

SUMMARY
MCL-1 is a high-priority target due to its dominant role in the pathogenesis and chemoresistance of cancer, yet clinical trials of MCL-1 inhibitors are revealing toxic side effects. MCL-1 biology is complex, extending beyond apoptotic regulation and confounded by its multiple isoforms, its domains of unresolved structure and function, and challenges in distinguishing noncanonical activities from the apoptotic response. We find that, in the presence or absence of an intact mitochondrial apoptotic pathway, genetic deletion or pharmacologic targeting of MCL-1 induces DNA damage and retards cell proliferation. Indeed, the cancer cell susceptibility profile of MCL-1 inhibitors better matches that of anti-proliferative than pro-apoptotic drugs, expanding their potential therapeutic applications, including synergistic combinations, but heightening therapeutic window concerns. Proteomic profiling provides a resource for mechanistic dissection and reveals the minichromosome maintenance DNA helicase as an interacting nuclear protein complex that links MCL-1 to the regulation of DNA integrity and cell-cycle progression.
Graphical abstract
In brief
Adhikary et al. report that MCL-1 regulates cell proliferation and DNA integrity independent from its role in apoptosis suppression. Genetic deletion or pharmacologic targeting has a striking anti-proliferative effect, informing opportunities to expand the utility of MCL-1 inhibitors in cancer as single agents and in combinations, but with heightened precautions.
INTRODUCTION
MCL-1 is a member of the anti-apoptotic subgroup of BCL-2 family proteins, which regulate the critical balance between cellular life and death.1,2 The canonical function of MCL-1 involves sequestering pro-apoptotic BCL-2 family proteins in heterodimeric interaction at the outer mitochondrial membrane. A surface groove on MCL-1 traps the critical killer motif, termed BCL-2 homology domain 3 (BH3), of pro-apoptotic members, which are subdivided into BH3-only and multi-domain proteins.3 The BH3-only proteins are a heterogeneous group of stress sensors that deploy their one conserved BH domain to block anti-apoptotic proteins and/or directly activate the multi-domain pro-apoptotic proteins, BAX and BAK.4,5 Once triggered, BAX and BAK transform from latent to activated monomers, which expose their BH3 helices, and then homo-oligomerize to permeabilize the outer mitochondrial membrane.6 The capture of BH3 helices by the grooves of BCL-2 family anti-apoptotic proteins prevents apoptosis induction,7 a mechanism that is hijacked by cancer cells to promote pathologic cell survival. The structure of the so called “BH3-in-groove” interaction formed the basis for a drug development strategy that relies on BH3 mimicry to block the anti-apoptotic groove and thereby release the brake on cancer cell apoptosis.8,9
There are six known anti-apoptotic proteins that exhibit overlapping and nonoverlapping functions. MCL-1 is unique among anti-apoptotic members on a variety of counts. First, it is larger than any other BCL-2 family protein, bearing an extended N terminus of unresolved structure and functional significance.2 Second, it is by far the most widely expressed anti-apoptotic protein across human cancers,10 raising the question of why it is the preferred anti-apoptotic member. Third, MCL-1 has distinct physiologic functions not shared with other anti-apoptotic BCL-2 proteins, as highlighted by the spectrum of pathologies that emerge upon MCL-1’s complete or tissue-specific deletion, including defective embryonic implantation,11 hematopoiesis,12 neurogenesis,13 immunity,14 and cardiac function.15 At the cellular level, MCL-1 and its multiple isoforms have been implicated in a host of signaling pathways and related protein interactions, spanning apoptosis regulation (e.g., BAX, BAK), calcium homeostasis (e.g., IP3R, VDAC), autophagy (e.g., Beclin-1), fatty acid oxidation (e.g., VLCAD), mitochondrial fission/fusion (e.g., DRP-1, OPA-1), DNA damage response (e.g., CHK1, IEX-1, γH2AX, Ku), cell cycle (e.g., p18, PCNA, CDK1), and its proteasomal degradation (e.g., HUWE1, FBW7, USP9X).16 Given the interdependence of many of these pathways, such as apoptosis, cell cycle, and cellular energetics, teasing apart the many proposed noncanonical functionalities of MCL-1 is a major challenge. Predicting how these nonapoptotic functions of MCL-1 could be affected by pharmacologic targeting presents another quagmire, which has especially high stakes given the advancement of MCL-1 inhibitors to human testing in cancer.17
The oncogenic potency of MCL-1 and its reported links to at least three of the defined hallmarks of cancer,18 namely resisting cell death, sustaining proliferative signaling, and enabling replicative immortality, suggest that MCL-1 has independent yet synergistic roles in multiple oncogenic signaling networks. Given that the combination of proliferative drive and apoptotic blockade is so critical to cancer pathogenesis and chemoresistance, as demonstrated by the genetic characteristics of follicular lymphoma (MYC plus BCL-2 overexpression) and mantle cell lymphoma (cyclin D1 plus BIM loss),19 we sought to characterize the influence of MCL-1 on cell proliferation in the absence of BAX/BAK-mediated apoptosis, which to date has otherwise confounded the analysis. Here, we find that genetic deletion and pharmacologic inhibition of MCL-1 has an independent anti-proliferative effect, which is coincident with induction of DNA damage. The identification of MCL-1 in a series of nuclear complexes, with the minichromosome maintenance (MCM) protein complex emerging as a topmost hit, underscores the multifaceted nature of MCL-1 biology. Our results inform the expanded utility of MCL-1 inhibitors as single agents and in specific combinations for the treatment of cancer, raise therapeutic window concerns owing to potential noncanonical mechanisms of toxicity, and reveal MCM as a target of MCL-1 in regulating cell proliferation and maintaining DNA integrity.
RESULTS
Characterization of the anti-proliferative effect of Mcl-1 deletion
To precisely evaluate the effect of Mcl-1 deletion on cell proliferation, we first compared the growth rate of cultured wild-type and Mcl-1−/− mouse embryonic fibroblasts (MEFs) in the absence of any stimulus and observed a more than 2-fold decrease in Mcl-1−/− MEF cell count by day 2 of plating (Figure 1A). We corroborated these results by measuring BrdU incorporation, an orthogonal readout of cell proliferation (Figure S1A). Importantly, this effect was not the result of apoptosis induction, as cell viability was >94% for both wild-type and Mcl-1−/− MEFs throughout the duration of the experiment (Figure S2A). We further confirmed that our growth rate monitoring assay can detect cell death, as demonstrated by the apoptotic effect of staurosporine treatment on the MEFs (Figure S1B). We next examined whether this decrease in cell proliferation was recapitulated upon acute deletion of Mcl-1 using Mcl-1fl/flRosa-ERCreT2 MEFs and tamoxifen treatment.20 Indeed, we observed identical results with the inducible knock-out system, and restoration of cell growth upon MCL-1 reconstitution (Figures 1B and S2B). Given the discovery of a mitochondrial matrix-localized analog of MCL-1 (MCL-1Matrix),20,21 which could potentially influence cell growth through its involvement in mitochondrial energetics, we further examined the comparative impact on cell growth of reconstituting the acutely deleted Mcl-1−/− MEFs with either MCL-1Matrix or the canonical outer mitochondrial membrane isoform (MCL-1OMM) using our previously generated and validated cell line system.22 Interestingly, we found that expressing MCL-1OMM, but not MCL-1Matrix, increased cell growth (Figures 1C and S2C).
Figure 1. Genetic deletion or pharmacologic targeting of MCL-1 reduces cell proliferation.
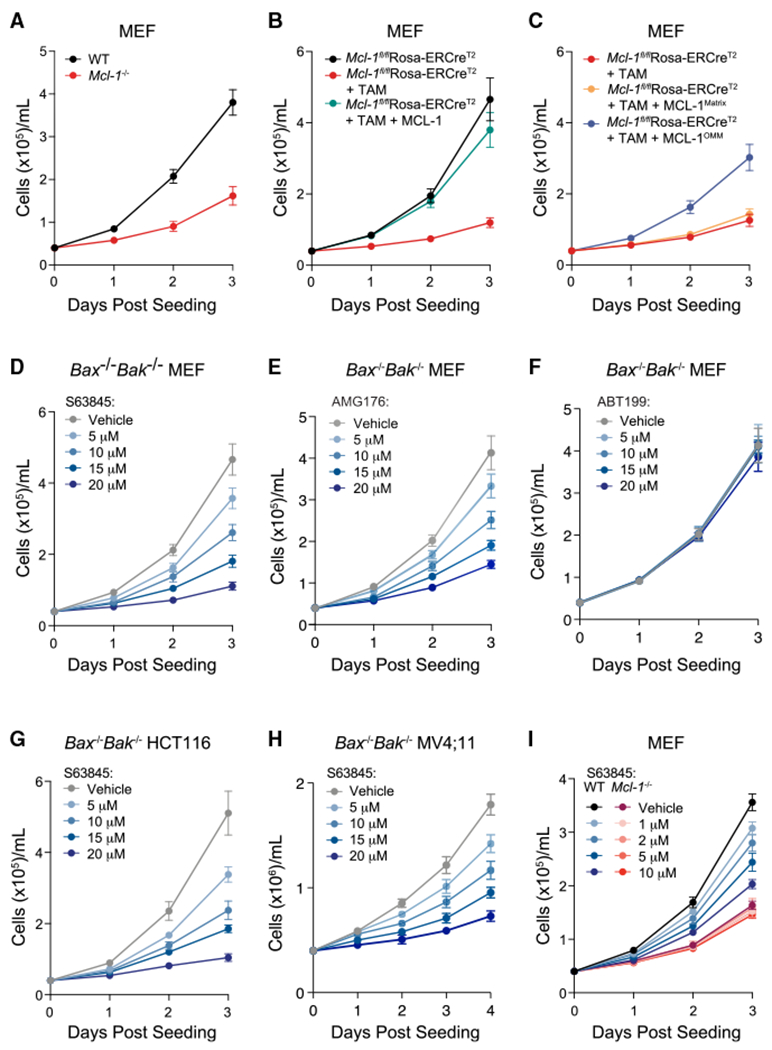
(A) Chronic deletion of Mcl-1 (red) in mouse embryonic fibroblasts (MEFs) resulted in decreased cell proliferation compared with wild-type cells (black), as monitored by trypan blue staining and cell count. Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells with similar results.
(B). Acute Mcl-1 deletion (red; tamoxifen-treated Mcl-1fl/flRosa-ERCreT2 MEFs) led to a similar decrease in cellular proliferation compared with the corresponding vehicle-treated MEFs (black), as monitored by trypan blue staining and cell count. Reconstitution of MCL-1 restored proliferation to wild-type levels (green). Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells with similar results.
(C) Reconstitution of the outer mitochondrial membrane isoform (MCL-1OMM) (blue) but not the matrix isoform (MCL-1Matrix) (yellow) of MCL-1 in tamoxifen-treated Mcl-1fl/flRosa-ERCreT2 MEFs (red) enhanced cell proliferation, as measured by trypan blue staining and cell count. Of note, the Mcl-1fl/flRosa-ERCreT2 MEF condition shown in (B) (black) was performed simultaneously with (C) experiments and thus serves as the positive control for both (B) and (C). Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells with similar results.
(D–F) Pharmacologic blockade of MCL-1 by S63845 (D) and AMG176 (E), but not BCL-2 by ABT-199 (Venetoclax) (F), caused a dose-responsive decrease in cell proliferation (blue) in Bax−/−Bak−/− MEFs, as measured by trypan blue staining and cell count. Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells and drug dilutions with similar results. (G–H) Pharmacologic blockade of MCL-1 by S63845 in Bax−/−Bak−/− HCT116 colon carcinoma cells (G) and Bax−/−Bak−/− MV4;11 acute myeloid leukemia cells (H) led to a similar dose- responsive decrease in cell proliferation (blue), as measured by trypan blue staining and cell count. Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells and S63845 dilutions with similar results.
(I) Treatment of wild-type MEFs with S63845 caused a dose-responsive reduction in cell proliferation (blue), whereas Mcl-1−/− MEFs that lack the MCL-1 target were unaffected (red). Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells and S63845 dilutions with similar results.
See also Figures S1 and S2.
To determine if the anti-proliferative consequences of Mcl-1 deletion occurred in the absence of mitochondrial apoptosis, which would further dissociate MCL-1’s role in cell proliferation from apoptosis regulation, we treated Bax−/−Bak−/− MEFs23 with the selective MCL-1 inhibitor molecule, S63845,24 applying doses that have no effect on cell viability, whether specific or nonspecific (Figure S2D). Strikingly, we found that S63845 dose-responsively inhibited cell proliferation in the absence of BAX/BAK and with little to no effect on cell viability (Figures 1D and S2D). To control for potential off-target side effects related to the structure of S63845, we repeated the experiment using an alternative selective MCL-1 inhibitor with a distinct chemical structure, AMG176, and observed the identical result (Figures 1E, S1C, and S2E–S2F). In contrast, the selective BCL-2 inhibitor ABT-199 (Venetoclax), administered at the same micromolar dosing levels, had no effect on either cell proliferation or cell viability (Figures 1F and S2G). To expand the analysis further, we treated Bax−/−Bak−/− HCT116 colon cancer cells25 and Bax−/−Bak−/− MV4;11 leukemia cells with S63845 and, in each case, again observed dose-responsive suppression of cell proliferation with little to no effect on cell viability (Figures 1G, 1H, S2H, and S2I).
Whereas single agent S63845 is insufficient at triggering cell death in MEFs (Figures S2J and S2K) due to the co-expression of other anti-apoptotic BCL-2 proteins, the micromolar-range dosing used to elicit the observed anti-proliferative effect is higher than the nanomolar concentrations that trigger apoptosis in MCL-1-dependent cancer cells.24 Therefore, we further investigated the MCL-1 dependence of the anti-proliferative effect of S63845 by comparing the response to treatment in wild-type and Mcl-1−/− MEFs. We observed dose-responsive suppression of cell growth in wild-type cells but no effect in the absence of the MCL-1 target, confirming that the anti-proliferative activity of S63845 is specific in that it requires the presence of MCL-1 (Figures 1I, S1D, S2J, and S2K). We further corroborated these results by genetic means, monitoring comparative cell proliferation in Bax−/−Bak−/− vs. Mcl-1−/−Bax−/−Bak−/− MEFs, and again observed decreased cell proliferation in the absence of MCL-1 in this non-apoptotic (i.e., BAX BAK-null) context (Figures S1E, S2L, and S2M). Taken together, these data indicate that genetic deletion of Mcl-1 significantly impairs cell growth, involves the canonical MCL-1OMM isoform of MCL-1, and can be recapitulated by pharmacologic or genetic targeting of MCL-1 in the presence and absence of an intact mitochondrial apoptotic pathway.
Correlation between MCL-1 and cell proliferation across cell and tissue datasets
To determine if our findings in genetically defined MEFs, HCT116, and MV4;11 cells extended to a broader context, we conducted a series of big data analyses evaluating the effects of genetic deletion and pharmacologic inhibition of MCL-1 on cells and tissues. We first analyzed and compared gene expression data from a previously reported dataset comparing wild-type and Mcl-1-deleted thymus, a notably proliferative tissue that is specifically dependent on MCL-1 for homeostasis26 (Figure 2A). Thymic epithelial cells lacking MCL-1 demonstrate upregulation of a series of cell proliferation genes, such as Cdk2, Cdk4, Ccnd2, Bub1b, Plk1, and the Mcm genes, which play critical roles during the cell cycle (Figures 2B and 2C). Kyoto Encyclopedia of Genes and Genomes analysis revealed ribosome biogenesis, cell cycle, and DNA replication as the most enriched transcriptomic pathways upon Mcl-1 deletion (Figure 2D). Although these results could reflect an attempted proliferative response to apoptosis induction (upon loss of the canonical function of MCL-1), the co-expression of BCL-2 and BCL-XL do not compensate for MCL-1 loss in this context and thymic atrophy ensues.26 Correspondingly, MCL-1 but not BCL-XL is required for the growth of thymic lymphoma in p53-deficient mice.27 Based on our findings of reduced cell proliferation in MEFs, HCT116, and MV4;11 cells upon Mcl-1 deletion and/or pharmacologic MCL-1 targeting (Figures 1 and S1), the transcriptomic findings in thymic epithelial cells could alternatively represent, at least in part, an attempt to compensate for a block in proliferation upon Mcl-1 deletion (due to loss of a non-canonical function of MCL-1) by upregulating the machinery needed for cell proliferation and tissue growth, namely ribosomal, cell-cycle, and DNA replication proteins.
Figure 2. Correlation between MCL-1 and cell-cycle perturbations across transcriptomic and pharmacologic datasets.
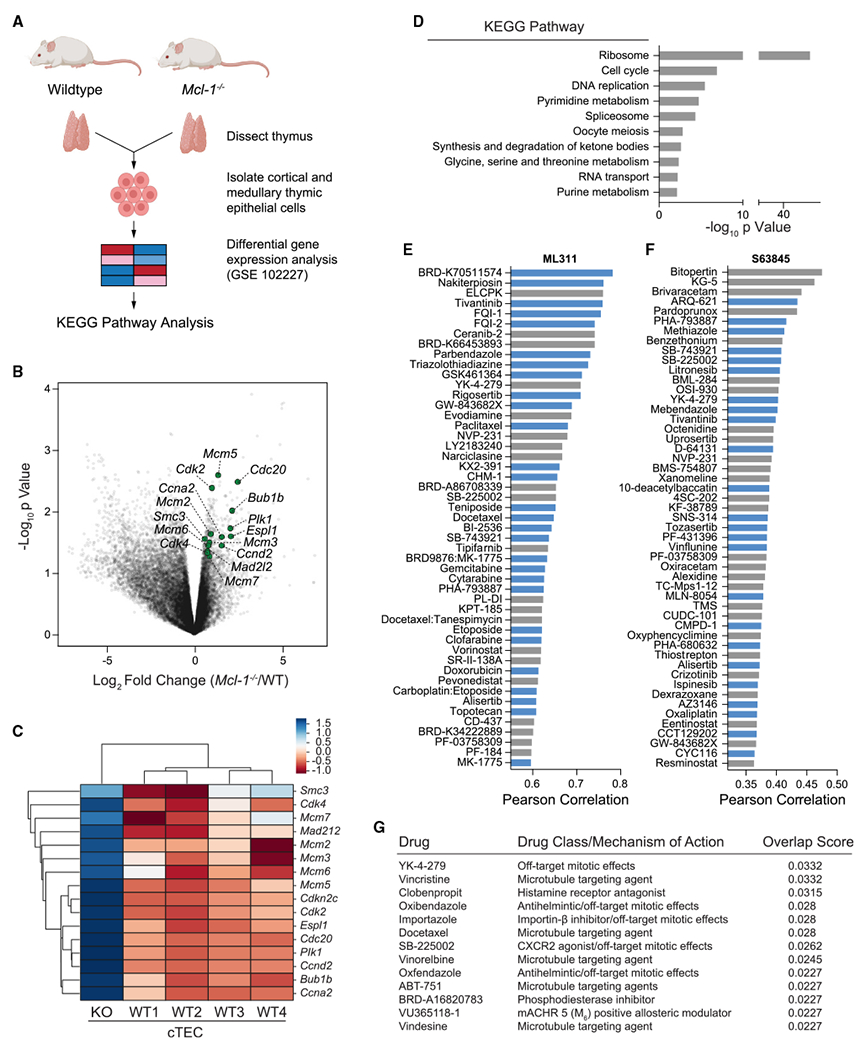
(A) Gene expression analysis workflow for comparative analysis of wild-type and Mcl-1 conditionally deleted thymic epithelial cells (GEO dataset GSE102227).
(B) Differential gene expression analysis of wild-type and Mcl-1−/− murine cortical thymic epithelial cells, as quantified by log2 fold changes (x axis) and significance (y axis). DNA replication and cell-cycle-related genes with the most differential upregulation upon conditional Mcl-1 deletion are highlighted in green.
(C) Clustermap showing the comparative gene expression of select DNA replication and cell-cycle genes in wild-type (WT1-4) vs. Mcl-1−/− (KO) cortical thymic epithelial cells (cTECs).
(D) Kyoto Encyclopedia of Genes and Genomes analysis revealed ribosome biogenesis, cell cycle, and DNA replication as the most significantly enriched transcriptomic pathways upon Mcl-1 deletion in murine thymic epithelial cells.
(E and F) Selective small-molecule inhibition of MCL-1 by ML311 (E) or S63845 (F) correlates with the pharmacologic profiles of anti-proliferative drugs based on an analysis of sensitivity data of ~500 drugs across the Cancer Cell Line Encyclopedia (CTRP v2.0, Broad Institute). Compounds with established anti-proliferative and anti-mitotic effects are colored in blue.
(G) The gene expression signature of conditional Mcl-1 deletion in murine thymic epithelial cells (GEO dataset GSE102227) correlates with that of pharmacologic treatment with microtubule-targeting and anti-mitotic agents (L1000CDS2 database).
See also Figure S3.
Given the critical role of drugs that target apoptosis and cell-cycle progression in cancer treatment, we next examined whether the susceptibility profile to pharmacologic inhibition of MCL-1 correlated with particular drug classes from two independent drug sensitivity databases (Cancer Therapeutics Response Portal28 and PRISM29) across the Cancer Cell Line Encyclopedia.30 Interestingly, sensitivity to two MCL-1 inhibitors, ML31131 and S63845,24 showed the highest correlation with cancer cell response profiles to anti-proliferative drugs, including inhibitors of mitosis and DNA synthesis such as BRD-K70511574 (Plk1 inhibitor), paclitaxel (microtubule targeting agent [MTA]), parbendazole (anti-mitotic drug), doxorubicin (topoisomerase II inhibitor), gemcitabine (DNA synthesis inhibitor), and etoposide (topoisomerase II inhibitor) (Figures 2E, 2F, and S3A–S3C). In contrast, pharmacologic targeting of BCL-XL or BCL-2 correlates with cancer cell response profiles to proapoptotic agents (including inhibitors of BCL-2 anti-apoptotic homologs and drug combinations incorporating BCL-2 protein-targeted agents) rather than anti-proliferative drugs, as would otherwise be expected (Figure S3D and E). Intrigued by these MCL-1-distinguishing results, we queried the RNA sequencing dataset of wild-type vs. Mcl-1−/− thymic epithelial cells against the Library of Integrated Network-Based Cellular Signatures L1000 dataset, which contains >106 gene expression profiles of chemically perturbed human cell lines (L1000CDS2). Remarkably, we found that the gene expression signature of Mcl-1 deletion in murine thymic epithelial cells most resembles that of treatment with MTAs (Figure 2G). Taken together, the comparative analyses of gene expression profiles upon genetic deletion and pharmacologic inhibition of MCL-1 consistently connect MCL-1 perturbation to the cell cycle and DNA replication.
Selective sensitivity to MTAs upon Mcl-1 deletion
Based on the observed link between MCL-1 targeting and an anti-proliferative response, we sought to determine if and how genetic deletion of Mcl-1, in the absence of dependency on MCL-1 for survival, would influence cell susceptibility to distinct classes of chemotherapy. We screened a panel of anti-cancer drugs in wild-type and Mcl-1−/− MEFs and ranked the agents by relative toxicity in the absence vs. presence of MCL-1. Although one might expect the majority of anti-cancer drugs to demonstrate a more potent cytotoxic effect in the absence of an anti-apoptotic protein, this was not the case. Instead, only five of the agents exhibited a greater than 10-fold differential potency in the absence of MCL-1, three of which were the MTAs vinorelbine, vincristine, and paclitaxel (Figure 3A). Importantly, ABT-737, the selective small-molecule inhibitor of BCL-2, BCL-XL, and BCL-w, but not MCL-1, was also in the top group, consistent with MCL-1 serving as a resistance factor for ABT-737-induced apoptosis.32 Bortezomib was also notably more cytotoxic in the absence of MCL-1, consistent with prior reports33–35 and the capacity of proteosomal inhibition to prolong the half-life and thus anti-apoptotic activity of MCL-1, which otherwise undergoes rapid turnover. To confirm the findings, we performed serial-dilution MTA treatments of wild-type and Mcl-1−/− MEFs, and indeed observed markedly enhanced cytotoxicity in the absence of MCL-1 (Figures 3B–3D). To further link the effect to MCL-1 expression, we reconstituted Mcl-1−/− MEFs with MCL-1 and relative drug resistance was restored (Figures 3E–3G). To probe the anti-apoptotic selectivity of the phenomenon, we compared the relative susceptibility of wild-type and Bcl-xL−/− MEFs to MTA treatments and observed no increased sensitivity in the absence of BCL-XL (Figures S4A–S4C).
Figure 3. Hypersensitivity of Mcl-1−/− MEFs to microtubule-targeting agents.
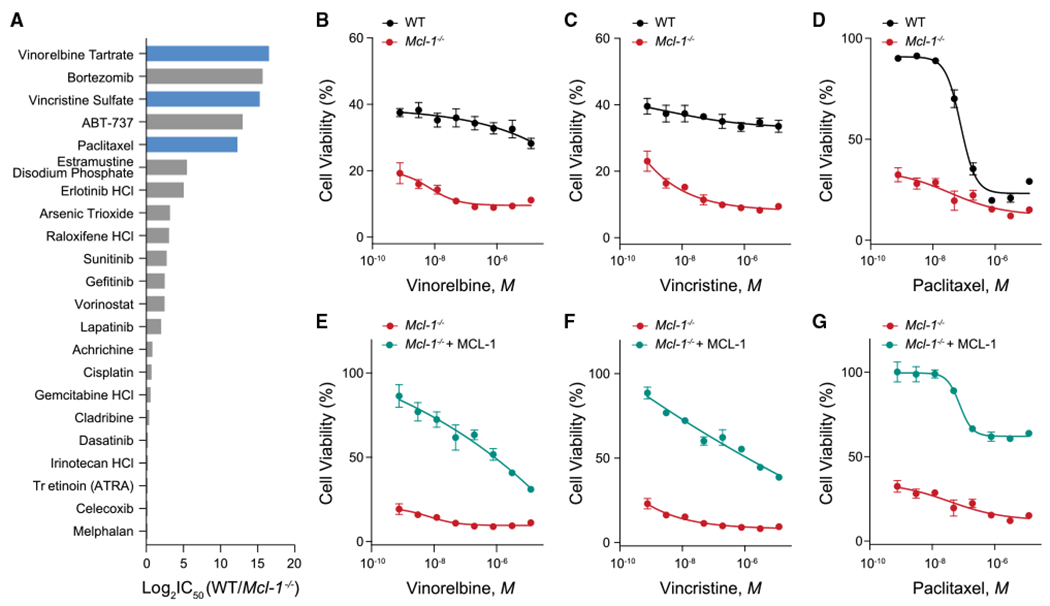
(A) The microtubule-targeting agents vinorelbine, vincristine, and paclitaxel (blue) were among the top hits in a screen of FDA-approved chemotherapeutics that demonstrated selectively increased cytotoxicity upon Mcl-1 deletion, clustering with the positive control compoundsABT-737 and bortezomib. Log2 fold change values were generated from the ratio of the mean of technical replicates for cell viability measurements of WT vs. Mcl-1−/− MEFs in response to the indicated chemotherapeutics.
(B–D) Heightened susceptibility of Mcl-1−/− MEFs to vinorelbine (B), vincristine (C), and paclitaxel (D) compared with wild-type MEFs by cell viability (Cell TiterGlo) assay measured after 48 h of drug treatments. Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells and drugs with similar results.
(E–G) Reconstitution of Mcl-1−/− MEFs with MCL-1 restored the relative resistance to MTAs seen in wild-type MEFs, as measured by cell viability (Cell TiterGlo) assay after 48 h of drug treatments. Data are mean ± SD for experiments performed in technical triplicate and conducted twice using independent preparations of cells and drugs with similar results.
See also Figure S4.
To examine the potential therapeutic relevance of the heightened MTA sensitivity observed in the absence of MCL-1, we tested the combination of the selective MCL-1 inhibitor S63845 and the MTA vincristine. Consistent with the results of the cancer drug screen in MEFs, the combination of S63845 and vincristine caused synergistic cytotoxicity in wild-type MEFs (BAX/BAK present), as determined by Calcusyn analysis (Figures 4A and 4B). Cell-cycle analysis revealed that the underlying synergy corresponded to pronounced mitotic arrest (Figures 4C and S5A–S5D). Importantly, this M phase arrest was also observed in the absence of BAX/BAK, where there was little to no change in cell viability (Figures 4D, S2N, and S5E–S5H), dissociating MCL-1’s role in cell-cycle progression from apoptosis regulation. As a further measure of validation, we examined the effect of an alternative selective MCL-1 inhibitor, AMG176, and a selective BCL-2 inhibitor, ABT-199 (Venetoclax), on the cell cycle using the distinct Bax−/−Bak−/− HCT116 cell line and again observed mitotic arrest upon treatment with the selective MCL-1 inhibitor but not the BCL-2 inhibitor (Figures S6A, S6B, and S5I–S5N). Thus, whereas a role for MCL-1 in mediating susceptibility to anti-mitotic drugs has been previously ascribed to an apoptosis mechanism of action,36 the data presented here suggest that a noncanonical role for MCL-1 in regulating cell-cycle progression could be contributory, particularly given that dual MCL-1 and MTA targeting triggers heightened mitotic arrest in the presence or absence of an intact apoptotic pathway.
Figure 4. Combinatorial targeting of MCL-1 and microtubules causes synergistic cytotoxicity that correlates with mitotic arrest independent of apoptosis induction.
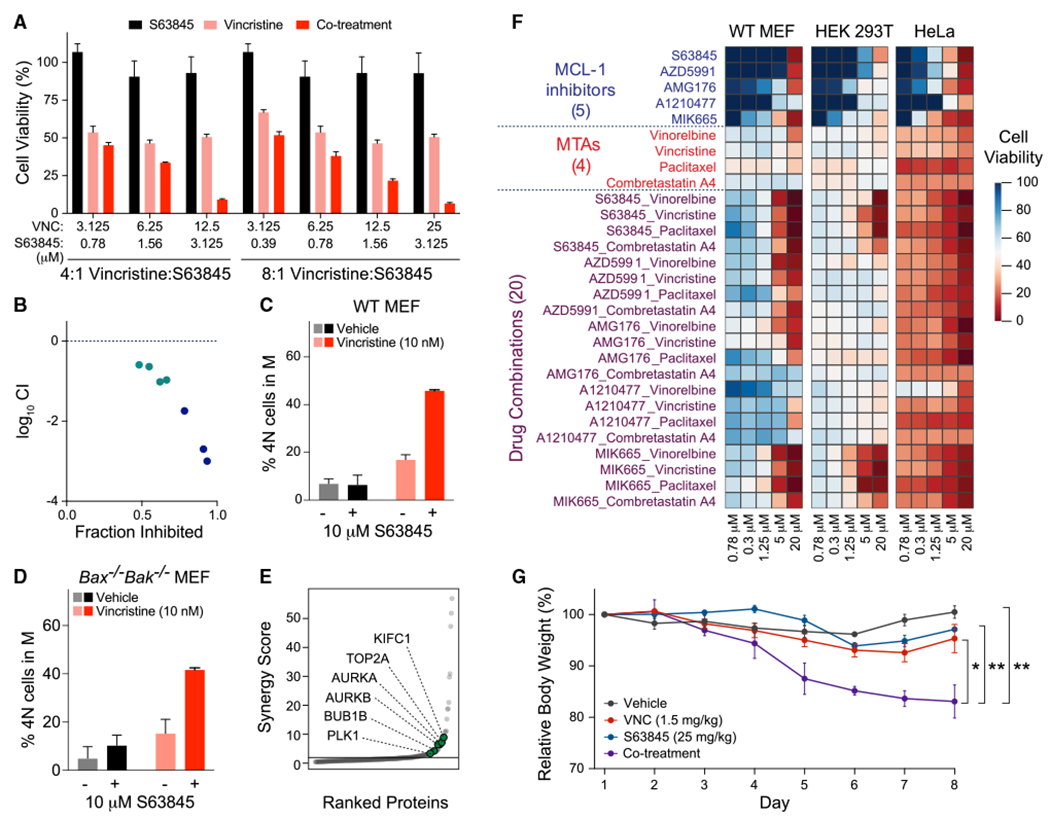
(A and B) Pharmacologic inhibition of MCL-1 by S63845 phenocopies Mcl-1 deletion in sensitizing wild-type MEFs to vincristine treatment (A), with Calcusyn analysis documenting a synergistic response (B). Data are mean ± SD for cell viability experiments (Cell TiterGlo assay) performed after 48 h of drug treatments in technical triplicate and conducted twice using independent preparations of cells and drugs with similar results.
(C and D) S63845 and vincristine combination treatment (16 h) caused mitotic arrest in wild-type MEFs (C), an effect that was independent of apoptosis, as demonstrated by replication of the finding in Bax−/−Bak−/− MEFs (D). Data are mean ± SD for experiments performed in technical duplicate and conducted twice using independent preparations of cells and drugs with similar results.
(E) Synergy scoring of protein abundances in wild-type MEFs treated with the S63845-vincristine combination for 16 h revealed selective upregulation of key proteins involved in the M phase, including KIFC1, TOP2A, AURKA, AURKB, BUB1, and PLK1.
(F) Enhanced cytotoxicity, as measured by Cell TiterGlo assay, upon combining a series of MCL-1 inhibitors and MTAs for 48 h of treatment, with relatively increased susceptibility in HeLa (cancer) cells compared with MEFs and HEK293T (non-cancer) cells. The heatmap was generated based on percent cell viability at the indicated doses, with experiments performed in technical triplicate.
(G) C57BL/6J 8-week-old female mice (n = 4 per arm) were treated intraperitoneally with either vehicle (PBS), S63845 (25 mg/kg) administered daily for 5 consecutive days, vincristine (1.5 mg/kg) administered once every 3 days (twice weekly), or the drug combination. Mice that received the drug combination demonstrated a statistically significant reduction in body weight compared with mice receiving vehicle or single-agent treatments. Error bars are mean ± SEM of body weights measured in four mice per treatment arm.
See also Figures S2N, S5, and S6.
To further characterize the cellular perturbation upon combined targeting of MCL-1 and the microtubule network, we performed comparative global proteomic profiling of wild-type MEFs treated with vehicle, S63845, vincristine, or the drug combination. Of the alterations in protein levels upon combination treatment, synergistic upregulation was observed for a series of mitotic proteins, including ESPL1, KIFC1, PRC1, AURKB, AURKA, BUB1B, and PLK1 (Figure 4E), several of which were also upregulated at the transcriptional level upon Mcl-1 deletion alone in thymic epithelial cells. These data further implicate a distinguishing role for MCL-1 in cell-cycle progression, providing a mechanistic basis beyond reactivating apoptosis for combining MCL-1 and microtubule targeting to arrest cancer growth.
To evaluate this therapeutic rationale further, we markedly expanded our cell viability analyses to test a series of five MCL-1 inhibitors and four MTAs, singly and in all 20 combinations, in MEF, HEK293T, and HeLa cells (BAX/BAK present). In nearly all instances, we observed synergistic cytotoxicity upon drug combination, with HeLa cells showing the most pronounced sensitivity, consistent with a potential therapeutic window for cancer vs. non-cancerous cells (Figure 4F). However, given the evidence of synergistic, albeit lower, cytotoxicity in MEFs and HEK293T cells, we advanced the S63845 and vincristine combination to in vivo toxicity testing in C57BL/6J mice at the standard dosing used for each agent in treating mouse cancer models, specifically 1.5 mg/kg biw for vincristine and 25 mg/kg qd for S63845. Notably, by day 5 of treatment, mice in the combined treatment arm exhibited a statistically significant loss of body weight compared with the vehicle and single-agent treatment arms, with continued progression of weight loss for the duration of the 8-day study (Figure 4G). Thus, clinical studies that test the combination of small-molecule MCL-1 inhibitors with MTAs require careful therapeutic window consideration given the potential physiologic implications of disrupting not only the role of MCL-1 in apoptosis regulation but also in cell-cycle progression.
Genetic deletion and pharmacologic inhibition of MCL-1 causes DNA damage
Given the intimate relationship between surveillance of the cell-cycle and DNA integrity, and the capacity of defects in either to trigger apoptosis, a variety of BCL-2 members have been linked to these physiologic processes as sensors, switches, and effectors–including MCL-1. For example, MCL-1 is induced upon DNA damage, is believed to facilitate DNA repair and genomic stability through interactions with CHK1, IEX-1, and Ku, and promotes homologous recombination over non-homologous end joining to repair double-strand breaks (DSBs) in DNA.16,37 How MCL-1 accomplishes and integrates these tasks at the biochemical and cellular levels remains an unresolved structure-function puzzle. Nevertheless, selective MCL-1 inhibitors are being tested in humans and toxicities are emerging, compelling the need to rigorously examine how such compounds could influence non-canonical roles of MCL-1, especially if they are mediated by protein interactions that involve the canonical BH3-binding pocket. We started our analysis by examining the impact of S63845 treatment at dosing that impairs proliferation on γH2AX levels, a sensitive indicator of DNA damage, and specifically DSBs. Wild-type MEFs demonstrated a dose-responsive increase in γH2AX levels upon micromolar S63845 treatment (Figure 5A). This phenomenon was not observed in Mcl-1−/− MEFs and re-emerged upon reconstituting Mcl-1−/− MEFs with MCL-1 (Figures 5B and 5C), highlighting a specific role for MCL-1 in maintaining DNA integrity that is specifically disrupted upon selective pharmacologic targeting of MCL-1. Acute knockout of Mcl-1 followed by reconstitution with full-length MCL-1 or its distinct isoforms, again showed that MCL-1OMM, rather than MCL-1Matrix, restored the γH2AX response to S63845 treatment (Figures 5D–5F). Although micromolar dosing of S63845 does not impair the cell viability of MEFs (Figures S2J and S2K), such experiments cannot definitively rule out a role for MCL-1 regulation of apoptosis in the underlying mechanism. Therefore, we repeated the experiment in Bax−/−Bakr−/− MEFs and HCT116 cells and observed dose-responsive increases in γH2AX levels upon S63845 treatment, even in the absence of an intact mitochondrial apoptosis pathway (Figures 5G, 5H, S7A, and S7B). The observed increases in γH2AX levels by western analysis upon S63845 treatment of Bax−/−Bak−/− cells correlated with an increase in the number and size of DNA repair foci, as quantified by anti-γH2AX immunofluorescence microscopy (Figures S8A–S8C).
Figure 5. Genetic deletion or pharmacologic targeting of MCL-1 induces DNA damage independent of apoptosis resulting in a growth disadvantage in vivo.
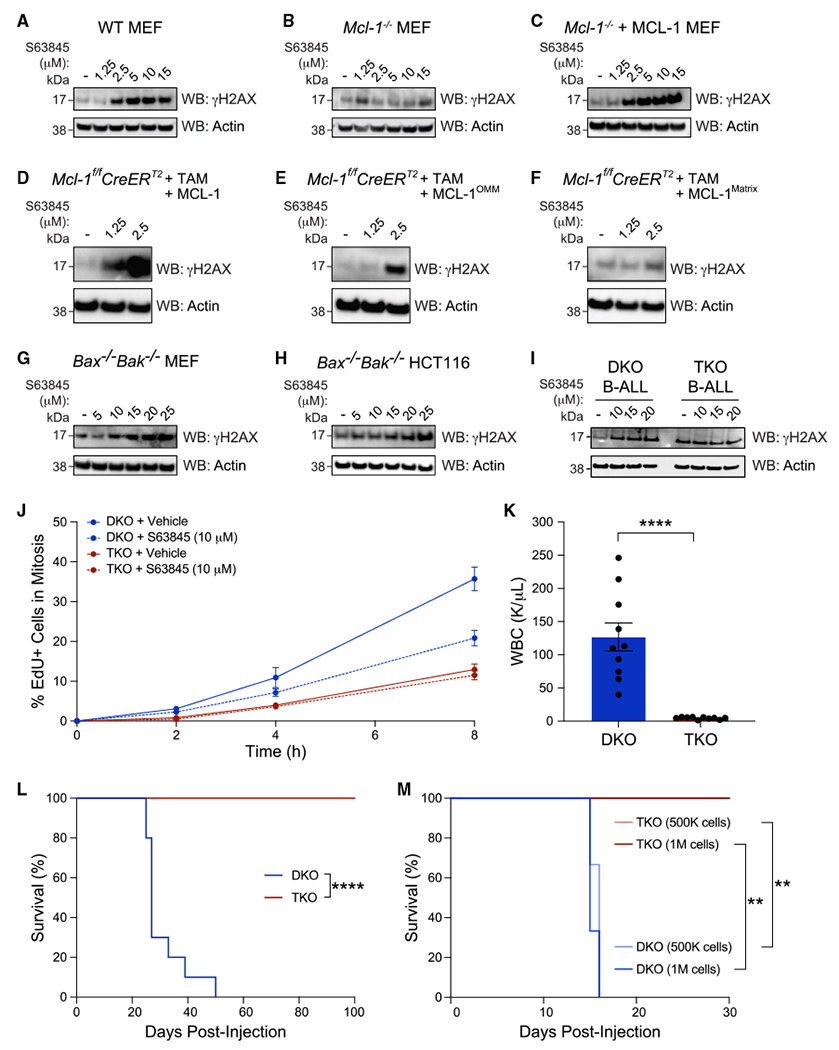
(A–C) Treatment with the selective MCL-1 inhibitor S63845 dose-responsively induced gH2AX levels in wild-type MEFs (A) but had no such effect in the absence of the MCL-1 target (B). Reconstitution of Mcl-1−/− MEFs with wild-type MCL-1 restored dose-responsive γH2AX induction upon S63845 treatment (C). The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(D–F) S63845 induced a dose-responsive increase of γH2AX levels in tamoxifen-treated Mcl-1fl/flRosa-ERCreT2 MEFs reconstituted with either wild-type MCL-1 or its OMM isoform (E), whereas no such effect was observed upon reconstitution with the matrix isoform (F). The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(G–H) S63845 likewise induced a dose-responsive increase in γH2AX levels in the absence of an intact mitochondrial apoptosis pathway, as demonstrated in Bax−/−Bak−/− MEFs (G) and Bax−/−Bak−/− HCT116 cells (H). The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(I) γH2AX levels were induced upon S63845 treatment of Bax−/−Bak−/− B-ALL (DKO) cells. Upon further deletion of Mcl-1 (TKO), basal levels of γH2AX were markedly increased compared with DKO cells and S63845 treatment had no effect, consistent with the absence of the MCL-1 drug target. The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(J) Genetic deletion of Mcl-1 impeded S-to-M progression independent of apoptosis (TKO) (compare solid red and blue lines). Treatment of DKO cells with the selective MCL-1 inhibitor S63845 phenocopied this effect (compare solid blue and dotted blue lines). As a control, S63845 treatment had no effect on the TKO cells that lack the MCL-1 drug target (compare solid red and dotted red lines). Data are mean ± SD for experiments performed in technical duplicate and conducted twice using independent preparations of cells and drugs with similar results.
(K) DKO or TKO B-ALL cells (2.5 × 105) were injected by tail vein into C57BL/6J 8-week-old female mice (n = 10 per arm) and complete blood counts (CBCs) performed on terminal bleeds at the time of euthanasia (period of 25–50 days post injection for DKO and day 105 for TKO), revealing markedly elevated white blood cell counts in DKO compared with TKO mice (n = 10 mice per arm, p < 0.0001).
(L) Kaplan-Meier plots demonstrated that mice injected with DKO B-ALL cells (250,000 cells) died between 25 and 50 days post injection, whereas no evidence of leukemia was detected in mice injected with TKO B-ALL cells even after 15 weeks, as monitored by weekly CBC (n = 10 mice per arm, p < 0.0001).
(M) Injection with higher doses of leukemia cells (5 × 105 and 1 × 106) resulted in an even earlier onset of leukemic death for mice injected with DKO B-ALL cells, whereas no leukemia was evident by 30 days in mice injected with TKO B-ALL cells, as monitored by weekly CBC (n = 3 mice per arm, p < 0.01).
See also Figures S7 and S8.
To further examine the impact of both genetic deletion and pharmacologic inhibition of MCL-1 on γH2AX levels in the absence of BAX and BAK, we generated MCL-1-dependent murine p185+Arf−/− B-ALL cells38–40 lacking either BAX and BAK (p185+Arf−/−Bax−/−Bak−/−) or BAX, BAK, and MCL-1 (p185+Arf−/−Bax−/−), hereafter referred to as DKO and TKO cells, respectively (Figure S7C). In this rigorously controlled cell system, not only do we find a higher basal level of γH2AX in the absence of MCL-1, but also observe an increase in γH2AX levels in response to treatment of DKO cells with S63845, with no such effect in TKO cells that lack the S63845 target (Figures 5I and S7D). Comparative cell-cycle analysis further revealed that TKO cells exhibit a markedly slower progression from S to M phase, a phenotype that was mimicked upon treatment of DKO cells with S63845; importantly, S63845 treatment had no effect on the cell-cycle progression of TKO cells, which lack the S63845 target (Figure 5J). We hypothesized that a baseline elevation in DNA damage coupled with slower progression through the cell cycle would confer a relative growth disadvantage of TKO vs. DKO cells in vivo—an experiment that would further probe a noncanonical role for MCL-1 in cell proliferation independent from apoptosis regulation. Whereas tail vein injection of 250,000 DKO cells into C57BL/6J mice resulted in profound leukocytosis and 100% leukemic fatality by day 50, none of the TKO-injected mice developed leukemia (Figure 5K and 5L). Even escalating the number of injected cells to 500,000 and 1,000,000 produced no leukemic TKO mice, whereas all DKO mice succumbed to leukemia by day 15 of the experiment (Figure 5M). Reconstitution of the TKO B-ALL cells with MCL-1 restored their capacity to robustly engraft, resulting in fatal leukemia (Figure S7E). Taken together, these data demonstrate that genetic deletion or pharmacologic inhibition of MCL-1 causes DNA damage, which could explain the decrease in cell proliferation that occurs even in the absence of mitochondrial apoptosis.
MCL-1 targeting increases the DNA damage induced by chemotherapy
The current indication for MCL-1 inhibitor treatment is the presence of anti-apoptotic dependency on MCL-1, such that blocking the capacity to sequester pro-apoptotic proteins in its BH3 groove can restore BAX/BAK-mediated apoptosis. However, our results suggest that MCL-1 inhibitors are capable of disrupting an alternative function of MCL-1, one that is also critical and constitutive, and occurs in the absence of selective anti-apoptotic dependency on MCL-1. Indeed, targeted inhibition of MCL-1’s role in maintaining DNA integrity and cell-cycle progression could expand the pharmacologic utility of MCL-1 as a single agent and in combination. For example, we found that TKO cells sustained more DNA damage upon treatment with hydroxyurea or camptothecin compared with DKO cells, highlighting the mechanistic potential to exacerbate chemotherapy-induced DNA damage as a consequence of MCL-1 targeting separable from apoptosis induction (Figures 6A and 6B). Reconstitution of TKO cells with MCL-1 restored the relative protection of DKO vs. TKO cells from camptothecin-induced DNA damage (Figure S7F). Comparative phosphorylation profiling of DKO vs. TKO cells treated with hydroxyurea revealed, for example, increased relative abundance in TKO of CHK1 pS317 and pS345, two key phosphoresidues within the CHK1 C-terminal domain known to be upregulated in response to DNA damage or replication stress,41 and of p53 pS15 and pS46, sites that are also known to be phosphorylated in response to DNA damage42,43 (Figure 6C). These data demonstrate a heightened vulnerability of the leukemic cells to DNA damage in the absence of MCL-1 and as a consequence independent from its anti-apoptotic role. Likewise, combined treatment of cancer cells lacking anti-apoptotic dependency on MCL-1 (BAX/BAK present) with an MCL-1 inhibitor and vincristine or camptothecin, resulted in enhanced γH2AX levels compared with single-agent treatment (Figures 6D–6G) and produced enhanced cytotoxicity across MCL-1 inhibitor and vincristine or camptothecin combinations (Figures 6H–6K). These data indicate that, in the context of relapsed and refractory cancers that exhibit formidable and MCL-1-independent apoptotic blockades, pharmacologic MCL-1 targeting, with or without an anti-mitotic or DNA-damaging agent, has the potential to achieve therapeutic benefit by a mechanism independent from MCL-1’s role in apoptosis regulation.
Figure 6. Chemotherapy-induced DNA damage is compounded by genetic deletion or pharmacologic targeting of MCL-1.
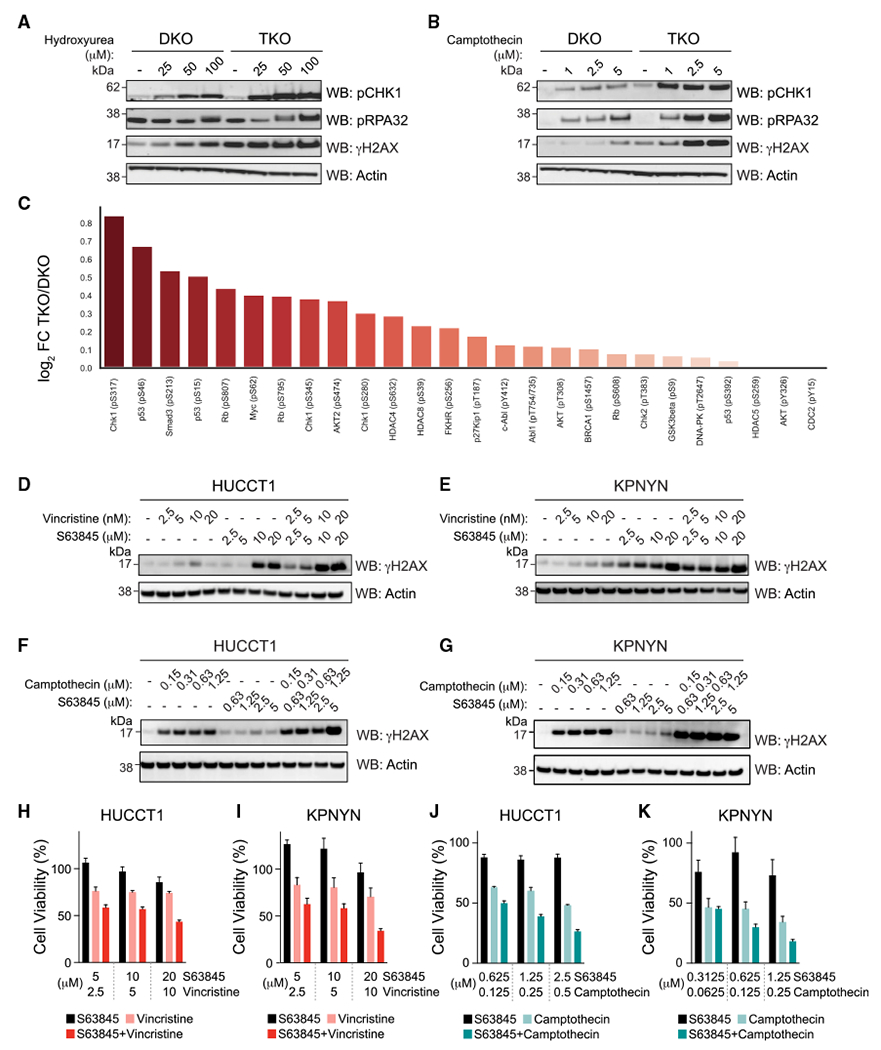
(A and B) p185+Arf−/−Bax−/−Bak−/−Mcl-1−/− B-ALL cells (TKO) were more susceptible than p185+Arf−/−Bax−/−Bak−/− B-ALL cells (DKO) to DNA damage upon treatment with hydroxyurea (A) and camptothecin (B) for 24 h, as demonstrated by relatively increased levels of pRPA32, pCHK1, and γH2AX levels. The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(C) Comparative phosphorylation profiling of DKO vs. TKO cells treated with 50 μM hydroxyurea for 8 h. TKO samples were notably enriched for phosphopeptides that correspond to phosphorylation sites on key proteins that respond to DNA damage and replicative stress, such as CHK1 and p53. The experiment was performed in technical sextuplicate.
(D and E) Pharmacologic inhibition of MCL-1 in combination with vincristine for 16 h increased γH2AX levels compared with single-agent treatments in HUCCT1 (D) and KPNYN (E) cancer cells, which express MCL-1 but are not exclusively dependent on MCL-1 for survival. The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(F and G) Pharmacologic inhibition of MCL-1 in combination with camptothecin for 16 h likewise increased gH2AX levels compared with single-agent treatments in HUCCT1 (F) and KPNYN (G) cancer cells. The experiment was conducted twice using independent preparations of cells and drugs with similar results.
(H and I) Combination treatment with S63845 and vincristine for 48h resulted in enhanced cytotoxicity compared with single-agent treatments of HUCCT1 (H) and KPNYN (I) cell lines. Data are mean ± SD for cell viability (Cell TiterGlo assay) experiments performed in technical triplicate and conducted twice using independent preparations of cells and drugs with similar results.
(J and K) Combination treatment with S63845 and camptothecin for 48 h resulted in enhanced cytotoxicity compared with single-agent treatments of HUCCT1 (J) and KPNYN (K) cell lines. Data are mean ± SD for cell viability (Cell TiterGlo assay) experiments performed in technical triplicate and conducted twice using independent preparations of cells and drugs with similar results.
See also Figure S7.
Interaction with the MCM complex provides a mechanistic link between MCL-1 and regulation of the cell cycle and DNA homeostasis
Deconvoluting the protein targets and mechanisms that underlie MCL-1 regulation of the cell cycle and DNA homeostasis is a formidable task that requires long-term study. To determine candidate protein interaction networks engaged by MCL-1 that could exert such regulatory effects, we undertook whole-cell, stage-specific proteomic profiling by affinity enrichment-mass spectrometry (AE-MS) (Figure S9A). Specifically, we expressed FLAG-MCL-1 (full length) in 293T cells (Figures S9B and S9C) followed by double-thymidine block synchronization, and then harvested the cells under discrete conditions at specific time points (see STAR Methods) to isolate G1, S, G2, and M populations. In accordance with established proteomic methods,44–46 stage-specific cell lysates were subjected to anti-FLAG immunoprecipitation and MS, and the data processed as follows: (1) the normalized spectral abundance factor was determined for each protein prey detected, (2) mean scale abundance of prey protein was calculated for each cell-cycle phase, (3) k-means clustering (k = 4) was used to identify proteins that bound to MCL-1 at each stage of the cell cycle, and (4) the protein complexes enriched within these clusters were identified and rank ordered (Figures S10 and S11). Importantly, our methodology accurately identified a series of well-established MCL-1 interactors, including BAX and BAK (apoptosis regulation) and PCNA and CDK1 (cell-cycle regulation) (Figures 7A and S11A). A series of notable complexes were also top-ranked hits, including (1) CDK2, emerin, and spliceosome complexes in G1, (2) PRC2, DCS, NURF, and MCM complexes in S, (3) PCNA, emerin, and MCM complexes in G2, and (4) spliceosome, toposome, TREX, and CDC5L complexes in M (Figures 7B and S11B). These data are consistent with roles for MCL-1 not only in apoptosis regulation but also in critical cellular processes involving cell proliferation, DNA replication, and RNA processing.
Figure 7. The MCL-1 interactome across stages of the cell cycle reveals candidate complexes that regulate DNA replication and cell division.
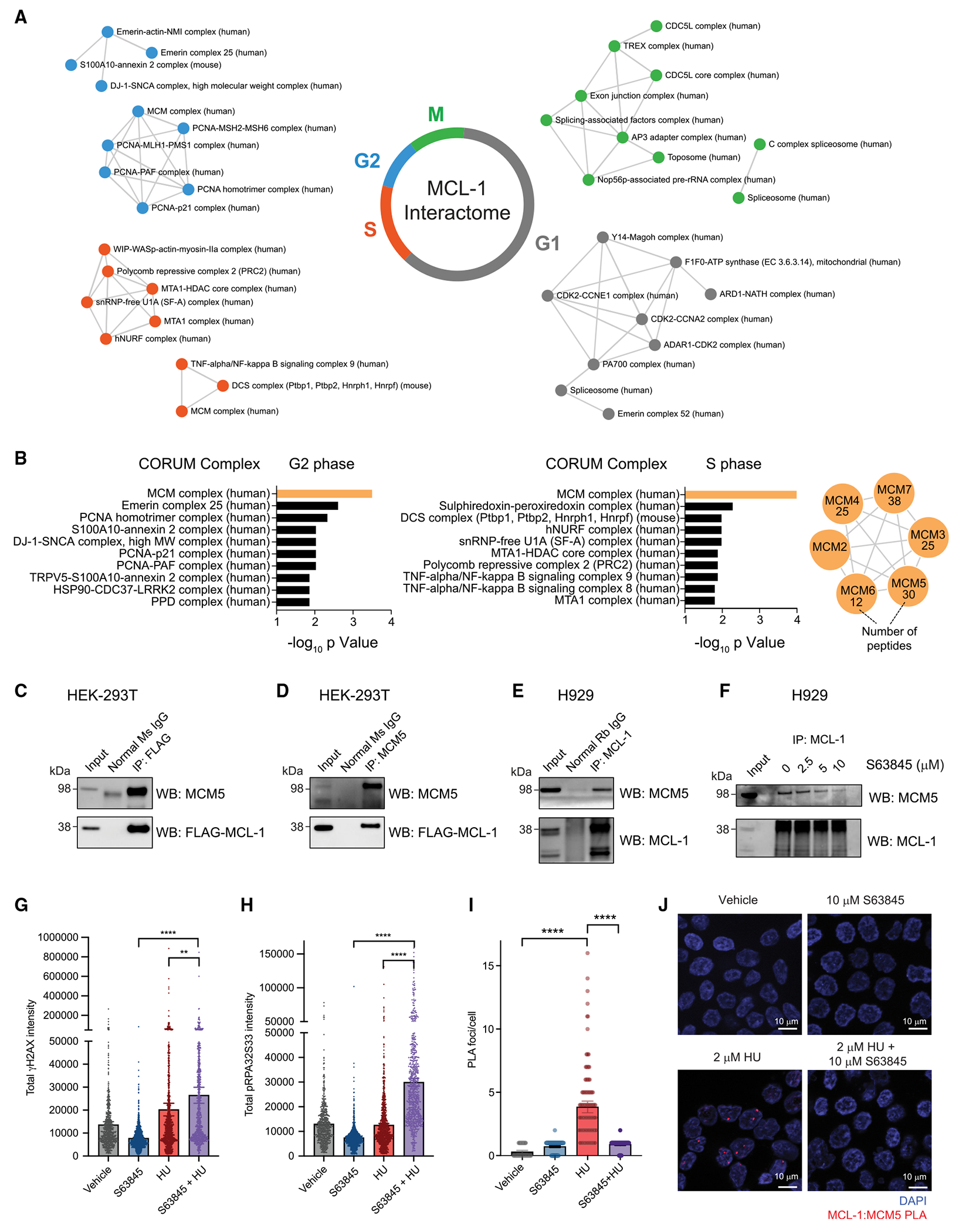
(A) Analysis of AE-MS data identified established MCL-1 interactors such as BAX, BAK, PCNA, and CDK1 (Figure S9A), as well as a series of unanticipated MCL-1 interactors, including nuclear complexes, across distinct stages of the cell cycle. Four biological replicates of the AE-MS experiments were performed.
(B) Cross-referencing AE-MS interaction data for each cell-cycle stage with the CORUM complex database revealed the minichromosome maintenance (MCM) complex as one of the most enriched complexes associated with MCL-1 in the S and G2 phases. All six members of the MCM complex (MCM27) co-immunoprecipitated with FLAG-MCL-1 from HEK293T cellular lysates.
(C and D) Reciprocal co-immunoprecipitation of FLAG-MCL-1 and MCM5 from HEK293T lysates using either FLAG (D) or MCM5 (E) antibodies.
(E) Co-immunoprecipitation of native MCL-1 and MCM5 from lysates of MCL-dependent H929 cells using an MCL-1 antibody.
(F) S36845 treatment dose-responsively disrupted co-immunoprecipitation of native MCL-1 and MCM5 from lysates of H929 cells using an MCL-1 antibody.
(G and H) S63845 and hydroxyurea co-treatment exacerbated replicative stress independent of apoptosis, as demonstrated by increased induction of γH2AX (E) and S33 phoshorylation of RPA32 (H) relative to single-agent treatments in Bax−/−Bak−/− HCT116 cells. Data are mean ± 95% CI for gH2AXand pRPA32S33 intensity values from >500 individual cells per treatment condition. The experiment was conducted twice using independent preparations of cells and drugs, with similar results.
(I) MCL1 interacts with MCM5 upon hydroxyurea treatment of Bax−/−Bak−/− HCT116 cells, as demonstrated by increased number of MCL-1:MCM5 proximity ligation assay (PLA) foci. The interaction was blocked upon pharmacologic inhibition of MCL-1 by S63845, resulting in ~80% reduction in the average number of PLA foci per cell. Data are mean ± 95% CI for γH2AX and pRPA32S33 intensity values from >50 individual cells per treatment condition.
(J) Representative images of MCL-1:MCM5 PLA foci (red) in Bax−/−Bak−/− HCT116 cells subjected to vehicle (0.1% DMSO), 10 μM S63845, 2 μM hydroxyurea, or co-treatment with 10 μM S63845 and 2 μM hydroxyurea.
See also Figures S9–S13.
Particularly intriguing for the current work is the identification of MCM as the highest-ranking protein complex hit in both S and G2 phases (Figures 7B and S11B). We validated this finding by proteomic analysis of endogenous MCL-1 protein interactors in wild-type MEFs, again identifying members of the MCM complex as prominent hits (Figures S9D and S12). The MCM complex plays a critical role in genome duplication in proliferating cells. During late M to early G1, MCM complexes prime the chromatin for DNA replication, and during S phase unwind double-stranded DNA at replication origins, recruiting DNA polymerases to synthesize DNA. MCM complexes then migrate from the replication origins as a component of the DNA replication fork, functioning as DNA helicases. Given their essential role in DNA replication and cell proliferation, it is not surprising that MCM proteins are highly expressed in human cancers, including upon malignant transformation of cells, and predicts poor outcomes.47,48 To validate the interaction between MCL-1 and MCM complex proteins, we performed a series of co-immunoprecipitations, first confirming that MCM5, for example, co-precipitated with MCL-1 when using either anti-FLAG or anti-MCM5 antibodies (Figures 7C and 7D). MCM5 also co-immunoprecipitated with native MCL-1 from lysates of MCL-1-dependent and -independent hematologic cancer cells, H929 and K562, respectively (Figures 7E and S9E). Treatment of H929 cells with the selective MCL-1 inhibitor, S63845, dose-responsively disrupted MCL-1/MCM5 co-immunoprecipitation (Figure 7F) and at dosing levels that induced DNA damage across a variety of cell lines, as assessed by γH2AX analysis (Figures 5A–5I). We further found that treating Bax−/−Bak−/− HCT116 cells with S63845 in combination with hydroxyurea exacerbated replicative stress compared with single-agent treatment, as demonstrated by increased levels of γH2AX as well as the replicative stress marker and ATR substrate, pRPA2S33 (Figures 7G and 7H). To directly link the heightened replicative stress induced by S63845 co-treatment in a non-apoptotic context to MCL-1 regulation of MCM, we performed a proximity ligation assay, which monitors endogenous protein interactions in situ. Strikingly, treatment of Bax−/−Bak−/− HCT116 cells with hydroxyurea induced the MCL-1/MCM interaction, which was specifically blocked by treatment with S63845 (Figures 7I, 7J, and S13A–S13D). These data support a mechanism by which MCL-1 engages the MCM complex in response to stress stimuli to maintain DNA integrity and cell-cycle progression, which is otherwise disrupted by selective pharmacologic targeting of the MCL-1 canonical groove. Taken together, our AE-MS data revealed both established and unanticipated MCL-1 interactors, including a series of nuclear protein complexes across each stage of the cell cycle. The MCM complex emerged as one of the most enriched MCL-1-interacting protein complexes of S and G2, and plays an essential role, potentially in concert with MCL-1, in maintaining the faithful DNA replication of proliferating cells during homeostasis and cancer.
DISCUSSION
MCL-1 is a prominent anti-apoptotic member of the BCL-2 family with far-reaching oncogenic activity, which has compelled the urgent need to develop and clinically test MCL-1 inhibitors for cancer therapy. Although discovered nearly three decades ago, MCL-1 is a complex and elusive protein, with major facets of its structure and function yet to be determined. MCL-1 has multiple isoforms, cellular localizations, and protein interactions that span a swath of cellular processes, many of which have not been studied in the absence of an intact mitochondrial pathway, further complicating whether potential noncanonical roles are distinct or intertwined with MCL-1’s established anti-apoptotic function. The motivation for this study was to determine whether MCL-1’s unique oncogenic supremacy could be explained, at least in part, by a functionality distinct from apoptotic suppression. We determined that genetic deletion or selective pharmacologic inhibition of MCL-1 impairs cell proliferation as a separable consequence, occurring whether BAX/BAK-mediated apoptosis is intact or not. The phenomenon is rescued by the outer mitochondrial membrane isoform of MCL-1, rather than its mitochondrial matrix-localized variant. Interestingly, conditional deletion of Mcl-1 in thymic epithelial cells provokes transcriptional upregulation of the machinery for ribosomal biogenesis, cell-cycle progression, and DNA replication in the face of organ atrophy, which is otherwise not salvaged by co-expressed anti-apoptotic homologs. What’s more, the transcriptional response of Mcl-1-deleted thymic epithelial cells phenocopies treatment with MTAs, which can induce DNA damage and cell-cycle arrest. Among cancer chemotherapies that are rendered more toxic by Mcl-1 deletion, MTAs emerged as the most prominent drug class. A common mechanistic theme underlying the observed defects in cell proliferation and cell-cycle progression is our finding that genetic deletion or pharmacologic inhibition of MCL-1 itself increases γH2AX levels, reflective of DNA damage. This phenomenon is rescued in an MCL-1OMM isoform-specific fashion and occurs in the presence or absence of an MCL-1-dependent (i.e., MCL-1 expressed but cell survival not selectively dependent on MCL-1) or BAX/BAK-dependent apoptotic context.
Dissecting nonapoptotic roles for MCL-1 requires careful genetic studies that examine the implications of Mcl-1 deletion in the absence of apoptosis, making the B-ALL cell system employed here especially relevant and informative. Indeed, experiments in this Bax−/−Bak−/− context were essential to demonstrating that (1) genetic deletion of Mcl-1 impairs leukemic engraftment and growth in vivo in the absence of apoptosis, (2) selective pharmacologic inhibition of the MCL-1 BH3-binding pocket slows cell-cycle progression in a target-dependent fashion, and that (3) baseline γH2AX levels are elevated in the absence of MCL-1 and the impact of MTAs and DNA-damaging agents is compounded by Mcl-1 deletion or pharmacologic targeting. The data derived from this Bax−/−Bak−/− context collectively indicate that the role of MCL-1 in regulating cell-cycle progression and DNA homeostasis is distinctly independent from the established function of MCL-1 in suppressing BAX/ BAK-mediated apoptosis.
We took an unbiased approach to begin the hypothesisgenerating process for determining explicit binding partners and mechanisms that underlie MCL-1’s noncanonical roles in cell-cycle progression and DNA homeostasis. As expected, the candidates are numerous, but a clear theme emerged: MCL-1 exerts a remarkably broad effect on tissue homeostasis and cancer pathogenesis through its involvement in seminal signaling networks across each stage of the cell cycle. An exemplary high-ranking target, the MCM complex, which plays crucial roles in DNA replication and genomic integrity, foretell lynch pin functions for MCL-1 beyond apoptosis and particularly in regulating DNA fidelity and cell proliferation. Indeed, we observed that treatment with DNA-damaging agents induced MCL-1 engagement of MCM, and selective pharmacologic inhibition of MCL-1 blocked the interaction, coincident with heightened replicative stress and DNA damage.
How the interactions of MCL-1 with multiple protein complexes are integrated to accomplish coordinated roles in the life and death of the cell will keep scientists busy for years if not decades to come. Nevertheless, the clinical implications of this work are compelling and immediately relevant: (1) by slowing cell-cycle progression, pharmacologic inhibition of MCL-1 may be of clinical benefit beyond the current standard indication for reactivating apoptosis in MCL-1-dependent cancers, (2) there is a distinct mechanistic basis for combining selective MCL-1 inhibitors with MTAs and DNA-damaging agents to achieve enhanced anti-cancer activity, (3) such drug combinations require rigorous toxicologic study given the potential to inflict DNA damage on normal tissues, and (4) as additional MCL-1 phenotypes and binding partners are identified and validated, the implications of disrupting homeostatic processes must be considered as potential liabilities for selective pharmacologic targeting of MCL-1. To the latter point, conditional deletion of Mcl-1 in cardiomyocytes was found to induce a rapid and fatal cardiomyopathy in mice,15 suggesting that pharmacologic targeting of MCL-1 in cancer could carry a cardiac risk, which has now been borne out in several clinical trials. Here, we find that among the MCL-1-binding complexes is the emerin family of proteins, which play a critical role in sarcomere physiology and, upon mutagenesis, can cause cardiomyopathy. Whether or not this circumstantial link between MCL-1, emerins, cardiomyopathy, and inhibitor-induced cardiotoxicity ultimately reveals a definitive physiologic and toxicologic mechanism, we must keep an open mind to bona fide noncanonical roles for MCL-1 outside of apoptosis regulation, given the incredibly high stakes, including the attendant benefits and risks, of treating cancer patients with MCL-1 inhibitors.
Limitations of the study
MCL-1 biology and the implications of its pharmacologic targeting are complex. Indeed, the continued emergence of non-canonical roles for MCL-1 in homeostatic processes could influence the therapeutic applications of MCL-1 inhibitors for the better (e.g., expanding treatment indications) or for the worse (e.g., increasing the risk of toxicities). Selective inhibition of MCL-1 by small-molecule inhibitors such as S63845 can reactivate apoptosis of MCL-1-dependent cancers at the low nanomolar range, whereas the perturbations in DNA integrity and cell-cycle progression identified here occur upon micromolar treatment. At face value, this difference in dosing raises questions regarding physiologic relevance and the potential contribution of such non-canonical perturbations to the toxicity observed in patients. However, in addition to having validated the observed phenomena using a series of genetically and pharmacologically controlled experimental systems, it is important to underscore that displacement of pro-apoptotic proteins from MCL-1 by nanomolar S63845 effectively triggers a catalytic and thus amplified cascade in the context of apoptosis induction,24 whereas the independent anti-proliferative activity identified here is coincident with stripping MCL-1 from a protein complex in stoichiometric fashion, which by definition necessitates higher dosing. A similar distinction is observed, for example, in the context of catalytic vs. stoichiometric FK506 activity in targeting calcineurin to achieve therapeutic immunosuppression49 and the IP3 receptor to modulate intracellular calcium levels,50 respectively. From a clinical standpoint, we do not yet know the full spectrum of sensitivities and cumulative effects of MCL-1 inhibitor treatment across the diversity of human tissues, or whether they derive from targeting canonical and/or noncanonical mechanisms. We hope that this study will inspire such analyses to be conducted in the context of preclinical and clinical investigations to further our understanding about how to best apply MCL-1 inhibitors for therapeutic benefit.
Our proteomic analyses across each stage of the cell cycle provided a starting point for dissecting the mechanistic basis for MCL-1’s role in regulating DNA integrity, cell-cycle progression, and proliferation, as identified and characterized here in the presence and absence of a BAX/BAK-competent apoptotic pathway. Whereas the MCM complex emerged as a validated target, how MCL-1 binds to MCM and modulates its critical functionality remains to be determined. Indeed, the spectrum of MCL-1 interacting complexes revealed by these studies provides a robust resource for future work. Ideally, the fruits of such inquiries will not only expand our knowledge of MCL-1 biology but also lead to optimized strategies for targeting MCL-1 to combat human disease in the most safe and effective manner.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled or facilitated by the lead contact Loren Walensky (loren_walensky@dfci.harvard.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request to the lead contact. Proprietary materials, such as cell lines generated by the Joseph T. Opferman laboratory of St. Jude Children’s Research Hospital, may require a material transfer agreement.
Data and code availability
The data supporting the findings of this study are available within the article and its supplementary materials. Proteomic datasets were submitted to the ProteomeXchange Consortium via PRIDE51 with identifier PXD036327.
This study did not generate code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
Murine Bax−/−Bak−/− (DKO), Bax−/−Bak−/−Mcl-1−/− (TKO), and Bax−/−Bak−/−Mcl-1−/− hMcl-1−/− (TKO + MCL-1) p185+ Arf−/− B-ALL cell lines38,39; Bax−/−Bak−/− MV4;11 cells; and Baxfl/flBak−/− (DKO after 4-OHT treatment) and Mcl-1fl/flBaxfl/flBak−/− (TKO after 4-OHT treatment) MEFs were provided by Joseph Opferman (St. Jude Children’s Research Hospital). DKO, TKO, and TKO + MCL-1 p185+ Arf−/− B-ALL cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Thermo Fisher Scientific, catalog #21870-076) supplemented with 10% (v/v) fetal bovine serum, 100 mg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco), 2 mM glutamine, 10 mM HEPES, 100 μM Minimum Essential Medium (MEM) non-essential amino acids, and 55 μM 2-mercaptoethanol. Human hematologic cancer cell lines (H929, K562, Bax−/−Bak−/− MV4;11 cells) were cultured in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum, 100 μg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco), and 2 mM glutamine. HEK-293T, HeLa, KPNYN and HUCCT1 cells were cultured in DMEM (Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 μg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco). SV40-transformed NIH 3T3 mouse embryonic fibroblast cells (MEFs), including wild-type, Mcl-1−/−,Bax−/−Bak−/−, Mcl-1fl/flRosa-ERCreT2 expressing either wild-type MCL-1 or the mitochondrial outer membrane (MCL-1OMM) or matrix (MCL-1Matrix) specific isoforms, and Baxfl/flBak−/− (DKO after 4-OHT treatment) and Mcl-1fl/flBaxfl/flBak−/− (TKO after 4-OHT treatment) MEFs were cultured in DMEM (Gibco) supplemented with 10% (v/v) FBS, 100 mg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco). All cell lines were maintained at 37°C with 5% CO2. Cells were verified as mycoplasma negative using the MycoAlert mycoplasma detection kit (Lonza Biologics, catalog #LT07-218) prior to experimental studies.
Animal models
For all animal experiments, C57BL/6J wildtype 8-week-old female mice (Jackson Laboratory) were used and housed in groups of 4–5 in a pathogen-free animal facility at 22°C with a 12-h light/dark cycle and fed normal chow ad libitum. All mouse experiments, including euthanasia, were performed in accordance with Dana-Farber Cancer Institute Animal Care and Use Committee approved protocol #06-004.
METHOD DETAILS
Cell proliferation assay
The indicated cell lines were seeded in 6-well plates at 40,000 cells/well. Cellular proliferation was determined by measuring total cell counts over the course of 4 days by Trypan-Blue exclusion using a TC10 Automated Cell Counter (Biorad).
BrdU incorporation assay
BrdU incorporation in WT and Mcl-1−/− MEFs was measured by using the BrdU Cell Proliferation Assay Kit(Cell Signaling Technology Cat# 6813) according to the manufacturer’s protocol. Briefly, cells were grown in the presence of 10 μM BrdU for 8 h, followed by fixation and denaturation. Fixed cells were labeled with the provided detection antibody and then incubated with an anti-mouse IgG HRP-linked secondary antibody, followed by addition of the TMB substrate for 10 min. The reaction was stopped using the STOP solution and absorbance immediately read at 450 nm using a Spectramax M5 microplate reader.
Differential gene expression analysis
Expression data from GEO dataset GSE102227 was quantified as gene-level counts using the ARCHS pipeline.52 To determine the pattern of gene expression upon genetic deletion of Mcl-1, differential gene expression analysis was performed and the data represented as volcano plots by quantifying log2 fold changes and statistical significance. The most differentially expressed genes upon genetic deletion of Mcl-1 were then subjected to pathway enrichment analysis (KEGG).53
ML311 drug sensitivity analysis
Drug sensitivity data for MCL-1 inhibitor ML311 was obtained from the Cancer Therapeutics Research Portal v2 (Broad Institute). Pearson correlations of ML311 sensitivity data with an informer dataset of 481 small molecules across 860 cancer cell lines were computed and visualized on the DepMap portal. The top 50 drugs exhibiting highest correlation with ML311 were manually queried for molecular targets and mechanisms of action.
Cancer cell line S63845 susceptibility screening by PRISM assay
S63845 was screened at an 8-point dosing regimen (3-fold dilution) against 930 genomically characterized cancer cell lines by Profiling Relative Inhibition Simultaneously in Mixtures (PRISM) assay.54 Cells were treated for 5 days in triplicate, with each treatment plate containing positive (bortezomib) and negative (0.1% DMSO) controls. For correlation of sensitivity, univariate associations between the PRISM sensitivity profile of S63845 and other drugs in the Drug Repurposing Hub (Broad Institute) were determined by computing Pearson correlations and associated p values. Compounds displaying the most positive correlation were tabulated as a rank ordered list.
LINCS1000 query of differentially expressed genes
Expression data from GEO dataset GSE102227 were quantified as gene-level counts using the ARCHS pipeline.52 The most differentially expressed genes upon genetic deletion of Mcl-1 were queried against drug-treated gene expression profiles using the L1000CDS2 database,55 a web-based tool for analyzing gene expression signatures against signatures created from human cell lines treated with over 20,000 small molecules and drugs (LINCS project, Broad Institute).
Chemotherapeutic drug screen in MEFs
Wild-type and Mcl-1−/− MEFs were seeded in 96-well plates (5 × 103 cells per well) the day prior to drug treatment. Cells were treated with a panel of FDA-approved anti-cancer drugs (AODX, National Cancer Institute, Division of Cancer Treatment and Diagnosis) for 72 h and viability measured by CellTiter-Glo assay (Promega), with luminescence detected by a Spectramax M5 microplate reader. To determine the relative sensitivity of cells upon Mcl-1 deletion, viability data were plotted as a function of drug concentration using Prism software (GraphPad), and the respective IC50 values computed for each drug against wild-type and Mcl-1−/− MEFs.
Dynamic assessment of endogenous protein levels
To monitor protein levels in cells subjected to drug treatments, cells were cultured in the corresponding culture medium containing vehicle or drug for 24 h. The cells were then harvested and lysed in 1% CHAPS lysis buffer (1% CHAPS, 150 mM NaCl, 50 mM Tris pH 7.4, phenylmethylsulfonyl fluoride [PMSF] and PhosSTOP [Sigma-Aldrich]). Cell lysates were subjected to electrophoresis and western blotting for phospho-H2AX S139 (Millipore Cat# 05–636, RRID:AB_309864), phospho-CHK1 S345 (Abcam Cat# ab58567, RRID:AB_10563825), phospho-RPA32 S33 (Bethyl Cat# A300-246A, RRID:AB_2180847), MCL-1 (Rockland Cat# 600-401-394S, RRID:AB_11179937) and actin (Cell Signaling Technology Cat# 5125, RRID:AB_1903890).
Immunofluorescence microscopy
For imaging of DNA repair nuclear foci, Bax−/−Bak−/− MEFs were grown in 96-well high content imaging plates (Corning #CLS4580) and exposed to the indicated doses of S63845 or vehicle (0.1% DMSO) for 24 h. Cells were then washed 2× with PBS and fixed in freshly prepared 4% paraformaldehyde in PBS for 20 min at 4°C. Fixed cells were washed 2× in cold PBS, permeabilized using 0.1% Triton X-100, blocked with 1% BSA in PBS, and labeled with primary antibody to phospho-H2AX (S139) (Millipore Cat#05–636, RRID:AB_309864) for 8 h at 4°C (1:500 dilution in 1% BSA in PBS). Cells were then washed with PBS and incubated with anti-rabbit Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific Cat# A-11012, RRID:AB_2534079) for 60 min (1:200 dilution in 1% BSA in PBS) at room temperature in the dark. Antibody-labeled cells were washed and stained in 0.1 μg/mL DAPI (Cell Signaling Technology, Cat# 4083) in PBS at room temperature in the dark for 10 min. All images were collected with a Leica Thunder Imager widefield microscope equipped with HC PL FLUOTAR 10x/0.32 NA objective and auto-focus to maintain samples at the focal plane over time. Immunofluorescence images were captured using LAS X Software (Leica). Subsequent analysis of fixed-cell images to quantify γH2AX foci size and intensity was performed using custom ImageJ/FIJI macros and the data plotted in Prism Software 9.0 (GraphPad).
For confocal microscopy of MCL-1 localization in cells, HEK 293T cells, either untransfected or transfected with FLAG-MCL-1, were grown overnight on No. 1.5 coverslips in 6 well 10 mm glass diameter uncoated plates (Mattek Cat #P06G-1.5-10-F). Cells were labeled with 100 nM MitoTrackerTM Deep Red FM (Thermo Fisher Scientific Cat# M22426) at 37°C for 5 min, washed 2x with PBS, and then fixed in freshly prepared 4% paraformaldehyde in PBS for 20 min at 4°C. Fixed cells were washed 2x in cold PBS, permeabilized using 0.1% Triton X-100 blocked with 1% BSA in PBS, and labeled with either primary antibody to MCL-1 (Proteintech Cat#66026-1-Ig, RRID:AB_11041711) for untransfected cells or to FLAG (Sigma-Aldrich Cat# A8592, RRID:AB_439702) for cells transiently transfected with FLAG-MCL-1, for 8 h at 4°C (1:500 dilution in 1% BSA in PBS). The cells were then washed with PBS and incubated with anti-mouse Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific Cat# A28175, RRID:AB_2536161) for 60 min (1:200 dilution in 1% BSA in PBS) at room temperature in the dark. Antibody-labeled cells were then washed and stained in 0.1 μg/mL DAPI (Cell Signaling Technology, Cat# 4083) in PBS at room temperature in the dark for 10 min. Images were collected with a fully automated multimodal Zeiss 980 microscope equipped with 63x/1.4 NA objective and Airyscan 2 detectors. Immunofluorescence images were captured using Zen Software (Zeiss), exported to TIF, and processed in FIJI (ImageJ) software.
Quantitative proteomics
Sample preparation
Quantitative proteomics was performed on whole cell extracts obtained from wild-type MEFs treated with either vehicle (0.025% DMSO), S63845 (10 μM, 16 h), vincristine (10 nM, 16 h) or the combination. Lysates were quantified by the Bradford assay and subsequently reduced, alkylated, and subjected to chloroform/methanol precipitation.
Trypsin and LysC digestion
Protein precipitates were resuspended in 200 mM EPPS, pH 8.5 and digested at room temperature for 14 h with LysC protease (Promega) at a 100:1 protein-to-protease ratio. Trypsin (Promega) was then added at a 100:1 protein-to-protease ratio and the reaction was incubated for 6 h at 37°C.
Tandem mass tag labeling
TMT11 reagents (0.8 mg) were dissolved in anhydrous acetonitrile (40 ¼L) of which 5 ¼L was added to the peptides (50 ¼g) with 15 ¼L of acetonitrile to achieve a final concentration of ~30% (v/v). Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.3% (v/v). TMT-labeled samples were pooled at a 1:1 ratio across all samples. For each experiment, the pooled sample was vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (SPE) (Sep-Pak, Waters).
Offline Basic pH reversed-phase (BPRP) Fractionation
The pooled, labeled peptide samples were fractionated by BPRP HPLC56 using an Agilent 1260 pump equipped with a degasser and detector (set at 220 and 280 nm wavelengths). Peptides were subjected to a 50-min linear gradient from 5% to 35% acetonitrile in 10 mM ammonium bicarbonate pH 8 at a flow rate of 0.6 mL/min over an Agilent 300Extend C18 column (3.5 μm particles, 4.6 mm ID and 250 mm in length). The peptide mixture was fractionated into a total of 96 fractions, which were consolidated into 24 super-fractions.57 We analyzed non-adjacent samples for a total of 12 samples. Samples were subsequently acidified with 1% formic acid and vacuum centrifuged to near dryness. Each consolidated fraction was desalted by StageTip, dried again by vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid chromatography and tandem mass spectrometry
Mass spectrometric data were collected on an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific, San Jose, CA) coupled to a Proxeon EASY-nLC 1000 liquid chromatograph (LC) (ThermoFisher Scientific, San Jose, CA). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ~35 cm of Accucore150 resin (2.6 ~m, 150 Å, ThermoFisher Scientific, San Jose, CA). For each analysis, 1 to 2 ¼g of peptide was loaded onto the column and fractionated over a 90 min gradient of 3–26% acetonitrile in 0.125% formic acid at a flow rate of ~450 nL/min. Mass spectrometric data were collected using an RTS-MS358 method with high-field asymmetric-waveform ion mobility spectrometry (FAIMS). The scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution, 120,000; mass range, 400–1400 Th; automatic gain control (AGC) target 4 × 105 maximum injection time, 50 ms). Precursors were then selected for MS2/MS3 analysis.59 MS2 analysis consisted of collision-induced dissociation (CID) with quadrupole ion trap analysis using the following parameters: scan speed, turbo; AGC target, 2 × 104; NCE, 35; q-value, 0.25; maximum injection time, 35 ms; and isolation window, 0.7 Th. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap with the following parameters: resolution, 50,000; NCE, 55; AGC, 200,000; maximum injection time, 150 ms; maximum synchronous precursor selection (SPS) ions, 10; and isolation window, 1.2 Th. Data were collected with the “close-out” parameter set to 2.
Data analysis
Mass spectra were processed using a Comet-based software pipeline.60,61 Database searching included all entries from the mouse UniProt database (March 20, 2019). This database was concatenated with a reverse database composed of all protein sequences in reversed order. Searches were performed using a 50-ppm precursor ion tolerance and the product ion tolerance was set to 0.9 Da for SPS-MS3. Enzyme specificity was assigned as trypsin. TMT labels on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR).62,63 PSM filtering was performed using a linear discriminant analysis as described previously,46 while considering the following parameters: XCorr, peptide length, ΔCn, charge state, missed cleavages, and mass accuracy of the precursor. For TMT-based reporter ion quantitation, we extracted the signal-to-noise (S:N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. PSMs were identified, quantified, and collapsed to a 1% peptide FDR and then collapsed further to a final protein-level FDR of 1%. Peptide intensities were quantified by summing reporter ion counts across all matching PSMs to give greater weight to more intense ions.64,65
Cell cycle analysis
Wild-type and Bax−/−Bak−/− MEFs were treated with the indicated concentrations of S63845, AMG176, ABT-199 (Venetoclax), vinorelbine, vincristine, or the indicated combinations for 16 h and then harvested and fixed in 70% ethanol at −20°C for 30 min. Fixed cells were washed in cold PBS, permeabilized using 0.1% Triton X-100, blocked with 1% BSA in PBS, and labeled with primary antibody to phospho-histone H3 (S10) (Cell Signaling Technology Cat# 53348, RRID:AB_2799431) for 90 min at room temperature (1:500 dilution in 1% BSA in PBS). Cells were then centrifuged, washed with PBS, and incubated with anti-rabbit Alexa Fluor 594 secondary antibody (Thermo Fisher Scientific Cat# A-11012, RRID:AB_2534079) for 60 min (1:200 dilution in 1% BSA in PBS) at room temperature in the dark. Antibody conjugated cells were washed and stained in 25 mg/mL propidium iodide and 0.2 mg/mL RNAse A in PBS at 37°C in the dark for 1 h. DNA content was determined using a FACSCalibur (Becton Dickinson), and cell-cycle distribution was analyzed using FlowJo software (Becton Dickinson).
Evaluation of drug tolerance in vivo
C57BL/6J 8-week-old female mice (n = 4 per arm) were treated with either vincristine (1.5 mg/kg IP) twice weekly, S63845 (25 mg/kg IP) daily for 5 days, or the combination and body weight recorded daily.
S-to-M cell cycle progression
A pulse-chase strategy with EdU was employed to define the population of EdU-labelled cells that transition into G2/M over time. Briefly, cells were pulsed with EdU (10 μM) for 20 min, washed twice with PBS, and then incubated in the presence of nocodazole (50 ng/mL). Over a 0–10 h time frame, cells were fixed and processed for flow cytometry analysis using FxCycleViolet for total nuclear staining, EdU (subsequently click-coupled with Alexa Fluor 488 azide) as a marker for cells in S-phase, and phosphorylated histone H3 Ser10 (H3-pS10) as a marker for mitosis. Following flow cytometry analysis, fraction of S-gated or G2/M-gated cells in mitosis were plotted as a function of time. For experiments designed to assess the effect of MCL-1 inhibition on S-to-M progression, cells were treated with 10 μM S63845 for 12 h prior to EdU labeling.
B-ALL growth in vivo
C57BL/6J 8-week-old female mice were injected intravenously with Bax−/−Bak−/− (DKO) or Bax−/−Bak−/−Mcl-1−/− (TKO) murine B-ALL cells (250,000 cells; n = 10 mice per arm) and monitored for 15 weeks. Leukemia progression was assessed by phlebotomy and CBC analysis. For the experiment using higher cell counts, C57BL/6J 8-week-old female mice were injected with either 500,000 or 1,000,000 DKO or TKO murine B-ALL cells (n = 3 mice per arm) and monitored for 5 weeks. To evaluate the impact of MCL-1 reconstitution on TKO cell engraftment, mice were injected intravenously with Bax−/−Bak−/−Mcl-1−/− (TKO) or Bax−/−Bak−/−Mcl-1−/− hMCL-1 (TKO + MCL-1) murine B-ALL cells (500,000 cells; n = 10 mice per arm) and monitored for 5 weeks.
Phosphorylation profiling
Total protein and phosphorylation changes in cellular lysates from DKO and TKO B-ALL cells treated with 50 μM hydroxyurea for 24 h were interrogated using the Cell Cycle Control Phospho Antibody Array (Full Moon Biosystems), which features 238 highly specific antibodies to proteins and phosphoproteins involved in cell cycle control and DNA damage/repair pathways. Lysate samples were subjected to biotin labeling followed by conjugation with Cy3-streptavidin according to the manufacturer’s protocol and then shipped to Full Moon Biosystems for array scanning and data acquisition.
Cell viability assays for drug combination treatments
Wild-type MEFs, HEK 293T, HeLa, HUCCT1, and KPNYN cells cultured in DMEM containing 10% FBS and penicillin-streptomycin were plated in 96-well plates (5 × 103 cells per well) and, after overnight incubation, treated with the indicated concentrations of S63845, AZD5991, AMG176, A1210477, MIK665 and/or vinorelbine, vincristine, paclitaxel, combretastatin A4, camptothecin (all from Selleck Chemicals except AMG176 from MedChemExpress) for the indicated durations. Cell viability was measured using CellTiter-Glo (Promega) and luminescence detected by a Spectramax M5 microplate reader.
Affinity enrichment mass spectrometry (AE-MS) and affinity purification mass spectrometry (AP-MS)
Sample preparation for AE-MS
Cell lines expressing FLAG-tagged MCL-1 were generated by transient transfection using Lipofectamine LTX reagent, in accordance with the manufacturer’s protocol (ThermoFisher). Briefly, plasmid DNA was transfected into low passage HEK 293T cells (ATCC) cultured in DMEM (Gibco), supplemented with 10% (v/v) FBS, 100 μg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco). Following transfection at 50–70% confluence, cells were subjected to cell cycle synchronization to yield populations in G1, S, G2 and M phases. Cells were treated with a double thymidine block, washed with PBS (×2) and either harvested to isolate the G1 cell population or resuspended in fresh medium for 6 additional hours of culture for collection of cells in S phase.66 Asynchronized cells were cultured in medium containing 9 μM of RO-3306 (Sigma-Aldrich) for 18 h, washed in PBS (2x) and either harvested to isolate the G2 cell population or released into fresh medium containing 100 nM of nocodazole (Sigma-Aldrich) for 2 h for collection of cells in M phase.67 Cell pellets from the synchronized cell populations were lysed in the presence of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% (v/v) CHAPS, followed by centrifugation and filtration to remove debris. The clarified lysate was subjected to immunoprecipitation by incubation with immobilized and pre-washed mouse monoclonal anti-FLAG M2 magnetic beads (Sigma-Aldrich) at 4°C overnight, followed by removal of supernatant and washing of the beads with lysis buffer (×4) and PBS pH 7.2 (×2). Protein complexes were eluted in two steps using 3× FLAG peptide in PBS at 4°C for 1 h and the resulting eluates subjected to trichloroacetic acid (TCA) precipitation. Precipitated samples were resuspended in 100 ¼L of 100 mM EPPS, pH 8.5 and digested overnight at 37°C with trypsin at a 100:1 protein-to-protease ratio. The samples were desalted by StageTip, dried by vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for analysis by LC-MS/MS.
Sample preparation for AP-MS
SV40-transformed WT NIH 3T3 MEFs were cultured in DMEM (Gibco), supplemented with 10% (v/v) FBS, 100 μg/mL streptomycin (Gibco) and 100 U/mL penicillin (Gibco). Cells were harvested and lysed in the presence of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% (v/v) CHAPS, followed by centrifugation and filtration to remove debris. The clarified lysate was subjected to immunoprecipitation by incubation with immobilized and pre-washed rabbit polyclonal anti-MCL-1 antibody conjugated to Protein A magnetic beads (Sigma-Aldrich) at 4°C overnight, followed by removal of supernatant and washing of the beads with cold lysis buffer (4x) and PBS pH 7.2 (2x). Protein complexes were eluted in 4 bead volumes of 500 mM NH4OH at 37°C for 20 min and samples were dried overnight using a SpeedVac concentrator. Dry eluted samples were resuspended in 100 ¼L of 100 mM EPPS, pH 8.5 and subjected to in solution digestion with trypsin at a 100:1 protein-to-protease ratio. The samples were then desalted by StageTip, dried by vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid chromatography and tandem mass spectrometry
Mass spectrometry data were collected using a Q Exactive mass spectrometer (Thermo Fisher Scientific) coupled with a Famos Autosampler (LC Packings) and an Accela600 liquid chromatography (LC) pump (Thermo Fisher Scientific). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ~20 cm of Accucore C18 resin (2.6 μm, 150 Å, Thermo Fisher Scientific). For each analysis, we loaded ~1 ¼g onto the column. Peptides were separated using a 60 min method of 4–24% acetonitrile in 0.125% formic acid with a flow rate of ~300 nL/min. The scan sequence began with an Orbitrap MS1 spectrum using the following parameters: resolution 70,000, scan range 300–1500 Th, automatic gain control (AGC) target 1 × 105, maximum injection time 250 ms, and centroid spectrum data type. The top twenty precursors were selected for MS2 analysis that consisted of HCD high-energy collision dissociation using the following parameters: resolution 17,500, AGC 1 × 105, maximum injection time 60 ms, isolation window 1.6 Th, normalized collision energy (NCE) 25, centroid spectrum data type and a 1 × 104 intensity threshold. Unassigned and singly charged species were excluded from MS2 analysis and dynamic exclusion was set to automatic.
Data analysis
Mass spectra were processed using a Sequest-based in-house software pipeline. MS spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the human Uniprot database (AE-MS experiment in FLAG-MCL-1 transfected HEK 293T cells) or mouse Uniprot database (AP-MS experiment in WT MEFs), which was concatenated with a reverse database composed of all protein sequences in reversed order. Searches were performed using a 50 ppm precursor ion tolerance. Product ion tolerance was set to 0.003 Th. Carbamidomethylation of cysteine residues (57.02146 Da) was set as a static modification, while oxidation of methionine residues (+15.99492 Da) was set as a variable modification. Peptide spectral matches (PSMs) were altered to a 1% FDR.62,63 PSM filtering was performed using a linear discriminant analysis as described previously,46 while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state, and precursor mass accuracy. Peptide-spectral matches were identified, quantified, and collapsed to a 1% FDR and then further collapsed to a final protein-level FDR of 1%. Protein assembly was guided further by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides.
Identification of interacting protein complexes by AE-MS
To identify protein complexes bound by MCL-1, the normalized spectral abundance factor (NSAF) values for each protein prey detected by the affinity enhancement mass spectrometry experiment was computed. NSAF values were corrected for background by subtracting abundance values of lysate immunoprecipitates from HEK 293T cells that underwent mock transfections and only proteins with greater than 5 peptides detected were filtered for further analyses. Mean scaled abundance values were computed for all filtered proteins detected in each cell cycle phase. K-means clustering (k = 4) was performed to identify proteins that predominantly bound to MCL-1 in a specific cell cycle phase, and enriched protein complexes within each cluster determined by cross-referencing the list of bound proteins with CORUM,68 a database for experimentally verified mammalian protein interactions through the Enrichr web platform.69
Co-immunoprecipitation assays
Cells transiently expressing FLAG-tagged MCL-1 were subjected to lysis in the presence of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% (v/v) CHAPS, and subsequently clarified by centrifugation and filtration to eliminate cellular debris. Clarified lysates were subjected to overnight incubation at 4°C with either anti-FLAG M2 magnetic beads (Sigma-Aldrich) or Protein A/G magnetic beads coupled to an MCM5 antibody (Santa Cruz Biotechnology Cat# sc-165994, RRID:AB_2142526). For immunoprecipitation of endogenous MCL-1, a rabbit polyclonal antibody against MCL-1 (Rockland Cat# 600-401-394S, RRID:AB_11179937) was immobilized on Protein A magnetic beads, which were used for capture of protein complexes from clarified cellular lysate. The beads were subsequently washed 4x with lysis buffer followed by 2x with PBS pH 7.2. Bead-bound proteins were eluted by boiling in 2× LDS loading buffer containing 100 mM DTT for 10 min and then subjected to gel electrophoresis. Proteins were transferred onto nitrocellulose membranes using the iBlot 7-Minute Blotting System, blocked in 5% milk in PBS-Tween (PBST), and incubated at 1:1000 with the corresponding primary antibody (FLAG: Sigma-Aldrich Cat# A8592, RRID:AB_439702; MCL-1: Rockland Cat# 600-401-394S, RRID:AB_11179937; MCM5: Santa Cruz Biotechnology Cat# sc-165994, RRID:AB_2142526) in 3% BSA in PBST. Secondary antibodies (1:2000) were diluted in 3% BSA in PBST and the Western blot developed using the ECL Prime Western Blotting Detection Reagent (Amersham).
Immunofluorescence analysis of replication stress and proximity ligation assays
Bax−/−Bak−/− HCT116 cells were grown on coverslips (Fisherbrand) overnight, followed by treatment with either vehicle (0.1% DMSO) or 10 μM S63845 for 16 h, and subsequently with either vehicle (0.02% DMSO) or 2 μM hydroxyurea for 2 h. Drug-treated cells were first pre-extracted with CSK buffer (10 mM PIPES pH 6.8, 100 mM NaCl, 1 mM MgCl2, 1 mM EGTA and 0.5% Triton X-100) for 4 min on ice, and then fixed in 4% paraformaldehyde in PBS (Sigma Aldrich Cat# 100496) for 15 min at room temperature (RT). Fixed cells were subsequently permeabilized and blocked for 1 h at RT using a blocking buffer containing 0.5% Triton X-100, 10% FBS, and 1% BSA in PBS. Following permeabilization and blocking, cells were incubated overnight at 4°C in a moist chamber with primary antibodies to phospho-H2AX (S139) (Cell Signaling Technology Cat#9718, RRID: AB_2799949) and phospho-RPA32 (S33) (Bethyl Cat# A300-246A, RRID:AB_2180847) at 1:500 dilution in antibody dilution buffer (0.3% Triton X-100, 10% FBS, and 1% BSA in PBS). Following labeling with primary antibody, cells were washed 3× with PBST (0.05% Tween 20 in PBS) and incubated with anti-mouse Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific Cat# A-11001, RRID:AB_2534069) and anti-rabbit Alexa Fluor 568 secondary antibody (Thermo Fisher Scientific Cat# A-11011, RRID:AB_143157) at 1:500 dilution for 1 h at RT. Cells were subsequently washed in PBST and mounted using VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories Cat# H-1200-10). Proximity ligation assay (PLA) was performed after the fixation and blocking step using the Duolink Proximity Ligation Assay (Sigma Aldrich) following the manufacturer’s protocol. For the PLA experiment, primary antibodies to MCL-1 (Proteintech Cat# 66026-1-Ig, RRID:AB_11041711) and MCM5 (Bethyl Cat# A300-195A, RRID:AB_185552) were used at 1:50 and 1:100 dilutions respectively. PLA images were acquired using a Zeiss Axio Observer microscope equipped with 63x/1.4 NA oil immersion objective, and quantified using FIJI (ImageJ) software. Quantification was performed by counting at least 50 cells per experiment, and the number of foci within individual cells was plotted using Prism software 9.0 (GraphPad).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as mean ± S.D. unless otherwise noted. Exact numbers of biological and technical replicates for each experiment are reported in the Figure Legends. p values less than 0.05 were considered statistically significant by unpaired, two-tailed Student’s t test. For all data, *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p <0.0001. Data were analyzed using Prism Software 9.0 (GraphPad) and Python 3 in Jupyter Notebook environment.
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-MCL-1 | Rockland Immunochemicals | Cat# 600-401-394S, RRID:AB_11179937 |
| Rabbit monoclonal anti-actin (HRP) | Cell Signaling Technology | Cat #5125, RRID: AB_1903890 |
| Mouse monoclonal phospho-H2AX S139 | Millipore | Cat# 05–636, RRID:AB_309864 |
| Rabbit polyclonal anti-phospho-CHK1 S345 | Abcam | Cat# ab58567, RRID:AB_10563825 |
| Rabbit polyclonal anti-phospho-RPA32 S33 | Bethyl | Cat# A300-246A, RRID:AB_2180847 |
| Goat Anti-rabbit AlexaFluor® 488 secondary antibody | Thermo Fisher Scientific | Cat# A-11034, RRID:AB_2576217 |
| Goat Anti-mouse AlexaFluor® 488 secondary antibody | Thermo Fisher Scientific | Cat# A-11001, RRID:AB_2534069 |
| Goat Anti-rabbit AlexaFluor® 568 secondary antibody | Thermo Fisher Scientific | Cat# A-11011, RRID:AB_143157 |
| Goat Anti-rabbit AlexaFluor® 594 secondary antibody | Thermo Fisher Scientific | Cat# A-11012, RRID:AB_2534079 |
| Mouse monoclonal anti-MCL-1 | Proteintech | Cat# 66026-1-Ig, RRID:AB_11041711 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# A8592, RRID:AB_439702 |
| Rabbit monoclonal anti-phospho-histone H3 S10 | Cell Signaling Technology | Cat# 53348, RRID:AB_2799431 |
| Mouse monoclonal anti-MCM5 | Santa Cruz Biotechnology | Cat# sc-165994, RRID:AB_2142526 |
| Rabbit polyclonal anti-MCM5 | Bethyl | Cat# A300-195A, RRID:AB_185552 |
| Chemicals, peptides, and recombinant proteins | ||
| Complete protease inhibitor cocktail tablets | Sigma-Aldrich | Cat# 05056489001 |
| Propidium iodide | Thermo Fisher Scientific | Cat# P3566 |
| DAPI | Cell Signaling Technology | Cat# 4083 |
| MitoTrackerTM Deep Red FM | Thermo Fisher Scientific | Cat# M22426 |
| EPPS | Thermo Fisher Scientific | Cat# J61296.AE |
| Trypsin | Promega | Cat# VA9000 |
| LysC | Promega | Cat# VA1170 |
| TMTpro™ 16plex Label Reagent Set | Thermo Fisher Scientific | Cat# A44522 |
| FxCycleViolet | Thermo Fisher Scientific | Cat# R37166 |
| Lipofectamine™ LTX Reagent with PLUS™ Reagent | Thermo Fisher Scientific | Cat# 15338100 |
| S63845 | Selleck Chemicals | Cat# S8383 |
| ABT-199 | Selleck Chemicals | Cat# S8048 |
| Vincristine | Selleck Chemicals | Cat# S9555 |
| Nocodazole | Selleck Chemicals | Cat# S2775 |
| Vinorelbine | Selleck Chemicals | Cat# S4269 |
| Paclitaxel | Selleck Chemicals | Cat# S1150 |
| Combretastatin A4 | Selleck Chemicals | Cat# S7783 |
| Hydroxyurea | Selleck Chemicals | Cat# S1896 |
| Camptothecin | Selleck Chemicals | Cat# S1288 |
| AZD5991 | Selleck Chemicals | Cat# S8643 |
| AMG176 | MedChemExpress | Cat# HY-101565 |
| A1210477 | Selleck Chemicals | Cat# S7790 |
| MIK665 | Selleck Chemicals | Cat# S8836 |
| RO-3306 | Sigma-Aldrich | Cat# SML0569 |
| Anti-FLAG M2 Magnetic Beads | Sigma-Aldrich | Cat# M8823 |
| Pierce™ Protein A Magnetic Beads | Sigma-Aldrich | Cat# 88845 |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories | Cat# H-1200-10 |
| Critical commercial assays | ||
| MycoAlert Mycoplasma Detection Kit | Lonza Biologics | Cat #LT07-218 |
| Pierce Bradford Protein Assay Kit | Thermo Fisher Scientific | Cat# 23238 |
| CellTiter-Glo© Luminescent Cell Viability Assay | Promega | Cat #G7571 |
| Click-iT™ EdU Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C10337 |
| Cell Cycle Control Phospho Antibody Array | Full Moon Biosystems | Cat# PCC238 |
| Duolink® Proximity Ligation Assay | Sigma-Aldrich | Cat# DUO92105 |
| BrdU Cell Proliferation Assay Kit | Cell Signaling Technology | Cat# 6813 |
| Deposited data | ||
| Proteomic Data | This paper | ProteomeXchange via the PRIDE database PXD036327 |
| RNA-seq Data | Jain et al.26 | NCBI GEO GSE102227 |
| Experimental models: Cell lines | ||
| Bax−/−Bak−/− (DKO) p185+Arf−/− B-ALL | Koss et al.38 | N/A |
| Bax−/−Bak−/−Mcl-1−/− (TKO) p185+Arf−/− B-ALL | Koss et al.38; Prew et al.70 | N/A |
| Bax−/−Bak−/−Mcl-1−/−hMCL-1 (TKO+MCL-1) p185+Arf−/− B-ALL | Prew et al.70 | N/A |
| Baxfl/flBak−/− MEF | This paper | N/A |
| Mcl-1fl/flBaxfl/flBak−/− MEF | This paper | N/A |
| WT MEF | Perciavalle et al.20 | N/A |
| Mcl-1−/− MEF | Opferman et al.14 | N/A |
| Bax−/−Bak−/− MEF | ATCC | Cat# CRL-2913 |
| Mcl-1fl/flRosa-ERCreT2 | Perciavalle et al.20 | N/A |
| Mcl-1fl/flRosa-ERCreT2 + MCL-1 | Perciavalle et al.20 | N/A |
| Mcl-1fl/flRosa-ERCreT2 + MCL-1Matrix | Perciavalle et al.20 | N/A |
| Mcl-1fl/flRosa-ERCreT2+ MCL-1OMM | Perciavalle et al.20 | N/A |
| Bax−/−Bak−/− (DKO) MV4;11 | This paper; Garciaz et al.71 | N/A |
| HCT116 | ATCC | Cat #CCL-247 |
| Bax−/−Bak−/− (DKO) HCT116 | Wang et al.72 | N/A |
| H929 | ATCC | Cat #CRL-9068 |
| K562 | ATCC | Cat #CCL-243 |
| 293T | ATCC | Cat #CRL-3216 |
| HeLa | ATCC | Cat #CCL-2 |
| KPNYN | JCRB Cell Bank | Cat# IFO50431 |
| HUCCT1 | JCRB Cell Bank | Cat# JCRB0425 |
| Experimental models: Organism/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain #:000664 RRID:IMSR_JAX:000664 |
| Software and algorithms | ||
| Prism v9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| LAS X | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| FIJI | ImageJ | https://imagej.net/software/fiji/ |
| FlowJo | BD and Company | https://www.fiowjo.com/solutions/fiowjo |
| Enrichr | Chen et al.69 | https://maayanlab.doud/Enrichr/ |
| Other | ||
| DMEM | Thermo Fisher Scientific | Cat# 12491015 |
| RPMI 1640 | Thermo Fisher Scientific | Cat #21870-076 |
| Fetal Bovine Serum | GeminiBio | Cat #100–106, Lot #A33H00L |
| Penicillin and Streptomycin | Gibco | Cat# 15070063 |
| Glutamine | Gibco | Cat# 25030081 |
Highlights.
Targeting MCL-1 induces DNA damage and impairs cell proliferation
MCL-1 regulates DNA integrity and cell-cycle progression independent from apoptosis
Replicative stress induces MCL-1 interaction with minichrosome maintenance complex
Noncanonical roles of MCL-1 expand the benefits and risks of therapeutic targeting
ACKNOWLEDGMENTS
We thank E. Smith for graphics support, J. Tepper for technical assistance with the chemotherapeutics screen of wild-type and Mcl-1−/− MEFs, and D. Neuberg for biostatistics consultation. This work was supported by NIH grant R35CA197583 and a Leukemia and Lymphoma Society Translational Research Program grant (to L.D.W.), a Landry Cancer Biology Research Fellowship and Chleck Family Foundation Fellowship (to U.A.), a National Science Foundation Predoctoral Award and a Landry Cancer Biology Research Fellowship (to M.S.P.), a Leukemia and Lymphoma Society Fellowship award (to Y.B.-N.), and NIH grants T32GM144273 and 1F30CA275293 (to E.W.Y.), R01GM132129 (to J.A.P.), R01GM67945 (to S.P.G.), and R01CA201069 (to J.T.O.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113176.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Green DR (2022). The Mitochondrial Pathway of Apoptosis Part II: The BCL-2 Protein Family. Cold Spring Harb. Perspect. Biol 14, a041046. 10.1101/cshperspect.a041046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reynolds JE, Yang T, Qian L, Jenkinson JD, Zhou P, Eastman A, and Craig RW (1994). Mcl-1, a member of the Bcl-2 family, delays apoptosis induced by c-Myc overexpression in Chinese hamster ovary cells. Cancer Res. 54, 6348–6352. [PubMed] [Google Scholar]
- 3.Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DCS, and Hinds MG (2005). Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J. Biol. Chem 280, 4738–4744. 10.1074/jbc.M411434200. [DOI] [PubMed] [Google Scholar]
- 4.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, and Korsmeyer SJ (2001). BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711. 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 5.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, and Korsmeyer SJ (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 6.Walensky LD, and Gavathiotis E (2011). BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem. Sci 36, 642–652. 10.1016/j.tibs.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. (1997). Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986. 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 8.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. (2005). An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 9.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, and Korsmeyer SJ (2004). Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470. 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rinkenberger JL, Horning S, Klocke B, Roth K, and Korsmeyer SJ (2000). Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 14, 23–27. [PMC free article] [PubMed] [Google Scholar]
- 12.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S.i., Akashi K, and Korsmeyer SJ (2005). Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 307, 1101–1104. 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 13.Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung ECC, Kelly MA, MacKenzie AE, Park DS, Opferman JT, and Slack RS (2008). Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci 28, 6068–6078. 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, and Korsmeyer SJ (2003). Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426, 671–676. 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, and Opferman JT (2013). Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 27, 1351–1364. 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Widden H, and Placzek WJ (2021). The multiple mechanisms of MCL1 in the regulation of cell fate. Commun. Biol 4, 1029. 10.1038/s42003-021-02564-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Guo M, Wei H, and Chen Y (2021). Targeting MCL-1 in cancer: current status and perspectives. J. Hematol. Oncol 14, 67. 10.1186/s13045-021-01079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, and Seto M (2005). Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 24, 1348–1358. 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 20.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, et al. (2012). Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol 14, 575–583. 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang CR, and Yang-Yen HF (2010). The fast-mobility isoform of mouse Mcl-1 is a mitochondrial matrix-localized protein with attenuated anti-apoptotic activity. FEBS Lett. 584, 3323–3330. 10.1016/j.febslet.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Escudero S, Zaganjor E, Lee S, Mill CP, Morgan AM, Crawford EB, Chen J, Wales TE, Mourtada R, Luccarelli J, et al. (2018). Dynamic Regulation of Long-Chain Fatty Acid Oxidation by a Noncanonical Interaction between the MCL-1 BH3 Helix and VLCAD. Mol. Cell 69, 729–743.e7. 10.1016/j.molcel.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. (2000). The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399. 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, and Youle RJ (2012). Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene 31, 3177–3189. 10.1038/onc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain R, Sheridan JM, Policheni A, Heinlein M, Gandolfo LC, Dewson G, Smyth GK, Sansom SN, Fu NY, Visvader JE, et al. (2017). A critical epithelial survival axis regulated by MCL-1 maintains thymic function in mice. Blood 130, 2504–2515. 10.1182/blood-2017-03-771576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grabow S, Delbridge ARD, Valente LJ, and Strasser A (2014). MCL-1 but not BCL-XL is critical for the development and sustained expansion of thymic lymphoma in p53-deficient mice. Blood 124, 3939–3946. 10.1182/blood-2014-09-601567. [DOI] [PubMed] [Google Scholar]
- 28.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, Taylor BR, Crenshaw A, Gould JZ, Tamayo P, et al. (2016). Highthroughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol 34, 419–423. 10.1038/nbt.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehá r J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bannister T, Koenig M, He Y, Mishra J, Spicer T, Minond D, Saldanha A, Mercer BA, Cameron M, Lena R, et al. (2010). ML311: A Small Molecule that Potently and Selectively Disrupts the Protein-Protein Interaction of Mcl-1 and Bim: A Probe for Studying Lymphoid Tumorigenesis. In Probe Reports from the NIH Molecular Libraries Program. [PubMed] [Google Scholar]
- 32.Yecies D, Carlson NE, Deng J, and Letai A (2010). Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313. 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosman MCJ, Schuringa JJ, Quax WJ, and Vellenga E (2013). Bortezomib sensitivity of acute myeloid leukemia CD34(+) cells can be enhanced by targeting the persisting activity of NF-kappaB and the accumulation of MCL-1. Exp. Hematol 41, 530–538.e1. 10.1016/j.exphem.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Zhu X, Hou K, Zhao J, Han Z, and Zhang X (2015). Mcl-1 downregulation sensitizes glioma to bortezomib-induced apoptosis. Oncol. Rep 33, 2277–2284. 10.3892/or.2015.3875. [DOI] [PubMed] [Google Scholar]
- 35.Zhou W, Hu J, Tang H, Wang D, Huang X, He C, and Zhu H (2011). Small interfering RNA targeting mcl-1 enhances proteasome inhibitor-induced apoptosis in various solid malignant tumors. BMC Cancer 11, 485. 10.1186/1471-2407-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. (2011). Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114. 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 37.Chen G, Magis AT, Xu K, Park D, Yu DS, Owonikoko TK, Sica GL, Satola SW, Ramalingam SS, Curran WJ, et al. (2018). Targeting Mcl-1 enhances DNA replication stress sensitivity to cancer therapy. J. Clin. Invest 128, 500–516. 10.1172/JCI92742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koss B, Morrison J, Perciavalle RM, Singh H, Rehg JE, Williams RT, and Opferman JT (2013). Requirement for antiapoptotic MCL-1 in the survival of BCR-ABL B-lineage acute lymphoblastic leukemia. Blood 122, 1587–1598. 10.1182/blood-2012-06-440230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koss B, Ryan J, Budhraja A, Szarama K, Yang X, Bathina M, Cardone MH, Nikolovska-Coleska Z, Letai A, and Opferman JT (2016). Defining specificity and on-target activity of BH3-mimetics using engineered B-ALL cell lines. Oncotarget 7, 11500–11511. 10.18632/oncotarget.7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee T, Christov PP, Shaw S, Tarr JC, Zhao B, Veerasamy N, Jeon KO, Mills JJ, Bian Z, Sensintaffar JL, et al. (2019). Discovery of Potent Myeloid Cell Leukemia-1 (Mcl-1) Inhibitors That Demonstrate in Vivo Activity in Mouse Xenograft Models of Human Cancer. J. Med. Chem 62, 3971–3988. 10.1021/acs.jmedchem.8b01991. [DOI] [PubMed] [Google Scholar]
- 41.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 42.Shieh SY, Ahn J, Tamai K, Taya Y, and Prives C (2000). The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- 43.Smeenk L, van Heeringen SJ, Koeppel M, Gilbert B, Janssen-Megens E, Stunnenberg HG, and Lohrum M (2011). Role of p53 serine 46 in p53 target gene regulation. PLoS One 6, e17574. 10.1371/journal.pone.0017574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e28. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gou K, Liu J, Feng X, Li H, Yuan Y, and Xing C (2018). Expression of Minichromosome Maintenance Proteins (MCM) and Cancer Prognosis: A meta-analysis. J. Cancer 9, 1518–1526. 10.7150/jca.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsu EC, Shen M, Aslan M, Liu S, Kumar M, Garcia-Marques F, Nguyen HM, Nolley R, Pitteri SJ, Corey E, et al. (2021). MCM2-7 complex is a novel druggable target for neuroendocrine prostate cancer. Sci. Rep 11, 13305. 10.1038/s41598-021-92552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu J, Farmer JD Jr., Lane WS, Friedman J, Weissman I, and Schreiber SL (1991). Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815. 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 50.Cameron AM, Nucifora FC Jr., Fung ET, Livingston DJ, Aldape RA, Ross CA, and Snyder SH (1997). FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400-1401) and anchors calcineurin to this FK506-like domain. J. Biol. Chem 272, 27582–27588. 10.1074/jbc.272.44.27582. [DOI] [PubMed] [Google Scholar]
- 51.Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, et al. (2022). The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, and Ma’ayan A (2018). Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9, 1366. 10.1038/s41467-018-03751-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, and Kanehisa M (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34. 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA, et al. (2020). Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 1, 235–248. 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duan Q, Reid SP, Clark NR, Wang Z, Fernandez NF, Rouillard AD, Readhead B, Tritsch SR, Hodos R, Hafner M, et al. (2016). L1000CDS(2): LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl 2, 16015. 10.1038/npjsba.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T, et al. (2011). Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026. 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, and Gygi SP (2016). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteomics 148, 85–93. 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schweppe DK, Eng JK, Yu Q, Bailey D, Rad R, Navarrete-Perea J, Huttlin EL, Erickson BK, Paulo JA, and Gygi SP (2020). Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res 19, 2026–2034. 10.1021/acs.jproteome.9b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eng JK, Hoopmann MR, Jahan TA, Egertson JD, Noble WS, and MacCoss MJ (2015). A deeper look into Comet–implementation and features. J. Am. Soc. Mass Spectrom 26, 1865–1874. 10.1007/s13361-015-1179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eng JK, Jahan TA, and Hoopmann MR (2013). Comet: an opensource MS/MS sequence database search tool. Proteomics 13, 22–24. 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 62.Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 63.Elias JE, and Gygi SP (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol 604, 55–71. 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, and Gygi SP (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem 84, 7469–7478. 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen G, and Deng X (2018). Cell Synchronization by Double Thymidine Block. Bio. Protoc 8, e2994. 10.21769/BioProtoc.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, and Chen L (2006). Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 103, 10660–10665. 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Mewes HW (2010). CORUM: the comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res. 38, D497–D501. 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prew MS, Adhikary U, Choi DW, Portero EP, Paulo JA, Gowda P, Budhraja A, Opferman JT, Gygi SP, Danial NN, et al. (2022). MCL-1 is a master regulator of cancer dependency on fatty acid oxidation. Cell Rep 41, 111445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garciaz S, Guirguis AA, Muller S, Brown FC, Chan YC, Motazedian A, Rowe CL, Kuzich JA, Chan KL, Tran K, et al. (2022). Pharmacologic reduction of mitochondrial iron triggers a noncanonical BAX/BAK-dependent cell death. Cancer Discov 12, 774–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang C, and Youle RJ (2012). Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene 31, 3177–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials. Proteomic datasets were submitted to the ProteomeXchange Consortium via PRIDE51 with identifier PXD036327.
This study did not generate code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.