

Abstract
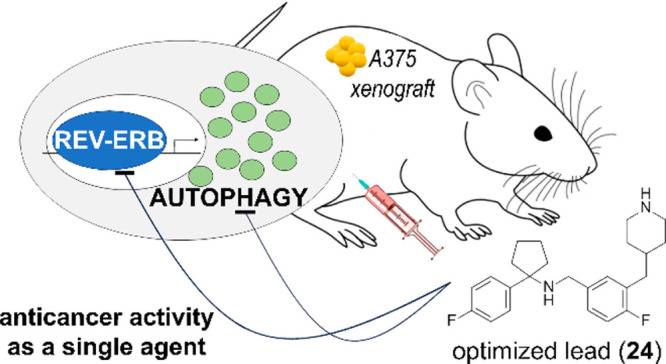
The autophagy process appears as a promising target for anticancer interventions. Chloroquine (CQ) and its derivative hydroxychloroquine (HCQ) are the only FDA-approved autophagy flux inhibitors. Although diverse anticancer clinical trials are providing encouraging results, several limitations associated with the need of high dosage and long-term administration of these autophagy inhibitors are also emerging. We showed that the inhibition of REV-ERB, a nuclear receptor regulating circadian rhythm and metabolism, enhances CQ-mediated cancer cell death and identified a class of dual inhibitors of autophagy and REV-ERB displaying an in vitro anticancer activity against diverse tumor cells greatly higher than CQ. Herein, we describe our lead optimization strategy that led to the identification of compound 24 as a dual autophagy and REV-ERB inhibitor, showing improved potency in blocking autophagy, enhanced toxicity against cancer cells, optimal drug-like properties, and efficacy in a mouse xenograft model of melanoma as a single anticancer agent.
Introduction
Macro-autophagy, generally simply called autophagy, is a multistep process driving the degradation of damaged and excess proteins and organelles to generate macromolecular building blocks and fuel metabolic pathways.1,2 The autophagy pathway involves diverse protein complexes coordinating the formation of double-membrane vesicles, named autophagosomes, which encapsulate intracellular substrates, referred to as cargo. Upon the fusion of autophagosomes with lysosomes, the cargo is degraded in the resulting autophagolysosomes.3
Autophagy has critical roles in many biological processes, such as protein homeostasis, stress response, metabolism, and cell death. Accordingly, altered autophagic degradation has been linked to diverse pathologies.2 In cancer, autophagy presents a double-edged sword effect on tumor development and progression. Although functional autophagic degradation has a protective role against the development of multiple tumor types, after a tumor is fully established, autophagy takes on a pro-survival role for cancer cells.4,5 Multiple cancers hijack autophagy to reprogram their proteome and cope with cancer metabolic needs and the activity of many chemotherapeutic agents.6 Accordingly, the autophagy–lysosome system appears as a promising target for anticancer interventions.
Chloroquine (CQ) and its derivative hydroxychloroquine (HCQ) are the only FDA-approved autophagy flux inhibitors (Figure 1).7
Figure 1.

Structures of known autophagy inhibitors (chloroquine (CQ), hydroxychloroquine (HCQ), and Lys05, top) and known antagonist ligands of REV-ERB (SR8278 and GSK1362, bottom).
Both CQ and HCQ target autophagy at the late stage by preventing the final maturation of autophagolysosomes, thus interrupting the autophagic flux and preventing autophagy-mediated degradation.8
Because of their long history of use in humans for the treatment of malaria, these agents have been repurposed in numerous clinical trials for the treatment of diverse cancers.
However, both CQ and HCQ presented several limitations in the treatment of tumors, especially for their use as single anticancer agents. High concentrations of these compounds are usually required for inhibiting the growth of cancer cells already in vitro and in animals,8 which implies the use of high dosages and long-term administration that could lead to severe toxicity. Indeed, adverse effects such as cardiac disorders and retinopathy have been associated with CQ and HCQ long-term use in humans.9−12 Moreover, a recent study indicated that CQ and HCQ can block the activity of the human ether-a-go-go-related gene (hERG) potassium channel at concentrations in the single-digit μM value,13 and regular 6-month cardiac monitoring tests are advised for patients taking a high dosage of either drugs.14 Accordingly, in recent years, different drug discovery projects have been focused on the identification of more potent autophagy inhibitory compounds for reducing the doses to be used in patients.15−17
An additional strategy to overcome the limitations deriving from the need of high doses of CQ and HCQ for inhibiting tumor growth is represented by a multitarget approach targeting both autophagy and pathways associated with autophagy-dependent cancer cell death. We previously reported that REV-ERB, a nuclear receptor regulating circadian rhythm and metabolism,18−20 plays a role in sustaining cancer cell survival when the autophagy flux is compromised.21 Indeed, pharmacological inhibition of REV-ERB significantly enhanced the cytotoxic activity of CQ against breast cancer cells, and cells from different cancer types knocked-down for REV-ERB were more sensitive to CQ-induced cell death.21,22 In addition, the knockdown of the essential autophagy gene ATG5 in breast cancer cells enhanced the toxicity of the REV-ERB antagonist, SR8278 (Figure 1), further indicating that REV-ERB inhibition induces cell death when autophagy is inhibited.21
An in house drug discovery screening campaign led to the identification of the hit compound 4-[[[1-(2-fluorophenyl)cyclopentyl]amino]methyl]-2-[(4-methylpiperazin-1-yl)methyl]phenol (1a, ARN5187, Figure 2) that inhibits both REV-ERB and autophagy.
Figure 2.
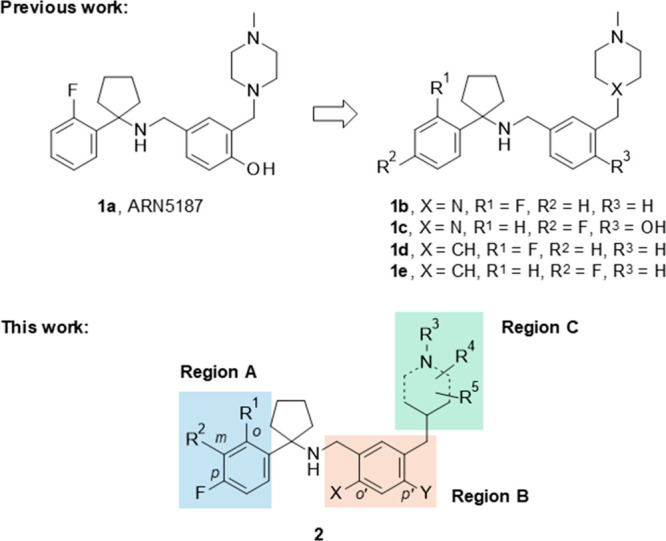
(A) Structures of known dual autophagy and REV-ERB antagonists 1a–e (top) and our planned chemical exploration around the scaffold of this class of molecules 2 (bottom).
Specifically, 1a disrupts lysosomal function and blocks the autophagy process at the late stage.21 In addition, 1a directly binds REV-ERB ligand binding domain (LBD) in fluorine magnetic resonance (NMR)-based assays and relieves REV-ERB-mediated transcriptional repression enhancing the expression of REV-ERB target genes in breast cancer cells.21 Notably, the single inhibition of autophagy by CQ does not affect REV-ERB-mediated transcription, indicating that the effect of 1a on REV-ERB transcriptional repression does not depend on its autophagy inhibitory activity.22
Preliminary structure–activity relationship (SAR) studies resulted in the identification of the lead compound, 1e, showing a greater REV-ERB inhibitory activity than 1a and a potency in inhibiting autophagy comparable to CQ.22 Of note, the in vitro anticancer activity of 1e against several tumor cell lines was up to 50 times higher than CQ. In addition, 1e did not affect the viability of mammary epithelial cells from healthy donors, supporting the development of this class of dual autophagy and REV-ERB inhibitors as novel anticancer agents with a good therapeutic index.22
However, additional investigation on the drug-like properties of 1e suggested further chemical optimization to be performed for advancing this class of molecules in the drug discovery pipeline.
Herein, we describe our lead optimization strategy that resulted in the identification of compound 24, which shows improved biological activity, optimal drug-like properties, and efficacy in a mouse xenograft model of melanoma as a single anticancer agent.
Chemistry
The preparation of all target compounds 3–26 has been accomplished through a convergent approach, involving the synthesis of amines 30a–i (Schemes 1–3) and aldehydes 40a–n (Schemes 4–7), followed by their condensation via reductive amination under standard reaction conditions,22 as depicted in Schemes 5 and 8. An additional step of N-Boc removal was performed for the preparation of compounds 17 and 24 and 25, as illustrated in Scheme 5.
Scheme 1. Synthesis of 30a–d.

Reagents and conditions: for 28a,b: (a) n-BuLi, anhydrous THF, −78 °C, 1 h, then, cyclopentanone, −78 °C to rt, 3 h, for 28c,d, cyclopentanone, anhydrous THF, 0 °C to rt, 3 h; (b) NaN3, TFA, anhydrous CH2Cl2, −5 °C to rt, (4–11% over two steps); (c) LiAlH4, anhydrous THF, 0 °C to rt.
Scheme 3. Synthesis of 30h–i.

Reagents and conditions: (a) Br(CH2)4Br, LiHMDS, anhydrous THF, 0 °C to rt, on (69–77%); (b) H2SO4, THF/H2O, 120 °C (42% for 34b); (c) for 30h: (i) ClCO2CH2CH3, NaN3, Et3N, acetone, (ii) anhydrous toluene, reflux, 1 h, (iii) 8 N HCl, 70 °C, 3 h; for 30i, (i) SOCl2, anhydrous toluene, rt, 2 h, (ii) NaN3, anhydrous toluene, rt, (iii) BnOH, reflux, (iv) Et3SiH, 10% Pd/C, MeOH, 90 °C, MW, 30 min.
Scheme 4. Synthesis of 40a–e.

Reagents and conditions: (a) P(OEt)3 or PPh3, anhydrous toluene, reflux, on (39–99%); (b) LiHMDS or t-BuOK, 1-Boc-4-piperidone, anhydrous THF, 0 °C to rt, (20–58%); (c) Et3SiH, 10% Pd/C, MeOH, rt, 7 h; (d) for 39a,b,d,e, LiAlH4, anhydrous THF, 0 °C to rt; for 39c, (i) 4 M HCl in dioxane, CH2Cl2, 0 °C to rt, (quant), (ii) Et3N, formaldehyde 37 wt % in H2O, NaBH(OAc)3, 0 °C to rt, (iii) DIBALH, anhydrous THF, 0 °C to rt; (e) MnO2, anhydrous Et2O, rt, 8 h.
Scheme 7. Synthesis of 40k–n.

Reagents and conditions: (a) LiHMDS or t-BuOK, cyclic amino ketone, anhydrous THF, 0 °C to rt or 80 °C, (83–34%); (b) Et3SiH, 10% Pd/C, MeOH, rt, 7 h; (c) LiAlH4, anhydrous THF, 0 °C to rt; (d) MnO2, anhydrous Et2O, rt, 8 h.
Scheme 5. Synthesis of 41f–h and 17, 24, and 25.
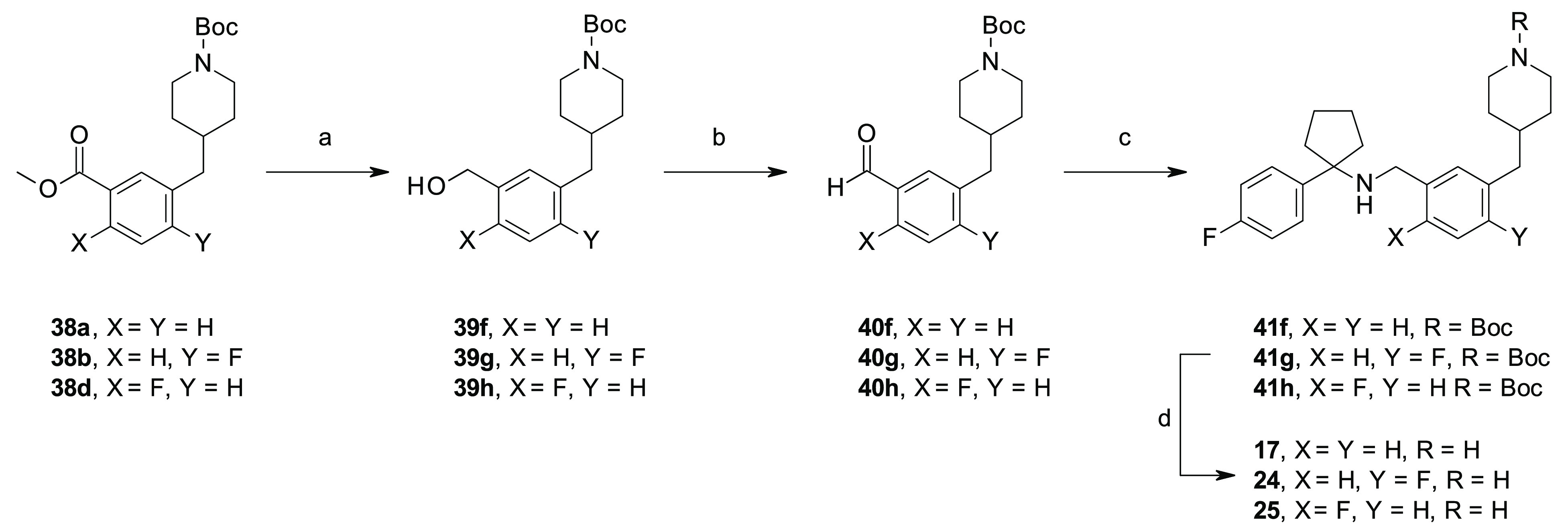
Reagents and conditions: (a) DIBALH, anhydrous THF, 0 °C to rt, (60–89%); (b) MnO2, anhydrous Et2O, rt, 8 h (74% for 40g); (c) 30e, NaBH(OAc)3, anhydrous CH2Cl2 0 °C to rt, rt (13–84%); (d) 4 M HCl in 1,4-dioxane, CH2Cl2, 0 °C to rt, (quant).
Scheme 8. Synthesis of 3–16, 18–23, and 26.

Reagents and conditions: (a) NaBH(OAc)3, anhydrous CH2Cl2, rt, on (11–60% over four steps).
Our versatile synthetic strategy allowed for rapid exploration of the targeted chemical modifications in the regions A, B, and C of scaffold 2 (Figure 2).
To overcome the issues associated with the limited commercial availability of some building blocks or the low yields of some synthetic routes, different procedures for the preparation of the key amines 30a–i were exploited, as illustrated in Schemes 1–3, which allowed the insertion of different substituents at the ortho- and meta-positions of the para-fluoro phenyl ring of our lead compound 1e (Figure 2). In the specific, the amines 30a–d were prepared through a previously reported three-step synthetic procedure,23,24 consisting of a nucleophilic addition of the appropriate in situ generated organolithium or commercially available Grignard reagents to cyclopentanone, followed by the treatment of the corresponding phenyl cyclopentyl alcohols 28a–d with sodium azide (NaN3) under acidic conditions to afford the azides 29a–d. Subsequent reduction of the azides 29a–d in the presence of lithium aluminum hydride (LiAlH4) gave the targeted amines 30a–d (Scheme 1). For the synthesis of the amines 30e–g, we exploited an alternative procedure, as previously reported by Tomashenko and co-workers,25 consisting of a one-step conversion of the nitriles 31a–c into the targeted amines 30e–g by reaction with 1,4-bis(bromomagnesio)butane, in situ generated from 1,4-dibromobutane and metallic magnesium (Mg) in anhydrous diethyl ether (Et2O), followed by the addition of titanium tetraisopropoxide Ti(iPrO)4 (Scheme 2). An alternative three-step procedure was exploited for the synthesis of amines 30h,i, starting from the phenylacetonitriles 32a,b, which were reacted with 1,4-dibromobutane in the presence of lithium bis(trimethylsilyl)amide (LiHMDS), followed by acid hydrolysis of the nitriles 33a,b to the corresponding carboxylic acids 34a,b under standard conditions (Scheme 3).26 Then, 34a was reacted with ethylchloroformate (ClCO2CH2CH3) in the presence of NaN3, followed by acidic hydrolysis, under thermal conditions, of the in situ generated isocyanate intermediate by Curtius rearrangement to afford the targeted amine 30h.27 Alternatively, 34b was reacted with thionylchloride (SOCl2) in the presence of NaN3, followed by Curtius rearrangement in the presence of BnOH which afforded the amine 30i, upon N-Cbz removal using triethylsilane (Et3SiH) and Pd/C under microwave (MW) conditions (Scheme 3).
Scheme 2. Synthesis of 30e–g.

Reagents and conditions: (a) Mg, Br(CH2)4Br, anhydrous Et2O, 40 °C; then, Ti(iPrO)4, anhydrous Et2O, rt, 24 h (20–26%).
The aldehydes 40a–e were prepared following a slightly modified five-step synthetic procedure, as previously reported for the preparation of compound 1e,22 which involved an olefination reaction between the activated intermediates 36a–f, obtained from the corresponding commercially available benzyl bromides 35a–e under standard conditions, and the appropriate cyclic amino ketones under Horner–Wadsworth–Emmons or Wittig reaction conditions to obtain the olefins 37a–e, which, upon hydrogenation reaction with Et3SiH and Pd/C and a reduction–oxidation procedure by the use of LiAlH4 and MnO2, were converted to the corresponding aldehydes 40a–e (Scheme 4). Alternatively, a chemo-selective reduction of the methylester functionality of intermediates 38a,b and 38d was used for the preparation of benzyl alcohols 39f–h, which, upon oxidation with MnO2, afforded the aldehydes 40f–h (Scheme 5). Same procedure was adopted for the preparation of compounds 40i,j and 40k–n (Schemes 6,7). Notably, for the synthesis of the aldehyde 40j, an alternative synthetic approach was exploited which involved a Pd cross-coupling reaction by the use of the commercially available phenylbromide 35i and tert-butyl 4-[(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methylidene]piperidine-1-carboxylate 42 that afforded the key olefin 37g in good yield (Scheme 6).
Scheme 6. Synthesis of 40i,j.
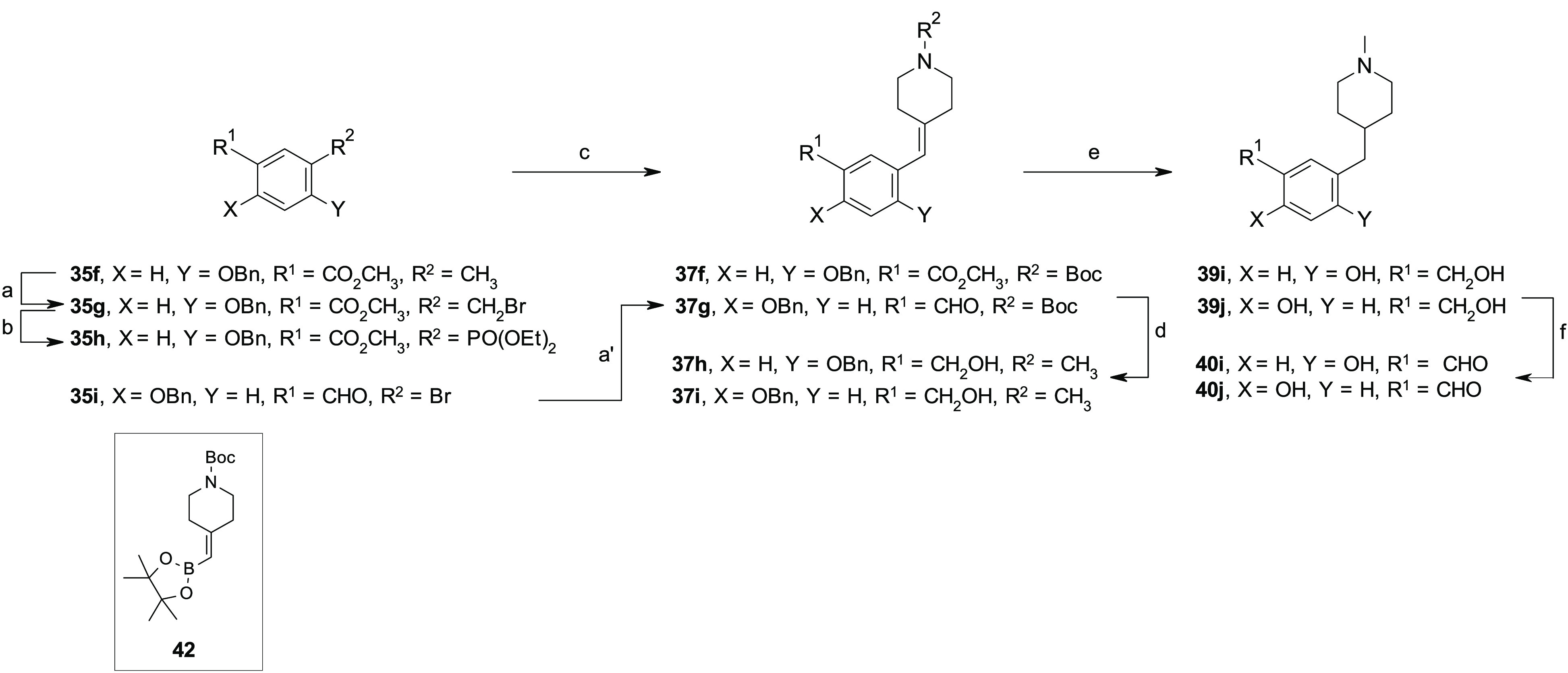
Reagents and conditions: for 37f: (a) NBS, AIBN, anhydrous CH3CN, 80 °C, 24 h; (b) P(OEt)3, anhydrous toluene, 120 °C, 20 h (20% over two steps); (c) t-BuOK, 1-Boc-4-piperidone, anhydrous THF, 0 °C to rt, (67%), for 37g: (a′) 42, Xphos, Pd2dba3, K3PO4, 1,4-dioxane/H2O, 100 °C, on (75%); (d) LiAlH4, anhydrous THF, 0 °C to rt, on; (e) Et3SiH, 10% Pd/C, MeOH, rt, 7 h; (f) MnO2, anhydrous CH2Cl2 or anhydrous CH2Cl2/iPrOH, rt, 8 h.
Results and Discussion
Our previous studies reported the identification of 1a as the first dual autophagy and REV-ERB inhibitor.21 Preliminary SAR studies around the 1a scaffold led to the discovery of new analogues 1b–e (Figure 2) with a good cytotoxicity profile against breast cancer BT-474 and selectivity against noncancerous human mammary epithelial cells (HMECs). In addition, 1e efficiently killed different cancer cells with diverse origins.22 Despite the enhanced potency and improved drug-likeness of 1e compared to the initial hit 1a (e.g., IC50 = 2.10 μM and IC50 = 30.14 μM, respectively, after 72 h treatment;22m-liver microsomal stability, t1/2 95 and 12 min, respectively), its moderate metabolic stability limits its use in in vivo studies. To address this issue, we performed a focused lead optimization strategy by exploring additional structural modifications on the three main regions of the scaffold of our class of molecules (regions A, B, and C of 2, Figure 2). The new synthesized compounds were first tested for their anticancer activity against BT-474 cells and toxicity against HMECs after 48 h treatment.
We started investigating modifications at the para-fluoro phenyl group of 1e with the aim of blocking or modifying potential metabolic “soft spots” present on the aromatic ring (region A) (Table 1).
Table 1. Cancer Cell Cytotoxicity of Compounds 1e–10 against Human Breast Cancer BT-474 and Noncancer HMEC Cells.


Concentration–response plots of indicated compounds cytotoxicity against human breast cancer BT-474 cells and nontumorigenic human mammary epithelial cells (HMECs) were used to calculate the concentration producing 50% cytotoxicity (CC50), as described in the Experimental Section. Shown as mean CC50 ± SEM (n ≥ 3).
For compounds showing a reduction in cell viability less than 50% of the vehicle treatment at the maximum concentration tested (30 μM), the CC50 value is indicated as >30 μM.
We were pleased to observe that, except for the phenolic derivative 6, the insertion of different substituents at the ortho-position of the para-fluoro phenyl ring (both electron-withdrawing and electron-donating groups, such as F, Cl, and CH3O) was tolerated (Table 1). Compounds 3–5 showed almost equipotent cytotoxicity activity on the BT-474 cells compared to the reference compound 1e. A similar trend was observed with compounds 7–10, bearing the same substituents at the meta-position of the para-fluoro phenyl ring, showing a slightly improved activity against BT-474 cells. Notably, the two chloro, fluoro regioisomers 4 and 8 also showed a double-digit μM toxic effect in HMECs.
We then continued the SAR exploration by modifying the scaffold with the insertion of different substituents on the phenyl ring at region B (Table 2).
Table 2. Cancer Cell Cytotoxicity of Compounds 11–16 against Human Breast Cancer BT-474 and Noncancer HMEC Cells.


Concentration–response plots of indicated compounds cytotoxicity against human breast cancer BT-474 cells and nontumorigenic human mammary epithelial cells (HMECs) were used to calculate the concentration producing 50% cytotoxicity (CC50), as described in the Experimental Section. Shown as mean CC50 ± SEM (n ≥ 3).
For compounds showing a reduction in cell viability less than 50% of the vehicle treatment at the maximum concentration tested (30 μM), the CC50 value is indicated as >30 μM.
In the specific, we first prepared analogues 11–13, bearing a F, OH, and Cl atom, respectively at the para-position with respect to the benzylic amine of 1e. These compounds were equipotent to 1e, except for the chloro analogue 13, which resulted to be more potent against BT-474 cells with undesired toxicity against HMECs (CC50 = 2.34 μM and CC50 = 15.38 μM, respectively). We applied the same strategy with the preparation of analogues 14 and 15, by blocking the ortho-position with respect to the benzylic amine on the phenyl ring of 1e at region B. Both compounds showed a similar profile, as reference 1e, and selectivity against BT-474 cells (Table 2). Interestingly, the di-F derivative 16, although showing improved cytotoxicity in cancer cells (CC50 = 3.11 μM), elicited an undesired effect in HMECs (CC50 = 20.39 μM).
We then moved our attention to the right-hand side (region C) of the scaffold by evaluating the effect of different substituents on the piperidine ring or alternative heterocycles (e.g., azetidine, spiro-bicycle) (Table 3), with the aim of improving the physicochemical and metabolic properties of our series.
Table 3. Cancer Cell Cytotoxicity of Compounds 17–21 against Human Breast Cancer BT-474 and Noncancer HMEC Cells.


Concentration– response plots of indicated compounds cytotoxicity against human breast cancer BT-474 cells and nontumorigenic human mammary epithelial cells (HMECs) were used to calculate the concentration producing 50% cytotoxicity (CC50), as described in the Experimental Section. Shown as mean CC50 ± SEM (n ≥ 3).
For compounds showing a reduction in cell viability less than 50% of the vehicle treatment at the maximum concentration tested (30 μM), the CC50 value is indicated as >30 μM.
A loss in potency was observed with the difluorinated analogue 19 and the azetidine 20 (CC50 = 21.86 and 13.39 μM, respectively), while the des-methylated piperidine 17 and the gem-dimethyl 18 resulted more and almost equipotent potent, respectively, compared to 1e (CC50 = 2.74 and 6.46 μM, respectively). The chemical exploration continued with the preparation of the spiro-bicycle 21 which resulted in a valid bioisosteric replacement of the piperidine ring, being slightly more potent (CC50 = 4.83 μM) than 1e, without showing toxicity against HMECs (Table 3).
With these results in hand, a comparative analysis of some selected analogues in terms of aqueous kinetic solubility (PBS, pH 7.4) and in vitro metabolism (t1/2, liver microsomes, in the presence of NADPH or UDPGA cofactors) was performed and reported in Tables 4 and 5.
Table 4. Aqueous Kinetic Solubility and in Vitro Metabolism of Compounds 1e–10.
| compd | solubility (μM)a(PBS, pH 7.4) | m-LMbt1/2 (min) [% at 60, 120 min] | h-LMct1/2 (min) [% at 60, 120 min] | m,h-LMdt1/2 (min) [% at 120 min] |
|---|---|---|---|---|
| 1ee | 235 | 95 [66%, 39%] | >120 [79%, 50%] | >120 [>95%] |
| 3 | 209 | <30 [19%, 5%] | ||
| 4 | 220 | 11 ± 1 | ||
| 5 | 247 | 98 [64%, 42%] | 92 [70%, 50%] | >120 [>95%] |
| 6 | 27 | 42 [34%, 13%] | >120 [>95%] | |
| 7 | 245 | 70 [56%, 31%] | >120 [75%, 53%] | >120 [>95%] |
| 8 | 249 | >60 [53%] | ||
| 9 | 219 | 91 [62%, 40%] | >120 [80%, 55%] | >120 [>95%] |
| 10 | >250 | >120 [80%, 63%] | >120 [84%, 71%] | >120 [>95%] |
Aqueous kinetic solubility in phosphate buffered saline (PBS). Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Mouse liver microsomes in the presence of NADPH cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Human liver microsomes in the presence of NADPH cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Mouse and human liver microsomes in the presence of UDPGA cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Rat liver microsomes in the presence of NADPH cofactor: t1/2 44 ± 4, dog liver microsomes in the presence of NADPH cofactor: t1/2 56 ± 1. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Table 5. Aqueous Kinetic Solubility and in Vitro Metabolism of Compounds 11–21.
| compd | solubility (μM)a (PBS, pH 7.4) | m-LMbt1/2 (min) [% at 60, 120 min] | h-LMct1/2 (min) [% at 60, 120 min] | m,h--LMdt1/2 (min) [% at 120 min] |
|---|---|---|---|---|
| 11 | >250 | 91 [60%, 39%] | >120 [84%, 71%] | >120 [95%] |
| 12 | >250 | 87 [61%, 43%] | >120 [83%, 63%] | >120 [95%] |
| 13 | 216 | 56 [51%, 29%] | ||
| 14 | 246 | 107 [68%, 46%] | >120 [75%, 53%] | >120 [95%] |
| 15 | 246 | 82 [79%, 61%] | >120 [74%, 54%] | >120 [95%] |
| 16 | 228 | 80 [58%, 34%] | >120 [76%, 52%] | >120 [95%] |
| 17 | 248 | >120 [85%, 71%] | >120 [80%, 62%] | >120 [95%] |
| 18 | 239 | >60 [58%] | ||
| 19 | 41 | <5 | ||
| 20 | >250 | >60 [63%] | ||
| 21e | 232 | 115 [70%, 49%] | >120 [82%, 66%] | >120 [95%] |
Aqueous kinetic solubility in phosphate buffered saline (PBS). Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Mouse liver microsomes in the presence of NADPH cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Hhuman liver microsomes in the presence of NADPH cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Mouse and human liver microsomes in the presence of UDPGA cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Rat liver microsomes in the presence of NADPH cofactor: t1/2 > 60 min, 64% compound remaining, dog liver microsomes in the presence of NADPH cofactor: t1/2 53 ± 4. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
We focused the in vitro ADME compound profiling on phase I oxidative and phase II conjugative metabolic processes in liver microsomes, considering that our class of molecules does not contain groups that may be susceptible to plasma enzyme hydrolysis and/or chemical instability at certain pH values. In general, we observed that our compounds showed high aqueous kinetic solubility, except for compound 6 (Table 4). Notably, the insertion of different substituents both in ortho- and meta- positions of the para-fluoro phenyl ring (region A) modulated the metabolic stability of our compounds both in mouse and human liver microsomes, in the presence of the NADPH cofactor. In general, meta,para- disubstituted phenyl cyclopentyl analogues showed a better liver microsomal stability profile compared to the corresponding ortho,para- disubstituted phenyl cyclopentyl analogues. As a representative example, compound 7 showed improved metabolic stability compared to 3. A similar trend was observed for analogues 8 and 10 in comparison to 4 and 6. On the contrary, compound 5 showed a similar metabolic profile compared to the corresponding meta,para- disubstituted phenyl cyclopentyl analogue 9 and our reference 1e (Table 4). Selected compounds were also evaluated in mouse and human liver microsomal stability assay in the presence of the UDPGA cofactor, showing t1/2 > 120 min with compound remaining >95% in both species, as reported in Table 4.
We then continued with the structure–property relationship (SPR) evaluation of some compounds from the chemical exploration of regions B and C, as reported in Table 5. In general, all the targeted compounds showed high solubility in aqueous media, except for the difluoro piperidine 19, which also suffered from a high metabolic instability in mouse liver microsomes in the presence of NADPH cofactor (Table 5). Analogues bearing F or OH groups on the central phenyl ring at region B showed a better overall metabolic profile compared to 1e, both in mouse and human microsomes, as for example, the fluorinated analogues 11 and 14. On the other hand, the insertion of a Cl atom, as for 13, did not give any beneficial effect in terms of metabolic stability, showing a half-life (t1/2) of 56 min in mouse liver microsomes (Table 5). Some important differences were observed with the chemical exploration of region C (Table 5). Notably, while we were pleased to observe that the des-methylated piperidine 17 and the spiro-bicycle 21 showed improved metabolic stability compared to 1e both in mouse and human microsomes, the gem-dimethyl piperidine 18 and the azetidine 20 were less stable with an t1/2 of almost 1 h in mouse microsomes in the presence of NADPH cofactor.
With these results in hand, we were finally interested in studying the effect of the combinations of some representative moieties identified during our SAR/SPR explorations of the three main regions A, B, and C. With this aim, we prepared and evaluated analogues 22–26 as reported in Table 6.
Table 6. Cancer Cell Cytotoxicity of Compounds 22–26 against Human Breast Cancer BT-474 and Noncancer HMEC Cells.


Concentration–response plots of indicated compounds cytotoxicity against human breast cancer BT-474 cells and nontumorigenic human mammary epithelial cells (HMECs) were used to calculate the concentration producing 50% cytotoxicity (CC50), as described in the Experimental Section. Shown as mean CC50 ± SEM (n ≥ 3).
For compounds showing a reduction in cell viability less than 50% of the vehicle treatment at the maximum concentration tested (30 μM), the CC50 value is indicated as >30 μM.
We were pleased to observe that, except for analogue 22, all the targeted compounds were more potent than the reference 1e against the BT-474 cells, with compounds 25 and 26 showing an undesired toxicity in HMECs. The new synthesized analogues were evaluated in kinetic solubility and liver microsomal stability assays (Table 7). All the targeted compounds 22–26 showed high solubility in aqueous media. Moreover, analogues 24 and 25 resulted in an improved microsomal stability profile with t1/2 > 2 h, both in mouse and human microsomes in the presence of NADPH and UDPGA cofactors. On the other hand, the spiro-cycle 26 showed an overall metabolic profile similar to the corresponding piperidine 7, with a slightly improved stability in human microsomes in the presence of NADPH cofactor (Table 7).
Table 7. Aqueous Kinetic Solubility and in Vitro Metabolism of Some Selected Compounds.
| compd | solubility (μM)a (PBS, pH 7.4) | m-LMbt1/2 (min) [% at 60, 120 min] | h-LMct1/2 (min) [% at 60, 120 min] | m,h-LMdt1/2 (min) [% at 120 min] |
|---|---|---|---|---|
| 22 | >250 | 69 [55%, 31%] | >120 [75%, 58%] | >120 [95%] |
| 23 | 231 | 58 [48%, 21%] | >120 [77%, 56%] | |
| 24e | >250 | >120 [84%, 72%] | >120 [81%, 68%] | >120 [95%] |
| 25e | 245 | >120 [82%, 74%] | >120 [75%, 54%] | >120 [95%] |
| 26 | 226 | 75 [56%, 32%] | >120 [84%, 65%] |
Aqueous kinetic solubility in phosphate buffered saline (PBS). Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Mouse liver microsomes in the presence of NADPH cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Human liver microsomes in the presence of NADPH cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Mouse and human liver microsomes in the presence of UDPGA cofactor. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Rat liver microsomes in the presence of NADPH cofactor: t1/2 > 60 min, 98% and 75% compound remaining for 24 and 25, respectively, dog liver microsomes in the presence of NADPH cofactor: t1/2 > 60 min, 89% and 83% compound remaining for 24 and 25, respectively. Values are reported as the mean of at least two independent experiments, performed in two technical replicates.
Because of the overall property profiles, compounds 21, 24, and 25 were profiled in metabolic stability assays using rat and dog liver microsomes in the presence of NADPH cofactor (Tables 5 and 7). The experiments revealed that compound 24 was more stable than compounds 21 and 25, with t1/2 > 60 min in both species with higher percentage of compound remaining after 1 h incubation (t1/2 > 60 min, 98% and 89% compound remaining in rat and dog microsomes, respectively). More pronounced differences were observed by comparing 24 to 1e in rat and dog liver microsomal stability properties (Tables 4 and 7).
Because of the overall property profile, compound 24 was selected for additional studies.
We previously showed that the cytotoxicity of our class of compounds is mediated by the induction of apoptosis, as indicated by caspase activation and enhanced cleaved PARP levels in BT-474 cells treated with 1a.21 Indicating that 24 preserved this mechanism of action of cell death, BT-474 treated with different concentrations of 24 showed a dose-dependent induction of both caspase-activity and cleaved PARP protein levels (Supporting Information, Figure S1).
We then aimed to compare 24 biological activity with the lead compound 1e. First, we evaluated the potency of 24 and 1e in reducing the viability of different human cancer cells with diverse origins. Specifically, we tested the in vitro anticancer activity of compounds against an additional breast cancer cell line (MDA-MD-231), and cells deriving from melanoma (A375), uveal melanoma (OMM-1 and UPMM-2), pancreatic (CAPAN-2), hepatocellular carcinoma (HEPG2), and colorectal (HT-29 and HCT116) tumors (Table 8).
Table 8. In Vtro Anticancer Activity of Compounds 1e and 24 against Cancer Cell Lines with Diverse Origins.
| cell line | origin | 1ea,b CC50 (μM) ± SEM | 24a,b CC50 (μM) ± SEM | P valuec |
|---|---|---|---|---|
| BT-474 | breast ductal carcinoma | 5.93 ± 0.19 | 2.31 ± 0.11 | <0.001 |
| MDA-MB-231 | breast adenocarcinoma | 7.42 ± 0.15 | 2.97 ± 0.07 | <0.001 |
| A375 | melanoma | 5.67 ± 0.11 | 2.56 ± 0.08 | <0.001 |
| OMM-1 | uveal melanoma | 2.28 ± 0.11 | 1.14 ± 0.24 | <0.05 |
| UPMM-2 | uveal melanoma | 1.91 ± 0.12 | 1.09 ± 0.16 | <0.05 |
| CAPAN-2 | pancreatic adenocarcinoma | >30 | 6.86 ± 0.65 | not applicable |
| HEPG2 | liver hepatocellular carcinoma | 2.80 ± 0.27 | 1.49 ± 0.16 | <0.01 |
| HT-29 | colorectal adenocarcinoma | 18.88 ± 0.36 | 4.74 ± 0.77 | <0.01 |
| HCT116 | colorectal carcinoma | 13.51 ± 1.09 | 4.56 ± 1.06 | <0.01 |
Concentration– response plots of 1e and 24 cytotoxicity against the indicated cancer cell lines were used to calculate the concentration producing 50% cytotoxicity (CC50), as described in the Experimental Section. Shown as mean CC50 ± SEM (n ≥ 6).
For compounds showing a reduction in cell viability equal or greater than 50% at the maximum concentration tested (30 μM), the CC50 value is indicated as >30 μM.
P values were calculated with a two-tailed unpaired t test comparison between CC50 of compound 1e and 24.
The result of this analysis indicated that 24 has significantly improved in vitro anticancer activity compared to 1e against all the cancer cells tested. Of note, compound 24 showed a comparable cytotoxicity against the ERBB2-positive BT-474 and the triple-negative MDA-MB-231 breast cancer cells, which is in line with our previous analysis indicating that the anticancer activity of this class of compounds is independent of ERBB2 expression.21 Remarkably, uveal melanoma cells showed a high sensitivity to compound toxicity. This result is particularly interesting considering that the cells adopted in our analysis present a loss of chromosome 3. Uveal melanoma is the most common primary intraocular malignant tumor in adults and has a predilection for hematogenous dissemination to the liver. Total or partial deletions of chromosome 3 in uveal melanoma are associated with a high incidence of mortality mainly due to liver metastasis, for which there is no effective treatment.28,29
We then compared the ability of 1e and 24 to inhibit REV-ERB and autophagy. For evaluating the effect of the compounds on REV-ERB transcriptional repression activity, we adopted a luciferase cell-based assay consisting of a reporter vector expressing a Cypridina luciferase gene under the control of a minimal SV40 promoter containing a REV-ERB responsive element (RevRE) (Figure 3A).21 In addition, a reporter bearing a mutated RevRE that is not able to bind REV-ERB protein was used for assessing the specificity of the luminescence response (Figure 3B).
Figure 3.

Comparison of the REV-ERB inhibitory activity of compounds 1e and 24. (A) Cotransfection assay in 1e- and 24-treated HEK-293 cells with REV-ERBβ and a luciferase REV-ERB-responsive reporter driven by two repetitions of a REV-ERB element (RevRE) consensus. A schematic representation of the reporters is shown at the top. Data are expressed as fold increase of luciferase activity versus vehicle (DMSO). Shown as mean ± SEM, n = 6. EC50 values for 1e and 24 antagonism versus REV-ERBβ were 3.61 ± 0.82 μM and 3.78 ± 0.94 μM, respectively. (B) Cotransfection assay in 1e- and 24-treated HEK-293 cells with REV-ERBβ and a REV-ERB-unresponsive reporter bearing a mutated RevRE that is not recognized by REV-ERB DNA binding domain. Shown as mean ± SEM, n = 6.
Indicating that 24 preserved the REV-ERB inhibitory activity, this compound activated the RevRE reporter in a concentration-dependent manner with an average EC50 value comparable to the one of compound 1e (3.61 ± 0.82 μM and 3.78 ± 0.94 μM, respectively) (Figure 3A), while it had no effects on the expression of the mutated RevRE (Figure 3B). To further validate the REV-ERB inhibitory activity of 24, we tested its effect on the expression of a REV-ERB target gene, BMAL1, in BT-474 knocked down for REV-ERBβ, which we previously demonstrated to be the predominantly functional REV-ERB variant expressed in this cell line.21 In line with our luciferase reporter assay, REV-ERBβ silencing abolished the 24-mediated induction of BMAL1 expression (Supporting Information, Figure S2).
We then compared the effect of the two compounds on the inhibition of autophagy. To this aim, we generated a fluorescent reporter cell-based assay in which melanoma A375 cells stably express a chimeric autophagy marker LC3B fused with an enhanced green fluorescence protein (GFP-LC3). During autophagosome maturation, a lipid conjugated version of LC3 protein (LC3-II) is recruited on the membrane of autophagosomes.3 Accordingly, GFP-LC3-containing autophagosomes will appear as fluorescent perinuclear dots, differing from a diffuse fluorescence signal deriving from the unconjugated GFP-LC3 protein (Figure 4A).30
Figure 4.

Comparison of the autophagy inhibitory activity of compounds 1e and 24. (A) Representative fluorescent images of melanoma A375 cells stably expressing a chimeric autophagy marker LC3B fused with a green fluorescence protein (GFP-LC3) treated with 5 μM of 1e, 24, or vehicle (DMSO) for 24 h. Nuclei were stained with Hoechst 33342 (blue) after fixation with 4% paraformaldehyde (PFA). White scale bar = 20 μm. (B) Melanoma A375 cells expressing GFP-LC3 reporter were treated with the indicated concentrations of 1e or 24 for 24 h. After fixation with 4% PFA and nuclear staining, fluorescent signals were acquired with a fluorescent microscope and images were used for quantifying GFP-LC3-positive fluorescent dots per cell as described in the Experimental Section. Shown as average number of fluorescent dots per cell ± SEM, n = 10. *P < 0.05, **P < 0.01, and ***P < 0.001, 24 vs 1e (two-way ANOVA with Bonferroni’s multiple comparison test). (C) Immunoblot analysis of total, cytosolic and organelle-enriched fractions from cells treated 24 h with vehicle (DMSO), 2.5 μM of 1e or 2.5 μM of 24. Lysosomal LAMP1 was used to confirm the enrichment in autophagolysosomes in organelle-enriched fractions, while cytoplasmic GAPDH protein was adopted to evaluate potential cytosolic contaminations. Immunoblot with antibodies against the autophagic receptors, SQSTM1 (also known as p62), and the autophagy marker LC3 were used to evaluate the effects of the treatments on autophagy. (D) Quantification of immunoblot analysis from organelle-enriched fractions of cells treated as in C. Relative LC3-I and LC3-II expression were calculated normalizing the optical density of LC3-II signals with the optical density of the Ponceau staining. Shown as mean ± SEM, n = 3. *P < 0.05 and **P < 0.01, 24 vs 1e (one-way ANOVA with Tukey’s multiple comparison test).
We thus treated GFP-LC3 A375 cells with different concentrations of 1e and 24 and evaluated the average number of fluorescent perinuclear dots per cell (Figure 4B). Consistent with the ability of both compounds to inhibit autophagy at the late stage, cells treated with 1e or 24 showed a dose-dependent accumulation of GFP-LC3-positive autophagosomes. However, compound 24 generated a significantly higher increase in fluorescent dots compared with 1e at all the doses tested (Figure 4B), indicating an improved autophagy inhibitory activity of this analogue.
To confirm this result with an independent approach, we prepared total protein extracts, cytosolic and organelle-enriched fractions from A375 cells treated with an equal dose of 1e or 24 (2.5 μM). Consistent with our fluorescence microscopy analysis, immunoblot with a specific LC3B antibody showed higher increasing in LC3-II protein levels in total and autophagosome-containing organelle-enriched fractions from cells treated with 24 compared to 1e-treated cells (Figure 4C,D). Furthermore, the treatment with 24 resulted in increased protein levels of the autophagic receptor SQSTM1 (also known as p62) in organelle-enriched fraction compared to 1e. SQSTM1 protein is incorporated into the completed autophagosome and is degraded in autolysosomes, thus serving as an index of autophagic degradation.30 Accordingly, the higher accumulation of SQSTM1 in organelle-enriched fraction from cells treated with 24 compared to 1e-treated cells indicates that this analogue is more potent in inhibiting the autophagic flux than the lead.
Collectively, our analyses indicate that compound 24 retains the REV-ERB inhibitory activity of 1e and improves the ability of blocking autophagy.
We then evaluated the plasma concentration–time profile of 24 following a single intraperitoneal (ip) dose of 10 mg/kg in female CD1 mice. This compound presented a biphasic elimination profile with a long half-life (9.67 h) and a large volume of distribution (106.07 L/kg) estimated through a noncompartmental analysis (NCA) of the plasma concentrations. Of note, a similar profile has also been reported for CQ, and it could represent the general pharmacokinetic (PK) behavior of lysosomotropic agents.31 The Cmax of 24 was 280.23 μg/L and resulted about 3-fold higher than the Cmax of 1e assessed in similar conditions.
Based on the observed biphasic elimination profiles, we used the Akaike Information Criterion (AIC) for comparing how one- and two-compartment models with a different weight fit to the plasma concentrations of 24 and 1e. A two-compartment model provided the best fit to the data, and it indicated that 24 has a half-life of distribution (t1/2α) 3.6-fold higher than 1e (12.78 and 46.40 min, respectively), which is consistent with the improved stability of 24 compared to 1e showed in mouse liver microsomal studies.
We then evaluated the tolerability of 24 in mice. Except for certain models of pancreatic cancer,32 the use of CQ as a single anticancer agent does not impair tumor growth in many animal models. In addition, doses of CQ lower than 50 mg/kg (ip daily) are completely ineffective in xenograft models.33 This dose of CQ corresponds to 138 μmol/kg. Of note, a similar dose of Lys05 (Figure 1), a CQ derivative compound showing a higher autophagy inhibitory activity than CQ, has been reported to not be tolerated by animals, producing arched backs and lethargy after 2 days of an ip treatment.34
Therefore, we first tested the tolerability of 24 in CD1 female mice treated with daily ip injections at the dose of 138 μmol/kg of compound, corresponding to 62 mg/kg. In contrast to the reported observation on Lys05, we did not observe clear signs of arched backs and lethargy in mice treated with this dose of 24. However, after 2 days of treatment, animal body weight was significantly reduced (data not shown). We thus evaluated the tolerability in mice treated with 138 μmol/kg (62 mg/kg) every other day or with 69 μmol/kg (31 mg/kg) daily over an 8-day period. Both treatments displayed only a minor reduction in body weight after 8 days (≤5%; Supporting Information, Figure S3). To identify a treatment schedule producing negligible effects on the weight of mice for avoiding potential confounding factors in our in vivo efficacy study, we thus evaluated the tolerability of 24 in mice treated with 69 μmol/kg (31 mg/kg) of compound dosed for 3 days of daily treatment with 2 days off treatment (3/5 d schedule) (Figure 5A). This schedule was well tolerated by animals over a 15-day period and treated mice did not show significant differences in body weight, water, and food intake compared to saline-treated animals (Figure 5B–D). Therefore, this treatment schedule was adopted for the in vivo efficacy study of 24.
Figure 5.

Tolerability of 24 in female CD1 mice. (A) Female CD1 mice were treated with 69 μmol/kg (31 mg/kg) of 24 dosed for 3 days of daily treatment with 2 days off treatment (3/5 d schedule). (B) Animal body weight, (C) food intake and (D) water intake were monitored daily over a 15-day period. The weight of mice the day before starting the treatment was set as 100% and used for calculating the relative body weight (%) and is shown as mean ± SEM, n = 3.
To this aim, we decided to take advantage of our GFP-A375 reporter cells. In fact, the expression of the exogenous LC3 chimeric protein permits the specific evaluation of tumor autophagy by immunoblotting from tumor protein extracts. We thus tested whether an acute treatment of 24 could affect autophagy and circadian gene expression in A375 flank xenografts. Accordingly, female NMRI-Foxn1nu mice were subcutaneously injected in the flank with reporter A375 cells and xenografts matched for tumor size received an ip injection of 24 (30 mg/kg) or saline for 2 days. After 48 h of treatment, mice were euthanized, and tumors were processed for immunoblot and qRT-PCR analyses.
Indicating that 24 affected tumor autophagy in vivo, 24-treated tumors showed increased GFP-LC3-II levels and LC3-II/LC3-I ratio compared with saline treated tumors (Figure 6A,B). In addition, RNA samples from tumors treated with 24 showed a significantly higher expression of the REV-ERB target genes, BMAL1 and PEPCK, compared with control tumors (Figure 6C).
Figure 6.

In vivo anticancer activity of 24 in the A375 xenograft tumor model. (A) Female NMRI-Foxn1nu mice were subcutaneously injected in the flank with reporter A375 cells and xenografts matched for tumor size received an ip injection of 24 (30 mg/kg) or saline for 2 days. After 48 h from the first injection, mice were euthanized, and tumors were collected and processed for immunoblot with an anti-LC3B antibody to evaluate the levels of GFP-LC3-I and GFP-LC3-II proteins. GAPDH was used as a loading control. (B) Quantification of immunoblot analysis from tumors collected as in A. GFP-LC3-I and GFP-LC3-II expression were normalized by GAPDH levels and are shown as mean ± SEM, n = 3, **P < 0.01 and ***P < 0.001, 24 versus saline (unpaired two-tail t test). (C) Expression of REV-ERB-regulated BMAL1 and PEPCK genes in tumors collected as in A. The HPRT1 gene was used as representative of a REV-ERB independent gene. Shown as relative expression normalized by GAPDH. The value in the vehicle sample was set to 1. Reported as mean ± SEM, n = 3, *P < 0.05; **P < 0.01 24 versus saline, unpaired two-tail t test. (D) Female NMRI-Foxn1nu mice bearing tumors matching for tumor volume were treated with 24 (30 mg/kg, ip) or saline dosed for 3 days of daily treatment with 2 days off treatment (3/5 d schedule). Tumor growth was monitored by digital caliper until several tumors in the saline-group reached a volume close to the defined human end point (day 15). Shown as mean tumor volume mean ± SEM, n = 8. ***P < 0.001, 24 versus saline, two-way ANOVA with Bonferroni posttest analysis. (E) On day 15, tumors were excised and tumor volumes were calculated according to the formula [(W × D × L)/2]. Shown as mean tumor volume mean ± SEM, n = 8. *P < 0.05, two-tailed Mann–Whitney t test. (F) The weight of excised tumors is shown as mean ± SEM, n = 8. *P < 0.05, two-tailed Mann–Whitney t test. (G) Animal body weight was monitored daily. The weight of mice the day before starting the treatment was set as 100% and used for calculating the relative body weight (%). Shown as mean ± SEM, n = 8.
We next evaluated the effects of 24 on tumor growth in A375 xenografts. Female NMRI-Foxn1nu mice bearing tumors were divided in two groups matching for tumor volume and treated with 24 (30 mg/kg, ip) or saline according to the 3/5d treatment schedule. Tumor growth was monitored by digital caliper until several tumors in the saline-group reached a volume close to the defined human end point (day 15, Figure 6D). Animals were sacrificed and tumors were excised and weighed. In addition, length, height, and depth of excised tumors were measured to obtain a more precise assessment of tumor volumes (Figure 6E).
Tumor growth curves indicated that the treatment with compound 24 produced a significant reduction of tumor growth compared to saline (Figure 6D; tumor growth inhibition = 55%).
On day 15, 24-treated excised tumors showed a significant 60% reduction in the average tumor volume compared with saline (307.7 vs 761.9 mm3; P = 0.0127 two-tailed Mann–Whitney t test, Figure 6E). In addition, 24-treated tumors showed a 56% significant reduction in the average tumor weight compared with controls (0.365 vs 0.836 g; P = 0.0127 two-tailed Mann–Whitney t test, Figure 6F). Consistent with our tolerability analysis in female CD1 mice, 24 treatment was tolerated by NMRI-Foxn1nu mice bearing A375 xenografts having negligible effects on body weight (Figure 6G).
Conclusions
Growing evidence on the role of autophagy in affecting cancer progression and response to chemotherapy has been making the pharmacological inhibition of this process a promising strategy for the development of novel anticancer therapies. We previously showed that when the autophagy flux is blocked in combination with the inhibition of the circadian nuclear receptor, REV-ERB, the autophagy-mediated cell death of cancer cells is augmented. We also identified the first class of dual REV-ERB and autophagy inhibitors showing a greater in vitro anticancer activity than the single autophagy inhibitor, CQ, against different cancer cells with diverse origins. In this study, we described the lead optimization strategy around the early lead 1e that resulted in the identification of analogues with improved biological and drug-like profiles, such as compounds 21, 24, and 25 (please compare Table 1 with Tables 3 and 6, and Table 4 with Tables 5 and 7).
The most promising candidate, 24, shows optimal physicochemical and metabolic stability properties in liver microsomes from different species (Table 7).
Compound 24 presents an improved ability to block the autophagic flux (Figure 4) and it has a greater in vitro anticancer activity than 1e against different cancer cells with diverse origins (Table 8).
Our data indicate that compound 24 inhibits REV-ERB transcriptional activity in the low μM range; this potency, on the one hand is comparable to that of 1e (Figure 3), and on the other hand is similar to those of the single REV-ERB inhibitors SR8278 and GSK1362 (Figure 1).35,36 For example, in luciferase transcriptional reporter assays GSK1362’s EC50 is 2.5 μM36 compared to 24’s EC50 of 3.6 μM (Figure 3). However, both SR8278 and GSK1362 were reported to have unfavorable PK profiles that limit their usage in vivo.20 In marked contrast, 24 shows desirable PK profile in mice following ip administration and it can block tumor autophagy and REV-ERB transcriptional activity in mice bearing melanoma A375 tumors (Figure 6).
Moreover, 24 shows a significant anticancer activity as a single agent in melanoma xenograft mouse model at a dose that was completely tolerated by both CD1 mice and NMRI-Foxn1nu mice bearing A375 xenografts (Figures 5 and 6). Nonetheless, future studies will focus on investigating additional treatment regimens for improving the in vivo anticancer activity of our compound. In this respect, it is worthy notice that because 24 targets a nuclear receptor with a different circadian expression pattern in cancerous and noncancerous cells,37 this compound represents an optimal candidate for evaluating the efficacy of a chronotherapeutic approach to maximize the efficacy and minimize possible adverse effects.38,39
In addition, because both the circadian cycle and autophagy have been related to cancer drug resistance,30,40−4224 constitutes a valuable experimental candidate in dedicated preclinical studies for identifying improved drug combination therapies.
Experimental Section
Chemistry
Synthetic Materials and Methods
Solvents and reagents were obtained from commercial suppliers and were used without further purification. Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash Rf 200) with prepacked SiO2 columns of different sizes (from 4 g to 80 g). Mixtures of increasing polarity of Cy and EtOAc or CH2Cl2 and MeOH were used as eluents. TLC analyses were performed using a precoated TLC sheets ALUGRAM Xtra SIL G/UV254 from Macherrey-Nagel. The visualization was done by UV light (254 nm) or staining with KMnO4 or H3[Mo12PO40]·12H2O. NMR experiments of all the intermediates and final compounds were run on a Bruker Avance III 400 system (400.13 MHz for 1H NMR and 100.62 MHz for 13C NMR) equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K using deuterated dimethyl sulfoxide (DMSO-d6) or deuterated chloroform (CDCl3) as solvent. Chemical shifts for 1H and 13C NMR spectra were recorded in parts per million using the residual nondeuterated solvent as the internal standard (for DMSO-d6: 2.50 ppm, 1H NMR, 39.52 ppm; 13C NMR, for CDCl3, 7.26 ppml 1H and 77.16 ppm, 13C NMR). Data are reported as follows: chemical shift (ppm), multiplicity (indicated as bs, broad singlet; s, singlet; d, doublet; t, triplet; q, quartet; p, quintet, m, multiplet, and combinations thereof), coupling constants (J) in hertz (Hz), and integrated intensity. Quantitative 1H NMR analyses of the freshly prepared 10 mM DMSO-d6 stock solutions (used for biological screenings) of the final compounds were performed using the PULCON method (Pulse Length Based Concentration determination, Bruker software, topspin 3.0. Refs: (a) ref (46) and (b) ref (47). UPLC/MS analyses of all the intermediates and final compounds were performed on Waters Acquity UPLC/MS system consisting of a single quadrupole detector (SQD) mass spectrometer (MS) equipped with an electrospray ionization (ESI) interface and a photodiode array detector (PDA). The PDA range was 210–400 nm. Analyses were performed on an Acquity UPLC BEH C18 column (50 mm × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 mm × 2.1 mm ID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN/H2O (95:5) at pH 5 (B). ESI in both positive and negative modes was used in the mass scan range of 100–650 Da. Analyses were performed with methods A, B, C, or D. Method A: gradient 0–100% B over 3.0 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method B: gradient 5–100% B over 3.0 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method C: gradient 50–100% B over 3.0 min. Flow rate 0.5 mL min–1. Temperature 40 °C. Method D: gradient 80–100% B over 3.0 min. Flow rate 0.5 mL min–1. Temperature 40 °C. UPLC/MS analyses of the final compounds were performed using freshly prepared 10 mM DMSO-d6 stock solutions (used for biological screenings), diluted 20-fold or 100-fold in MeCN/H2O (1:1) and directly analyzed. The analysis was performed on an Acquity UPLC BEH C18 column (100 mm × 2.1 mm ID, particle size: 1.7 μm) with a VanGuard BEH C18 precolumn (5 mm × 2.1 mm ID, particle size: 1.7 μm) at 40 °C using 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5 (B) as mobile phase at 0.5 mL/min. Method E: gradient 10–90% B over 6.0 min. Flow rate 0.5 mL min–1. Temperature 40 °C. The detection wavelength (λ) was set at 215 nm for relative purity determination. Rt (min) of the final compounds under method E UPLC/MS analytical conditions are reported in Supporting Information, Table S2. Accurate mass measurements were performed on a SCIEX TripleTOF high-resolution LC-MS using a Waters UPLC Acquity chromatographic system (Waters, Milford, MA, USA) coupled to a TripleTOF 5600+ mass apectrometer (Sciex, Warrington, UK) equipped with a NanoSpray III Ion source. The analyses were run on an Acquity UPLC BEH C18 column (50 mm × 2.1 mm ID, particle size 1.7 μm), using H2O + 0.1% HCOOH (A) and CH3CN + 0.1% HCOOH as mobile phase. All final compounds displayed ≥95% purity as determined by NMR and UPLC/MS analysis.
General Procedure for Organometallic Nucleophilic Addition Reaction (Procedure A)
Method A: To a solution of the appropriate aryl bromide (1.0 equiv) in anhydrous THF (0.5 M) at −78 °C under N2 atmosphere, a solution of n-BuLi (2.5 M in hexane, 1.0 equiv) was added dropwise. The reaction mixture was stirred for 1 h at the same temperature, and then the cyclopentanone (1.3 equiv) was added. The reaction mixture was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The crude was used in the next step without further purification. Method B: To a solution of cyclopentanone (1.0 equiv) in anhydrous THF (5.0 M), the appropriate Grignard reagent (1.2 equiv) was added dropwise at 0 °C under N2 atmosphere. The reaction mixture was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layers were combined, washed with brine, dried over Na2SO4, filtered. and the solvent was evaporated under reduced pressure. The crude was used in the next step without further purification.
General Procedure for Nucleophilic Substitution Reaction (SN1) (Procedure B)
To a solution of the appropriate alcohol (1.0 equiv) in anhydrous CH2Cl2 (0.4 M), NaN3 (2.2 equiv) was added under N2 atmosphere. The reaction was stirred at −5 °C, and then a solution of TFA (8.2 equiv) in anhydrous CH2Cl2 was slowly added dropwise. The resulting suspension was stirred at 0 °C for 1 h. To the cold solution, distilled H2O was added dropwise, followed by the addition of 1:1 mixture of distilled H2O and 28% aqueous NH4OH solution dropwise. After 30 min, the reaction mixture was extracted with CH2Cl2, and the organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by column chromatography, eluting with Cy.
General Procedure for Azide Reduction Reaction (Procedure C)
To a solution of the appropriate azide (1.0 equiv) in anhydrous THF (0.25 M), LiAlH4 (2.0 M in THF, 1.0 equiv) was added dropwise at 0 °C under N2 atmosphere. The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The reaction was quenched with a solution of NaOH (1N) and stirred for 30 min. The mixture was extracted with CH2Cl2, the organic layers were combined, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was used in the next step without further purification.
General Procedure for the Synthesis of 1-Substituted-Phenyl Cyclopentylamines (Procedure D)
1,4-Dibromobutane (2.0 equiv) dissolved in anhydrous Et2O (0.4 M) was added dropwise to Mg turnings (4.4 equiv) covered with 1 mL of the dibromide solution at 40 °C. After the formation of the Grignard reagent, a solution of the appropriate aryl nitrile in anhydrous Et2O (1.0 M) was added at rt. After stirring for an additional 30 min, a solution of Ti(OiPr)4 in anhydrous Et2O or THF (2.0 M) was added dropwise. The reaction was stirred at rt for 24 h, and then 10% aqueous solution of NaOH was added. The mixture was filtered and the filtrate was extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc as indicated in each case.
General Procedure for Cyclization Reaction (Procedure E)
To a solution of 1,4-dibromobutane (1.5 equiv) and the appropriate benzyl nitrile in anhydrous THF (0.15 M), LiHMDS (1.0 M in THF, 1.2 equiv) was added dropwise at 0 °C. The reaction was stirred at rt for 2 h. Then, LiHMDS (1.0 M in THF, 1.2 equiv) was added dropwise at 0 °C. The reaction mixture was warmed up to rt and stirred on. The reaction was quenched with brine and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and the solvent was concentrated under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc as indicated in each case.
General Procedure for Hydrolysis Reaction (Procedure F)
To the appropriate nitrile (1.0 equiv) H2O and H2SO4 (ratio 1:1, 0.7 M) was added. The reaction was stirred at 120 °C on in a sealed tube. The mixture was poured onto ice-H2O and then extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and the solvent was concentrated under reduced pressure. The crude was used in the next step without further purification.
General Procedure for the Synthesis of Phosphonium Salts (Procedure G)
To a solution of the appropriate alkyl bromide (1.0 equiv) in anhydrous toluene (0.2 M) was added PPh3 (1.0 equiv) under N2 atmosphere. The reaction was stirred at reflux. The reaction was cooled to rt, and the solid was filtered off and washed with cold toluene. The solid was used in the next step without further purification.
General Procedure for the Synthesis of Phosphonates (Michaelis–Arbuzov Reaction) (Procedure H)
To a solution of the appropriate alkyl bromide (1.0 equiv) in anhydrous toluene (0.8 M) was added P(OEt)3 (3.0 equiv). The reaction was stirred at reflux in a sealed tube. The reaction was cooled to rt, and the solvent was evaporated under reduced pressure. The crude was purified by column chromatography eluting with Cy/EtOAc or used as crude, in the next step without further purification, as indicated in each case.
General Procedure for Wittig Reaction (Procedure I)
To a suspension of the appropriate phosphonium salt (1.0 equiv) in anhydrous THF (0.3 M), LiHMDS (1.0 M in THF, 1.5 equiv) was added dropwise at 0 °C under N2 atmosphere. After 15 min, a solution of the appropriate ketone (1.5 equiv) in anhydrous THF (1.0 M) was added dropwise. The reaction was stirred at rt or at 80 °C, as indicated in each case. The reaction mixture was quenched with H2O and extracted with EtOAc. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc as indicated in each case.
General Procedure for Horner–Wadsworth–Emmons Reaction (Procedure J)
Method A: To a solution of the appropriate phosphonate (1.0 equiv) in anhydrous THF (0.3 M), LiHMDS (1.0 M in THF, 1.2 equiv) was added dropwise at −40 °C under N2 atmosphere. After 15 min, a solution of the appropriate ketone in anhydrous THF (1.0 M) was added dropwise. The reaction was stirred at rt. The reaction mixture were quenched with H2O and extracted with EtOAc. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc as indicated in each case. Method B: To a solution of the appropriate phosphonate (1.0 equiv) in anhydrous THF (0.3 M), KOtBu (1 or 3 equiv, as indicated in each case) was added dropwise at 0 °C under N2 atmosphere. After 15 min, a solution of the appropriate ketone in anhydrous THF (1.0 M) was added dropwise. The reaction was stirred at rt. The reaction mixture were quenched with H2O and extracted with EtOAc. The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc as indicated in each case.
General Procedure for Pd Catalyzed Hydrogenation Reaction (Procedure K)
Method A: To a solution of the appropriate alkene (1.0 equiv) in MeOH (0.4 M) were added 10% Pd/C (0.15 equiv) and cyclohexene (25 equiv). The reaction mixture was stirred at reflux until the disappearance of the starting material, as indicated by UPLC/MS analysis. The suspension was filtered through a pad of Celite, and the filtrate was quickly evaporated under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc, or used in the next step without further purification, as indicated in each case. Method B: To a solution of the appropriate alkene (1.0 equiv) in MeOH (0.15 M) were added Et3SiH (10 equiv) and 10% Pd/C (0.02 equiv). The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The suspension was filtered through a pad of Celite, and the filtrate was quickly evaporated under reduced pressure. The crude was used in the next step without further purification.
General Procedure for Carbonyl Compounds Reduction Reaction (Procedure L)
Method A: To a solution of appropriate carbonyl compound (1.0 equiv) in anhydrous THF (0.2 M) LiAlH4 (2 M in THF, 2 equiv) was added dropwise at 0 °C under N2 atmosphere. The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The reaction mixture was quenched with a solution of NaOH (1N), and it was stirred for 15 min. Then, the mixture was extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was used in the next step without further purification. Method B: To a solution of the appropriate carbonyl compound (1.0 equiv) in anhydrous THF (0.2 M), DIBALH (1 M in hexane, 1.5 equiv) was added dropwise at 0 °C under N2 atmosphere. The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The reaction mixture was quenched with H2O and the extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by column chromatography, eluting with Cy/EtOAc, or used in the next step without further purification, as indicated in each case.
General Procedure for Oxidation Reaction (Procedure M)
To a solution of the appropriate alcohol (1.0 equiv) in Et2O or CH2Cl2 or CH2Cl2/iPrOH (0.1 M) was added activated MnO2 (10.0 equiv). The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The suspension was filtered through a pad of Celite, and the filtrate was quickly evaporated under reduced pressure. The crude was used in the next step without further purification.
General Procedure for Reductive Amination Reaction (Procedure N)
To a solution of the appropriate aldehyde (1.0 equiv) in anhydrous CH2Cl2 (0.1 M), a solution of the appropriate primary amine (1.0 equiv) in anhydrous CH2Cl2 (0.1 M) was added. The reaction was stirred at rt on. Then, NaBH(OAc)3 (2.0 equiv) was added, and the reaction was stirred at rt. The reaction mixture was quenched with 10% aqueous solution of K2CO3 and extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by column chromatography, eluting with CH2Cl2/MeOH as indicated in each case. The free base was dissolved in CH2Cl2 (0.1 M) and HCl (4 M in 1,4-dioxane, 19.0 equiv) was added. Evaporation of the solvent afforded the desired compound. Synthesis of 1-(2-chloro-4-fluoro-phenyl)cyclopentanol (28a). Compound 28a was prepared according to general procedure A (method A) using compound 27a (0.58 mL, 4.77 mmol), n-BuLi (2.07 mL, 4.77 mmol), and cyclopentenone (0.52 mL, 6.21 mmol) in anhydrous THF (9.54 mL). The crude was used in the next step without further purification. UPLC/MS (method B) Rt 2.29 min. MS (ES): C11H12ClFO, no ionization. 1H NMR (400 MHz, CDCl3) δ 7.62–7.54 (m, 1H), 7.16–7.09 (m, 1H), 7.00–6.91 (m, 1H), 2.25–2.03 (m, 2H), 2.00–1.73 (m, 6H).
Synthesis of 1-(4-Fluoro-2-methoxy-phenyl)cyclopentanol (28b)
Compound 28b was prepared according to general procedure A (method A) using compound 27b (0.62 mL, 4.88 mmol), n-BuLi (2.93 mL, 7.32 mmol), and cyclopentenone (0.66 mL, 7.80 mmol) in anhydrous THF (9.76 mL). The crude was used in the next step without further purification. UPLC/MS (method B) Rt 2.21 min. MS (ES): C12H15FO2 requires, 210 m/z; found, 193 m/z [M – H2O]+. 1H NMR (400 MHz, CDCl3) δ 7.20 (dd, J = 6.9, 9.2 Hz, 1H), 6.63–6.57 (m, 1H), 6.37–6.34 (m, 1H), 3.85 (s, 3H), 1.85–1.67 (m, 2H), 1.67–1.50 (m, 6H).
Synthesis of 1-(3,4-Difluorophenyl)cyclopentanol (28c)
Compound 28c was prepared according to general procedure A (method B) using compound 27c (28.54 mL, 14.27 mmol) and cyclopentenone (0.63 mL, 11.89 mmol) in anhydrous THF (2.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B) Rt 2.32 min. MS (ES): C11H12F2O no ionization. 1H NMR (400 MHz, CDCl3) δ 7.35–7.27 (m, 1H), 7.21–7.15 (m, 1H), 7.14–7.05 (m, 1H), 2.06–1.90 (m, 2H), 1.89–1.75 (m, 6H).
Synthesis of 1-(3-Chloro-4-fluoro-phenyl)cyclopentanol (28d)
Compound 28d was prepared according to general procedure A (method B) using 27d (28.54 mL, 14.27 mmol) and cyclopentenone (0.63 mL, 11.89 mmol) in anhydrous THF (2.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B) Rt 2.16 min. MS (ES): C11H12ClFO no ionization. 1H NMR (400 MHz, CDCl3) δ 7.42–7.35 (m, 1H), 7.25–7.19 (m, 1H), 7.11–7.04 (m, 1H), 2.03–1.91 (m, 2H), 1.90–1.78 (m, 4H).
Synthesis of 1-(1-Azidocyclopentyl)-2-chloro-4-fluoro-benzene (29a)
Compound 29a was prepared according to general procedure B using alcohol 28a (0.79 g, 3.68 mmol), NaN3 (0.53 g, 8.10 mmol) and TFA (2.31 mL, 30.20 mmol) in anhydrous CH2Cl2 (9.20 mL). The crude was purified by column chromatography (SiO2), eluting with Cy to afford 29a as yellow oil (0.054 g, 5% over 2 steps). UPLC/MS (method C) Rt 1.58 min. MS (ES): C11H11ClFN3 requires, 239 m/z; found, 211 m/z [M – N2]+. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 6.0, 8.8 Hz, 1H), 7.20 (dd, J = 2.7, 8.4 Hz, 1H), 6.96 (ddd, J = 2.7, 7.5, 8.9 Hz, 1H), 2.52–2.39 (m, 2H), 2.15–2.03 (m, 2H), 1.96–1.76 (m, 4H).
Synthesis of 1-(1-Azidocyclopentyl)-4-fluoro-2-methoxy-benzene (29b)
Compound 29b was prepared according to general procedure B using alcohol 28b (0.92 g, 4.37 mmol), NaN3 (0.62 g, 9.62 mmol) and TFA (2.74 mL, 35.84 mmol) in anhydrous CH2Cl2 (10.90 mL). The crude was purified by column chromatography (SiO2), eluting with Cy to afford 29b as a brown oil (0.123 g, 4% over 2 steps). UPLC/MS (method B) Rt 1.90 min. MS (ES): C12H14FN3O no ionization. 1H NMR (400 MHz, CDCl3) δ 7.27 (dd, J = 6.8, 8.6 Hz, 1H), 6.68–6.58 (m, 2H), 3.85 (s, 3H), 2.41–2.33 (m, 2H), 2.03–1.75 (m, 6H).
Synthesis of 4-(1-Azidocyclopentyl)-1,2-difluoro-benzene (29c)
Compound 29c was prepared according to general procedure B using alcohol 28c (1.14 g, 5.76 mmol), NaN3 (0.82 g, 12.67 mmol), and TFA (3.62 mL, 47.23 mmol) in anhydrous CH2Cl2 (14.40 mL). The crude was purified by column chromatography (SiO2), eluting with Cy to afford 29c as yellow oil (0.172 g, 7% over 2 steps). UPLC/MS (method C) Rt 2.11 min. MS (ES): C11H11F2N3 no ionization. 1H NMR (400 MHz, CDCl3) δ 6.94–6.86 (m, 1H), 6.85–6.78 (m, 2H), 1.91–1.78 (m, 2H), 1.66–1.46 (m, 6H).
Synthesis of 4-(1-Azidocyclopentyl)-2-chloro-1-fluoro-benzene (29d)
Compound 29d was prepared according to general procedure B using alcohol 28d (1.23 g, 5.73 mmol), NaN3 (0.82 g, 12.61 mmol), and TFA (3.60 mL, 46.99 mmol) in anhydrous CH2Cl2 (14.30 mL). The crude was purified by column chromatography (SiO2), eluting with Cy to afford 29d as a colorless oil (0.220 g, 11% over 2 steps). UPLC/MS (method B) Rt 1.87 min. MS (ES): C11H11ClFN3 no ionization. 1H NMR (400 MHz, CDCl3) δ 7.46 (dd, J = 2.4, 6.9 Hz, 1H), 7.29 (ddd, J = 2.4, 4.5, 8.6 Hz, 1H), 7.14 (t, J = 8.7 Hz, 1H), 2.25–2.12 (m, 2H), 1.99–1.82 (m, 6H).
Synthesis of 1-(2-Chloro-4-fluoro-phenyl)cyclopentanamine (30a)
Compound 30a was prepared according to the general procedure C using azide 29a (0.054 g, 0.24 mmol), LiAlH4 (0.12 mL, 0.24 mmol), in anhydrous THF (1.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.58 min. MS (ES): C11H13ClFN requires, 213 m/z; found, 214 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 6.2, 8.9 Hz, 1H), 7.12 (dd, J = 2.7, 8.4 Hz, 1H), 6.91 (ddd, J = 2.7, 7.7, 8.8 Hz, 1H), 2.19–1.86 (m, 6H), 1.81–1.69 (m, 2H).
Synthesis of 1-(4-Fluoro-2-methoxy-phenyl)cyclopentanamine (30b)
Compound 30b was prepared according to the general procedure C using azide 29b (0.123 g, 0.52 mmol), LiAlH4 (0.31 mL, 0.63 mmol), in anhydrous THF (2.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.54 min. MS (ES): C12H16FNO requires, 209 m/z; found, 210 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.23 (dd, J = 6.8, 8.5 Hz, 1H), 6.65–6.55 (m, 2H), 3.84 (s, 3H), 2.09–1.82 (m, 6H), 1.76–1.65 (m, 2H).
Synthesis of 1-(3,4-Difluorophenyl)cyclopentanamine (30c)
Compound 30c was prepared according to the general procedure C using azide 29c (0.172 g, 0.77 mmol), LiAlH4 (0.72 mL, 1.54 mmol), in anhydrous THF (3.08 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.27 min. MS (ES): C11H13F2N requires, 197 m/z; found, 198 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.34–7.29 (m, 1H), 7.21–7.14 (m, 1H), 7.12 (dd, J = 8.2, 10.1 Hz, 1H), 2.09–1.62 (m, 8H).
Synthesis of 1-(3-Chloro-4-fluoro-phenyl)cyclopentanamine (30d)
Compound 30d was prepared according to the general procedure C using azide 29d (0.22 g, 0.92 mmol), LiAlH4 (0.46 mL, 0.92 mmol), in anhydrous THF (3.70 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.44 min. MS (ES): C11H13ClFN requires, 213 m/z; found, 214 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.52 (dd, J = 2.4, 7.2 Hz, 1H), 7.33 (ddd, J = 2.4, 4.6, 8.7 Hz, 1H), 7.07 (t, J = 8.7 Hz, 1H), 2.01–1.82 (m, 8H).
Synthesis of 1-(4-Fluorophenyl)cyclopentanamine (30e)
Compound 30e was prepared according to procedure D using 1,4-dibromobutane (1.08 g, 5.00 mmol), Mg turnings (0.27 g, 11.0 mmol) in anhydrous Et2O (12.50 mL), nitrile 31a (0.30 g, 2.50 mmol) in anhydrous Et2O (2.50 mL), Ti(OiPr)4 (0.75 mL, 2.05 mmol) in anhydrous Et2O (1.50 mL). The crude was purified by column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 95:5 to afford 30e as a yellowish oil (0.101 g, 22%). UPLC/MS (method B): Rt 1.30 min. MS (ES): C11H14FN requires, 179 m/z; found, 197 m/z [M+NH4]+. 1H NMR (400 MHz, CDCl3) δ 7.46–7.39 (m, 2H), 7.03–6.95 (m, 2H), 2.00–1.74 (m, 8H).
Synthesis of 2-(1-Aminocyclopentyl)-5-fluoro-phenol (30f)
Compound 30f was prepared according to the general procedure D using 1,4-dibromobutane (1.08 g, 5.00 mmol), Mg turnings (0.27 g, 11.00 mmol), in anhydrous Et2O (12.50 mL), nitrile 31b (0.57 g, 2.50 mmol) in anhydrous Et2O (2.50 mL), Ti(OiPr)4 (0.75 mL, 2.50 mmol) in anhydrous Et2O (1.25 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 50:50 to afford 30f as a brownish solid (0.99 g, 20%). UPLC/MS (method B): Rt 1.33 min. MS (ES): C11H14FNO requires, 195 m/z; found, 196 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.06–6.98 (m, 1H), 6.56–6.51 (m, 1H), 6.49–6.40 (m, 1H), 5.27 (br s, 1H), 2.12–2.05 (m, 2H), 1.90–1.76 (m, 6H).
Synthesis of 5-(1-Aminocyclopentyl)-2-fluoro-phenol (30g)
Compound 30g was prepared according to procedure D using 1,4-dibromobutane (1.33 g, 6.16 mmol), Mg turnings (0.33 g, 13.50 mmol) in anhydrous Et2O (15.40 mL), nitrile 31c (0.70 g, 3.08 mmol) in anhydrous Et2O (3.00 mL), Ti(OiPr)4 (2.31 mL, 3.08 mmol) in anhydrous Et2O (1.50 mL). The crude was purified by column chromatography (SiO2), eluting with CH2Cl2/MeOH from 95:5 to 80:20 to afford 30g as a white solid (0.154 g, 26%). UPLC/MS (method A): Rt 1.18 min. MS (ES): C11H14FNO requires, 195 m/z; found, 196 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.06 (dd, J = 2.3, 8.9 Hz, 1H), 6.99 (dd, J = 8.5, 11.2 Hz, 1H), 6.85 (ddd, J = 2.4, 4.3, 8.5 Hz, 1H), 1.90–1.60 (m, 8H).
Synthesis of 1-(2,4-Difluorophenyl)cyclopentanamine (30h)
To a solution of compound 34a (0.112 g, 0.50 mmol, 1.0 equiv) in acetone (0.50 mL), Et3N (0.08 mL, 0.55 mmol, 1.1 equiv) was added. The reaction was stirred at −5 °C and ethyl chloroformate (0.05 mL, 0.54 mmol, 1.1 equiv) was added slowly. The reaction was stirred for 15 min at the same temperature. A solution of NaN3 (0.065 g, 1.00 mmol, 2.0 equiv) in H2O (0.2 mL) was added. The reaction was stirred for additional 30 min at the same temperature. Then, the reaction mixture was poured into ice–H2O and extracted with toluene. The organic layers were combined, dried over Na2SO4, and transferred to two neck round-bottom flask. The reaction mixture was heated cautiously under reflux for 1 h. Then, the solvent was removed under reduced pressure and 8 N HCl (0.75 mL) was added at 0 °C. The reaction was gradually heated up to 70 °C. The reaction was cooled at rt and poured into ice–H2O. A solution of NaOH (1N) was added up to pH 9. The mixture was extracted with CH2Cl2, dried over Na2SO4, filtered, and concentrated under reduced pressure. Then, the crude was solubilized with CH2Cl2, washed with a solution of HCl (0.1 N), and extracted with CH2Cl2. The aqueous phase was treated with a solution of NaOH (0.1 N) up to pH 9 and extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The product was used in the next step without further purification. UPLC/MS (method A): Rt 1.22 min. MS (ES): C11H13F2N requires, 197 m/z; found, 198 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.39–7.28 (m, 1H), 6.83–6.72 (m, 2H), 2.05–1.86 (m, 4H), 1.81–1.64 (m, 4H).
Synthesis of 1-(4-Fluoro-3-methoxy-phenyl)cyclopentanamine (30i)
To a solution of compound 34b (0.216 g, 0.91 mmol, 1.0 equiv) in anhydrous toluene (5.00 mL), SOCl2 (0.33 mL, 4.53 mmol, 5.0 equiv) was added. The reaction was stirred at room temperature until the consumption of the starting material. The solvent was removed under reduced pressure. The oil residue was solubilized in anhydrous toluene (5.00 mL) and NaN3 (0.118 g, 1.81 mmol, 2.0 equiv) was added. The reaction was stirred. The reaction mixture was cooled down and concentrated under reduced pressure. The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 100 to afford the desired intermediate benzyl N-[1-(4-fluoro-3-methoxy-phenyl)cyclopentyl]carbamate (0.048 g, 15%). UPLC/MS (method A): Rt 1.18 min. MS (ES): C20H22FNO3 requires, 343 m/z; found, 344 m/z [M + H]+. The protecting group was removed using Et3SiH (0.19 mL, 0.73 mmol, 10 equiv), 10% Pd/C (0.001 g, 0.02 equiv), and MeOH (1.00 mL). The reaction was stirred at 90 °C under microwave irradiation for 30 min. The crude was used for the next step without further purification. UPLC/MS (method B): Rt 1.38 min. MS (ES): C12H16FNO requires, 209 m/z; found, 210 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.11 (dd, J = 8.3, 2.2 Hz, 1H), 7.02–6.92 (m, 2H), 3.90 (s, 3H), 2.04–1.76 (m, 8H).
Synthesis of 1-(2,4-Difluorophenyl)cyclopentanecarbonitrile (33a)
Compound 33a was prepared according to the general procedure E using benzyl nitrile 32a (0.50 g, 3.26 mmol), 1,4-dibromobutane (1.57 g, 4.90 mmol), and LiHMDS (3.91 mL, 3.91 mmol) in anhydrous THF (22.00 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 90:10 to afford 33a as a colorless oil (0.46 g, 69%). UPLC/MS (method A): Rt 1.03 min. MS (ES): C12H11F2N no ionization. 1H NMR (400 MHz, CDCl3) δ 7.44–7.36 (m, 1H), 6.92–6.84 (m, 2H), 2.61–2.46 (m, 2H), 2.17–1.84 (m, 6H).
Synthesis of 1-(4-Fluoro-3-methoxy-phenyl)cyclopentanecarbonitrile (33b)
Compound 33b was prepared according to the general procedure E using benzyl nitrile 32b (0.50 g, 3.03 mmol), 1,4-dibromobutane (0.88 g, 4.54 mmol), LiHMDS (3.64 mL, 3.64 mmol) in anhydrous THF (20.00 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 92:8 to afford 33b as a colorless oil (0.51 g, 77%). UPLC/MS (method A): Rt 2.14 min. MS (ES): C13H14FNO no ionization. 1H NMR (400 MHz, CDCl3) δ 7.09–7.03 (m, 2H), 6.94 (ddd, J = 2.3, 4.1, 8.5 Hz, 1H), 3.92 (s, 3H), 2.55–2.40 (m, 2H), 2.20–1.91 (m, 6H).
Synthesis of 1-(2,4-Difluorophenyl)cyclopentanecarboxylic Acid (34a)
Compound 34a was prepared according to the general procedure F using nitrile 32a (0.58 g, 2.80 mmol), H2O and H2SO4 (4.00 mL). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 1.74 min. MS (ES): C12H12F2O2 requires, 226 m/z; found, 227 m/z [M + H]+, 225 m/z [M – H]−. 1H NMR (400 MHz, CDCl3) δ 7.37–7.27 (m, 1H), 6.91–6.75 (m, 2H), 2.59–2.45 (m, 2H), 2.05–1.93 (m, 2H), 1.89–1.77 (m, 2H), 1.76–1.61 (m, 2H).
Synthesis of 1-(4-Fluoro-3-methoxy-phenyl)cyclopentanecarboxylic Acid (34b)
Compound 34b was prepared according to the general procedure F using nitrile 33b (0.51 g, 2.17 mmol), H2O and H2SO4 (3.10 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 50:50 to afford 34b as white solid (0.216 g, 42%). UPLC/MS (method A): Rt 1.68 min. MS (ES): C13H15FO3 requires, 238 m/z; found, 239 m/z [M + H]+, 237 m/z [M – H]−. 1H NMR (400 MHz, DMSO-d6) δ 7.16–7.02 (m, 1H), 6.91–6.85 (m, 2H), 3.83 (s, 3H), 1.93–1.47 (m, 8H).
Synthesis of Methyl 4-Benzyloxy-3-(diethoxyphosphorylmethyl)benzoate (35h)
Step 1: compound 35f (1.00 g, 3.90 mmol, 1.0 equiv) and N-bromosuccinimide (0.73 g, 4.09 mmol, 1.05 equiv) was suspended in anhydrous acetonitrile (4.00 mL). Then, AIBN (0.03 g, 0.19 mmol, 0.05 equiv) was added. The reaction was stirred at 80 °C for 24 hs. The reaction was cooled to rt, and the precipitate was filtered off. The filtrate was diluted with CH2Cl2 and washed with a saturated solution of NaHCO3. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude of compound 35g was used in the next step without further purification. UPLC/MS (method C): Rt 1.92 min. MS (ES): C16H15BrO3 no ionization. Step 2: to a solution of compound 35g (1.31 g, 3.90 mmol, 1.0 equiv) in anhydrous toluene (4.90 mL) was added P(OEt)3 (1.34 mL, 7.8 mmol, 2.0 equiv). The reaction was stirred at 120 °C under N2 atmosphere for 20 h. The reaction was cooled to rt, and the solvent was evaporated under reduced pressure. The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 90:10 to afford 35h as a colorless oil (0.28 g, 20% over 2 steps). UPLC/MS (method B): Rt 2.24 min. MS (ES): C20H25O6P requires, 392 m/z; found 393 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 8.02 (t, J = 2.6 Hz, 1H), 7.92 (dt, J = 2.2, 8.6 Hz, 1H), 7.49–7.32 (m, 5H), 6.95 (d, J = 8.6 Hz, 1H), 5.16 (s, 2H), 4.07–3.95 (m, 4H), 3.88 (s, 3H), 3.38–3.26 (m, 2H), 1.22 (t, J = 7.0 Hz, 6H).
Synthesis of Methyl 3-(Diethoxyphosphorylmethyl)benzoate (36a)
Compound 36a was prepared using compound 35a (3.00 g, 13.09 mmol) and P(OEt)3 (6.74 mL, 39.29 mmol) in anhydrous toluene (13.00 mL), according to the procedure as previously described.22
Synthesis of (3-Methoxycarbonylphenyl)methyl-triphenyl-phosphonium Bromide (36b)
Compound 36b was prepared using compound 35a (2.00 g, 8.73 mmol) and PPh3 (2.29 g, 8.73 mmol) in anhydrous toluene (44.00 mL) according to the general procedure G. The resulting solid was collected by filtration to afford 36b as a white solid (4.03 g, 94%). UPLC/MS (method C): Rt 0.89 min. MS (ES): C27H24O2P requires, 411 m/z; found, 412 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.81–7.67 (m, 12H), 7.66–7.57 (m, 6H), 7.39 (d, J = 2.5 Hz, 1H), 5.55 (d, J = 14.6 Hz, 2H), 3.77 (s, 3H).
Synthesis of Methyl 3-(Diethoxyphosphorylmethyl)-4-fluoro-benzoate (36c)
Compound 36c was prepared using compound 35b (7.78 g, 31.49 mmol) and P(OEt)3 (16.02 mL, 94.47 mmol) in anhydrous toluene (39.40 mL) according to the general procedure H. The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 67:33 to afford 36c as a colorless oil (10.03 g, 99%). UPLC/MS (method B): Rt 1.89 min. MS (ES): C13H18FO5P requires, 304 m/z; found, 305 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 8.06 (td, J = 2.6, 7.3 Hz, 1H), 7.96–7.90 (m, 1H), 7.10 (t, J = 8.9 Hz, 1H), 4.12–4.02 (m, 4H), 3.89 (s, 3H), 3.27–3.14 (m, 2H), 1.26 (t, J = 7.0 Hz, 6H).
Synthesis of (2-Chloro-5-methoxycarbonyl-phenyl)methyl-triphenyl-phosphonium Bromide (36d)
Compound 36d was prepared using compound 35c (1.42 g, 3.23 mmol) and PPh3 (0.85 g, 3.23 mmol) in anhydrous toluene (16.15 mL) according to the general procedure G. The resulting solid was collected by filtration to afford 36d as a gray solid (1.12 g, 39%). UPLC/MS (method C): Rt 1.91 min. MS (ES): C27H23ClO2P requires, 445 m/z; found, 446 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.96 (s, 1H), 7.89–7.83 (m, 1H), 7.82–7.68 (m, 10H), 7.68–7.60 (m, 6H), 5.75 (d, J = 14.7 Hz, 2H), 3.77 (s, 3H).
Synthesis of (4-Fluoro-3-methoxycarbonyl-phenyl)methyl-triphenyl-phosphonium Bromide (36e)
Compound 36e was prepared using compound 35d (2.52 g, 10.20 mmol) and PPh3 (2.67 g, 10.20 mmol) in anhydrous toluene (51.00 mL) according to the general procedure G. The resulting solid was collected by filtration to afford 36e as a white solid (3.48 g, 67%). UPLC/MS (method B): Rt 1.91 min. MS (ES): C27H23FO2P requires, 429 m/z; found, 430 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.86–7.72 (m, 10H), 7.69–7.56 (m, 6H), 7.33–7.28 (m, 1H), 6.99–6.86 (m, 1H), 5.64 (d, J = 14.4 Hz, 2H), 3.78 (s, 3H).
Synthesis of (2,4-Difluoro-5-methoxycarbonyl-phenyl)methyl-triphenyl-phosphonium Bromide (36f)
Compound 36f was prepared using compound 35e (1.53 g, 4.61 mmol) and PPh3 (1.21 g, 4.61 mmol) in anhydrous toluene (23.00 mL) according to the general procedure G. The resulting solid was collected by filtration to afford 36f as a white solid (1.82 g, 60%). UPLC/MS (method C): Rt 0.67 min. MS (ES): C27H22F2O2P requires, 447 m/z; found, 448 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 8.00–7.89 (m, 10H), 7.93–7.74 (m, 6H), 6.67 (t, J = 9.8 Hz, 1H), 5.68 (d, J = 13.8 Hz, 2H), 3.80 (s, 3H).
Synthesis of tert-Butyl 4-[(3-Methoxycarbonylphenyl)methylene]piperidine-1-carboxylate (37a)
Compound 37a was prepared using compound 36a (1.29 g, 4.51 mmol), NaH (0.72 g, 18.03 mmol), and N-Boc-4-piperidone (1.35 g, 6.76 mmol) in anhydrous THF (47.00 mL), according to the procedure as previously described.22
Synthesis of tert-Butyl 4-[(2-Fluoro-5-methoxycarbonyl-phenyl)methylene]piperidine-1-carboxylate (37b)
Compound 37b was prepared according to the general procedure K (method A) using compound 36c (2.67 g, 8.78 mmol), LiHMDS (10.53 mL, 10.53 mmol) in anhydrous THF (29.00 mL), and N-Boc-4-piperidone (2.62 g, 13.2 mmol). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 80:20 to afford 37b as a white solid (1.77 g, 58%). UPLC/MS (method B): Rt 1.93 min. MS (ES): C19H24FNO4 requires, 349 m/z; found 350 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.94–7.86 (m, 2H), (t, J = 8.9 Hz, 1H), 6.26 (s, 1H), 3.91 (s, 3H), 3.56–3.40 (m, 2H), 3.45–3.37 (m, 2H), 2.40–2.30 (m, 4H), 1.48 (s, 9H).
Synthesis of tert-Butyl 4-[(2-Chloro-5-methoxycarbonyl-phenyl)methylene]piperidine-1-carboxylate (37c)
Compound 37c was prepared according to the general procedure I using compound 36d (1.1 g, 2.09 mmol), LiHMDS (3.14 mL, 3.14 mL) in anhydrous THF (7.00 mL), and N-Boc-4-piperidone (0.62 g, 3.14 mmol) in anhydrous THF (3.14 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 90:10 to afford 37c as a pale-yellow oil (0.15 g, 20%). UPLC/MS (method C): Rt 1.92 min. MS (ES): C19H24ClNO4 requires, 365 m/z; found 366 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.87–7.83 (m, 1H), 7.83 (d, J = 2.1 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 6.34 (s, 1H), 3.91 (s, 3H), 3.56–3.51 (m, 2H), 3.44–3.39 (m, 2H), 2.41–2.35 (m. 2H), 2.33–2.28 (m, 2H), 1.48 (s, 9H).
Synthesis of tert-Butyl 4-[(4-Fluoro-3-methoxycarbonyl-phenyl)methylene]piperidine-1-carboxylate (37d)
Compound 37d was prepared according to the general procedure I using compound 36e (0.70 g, 1.37 mmol), LiHMDS (2.06 mL, 2.06 mL) in anhydrous THF (4.60 mL) and N-Boc-4-piperidone (0.41 g, 2.06 mmol) in anhydrous THF (2.06 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 90:10 to afford 37d as a colorless oil (0.17 g, 35%). UPLC/MS (method C): Rt 1.52 min. MS (ES): C19H24FNO4 requires, 349 m/z; found, 350 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.74 (dd, J = 2.4, 6.9 Hz, 1H), 7.31 (ddd, J = 2.5, 4.7, 2.5 Hz, 1H), 7.09 (dd, J = 8.5, 10.5 Hz, 1H), 6.31 (s, 1H), 3.93 (s, 3H), 3.54–3.47 (m, 2H), 3.43–3.37 (m, 2H), 2.43–2.37 (m, 2H), 2.36–2.30 (m, 2H), 1.48 (s, 9H).
Synthesis of tert-Butyl 4-[(2,4-Difluoro-5-methoxycarbonyl-phenyl)methylene]piperidine-1-carboxylate (37e)
Compound 37e was prepared according to the general procedure I using compound 36f (1.01 g, 1.91 mmol), LiHMDS (2.87 mL, 2.87 mL) in anhydrous THF (6.40 mL) and N-Boc-4-piperidone (0.57 g, 2.87 mmol) in anhydrous THF (2.87 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 90:10 to afford 37e as a colorless oil (0.37 g, 52%). UPLC/MS (method B): Rt 1.64 min. MS (ES): C19H23F2NO4 requires, 367 m/z; found, 368 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.80 (t, J = 8.3 Hz, 1H), 6.87 (t, J = 9.9 Hz, 1H), 6.17 (s, 1H), 3.92 (s, 3H), 3.52 (t, J = 5.8 Hz, 2H), 3.41 (t, J = 5.9 Hz, 2H), 2.36 (t, J = 5.8 Hz, 2H), 2.29 (t, J = 5.9 Hz, 2H), 1.45 (s, 9H).
Synthesis of 4-Benzyloxy-3-[(1-tert-butoxycarbonyl-4-piperidylidene)methyl]benzoic Acid (37f)
Compound 37f was prepared according to procedure J (method B) using compound 35h (0.28 g, 0.71 mmol), t-BuOK (0.080 g, 0.71 mmol), and N-Boc-4-piperidone (0.21 g, 1.07 mmol) in anhydrous THF (3.40 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 80:20 to 20:80 to afford 37f as a white solid (0.20 g, 67%). UPLC/MS (method B): Rt 2.24 min. MS (ES): C26H31NO5 requires, 437 m/z; found 438 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.96 (dd, J = 2.2, 8.6 Hz, 1H), 7.89 (d, J = 2.2 Hz, 1H), 7.44–7.30 (m, 5H), 6.96 (d, J = 8.6 Hz, 1H), 6.39 (s, 1H), 5.18 (s, 2H), 3.87 (s, 3H), 3.55–3.48 (m, 2H), 3.45–3.37 (m, 2H), 2.48–2.33 (m, 4H), 1.49 (s, 9H).
Synthesis of tert-Butyl 4-[(4-Benzyloxy-3-formyl-phenyl)methylene]piperidine-1-carboxylate (37g)
Under argon atmosphere, a mixture of compound 35i (0.200 g, 0.69 mmol, 1.0 equiv), XPhos (0.003 g, 0.01 mmol, 1 mol %), tert-butyl 4-[(tetramethyl-1,3,2-dioxaborolan-2-yl)methylidene]piperidine-1-carboxylate (0.220 g, 0.69 mmol, 1.0 equiv), Pd2dba3 (0.031 g, 0.03 mmol, 5 mol %) was solubilized in 1,4-dioxane (3.96 mL, previously degassed under a N2 atmosphere). Then, an aqueous solution of K3PO4 (0.220 g, 1.03 mmol, 1.5 equiv, 5.0 M, previously degassed under a N2 atmosphere) was added. The mixture was stirred at 100 °C. The reaction was cooled down at rt, filtered through a pad of Celite using EtOAc. The filtrate was washed with H2O, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 80:20 to afford 37g as a yellowish oil (0.212 g, 75%). UPLC/MS (method C): Rt 2.22 min. MS (ES): C25H29NO4 requires, 407 m/z; found, 408 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.55 (s, 1H), 7.68 (d, J = 2.4 Hz, 1H), 7.54–7.33 (m, 6H), 7.01 (d, J = 8.6 Hz, 1H), 6.28 (s, 1H), 5.19 (s, 2H), 3.49 (t, J = 5.8 Hz, 2H), 3.39 (t, J = 5.9 Hz, 2H), 2.45–2.36 (m, 2H), 2.32 (t, J = 5.9 Hz, 2H), 1.47 (s, 9H).
Synthesis of [4-Benzyloxy-3-[(1-methyl-4-piperidylidene)methyl]phenyl]methanol (37h)
Compound 37h was prepared according to the general procedure L (method A) using compound 37f (0.23 g, 0.40 mmol) and LiAlH4 (0.40 mL, 0.81 mmol) in anhydrous THF (2.00 mL). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 1.62 min. MS (ES): C21H25NO2 requires, 323 m/z; found, 324 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.44–7.27 (m, 5H), 7.20–7.13 (m, 2H), 6.91–6.87 (m, 1H), 6.35 (s, 1H), 5.09 (s, 2H), 4.60 (s, 2H), 2.78–2.67 (m, 2H), 2.49–2.41 (m, 2H), 2.29 (s, 3H), 2.23–2.10 (m, 1H), 1.98–1.87 (m, 1H), 1.72–1.55 (m, 2H).
Synthesis of [2-Benzyloxy-5-[(1-methyl-4-piperidylidene)methyl]phenyl]methanol (37i)
Compound 37i was prepared according to the general procedure N (method A) using compound 37g (0.212 g, 0.52 mmol) and LiAlH4 (0.77 mL, 1.55 mmol) in anhydrous THF (2.60 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.78 min. MS (ES): C21H27NO2 requires, 323 m/z; found, 324 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.43–7.33 (m, 5H), 7.08–7.04 (m, 1H), 6.93–6.89 (m, 2H), 6.36 (s, 1H), 5.13 (s, 2H), 4.71 (br s, 2H), 2.90–2.78 (m, 2H), 2.72–2.30 (m, 7H), 1.69–1.50 (m, 4H).
Synthesis of tert-Butyl (4E)-4-[(3-Methoxycarbonylphenyl)methylene]-2,2-dimethylpiperidine-1-carboxylate and tert-Butyl (4Z)-4-[(3-Methoxycarbonylphenyl)methylene]-2,2-dimethylpiperidine-1-carboxylate (37k)
Compound 37k was prepared according to procedure J (method B) using compound 36a (0.50 g, 1.75 mmol), t-BuOK (0.59 g, 5.24 mmol, 3 equiv), and tert-butyl-2,2-dimethylpiperidine-1-carboxylate (0.60 g, 2.62 mmol) in anhydrous THF (8.45 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 90:10 to afford 37k as a colorless oil (0.39 g, 62%) and as a mixture of E and Z isomers. UPLC/MS (method C): Rt 2.03 min. MS (ES) C21H29NO4 requires, 359 m/z; found 360 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.98 (t, J = 1.8 Hz, 1H), 7.88–7.83 (m, 1H), 7.47 (td, J = 1.8, 7.7 Hz, 1H), 7.39 (d, J = 7.7 Hz, 1H), 6.45 (s, 1H, distinct signal of one isomer), 6.33 (s, 1H), 3.92 (s, 3H), 3.70–3.64 (m, 2H), 2.74–2.68 (m, 2H), 2.50–2.46 (m, 2H), 1.47 (s, 9H), 1.44 (s, 6H).
Synthesis of tert-Butyl (4Z)-3,3-Difluoro-4-[(3-methoxycarbonylphenyl)methylene]piperidine-1-carboxylate (37l)
Compound 37l was prepared according to general procedure I using compound 36b (0.70 g, 1.42 mmol), LiHMDS (2.14 mL, 2.12 mL) in anhydrous THF (4.70 mL) and tert-butyl 3,3-difluoro-4-oxo-piperidine-1-carboxylate (0.50 g, 2.14 mmol) in anhydrous THF (2.14 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 95:5 to afford 37l as a colorless oil (0.19 g, 36%). UPLC/MS: (method B): Rt 1.62; C19H23F2NO4 requires 367 m/z, found 368 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.97 (td, J = 1.8, 7.3 Hz, 1H), 7.92 (s, 1H), 7.49–7.40 (m, 2H), 7.00 (s, 1H), 3.93 (s, 3H), 3.86–3.74 (m, 2H), 3.50–3.41 (m, 2H), 2.69–2.61 (m, 2H), 1.49 (s, 9H).
Synthesis of tert-Butyl 3-[(3-Methoxycarbonylphenyl)methyl]azetidine-1-carboxylate (37m)
Compound 37m was prepared according to general procedure I using compound 36b (0.70 g, 1.42 mmol), LiHMDS (2.14 mL, 2.12 mL) in anhydrous THF (4.70 mL), and 1-Boc-3-azetidinone (0.37 g, 2.14 mmol) in anhydrous THF (2.14 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 90:10 to afford 37m as a colorless oil (0.36 g, 83%). UPLC/MS (method B): Rt 2.42 min. MS (ES): C17H21NO4 requires, 303 m/z; found 304 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.89 (td, J = 1.4, 7.7 Hz, 1H), 7.78 (t, J = 1.4 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.28 (td, J = 1.4, 7.7 Hz, 1H), 6.32–6.27 (m, 1H), 4.88–4.84 (m, 2H), 4.73–4.59 (m, 2H), 3.92 (s, 3H), 1.48 (s, 9H).
Synthesis of tert-Butyl 6-[(3-Methoxycarbonylphenyl)methylene]-2-azaspiro[3.3]heptane-2-carboxylate (37n)
Compound 37n was prepared according to the general procedure I using compound 36b (1.00 g, 2.03 mmol), LiHMDS (3.05 mL, 3.05 mL) in anhydrous THF (7.0 mL), and tert-butyl 6-oxo-2-azaspiro[3.3]heptane-2-carboxylate (0.64 g, 3.05 mmol) in anhydrous THF (3.05 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 90:10 to afford 37n as a white solid (0.24 g, 34%). UPLC/MS (method C): Rt 1.82 min. MS (ES): C20H25NO4 requires, 343 m/z; found, 344 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.87–7.81 (m, 2H), 7.40–7.32 (m, 2H), 6.21–6.17 (m, 1H), 4.00–3.98 (m, 4H), 3.91 (s, 3H), 3.26–3.22 (m, 2H), 3.08–3.04 (m, 2H), 1.43 (s, 9H).
Synthesis of tert-Butyl 4-[(3-Methoxycarbonylphenyl)methyl]piperidine-1-carboxylate (38a)
Compound 38a was prepared using compound 37a (1.24 g, 3.59 mmol), Et3SiH (5.73 mL, 35.90 mmol). and 10% Pd/C (0.036 g) in MeOH (28.00 mL), according to the procedure as previously described.22
Synthesis of tert-Butyl 4-[(2-Fluoro-5-methoxycarbonyl-phenyl)methyl]piperidine-1-carboxylate (38b)
Compound 38b was prepared according to the general procedure K (method B) using alkene 37b (1.77 g, 5.06 mmol), Et3SiH (8.07 mL, 50.60 mmol), and 10% Pd/C (0.035 g) in MeOH (33.70 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.93 min. MS (ES): C19H26FNO4 requires, 351 m/z; found, 352 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.92–7.83 (m, 2H), 7.06 (t, J = 8.9 Hz, 1H), 4.07 (d, J = 13.4 Hz, 2H), 3.90 (s, 3H), 2.69–2.57 (m, 4H), 1.77–1.66 (m, 1H), 1.60 (d, J = 13.2 Hz, 2H), 1.44 (s, 9H), 1.18 (dq, J = 4.4, 12.5 Hz, 2H).
Synthesis of Methyl 4-Chloro-3-[(1-methyl-4-piperidyl)methyl]benzoate (38c)
Compound 38c was prepared according to the general procedure K (method B) using alkene 37c (0.15 g, 0.41 mmol), Et3SiH (0.65 mL, 4.10 mmol), and 10% Pd/C (0.009 g) in MeOH (2.70 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.55 min. MS (ES): C15H20ClNO2 requires, 281 m/z; found, 282 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.84 (dd, J = 2.1 Hz, 1H), 7.80 (dd, J = 2.1, 8.3 Hz, 1H), 7.41 (d, J = 8.3, 1H), 3.91 (s, 3H), 3.00–2.87 (m, 2H), 2.73 (d, J = 6.5 Hz, 2H), 2.33 (s, 3H), 2.08–1.92 (m, 2H), 1.72–1.42 (m, 5H).
Synthesis of tert-Butyl 4-[(4-Fluoro-3-methoxycarbonyl-phenyl)methyl]piperidine-1-carboxylate (38d)
Compound 38d was prepared according to the procedure K (method A) using alkene 37d (0.34 g, 0.97 mmol), cyclohexene (2.46 mL, 24.29 mmol), 10% Pd/C (0.051 g) in MeOH (2.40 mL). The crude was purified by flash column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 85:15 to afford 38d as colorless oil (0.296 g, 87%). UPLC/MS (method C): Rt 1.53 min. MS (ES): C19H26FNO4 requires, 351 m/z; found, 352 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.69 (dd, J = 2.4, 6.9 Hz, 1H), 7.29–7.24 (m, 1H), 7.05 (dd, J = 8.4, 10.6 Hz, 1H), 4.08 (d, J = 13.2 Hz, 2H), 3.93 (s, 3H), 2.68–2.57 (m, 2H), 2.54 (d, J = 7.0 Hz, 2H), 1.71–1.52 (m, 3H), 1.45 (s, 9H), 1.14 (qd, J = 4.2, 12.4 Hz, 2H).
Synthesis of tert-Butyl 4-[(2,4-Difluoro-5-methoxycarbonyl-phenyl)methyl]piperidine-1-carboxylate (38e)
Compound 38e was prepared according to the general procedure K (method B) using alkene 37e (0.31 g, 0.83 mmol), Et3SiH (1.33 mL, 8.33 mmol), and 10% Pd/C (0.006 g) in MeOH (5.53 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.64 min. MS (ES): C19H25F2NO4 requires, 369 m/z; found, 370 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.76 (t, J = 8.2 Hz, 1H), 6.84 (t, J = 10.0 Hz, 1H), 4.08 (d, J = 13.2 Hz, 2H), 3.92 (s, 3H), 2.64 (t, J = 12.8 Hz, 2H), 2.57 (d, J = 7.1 Hz, 2H), 1.75–1.63 (m, 1H), 1.59 (d, J = 13.3 Hz, 2H), 1.45 (s, 9H), 1.25–1.11 (m, 2H).
Synthesis of tert-Butyl 3,3-Difluoro-4-[(3-methoxycarbonylphenyl)methyl]piperidine-1-carboxylate (38l)
Compound 38l was prepared according to the procedure K (method A) using alkene 37l (0.19 g, 0.52 mmol), cyclohexene (1.57 mL, 15.51 mmol), 10% Pd/C (0.038 g) in MeOH (1.30 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.55 min. MS (ES): C19H25F2NO4 requires, 369 m/z; found, 370 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.93–7.88 (m, 1H), 7.85 (s, 1H), 7.42–7.34 (m, 2H), 4.52–3.98 (m, 2H), 3.92 (s, 3H), 3.25 (dd, J = 3.6, 13.7 Hz, 1H), 3.07–2.83 (m, 1H), 2.73–2.55 (m, 1H), 2.51 (dd, J = 10.5, 13.7 Hz, 1H), 2.16–1.95 (m, 1H), 1.61–1.50 (m, 2H), 1.46 (s, 9H).
Synthesis of tert-Butyl 3-[(3-Methoxycarbonylphenyl)methyl]azetidine-1-carboxylate (38m)
Compound 38m was prepared according to the procedure K (method A) using alkene 37m (0.36 g, 1.19 mmol), cyclohexene (3.61 mL, 35.60 mmol), 10% Pd/C (0.072 g) in MeOH (3.00 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.19 min. MS (ES): C17H23NO4 requires, 305 m/z; found, 306 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 6.9 Hz, 1H), 7.83 (s, 1H), 7.40–7.32 (m, 2H), 4.04–3.95 (m, 2H), 3.92 (s, 3H), 3.69–3.61 (m, 2H), 2.95 (d, J = 7.7 Hz, 2H), 2.89–2.76 (m, 1H), 1.44 (s, 9H).
Synthesis of tert-Butyl 6-[(3-Methoxycarbonylphenyl)methyl]-2-azaspiro[3.3]heptane-2-carboxylate (38n)
Compound 38n was prepared according to the procedure K (method A) using alkene 37n (0.24 g, 0.70 mmol), cyclohexene (1.76 mL, 17.40 mmol), 10% Pd/C (0.036 g) in MeOH (1.75 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.88 min. MS (ES): C20H27NO4 requires, 345 m/z; found, 346 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 7.5 Hz, 1H), 7.79 (s, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 3.91 (s, 3H), 3.89 (s, 2H), 3.81 (s, 2H), 2.70 (d, J = 7.5 Hz, 2H), 2.48–2.36 (m, 1H), 2.28–2.19 (m, 2H), 1.93–1.81 (m, 2H), 1.42 (s, 9H).
Synthesis of [3-[(1-Methyl-4-piperidyl)methyl]phenyl]methanol (39a)
Compound 39a was prepared using compound 38a (1.10 g, 3.17 mmol) and LiAlH4 (3.17 mL, 6.33 mmol) in anhydrous THF (15.85 mL), according to the procedure as previously described.22
Synthesis of [4-Fluoro-3-[(1-methyl-4-piperidyl)methyl]phenyl]methanol (39b)
Compound 39b was prepared according to the general procedure L (method A) using compound 38b (0.15 g, 0.40 mmol) and LiAlH4 (0.40 mL, 0.81 mmol) in anhydrous THF (2.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.24 min. MS (ES): C14H20FNO requires, 237 m/z; found, 238 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.15–7.04 (m, 2H), 6.91 (t, J = 9.2 Hz, 1H), 4.52 (s, 2H), 2.75–2.63 (m, 2H), 2.51 (d, J = 6.6 Hz, 2H), 2.09 (s, 3H), 1.84–1.61 (m, 2H), 1.57–1.40 (m, 3H), 1.32–1.16 (m, 2H).
Synthesis of [4-Chloro-3-[(1-methyl-4-piperidyl)methyl]phenyl]methanol (39c)
To a solution of compound 38c (0.160 g, 0.43 mmol, 1.0 equiv) in CH2Cl2 (0.5 mL) was added HCl (4 M in 1,4-dioxane, 2.00 mL, 19.0 equiv). The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. Then, the mixture was concentrated under reduced pressure. After evaporation of the solvent, the crude was suspended in CH2Cl2 (2.30 mL) and Et3N (0.06 mL, 0.43 mmol, 1.0 equiv) was added at 0 °C. The reaction was stirred at rt for 10 min and then formaldehyde 37 wt % in H2O (0.06 mL, 2.15 mmol, 5.0 equiv) was added. The reaction was stirred at the same temperature and after 30 min NaBH(OAc)3 (0.456 g, 2.15 mmol, 5.0 equiv) was added. The reaction mixture was quenched with 10% aqueous solution of K2CO3 and extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.57 min. MS (ES): C15H20ClNO2 requires, 281 m/z; found, 282 m/z [M + H]+. Compound 39c was prepared according to the general procedure L (method B) using the intermediate methyl 4-chloro-3-[(1-methyl-4-piperidyl)methyl]benzoate (0.09 g, 0.31 mmol) and DIBALH (0.46 mL, 0.46 mmol) in anhydrous THF (1.55 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.24 min. MS (ES): C14H20ClNO requires, 253 m/z; found, 254 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.33–7.28 (m, 1H), 7.16 (s, 1H), 7.13 (d, J = 8.1 Hz, 1H), 4.63 (s, 2H), 2.95–2.82 (m, 2H), 2.66 (d, J = 6.7 Hz, 2H), 2.30 (s, 3H), 2.05–1.91 (m, 2H), 1.71–1.59 (m, 3H), 1.55–1.34 (m, 2H).
Synthesis of [2-Fluoro-5-[(1-methyl-4-piperidyl)methyl]phenyl]methanol (39d)
Compound 39d was prepared according to the general procedure L (method A) using compound 38d (0.31 g, 0.88 mmol) and LiAlH4 (0.88 mL, 1.76 mmol) in anhydrous THF (4.40 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.16 min. MS (ES): C14H20FNO requires, 237 m/z; found, 238 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.19 (dd, J = 2.3, 7.2 Hz, 1H), 7.02 (ddd, J = 2.3, 5.2, 7.7 Hz, 1H), 6.94 (dd, J = 8.3, 9.9 Hz, 1H), 4.73 (s, 2H), 2.89–2.80 (m, 2H), 2.51 (d, J = 7.0 Hz, 2H), 2.26 (s, 3H), 1.95–1.84 (m, 2H), 1.68–1.58 (m, 2H), 1.51–1.42 (m, 1H), 1.41–1.26 (m, 2H).
Synthesis of [2,4-Difluoro-5-[(1-methyl-4-piperidyl)methyl]phenyl]methanol (39e)
Compound 39e was prepared according to the general procedure L (method A) using compound 38e (0.19 g, 0.51 mmol) and LiAlH4 (0.51 mL, 1.02 mmol) in anhydrous THF (2.55 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.19 min. MS (ES): C14H19F2NO requires, 255 m/z; found, 256 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.20 (t, J = 8.4 Hz, 1H), 6.76 (t, J = 9.8 Hz, 1H), 4.68 (s, 2H), 2.87–2.76 (m, 2H), 2.53 (d, J = 6.7 Hz, 2H), 2.23 (s, 3H), 1.94–1.82 (m, 2H), 1.66–1.55 (m, 2H), 1.55–1.45 (m, 1H), 1.40–1.26 (m, 2H).
Synthesis of tert-Butyl 4-[[3-(Hydroxymethyl)phenyl]methyl]piperidine-1-carboxylate (39f)
Compound 39f was prepared according to the general procedure L (method B) using compound 38a (0.07 g, 0.20 mmol) and DIBALH (0.40 mL, 0.40 mmol) in anhydrous THF (1.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 2.24 min. MS (ES): C18H27NO3 requires, 305 m/z; found, 306 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.25–7.17 (m, 2H), 7.15 (s, 1H), 7.08–7.03 (m, 1H), 4.67 (s, 2H), 4.14–3.98 (m, 1H), 3.53–3.44 (m, 1H), 2.68–2.56 (m, 2H), 2.54 (d, J = 7.0 Hz, 2H), 1.79–1.58 (m, 3H), 1.45 (s, 9H), 1.22–1.07 (m, 2H).
Synthesis of tert-Butyl 4-[[2-Fluoro-5-(hydroxymethyl)phenyl]methyl]piperidine-1-carboxylate (39g)
Compound 39g was prepared according to the general procedure L (method B) using compound 38b (1.06 g, 3.01 mmol) and DIBALH (6.02 mL, 6.02 mmol) in anhydrous THF (15.00 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 70:30 to afford 39g as a colorless oil (1.46 g, 89% over 2 steps). UPLC/MS (method B): Rt 1.22 min. MS (ES): C18H26FNO3 requires, 323 m/z; found, 324 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.21–7.11 (m, 2H), 6.99 (t, J = 8.3 Hz, 1H), 4.64 (s, 2H), 4.07 (d, J = 12.6 Hz, 2H), 2.63 (dt, J = 2.7, 12.9 Hz, 2H), 2.57 (d, J = 6.5 Hz, 2H), 1.75–1.64 (m, 1H), 1.61 (d, J = 12.9 Hz, 2H), 1.44 (s, 9H), 1.17 (dq, J = 4.2, 12.6 Hz, 2H).
Synthesis of tert-Butyl 4-[[4-Fluoro-3-(hydroxymethyl)phenyl]methyl]piperidine-1-carboxylate (39h)
Compound 39h was prepared according to the general procedure L (method B) using compound 38d (0.21 g, 0.60 mmol) and DIBALH (0.89 mL, 0.89 mmol) in anhydrous THF (3.00 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc from 100% to 60:40 to afford 39h as a colorless oil (0.263 g, 83%). UPLC/MS (method A): Rt 2.22 min. MS (ES): C18H26FNO3 requires, 323 m/z; found, 324 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.18 (dd, J = 2.3, 7.2 Hz, 1H), 7.02 (ddd, J = 2.3, 5.2, 7.7 Hz, 1H), 6.95 (dd, J = 8.3, 9.8 Hz, 1H), 4.74 (s, 2H), 4.07 (td, J = 2.0, 11.1 Hz, 2H), 2.62 (dt, J = 2.4, 12.8 Hz, 2H), 2.50 (d, J = 6.8 Hz, 2H), 1.68–1.60 (m, 4H), 1.44 (s, 9H), 1.30 (dq, J = 4.3, 12.5 Hz, 2H).
Synthesis of 4-(Hydroxymethyl)-2-[(1-methyl-4-piperidyl)methyl]phenol (39i)
Compound 39i was prepared according to the general procedure K (method B) using alkene 37f (0.15 g, 0.46 mmol), Et3SiH (0.74 mL, 4.61 mmol), and 10% Pd/C (0.010 g) in MeOH (3.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 0.91 min. MS (ES): C14H21NO2 requires, 235 m/z; found, 236 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.06–697 (m, 2H), 6.68 (t, J = 8.6 Hz, 1H), 4.66–4.53 (m, 2H), 2.98–2.84 (m, 2H), 2.58–2.48 (m, 2H), 2.31 (s, 3H), 2.08–1.93 (m, 2H), 1.71–1.54 (m, 5H).
Synthesis of 2-(Hydroxymethyl)-4-[(1-methyl-4-piperidyl)methyl]phenol (39j)
Compound 39j was prepared according to the general procedure K (method B) using alkene 37i (0.10 g, 0.30 mmol), Et3SiH (0.48 mL, 3.00 mmol), and 10% Pd/C (0.002 g) in MeOH (2.00 mL). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 0.96 min. MS (ES): C14H21NO2 requires, 235 m/z; found, 236 m/z [M + H]+; 237 m/z [M – H]−. 1H NMR (400 MHz, CDCl3) δ 6.96–6.90 (m, 1H), 6.81–6.74 (m, 2H), 4.80 (s, 2H), 2.84–2.75 (m, 1H), 2.43 (d, J = 7.0 Hz, 2H), 2.31–2.21 (m, 1H), 2.25 (s, 3H), 1.97–1.86 (m, 2H), 1.64–1.54 (m, 2H), 1.50–1.20 (m, 3H).
Synthesis of [3-[(1,2,2-Trimethyl-4-piperidyl)methyl]phenyl]methanol (39k)
Step 1: to a solution of compound 37k (0.39 g, 1.09 mmol, 1 equiv) in MeOH (7.26 mL) were added Et3SiH (1.74 mL, 10.9 mmol, 10 equiv) and 10% Pd/C (0.008 g). The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The suspension was filtered through a pad of Celite, and the filtrate was quickly evaporated under reduced pressure. The crude of compound 38k was used in the next without further purification. Step 2: compound 38k was dissolved in anhydrous THF (5.45 mL) LiAlH4 (1.09 mL, 2.18, 2.0 equiv) was added dropwise at 0 °C under N2 atmosphere. The reaction mixture was stirred at rt until the disappearance of the starting material, as indicated by UPLC/MS analysis. The reaction mixture was quenched with a solution of NaOH (1N) and it was stirred for 15 min. Then, the mixture was extracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.17 min. C16H25NO requires, 247 m/z; found, 248 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.27 (t, J = 7.7 Hz, 1H), 7.20 (d, J = 7.7 Hz, 1H), 7.15 (s, 1H), 7.05 (d, J = 7.7 Hz, 1H), 4.67 (s, 2H), 3.06–2.81 (m, 1H), 2.69–2.43 (m, 3H), 2.44 (s, 3H), 1.99–1.77 (m, 2H), 1.78–1.48 (m, 4H), 1.35 (s, 3H), 1.07 (s, 3H).
Synthesis of [3-[(3,3-Difluoro-1-methyl-4-piperidyl)methyl]phenyl]methanol (39l)
Compound 39l was prepared according to the general procedure N (method A) using compound 38l (0.18 g, 0.59 mmol) and LiAlH4 (0.59 mL, 1.18 mmol) in anhydrous THF (2.95 mL). The crude was used in the next step without further purification. UPLC/MS (method A): Rt 1.40 min. MS (ES): C14H19F2NO requires, 255 m/z; found, 256 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 7.5 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 7.19 (s, 1H), 7.11 (d, J = 7.3 Hz, 1H), 4.69 (s, 2H), 3.22 (dd, J = 3.5, 13.7 Hz, 1H), 3.15–3.06 (m, 1H), 2.86–2.78 (m, 1H), 2.46 (dd, J = 10.6, 13.7 Hz, 1H), 2.35 (s, 3H), 2.32–2.17 (m, 1H), 1.99–1.80 (m, 2H), 164–1.54 (m, 2H).
Synthesis of [3-[(1-Methylazetidin-3-yl)methyl]phenyl]methanol (39m)
Compound 39m was prepared according to the general procedure L (method A) using compound 38m (0.21 g, 0.69 mmol) and LiAlH4 (0.69 mL, 1.37 mmol) in anhydrous THF (3.50 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 0.90 min. MS (ES): C12H17NO requires, 191 m/z; found, 192 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.26 (t, J = 8.2 Hz, 1H), 7.20 (d, J = 7.7 Hz, 1H), 7.14 (s, 1H), 7.04 (d, J = 7.3 Hz, 1H), 4.66 (s, 2H), 3.43 (t, J = 7.5 Hz, 2H), 2.97 (t, J = 7.7 Hz, 2H), 2.86 (d, J = 7.8 Hz, 2H), 2.71 (h, J = 7.1 Hz, 1H), 2.34 (s, 3H).
Synthesis of [3-[(2-Methyl-2-azaspiro[3.3]heptan-6-yl)methyl]phenyl]methanol (39n)
Compound 39n was prepared according to the general procedure N (method A) using compound 38n (0.23 g, 0.66 mmol) and LiAlH4 (0.66 mL, 1.32 mmol) in anhydrous THF (3.30 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.24 min. MS (ES): C15H21NO requires, 231 m/z; found, 232 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.86 (t, J = 7.5 Hz, 1H), 7.17 (d, J = 7.5 Hz, 1H), 7.12 (s, 1H), 7.03 (d, J = 7.5 Hz, 1H), 4.66 (s, 2H), 3.28 (s, 2H), 3.18 (s, 2H), 2.65 (d, J = 7.4 Hz, 2H), 2.44–2.34 (m, 1H), 2.30 (s, 3H), 2.26–2.18 (m, 2H), 1.88–1.80 (m, 2H).
Synthesis of 3-[(1-Methyl-4-piperidyl)methyl]benzaldehyde (39a)
Compound 39a was prepared using compound 38a (0.66 g, 3.00 mmol) and MnO2 (1.57 g, 18.06 mmol) in Et2O (13.63 mL), according to the procedure as previously described.22
Synthesis of 4-Fluoro-3-[(1-methyl-4-piperidyl)methyl]benzaldehyde (40b)
Compound 40b was prepared according to the general procedure M using alcohol 39b (0.093 g, 0.39 mmol) and MnO2 (0.34 g, 3.92 mmol) in Et2O (3.90 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.34 min. MS (ES): C14H18FNO requires, 235 m/z; found, 236 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.77 (ddd, J = 2.2, 5.0, 8.4 Hz, 1H), 7.74 (dd, J = 2.2, 7.3 Hz, 1H), 7.20 (t, J = 8.8 Hz, 1H), 3.11–2.95 (m, 2H), 2.71 (d, J = 5.2 Hz, 2H), 2.42 (s, 3H) 2.26–2.01 (m, 2H), 1.80–1.53 (m, 5H).
Synthesis of 4-Chloro-3-[(1-methyl-4-piperidyl)methyl]benzaldehyde (40c)
Compound 40c was prepared according to the general procedure M using alcohol 39c (0.056 g, 0.22 mmol) and MnO2 (0.19 g, 2.19 mmol) in CH2Cl2 (2.20 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.46 min. MS (ES): C14H18ClNO requires, 251 m/z; found, 252 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.95 (s, 1H), 7.66 (s, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.50 (t, J = 8.2 Hz, 1H), 2.88–2.77 (m, 2H), 2.74 (d, J = 6.6 Hz, 2H), 2.24 (s, 3H) 1.93–1.82 (m, 2H), 1.68–1.56 (m, 3H), 1.49–1.32 (m, 2H).
Synthesis of 2-Fluoro-5-[(1-methyl-4-piperidyl)methyl]benzaldehyde (40d)
Compound 40d was prepared according to the general procedure M using alcohol 39d (0.19 g, 0.80 mmol) and MnO2 (0.69 g, 8.00 mmol) in Et2O (8.00 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.25 min. MS (ES): C14H18FNO requires, 235 m/z; found 236 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.35 (s, 1H), 7.63 (dd, J = 2.4, 6.6 Hz, 1H), 7.36 (ddd, J = 2.4, 5.1, 7.9 Hz, 1H), 7.08 (dd, J = 8.5, 10.2 Hz, 1H), 2.89–2.81 (m, 2H), 2.56 (d, J = 6.9 Hz, 2H), 2.26 (s, 3H), 1.95–1.85 (m, 2H), 1.65–1.57 (m, 2H), 1.55–1.43 (m, 1H), 1.41–1.27 (m, 2H).
Synthesis of 2,4-Difluoro-5-[(1-methyl-4-piperidyl)methyl]benzaldehyde (40e)
Compound 40e was prepared according to the general procedure M using alcohol 39e (0.104 g, 0.41 mmol) and MnO2 (0.35 g, 4.07 mmol) in Et2O (4.10 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.30 min. MS (ES): C14H17F2NO requires 253 m/z, found 254 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.25 (s, 1H), 7.68 (t, J = 8.1 Hz, 1H), 6.85 (t, J = 9.8 Hz, 1H), 2.87–2.76 (m, 2H), 2.57 (d, J = 6.9 Hz, 2H), 2.23 (s, 3H), 1.92–1.79 (m, 2H), 1.64–1.43 (m, 3H), 1.40–1.24 (m, 2H).
Synthesis of tert-Butyl 4-[(3-Formylphenyl)methyl]piperidine-1-carboxylate (40f)
Compound 40f was prepared according to the general procedure M using alcohol 39f (0.044 g, 0.14 mmol) and MnO2 (0.13 g, 1.44 mmol) in Et2O (1.40 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.40 min. MS (ES): C18H25NO3 requires, 303 m/z; found, 304 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 7.71 (d, J = 7.4 Hz, 1H), 7.66 (s, 1H), 7.45 (t, J = 7.4 Hz, 1H), 7.40 (d, J = 7.4 Hz, 1H), 4.18–3.99 (m, 1H), 3.15–2.97 (m, 1H), 2.71–2.56 (m, 3H), 2.49–2.36 (m, 1H), 1.80–1.52 (m, 3H), 1.45 (s, 9H), 1.16 (qd, J = 12.4, 4.1 Hz, 2H).
Synthesis of tert-Butyl 4-[(2-Fluoro-5-formyl-phenyl)methyl]piperidine-1-carboxylate (40g)
Compound 40g was prepared according to the general procedure M using alcohol 39g (2.39 g, 7.39 mmol) and MnO2 (6.45 g, 74.19 mmol) in Et2O (74.00 mL). The crude was purified by column chromatography (SiO2), eluting with Cy/EtOAc (from 100% to 80:20) to afford 40g as a colorless oil (2.88 g, 74%). UPLC/MS (method C): Rt 1.61 min. MS (ES): C18H24FNO3 requires, 321 m/z; found, 322 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.94 (s, 1H), 7.78–7.68 (m, 2H), 7.18 (t, J = 8.7 Hz, 1H), 4.08 (d, J = 13.1 Hz, 2H); 2.71–2.57 (m, 4H), 1.80–1.68 (m, 1H), 1.60 (d, J = 13.0 Hz, 2H), 1.45 (s, 9H), 1.19 (dq, J = 4.2, 12.3 Hz, 2H).
Synthesis of tert-Butyl 4-[(4-Fluoro-3-formyl-phenyl)methyl]piperidine-1-carboxylate (40h)
Compound 40h was prepared according to the general procedure M using alcohol 39h (0.263 g, 0.81 mmol) and MnO2 (0.71 g, 8.13 mmol) in Et2O (8.10 mL). The crude was used in the next step without further purification. UPLC/MS (method C): Rt 1.66 min. MS (ES): C18H24FNO3 requires, 321 m/z; found, 322 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.36 (s, 1H), 7.63 (dd, J = 2.4, 6.6 Hz, 1H), 7.36 (ddd, J = 2.4, 5.1, 8.1 Hz, 1H), 7.09 (dd, J = 8.4, 10.1 Hz, 1H), 4.08 (d, J = 13.1 Hz, 2H), 2.63 (t, J = 12.9 Hz, 2H), 2.56 (d, J = 7.1 Hz, 2H), 1.71–1.59 (m, 1H), 1.58 (d, J = 13.5 Hz, 2H), 1.44 (s, 9 H), 1.14 (dq, J = 4.5, 12.5 Hz, 2H).
Synthesis of 4-Hydroxy-3-[(1-methyl-4-piperidyl)methyl]benzaldehyde (40i)
Compound 40i was prepared according to the procedure M using alcohol 39i (0.112 g, 0.48 mmol) and MnO2 (0.41 g, 4.76 mmol) in CH2Cl2/iPrOH (4.80 mL). UPLC/MS (method A): Rt 1.07 min. MS (ES): C14H19NO2 requires, 233 m/z; found, 234 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.78 (s, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 2.1, 8.2 Hz, 1H), 7.09 (d, J = 8.2 Hz, 1H), 3.39–3.26 (m, 2H), 3.13–3.01 (m, 2H), 2.65 (s, 3H), 2.25–2.11 (m, 2H), 1.91–1.68 (m, 5H).
Synthesis of 2-Hydroxy-5-[(1-methyl-4-piperidyl)methyl]benzaldehyde (40j)
Compound 40j was prepared according to the procedure O using alcohol 39j (0.080 g, 0.34 mmol) and MnO2 (0.295 g, 3.40 mmol) in CH2Cl2 (3.40 mL). The crude was purified by column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100% to 80:20 to afford 40j as a colorless oil (0.025 g, 20% over 3 steps). UPLC/MS (method A): Rt 2.30 min. MS (ES): C14H19NO2 requires, 233 m/z; found, 234 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1H), 7.34–7.26 (m, 2H), 6.96–6.87 (m, 1H), 2.96–2.88 (m, 2H), 2.58–2.49 (m, 2H), 2.32 (s, 3H), 2.03–1.93 (m, 2H), 1.70–1.60 (m, 2H), 1.54–1.33 (m, 3H).
Synthesis of 3-[(1,2,2-Trimethyl-4-piperidyl)methyl]benzaldehyde (40k)
Compound 40k was prepared according to the procedure M using alcohol 39k (0.300 g, 1.26 mmol) and MnO2 (1.09 g, 12.64 mmol) in CH2Cl2 (12.60 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.31 min. MS (ES): C16H23NO requires, 245 m/z; found, 246 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 7.71 (td, J = 1.6, 7.4 Hz, 1H), 7.66 (t, J = 1.6 Hz, 1H), 7.45 (t, J = 7.4, 1H), 7.40 (td, J = 1.6, 7.4 Hz, 1H), 2.84–2.69 (m, 1H), 2.67–2.45 (m, 3H), 2.34 (s, 3H), 1.92–1.76 (m, 1H), 1.67–1.38 (m, 3H), 1.34–1.17 (m, 4H), 0.97 (s, 3H).
Synthesis of 3-[(3,3-Difluoro-1-methyl-4-piperidyl)methyl]benzaldehyde (40l)
Compound 40l was prepared according to the general procedure M using alcohol 39l (0.125 g, 0.53 mmol) and MnO2 (0.46 g, 5.27 mmol) in Et2O (5.30 mL). UPLC/MS (method B): Rt 1.68 min. MS (ES): C14H17F2NO requires, 253 m/z; found, 254 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 10.01 (s, 1H), 7.75 (d, J = 7.1 Hz, 1H), 7.71 (d, J = 1.8 Hz, 1H), 7.52–7.43 (m, 2H), 3.35–3.15 (m, 2H), 3.05–2.87 (m, 1H), 2.64–2.53 (m, 1H), 2.46 (s, 3H), 2.23–1.88 (m, 2H), 1.79–1.55 (m, 3H).
Synthesis of 3-[(1-Methylazetidin-3-yl)methyl]benzaldehyde (40m)
Compound 40m was prepared according to the procedure M using alcohol 39m (0.13 g, 0.68 mmol) and MnO2 (0.59 g, 6.79 mmol) in CH2Cl2 (6.00 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.01 min. MS (ES): C12H15NO requires, 189 m/z; found, 190 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.99 (s, 1H), 7.71 (d, J = 7.3 Hz, 1H), 7.65 (s, 1H), 7.49–7.50 (m, 2H), 3.47–3.38 (m, 2H), 3.00–2.91 (m, 4H), 2.80–2.69 (m, 1H), 2.34 (s, 3H).
Synthesis of 3-[(2-Methyl-2-azaspiro[3.3]heptan-6-yl)methyl]benzaldehyde (40n)
Compound 40n was prepared according to the procedure M using alcohol 39n (0.129 g, 0.56 mmol) and MnO2 (0.48 g, 5.58 mmol) in Et2O (5.60 mL). The crude was used in the next step without further purification. UPLC/MS (method B): Rt 1.40 min. MS (ES): C15H19NO requires, 229 m/z; found, 230 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 7.69 (dd, J = 1.6, 7.4 Hz, 1H), 7.62 (br s, 1H), 7.42 (t, J = 7.4 Hz, 1H), 7.37 (dd, J = 1.6, 7.4 Hz, 1H), 3.20 (br s, 2H), 3.12 (br s, 2H), 2.73 (d, J = 7.5 Hz, 2H), 2.48–2.36 (m, 1H), 2.29–2.16 (m, 5H), 1.87–1.78 (m, 2H).
Synthesis of tert-Butyl 4-[[3-[[[1-(4-Fluorophenyl)cyclopentyl]amino]methyl]phenyl]methyl]piperidine-1-carboxylate (41f)
Compound 41f was prepared according to the general procedure N using aldehyde 40f (0.030 g, 0.10 mmol), amine 30e (0.18 g, 0.10 mmol), NaBH(OAc)3 (0.42 g, 0.20 mmol) in anhydrous CH2Cl2 (1.00 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 95:5 to afford 41f as yellowish oil (0.021 g, 17% over 4 steps). UPLC/MS (method C): Rt 2.35 min. MS (ES): C29H39FN2O2 requires, 466 m/z; found, 467 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.54–7.50 (m, 2H), 7.23–7.14 (m, 2H), 7.10–6.94 (m, 4H), 4.14–3.97 (m, 2H), 3.42–3.30 (m, 2H), 2.71–2.57 (m, 2H), 2.48 (d, J = 6.8 Hz, 2H), 2.15–2.17 (m, 5H), 1.74–1.53 (m, 6H), 1.45 (s, 9H), 1.18–1.06 (m, 2H).
Synthesis of tert-Butyl 4-[[2-Fluoro-5-[[[1-(4-fluorophenyl)cyclopentyl]amino]methyl]phenyl]methyl]piperidine-1-carboxylate (41g)
Compound 41g was prepared according to the general procedure N using aldehyde 40g (0.56 g, 1.74 mmol), amine 30e (0.312 g, 1.74 mmol), NaBH(OAc)3 (0.736 g, 3.48 mmol) in anhydrous CH2Cl2 (17.4 mL). The crude was purified by flash column chromatography (SiO2), eluting with Cy/EtOAc from 100 to 80:20 to afford 41g as colorless oil (0.713 g, 84%). UPLC/MS (method C): Rt 2.50 min. MS (ES): C29H38F2N2O2 requires, 484 m/z; found, 485 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.51–7.46 (m, 2H), 7.16–6.96 (m, 5H), 3.95–3.84 (m, 2H), 3.25–3.16 (m, 2H), 2.56–2.45 (m, 4H overlapped with DMSO signal), 2.06–1.95 (m, 2H), 1.89–1.70 (m, 4H), 1.67–1.57 (m, 3H), 1.56–1.46 (m, 2H), 1.38 (s, 9H), 1.11–0.94 (m, 2H).
Synthesis of tert-Butyl 4-[[4-Fluoro-3-[[[1-(4-Fluorophenyl)cyclopentyl]amino]methyl]phenyl]methyl]piperidine-1-carboxylate (41h)
Compound 41h was prepared according to the general procedure N using aldehyde 40h (0.100 g, 0.31 mmol), amine 30e (0.056 g, 0.31 mmol), NaBH(OAc)3 (0.131 g, 0.62 mmol) in anhydrous CH2Cl2 (3.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 95:5 to afford 41h as yellowish oil (0.055 g, 13% over 2 steps). UPLC/MS (method D): Rt 1.72 min. MS (ES): C29H38F2N2O2 requires, 484 m/z; found, 485 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.46–7.40 (m, 2H), 7.05–6.97 (m, 3H), 6.94–6.83 (m, 2H), 4.18–3.98 (m, 2H), 3.42–3.33 (m, 2H), 2.71–2.54 (m, 2H), 2.45 (d, J = 6.8 Hz, 2H), 2.09–1.78 (m, 5H), 1.77–1.52 (m, 6H), 1.45 (s, 9H), 1.19–1.01 (m, 2H).
Synthesis of 1-(2,4-Difluorophenyl)-N-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (3)
Compound 3 was prepared according to the general procedure N using aldehyde 40a (0.023 g, 0.11 mmol), amine 30h (0.021 g, 0.11 mmol), and NaBH(OAc)3 (0.045 g, 0.21 mmol) in anhydrous CH2Cl2 (1.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 97:3 to afford 3 as colorless oil (0.015 g, 2% over 3 steps). UPLC/MS (method B): Rt 2.31 min. C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.33–7.25 (m, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.03 (dt, J = 7.7, 1.4 Hz, 1H), 6.99–6.33 (m, 2H), 6.86–6.76 (m, 2H), 3.32 (s, 2H), 2.83 (d, J = 11.9 Hz, 1H), 2.48 (d, J = 7.0 Hz, 2H), 2.26 (s, 3H), 2.19–2.09 (m, 2H), 1.98–1.82 (m, 7H), 1.75–1.56 (m, 5H), 1.52–1.39 (m, 1H), 1.33 (qd, J = 3.7, 12.3 Hz, 1H). The free base was dissolved in CH2Cl2 (0.40 mL) and HCl [4 M in dioxane, 0.20 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.11 min. C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (br s, 1H), 9.59 (br s, 2H), 7.71–7.61 (m, 1H), 7.35–7.11 (m, 6H), 3.87–3.80 (m, 2H), 3.38–3.27 (m, 2H overlapped with H2O signal), 2.83 (q, J = 11.6 Hz, 2H), 2.51–2.42 (m, 2H overlapped with DMSO signal), 2.66 (d, J = 5.2 Hz, 3H), 2.46 (d, J = 7.0 Hz, 2H), 2.38–2.28 (m, 2H), 1.96–1.84 (m, 2H), 1.80–1.63 (m, 3H), 1.63–1.43 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 162.7 (dd, 3JC–F = 13 Hz, 1JC–F = 244 Hz), 160.7 (dd, 3JC–F = 9 Hz, 1JC–F = 245 Hz), 139.7, 131.8, 131.6, 129.3, 128.4, 127.9, 120.9, 111.9 (dd, 4JC–F = 3 Hz, 2JC–F = 21 Hz), 105.1 (t, 2JC–F = 27 Hz), 69.7, 53.3 (2C), 47.4, 42.5, 41.3, 40.1, 35.6, 35.6, 34.3, 28.8 (2C), 22.2 (2C). HRMS C25H32F2N2 [M + H]+ calculated 399.2606; measured 399.2614, Δppm 1.9.
Synthesis of 1-(2-Chloro-4-fluoro-phenyl)-N-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (4)
Compound 4 was prepared according to the general procedure N using aldehyde 40a (0.11 g, 0.51 mmol), amine 30a (0.10 g, 0.51 mmol), and NaBH(OAc)3 (0.22 g, 1.02 mmol) in anhydrous CH2Cl2 (5.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 83:7 to afford 4 as yellow oil (0.090 g, 42% over 2 steps). UPLC/MS (method B): Rt 2.56 min. MS (ES) C25H32ClFN2 requires, 414 m/z; found, 415 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.38 (dd, J = 6.2, 8.8 Hz, 1H), 7.20–17.12 (m, 2H), 7.05 (d, J = 7.9 Hz, 1H), 6.99–6.93 (m, 3H), 3.21 (s, 2H), 3.05–2.91 (m, 2H), 2.51 (d, J = 5.8 Hz, 2H), 2.37 (s, 3H), 2.31–2.18 (m, 2H), 2.13–1.87 (m, 7H), 1.74–1.61 (m, 4H), 1.58–1.41 (m, 2H). The free base was dissolved in CH2Cl2 (2.0 mL) and HCl [4 M in dioxane, 1.0 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.55 min. MS (ES): C25H32ClFN2 requires, 414 m/z; found, 415 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (br s, 1H), 9.38 (br s, 2H), 7.66 (dd, J = 5.9, 9.1 Hz, 1H), 7.49 (dd, J = 2.8, 8.5 Hz, 1H), 7.35–7.28 (m, 1H), 7.28–7.19 (m, 1H), 7.19–7.09 (m, 3H), 3.84 (br s, 2H), 3.37–3.30 (m, 2H overlapping with H2O signal), 2.82 (q, J = 11.6 Hz, 2H), 2.67 (d, J = 4.7 Hz, 3H), 2.61–2.50 (m, 4H overlapping with DMSO signal), 2.46 (d, J = 6.8 Hz, 2H), 1.99–1.83 (m, 2H), 1.84–1.63 (m, 3H), 1.60–1.46 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 161.9 (d, 1JC–F = 250.0 Hz), 139.6, 133.5 (d, 3JC–F = 10.4 Hz), 132.7, 131.4, 130.9, 130.4, 129.4, 128.3, 128.0, 118.8 (d, 2JC–F = 24.8 Hz), 114.6 (d, 2JC–F = 20.3 Hz), 71.6, 53.2 (2C), 47.8, 42.5, 41.3, 40.2, 35.9, 34.3, 28.8 (2C), 22.6 (2C). HRMS C25H32ClFN2 [M + H]+ calculated 415.2311; measured 415.23121, Δppm 0.3.
Synthesis of 1-(4-Fluoro-2-methoxy-phenyl)-N-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (5)
Compound 5 was prepared according to the general procedure N using aldehyde 40a (0.10 g, 0.49 mmol), amine 30b (0.10 g, 0.49 mmol), NaBH(OAc)3 (0.21 g, 0.98 mmol) in anhydrous CH2Cl2 (4.90 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 90:10 to afford 5 as yellow oil (0.023 g, 11% over 2 steps). UPLC/MS (method B): Rt 2.26 min. MS (ES): C26H35FN2O requires, 410 m/z; found, 411 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.23–7.11 (m, 2H), 6.99–6.93 (m, 2H), 6.90 (bs, 1H), 6.68–6.57 (m, 2H), 3.79 (s, 3H), 3.25 (s, 2H), 2.86 (d, J = 11.2 Hz, 2H), 2.47 (d, J = 6.9 Hz, 2H), 2.28 (s, 3H), 2.18–2.06 (m, 2H), 1.98–1.85 (m, 6H), 1.68–1.56 (m, 4H), 1.51–1.40 (m, 1H), 1.40–1.27 (m, 2H). The free base was dissolved in CH2Cl2 (0.56 mL) and HCl [4 M in dioxane, 0.3 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.42 min. MS (ES): C26H35FN2O requires, 410 m/z; found, 411 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.49 (br s, 1H), 9.14 (br s, 2H), 7.36–7.22 (m, 2H), 7.18–7.09 (m, 2H), 7.03 (s, 1H), 6.99–6.93 (m, 1H), 6.84 (t, J = 8.4 Hz, 1H), 3.83 (s, 3H), 3.78–3.68 (m, 2H), 3.41–3.31 (m, 2H overlapped with H2O), 2.89–2.71 (m, 2H), 2.67 (s, 3H), 2.51–2.42 (m, 2H overlapped with DMSO), 2.40–2.23 (m, 4H), 1.91–1.78 (m, 2H), 1.78–1.61 (m, 3H), 1.60–1.38 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.4 (d, 1JC–F = 245.0 Hz), 158.1 (d, 3JC–F = 10 Hz), 139.6, 131.9, 130.9, 129.7 (d, 3JC–F = 10 Hz), 129.2, 128.2, 127.8, 120.8, 106.6 (d, 2JC–F = 21 Hz), 100.4 (d, 2JC–F = 26 Hz), 70.4, 55.9, 53.2, 47.6, 42.5, 41.3, 40.2, 35.1 (2C), 34.3, 28.8 (2C), 22.6 (2C). HRMS C26H35FN2O [M + H]+ calculated 411.2806; measured 411.2808, Δppm 0.4.
Synthesis of 5-Fluoro-2-[1-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methylamino]cyclopentyl]phenol Dihydrochloride (6)
Compound 6 was prepared according to the general procedure N using aldehyde 40a (0.023 g, 0.11 mmol), amine 30f (0.021 g, 0.11 mmol), and NaBH(OAc)3 (0.045 g, 0.22 mmol) in anhydrous CH2Cl2 (1.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 90:10 to afford 6 as yellowish oil (0.017 g, 38%). UPLC/MS (method B): Rt 1.98 min. C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+, 395 m/z [M – H]−. 1H NMR (400 MHz, CDCl3) δ 7.30–7.23 (m, 1H), 7.14 (d, J = 7.5 Hz, 1H), 7.09–6.98 (m, 3H), 6.62 (dd, J = 2.7, 10.5 Hz, 1H), 6.52 (td, J = 2.7, 2.7 Hz, 1H), 3.53 (s, 2H), 2.97 (d, J = 11.3 Hz, 2H), 2.56 (d, J = 6.8 Hz, 2H), 2.35 (s, 3H), 2.30–2.14 (m, 2H), 2.10–1.87 (m, 5H), 1.88–1.61 (m, 5H), 1.61–1.37 (m, 3H). The free base was dissolved in CH2Cl2 (0.40 mL) and HCl [4 M in dioxane, 2.0 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.01 min. C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (br s, 1H), 9.11 (br s, 2H), 7.31–7.22 (m, 2H), 7.21–7.10 (m, 3H), 6.96 (dd, J = 2.7, 10.6 Hz, 1H), 6.71 (td, J = 2.7, 8.5 Hz, 1H), 3.78–3.77 (m, 2H), 3.33 (d, J = 12.2 Hz, 2H), 2.88 (q, J = 11.5 Hz, 2H), 2.66 (d, J = 4.6 Hz, 3H), 2.53–2.44 (m, 2H overlapped with DMSO signal), 2.39–2.22 (m, 4H), 1.91–1.64 (m, 5H), 1.62–1.42 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.2 (d, 1JC–F = 245 Hz), 156.4, 139.9, 132.0, 130.5, 129.6, 129.4, 128.4, 127.7, 119.6, 105.9 (d, 2JC–F = 21 Hz), 103.4 (d, 2JC–F = 24 Hz), 70.6, 53.2 (2C), 47.5, 42.5, 41.3, 40.3 (overlapped with DMSO signal), 34.9, 34.3, 28.8 (2C), 22.6 (2C). HRMS C25H33FN2O [M + H]+ calculated 397.265; measured 397.2648, Δppm −0.4.
Synthesis of 1-(3,4-Difluorophenyl)-N-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine;dihydrochloride (7)
Compound 7 was prepared according to the general procedure R using aldehyde 40a (0.065 g, 0.30 mmol), amine 30c (0.059 g, 0.30 mmol), and NaBH(OAc)3 (0.127 g, 0.60 mmol) in anhydrous CH2Cl2 (3.00 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 7 as yellow oil (0.056 g, 18% over 2 steps). UPLC/MS (method B): Rt 1.78 min. MS (ES): C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.30 (ddd, J = 2.2, 7.8, 12.2 Hz, 2H), 7.21–7.05 (m, 4H), 7.01–6.97 (m, 2H), 3.35 (br s, 2H), 2.84 (d, J = 11.9 Hz, 1H), 2.50 (d, J = 7.0 Hz, 2H), 2.26 (s, 3H), 2.04–1.81 (m, 8H), 1.78–1.68 (m, 2H), 1.66–1.58 (m, 2H), 1.54–1.41 (m, 1H), 1.32 (qd, J = 12.1, 3.9 Hz, 2H). The free base was dissolved in CH2Cl2 (1.40 mL) and HCl [4 M in dioxane, 0.67 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.96 min. MS (ES) C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (br s, 1H), 10.04 (br s, 2H), 7.91 (ddd, J = 2.1, 7.6, 12.6 Hz, 1H), 7.63–7.51 (m, 2H), 7.31–7.14 (m, 4H), 3.74–3.64 (m, 2H), 3.37–3.28 (m, 2H overlapped with H2O signal), 2.82 (q, J = 11.5 Hz, 2H), 2.66 (d, J = 4.5 Hz, 3H), 2.50–2.45 (m, 2H overlapped with DMSO signal), 2.44–2.35 (m, 4H), 1.93–1.64 (m, 5H), 1.61–1.41 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 149.5 (d, 1JC–F = 244 Hz), 149.3 (d, 1JC–F = 244 Hz), 139.8, 135.5, 131.8, 130.9, 129.4, 128.4, 128.1, 124.9, 117.8 (d, 2JC–F = 17 Hz), 117.5 (d, 2JC–F = 18 Hz), 70.6, 53.3 (2C), 47.3, 42.5, 41.2, 35.4 (2C), 34.3, 28.8 (2C), 21.8 (2C). HRMS C25H32F2N2 [M + H]+ calculated 399.2606; measured 399.2612, Δppm 1.4.
Synthesis of 1-(3-Chloro-4-fluoro-phenyl)-N-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (8)
Compound 8 was prepared according to the general procedure N using aldehyde 40a (0.064 g, 0.29 mmol), amine 30d (0.063 g, 0.29 mmol), NaBH(OAc)3 (0.125 g, 0.59 mmol) in anhydrous DCM (2.90 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 8 as yellow oil (0.077 g, 19% over 2 steps). UPLC/MS (method B): Rt 2.16 min. MS (ES): C25H32ClFN2 requires, 414 m/z; found, 415 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.51 (dd, J = 2.3, 7.2 Hz, 1H), 7.33 (ddd, J = 2.3, 4.6, 8.6 Hz, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.12–7.04 (m, 2H), 7.01–6.96 (m, 2H), 3.35 (br s, 2H), 2.91–2.81 (m, 2H), 2.50 (d, J = 6.9 Hz, 2H), 3.09 (br s, 3H), 2.05–1.84 (m, 8H), 1.77–1.69 (m, 2H), 1.68–1.59 (m, 2H), 1.55–1.44 (m, 1H), 1.43–1.29 (m, 2H). The free base was dissolved in CH2Cl2 (1.85 mL) and HCl [4 M in dioxane, 0.88 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method A): Rt 2.18 min. MS (ES): C25H32ClFN2 requires, 414 m/z; found, 415 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.51 (br s, 1H), 10.02 (br s, 2H), 8.00 (dd, J = 2.5, 7.0 Hz, 1H), 7.77 (ddd, J = 2.4, 4.5, 8.8 Hz, 1H), 7.53 (t, J = 8.9 Hz, 1H), 7.31–7.13 (m, 4H), 3.75–3.64 (m, 2H), 3.33 (d, J = 12.2 Hz, 2H), 2.90–2.75 (m, 2H), 2.66 (d, J = 4.7 Hz, 3H), 2.49–2.45 (m, 2H overlapped with DMSO signal), 2.44–2.37 (m, 4H), 1.91–1.81 (m, 2H), 1.79–1.68 (m, 3H), 1.57–1.43 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 157.2 (d, 1JC–F = 248 Hz), 139.8, 135.5, 131.8, 130.9, 130.3, 129.4, 128.8 (d, 3JC–F = 7 Hz), 128.4, 128.0, 120.0 (d, 2JC–F = 18 Hz), 117.2 (d, 2JC–F = 21 Hz), 70.6, 53.3 (2C), 47.2, 42.5, 41.2, 40.2, 35.3, 34.3, 28.8 (2C), 21.8 (2C). HRMS C25H32ClFN2 [M + H]+ calculated 415.2311; measured 415.2312, Δppm 0.3.
Synthesis of 1-(4-Fluoro-3-methoxy-phenyl)-N-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (9)
Compound 9 was prepared according to the general procedure N using aldehyde 40a (0.013 g, 0.06 mmol), amine 30i (0.013 g, 0.06 mmol), and NaBH(OAc)3 (0.026 g, 0.12 mmol) in anhydrous CH2Cl2 (1.00 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 9 as yellowish oil (0.005 g, 3% over 2 steps). UPLC/MS (method B): Rt 1.68 min. C26H35FN2O requires, 410 m/z; found, 411 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.21–7.13 (m, 2H), 7.09–7.04 (m, 1H), 7.00–6.91 (m, 4H), 3.89 (s, 3H), 3.36 (s, 2H), 2.97 (d, J = 11.4 Hz, 2H), 2.50 (d, J = 6.3 Hz, 2H), 2.36 (s, 3H), 2.11–1.79 (m, 8H), 1.78–1.61 (m, 4H), 1.54–1.41 (m, 3H). The free base was dissolved in CH2Cl2 (0.12 mL) and HCl [4 M in dioxane, 0.60 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.63 min. C26H35FN2O requires, 410 m/z; found, 411 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.81 (br s, 3H), 7.67 (s, 1H), 7.33–7.25 (m, 2H), 7.23–7.13 (m, 4H), 3.90 (s, 3H), 3.74–3.61 (m, 2H), 3.34–3.19 (m, 2H overlapped with H2O signal), 2.90–2.73 (m, 2H), 2.73–2.62 (m, 2H), 2.46–2.24 (m, 7H), 1.91–1.79 (m, 2H), 1.77–1.65 (m, 3H), 1.62–1.36 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 157.7 (1JC–F = 245 Hz), 153.0, 140.2, 134.9, 132.4, 131.4, 129.9, 128.9 (2C), 128.5, 120.5, 114.0, 71.7, 56.8, 53.8 (2C), 47.7, 43.0, 41.7, 35.9 (2C), 34.8, 29.3 (2C), 22.4 (2C). HRMS C26H35FN2O [M + H]+ calculated 411.2806; measured 411.2811, Δppm 1.2.
Synthesis of 2-Fluoro-5-[1-[[3-[(1-methyl-4-piperidyl)methyl]phenyl]methylamino]cyclopentyl]phenol Dihydrochloride (10)
Compound 10 was prepared according to the general procedure N using aldehyde 40a (0.100 g, 0.46 mmol), amine 30g (0.013 g, 0.06 mmol), and NaBH(OAc)3 (0.195 g, 0.92 mmol) in anhydrous CH2Cl2 (4.6 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 90:10 to afford 10 as colorless oil (0.055 g, 30%). UPLC/MS (method B): Rt 1.68 min. C25H33FN2O requires, 396 m/z; found, 395 m/z [M – H]−. 1H NMR (400 MHz, CDCl3) δ 7.14 (t, J = 7.5 Hz, 1H), 7.07–7.00 (m, 2H), 7.01–6.88 (m, 3H), 6.83 (ddd, J = 2.3, 4.4, 8.5 Hz, 1H), 3.37 (br s, 2H), 2.96–2.87 (m, 2H), 2.46 (d, J = 6.7 Hz, 2H), 2.28 (s, 3H), 2.10–1.79 (m, 8H), 1.76–1.64 (m, 2H), 1.64–1.54 (m, 2H), 1.54–1.42 (m, 1H), 1.42–1.29 (m, 2H). The free base was dissolved in CH2Cl2 (0.80 mL) and HCl [4 M in dioxane, 0.36 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.65 min. C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (br s, 1H), 9.70 (br s, 2H), 7.33–7.22 (m, 4H), 7.21–7.15 (m, 3H), 3.72–3.63 (m, 2H), 3.38–3.30 (m, 2H overlapped with H2O signal), 2.83 (q, J = 11.7 Hz, 2H), 2.67 (d, J = 4.5 Hz, 2H), 2.51–2.44 (m, 2H overlapped with DMSO signal), 2.40–2.28 (m, 6H), 1.89–1.81 (m, 2H), 1.79–1.69 (m, 3H), 1.59–1.37 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 150.9 (d, 1JC–F = 243 Hz), 144.9 (d, 3JC–F = 12 Hz), 139.8, 134.2, 131.9, 130.8, 129.5, 128.5, 127.9, 118.7, 117.5, 116.4 (d, 2JC–F = 18 Hz), 70.8, 53.3 (2C), 47.1, 42.5, 41.2, 35.3, 34.3, 28.8, 21.9 (2C), 21.5 (2C). HRMS C25H33FN2O [M + H]+ calculated 397.265; measured 397.265, Δppm 0.1.
Synthesis of N-[[4-Fluoro-3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]-1-(4-fluorophenyl)cyclopentanamine Dihydrochloride (11)
Compound 11 was prepared according to the general procedure N using aldehyde 40b (0.074 g, 0.31 mmol), amine 30e (0.06 g, 0.31 mmol), and NaBH(OAc)3 (0.13 g, 0.62 mmol) in anhydrous CH2Cl2 (3.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH3Cl/MeOH from 98:2 to 92:8 to afford 11 as colorless oil (0.054 g, 32% over 3 steps). UPLC/MS (method A): Rt 1.90 min. MS (ES): C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.45–7.38 (m, 2H), 7.06–6.98 (m, 3H), 6.95 (dd, J = 2.2, 7.4 Hz, 1H), 6.89 (dd, J = 8.3, 9.8 Hz, 1H), 3.29 (s, 2H), 2.89 (d, J = 11.2 Hz, 2H), 2.53 (d, J = 6.8 Hz, 2H), 2.30 (s, 3H), 2.06–1.82 (m, 8H), 1.78–1.59 (m, 4H), 1.58–1.47 (m, 1H), 1.46–1.34 (m, 2H). The free base was dissolved in DCM (1.35 mL) and HCl [4 M in dioxane, 0.60 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.79 min. MS (ES): C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (br s, 1H), 10.01 (br s, 2H), 7.85–7.76 (m, 2H), 7.37–7.21 (m, 4H), 7.15 (dd, J = 8.4, 9.9 Hz, 1H), 3.66 (m, 2H), 3.38–3.41 (m, 2H overlapped with H2O signal), 2.84 (q, J = 11.5 Hz, 2H), 2.66 (s, 3H), 2.51–2.42 (m, 2H overlapped with DMSO signal), 2.46–2.31 (m, 4H), 1.91–1.80 (m, 2H), 1.80–1.67 (m, 3H), 1.59–1.43 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 162.1 (d, 1JC–F = 246 Hz), 160.7 (d, 1JC–F = 245 Hz), 133.8, 130.5 (d, 3JC–F = 9 Hz), 129.9 (d, 3JC–F = 7 Hz, 2C), 128.9, 128.0 (d, 4JC–F = 3.4 Hz), 126.1 (d, 2JC–F = 17 Hz), 115.7 (d, 2JC–F = 21 Hz, 2C), 115.2 (d, 2JC–F = 23 Hz), 70.7, 53.2 (2C), 46.4, 42.5, 40.0, 35.4 (2C), 34.3, 28.8 (2C), 21.9 (2C). HRMS C25H32F2N2 [M + H]+ calculated 399.2606; measured 399.2614, Δppm 1.9.
Synthesis of 4-[[[1-(4-Fluorophenyl)cyclopentyl]amino]methyl]-2-[(1-methyl-4-piperidyl)methyl]phenol Dihydrochloride (12)
Compound 12 was prepared according to the general procedure N using aldehyde 40i (0.067 g, 0.29 mmol), amine 30e (0.052 g, 0.29 mmol), and NaBH(OAc)3 (0.123 g, 0.58 mmol) in anhydrous CH2Cl2/iPrOH (2.90 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 98:2 to 90:10 to afford 12 as yellowish oil (0.022 g, 12% over 4 steps). UPLC/MS (method B): Rt 1.44 min. MS (ES) C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+, 395 m/z [M – H]−. 1H NMR (400 MHz, CDCl3) δ 7.47–7.38 (m, 2H), 7.07–6.98 (m, 2H), 6.92–6.82 (m, 2H), 6.69–6.57 (m, 1H), 3.25 (br s, 2H), 3.11–2.29 (m, 2H), 2.51 (d, J = 5.2 Hz, 2H), 2.44–2.31 (m, 3H), 2.08–1.81 (m, 8H), 1.76–1.47 (m, 7H). The free base was dissolved in CH2Cl2 (0.55 mL) and HCl [4 M in dioxane, 0.3 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.71 min. MS (ES): C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+, 395 m/z [M – H]−. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (br s, 1H), 9.69 (br s, 2H), 7.82–7.75 (m, 2H), 7.37–7.29 (m, 2H), 6.99–6.93 (m, 2H), 6.79 (d, J = 8.0 Hz, 1H), 3.58–3.49 (m, 2H), 3.40–3.27 (m, 2H overlapped with H2O signal), 2.82 (q, J = 11.7 Hz, 2H), 2.66 (s, 3H), 2.40 (d, J = 6.8 Hz, 2H), 2.38–2.31 (m, 4H), 1.89–1.81 (m, 2H), 1.78–1.63 (m, 3H), 1.58–1.43 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 161.9 (d, 1JC–F = 246 Hz), 155.9, 133.9, 132.9, 129.9 (d, 3JC–F = 8.5 Hz, 2C), 129.3, 125.6, 121.7, 115.7 (d, 2JC–F = 21.4 Hz, 2C), 114.8, 70.5, 53.4 (2C), 49.5, 46.9, 42.5, 40.2, 35.8, 35.4, 32.9, 29.0, 21.8 (2C). HRMS C25H33FN2O [M + H]+ calculated 397.265; measured 397.2649, Δppm −0.2.
Synthesis of N-[[4-Chloro-3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]-1-(4-fluorophenyl)cyclopentanamine Dihydrochloride (13)
Compound 13 was prepared according to the general procedure N using aldehyde 40c (0.035 g, 0.14 mmol), amine 30e (0.025 g, 0.14 mmol), and NaBH(OAc)3 (0.059 g, 0.28 mmol) in anhydrous CH2Cl2 (1.40 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 92:8 to afford 13 as yellow oil (0.024 g, 15% over 4 steps). UPLC/MS (method B): Rt 2.13 min. MS (ES): C25H32ClFN2 requires, 414 m/z; found, 415 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.46–7.41 (m, 2H), 7.25 (d, J = 8.0 Hz, 1H), 7.08–7.00 (m, 4H), 3.33 (s, 2H), 2.91 (d, J = 11.3 Hz, 2H), 2.65 (d, J = 6.5 Hz, 2H), 2.32 (s, 3H), 2.08–1.84 (m, 8H), 1.81–1.70 (m, 2H), 1.69–1.56 (m, 3H), 1.52–1.37 (m, 2H). The free base was dissolved in CH2Cl2 (0.57 mL) and HCl [4 M in dioxane, 0.3 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.15 min. MS (ES): C25H32ClFN2 requires, 414 m/z; found, 415 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (br s, 1H), 10.06 (br s, 2H), 7.84–7.76 (m, 2H), 7.42 (dd, J = 2.6, 8.2 Hz, 1H), 7.38–7.28 (m, 3H), 7.27–7.02 (m, 1H), 3.71–3.61 (m, 2H), 3.36–3.27 (m, 2H overlapped with H2O signal), 2.85 (q, J = 11.6 Hz, 2H), 2.70–2.62 (m, 3H), 2.59 (d, J = 7.0 Hz, 2H), 2.47–2.31 (m, 4H), 1.92–1.68 (m, 5H), 1.64–1.44 (m, 4H). 13C NMR (101 MHz, DMSO) δ 162.0 (d, 1JC–F = 246 Hz), 136.9, 133.8 (d, 4JC–F = 3 Hz), 133.5, 130.9, 130.0 (d, 3JC–F = 8 Hz, 2C), 130.0, 129.9, 129.3, 115.7 (d, 2JC–F = 21 Hz, 2C), 70.8, 53.2 (2C), 46.4, 42.5, 38.7, 35.4 (2C), 33.1, 28.8 (2C), 21.9 (2C). HRMS C25H32ClFN2 [M + H]+ calculated 415.2311; measured 415.2311, Δppm 0.0.
Synthesis of N-[[2-Fluoro-5-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]-1-(4-fluorophenyl)cyclopentanamine Dihydrochloride (14)
Compound 14 was prepared according to the general procedure N using aldehyde 40d (0.058 g, 0.24 mmol), amine 30e (0.044 g, 0.24 mmol), and NaBH(OAc)3 (0.104 g, 0.49 mmol) in anhydrous CH2Cl2 (2.40 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 14 as yellow oil (0.044 g, 11% over 4 steps). UPLC/MS (method B): Rt 1.83 min. MS (ES): C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.45–7.39 (m, 2H), 7.05–6.96 (m, 3H), 6.95–6.82 (m, 2H), 3.37 (s, 2H), 2.87–2.81 (m, 2H), 2.45 (d, J = 6.9 Hz, 2H), 2.26 (s, 3H), 2.07–1.81 (m, 8H), 1.78–1.66 (m, 2H), 1.65–1.55 (m, 2H), 1.49–1.36 (m, 1H), 1.32 (qd, J = 3.8, 12.3 Hz, 2H). The free base was dissolved in CH2Cl2 (1.10 mL) and HCl [4 M in dioxane, 0.52 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.05 min. MS (ES): C25H32F2N2 requires, 398 m/z; found, 399 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.63 (br s, 1H), 10.07 (br s, 2H), 7.46 (dd, J = 2.3, 7.3 Hz, 1H), 7.35–7.27 (m, 2H), 7.22 (ddd, J = 2.3, 5.1, 7.7 Hz, 1H), 7.14–7.06 (m, 2H), 3.76–3.59 (m, 2H), 3.36–3.29 (m, 2H), 2.83 (q, J = 11.7 Hz, 2H), 2.66 (d, J = 4.7 Hz, 4H), 2.47 (d, J = 6.8 Hz, 1H), 2.46–2.37 (m, 5H), 1.93–1.81 (m, 2H), 1.80–1.67 (m, 3H), 1.58–1.42 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 246 Hz), 159.2 (d, 1JC–F = 246 Hz), 135.5 (d, 4JC–F = 3 Hz), 133.7 (d, JC–F = 3 Hz), 133.5 (d, JC–F = 3 Hz), 131.7 (d, 3JC–F = 8 Hz), 130.0 (d, 3JC–F = 8 Hz, 2C), 118.7 (d, 2JC–F = 15 Hz), 115.6 (d, 2JC–F = 21.3 Hz, 2C), 115.1 (d, 2JC–F = 21 Hz), 70.8, 53.3 (2C), 42.5, 40.4, 35.2 (3C), 34.3, 28.7 (2C), 21.9 (2C). HRMS C25H32F2N2 [M + H]+ calculated 399.2606; measured 399.2614, Δppm 1.9.
Synthesis of 2-[[[1-(4-Fluorophenyl)cyclopentyl]amino]methyl]-4-[(1-methyl-4-piperidyl)methyl]phenol Dihydrochloride (15)
Compound 15 was prepared according to the general procedure N using aldehyde 40j (0.025 g, 0.11 mmol), amine 30e (0.019 g, 0.11 mmol), and NaBH(OAc)3 (0.046 g, 0.22 mmol) in anhydrous CH2Cl2 (1.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 92:8 to afford 15 as colorless oil (0.019 g, 43%). UPLC/MS (method B): Rt 2.06 min. MS (ES): C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+, 395 m/z [M – H]−. 1H NMR (400 MHz, CDCl3) δ 7.37–7.31 (m, 2H), 7.07–7.01 (m, 2H), 6.87 (dd, J = 2.2, 8.2 Hz, 1H), 6.70 (d, J = 8.2 Hz, 1H), 6.58 (d, J = 2.2 Hz, 1H), 2.86–2.86 (m, 2H), 2.34 (d, J = 6.8 Hz, 2H), 2.23 (s, 3H), 2.20–2.11 (m, 4H), 2.03–1.75 (m, 8H), 1.63–1.53 (m, 2H), 1.40–1.15 (m, 3H). The free base was dissolved in CH2Cl2 (0.50 mL) and HCl [4 M in dioxane, 0.25 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.96 min. MS (ES): C25H33FN2O requires, 396 m/z; found, 397 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.47 (br s, 1H), 9.89 (s, 1H), 9.35 (br s, 2H), 7.84–7.76 (m, 2H), 7.36–7.26 (m, 2H), 7.07 (d, J = 2.2 Hz, 1H), 6.98 (dd, J = 2.2, 8.2 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H), 3.59 (s, 2H), 3.37–3.28 (m, 2H), 2.82 (q, J = 11.6 Hz, 2H), 2.67 (d, J = 4.8 Hz, 3H), 2.44–2.34 (m, 6H), 1.91–1.81 (m, 2H), 1.75–1.59 (m, 3H), 1.61–1.39 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 246 Hz), 154.3, 133.9, 132.5, 130.8, 130.1 (d, 3JC–F = 8.4 Hz, 2C), 129.54, 117.9, 115.5 (d, 2JC–F = 21 Hz, 2C), 115.1, 70.7, 53.4 (2C), 42.5, 42.3, 40.6, 39.8 (overlapped with DMSO signal), 35.3, 34.6, 28.8 (2C), 21.9 (2C). HRMS C25H33FN2O [M + H]+ calculated 397.265; measured 397.2655, Δppm 1.3.
Synthesis of N-[[2,4-Difluoro-5-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]-1-(4-fluorophenyl)cyclopentanamine Dihydrochloride (16)
Compound 16 was prepared according to the general procedure N using aldehyde 40e (0.071 g, 0.28 mmol), amine 30e (0.050 g, 0.28 mmol), and NaBH(OAc)3 (0.119 g, 0.56 mmol) in anhydrous CH2Cl2 (2.28 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 16 as colorless oil (0.070 g, 20% over 4 steps). UPLC/MS (method B): Rt 2.06 min. MS (ES): C25H31F3N2 requires, 416 m/z; found, 417 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.46–7.36 (m, 2H), 7.07–6.94 (m, 3H), 6.67 (t, J = 9.7 Hz, 1H), 3.33 (br s, 2H), 2.92–2.81 (m, 2H), 2.49 (d, J = 6.8 Hz, 2H), 2.27 (s, 3H), 2.06–1.80 (m, 8H), 1.77–1.67 (m, 2H), 1.65–1.55 (m, 2H), 1.53–1.28 (m, 3H). The free base was dissolved in CH2Cl2 (1.68 mL) and HCl [4 M in dioxane, 0.80 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.08 min. MS (ES): C25H31F3N2 requires, 416 m/z; found, 417 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.52 (br s, 1H), 10.09 (br s, 2H), 7.84–7.77 (m, 2H), 7.59 (t, J = 8.6 Hz, 1H), 7.35–7.27 (m, 2H), 7.22 (t, J = 9.9 Hz, 1H), 3.72–3.63 (m, 2H), 3.33 (d, J = 12.0 Hz, 2H), 2.84 (q, J = 11.7 Hz, 2H), 2.67 (d, J = 4.8 Hz, 3H), 2.50–2.46 (m, 2H overlapped with DMSO signal), 2.45–2.32 (m, 4H), 1.93–1.82 (m, 2H), 1.81–1.69 (m, 3H), 1.59–1.44 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 246 Hz), 160.8 (dd, 1JC–F = 247 Hz, 3JC–F = 12 Hz), 158.8 (dd, 1JC–F = 248 Hz, 3JC–F = 12 Hz), 135.7, 133.7 (d, 4JC–F = 3 Hz), 130.0 (d, 3JC–F = 8 Hz, 2C) 122.1 (dd, JC–F = 4 Hz, JC–F = 17 Hz), 115.6 (d, 2JC–F = 21 Hz, 2C), 115.2 (dd, 4JC–F = 4 Hz, 3JC–F = 15 Hz), 103.6 (t, 2JC–F = 27 Hz), 70.8, 53.2 (2C), 42.5, 40.2, 35.3 (2C), 33.6, 33.3, 28.7 (2C), 21.9 (2C). HRMS C25H31F3N2 [M + H]+ calculated 417.2512; measured 417.2516, Δppm 0.9.
Synthesis of 1-(4-Fluorophenyl)-N-[[3-(4-piperidylmethyl)phenyl]methyl]cyclopentanamine Dihydrochloride (17)
Compound 41f was dissolved in CH2Cl2 (0.50 mL) and HCl [4 M in dioxane, 0.23 mL] was added. Evaporation of solvent afforded compound 17 as whitish solid. UPLC/MS (method B): Rt 1.70 min. MS (ES): C24H31FN2 requires, 366 m/z; found, 367 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.91 (br s, 2H), 8.95 (br s, 1H), 8.74 (br s, 1H), 7.84–7.77 (m, 2H), 7.46–7.24 (m, 3H), 7.23–7.13 (m, 3H), 3.70–3.62 (m, 2H), 3.26–3.16 (m, 2H), 2.84–2.70 (m, 2H), 2.45–2.36 (m, 4H), 1.94–1.75 (m, 5H), 1.74–1.64 (m, 2H), 1.55–1.44 (m, 2H), 1.43–1.27 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 245 Hz), 139.8, 133.9 (d, 4JC–F = 3 Hz), 131.9, 131.0, 130.0 (d, 3JC–F = 8 Hz, 2C), 129.4, 128.4, 128.0, 115.7 (d, 2JC–F = 21 Hz, 2C), 70.8, 47.2, 42.9 (2C), 41.4, 35.3 (2C), 34.7, 28.1 (2C), 21.8 (2C). HRMS C24H31FN2 [M + H]+ calculated 367.2544; measured 367.2551, Δppm 1.9.
Synthesis of rac-1-(4-Fluorophenyl)-N-[[3-[[1,2,2-trimethyl-4-piperidyl]methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (18)
Compound 18 was prepared according to the general procedure N using aldehyde 40k (0.056 g, 0.23 mmol), amine 30e (0.041 g, 0.23 mmol), and NaBH(OAc)3 (0.098 g, 0.46 mmol) in anhydrous CH2Cl2 (2.30 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 96:4 to afford 18 as yellow oil (0.049 g, 11% over 4 steps). UPLC/MS (method B): Rt 1.83 min. MS (ES): C27H37FN2 requires, 408 m/z; found, 409 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.46–7.38 (m, 2H), 7.18 (t, J = 7.8 Hz, 1H), 7.09–6.95 (m, 5H), 3.34 (br s, 2H), 2.66 (d, J = 12.0 Hz, 1H), 2.46–2.35 (m, 3H), 2.26 (br s, 3H), 2.07–1.83 (m, 6H), 1.78–1.68 (m, 3H), 1.63–1.55 (m, 1H), 1.47–1.40 (m, 1H), 1.38–1.17 (m, 2H), 1.15 (s, 3H), 0.90 (s, 3H). The free base was dissolved in CH2Cl2 (1.20 mL) and HCl [4 M in dioxane, 0.52 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.83 min. MS (ES): C27H37FN2 requires, 408 m/z; found, 409 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (br s, 1H), 10.06 (br s, 1H), 9.89 (br s, 1H), 7.85–7.79 (m, 2H), 7.36–7.30 (m, 2H), 7.28 (t, J = 7.6 Hz, 1H), 7.22 (br s, 1H), 7.20–7.13 (m, 2H), 3.70–3.61 (m, 1H), 3.15–2.95 (m, 2H), 2.56 (d, J = 4.9 Hz, 3H), 2.50–2.46 (m, 2H overlapped with DMSO signal), 2.44–2.34 (m, 4H), 2.11–1.98 (m, 1H), 1.92–1.77 (m, 2H), 1.73–1.51 (m, 6H), 1.37 (s, 3H), 1.25 (d, J = 12.6 Hz, 1H), 1.21 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 161.9 (d, 1JC–F = 245 Hz), 139.6, 133.9 (d, 4JC–F = 3 Hz), 131.8, 130.9, 130.0 (d, 3JC–F = 8 Hz, 2C), 129.4, 128.3, 128.0, 115.7 (d, 2JC–F = 21 Hz, 2C), 70.7, 60.5, 49.9, 47.2, 42.7, 41.4, 40.1, 35.7, 30.9, 28.8, 25.9, 21.9, 21.8 (2C), 17.2. HRMS C27H37FN2 [M + H]+ calculated 409.3014; measured 409.3016, Δppm 0.6.
Synthesis of 1-(4-Fluorophenyl)-N-[[3-[[rac-3,3-difluoro-1-methyl-4-piperidyl]methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (19)
Compound 19 was prepared according to the general procedure N using aldehyde 40l (0.032 g, 0.13 mmol), amine 30e (0.023 g, 0.13 mmol), and NaBH(OAc)3 (0.054 g, 0.26 mmol) in anhydrous CH2Cl2 (1.30 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 97:3 to afford 19 as yellowish oil (0.032 g, 15% over 4 steps). UPLC/MS (method A): Rt 2.28 min. MS (ES): C25H31F3N2 requires, 416 m/z; found, 417 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.47–7.40 (m, 2H), 7.20 (t, J = 7.8 Hz, 1H), 7.09 (d, J = 7.7 Hz, 1H), 7.06–6.98 (m, 4H), 3.36 (br s, 2H), 3.16 (dd, J = 3.4, 13.7 Hz, 1H), 3.07 (dddd, J = 1.6, 5.1, 10.0, 11.7 Hz, 1H), 2.78 (d, J = 11.8 Hz, 2H), 2.40 (dd, J = 10.7, 13.7 Hz, 1H), 2.32 (s, 3H), 2.17 (dd, J = 2.3, 11.9 Hz, 1H), 2.08–1.81 (m, 6H), 1.77–1.67 (m, 2H), 1.63–1.44 (m, 3H). The free base was dissolved in CH2Cl2 (0.77 mL) and HCl [4 M in dioxane, 0.36 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.33 min. MS (ES): C25H31F3N2 requires, 416 m/z; found, 417 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.16 (br s, 1H), 10.06 (br s, 1H), 9.96 (br s, 1H), 7.85–7.79 (m, 2H), 7.33–7.26 (m, 4H), 7.25–7.18 (m, 2H), 3.96 (t, J = 11.9 Hz, 1H), 3.74–3.72 (m, 2H), 3.33 (d, J = 12.4 Hz, 1H), 3.08–2.96 (m, 2H), 2.79 (br s, 3H), 2.47–2.31 (m, 7H), 1.93–1.60 (m, 4H), 1.57–1.41 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 246 Hz), 138.2, 133.8 (d, 4JC–F = 3.1 Hz), 132.0, 131.0, 130.0 (d, 3JC–F = 8.3 Hz, 2C), 129.6, 128.6, 128.4, 119.6, 115.7 (d, 2JC–F = 21.3 Hz, 2C), 70.8, 52.4 (2C), 47.1, 42.9, 40.1, 35.4, 35.3 (3C), 21.9 (2C). HRMS C25H31F3N2 [M + H]+ calculated 417.2512; measured 417.2517, Δppm 1.2.
Synthesis of 1-(4-Fluorophenyl)-N-[[3-[(1-methylazetidin-3-yl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (20)
Compound 20 was prepared according to the general procedure N using aldehyde 40m (0.046 g, 0.24 mmol), amine 30e (0.044 g, 0.24 mmol), and NaBH(OAc)3 (0.103 g, 0.49 mmol) in anhydrous CH2Cl2 (2.40 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 90:10 to afford 20 as yellow oil (0.028 g, 6% over 4 steps). UPLC/MS (method B): Rt 1.70 min. MS (ES): C23H29FN2 requires, 352 m/z; found, 353 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.45–7.40 (m, 2H), 7.18 (t, J = 7.5 Hz, 1H), 7.09–6.94 (m, 5H), 3.59–3.46 (m, 2H), 3.33 (s, 2H), 3.13–3.00 (m, 2H), 2.88–2.73 (m, 3H), 2.43–2.36 (m, 3H), 2.06–1.81 (m, 6H), 1.78–1.66 (m, 2H). The free base was dissolved in CH2Cl2 (0.79 mL) and HCl [4 M in dioxane, 0.38 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.52 min. MS (ES): C23H29FN2 requires, 352 m/z; found, 353 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (br s, 1H), 10.91 (br s, 1H), 7.85–7.77 (m, 2H), 7.36–7.25 (m, 4H), 7.24–7.14 (m, 2H), 4.16–4.04 (m, 1H), 3.98–3.83 (m, 1H), 3.82–3.72 (m, 1H), 3.69–3.62 (m, 3H), 3.05–2.90 (m, 1H), 2.90–2.83 (m, 1H), 2.81–2.72 (m, 3H), 2.46–2.33 (m, 4H), 1.93–1.77 (m, 2H), 1.57–1.40 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 245 Hz), 138.9, 133.8, 132.1, 130.5, 129.9 (d, 3JC–F = 8 Hz, 2C), 129.1, 128.7, 128.4, 115.7 (d, 2JC–F = 21 Hz, 2C), 70.8, 60.0, 58.8, 47.1, 40.1, 37.6, 35.4 (2C), 30.0, 21.9 (2C). HRMS C23H29FN2 [M + H]+ calculated 353.2388; measured 353.2395, Δppm 2.1.
Synthesis of 1-(4-Fluorophenyl)-N-[[3-[(2-methyl-2-azaspiro[3.3]heptan-6-yl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (21)
Compound 21 was prepared according to the general procedure N using aldehyde 40n (0.031 g, 0.13 mmol), amine 30e (0.024 g, 0.13 mmol), and NaBH(OAc)3 (0.057 g, 0.27 mmol) in anhydrous CH2Cl2 (1.30 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 95:5 to afford 21 as yellowish oil (0.016 g, 6% over 4 steps). UPLC/MS (method B): Rt 1.80 min. MS (ES): C26H33FN2 requires, 392 m/z; found, 393 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.45–7.39 (m, 2H), 7.16 (t, J = 7.8 Hz, 1H), 7.07–6.98 (m, 3H), 6.96–6.92 (m, 2H), 3.33 (s, 2H), 3.26 (s, 1H), 3.18 (s, 1H), 2.61 (d, J = 7.5 Hz, 2H), 2.41–2.31 (m, 2H), 2.30 (s, 3H), 2.25–2.16 (m, 2H), 2.06–1.78 (m, 9H), 1.76–1.69 (m, 2H). The free base was dissolved in CH2Cl2 (0.40 mL) and HCl [4 M in dioxane, 0.20 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.82 min. C26H33FN2 requires, 392 m/z; found, 393 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.81 (br s, 1H), 9.91 (br s, 2H), 7.84–7.76 (m, 2H), 7.37–7.29 (m, 2H), 7.25 (t, J = 7.5 Hz, 1H), 7.18–7.01 (m, 3H), 4.18–4.10 (m, 1H), 4.06–3.97 (m, 1H), 3.92–3.81 (m, 2H), 3.68–3.59 (m, 2H), 2.71 (s, 3H), 2.65–2.55 (m, 2H), 2.42–2.13 (m, 7H), 1.95–1.81 (m, 4H), 1.55–1.44 (m, 2H). 13C NMR (151 MHz, DMSO-d6) 162.0 (d, 1JC–F = 245 Hz), 140.5, 130.4, 130.0 (2C), 128.8, 128.4, 127.8, 115.7 (d, 2JC–F = 22 Hz, 2C), 70.8, 66.2, 65.1, 47.2, 41.3, 40.7, 40.0, 35.4, 33.9, 30.2, 21.9 (2C). HRMS C26H33FN2 [M + H]+ calculated 393.2701; measured 393.2706, Δppm 1.4.
Synthesis of 1-(4-Fluoro-2-methoxy-phenyl)-N-[[4-fluoro-3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (22)
Compound 22 was prepared according to the general procedure N using aldehyde 40b (0.073 g, 0.31 mmol), amine 30b (0.073 g, 0.31 mmol), and NaBH(OAc)3 (0.131 g, 0.62 mmol) in anhydrous CH2Cl2 (3.10 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 85:15 to afford 22 as yellowish oil (0.105 g, 60% over 3 steps). UPLC/MS (method B): Rt 1.80 min. MS (ES): C26H34F2N2O requires, 428 m/z; found, 429 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.19 (dd, J = 6.8, 8.3 Hz, 1H), 6.98–6.84 (m, 3H), 6.67–6.84 (m, 2H), 3.80 (s, 3H), 3.22 (s, 2H), 3.05–2.89 (m, 2H), 2.53 (d, J = 6.3 Hz, 2H), 2.35 (br s, 3H), 2.19–1.83 (m, 8H), 1.71–1.40 (m, 7H). The free base was dissolved in CH2Cl2 (2.45 mL) and HCl [4 M in dioxane, 1.16 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.55 min. C26H34F2N2O requires, 428 m/z; found, 429 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (br s, 1H), 9.18 (br s, 2H), 7.30 (dd, J = 6.6, 8.8 Hz, 1H), 7.23–7.18 (m, 1H), 7.17–7.07 (m, 2H), 6.93 (dd, J = 2.6, 11.0 Hz, 1H), 6.82 (td, J = 2.6, 8.4 Hz, 1H), 3.82 (s, 3H), 3.77–3.71 (m, 2H), 3.37–3.28 (m, 2H), 2.91–2.77 (m, 2H), 2.66 (d, J = 4.7 Hz, 3H), 2.47 (d, J = 6.7 Hz, 2H), 2.43–2.26 (m, 4H), 1.89–1.79 (m, 2H), 1.78–1.65 (m, 3H), 1.62–1.43 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 163.3 (d, 1JC–F = 245 Hz) 160.6 (d, 1JC–F = 245 Hz), 158.0 (d, 3JC–F = 13 Hz), 133.9 (d, 3JC–F = 5.3 Hz), 130.3 (d, 3JC–F = 8.7 Hz), 129.6 (d, 3JC–F = 10.3 Hz), 128.0 (d, 4JC–F = 3.3 Hz), 128.0 (d, 4JC–F = 3.3 Hz), 120.9 (d, 4JC–F = 3.0 Hz), 114.9 (d, 2J = 23 Hz), 106.5 (d, 2JC–F = 21 Hz), 100.3 (d, 2JC–F = 26 Hz), 70.3, 55.9, 53.1 (2C), 46.5, 42.4, 40.0, 35.0, 34.4, 33.5, 28.7 (2C), 22.54 (2C). HRMS C26H34F2N2O [M + H]+ calculated 429.2712; measured 429.2709, Δppm −0.7.
Synthesis of 1-(3,4-Difluorophenyl)-N-[[4-fluoro-3-[(1-methyl-4-piperidyl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (23)
Compound 23 was prepared according to the general procedure N using aldehyde 40b (0.045 g, 0.19 mmol), amine 30c (0.038 g, 0.19 mmol), and NaBH(OAc)3 (0.080 g, 0.38 mmol) in anhydrous CH2Cl2 (1.90 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 23 as yellowish oil (0.011 g, 6% over 3 steps). UPLC/MS (method B): Rt 2.11 min. MS (ES): C25H31F3N2 requires, 416 m/z; found, 417 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.31–7.25 (m, 1H overlapped with CHCl3 signal), 7.18–7.07 (m, 2H), 7.04 (ddd, J = 2.2, 5.0, 7.9 Hz, 1H), 6.98 (d, J = 7.2 Hz, 1H), 6.91 (dd, J = 8.3, 9.3 Hz, 1H), 3.30 (s, 2H), 2.93–2.80 (m, 2H), 2.53 (d, J = 6.8 Hz, 2H), 2.30 (s, 3H), 2.03–1.81 (m, 8H), 1.77–1.68 (m, 2H), 1.67–1.59 (m, 2H), 1.58–1.47 (m, 1H), 1.40 (qd, J = 3.8, 12.0 Hz, 2H). The free base was dissolved in CH2Cl2 (0.26 mL) and HCl [4 M in dioxane, 0.12 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 1.98 min. MS (ES): C25H31F3N2 requires, 416 m/z; found, 417 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (br s, 1H), 10.07 (br s, 2H), 7.95–7.84 (m, 1H), 7.63–7.49 (m, 2H), 7.37 (d, J = 7.3 Hz, 1H), 7.33–7.25 (m, 1H), 7.20–7.12 (m, 1H), 3.76–3.64 (m, 2H), 3.38–3.25 (m, 2H overlapped with H2O signal), 2.84 (q, J = 11.5 Hz, 2H), 2.73–2.63 (m, 3H), 2.54–2.45 (m, 2H, overlapped with DMSO signal), 2.44–2.34 (m, 4H), 1.92–1.66 (m, 5H), 1.61–1.42 (m, 4H). 13C NMR (151 MHz, DMSO-d6) δ 161.1 (d, 1JC–F = 245 Hz), 149.9 (d, 1JC–F = 245 Hz), 149.8 (d, 1JC–F = 245 Hz), 135.9, 134.4, 131.0 (d, 3JC–F = 8.5 Hz), 128.4, 126.5 (d, 2JC–F = 17 Hz), 125.4, 118.2 (d, 2JC–F = 17 Hz), 117.9 (d, 2JC–F = 18 Hz), 115.6 (d, 2JC–F = 23 Hz), 71.1, 53.6 (2C), 46.9, 42.9, 40.5, 35.9, 34.7, 33.9, 29.2 (2C), 22.3 (2C). HRMS C25H31F3N2 [M + H]+ calculated 417.2512; measured 417.2517, Δppm 1.2.
Synthesis of 1-(4-Fluorophenyl)-N-[[4-fluoro-3-(4-piperidylmethyl)phenyl]methyl]cyclopentanamine Dihydrochloride (24)
Compound 41g was dissolved in CH2Cl2 (14.71 mL) and HCl [4 M in dioxane, 7.00 mL] was added. Evaporation of solvent afforded compound 24 as whithish solid. UPLC/MS (method B): Rt 1.74 min. MS (ES): C24H30F2N2 requires, 384 m/z; found, 385 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.05 (br s, 2H), 9.07 (br s, 1H), 8.86 (br s, 1H), 7.84–7.77 (m, 2H), 7.36–7.29 (m, 3H), 7.25 (ddd, J = 2.2, 5.0, 7.6 Hz, 1H), 7.14 (dd, J = 8.4, 9.9 Hz, 1H), 3.69–3.61 (m, 2H), 3.24–3.15 (m, 2H), 2.85–2.71 (m, 2H), 2.54–2.49 (m, 2H overlapped with DMSO signal), 2.48–2.30 (m, 4H), 1.92–1.77 (m, 3H), 1.74–1.64 (m, 2H), 1.56–1.31 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.0 (d, 1JC–F = 246 Hz), 160.7 (d, 1JC–F = 245 Hz), 133.9, 130.4 (d, 3JC–F = 9 Hz), 129.9 (d, 3JC–F = 9 Hz, 2C), 127.9 (d, 4JC–F = 3 Hz), 126.1, 125.9, 115.7 (d, 2JC–F = 22 Hz, 2C), 115.2 (d, 2JC–F = 22 Hz), 70.7, 46.4, 42.9 (2C), 35.4 (2C), 34.3, 33.8, 28.1 (2C), 21.8 (2C). HRMS C24H30F2N2 [M + H]+ calculated 385.245; measured 385.2461, Δppm 2.9.
Synthesis of 1-(4-Fluorophenyl)-N-[[2-fluoro-5-(4-piperidylmethyl)phenyl]methyl]cyclopentanamine Dihydrochloride (25)
Compound 41h was dissolved in CH2Cl2 (1.13 mL) and HCl [4 M in dioxane, 0.54 mL] was added. Evaporation of solvent afforded compound 25 as whitish solid. UPLC/MS (method B): Rt 1.81 min. MS (ES): C24H30F2N2 requires, 384 m/z; found, 385 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (br s, 2H), 9.04 (br s, 1H), 8.83 (br s, 1H), 7.85–7.75 (m, 2H), 7.46 (d, J = 7.3 Hz, 1H), 7.36–7.26 (m, 2H), 7.25–7.18 (m, 1H), 7.15–7.06 (m, 1H), 3.77–3.63 (m, 2H), 3.27–3.14 (m, 2H), 2.86–2.71 (m, 2H), 2.57–2.34 (m, 6H overlapped with DMSO signal), 1.96–1.77 (m, 3H), 1.75–1.63 (m, 2H), 1.59–1.46 (m, 2H), 1.44–1.29 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.7 (d, 1JC–F = 244 Hz), 159.8 (d, 1JC–F = 246 Hz), 136.4 (2C), 133.8, 133.5 (d, 4JC–F = 3 Hz), 132.6 (d, 3JC–F = 8 Hz), 130.59 (d, 3JC–F = 8 Hz, 2C), 116.32 (d, 2JC–F = 21 Hz, 2C), 115.89 (d, 2JC–F = 21 Hz), 71.6, 43.8 (2C), 41.0, 40.9, 35.9 (2C), 35.2, 28.5 (2C), 22.4 (2C). HRMS C24H30F2N2 [M + H]+ calculated 385.245; measured 385.2452, Δppm 0.6.
Synthesis of 1-(3,4-Difluorophenyl)-N-[[3-[(2-methyl-2-azaspiro[3.3]heptan-6-yl)methyl]phenyl]methyl]cyclopentanamine Dihydrochloride (26)
Compound 26 was prepared according to the general procedure N using aldehyde 40n (0.087 g, 0.38 mmol), amine 30e (0.075 g, 0.38 mmol), and NaBH(OAc)3 (0.161 g, 0.76 mmol) in anhydrous CH2Cl2 (3.80 mL). The crude was purified by flash column chromatography (SiO2), eluting with CH2Cl2/MeOH from 100 to 93:7 to afford 26 as yellowish oil (0.050 g, 17% over 4 steps). UPLC/MS (method B): Rt 2.10 min. MS (ES): C26H32F2N2 requires, 410 m/z; found, 411 m/z [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.34–7.26 (m, 1H), 7.20–7.13 (m, 2H), 7.12–7.08 (m, 1H), 7.07–7.03 (m, 1H), 6.99–6.92 (m, 2H), 3.43–3.37 (m, 2H), 3.35–3.29 (m, 4H), 2.61 (d, J = 7.4 Hz, 2H), 2.42–2.33 (m, 4H), 2.30–2.18 (m, 2H), 2.04–1.79 (m, 8H), 1.78–1.68 (m, 2H). The free base was dissolved in CH2Cl2 (1.22 mL) and HCl [4 M in dioxane, 0.58 mL] was added. Evaporation of solvent afforded compound as whitish solid. UPLC/MS (method B): Rt 2.03 min. MS (ES): C26H32F2N2 requires, 410 m/z; found, 411 m/z [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 10.82 (br s, 1H), 10.04 (br s, 2H), 7.98–7.83 (m, 1H), 7.64–7.48 (m, 2H), 7.30–7.08 (m, 4H), 4.21–3.98 (m, 2H), 3.95–3.75 (m, 2H), 3.74–3.62 (m, 2H), 2.71 (s, 3H), 2.58 (d, J = 6.9 Hz, 2H), 2.44–2.18 (m, 6H), 1.97–1.78 (m, 5H), 1.58–1.41 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 149.4 (d, 1JC–F = 243 Hz), 149.3 (d, 1JC–F = 244 Hz), 148.6, 140.4, 135.5, 131.8, 130.5, 128.8, 128.3, 127.8, 124.9, 117.8 (d, 2JC–F = 16.5 Hz), 117.5 (d, 2JC–F = 18.2 Hz), 70.6, 66.0, 65.0, 47.3, 41.3, 40.7, 40.0, 35.3 (2C), 33.9, 30.2, 21.8 (2C). HRMS C26H32F2N2 [M + H]+ calculated 411.2606; measured 411.2613, Δppm 1.6.
Biology
Cell Lines
BT-474, MDA-MB-231, A375, CAPAN-2, HEPG2, HEK-293, HT-29, and HCT116 cells were acquired from the American Type Culture Collection and the National Collection of Type Cultures (ATCC). Uveal melanoma OMM-1 and UPMM-2 were kindly provided by Prof. Pfeffer Ulrich, University of Genoa (Italy). BT-474 cells were grown in DMEM high glucose (4.5 g/L d-glucose) containing 4 mM l-glutamine, 10% FBS, and 0.25 mM sodium pyruvate. All the other cancer cell lines were grown in DMEM high glucose containing 4 mM l-glutamine, 10% FBS, and 0.5 mM sodium pyruvate. primary human mammary epithelial cells (HMECs) were acquired from Invitrogen and growth in HuMEC basal serum-free medium supplemented with HuMEC supplement kit (12752010, Invitrogen). Cells were regularly tested for mycoplasma contamination by fluorescent Hoechst 33258 staining.43
Cytotoxicity Assay
Cell counting was performed with Countess II automated cell counter (Thermofisher) in the presence of Trypan blue dye to discriminate live and dead cells. Proliferation and viability were assessed by Cyquant assay (Invitrogen) as previously described.21 All concentration–response plots of compounds cytotoxicity against cultured cells were obtained by performing at least three independent experiments with 3 biological replicates for each concentration used. Concentration–response plots were used to calculate the concentration required to kill 50% of cells (cytotoxic concentration, CC50).
Generation of GFP-LC3 Reporter A375 Cells
EGFP-LC3 plasmid bearing a chimeric LC3B protein fused with an enhanced green fluorescent protein (GFP) was a gift from Karla Kirkegaard (Addgene plasmid no. 11546). For the generation of melanoma A375 cells stably expressing the chimeric LC3B, cells were transfected with X-tremeGene HP reagent (Roche). After 48 h, cells were split in selection media containing G418 (A1720, Sigma-Aldrich). Five diverse single clones from G418-resistant cells were tested for their ability to induce perinuclear fluorescent dots upon the treatment with 10 μM of the autophagy inhibitor, chloroquine (C6628, Sigma-Aldrich). All the tested clones generated a similar CQ-mediated response and clone no. 4 was adopted for evaluating the effects of compounds on autophagy by fluorescent microscopy.
Fluorescent Microscopy
Cells were seeded on 48-well plates with bottom glass (Mattek Corporation, P48G-1.5-6-F) and treated with diverse concentrations of compounds 1e or 24. After 24 h, cells were fixed with 4% paraformaldehyde (16005, Sigma-Aldrich) in PBS and nuclei were stained with Hoechst 33342 (14553, Sigma-Aldrich). The number of GFP-LC3B perinuclear fluorescent dots was assessed from images acquired with a fluorescent inverted microscope DM IL LED (Leica Microsystems). Ten different fields per each dose of compounds from four independent experimental replicates were used to obtain the quantification shown in Figure 4.
REV-ERB Luciferase Assay
REV-ERB inhibition was assessed as previously described.21 Briefly, a reporter vector that contains two repetitions of the REV-ERB-responsive element consensus (5′-AGA ATG TAG GTC ATC TAG AAT GTA GGT CA-3′) or a mutated version (5′-AGC CCG TAG GTC ATC TAG CCC GTA GGT CA-3′) that is no longer able to bind REV-ERB driving the expression of Cypridina luciferase gene was cotransfected with a plasmid expressing REV-ERBβ in HEK-293 cells. The following day, cells were treated with diverse doses of the compounds, and after 24 h luciferase activities were measured according to the manufacturer’s instructions. A vector with an SV40 promoter driven Gaussia luciferase was used for normalization. Dose–response values were used to calculate the concentration required to achieve 50% of the maximal effect on REV-ERB transcriptional derepression (EC50). EC50 values derived from the average of at least three independent experimental replicates.
Quantitative RT-PCR
RNA sample preparation and relative transcript expression levels were assessed as described previously.21 GAPDH transcript was used for normalization. Primer sequences were the following: BMAL1 (5′-CCA GAG GCC CCT AAC TCC TC-3′ and 5′-TGG TCT GCC ATT GGA TGA TCT-3′, forward and reverse, respectively); HRTP (5′-GTT ATG GCG ACC CGC AG-3′ and 5′-ACC CTT TCC AAA TCC TCA GC-3′, forward and reverse, respectively); GAPDH (5′-AAG GTG AAG GTC GGA GTC AA-3′ and 5′-AAT GAA GGG GTC ATT GAT GG-3′, forward and reverse, respectively); PEPCK (5′-AAA ACG GCC TGA ACC TCT CG-3′ and 5′-ACA CAG CTC AGC GTT ATT CTC-3′, forward and reverse, respectively). Relative expression values derived from the average of at least three independent experimental replicates.
Immunoblot Analysis
Protein samples were extracted in RIPA buffer as described previously.44 LC3B, SQSTM1, LAMP1, cleaved-PARP, and GAPDH levels were analyzed with anti-LC3B (Cell Signaling, 3868), anti-SQSTM1/p62 (Santa Cruz Biotechnology, 28359), anti-LAMP1 (Cell Signaling, 9091), anticleaved PARP (Invitrogen, 44-698G), and anti-GADPH (Invitrogen, 398600) specific antibodies. Immunoblot experiments were performed in TBS-T buffer containing 5% bovine serum albumin (BSA). Anti-LC3B, anti-SQSTM1, anti-LAMP1, and anticleaved PARP antibodies were diluted 1:2000 while anti-GAPDH antibody was diluted 1:100000. Complementary HPR-conjugated secondary antibodies were diluted 1:10000. Upon reaction with ECL Star detection reagent (Euroclone, EMP001005), chemiluminescent signals were acquired with a ChemiDoc Imaging System (Biorad). Optical density of chemiluminescent signals was quantified using Adobe Photoshop CS6 (version 13.0). Quantifications derived from the average of three independent experimental replicates.
Autophagosome-Enriched Preparation
Autophagosome-enriched preparation has been performed as described previously.44 Briefly, cells treated 24 h with compound 1e or 24 were detached using trypsin and collected by low-speed centrifugation. Packed cell pellets were resuspended in phosphate-buffered saline (PBS) containing 100 μg/mL of digitonin (D141, Sigma-Aldrich), then cytosolic and organelle-bound proteins were separated by centrifugation at 7000 rpm for 8 min. Organelle-enriched pellets were solubilized in RIPA buffer, mixed with Laemmli buffer and DTT.
siRNA Transfection
For RNAi experiments, 30 nM siRNA sequences against REV-ERBβ (Dharmacon, Lafayette, CO, USA catalogue no. 9975) following the manufacturer’s protocol. As a control, cells were transfected with MISSION siRNA Universal Negative Control no. 1 (Sigma, catalogue no. SIC001).
Caspase Activity
Poly Caspases activity was evaluated with the Image-iT LIVE Red and Green Caspase Apoptosis Detection Kit (I35101, Invitrogen).
In Vitro ADME Assays
Aqueous Kinetic Solubility Assay
The aqueous kinetic solubility was determined from a 10 mM DMSO stock solution of test compound in phosphate buffered saline (PBS) at pH 7.4. The study was performed by incubation of an aliquot of 10 mM DMSO stock solution in PBS (pH 7.4) at a target concentration of 250 μM (2.5% DMSO). The incubation was carried out under shaking at 25 °C for 24 h followed by centrifugation at 21 100g for 30 min. The supernatant was analyzed by UPLC/MS for the quantification of dissolved compound (in μM) by UV at a specific wavelength (215 nm). The aqueous kinetic solubility (in μM) was calculated by dividing the peak area of the dissolved test compound (supernatant) by the peak area of the test compound in the reference (250 μM in CH3CN) and further multiplied by the target concentration and dilution factor. The UPLC/MS analyses were performed on a Waters Acquity UPLC/MS system consisting of a single quadrupole detector (SQD) mass spectrometer (MS) equipped with an electrospray ionization (ESI) interface and a photodiode array detector (PDA). The PDA range was 210–400 nm. ESI in positive mode was used in the mass scan range 100–650 Da. The analyses were run on an Acquity UPLC BEH C18 column (50 mm × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 mm × 2.1 mm ID, particle size 1.7 μm), using 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in CH3CN-H2O (95:5) at pH 5 (B) as mobile phase. Values are the mean of at least two independent experiments, performed in two technical replicates.
Liver Microsomal Stability Assay
Phase-I metabolism: Freshly prepared 10 mM DMSO stock solution of test compound was preincubated at 37 °C for 15 min with mouse, rat, dog, and human liver microsomes in 0.1 M Tris-HCl buffer (pH 7.5) with 10% DMSO. The final concentration was 4.6 μM. After preincubation, the cofactors (NADPH, G6P, G6PDH, and MgCl2 predissolved in 0.1 M Tris-HCl) were added to the incubation mixture, and the incubation was continued at 37 °C for 1 or 2 h. Phase-II metabolism: 10 mM DMSO stock solution of the test compound was preincubated at 37 °C for 15 min with mouse and human liver microsomes added alamethicin in 0.1 M Tris–HCl buffer (pH 7.5) with 10% DMSO. The final concentration was 4.6 μM. After preincubation, the cofactors (UDPGA, d-saccharic acid lactone, and MgCl2 predissolved in 0.1 M Tris-HCl) were added to the incubation mixture and the incubation was continued at 37 °C for 2 h. For both phase-I and phase-II metabolism: At each time point (for 1 h incubation time: 0, 5, 15, 30, 60 min; for 2 h incubation time: 0, 30, 60, 90, 120 min), A 30 μL of incubation mixture was diluted with 200 μL of cold CH3CN spiked with 200 nM of internal standard, followed by centrifugation at 3300g for 15 min. The supernatant was further diluted with H2O (1:1) for analysis. A reference incubation mixture (microsomes without cofactors) was prepared for each test compound and analyzed at t = 0 and 60 or 120 min in order to verify the compound’s stability in the matrix. The time points were diluted as for the time points of the incubation mixture above. The supernatant of test compound was analyzed by LC/MS-MS on a Waters Acquity UPLC/MS TQD system consisting of a triple quadrupole detector (TQD) MS equipped with an ESI interface. The analyses were run on an Acquity UPLC BEH C18 (50 mm × 2.1 mm ID, particle size 1.7 μm) with a VanGuard BEH C18 precolumn (5 mm × 2.1 mm ID, particle size 1.7 μm) at 40 °C. For each compound the appropriate mobile phase was chosen. ESI was applied in positive mode. The percentage of test compound remaining at each time point relative to t = 0 was calculated by the response factor on the basis of the internal standard peak area. The half-life (t1/2) of tested compound was estimated by a one-phase decay equation using a nonlinear regression of compound concentration versus time, fitted by GraphPad Prism (GraphPad Software, Version 5 for Windows, CA, USA, www.igraphpad.com). The t1/2 of tested compound was reported as mean value along with the standard deviation. Values are the average of at least two independent experiments, performed in two technical replicates.
In Vivo Pharmacology
Pharmacokinetic Studies
The plasma pharmacokinetic studies of single dose of compounds 1e and 24 were performed in compliance with the protocol approved by the Italian Ministry of Health Protocol no. 184 according to the DLgs 116/1992. Female CD1 mice between 8 and 9 weeks of age were acquired by Charles River Laboratory (Italy) and were housed in a temperature-controlled environment with a 12 h light/dark cycle. All mice received a standard diet and water ad libitum. After 4 days of acclimation, animals received one dose of 1e or 24 at 10 mg/kg of body weight intraperitoneally, and groups of 3 mice were sacrificed 0.25, 0.5, 1, 2, 4, 8, and 24 h later. Compounds were dissolved in saline and three control mice that received only saline were sacrificed at 24 h. Pharmacokinetic analysis was performed using PKSolver (https://www.sciencedirect.com/science/article/pii/S0169260710000209). Noncompartmental analysis of the plasma concentration–time data was used to estimate, Cmax (maximum plasma concentration), tmax (time of maximum plasma concentration), t1/2, and volume of distribution (V/F) of 1e and 24. A two-compartment model (weighting = 1/y2) provided the best fit to the data and was adopted to estimate pharmacokinetic descriptors for the observed biphasic elimination of 1e and 24 (t1/2α and t1/2β).
Tolerability Studies
Tolerability studies were performed in compliance with the protocol approved by the Italian Ministry of Health Protocol no. 184 according to the DLgs 116/1992. Female CD1 mice were acquired by Charles River Laboratory (Italy). Mice between 8 and 9 weeks of age were housed in a temperature-controlled environment with a 12-h light/dark cycle. All mice received a standard diet and water ad libitum. After 4 days of acclimation, mice were divided into two groups (3 animals per group): control animals, which received an ip injection of saline daily, and treated group, which received an ip injection of saline containing compound 24 daily. For the studies at the dose of 138 μmol/kg (62 mg/kg), mice received an ip injection of 24 daily or every other day. For the studies at the dose of 69 μmol/kg (31 mg/kg) animals received an ip injection of 24 daily or according to a 3/5 days treatment schedule (3 days of daily treatment followed by 2 days off treatment). General behavior, animal body weight, food consumption, and water consumption were monitored daily. For assessing the effect of the treatments on body weight, the average animal weight at the day before the start of the treatment was set to 100% and used to calculate the relative body weights of mice during the treatment.
A375 Subcutaneous Xenograft Model
Mouse xenograft studies were performed in compliance with the protocol approved by the Italian Ministry of Health Protocol no. 402 according to the DLgs 116/1992. Reporter A375 cells were maintained in the exponential growth phase prior to the injection. Cells were then collected for injection by trypsinization and resuspended in PBS at the concentration of 10 million cells per milliliter. The percentage of viable cells was determined using the trypan blue exclusion protocol,45 adopting a viability greater than 95% as a cutoff. Female NMRI-Foxn1nu mice were acquired from Charles River Laboratory (Italy). Animals between 8 and 9 weeks of age were housed in a temperature-controlled environment with a 12 h light/dark cycle. Mice received a standard diet and water ad libitum. After 4 days of acclimation, mice received a subcutaneous injection in the hind leg flank of 100 μL of cell suspension (i.e., 1 million cells). Three times weekly, the injection sites were palpated until tumors were established, and a digital caliper was used to measure tumor growth until an average size of 50–100 mm3 was reached. For the efficacy study, animals were then assigned to two groups: one group of 8 animals received a dose of 30 mg/kg of compound 24 (intraperitoneally) according to a 3/5 d treatment schedule, and control group of 7 animals received saline according to the same treatment schedule. Tumor growth and animal body weight were monitored three times weekly. After 15 days of treatment, several tumors in the saline-group reached a volume close to the defined human end point (2000 mm3). Animals were thus sacrificed, and tumors were excised and weighed. Length, height, and depth of excised tumors were measured to obtain a more precise assessment of tumor volumes according to the formula: tumor volume = (height × depth × length)/2.
For evaluating the effect of 24 on tumor autophagy and REV-ERB transcription, 6 mice bearing reporter A375 xenografts with an average tumor size of 100–200 mm3 were divided into two groups of 3 animals. One group received an ip injection of 24 at the dose of 30 mg/kg for 2 days while the control group received an ip dose of saline. After 48 h from the first injection, mice were sacrificed, and tumors were processed for immunoblot and qRT-PCR analyses.
Statistical Analysis
Log(inhibitor) versus response curves, one- and two-way ANOVA and two-tails t test were performed using GraphPad Prism Software (San Diego, CA, USA).
Acknowledgments
This work was funded by SOTIO Biotech through a Sponsored Research Agreement signed between Fondazione Istituto Italiano di Tecnologia (IIT) and SOTIO Biotech. We thank Silvia Venzano (Medicinal Chemistry and Technologies for Drug Discovery and Delivery Facility), Andrea Armirotti, Sine Mandrup, Giuliana Ottonello, Luca Goldoni (Analytical Chemistry Facility), and Rosalia Bertorelli and Maria Summa (Translational Pharmacology Facility) for their technical support.
Glossary
Abbreviations Used
- ADME
absorption, distribution, metabolism and excretion
- AIC
Akaike information criterion
- NH4Cl
ammonium chloride
- NH4OH
ammonium hydroxide
- BSA
bovine serum albumin
- BMAL1
brain and muscle ARNT-like protein-1
- HCl
chloridric acid
- CQ
chloroquine
- Cy
cyclohexane
- CDCl3
deuterated chloroform
- DMSO-d6
deuterated dimethyl sulfoxide
- CH2Cl2
dichloromethane
- Et2O)
diethyl ether
- DIBALH
diisobutylaluminum hydride
- DMSO
dimethyl sulfoxide
- Xphos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
- DTT
dithiothreitol
- DMEM
Dulbecco’s Modified Eagle’s Medium
- ESI
electrospray ionization
- equiv.
equivalent
- ERBB2
Erb-B2 receptor tyrosine kinase 2
- EtOAC
ethyl acetate
- FBS
fetal bovine serum
- FDA
Food and Drug Administration
- G418
geneticin
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFP
green fluorescence protein
- Hz
Hertz
- HRMS
high resolution mass spectroscopy
- HRP
horseradish peroxidase
- hERG
human ether-a-go-go related gene
- HMECs
human mammary epithelial cells
- HCQ
hydroxychloroquine
- t1/2
in vitro half-life
- CC50
cytotoxic concentration 50%
- i.p.
intraperitoneal
- iPrOH
2-propanol
- LBD
ligand binding domain
- LiAlH4
lithium aluminum hydride
- LIHMDS
lithium bis(trimethylsilyl)amide
- LAMP1
lysosomal-associated membrane protein 1
- MnO2
manganese oxide
- MS
mass spectrometer
- Cmax
maximum plasma concentration
- MeOH
methanol
- LC3B
microtubule-associated protein 1-light chain 3 beta
- n-BuLi
n-butyl lithium
- NADPH
nicotinamide adenine dinucleotide phosphate
- NCA
noncompartmental analysis
- NMR
nuclear magnetic resonance
- on
overnight
- Pd/C
palladium on carbon
- PFA
paraformaldehyde
- PK
pharmacokinetic
- PBS
phosphate buffered saline
- PEPCK
phosphoenolpyruvate carboxykinase
- PDA
photodiode array detector
- SV40
polyomavirus simian virus 40
- K2CO3
potassium carbonate
- KOtBu
potassium tert-butoxide
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- RIPA
radio-immunoprecipitation assay
- Rt
retention time
- RevRE
REV-ERB responsive element
- REV-ERB
reverse-Erb receptors
- RNA
ribonucleic acid
- rt
room temperature
- SQSTM1
sequestosome 1
- SiO2
silica gel
- SQD
single quadrupole detector
- NaN3
sodium azide
- NaOH
sodium hydroxide
- Na2SO4
sodium sulfate
- NaBH(OAc)3
sodium triacetoxyborohydride
- SAR
structure–activity relationship
- SPR
structure–property relationship
- H2SO4
sulfuric acid
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- tmax
time of maximum plasma concentration
- Ti(OiPr)4
titanium(IV) isopropoxide
- P(OEt)3
triethylphosphite
- Et3SiH
triethylsilane
- TFA
trifluoroacetic acid
- PPh3
triphenyl phosphine
- Pd2dba3
tris(dibenzylideneacetone)dipalladium(0)
- TBS-T
Tris buffered saline–0.1% Tween 20 detergent
- UPLC/MS
Ultra performance liquid chromatography–mass spectrometer
- UV
Ultraviolet
- UDPGA
uridine diphosphate glucuronic acid
- UM
Uveal melanoma
- H2O
water
- wt
weight
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01432.
1H NMR and 13C NMR spectra of the final compounds; UPLC/MS traces of the final compounds; retention times and UPLC analytical method of the final compounds; compound 24 induced apoptosis in BT-474 cells; abolishment of 24-mediated REV-ERB response in REV-ERBβ-silenced breast cancer cells; tolerability of 24 in CD1 mice (PDF)
Molecular formula strings (CSV)
Author Present Address
⊥ MP.: Istituto Italiano di Tecnologia, Genoa 16163, Italy
Author Present Address
# D.V.: Istituto Italiano di Tecnologia, Genoa 16163, Italy
Author Present Address
∇ G.A.: Istituto Italiano di Tecnologia, Genoa 16163, Italy
Author Present Address
○ V.C.: Istituto Italiano di Tecnologia, Genoa 16163, Italy
Author Present Address
◆ C.D.M.: Istituto Italiano di Tecnologia, Genoa 16163, Italy
Author Contributions
M.P. and D.V. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Benedetto Grimaldi and Rita Scarpelli are co-inventors in a patent that includes compounds here disclosed owned by Fondazione Istituto Italiano di Tecnologia (IIT). The remaining authors declare no conflict of interest.
Supplementary Material
References
- Dikic I.; Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19 (6), 349–364. 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- Yamamoto H.; Zhang S.; Mizushima N. Autophagy genes in biology and disease. Nat. Rev. Genet 2023, 24 (6), 382–400. 10.1038/s41576-022-00562-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Virgilio L.; Silva-Lucero M. D.; Flores-Morelos D. S.; Gallardo-Nieto J.; Lopez-Toledo G.; Abarca-Fernandez A. M.; Zacapala-Gomez A. E.; Luna-Munoz J.; Montiel-Sosa F.; Soto-Rojas L. O.; et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11 (15), 2262. 10.3390/cells11152262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F.; Bordi M.; Cianfanelli V.; Locatelli F.; Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26 (4), 690–702. 10.1038/s41418-019-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. C.; Guan K. L. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41 (13), e110031 10.15252/embj.2021110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi-Jeda A.; Ghabeshi S.; Gol Mohammad Pour Z.; Jazaeri E. O.; Araiinejad M.; Sheikholeslami F.; Abdoli M.; Edalat M.; Abdoli A. Autophagy Modulation and Cancer Combination Therapy: A Smart Approach in Cancer Therapy. Cancer Treat Res. Commun. 2022, 30, 100512. 10.1016/j.ctarc.2022.100512. [DOI] [PubMed] [Google Scholar]
- Baradaran Eftekhari R.; Maghsoudnia N.; Dorkoosh F. A. Chloroquine: a brand-new scenario for an old drug. Expert Opin Drug Deliv 2020, 17 (3), 275–277. 10.1080/17425247.2020.1716729. [DOI] [PubMed] [Google Scholar]
- De Sanctis J. B.; Charris J.; Blanco Z.; Ramirez H.; Martinez G. P.; Mijares M. R. Molecular Mechanisms of Chloroquine and Hydroxychloroquine Used in Cancer Therapy. Anticancer Agents Med. Chem. 2023, 23 (10), 1122–1144. 10.2174/1871520622666220519102948. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A. K.; Saadeldin M. K.; Salem A. H.; Ibrahim S. A.; Shouman S.; Abdel-Naim A. B.; Orecchia R. A Critical Review of Chloroquine and Hydroxychloroquine as Potential Adjuvant Agents for Treating People with Cancer. Future Pharmacology 2022, 2 (4), 431–443. 10.3390/futurepharmacol2040028. [DOI] [Google Scholar]
- Mercuro N. J.; Yen C. F.; Shim D. J.; Maher T. R.; McCoy C. M.; Zimetbaum P. J.; Gold H. S. Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA cardiology 2020, 5 (9), 1036–1041. 10.1001/jamacardio.2020.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oscanoa T. J.; Vidal X.; Kanters J. K.; Romero-Ortuno R. Frequency of Long QT in Patients with SARS-CoV-2 Infection Treated with Hydroxychloroquine: A Meta-analysis. Int. J. Antimicrob. Agents 2020, 56 (6), 106212. 10.1016/j.ijantimicag.2020.106212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seydi E.; Karbalaei Hassani M. K.; Naderpour S.; Arjmand A.; Pourahmad J. Cardiotoxicity of chloroquine and hydroxychloroquine through mitochondrial pathway. BMC Pharmacology Toxicology 2023, 24, 26. 10.1186/s40360-023-00666-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Sun L.; Chen C.; Xin J.; Zhang Y.; Bai Y.; Pan Z.; Zhang Y.; Li B.; Lv Y.; Yang B. Hydroxychloroquine induces long QT syndrome by blocking hERG channel. Frigid Zone Medicine 2023, 3, 105–113. 10.2478/fzm-2023-0014. [DOI] [Google Scholar]
- Gao X.; Jing X.; Wang J.; Zheng Y.; Qiu Y.; Ji H.; Peng L.; Jiang S.; Wu W.; Guo D. Safety considerations of chloroquine in the treatment of patients with diabetes and COVID-19. Chem. Biol. Interact 2022, 361, 109954. 10.1016/j.cbi.2022.109954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzun K.; Gornowicz A.; Lesyk R.; Bielawski K.; Bielawska A. Autophagy Modulators in Cancer Therapy. Int. J. Mol. Sci. 2021, 22 (11), 5804. 10.3390/ijms22115804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin E. Anticancer autophagy inhibitors attract ’resurgent’ interest. Nat. Rev. Drug Discov 2019, 18 (6), 408–410. 10.1038/d41573-019-00072-1. [DOI] [PubMed] [Google Scholar]
- Kondapuram S. K.; Sarvagalla S.; Coumar M. S. Targeting autophagy with small molecules for cancer therapy. J. Cancer Metastasis Treat. 2019, 5, 32. 10.20517/2394-4722.2018.105. [DOI] [Google Scholar]
- Adlanmerini M.; Lazar M. A. The REV-ERB Nuclear Receptors: Timekeepers for the Core Clock Period and Metabolism. Endocrinology 2023, 164 (6), bqad069. 10.1210/endocr/bqad069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomatou G.; Karachaliou A.; Veloudiou O. Z.; Karvela A.; Syrigos N.; Kotteas E. The Role of REV-ERB Receptors in Cancer Pathogenesis. Int. J. Mol. Sci. 2023, 24 (10), 8980. 10.3390/ijms24108980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriz-Huarte A.; Date A.; Ang H.; Ali S.; Brady H. J. M.; Fuchter M. J. The transcriptional repressor REV-ERB as a novel target for disease. Bioorg. Med. Chem. Lett. 2020, 30 (17), 127395. 10.1016/j.bmcl.2020.127395. [DOI] [PubMed] [Google Scholar]
- De Mei C.; Ercolani L.; Parodi C.; Veronesi M.; Vecchio C. L.; Bottegoni G.; Torrente E.; Scarpelli R.; Marotta R.; Ruffili R.; et al. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene 2015, 34, 2597–2608. 10.1038/onc.2014.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente E.; Parodi C.; Ercolani L.; De Mei C.; Ferrari A.; Scarpelli R.; Grimaldi B. Synthesis and in Vitro Anticancer Activity of the First Class of Dual Inhibitors of REV-ERBbeta and Autophagy. J. Med. Chem. 2015, 58 (15), 5900–5915. 10.1021/acs.jmedchem.5b00511. [DOI] [PubMed] [Google Scholar]
- Tagad H. D.; Hamada Y.; Nguyen J. T.; Hidaka K.; Hamada T.; Sohma Y.; Kimura T.; Kiso Y. Structure-guided design and synthesis of P1’ position 1-phenylcycloalkylamine-derived pentapeptidic BACE1 inhibitors. Bioorg. Med. Chem. 2011, 19 (17), 5238–5246. 10.1016/j.bmc.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Thurkauf A.; De Costa B.; Yamaguchi S.; Mattson M. V.; Jacobson A. E.; Rice K. C.; Rogawski M. A. Synthesis and anticonvulsant activity of 1-phenylcyclohexylamine analogs. J. Med. Chem. 1990, 33 (5), 1452–1458. 10.1021/jm00167a027. [DOI] [PubMed] [Google Scholar]
- Tomashenko O. A.; Rudenko A. E.; Sokolov V. V.; Tomashevskiy A. A.; de Meijere A. 1-Substituted Cyclopentylamines from Nitriles and Tetramethylenebismagnesium Dibromide in the Presence of Ti(OiPr)4. Eur. J. Org. Chem. 2010, 2010 (8), 1574–1578. 10.1002/ejoc.200900817. [DOI] [Google Scholar]
- Watanuki S.; Matsuura K.; Tomura Y.; Okada M.; Okazaki T.; Ohta M.; Tsukamoto S. Synthesis and Pharmacological Evaluation of 1-Alkyl-[(1)-1-(4-fluorophenyl)-2-methylpropyl]piperidine-4-carboxamide Derivatives as Novel Antihypertensive Agents. Chem. Pharm. Bull. 2011, 59 (11), 1376–1385. 10.1248/cpb.59.1376. [DOI] [PubMed] [Google Scholar]
- Kaiser C.; Weinstock J. Amines from mixed carboxylic-carbonic anhydrides: 1-phenylcyclopentylamine. Org. Synth 1971, 51, 48. 10.15227/orgsyn.051.0048. [DOI] [Google Scholar]
- Afshar A. R.; Damato B. E.; Stewart J. M.; Zablotska L. B.; Roy R.; Olshen A. B.; Joseph N. M.; Bastian B. C. Next-Generation Sequencing of Uveal Melanoma for Detection of Genetic Alterations Predicting Metastasis. Transl Vis Sci. Technol. 2019, 8 (2), 18. 10.1167/tvst.8.2.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit K. N.; Jager M. J.; de Klein A.; Kiliç E. Uveal melanoma: Towards a molecular understanding. Prog. Retin Eye Res. 2020, 75, 100800. 10.1016/j.preteyeres.2019.100800. [DOI] [PubMed] [Google Scholar]
- Klionsky D. J.; Abdel-Aziz A. K.; Abdelfatah S.; Abdellatif M.; Abdoli A.; Abel S.; Abeliovich H.; Abildgaard M. H.; Abudu Y. P.; Acevedo-Arozena A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17 (1), 1–382. 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B. R.; Page-Sharp M.; Stoney J. R.; Ilett K. F.; Jago J. D.; Batty K. T. Pharmacokinetics, pharmacodynamics, and allometric scaling of chloroquine in a murine malaria model. Antimicrob. Agents Chemother. 2011, 55 (8), 3899–3907. 10.1128/AAC.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Wang X.; Contino G.; Liesa M.; Sahin E.; Ying H.; Bause A.; Li Y.; Stommel J. M.; Dell’antonio G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25 (7), 717–729. 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Liao Z.; Zhang L. J.; Xiao H. T. The utility of chloroquine in cancer therapy. Curr. Med. Res. Opin 2015, 31 (5), 1009–1013. 10.1185/03007995.2015.1025731. [DOI] [PubMed] [Google Scholar]
- McAfee Q.; Zhang Z. H.; Samanta A.; Levi S. M.; Ma X. H.; Piao S. F.; Lynch J. P.; Uehara T.; Sepulveda A. R.; Davis L. E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (21), 8253–8258. 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin D.; Wang Y.; Kamenecka T. M.; Burris T. P. Identification of SR8278, a synthetic antagonist of the nuclear heme receptor REV-ERB. ACS Chem. Biol. 2011, 6 (2), 131–134. 10.1021/cb1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariollaud M.; Gibbs J. E.; Hopwood T. W.; Brown S.; Begley N.; Vonslow R.; Poolman T.; Guo B.; Saer B.; Jones D. H.; et al. Circadian clock component REV-ERBalpha controls homeostatic regulation of pulmonary inflammation. J. Clin Invest 2018, 128 (6), 2281–2296. 10.1172/JCI93910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercolani L.; Ferrari A.; De Mei C.; Parodi C.; Wade M.; Grimaldi B. Circadian clock: Time for novel anticancer strategies?. Pharmacol. Res. 2015, 100, 288–295. 10.1016/j.phrs.2015.08.008. [DOI] [PubMed] [Google Scholar]
- Amiama-Roig A.; Verdugo-Sivianes E. M.; Carnero A.; Blanco J. R. Chronotherapy: Circadian Rhythms and Their Influence in Cancer Therapy. Cancers 2022, 14 (20), 5071. 10.3390/cancers14205071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damato A. R.; Herzog E. D. Circadian clock synchrony and chronotherapy opportunities in cancer treatment. Semin Cell Dev Biol. 2022, 126, 27–36. 10.1016/j.semcdb.2021.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevinakoppamath S.; Ramachandra S. C.; Yadav A. K.; Basavaraj V.; Vishwanath P.; Prashant A. Understanding the Emerging Link Between Circadian Rhythm, Nrf2 Pathway, and Breast Cancer to Overcome Drug Resistance. Front Pharmacol 2022, 12, 719631. 10.3389/fphar.2021.719631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. Roles of circadian clocks in cancer pathogenesis and treatment. Exp Mol. Med. 2021, 53 (10), 1529–1538. 10.1038/s12276-021-00681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y.; Ashrafizadeh M.; Mongiardini V.; Grimaldi B.; Crea F.; Rietdorf K.; Gyorffy B.; Klionsky D. J.; Ren J.; Zhang W.; et al. Autophagy and cancer drug resistance in dialogue: Pre-clinical and clinical evidence. Cancer Lett. 2023, 570, 216307. 10.1016/j.canlet.2023.216307. [DOI] [PubMed] [Google Scholar]
- Chen T. R. In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 1977, 104 (2), 255–262. 10.1016/0014-4827(77)90089-1. [DOI] [PubMed] [Google Scholar]
- Allavena G.; Debellis D.; Marotta R.; Joshi C. S.; Mysorekar I. U.; Grimaldi B. A broad-spectrum antibiotic, DCAP, reduces uropathogenic Escherichia coli infection and enhances vorinostat anticancer activity by modulating autophagy. Cell Death Dis 2018, 9 (7), 780. 10.1038/s41419-018-0786-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001, 3, 3B. 10.1002/0471142735.ima03bs21. [DOI] [PubMed] [Google Scholar]
- Wider G.; Dreier L. Measuring Protein Concentrations by NMR Spectroscopy. J. Am. Chem. Soc. 2006, 128, 2571–2576. 10.1021/ja055336t. [DOI] [PubMed] [Google Scholar]
- Burton I. W.; Quilliam M. A.; Walter J. A. Quantitative 1 H NMR with External Standards: Use in Preparation of Calibration Solutions for Algal Toxins and Other Natural Products. Anal. Chem. 2005, 77, 3123–3131. 10.1021/ac048385h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.