs were associated with CK2 kinase and, to a lesser extent, with CK1 (Figures 6B and 6D) Although we detected some alterations in signaling involved in chromatin remodeling on the phosphoproteome level, once again mRNA processing clusters were the most significantly affected, followed by rRNA processing and DDR (Figure 6E, detailed characteristics of the main clusters available in Figures S6A–S6F). Similarly as in the initial analysis after transduction in control conditions (Figure 1), we noted considerably less alterations in phosphorylation sites in response to scAAV than to ssAAV. Then, we verified the impact of selected proteins with the most significant phosphorylation change on the AAV transduction, namely NCOA5, MATR3, and THRAP3 (silencing efficiency presented in Figure S7). We noticed impaired AAV9 transduction of iPSC-CFs with MATR3 and NCOA5 knockdown (Figures 6F and 6G). These results were partially specific to iPSC-CFs, as in iPSC-CMs the same effect was observed only for siMATR3 (Figures 6H and 6I; based on median fluorescence intensity [MFI]). Surprisingly, silencing of NCOA5 resulted in increased percentage of GFP+ iPSC-CMs (Figure 6H). In the case of THRAP3 knockdown the outcome was more consistent, with augmented AAV9 transduction in both iPSC-CFs (Figures 6F and 6G) and iPSC-CMs (Figures 6H and 6I). While analyzed factors are primarily linked to mRNA processing pathways, we also noticed that silencing of their expression may affect DDR (Figure 6J).
Figure 6.

The role of RNA processing pathway in AAV transduction
(A) Volcano plot representing detected phosphorylation sites in iPSC-CFs 16 h after ssAAV9-CMV-GFP transduction in the presence of silmitasertib vs. ssAAV9 only. Only proteins with phosphorylation fold change above 1.5 with p < 0.05 are marked in blue and were considered in any further analyses. Data normalized to full proteome analysis. (B) Differential phosphorylation motifs grouped by the putative kinases targeting particular site. (C) Volcano plot representing detected phosphorylation sites in iPSC-CFs 16 h after scAAV9-CMV-GFP transduction in the presence of silmitasertib vs. scAAV9 only. (D) Differential phosphorylation motifs grouped by the putative kinases targeting particular sites. (E) Dot plot showing results of GO analysis of differentially regulated phosphosites by silmitasertib and ssAVV vs. ssAAV only. Analysis of ssAAV9-CMV-GFP transduction efficiency: percentage of GFP+ cells and median fluorescence intensity in iPSC-CFs (F and G) and iPSC-CMs (H and I) after silencing of MATR3, THRAP3, and NCOA5 assessed 5 days after scAAV9 transduction. MOI was increased to 105 from standard 104 for better visualization of the silencing impact. Student’s t test vs. appropriate control ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (J) Western blot analysis of the ATM/ATR phosphorylation motif in iPSC-CFs 48 h after transfection with siNCOA5, siMATR3, or siTRHAP3; α-tubulin as a loading control.
Discussion
In the last couple of years, gene therapy has become a feasible alternative to regular treatment in numerous monogenic and multifactor disorders. However, even though cardiovascular disorders are still a leading cause of death worldwide,35 only less than 6% of all registered gene therapy clinical trials target cardiovascular system (http://www.wiley.co.uk/genmed/clinical database). One of the underlying obstacles is a complex etiology of these disorders, which impedes the selection of effective and versatile transgenes. Still, numerous studies in murine models have shown beneficial effects of delivery of various genes in alleviating the symptoms of heart failure (reviewed in Kieserman et al.36). Especially promising results were obtained with the use of AAV vectors, due to their high transduction efficiency in cardiac muscle and low immunogenicity. Unfortunately, to date, translation of these studies into the clinic remains out of reach, as the efficiency of AAV-based transgene delivery into the human heart has been insufficient to improve patients’ condition.4
Since low transduction efficiency appears to be a detrimental factor that restricts the application of AAV vectors for treatment of heart diseases,4 we decided to get a deeper insight into the interaction of AAVs with various types of human cardiac cells. But, due to limited access to human cardiac tissue as well as challenging maintenance of primary cardiomyocytes in the in vitro cultures, we utilized human iPSC-derived cells as a model for our studies. Apart from cardiomyocytes, which constitute approximately 35% of cells in the adult heart,22 we used cardiac fibroblasts. Although in a healthy heart they may constitute up to 12% of the cells,22 they can expand rapidly in response to damage, i.e., myocardial infarction. From three major populations of cardiac fibroblasts, the most abundant one is that of epicardial origin (80% of all cardiac fibroblasts).37,38
CK2 controls multiple signaling pathways and phosphorylates hundreds of downstream substrates.39 CK2 activity is involved in regulation of cell-cycle progression, cell survival, migration, and angiogenesis, and therefore is widely studied in the context of tumor development.40,41 In the case of AAVs, silmitasertib significantly improved the transduction efficiency in iPSC-CMs, iPSC-CFs, and even HAECs, regardless of the serotype used. What is important, similar results were obtained for both ssAAV and scAAV, proving that inhibition of CK2 activity may be a viable and versatile strategy to enhance AAV transduction.
While, to our knowledge, CK2 has not been associated with AAV transduction in any of the previous studies, we also analyzed the data provided for published siRNA and CRISPR screenings. Although CK2 silencing allowed for improvement in transduction of endothelial cells by approximately 3- to 4-fold,42 no effect of CRISPR-based CK2 disruption could be observed in Huh7 hepatocellular carcinoma.43 Still, it is challenging to compare these results with our study, as the efficiency of silencing and chemical inhibition in attenuating CK2 activity may differ. On the other hand, application of the CRISPR system may exert lethal effects, as it would cause permanent lack of CK2 in modified cells. Interestingly, both aforementioned screenings identified various isoforms of CK1 as transduction restricting factors, which supports our findings in iPSC-CMs and iPSC-CFs. However, we were not able to unravel the mechanism behind such effect due to the high toxicity of the CK1 inhibitor.
While analyzing the impact of CK2 on AAV transduction, we found that the AAV9 capsid is directly associated with CK2 kinase in the cytoplasm 2 h after transduction. Although interaction of CK2 and AAV9 can imply capsid phosphorylation, which is a prerequisite for proteasomal degradation,7 to further address this question it would be necessary to analyze site-specific phosphorylation at T561 (or T560 for AAV2). When it comes to internalization of AAVs, so far several routes have been reported, i.e., clathrin-dependent endocytosis,25 micropinocytosis,44 and CLIC-GEEC.26 Since CK2 was shown to participate in stabilization of caveolin-mediated transport and silmitasertib treatment was associated with reduced level of CAV1 protein in iPSC-CFs, we assessed transduction efficiency in cells with CAV1 knockdown. Interestingly, in such conditions, transduction rate was elevated in all cell types, suggesting that caveolin mediates an unproductive uptake/transport route. Such a conclusion would support the hypothesis presented by Nonnenmacher et al. that, even though AAV can utilize different entry pathways at the same time, not all of them lead to productive transduction.45
Monitoring of the transduction process at later time points unveiled that AAV9 capsids directly interact with BIP chaperone protein in iPSC-CFs, but not iPSC-CMs. Silencing of BIP or application of a small-molecule BIP inhibitor hampered AAV transduction in iPSC-CFs and reduced formation of the larger AAV foci in the cytoplasm 6 h after exposure to the vectors, suggesting that BIP may be an essential factor involved in vector trafficking through the ER. Although there are no studies describing the role of ER chaperone proteins in AAV processing, analogous conclusions can be inferred from studies where activation of UPR was linked with enhanced AAV transduction.46,47 As UPR activation results in elevated ER chaperone protein level, such data may indirectly point at their beneficial effect in AAV transduction.
Although CK2 inhibition may regulate AAV transduction process already at the step of trafficking through the cytoplasm, bioinformatic analysis of the CK2 interactome pointed to DDR as the main pathway targeted by this kinase. CK2 can phosphorylate multiple DDR-associated proteins, engaged in both homologous recombination (HR) and NHEJ pathways.48,49 What is more, recruitment of CK2 to the sites of DSBs promotes HR DNA repair through phosphorylation of MDC1,49 which stabilizes binding of MRN complex to DNA. Multiple studies have demonstrated that AAV transduction is heavily dependent on AAV genomes processing by DDR (reviewed in Adachi and Nakai24). While direct interaction of AAV genomes was identified for MRN, Ku86, Rad52, RPA, and DNA polymerase delta,50,51,52 a plethora of other DNA damage-associated proteins were shown to localize in AAV genome processing foci. So far, deficiency of several HR-related proteins, i.e., ATM, MRN complex, and MDC1 was described as beneficial for AAV transduction rate.50,52,53,54,55 These observations correlate with our data, where destabilization of HR-related signaling through CK2 inhibition resulted in significant improvement in transduction efficiency.
To our best knowledge, this is the first report showing potential strategy to rescue the expression from silent AAV genomes retained in transduced cells. Our observations imply that MRE11 may impede transgene expression process at a stage downstream to second strand synthesis and for some reason it does not execute its endonuclease function. Since reactivation of transgene expression from these genomes is feasible, defining transduced and untransduced cells unexpectedly gets complicated as multiple cell types may actually internalize and process AAVs but fail to produce delivered transgene on a detectable level. To some extent our data on maintenance of silent AAV genomes corroborate with recent RNA scope analysis of the vector genome content after in vivo administration of AAV9, where the authors confirmed transduction of heart, liver, and skeletal muscle but also kidney, spleen, adrenal glands, testis, ovaries, pancreas, lung, and bone marrow.56 This would suggest much wider biodistribution of AAV9 than previously assumed. At this point it remains to be elucidated whether these observations are caused by presence of active mechanisms that preclude transgene expression from successfully delivered AAV genomes. Nevertheless, our data imply that DDR and particularly the MRN complex may be engaged in such regulation independently of its role in ssDNA to dsDNA conversion.
While beneficial in terms of transduction efficiency, transient destabilization of DDR by CK2 inhibition may have serious consequences for AAV genome processing in the nuclei. Generation of DSBs in the host genome and concomitant release of vector DNA from the control of DDR machinery may exert unexpected effects related to the random integration of the vector DNA to the host DNA. Furthermore, it remains to be elucidated what the impact of CK2 inhibition on the long-term stability of the genomes is, as impaired activation of the NHEJ pathway in the presence of CK2 inhibitors may translate to hampered concatemerization or circularization of the AAV genomes53,57 and therefore instability of the delivered construct.
Apart from identification of CK1 and CK2 kinases as potential targets in modulation of AAV transduction, our phosphoproteome analysis pointed out a very significant activation of signaling pathways associated with mRNA processing. These findings corroborate with the AAV9 interactome data obtained by Chandran et al., where the authors observed enrichment in RNA binding proteins after a pull-down of AAVs from murine brain.23 A closer look into the identified proteins revealed further similarities, as binding to AAVs and elevated phosphorylation in response to AAV transduction could be confirmed for SRSF1 and HNRNP proteins (our analysis of the dataset from the aforementioned study is presented in Figure S8). The role of RNA splicing machinery in restricting AAV transduction process is additionally supported by another study by Schreibacher et al. that identified U2 snRNP and associated splicing factors as inhibitors of transgene expression due to direct binding of AAV capsid.15 Here, we assessed the role of MATR3, involved in assembly of HEXIM1-DNA-PK-paraspeckle component-ribonucleoprotein complex that regulates the activation of STING-cGAS pathway,58 as well as NCOA5, its direct partner.59 NCOA5 was also demonstrated to directly interact with MAPK14 kinase, which is a CK2 target.60 Furthermore, due to its role in promoting DDR,61 and direct association with CK2,60 we focused on THRAP3 splicing factor. Interestingly, we noticed that MATR3 was an essential protein for efficient AAV transduction, yet its exact role in that process remains unclear. On the other hand, THRAP3 silencing significantly improved transduction. While the interplay between splicing factors and AAV transduction is for now unexplored, we can speculate that THRAP3 may affect cellular response to AAV vectors due to its role in selective splicing and export of transcripts encoding DNA damage-associated proteins.61
What is interesting, in terms of phosphoproteome analysis, we noticed considerable discrepancies between ssAAV and scAAV vectors. Since the proposed mechanisms of CK2 action seem to affect both types of vectors similarly, we suggest that such an outcome may be related to different kinetics of AAV processing because, for scAAV, the synthesis of the second strand of the DNA is not needed and the cellular response can be resolved quicker. Still, this hypothesis would require further studies and analysis of the AAV capsid and genome trafficking at multiple time points after transduction.
In summary, we showed that CK2-related signaling acts on multiple levels of AAV processing in the cells, hampering the transduction process, and possibly also transgene expression itself through regulation of MRE11 stability. Our findings provide a deeper insight into the mechanisms behind vector-host interaction and offer a novel strategy to improve transduction efficiency in various cell types, even those initially refractory to AAVs.
Materials and methods
Cell culture
Human iPSC lines used in this study (abbreviated as DMB01, DMB02, and DMB04) correspond to those described previously (donors 1 to 3, respectively).62 Detailed procedure of iPSC generation and characteristics was described earlier by Stepniewski et al.63 and Martyniak et al.64 Presented results refer to the DMB01 line unless stated otherwise. Human iPSCs were cultured in Essential 8 medium (Gibco), which was replaced every day. Cells were passaged at 80%–90% confluence using 0.5 mM EDTA and seeded on a Geltrex (LDEV-Free, hESC-Qualified, Reduced Growth Factor Basement Membrane Matrix, Gibco)-coated surface. During seeding, 10 μM Y-27632 dihydrochloride (Abcam) was added to the medium.
Human iPSC-CMs were maintained in RPMI 1640 (Biowest/Lonza) with B-27 supplement (Gibco). Cells were passaged using 1× TrypLE (Gibco) supplemented with 0.5 mM EDTA and seeded on a Geltrex-coated surface.
Human iPSC-CFs were cultured in Fibroblast Growth Medium 3 (Promocell) in the presence of 10 μM TGF-β inhibitor (SB421543; Sellekchem). Medium was replaced every day to prevent activation of the cells during the culture. Cells were passaged using 1× TrypLE at confluence reaching approximately 70% and cultured until passage 5.
HAECs (Gibco) were cultured in EGM-2 Endothelial Cell Growth Medium-2 BulletKit medium (Lonza) and passaged at 80%–90% confluence using 0.25% trypsin-EDTA (Gibco) until passage 8.
AAV293 cells (Lonza) were cultured in Dulbecco’s modified Eagle’s medium containing 4.5 g/L glucose (DMEM HG, Biowest) supplemented with 10% fetal bovine serum (FBS) (Biowest) and antibiotics (penicillin 100 U/mL, streptomycin 10 μg/mL, Sigma-Aldrich) and passaged at 60%–70% confluence using 0.05% trypsin-EDTA. All cells were maintained under standard culture conditions (37°C, 5% CO2, humidified atmosphere).
Human aortic adventitial fibroblasts were purchased from Lonza (CC-7014), human cardiac fibroblasts were obtained from Sigma-Aldrich (306-05A) and cultured according to the manufacturers’ instructions.
Differentiation of human iPSC-CMs
Differentiation to cardiomyocytes was conducted using modified protocol by Lian et al.65 Human iPSCs were seeded on a 24-well plate (30,000 cells per well) and at approximately 90%–100% confluence, stimulated with 12 μM (DMB01 line) or 10 μM (DMB02 line) CHIR99021 (Sigma-Aldrich) in RPMI 1640 medium supplemented with B-27 minus insulin (Gibco) for 24 h (day 0). Then, the medium was replaced with fresh RPMI 1640 with B-27 minus insulin supplement and cells were cultured for another 48 h. On day 3, cells were stimulated with 5 μM IWR-1 (Sigma-Aldrich) in fresh RPMI 1640 with B-27 minus insulin, mixed in 1:1 ratio with conditioned medium collected from the differentiating cells on the same day. Fresh RPMI 1640 with B-27 minus insulin was added on day 5. From day 8 onward, cells were cultured in RPMI 1640 with B-27 supplement with medium changes every 3 days. Differentiated iPSC-CMs were used for experiments after day 23 of the procedure. To increase the purity of the iPSC-CM population, metabolic selection was applied.66 Between days 11–14 and days 17–20, cells were cultured in RPMI 1640 medium without glucose (Gibco) supplemented with B-27 and 4 mM sodium DL-lactate (Sigma-Aldrich).
Differentiation of human iPSCs to epicardial fibroblasts
Differentiation to fibroblasts was conducted using a modified protocol established by Zhang et al.67 Human iPSCs were seeded on a 24-well plate (30,000 cells per well) and, when they reached 90%–100% confluence, they were stimulated with 6 μM CHIR99021 (the same concentration for all cell lines) in RPMI with B-27 minus insulin for 48 h (day 0). On day 2, medium was replaced with fresh RPMI 1640 with B-27 minus insulin. The following day, cells were stimulated with 5 μM IWR-1 for 48 h and then they were reseeded on a Geltrex-coated surface (35,000 cells per cm2) in Advanced DMEM/F-12 medium (Gibco) with GlutaMAX I-CTS (Gibco) and 5 μM CHIR99021. On day 8, fresh Advanced DMEM/F-12 medium was added. On day 12, cells were once again reseeded (20,000 cells per cm2) in Fibroblast Growth Medium 3 supplemented with 20 ng/mL bFGF2 (PeproTech) and 10 μM SB421543. Medium was changed every day for another 6 days and such cells were cultured for further experiments.
Immunophenotyping of iPSC-CMs and iPSC-CFs
Detached cells were resuspended in 100 μL phosphate-buffered saline (PBS) (Lonza) and fixed by adding 25 μL of 10% ultra-pure methanol-free formaldehyde (Polysciences). After 10 min of incubation at room temperature (RT), 1 mL of 0.1% Triton X-100 (BioShop) was added and samples were incubated for another 15 min. Then, 2 mL of 4% FBS in PBS was added and tubes were centrifuged for 5 min, at 1,000 × g and RT. Supernatant was discarded and cells were resuspended in 100 μL of 2% FBS in PBS. At this point, primary antibodies were added—for iPSC-CMs, recognizing Cardiac Troponin T, and for iPSC-CFs, DDR2, collagen type I, and TCF21, in 1:100 dilution (Table S1). After 30 min of incubation at 4°C, 2 mL of 2% FBS in PBS was added, cells were centrifuged and resuspended in PBS containing appropriate secondary antibodies (Table S1) in 1:300 dilution. Staining was conducted for 30 min at RT and, after centrifugation, cells were resuspended in 250 μL of PBS containing 0.5 mM EDTA and 200 ng/mL DAPI (Sigma-Aldrich). Cells were analyzed on a BD LSRFortessa flow cytometer using FACS Diva software. As a negative control, cells incubated only with secondary antibodies were used.
Transfection with siRNA
Transfection was performed using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) and 10 pmol of Silencer Select siRNA (Thermo Fisher Scientific) according to the manufacturer’s instructions. In the study, the following siRNA sequences were used: negative control no. 1, siATM ID: s1710, siBIP ID: s6981, siCAV1 ID: s2448, siATG7 ID: s20652, siTHRAP3 ID: s107633, siMATR3 ID: s138540, siNCOA5 ID: s109312. After 24 h medium was changed and cells were further cultured for downstream applications. Silencing effect was assessed 48–72 h after transfection.
Immunofluorescent staining
Cells were seeded on glass coverslips and fixed with methanol for 10 min at −20°C or paraformaldehyde (PFA) for 15 min at RT. Then, wells (or slides with spheroid sections) were washed in PBS and incubated for 1 h (RT) with blocking solution (3% BSA). In the next step, primary antibodies diluted 100 times in blocking solution were added and slides were incubated overnight at 4°C. A detailed description of antibodies used in the study is presented in Table S1. The next day, cells were washed 3 times with PBS for 5 min and incubated with secondary antibodies (Table S1) conjugated with fluorochromes for 45 min at RT. Nuclei were counterstained with DAPI (final concentration: 200 ng/mL). Coverslips were sealed on glass slides in fluorescence mounting medium (Dako) and imaged using a laser scanning confocal microscope (LSM 880 Zeiss).
Proximity ligation assay
Proximity ligation assay (PLA) was performed using Duolink In Situ PLA probes (Sigma-Aldrich) appropriate for the host of primary antibody and Duolink In Situ Detection Orange (Sigma-Aldrich) reagents. The procedure was performed according to the manufacturer’s instructions, using primary antibodies (Table S1) in 1:100 dilution. Imaging of the cells was conducted using a LSM 880 Zeiss confocal microscope. Negative control for the reaction was prepared using only one primary antibody and both probes. For positive control, antibodies against MRE11 and NBS1 were used, as these proteins directly interact with each other as parts of MRN complex, which is present in the nuclei.
AAV vectors production
AAV vectors were produced using a helper-free three-plasmid transfection method. In brief, AAV293 cells were seeded on 15-cm plates and at 60% confluence, transfected with 20 μg of each plasmid: pHelper (Stratagene), pRC (pAAV2/2 or pAAV2/6 or pAAV2/9 depending on produced serotype; pAAV2/2 was obtained from Stratagene, pAAV2/9 was kindly provided by Prof. James Wilson, University of Pennsylvania; pAAV2/6 was constructed from pDGM6 obtained from Addgene) and pGOI (pAAV-CMV-GFP or pdAAV-CMV-GFP). In the case of pGOI, the abbreviations pAAV and pdAAV refer to plasmids suitable for production of ssAAV or scAAV, respectively. Plasmids were diluted in 750 μL of 150 mM NaCl and mixed with 60 μL of PEI MAX (2.58 mg/mL) diluted in 750 μL of 150 mM NaCl.
Isolation of vectors was conducted 3 days after transfection. Cells were scraped in culture medium, transferred to 50-mL conical tubes and centrifuged (350 × g, 10 min, 4°C). Then, all cells were pooled to one tube and washed with 10 mL of PBS. The pellet was resuspended in 6 mL of PBS with calcium and magnesium ions and cells were lysed through three freeze-thaw cycles in a liquid nitrogen/water bath at 37°C, with vigorous mixing between cycles. To digest nucleic acids, in the next step HS nuclease (MoBiTec) was added to the lysates in a final concentration of 50 U/mL. Samples were incubated for 1 h at 37°C and subsequently centrifuged twice to pellet cellular debris (4,000 × g, 30 min, 4°C). Supernatant was collected and stored at −20°C for further purification steps.
Purification of AAV vectors
AAV vectors were purified through ultracentrifugation on a discontinuous iodixanol gradient. Iodixanol solution (OptiPrep, Sigma-Aldrich; initial concentration: 60%) was diluted in PBS containing 0.5 mM magnesium chloride and 2.5 mM potassium chloride to 15%, 25%, 40%, and 54% solutions (v/v). In addition, to visualize phase boundaries, 10 μL of 0.5% (w/v) phenol red (Sigma-Aldrich) was added to 25% and 54% fractions. The gradient was built by adding solutions to 39.2 mL UltraClear tubes (Beckmann Coulter) in the following order: 19 mL of vector-containing lysate, 6 mL of 15% iodixanol, 4.5 mL of 25% iodixanol, 4.5 mL of 40% iodixanol, and 4.5 mL of 54% iodixanol. Then, if needed, PBS was added to the top layer to fill the tube and prevent formation of air bubbles. Tubes were sealed and equilibrated with counterweight, with 20 mg accepted mass difference between samples. Tubes were centrifuged at 300,000 × g for 2 h 15 min at 18°C using an Optima XPN-90 ultracentrifuge (Beckman Coulter).
Next, 40% iodixanol fraction was collected using a needle connected with a syringe, diluted to 50 mL final volume with PBS containing magnesium and potassium chloride and concentrated using Amicon ultra-15 Centrifugal Filters (100 kDa, Merck). Solutions were centrifuged at 4,000 × g for 10 min at 4°C until the volume of the vector preparation reached 500 μL. Such AAV vectors were stored in aliquots at −80°C.
Quantification of AAV vectors
AAV titration was based on the quantity of the genomes in the preparation. First, DNA was isolated from AAVs using phenol-chloroform extraction method. Standard curve for qPCR was prepared using 10-fold serial dilutions of linearized pdAAV-CMV-GFP plasmid. Digestion was performed using SmaI enzyme (New England Biolabs) for 3 h at 25°C. Next, the products were resolved on 1% agarose gel in Tris-acetate-EDTA (0.04 M Tris-acetate, 0.001 M EDTA) buffer and fragment corresponding to 2,282 bp was excised and purified using a Zymoclean Gel DNA Recovery Kit (ZymoResearch).
Detection of AAV genome copies was performed using a qPCR method with a TaqMan probe, recognizing fragment of GFP sequence. Reaction mix contained 10 μL of TaqMan Gene Expression Master Mix (Applied Biosystems), primers recognizing GFP sequence (For.: 5′-CCACGTGTTCACCATGGAGG-3′; Rev: 5′-AAGGCGAAGGGCAGGG-3′) in a final concentration of 250 nM and TaqMan probe (5′-6-FAM-GGGCAACATCCTGTTCGGCAACCAGC-BHQ-1-3′) in a final concentration of 100 nM. The volume was adjusted to 18 μL with nuclease-free water. For the reaction, 2 μL of standard or diluted vector DNA was used as a template. The following reaction conditions were applied: 50°C for 2 min, 95°C for 10 min, 40 cycles; 95°C for 15 s, 60°C for 1 min. Titer was calculated as a number of genome copies per 1 μL of concentrated AAV stock.
Transduction of cells with AAV vectors
For the transduction assays, cells were seeded at 75%–85% confluency. AAV vectors harboring GFP transgene were added in culture medium at a multiplicity of infection of 104 viral genomes/cell, unless specified otherwise. Compounds modulating investigated pathways were added together with AAV vectors. All the small-molecule inhibitors used in the study are listed in Table S2.
Assessment of transduction efficiency using flow cytometry
Transduction efficiency was analyzed using flow cytometry 5 days after addition of the vectors unless specified otherwise. Single-cell suspension was prepared through dissociation of cell cultures with 1× TrypLE solution supplemented with 0.5 mM EDTA. Samples were centrifuged and resuspended in 0.5 mM EDTA in PBS containing 200 ng/mL DAPI. Transduction efficiency was assessed as percentage of GFP+ cells and MFI of GFP+ population after exclusion of DAPI+ events. Data were analyzed only when the viability of treated cells (compounds from Table S2) reached at least 80% of control cells, based on DAPI staining, to exclude toxic compounds or compound concentrations.
Analysis of gene expression
Total RNA was isolated using the phenol-chloroform extraction method. Reverse transcription was performed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) with random hexamer primers according to the manufacturer’s instructions. The reaction mixture was diluted 5 times and used as a template for real-time PCR.
Quantitative real-time PCR was performed using a Step One Plus Real-Time PCR System device (Applied Biosystems). The reaction mix for one sample was prepared as follows: 7 μL of SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich), 0.5 μL of 10 μM forward primer, 0.5 μL of 10 μM reverse primer, 5 μL of nuclease-free water, and 2 μL of cDNA template. Primers used for the reaction as well as their annealing temperatures are listed in Table S3. Reaction was carried out under the following conditions: 95°C for 10 min (initial denaturation); 40 cycles: 95°C for 25 s (denaturation), 58°C–60°C for 60 s (primer annealing), 72°C for 45 s (extension); 72°C for 10 min (final extension). To ensure that the obtained products were specific, analysis of melt curves was conducted after every reaction. Normalization of the data was performed in comparison with expression of EEF2 or TBP. Results were calculated as 2−ΔCt where ΔCt = Cttarget − Cthousekeeping gene. Forward and reverse primers were always separated by at least one intron on the corresponding genomic DNA.
Isolation of protein and western blotting
Total protein was isolated using RIPA buffer (Thermo Fisher Scientific) with Halt protease inhibitor cocktail (Thermo Fisher Scientific) and PhosSTOP (Roche) phosphatase inhibitors. Cells were washed twice with PBS and then scraped from the surface in the lysis buffer. After 20 min incubation on ice, samples were centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was collected and stored at −20°C for further analysis.
Protein concentration was determined using DC Protein Assay (Bio-Rad) according to the manufacturer’s instructions. Samples containing 15–30 μg of total protein were prepared in reducing, denaturing conditions and loaded on polyacrylamide gel. After SDS-PAGE electrophoresis, proteins were transferred to 0.45-μm nitrocellulose membrane (Bio-Rad) for 2 h at 100 V or overnight at 40 V for proteins with mass exceeding 150 kDa. Next, membranes were incubated for 1 h at RT in blocking buffer (5% dry skimmed milk or 5% BSA in TBS containing 0.1% Tween 20). Then, membranes were transferred to primary antibody solution diluted in blocking buffer for overnight incubation at 4°C (details in Table S4). The following day, membranes were washed in TBS (5 times, 5 min), incubated for 45 min at RT with appropriate secondary antibodies (Table S4) and again washed with TBS buffer. In the last step, chemiluminescent substrate (Immobilon Western Chemiluminescent HRP substrate, Merck Millipore) was added to the membranes and the signal was measured using ChemiDoc Imaging System device (Bio-Rad).
Protein coIP
When indicated, to ensure detection of the interaction, cells before protein isolation were additionally treated with 1 mM dithiobis succinimidyl propionate for 45 min to allow protein-protein crosslinking. For protein coIP, fresh protein lysates prepared in immunoprecipitation (IP) lysis buffer (50 mM Tris-HCl [pH 7.4], 120 mM NaCl, 0.5% NP-40, 2 mM EDTA, and cOmplete protease inhibitors) and incubated overnight with protein G Dynabeads (Thermo Fisher Scientific) pre-coated with primary antibody against CK2 or PIH1D1 (1 h at 4°C, 4 μg of antibody per sample in 0.02% Tween 20 in PBS). The next day, beads were collected on the magnet and washed 3 times in 0.02% Tween 20 in PBS. Elution of the bound protein was carried out in 100 mM glycine solution (pH 2.0) for 10 min at RT. Such samples were then separated from the beads and incubated for 10 min at 95°C in western blot loading buffer.
Immunoprecipitation of AAV genomes
For the assessment of MRE11 interaction with scAAV genomes, iPSC-CFs were transduced with scAAV9 vectors. After 72 h, cells were washed 2 times with PBS and fixed using 1% PFA for 10 min at RT. Then glycine was added in a final concentration of 125 mM and cells were once again incubated for 5 min at RT to quench the crosslinking reaction. In the next step, cells were scraped and washed once in B1 buffer (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris-HCl [pH 8.0]), once in B2 buffer (1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl, 10 mM Tris-HCl [pH 8.0]), and subsequently lysed in RIPA buffer. Lysates were sonicated and after centrifugation (10,000 × g, 10 min, 4°C) used for the IP. For this purpose, 2 μg of anti-MRE11 antibody (Abcam, clone 12D7, IgG1) or anti-FLAG control antibody (Sigma-Aldrich, clone M2, IgG1) were added and incubated overnight with end-to-end mixing at 4°C. DNA-protein complexes were collected using protein G Dynabeads (Thermo Fisher Scientific) according to the manufacturer’s instructions. Then, beads were washed once with RIPA buffer, 2 times with PBS containing 0.02% Tween, and incubated overnight at 65°C in Tris-EDTA buffer (pH 8) with 0.2% SDS to revert crosslinking. The next day, 80 μg of proteinase K was added and samples were incubated for 1 h at 56°C. After that supernatants were separated from the beads and used for AAV genome quantification as described above.
Phosphopeptide enrichment and MS analysis
Nuclear fractions from iPSC-CFs were isolated through hypotonic lysis in buffer containing 20 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2 (pH 7.4). The pellet was washed 3 times in hypotonic buffer and lysed in 40 μL of 4% SDS in PBS. Then, after denaturation (10 min, 95°C, 1,200 rpm), protein lysates were sonicated (Bioruptor device, 10 cycles, 30 s each) and diluted to 200 μL. Cell debris was removed by centrifugation at 20,000 × g for 15 min at RT. Proteins were reduced and alkylated by adding TCEP and CAA in a final concentration of 5 and 15 mM, respectively. Proteins were acetone precipitated and protein pellet was reconstituted in 10 μL of 6 M guanidine hydrochloride (GuHCl). After dilution to 0.5 M GuHCl proteins were digested subsequentially with LysC (enzyme/substrate ratio 1:50) and trypsin (enzyme/substrate ratio 1:100) overnight at 37°C. Peptides were acidified with TFA (pH < 2) and desalted using Sep-Pak C18 1 cc Vac Cartridge, 50 mg (Waters, Milford, MA). Eluate was dried using a SpeedVec Concentrator Plus (Eppendorf). Peptides (10 μg) were used for proteomic analysis. Phosphopeptide enrichment was performed using MagReSyn TiO2 functional magnetic microparticles (ReSyn Biosciences) and desalted with double-layered SDB-RPS tips.
All samples were analyzed on an Orbitrap Eclipse Tribid Mass Spectrometer (Thermo Fisher Scientific) equipped with an FAIMS.Pro interface (Thermo Fisher Scientific) and coupled with an easy nLC 1200 (Thermo Fisher Scientific). An in-house packed C18 analytical column (30 cm, 150 μm inside diameter, and 1.9 μm ReproSil-Pur C18 beads; Dr. Maisch, Germany) was used for each sample with an integrated column oven (50°C; PRSO-V1, Biberach, Germany). Proteome samples were analyzed using a 90-min gradient with water, 0.1% formic acid as solvent A, and 80% acetonitrile, 0.1% formic acid as solvent B (0–65 min, 3%–23 % B; 65–78 min, 23%–55% B; 78–90 min, 95% B) with a flow rate of 250 nL/min. Peptides were analyzed in data-dependent acquisition (DDA) mode with a constant compensation voltage (CV) +of −50 of the FAIMS device. Full MS scan was set to 15,000 at a mass/charge ratio (m/z) of 350–1,800, with an AGC target at 300%. MS2 resolution was set to 30,000. Phosphopeptides were analyzed using a 120-min gradient (0–86 min, 3%–30% B; 86–111 min, 30%–50% B; 111–120 min, 50%–95% B). Peptides were analyzed in DDA mode with three CVs of −45, −60, and −75 of the FAIMS device. Full MS scan was set to 60,000 at m/z = 350–1,800, with AGC target at 300%. MS2 resolution was set to 15,000.
First, spectra were split according to their CV using a FAIMS-MzXML-Generator-master, followed by the analysis using MaxQuant (v.2.0.3.0) with phosphosite-serine-threonine-tyrosine as variable modification using a non-canonical human library. Identified phosphosites were normalized to protein abundances. Further analysis was performed in Perseus (v.1.6.5.0) and visualized in Instant Clue (v.0.10.10.2021)
Data analysis
Flow cytometry data were analyzed using FACS Diva software. Microscopy images were processed and analyzed using ImageJ and ZEN 3.3 software. Analysis of the presence of phosphorylation motifs in AAV capsids was performed using a NetPhos 3.1 online tool. Pathway enrichment analysis was carried out using a STRING protein-protein interaction networks functional enrichment analysis tool, v.11.5, available online. Figures were generated using CorelDRAW 2021 and BioRender software. Statistical analysis was performed in GraphPad 8 Software. All data are presented as mean ± SEM of three repetitions, unless stated otherwise, with individual values marked on the graphs.
Data and code availability
All original data are available from the authors without any restrictions. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD040035.
Acknowledgments
This work was supported by PRELUDIUM grant 2019/33/N/NZ1/03066 to I.K. from the National Science Centre. Human iPSC lines used in the study were generated in frame of MAESTRO grant 2018/30/A/NZ3/00412 to J.D. from the National Science Centre. The open-access publication has been supported by the Faculty of Biochemistry, Biophysics and Biotechnology under the Strategic Programme Excellence Initiative at Jagiellonian University in Krakow, Poland. The graphical abstract was created using biorender.com.
Author contributions
I.K. secured funding, designed and performed the experiments, and prepared the manuscript. K.S., K.A., and A.K. performed the experiments. K.S. contributed to data analysis and visualization. L.S. performed MS measurements and analyzed the data. M.K., J.D., and A.J.-K. provided expertise and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.11.010.
Contributor Information
Izabela Kraszewska, Email: izabela.kraszewska@doctoral.uj.edu.pl.
Agnieszka Jaźwa-Kusior, Email: agnieszka.jazwa@uj.edu.pl.
Supplemental information
References
- 1.Domenger C., Grimm D. Next-generation AAV vectors—do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019;28:R3–R14. doi: 10.1093/hmg/ddz148. [DOI] [PubMed] [Google Scholar]
- 2.Zhang H., Zhan Q., Huang B., Wang Y., Wang X. AAV-mediated gene therapy: Advancing cardiovascular disease treatment. Front. Cardiovasc. Med. 2022;9:952755. doi: 10.3389/fcvm.2022.952755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ylä-Herttuala S., Baker A.H. Cardiovascular Gene Therapy: Past, Present, and Future. Mol. Ther. 2017;25:1095–1106. doi: 10.1016/j.ymthe.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg B., Butler J., Felker G.M., Ponikowski P., Voors A.A., Desai A.S., Barnard D., Bouchard A., Jaski B., Lyon A.R., et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 5.Ertl H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022;13:975803. doi: 10.3389/fimmu.2022.975803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kishimoto T.K., Samulski R.J. Addressing high dose AAV toxicity – ‘one and done’ or ‘slower and lower’? Expert Opin. Biol. Ther. 2022;22:1067–1071. doi: 10.1080/14712598.2022.2060737. [DOI] [PubMed] [Google Scholar]
- 7.Zhong L., Zhao W., Wu J., Li B., Zolotukhin S., Govindasamy L., Agbandje-McKenna M., Srivastava A. A Dual Role of EGFR Protein Tyrosine Kinase Signaling in Ubiquitination of AAV2 Capsids and Viral Second-strand DNA Synthesis. Mol. Ther. 2007;15:1323–1330. doi: 10.1038/sj.mt.6300170. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell A.M., Samulski R.J. Mechanistic Insights into the Enhancement of Adeno-Associated Virus Transduction by Proteasome Inhibitors. J. Virol. 2013;87:13035–13041. doi: 10.1128/JVI.01826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelich J.M., Ma J., Dong B., Wang Q., Chin M., Magura C.M., Xiao W., Yang W. Super-resolution imaging of nuclear import of adeno-associated virus in live cells. Mol. Ther. Methods Clin. Dev. 2015;2 doi: 10.1038/mtm.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrari F.K., Samulski T., Shenk T., Samulski R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovric J., Mano M., Zentilin L., Eulalio A., Zacchigna S., Giacca M. Terminal Differentiation of Cardiac and Skeletal Myocytes Induces Permissivity to AAV Transduction by Relieving Inhibition Imposed by DNA Damage Response Proteins. Mol. Ther. 2012;20:2087–2097. doi: 10.1038/mt.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudek A.M., Pillay S., Puschnik A.S., Nagamine C.M., Cheng F., Qiu J., Carette J.E., Vandenberghe L.H. An Alternate Route for Adeno-associated Virus (AAV) Entry Independent of AAV Receptor. J. Virol. 2018;92 doi: 10.1128/JVI.02213-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudek A.M., Zabaleta N., Zinn E., Pillay S., Zengel J., Porter C., Franceschini J.S., Estelien R., Carette J.E., Zhou G.L., Vandenberghe L.H. GPR108 Is a Highly Conserved AAV Entry Factor. Mol. Ther. 2020;28:367–381. doi: 10.1016/j.ymthe.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meisen W.H., Nejad Z.B., Hardy M., Zhao H., Oliverio O., Wang S., Hale C., Ollmann M.M., Collins P.J. Pooled Screens Identify GPR108 and TM9SF2 as Host Cell Factors Critical for AAV Transduction. Mol. Ther. Methods Clin. Dev. 2020;17:601–611. doi: 10.1016/j.omtm.2020.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber C.A., Sakuma T., Izumiya Y., Holditch S.J., Hickey R.D., Bressin R.K., Basu U., Koide K., Asokan A., Ikeda Y. An siRNA Screen Identifies the U2 snRNP Spliceosome as a Host Restriction Factor for Recombinant Adeno-associated Viruses. Plos Pathog. 2015;11 doi: 10.1371/journal.ppat.1005082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frattini A., Fabbri M., Valli R., De Paoli E., Montalbano G., Gribaldo L., Pasquali F., Maserati E. High variability of genomic instability and gene expression profiling in different HeLa clones. Sci. Rep. 2015;5 doi: 10.1038/srep15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landry J.J.M., Pyl P.T., Rausch T., Zichner T., Tekkedil M.M., Stütz A.M., Jauch A., Aiyar R.S., Pau G., Delhomme N., et al. The Genomic and Transcriptomic Landscape of a HeLa Cell Line. G3 (Bethesda). 2013;3:1213–1224. doi: 10.1534/g3.113.005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stepanenko A.A., Dmitrenko V.V. HEK293 in cell biology and cancer research: phenotype, karyotype, tumorigenicity, and stress-induced genome-phenotype evolution. Gene. 2015;569:182–190. doi: 10.1016/j.gene.2015.05.065. [DOI] [PubMed] [Google Scholar]
- 19.Musunuru K., Sheikh F., Gupta R.M., Houser S.R., Maher K.O., Milan D.J., Terzic A., Wu J.C., American Heart Association Council on Functional Genomics and Translational Biology; Council on Cardiovascular Disease in the Young; and Council on Cardiovascular and Stroke Nursing Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2018;11:e000043. doi: 10.1161/HCG.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K., Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 22.Pinto A.R., Ilinykh A., Ivey M.J., Kuwabara J.T., D’Antoni M.L., Debuque R., Chandran A., Wang L., Arora K., Rosenthal N.A., Tallquist M.D. Revisiting Cardiac Cellular Composition. Circ. Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandran J.S., Sharp P.S., Karyka E., Aves-Cruzeiro J.M.d.C., Coldicott I., Castelli L., Hautbergue G., Collins M.O., Azzouz M. Site Specific Modification of Adeno-Associated Virus Enables Both Fluorescent Imaging of Viral Particles and Characterization of the Capsid Interactome. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-15255-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adachi K., Nakai H. DNA Repair and Human Health. InTech; 2011. The Role of DNA Repair Pathways in Adeno-Associated Virus Infection and Viral Genome Replication/Recombination/Integration. [Google Scholar]
- 25.Douar A.-M., Poulard K., Stockholm D., Danos O. Intracellular Trafficking of Adeno-Associated Virus Vectors: Routing to the Late Endosomal Compartment and Proteasome Degradation. J. Virol. 2001;75:1824–1833. doi: 10.1128/JVI.75.4.1824-1833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nonnenmacher M., Weber T. Adeno-Associated Virus 2 Infection Requires Endocytosis through the CLIC/GEEC Pathway. Cell Host Microbe. 2011;10:563–576. doi: 10.1016/j.chom.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan D., Li Q., Kao A.W., Yue Y., Pessin J.E., Engelhardt J.F. Dynamin is required for recombinant adeno-associated virus type 2 infection. J. Virol. 1999;73:10371–10376. doi: 10.1128/jvi.73.12.10371-10376.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galovic M., Xu D., Areces L.B., van der Kammen R., Innocenti M. Interplay between N-WASP and CK2 optimizes clathrin-mediated endocytosis of EGFR. J. Cell Sci. 2011;124:2001–2012. doi: 10.1242/jcs.081182. [DOI] [PubMed] [Google Scholar]
- 29.Sowa G., Pypaert M., Fulton D., Sessa W.C. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc. Natl. Acad. Sci. USA. 2003;100:6511–6516. doi: 10.1073/pnas.1031672100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen B.B., Svenstrup T.H., Guerra B. Downregulation of protein kinase CK2 induces autophagic cell death through modulation of the mTOR and MAPK signaling pathways in human glioblastoma cells. Int. J. Oncol. 2012;41:1967–1976. doi: 10.3892/ijo.2012.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balakrishnan B., Sen D., Hareendran S., Roshini V., David S., Srivastava A., Jayandharan G.R. Activation of the Cellular Unfolded Protein Response by Recombinant Adeno-Associated Virus Vectors. PLoS One. 2013;8:e53845. doi: 10.1371/journal.pone.0053845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carson C.T., Schwartz R.A., Stracker T.H., Lilley C.E., Lee D.V., Weitzman M.D. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J.-H., Paull T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science (80-. ) 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 34.von Morgen P., Burdova K., Flower T.G., O’Reilly N.J., Boulton S.J., Smerdon S.J., Macurek L., Hořejší Z. MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene. 2017;36:4943–4950. doi: 10.1038/onc.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth G.A., Mensah G.A., Johnson C.O., Addolorato G., Ammirati E., Baddour L.M., Barengo N.C., Beaton A.Z., Benjamin E.J., Benziger C.P., et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020;76:2982–3021. doi: 10.1016/j.jacc.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kieserman J.M., Myers V.D., Dubey P., Cheung J.Y., Feldman A.M. Current Landscape of Heart Failure Gene Therapy. J. Am. Heart Assoc. 2019;8:e012239. doi: 10.1161/JAHA.119.012239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quijada P., Misra A., Velasquez L.S., Burke R.M., Lighthouse J.K., Mickelsen D.M., Dirkx R.A., Small E.M. Pre-existing fibroblasts of epicardial origin are the primary source of pathological fibrosis in cardiac ischemia and aging. J. Mol. Cell. Cardiol. 2019;129:92–104. doi: 10.1016/j.yjmcc.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tallquist M.D., Molkentin J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017;14:484–491. doi: 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meggio F., Pinna L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 40.Loizou J.I., El-Khamisy S.F., Zlatanou A., Moore D.J., Chan D.W., Qin J., Sarno S., Meggio F., Pinna L.A., Caldecott K.W. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 41.Borgo C., D’Amore C., Sarno S., Salvi M., Ruzzene M. Protein kinase CK2: a potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021;6:183. doi: 10.1038/s41392-021-00567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallen A.J., Barker G.A., Fein D.E., Jing H., Diamond S.L. Enhancers of adeno-associated virus AAV2 transduction via high throughput siRNA screening. Mol. Ther. 2011;19:1152–1160. doi: 10.1038/mt.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madigan V.J., Tyson T.O., Yuziuk J.A., Pillai M., Moller-Tank S., Asokan A. A CRISPR Screen Identifies the Cell Polarity Determinant Crumbs 3 as an Adeno-associated Virus Restriction Factor in Hepatocytes. J. Virol. 2019;93:e009433-19. doi: 10.1128/JVI.00943-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanlioglu S., Benson P.K., Yang J., Atkinson E.M., Reynolds T., Engelhardt J.F. Endocytosis and Nuclear Trafficking of Adeno-Associated Virus Type 2 Are Controlled by Rac1 and Phosphatidylinositol-3 Kinase Activation. J. Virol. 2000;74:9184–9196. doi: 10.1128/jvi.74.19.9184-9196.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nonnenmacher M.E., Cintrat J.-C., Gillet D., Weber T. Syntaxin 5-Dependent Retrograde Transport to the trans -Golgi Network Is Required for Adeno-Associated Virus Transduction. J. Virol. 2015;89:1673–1687. doi: 10.1128/JVI.02520-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson J.S., Gentzsch M., Zhang L., Ribeiro C.M.P., Kantor B., Kafri T., Pickles R.J., Samulski R.J. AAV Exploits Subcellular Stress Associated with Inflammation, Endoplasmic Reticulum Expansion, and Misfolded Proteins in Models of Cystic Fibrosis. Plos Pathog. 2011;7 doi: 10.1371/journal.ppat.1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cui M., Zhao Q., Wang J., Si Y., Cheng S., Ding W. ATF6-Mediated Unfolded Protein Response Facilitates Adeno-associated Virus 2 (AAV2) Transduction by Releasing the Suppression of the AAV Receptor on Endoplasmic Reticulum Stress. J. Virol. 2022;96:e0110321. doi: 10.1128/JVI.01103-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olsen B.B., Fritz G., Issinger O.G. Characterization of ATM and DNA-PK wild-type and mutant cell lines upon DSB induction in the presence and absence of CK2 inhibitors. Int. J. Oncol. 2012;40:592–598. doi: 10.3892/ijo.2011.1227. [DOI] [PubMed] [Google Scholar]
- 49.Olsen B.B., Wang S.Y., Svenstrup T.H., Chen B.P.C., Guerra B. Protein kinase CK2 localizes to sites of DNA double-strand break regulating the cellular response to DNA damage. BMC Mol. Biol. 2012;13:7. doi: 10.1186/1471-2199-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cervelli T., Palacios J.A., Zentilin L., Mano M., Schwartz R.A., Weitzman M.D., Giacca M. Processing of recombinant AAV genomes occurs in specific nuclear structures that overlap with foci of DNA-damage-response proteins. J. Cell Sci. 2008;121:349–357. doi: 10.1242/jcs.003632. [DOI] [PubMed] [Google Scholar]
- 51.Jurvansuu J., Raj K., Stasiak A., Beard P. Viral Transport of DNA Damage That Mimics a Stalled Replication Fork. J. Virol. 2005;79:569–580. doi: 10.1128/JVI.79.1.569-580.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zentilin L., Marcello A., Giacca M. Involvement of Cellular Double-Stranded DNA Break Binding Proteins in Processing of the Recombinant Adeno-Associated Virus Genome. J. Virol. 2001;75:12279–12287. doi: 10.1128/JVI.75.24.12279-12287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi V.W., McCarty D.M., Samulski R.J. Host cell DNA repair pathways in adeno-associated viral genome processing. J. Virol. 2006;80:10346–10356. doi: 10.1128/JVI.00841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cataldi M.P., McCarty D.M. Differential Effects of DNA Double-Strand Break Repair Pathways on Single-Strand and Self-Complementary Adeno-Associated Virus Vector Genomes. J. Virol. 2010;84:8673–8682. doi: 10.1128/JVI.00641-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwartz R.A., Palacios J.A., Cassell G.D., Adam S., Giacca M., Weitzman M.D. The Mre11/Rad50/Nbs1 Complex Limits Adeno-Associated Virus Transduction and Replication ᰔ. J. Virol. 2007;81:12936–12945. doi: 10.1128/JVI.01523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao J., Yue Y., Patel A., Wasala L., Karp J.F., Zhang K., Duan D., Lai Y. High-Resolution Histological Landscape of AAV DNA Distribution in Cellular Compartments and Tissues following Local and Systemic Injection. Mol. Ther. Methods Clin. Dev. 2020;18:856–868. doi: 10.1016/j.omtm.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang J., Zhou W., Zhang Y., Zidon T., Ritchie T., Engelhardt J.F. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J. Virol. 1999;73:9468–9477. doi: 10.1128/jvi.73.11.9468-9477.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morchikh M., Cribier A., Raffel R., Amraoui S., Cau J., Severac D., Dubois E., Schwartz O., Bennasser Y., Benkirane M. HEXIM1 and NEAT1 Long Non-coding RNA Form a Multi-subunit Complex that Regulates DNA-Mediated Innate Immune Response. Mol. Cell. 2017;67:387–399.e5. doi: 10.1016/j.molcel.2017.06.020. [DOI] [PubMed] [Google Scholar]
- 59.Hein M.Y., Hubner N.C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I.A., Weisswange I., Mansfeld J., Buchholz F., et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 60.Golkowski M., Lius A., Sapre T., Lau H.-T., Moreno T., Maly D.J., Ong S.-E. Multiplexed kinase interactome profiling quantifies cellular network activity and plasticity. Mol. Cell. 2023;83:803–818.e8. doi: 10.1016/j.molcel.2023.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vohhodina J., Barros E.M., Savage A.L., Liberante F.G., Manti L., Bankhead P., Cosgrove N., Madden A.F., Harkin D.P., Savage K.I. The RNA processing factors THRAP3 and BCLAF1 promote the DNA damage response through selective mRNA splicing and nuclear export. Nucleic Acids Res. 2017;45:12816–12833. doi: 10.1093/nar/gkx1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kraszewska I., Tomczyk M., Andrysiak K., Biniecka M., Geisler A., Fechner H., Zembala M., Stępniewski J., Dulak J., Jaźwa-Kusior A. Variability in Cardiac miRNA-122 Level Determines Therapeutic Potential of miRNA-Regulated AAV Vectors. Mol. Ther. Methods Clin. Dev. 2020;17:1190–1201. doi: 10.1016/j.omtm.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stępniewski J., Tomczyk M., Andrysiak K., Kraszewska I., Martyniak A., Langrzyk A., Kulik K., Wiśniewska E., Jeż M., Florczyk-Soluch U., et al. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes, in Contrast to Adipose Tissue-Derived Stromal Cells, Efficiently Improve Heart Function in Murine Model of Myocardial Infarction. Biomedicines. 2020;8:578. doi: 10.3390/biomedicines8120578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martyniak A., Andrysiak K., Motais B., Coste S., Podkalicka P., Ferdek P., Stępniewski J., Dulak J. Generation of microRNA-378a-deficient hiPSC as a novel tool to study its role in human cardiomyocytes. J. Mol. Cell. Cardiol. 2021;160:128–141. doi: 10.1016/j.yjmcc.2021.07.007. [DOI] [PubMed] [Google Scholar]
- 65.Lian X., Zhang J., Azarin S.M., Zhu K., Hazeltine L.B., Bao X., Hsiao C., Kamp T.J., Palecek S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tohyama S., Hattori F., Sano M., Hishiki T., Nagahata Y., Matsuura T., Hashimoto H., Suzuki T., Yamashita H., Satoh Y., et al. Distinct Metabolic Flow Enables Large-Scale Purification of Mouse and Human Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 67.Zhang H., Tian L., Shen M., Tu C., Wu H., Gu M., Paik D.T., Wu J.C. Generation of Quiescent Cardiac Fibroblasts From Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis. Circ. Res. 2019;125:552–566. doi: 10.1161/CIRCRESAHA.119.315491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data are available from the authors without any restrictions. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD040035.