

Abstract
In the event of an outbreak due to an emerging pathogen, time is of the essence to contain or to mitigate the spread of the disease. Drug repositioning is one of the strategies that has the potential to deliver therapeutics relatively quickly. The SARS-CoV-2 pandemic has shown that integrating critical data resources to drive drug-repositioning studies, involving host-host, host-pathogen and drug-target interactions, remains a time-consuming effort that translates to a delay in the development and delivery of a life-saving therapy. Here, we describe a workflow we designed for a semi-automated integration of rapidly emerging datasets that can be generally adopted in a broad network pharmacology research setting. The workflow was used to construct a COVID-19 focused multimodal network that integrates 487 host-pathogen, 63,278 host-host protein and 1,221 drug-target interactions. The resultant Neo4j graph database named “Neo4COVID19” is made publicly accessible via a web interface and via API calls based on the Bolt protocol. Details as to accessing the database are provided on a landing page (https://neo4covid19.ncats.io/). We believe that our Neo4COVID19 database will be a valuable asset to the research community and will catalyze the discovery of therapeutics to fight COVID-19.
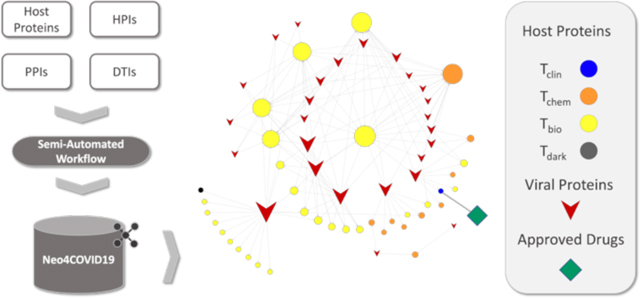
Graphical Abstract
INTRODUCTION.
The pandemic of the SARS-CoV-2 virus (also commonly referred to as COVID-19 pandemic by the name of the disease it induces) put a spotlight on the need for mechanisms that can rapidly identify and integrate relevant information to give a fighting chance to the medical and research community. The Ebola and Zika outbreaks presented similar challenges, and significant advances have been made in the past years in terms of digital, laboratory, and epidemiological techniques [1]–[3]. Although numerous resources covering many aspects of pertinent biomedical research have been developed and made publicly available, e.g. ChEMBL[4], [5], Reactome [6], Pharos [7], PathwayCommons [8], BioPlanet [9], and DrugCentral [10] to name a few, their on-demand integration has been a translational bottleneck to date.
In the case of the outbreak of an unknown pathogen, such as SARS-CoV-2, the first line of defense – absent viable therapeutic options – is containment. Should this fail, we have to resort to mitigation to slow down the spread of the pathogen. Delays of mere days in the early stages of containment and mitigation can lead to catastrophic outcomes regarding the number of infections and death toll [11]–[13]. Furthermore, the COVID-19 pandemic was reported to have a dramatic impact on primary healthcare providers, limiting them to essential clinical services, which eventually led to unforeseen delays in diagnosing highly critical diseases such as cancers [14], [15]. Therefore, it is imperative that we have computational workflows in place that can help researchers to connect and navigate the relevant information very fast – much faster than today. Multiple pertinent datasets subjected to such a workflow would produce a condensed, enriched starting point for timely hypothesis generation that would drive a successful containment or mitigation strategy, such as drug repositioning [16].
Indeed, a small number of publicly available databases and knowledge-graphs have been reported that connect various types of information related to COVID-19. Such resources are primarily limited to data extracted via text mining from literature and patents, reports, and experimental data [17]–[26]. While these resources are valuable for advancing our efforts toward a possible therapy for COVID-19, they suffer from certain limitations from a translational point of view.
First, hypothesis generation in the current drug discovery paradigm, i.e. network pharmacology [27]–[29], can be enhanced by normalizing the nature of interactions between protein target pairs, and between compounds and targets to reflect if they are engaged in a stimulatory or an inhibitory relationship. Although necessary information might be present in existing data sources, the interaction categories are typically encoded as separate relationships. This makes it difficult to readily assess the inhibitory and stimulatory relationships. For instance, both “antagonist” and “ion channel blocker” actions can be further reduced to an “inhibitory” relationship to aid the analysis of network perturbation. Naturally, the original relationships can also be preserved to avoid loss of information. A normalized relationship of protein-protein and drug-target interactions would consist of only three values: “stimulates”, “inhibits”, and “undefined” (or the equivalent of these phrases).
Next, the recent concept of “target development level (TDL)” [7] of protein targets is not captured in existing knowledge graphs related to COVID-19. The annotation of TDL category of targets makes it easy to identify those whose activity can be modulated by FDA-approved drugs or by small molecules. Such information therefore facilitates drug repositioning-oriented hypothesis generation.
Another category of limitations pertains to translational aspects: knowledge dissemination and real-time data integration. To our knowledge, no data source related to COVID-19 to date has been equipped with a mechanism to facilitate the data exploration and analysis for those without and with substantial bioinformatics background at the same time. However, in the case of a pandemic, it is imperative to disseminate data sources and data analysis tools to as broad a scientific community as possible and as soon as possible.
Finally, from a technical standpoint, it is of paramount importance that we have publicly available mechanisms and workflows for real-time integration of heterogeneous information. Typically, careful integration of databases can take months and even years, and oftentimes the integration workflow is quite specific for the knowledge base at hand [30], [31].
In this study, we describe such a workflow and utilize it to assemble, from several pertinent sources, a knowledge network aimed at defeating COVID-19 via enhanced hypothesis generation. The first building block is a list of pathogen–host protein interactions that was published in a preprint on March 27, 2020 by Krogan et al. [32], [33] within two months of the disclosure of the SARS-CoV-2 genetic sequence and within two weeks of the WHO declaring COVID-19 a pandemic [34], [35]. Further building blocks encompass interactions between FDA approved drugs and host protein targets (DTIs), host protein-protein and host–pathogen proteinprotein interactions (PPIs, and HPIs, respectively), and predicted drugs and host targets that will be introduced in this study. While this particular resultant Neo4j database is intended to catalyze COVID-19 research, the process of its creation can serve as a blueprint for inevitable future outbreaks, translating to saving precious time and, consequently, lives.
Related Work
The workflow presented in this study was inspired by earlier works, namely SmartGraph [36] and Hetionet [37], both of which are so-called multimodal networks designed to aid drug discovery efforts in a network pharmacology setting. SmartGraph is a computational platform that consist of a knowledge base and a web-based user interface. The knowledge base of SmartGraph integrates drug-target and protein-protein interactions. The user interface integrates complex but easy-to-execute workflows for the analysis of network perturbation, and for bioactivity prediction and drug repositioning. Relationships between proteins are labeled as stimulatory or inhibitory to reflect the nature of the interactions. Hetionet is an open-source resource that is of notable importance and relevance. A wide variety of publicly available biomedical, disease-specific, and pharmacological databases were integrated into a large network. Unlike SmartGraph, Hetionet annotates drug-target interactions in terms of pharmacological action. Nevertheless, neither SmartGraph nor Hetionet provide a normalized annotation of the stimulatory and inhibitory relationships between proteins or between drugs and targets.
In a recent effort, researchers from University of Minnesota, Hunan University, and Amazon’s AWS artificial intelligence laboratories have collectively built the COVID-19-related Drug Repurposing Knowledge Graph (DRKG) [25] that integrates Hetionet among other data sources. Although DRKG addresses, to some extent, the issue of knowledge dissemination, this particular resource is available only as data tables, a format that requires advanced bioinformatics skills for analyzing the data in terms of network perturbation caused by drugs. Furthermore, the workflow to assemble the DRKG is not publicly available. COVID-KG [24] is another resource that focuses on extracting multimedia knowledge elements from scientific literature to be used as a knowledge graph for querying and report generation. While researchers developing both DRKG and COVID-KG have addressed the issue of data dissemination to some extent, the format they chose for distribution (tab-separated flat files) restricts network-based data analytics absent advanced bioinformatics skills. Furthermore, neither DRKG nor COVID-KG are associated with a publicly available implementation reflective of their underlying data integration workflows.
Another COVID-19 related resource is the recent “COVID-19 Disease Map” that was created by the means of a collaborative effort. While this study addresses the importance of the standardization of file formats, it does not provide a flexible workflow (and implementation as source code) that could serve as the basis for an automated or semi-automated data integration process. Although at the time writing of this manuscript the COVID-19 Disease Map is not yet accessible, the description of the integration process indicates that much emphasis was put on the use of curated data sources. This study exemplifies that a collaborative data integration strategy can yield high quality data sources, however, at the sacrifice of time.
Apart from KGs and databases, several other studies identified potential targets and contributed useful datasets that can guide drug discovery efforts. For instance, Gil et al. [17] provided their perspective on the main targets that are involved in viral replication and control of host cellular processes. In parallel, structure-based efforts [19], [20] have been reported where the authors performed molecular docking simulations and virtual high-throughput screening to prioritize candidates for drug repurposing. Experimental high-throughput screening data have been made available by the National Center for Advancing Translational Sciences (NCATS/NIH) on the “OpenData Portal” [21].
RESULTS AND DISCUSSION.
Semi-Automated Data Integration Workflow.
In order to build a COVID-19 focused network, we needed to integrate data from multiple, diverse data sources. The bottleneck of the integration proved to be the consolidation of the differing data structures, exemplified by the lack of standardized data categories, preference of one protein identifier over the other, aggregating protein identifiers as delimiter separated list inside a column, to name a few. Also, some of the data originate from experiments whereas others from predictions. This made it necessary to keep track of this information as well as data provenance in a transparent manner. Here, we present a rigorous workflow that addresses the above challenges and can serve as a template for semi-automated integration of the future databases inspired by network pharmacology. Having such an integration workflow in place is key to the timely assembly of a network that is focused on a certain biological aspect, e.g., an infectious disease caused by an emerging pathogen.
The assembly of a COVID-19 focused network involved data sources that emerged over a matter of weeks since the start of the pandemic and others that were well-established long before. Considering that new information surfaced relatively quickly, we had to ensure the workflow we created was sufficiently flexible to accommodate new data. Here, we describe such a workflow (see: Fig 1) and a COVID-19 focused Neo4j database that was produced by it. Information regarding the reproduction of the workflow is provided in the “Reproducing the Integration Workflow” section in SI.
Figure 1. Resource integration logic.

The schema highlights the most important steps of data processing. Individual inputs are labeled with letters. PPIs: host–host protein interactions, HPIs: host–pathogen (here: SARS-CoV-2) protein interactions, DTIs: drug–target interactions, TDL: target development levels, T: True. Blue: data processing step, white: not yet aggregated data, yellow: input-independent data source and processing step, dark gray: experimental dataset, light gray: predicted/hypothesized dataset, green: final dataset. Solid triangle symbol indicates which proteins should be used as starting nodes in a SmartGraph analysis, whereas the solid diamond symbol indicates which host proteins should be used for integrating relevant PPIs from the STRING DB. The open triangle and diamond symbols indicate the destination of human proteins routed toward a specific resource, i.e. to SmartGraph as starting nodes and to STRING for PPI expension, respectively.
The building blocks of the COVID-19 focused network represent experimentally determined as well as predicted HPI, PPI and DTI data, and additionally, prioritized host targets. In these data sources, host targets are typically identified by their gene name, with some exceptions where UniProt ACs are used, such as TDLs from Pharos DB and PPIs from SmartGraph. The name of several viral proteins in the two HPI datasets were slightly different. Such differences were manually reconciled (see: “Mapping of Viral Protein Names in HPIs” section in SI).
The next stage required harmonization of the input data structure. For each input type (HPI, DTI and host protein), a data structure was defined that can accommodate all data of the respective type. Naturally, each dataset needed to be tailored individually to fit the respective data structure, which is the main reason why the overall workflow is called semi-automated instead of automatic. Nonetheless, the workflow was equipped with a data registry mechanism that will allow the integration of additional HPI, DTI and host protein data in a robust and facile manner.
The data registry mechanism consists of configuring a registry of input files with focus on the type of the input (HPI/PPI/host protein), data provenance, and whether proteins extracted from the input should be subject to STRING and SmartGraph analysis. Additionally, it can be configured what information, i.e. fields, of a given input file should be conserved as metadata, what should be extracted as associated score, where applicable, and how a given resource should be referenced in database for the sake of data filtering.
Recognizing that various resources come in various data structures, the data registry mechanism enables the facile extension of workflow with code snippets, tasked with internal data structure harmonization. This is achieved by first providing the name of the data harmonization method to be applied in the registry file. Subsequently, this name needs to be used to create new a condition in a particular function of workflow, which in turn will branch off to new function that the investigator needs to provide. This function needs to be added to a specific source code file, and it will implement the harmonization logic. The logic will convert the data structure into a pre-defined internal standard that we provide. For a detailed description of the data registry mechanism and how data converter functions can be added to the workflow please refer to section “Integration of Additional Datasets via Data Registry Mechanism”, and Table S2–S3 in SI.
With the help of the data registry mechanism, each resource is first harmonized to an internal standard format, see: Table S3 in SI. In this process individual inputs are deduplicated, and certain derivative data types are also created, such as pathogen proteins and drugs.
The final stage of the workflow integrates all internally harmonized inputs with the help of a well-defined aggregation strategy. This strategy focuses on preserving all the relevant data while tracking the data provenance in a transparent manner. Therefore, it gives the choice to the investigator on how to prioritize data in the light of a particular research setting, e.g. by prioritizing experimental vs. predicted data, or by prioritizing a certain data source over another if conflicting data is found.
The aggregation process employs concatenation, which assures that data entries of entities occurring in multiple resources is tracked in a transparent and obvious manner. For example, a particular host protein might appear in multiple inputs so that it is associated with different metadata in each of these inputs. During the aggregation process, the metadata and data source information will be stored in a delimiter separated string in their respective data fields, so that the order of individual metadata and data source records are kept in sync.
The workflow of this study was designed to allow for the flexible data extension of data for each of the interaction types while providing an option to the investigator to restrict which data segment is subject to the extension. Briefly, Fig 1 depicts the following mechanism. Host proteins of potential importance were collected from resources “A”, “B”, “C”, “D”, “E”, “F”, and “H”. The data structures of these input data sets were harmonized with respect to the data type. In this process, information specific to individual data sets, as well as metadata and data provenance is maintained in a transparent manner. After merging and deduplicating each data type, a set of unique host proteins was extracted from them.
The data registry file requires to indicate whether host proteins extracted from any given resource should be used to assemble PPI with the help of STRING and stringApp APIs [38]–[40] (resource “G”) and SmartGraph [36] (resource “I”). In the workflow, respective subsets of host proteins are routed to SmartGraph and/or STRING analysis. Note that host proteins extracted from SmartGraph were not subject to the further STRING-expansion, and vice versa, unless there was an overlap between the proteins of these PPIs and the set of unique proteins extracted upstream in the workflow. Once the induced PPI subnetworks were assembled by the STRING and SmartGraph analysis, these data sets were also subject to data structure harmonization and deduplication.
One of the final challenges that we had to address in the workflow was integration of PPIs from different sources. Typically, this procedure is rather challenging due to potentially conflicting and missing PPI information. For instance, PPIs from the STRING API are not annotated with the mode of regulation (whether a target up-regulates or down-regulates its interacting partner).
A viable strategy to resolve such issues is to utilize a comprehensive and preferably curated PPI. To this end, we decided to use the “functional interactions” subset of Reactome (RFI) to fulfill this role. Each PPI was cross-referenced to the RFI subset. This process resulted in the assignment of both the direction and mechanism of regulation for each PPI in the integrated database, as well as the respective confidence score for each RFI. If a given PPI was missing from the RFI subset or the mechanism or the direction of regulation was undefined, then the corresponding properties of that PPI were set to “unknown” and “undefined”, respectively.
Each hostprotein target was annotated by the respective TDL category extracted from the Pharos [7] database. However, as proteins are encoded with UniProt ACs [41], [42] both in Pharos and in SmartGraph, we had to resolve them to gene names and vice versa. We used a UniProt resource [42]–[45] to retrieve UniProt to gene name mapping. Considering that this mapping is a many-to-many relationship, the 1:1 mapping was achieved by retaining the highest TDL of any of the UniProt ACs in the case of Pharos data. The TDL annotation of targets enables investigators to instantly identify targets for which FDA-approved drugs exist. This information combined with pathway analysis provide the foundation for the formulation of drug repositioning hypotheses. Such hypotheses can mean the first steps towards the discovery of therapeutics.
Once the final set of unique host proteins is identified, the workflow identifies DTIs with the help of the DrugCentral [46] (resource “K”), which involve any of these host proteins. These DTIs are harmonized and merged with the other DTIs provided as input to the workflow. After aggregating the DTIs, the set of unique drugs is identified by the workflow.
Finally, the Neo4COVID19 database is built by integrating the unique set of HPIs, PPIs, DTIs, host and viral (pathogen) protein targets, and drugs into a Neo4j database [47].
Database Deployment and Dissemination.
The data integration scheme discussed above was implemented in Python [48] and is available as a source code repository at https://github.com/ncats/neo4covid19 [49]. The data integration script builds a Neo4j database [47] which can be accessed publicly via Neo4j Browser, a web-based graphical user interface (GUI) provided at https://aspire.covid19.ncats.io:7473 by Neo4j. When prompted for login credentials, please select “No authentication” from the “Authentication type” drop-down list. Programmatic access is also provided to the same database via various Neo4j API interfaces, e.g. “py2neo” [50], which make use of the Neo4j Bolt protocol [47]. For further information please refer to section “Sample Python Code Snippet to Access Neo4COVID19 Database via API” in SI. Detailed information on accessing the database is available on the Neo4COVID19 website at https://neo4covid19.ncats.iounderthe“ACCESS”tab.
As Neo4COVID19 is a graph-database, fundamentally, it is a collection of nodes and edges. In the database, there are 903 “HostProtein” nodes, 55 “PathogenProtein” and 635 “Drug” nodes. Three types of relationships are contained by the database: “HPI”, “PPI” and “DTI” defined between two “HostProtein”, a “PathogenProtein” and a “HostProtein”, and a “Drug” and a “HostProtein” nodes, respectively. The Neo4COVID19 database contains 487 HPI, 63,278 PPI and 1,221 DTI relationships. Further information regarding the node and edge types extracted from each data resource are provided in Table 1. Node and edge attributes shown in Table S1 in SI can be used to fine-tune the network in a way that matches the needs of the analysis at hand. For instance, one might decide to only consider nodes and edges of the COVID-19 focused network that represent experimental data. This can be achieved with ease by filtering the data with the help of Boolean fields, each representing a specific data source. Another example is application of a confidence threshold to PPI edges retrieved from stringApp API using the “source_specific_score” field value to filter the data. This data structure facilitates the versatile use of the Neo4COVID19 database in many research settings and the corroboration of information pertaining to various interactions.
Table 1. COVID-19 focused network statistics.
Shown are summary of individual data types integrated into the Neo4COVID-19 Neo4j database. Of note, overlap may exist between data types associated with the original data sources. HHIs: host–host protein interactions, HPIs: host–pathogen (here: SARS-CoV-2) protein interactions, DTIs: drug–target interactions.
| Dataset | Host Targets | Viral Targets | Drugs | HPIs | PPIs | DTIs |
|---|---|---|---|---|---|---|
| Proteomics Study | 102 | - | - | - | - | - |
| CRISPR | 105 | - | - | - | - | - |
| Meta Path AI/ML | 185 | - | - | - | - | - |
| STRING | 743 | - | - | - | 63,076 | - |
| SmartGraph / HATs | 148 | - | - | - | 225 | - |
| Interactome Study | 332 | 27 | - | 332 | - | - |
| P-HIPSter | 38 | 28 | - | 155 | - | - |
| Predicted DTIs | 46 | - | 31 | - | - | 86 |
| DrugCentral | 127 | - | 619 | - | - | 1,163 |
Importing Neo4COVID-19 Database into Cytoscape.
In order to facilitate the translational impact via dissemination [51] and flexible downstream analysis of the Neo4COVID19 focused network in the bioinformatics community, here we describe a simple procedure to import the database into the widely utilized Cytoscape application [52].
Importing the Neo4COVID19 database into Cytoscape v3.8.2 requires the installation of the “Cytoscape Neo4j Plugin” v0.4 [53] which can be easily achieved from within the Cytoscape application (see: Fig S1a in SI). Once the plugin is installed, a connection to the Neo4j database has to be established via the Bolt protocol, as shown in Fig S1b in SI.
After that, the successful establishment of database connection, the entire Neo4COVID19 database can be imported into Cytoscape with only a basic query statement written in the Cypher [47] language as shown in Fig S1c in SI. The statement is actually identical to the query provided by the plugin when removing the “LIMIT” clause from the default statement. Finally, a custom visualization style can be applied (see: Fig S2 and “Applying Custom Visual Style to the Imported Network in Cytoscape”, SI). The resultant network is shown in Fig S1d in SI.
Use Cases.
Here, we describe use cases to demonstrate how one can use the Neo4COVID19 for hypothesis generation in a network pharmacology setting. First, we examined the Tclin-designated HPI subset: Tclin designated proteins (according to TDL) from the Krogan dataset that have a fold change of 10 or higher following SARS-CoV-2 exposure. Out of 166 such HPIs, at least 63 occur between 27 human and 11 viral proteins. These 27 proteins are integral components of the mitochondrial respiratory chain complex I (GO:0005747) and are targeted by the antidiabetic drug metformin. Initially, we were excited to note that metformin (1) shares chemical similarity with an old antiviral drug moroxydine (2) (see: Fig 2). Shortly thereafter, as type 2 diabetes was identified as risk factor for severe COVID-19 [54], we had to briefly suspend further metformin research as we suspected its effect on the Tclin-designated HPI subset was indirect. However, recent reports show that metformin treatment is actually independently associated with a significant reduction in mortality in subjects with diabetes and COVID-19 [55]. Thus, while we cannot assume that the antiviral activity of moroxydine is similar, we are quite confident that metformin acts as a significant HPI perturbagen during SARS-CoV-2 infection and could serve as an adjuvant antiviral therapy under appropriate conditions.
Figure 2. Molecular structures of metformin and moroxydin.

Molecules were depicted with the help of ChemAxon’s MarvinSketch v17.15.0 [56].
Next, we investigated the interactions between host and virus proteins (the HPI relationships). With the help of node and edge attributes it is possible to construct a sub-network of the Neo4COVID19 network by retaining only virus and host targets, and the edges between them. This process gives rise to a bipartite network, in which host nodes are only connected to virus nodes and vice versa. In this network, a natural clustering emerges where several connected components exist involving many human proteins centered around a single virus protein (see: Fig 3a). The network topology also reveals that certain host targets might be thought of as the “Achilles’ heel” of the virus due to their connection structure. The peculiarity of these host proteins is that they are connected to many virus proteins hence they were named as “virus hubs”. An ideal strategy would be to target such virus hub that affects multiple biological processes of the virus while only causing a small perturbation in the regulatory network of the host.
Figure 3. Bipartite network of HPIs.

Human and virus proteins are depicted by circles and “v-like” shapes, respectively. The larger the node size, the higher the degree of the node connectivity. Color of the human proteins encode their TDL annotation: blue: Tclin, orange: Tchem, yellow: Tbio, gray: Tdark. A: The complete HPI bipartite network visualized in a “yFiles Circular” layout. B: The subnetwork of the largest connected component centered around the virus hub YWHAQ visualized in a “yFiles Radial” layout. network layouts were generated with the help of Cytoscape v3.8.2 [57] and yFiles modules [58].
As shown on Fig 3b, a few host targets are connected to a substantially higher number of nodes than others, as indicated a larger size of the corresponding nodes. One of such targets, YWHAQ (UniProt AC: P27348), is associated with “Tchem” TDL category, which means that there is a small molecule modulator for that target. Therefore, one might hypothesize that YWHAQ might be a potential host target to develop a drug against based on the existing modulator(s) as seed active molecule(s). However, the next step will require the analysis of the role of YWHAQ in the signal transduction network of the host (human). While such analysis is outside the scope of this work, it should be noted that the Pharos database indicates 260 PPIs associated with YWHAQ, which suggests that this particular target is involved in many biological processes of the host, hence its modulation will likely considerably perturb the network. It could be still possible that a redundant (parallel) path exists in the signal transduction network, which might mitigate the potential adverse effects of the perturbation.
While the aforementioned strategy might identify promising virus hubs, unfortunately, there are no Tclin targets among them. This indicates that at the time of this study we could not apply drug repositioning to target virus hubs. An alternative strategy could be to target multiple Tclin host targets (blue circles on Fig 3a) that appear in separate clusters. Drug compounds associated with Tclin targets can be easily imported into the network. This approach might form the basis for the engineering of a multi-agent therapy ideally employing approved drugs if they can effectively interfere with the implicated pathogen-host interactions. The detailed procedure to replicate the uses cases described above is provided in “Reproducing the Use Cases” section in SI.
Considerations Regarding the Continuous Integration and the Validity of the Data.
The data integration workflow presented in this study was designed to be widely adoptable in a network pharmacology research setting. In this sense, our workflow can facilitate any semi- automated integration of constantly evolving data – which happens to be the nature of many modern data sets, such as Reactome [6], DrugCentral [10], and Pharos [7], to name a few. However, the unprecedented pace of data influx we have witnessed since the beginning of the COVID-19 pandemic highlighted challenges with regards to continuous data integration and to data quality.
The workflow presented in this study demonstrates how essential data types can be integrated quickly with the help of the data registry mechanism. However, the rate limiting step of the integration remains the conversion and curation of data. While it would be desirable to integrate all emerging COVID-19 related HPI, PPI and DTI data [59]–[66] appropriately, this remains a significant challenge in the light of these considerations even with the help of the workflow of this study. Nonetheless, we are hopeful that the workflow can be the first step toward such a research effort, which may be realized in a form of a consortium. Also, we hope that our work will promote the effort of releasing data sets, even supplementary data, in a standardized format and/or via an API to facilitate the continuous integration of relevant data.
While we are hopeful that data integration efforts can be solved with the help of automated workflows and collaborative research, it is of outmost importance that the quality of source and integrated data is constantly monitored. The importance of this is aspect is reflected in the context of some early COVID-19 related results which have since been questioned, and manuscripts have been retracted [67], [68]. A specific controversy that emerged over time is associated with the efficacy of the lack thereof of Hydroxycholoroquine. Although the Neo4COVID-19 database includes data related to Hydroxycholoroquine we intended to keep those records to serve as example for historically data that needs to be taken account with care. Nevertheless, with the data structure of Neo4COVID-19 it is easy to filter out Hydroxycholoroquine related records.
We contemplate that the dichotomy between the need for relevant and bleeding edge information and the quality of the data will recreate this scenario in the case of a similar event in the future. Therefore, any data processing workflow should be dynamic, transparent, regularly reviewed, and constantly updated.
CONCLUSIONS.
Here, we describe a semi-automated workflow for the integration of data sources to produce a COVID-19 focused graph database. The workflow can be easily generalized to other drug discovery scenarios which can save precious time in the case of a pathogen outbreak. The workflow makes use of the state-of-the-art network pharmacology approaches and yields an interconnected network of host and viral protein targets and drugs, containing information on HPIs, PPIs, and DTIs. The workflow is flexible, which makes it possible to replace data sources and/or add new ones to it. The layered structure of the network and the underlying data schema allows researchers to filter data sources that they find relevant in their investigation. During the development of this workflow, we came across known bottlenecks related to data integration that we believe could be ameliorated to a great extent by following certain practices. For instance, the interaction types in a network pharmacology setting are well defined (PPI, HPI, DTI), and therefore, such data should be made available in a few well-established formats. This would allow for seamless integration of already existing and emerging datasets. Furthermore, providing a robust API for a dataset facilitates its integration and allows for programmatic updates. For instance, equipping SmartGraph with an API would allow for automating a significant part of the current workflow. If this API existed, then all manual intervention would be limited to configuring the data registry file and writing the data specific standardization snippets, allowing the rest of the workflow to be executed entirely automatically.
The Neo4j database generated by this workflow can be accessed via a web interface at https://aspire.covid19.ncats.io:7473/ to enable the exploration of data without much expertise in the bioinformatics field. In addition, it takes advantage of the Neo4j Bolt protocol and provides an API to facilitate the integration of the COVID-19 focused network into virtually any bioinformatics workflow. The Neo4COVID19 landing page (https://neo4covid19.ncats.io) provides detailed information under the “ACCESS” for those who would like to connect to the database in a programmatic manner. In this study, we provided use cases to show how the Neo4COVID-19 network can be utilized to generate hypotheses with focus on drug repositioning.
We believe that our Neo4COVID19 database will be a valuable asset to the research community and will catalyze the discovery of therapeutics to defeat COVID-19. Furthermore, the underlying flexible workflow can serve as a starting point for the integration of critical knowledge in the event of a potential future outbreak, which we all hope will never happen.
METHODS.
Assembly of a Multimodal Network.
In a drug discovery setting driven by network pharmacology, a multimodal network [36], [37], [69] needs to be assembled and tailored according to the disease context. In this study, we set forth to create a multimodal network focused on COVID-19 by integrating host and pathogen protein targets, drugs, and relations defined between them, such as PPIs, HPIs and DTIs. The respective information was derived from various data sources described in detail below. Furthermore, we use a “resource” alias (see: Fig 1) for each dataset throughout the text in order to concisely and unambiguously refer to individual input datasets. A pseudo-code of the data integration workflow is provided in “Pseudo-Code of the Data Integration Workflow” section in Supporting Information (SI).
Host Targets Implicated to Play Important Role in Pathogenesis.
Host cell translation changes [70] following SARS-CoV-2 infection were studied in Caco-2 cells using translatome [71] and proteome proteomics at four different time points (2, 6, 10, and 24 hours, respectively) after infection. This unbiased profile of the cellular response to SARS-CoV-2 infection was used to identify key determinants of the host cell response to infection. Extensive proteome modulation occurs 24 hours post infection, e.g., reduced expression of cholesterol metabolism proteins, and increased expression profile for carbon metabolism proteins and spliceosome components. Some pathways appear amenable to therapeutic intervention, e.g., along the proteostasis and nucleotide biosynthesis pathways. Given quantified translation data for 2,715 proteins (as documented in the Supplementary files), we selected proteins with P values below 0.05 at 24 hours (virus exposure compared to control), as follows: 75 proteins having lower translation values across all 4 time points (of these, 38 are involved in acetylation according to STRING); and 23 proteins having positive virus-induced translation values at 24 hours (of which, 12 are also involved in acetylation), respectively. These 98 host proteins were subject to further processing: one (UniProt AC: Q9N2J8) was removed as it cannot be mapped to a gene name, four (UniProt ACs: P84243, Q8IZP9, Q8IXH7, P63302) were mapped to multiples gene names, thus the respective records were replicated. This gave rise to 102 proteins in total, that were denoted as resource “A”.
Host genes essential for cell survival in response to SARS-CoV-2 infection were identified using two Cas9 Vero-E6 cell line constructs, using a genome-wide pooled CRISPR (clustered regularly interspaced short palindromic repeats) library [72], [73]. This CRISPR-Cas9 screen identified “pro-viral” and “anti-viral” genes, as follows. “Pro-viral” genes are involved in resistance, and their knockout confers resistance to virus-induced cell death. These genes include the ACE2 viral entry receptor, 11 genes from the SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex [74] and 7 genes associated with CDKN1A transcription upregulation via RUNX3 [75], respectively. “Anti-viral” genes are involved in sensitization, and their knockout sensitizes a cell to virus-induced cell death; these genes include HIRA (a subunit of the H3.3 histone chaperone complex), a set of 6 genes involved in viral translation, 8 genes associated with the SMN (survival of motor neurons) complex, and 5 components of the NURF (Nucleosome Remodeling Factor) complex, respectively. A set of 53 “pro-viral” and 52 “antiviral” host genes detected via this CRISPR-Cas9 screen [73] were incorporated, and assigned a resource label “B”.
Next, we describe an AI/ML framework that was utilized to predict host proteins that potentially interact with viral proteins. The Target Central Relational Database (TCRD) [7] aggregates protein-specific data from different sources (e.g., GTEx [76], LINCS [77], STRING [39], Reactome [6], [78]), and it was used to build the TCRD-KG knowledge graph. The TCRD-KG nodes can be proteins, diseases, or phenotypes, and edges can be pathways, protein-protein interactions, or other biological relationships among proteins and diseases. A Machine Learning (ML) framework based on the TCRD-KG meta paths [79] and XGBoost [80] classification algorithm was developed to predict disease-associated genes (proteins). The meta paths specify network paths that connect proteins to specific diseases in the TCRD-KG. The degree-weighted path count (DWPC) [81] metric is used to quantify the meta paths prevalence, transforming TCRD-KG data to feature vectors for ML model by meta paths matching, based on an input disease. The Python package to build TCRD-KG using TCRD database and XGBoost ML model is available on GitHub (https://github.com/unmtransinfo/ProteinGraphML) [82].
A training data set comprising 104 proteins as positive labels (known to be associated with SARS-CoV-2) and 114 proteins as negative labels (not associated with SARS-CoV-2) was used to train the ML model. A total of six different models were built, using slight variations of the input data (e.g., inclusion/deletion of human proteins identified by P-HIPSTER [83] to interact with SARS-CoV-2 proteins; and inclusion or absence of the LINCS [77] descriptors). Using 5-fold cross-validation on the training data, the area under the curve (AUC), accuracy, and Matthews Correlation Coefficient (MCC) were computed for each model. The ML models trained on 218 proteins were used to predict the association between 20,029 proteins and SARSCoV-2. Using XGBoost feature importance output, meta paths were sorted in decreasing order of “gain” score to understand node interactions. The features with higher “gain” were more dominant in predicting SARS-CoV-2 associated proteins. A total of 986 proteins were predicted with “high confidence” by the 6 models; of the 136 predicted by 3 or more models, 99 were not part of the “understudied” proteins [54] and were given preference for this study (denoted as resource “C”).
Virus-Implicated Host Proteins.
Host proteins that were either identified as interacting partners of SARS-CoV-2 virus proteins in experimental studies or were predicted to be of potential importance in terms of pathogenesis or therapy are referred to as virus-implicated host proteins (VIHPs) in this study. VIHPs were derived from experimental determined HPIs (resource “E”), and from host proteins implicated in experimental studies (resources “A”, “B”). This initial set of VIHPs was extended by predicted HPIs (resource “F”), DTIs (resource “D”) and host proteins of potential importance (resource “C”).
Host–Pathogen and Host–Host Interactions.
Experimentally determined host–pathogen interactome was published by Krogan et al. [32], [33] They identified 332 HPIs defined between 26 viral and 332 host proteins or host factors (resource “E”) that were determined via affinity purification mass-spectroscopy (AP-MS) [84]. A systematic analysis revealed 67 druggable human proteins and 69 compounds (including FDA-approved drugs and investigational drugs currently tested in clinical trials and preclinical studies) that were proposed to be evaluated for efficacy against SARS-CoV-2. While the host–pathogen interactome suggests potential pharmacological targets and possible interventions, the outcomes should be cautiously interpreted as it was mentioned that the identified agents could have either beneficial or detrimental effects (e.g., HDAC2 inhibitors).
Potential HPIs were predicted using the P-HIPSTer algorithm [83] giving rise to 155 HPIs involving 28 SARS-CoV-2 and 38 human proteins (resource “F”). P-HIPSTer offers a computational framework that utilizes sequence and structural information to infer HPIs, currently covering a total of 28 viral families (>900 human viruses) and > 5,000 human proteins accounting for 282,528 HPIs [85].
Experimentally Determined PPIs.
We collected PPIs from various data sources. The network assembly process involved a network expansion step with the help of the STRING and stringApp APIs (resource “G”) [39], [40]. In this step, experimentally determined and inferred PPIs were imported with the following parameter settings: maximum interactors = 100, alpha = 0.5. For more details on STRING expansion of the network please refer to section “Expansion of PPIs via StringApp API” in SI. Considering that interactions returned by the stringApp API are undirected, these edges were introduced into the network in both directions.
Due to the implication of histone acetyltransferases (HATs) in the pathogenesis, a set of HATs was prioritized (resource “H”), and in conjunction with virus-implicated host proteins (VIHPs) it was utilized to construct a subnetwork with the help of SmartGraph (resource “I”). In the SmartGraph analysis, the HATs and VIHPs were used as starting and end nodes, and vice versa. In either case, the maximum length of shortest paths between starting and end nodes was limited to 3. For more details on the assembly of the SmartGraph subnetwork please refer to section “Assembly of the SmartGraph Subnetwork”, SI. This input dataset contains 248 PPIs involving 108 host targets. The “Functional interactions” dataset (resource “J”), derived from Reactome database (version 2019) [6], [78] was used to cross-reference PPIs originating from various data sources.
Drug–Target Interactions.
Pertinent DTIs were primarily extracted from the DrugCentral database (version 2020, resource “K”) [10]. The DrugCentral database includes 4,642 drugs, of which 2,549 have regulatory approval dates, 3,082 have ATC codes [86], and 1,884 have INN stem annotations [87]. These drugs are associated with 110,577 formulations (drug labels) in total. Furthermore, the DrugCentral database also provides information on DTIs; 15,397 human DTIs and 4,910 non-human DTIs; of these 2,752 (2,328 human) are MoA drug-target associations.
For over 90% of the DTIs, the DTI therapeutic consequence is documented. We also collected the pharmacological action for each DTI which provides additional information about the potential intervention. The DTIs were originally extracted from scientific literature, drug labels and other data sources such as, ChEMBL [4], IUPHAR Guide2Pharmacology [88], WOMBAT-PK [89], DrugBank [90] and KEGG Drug [91].
To further enrich the network, we included both known and predicted DTIs for drugs and human endogenous metabolites found in the vicinities of the chemical space defined by three small molecules being investigated at the time for their antiviral activity against SARS-CoV-2 assays, namely, N4-hydroxycytidine (NHC) [92], hydroxychloroquine (HCQ) [67] and camostat (CAM) [93]. A set of 25 compounds was compiled around NHC [94], 5 around the chemical neighborhood of HCQ, and camostat. Processing those 31 small molecules against the CLARITY platform [95] returned a total of 86 DTIs, known (from public sources [5]) and/or predicted [96] to have activity against 46 unique protein targets (resource “D”).
Target Development Level Information.
Target development level (TDL) information for host proteins were imported from the Pharos portal (data track “L”) [7], [97].
Supplementary Material
Fig S1 – “Importing Neo4COVID19 graph database into Cytoscape – part 1.”
Fig S2 – “Importing Neo4COVID19 graph database into Cytoscape – part 2.”
Fig S3 – “Customizing network visualization.”
Table S1 – “Node and edge attributes of the Neo4COVID19 graph database.”
Table S2 – “Structure of data registry file.”
Table S3 – “Definition of mandatory and optional fields of internal data structure.”
ACKNOWLEDGEMENTS
The authors are thankful for the devoted work of Henderson Tozer, Kevin Diaz, James McWilliams and all members of the IT support team of the Information Resources Technology Branch of NCATS/NIH who enabled us to continue our research in unprecedented times. Furthermore, we would like to acknowledge Tim Willson, Dac-Trung Nguyen and Noel T. Southall, PhD, Cullen Kelin, PhD and Dave calabrese, PhD for their fruitful discussions. This research was supported in part by the Intramural research program of the NCATS, NIH and by the Illuminating the Druggable Genome-Knowledge Management Center NIH-U54 Grant (1U54CA189205-01, PI: Tudor I. Oprea, MD Ph.D.). This research was partly supported by a project from the Spanish Ministerio de Ciencia, Innovación y Universidades (SAF2017-83614-R, PI: Jordi Mestres).
LJJ is co-founder and scientific advisory board member of Intomics A/S. All other authors have no competing interests to declare.
ABBREVIATIONS
- API
Application Programming Interface
- ATC
Anatomical Therapeutic Chemical
- INN
International Nonproprietary Names
- MoA
Mechanism-of-Action
- FDA
U.S.Food and Drug Administration
Footnotes
ASSOCIATED CONTENT
Supporting Information
Python code to access Neo4COVID19 database via API, overview of data integration workflow, instructions for reproducing integration workflow, detailed steps for assembling SmartGraph subnetwork, expansion of host-host interactions via StringApp API, applying custom visual style to Cytoscape network, instructions to reproduce the use cases, additional figures and tables.
The instructions can be found in this file: https://github.com/ncats/neo4covid19/blob/master/README.md.
All other datasets and computational tools utilized in this study were described in detail in “Methods” section.
Data and Software Availability
The Neo4COVID19 database is publicly available at https://aspire.covid19.ncats.io:7473. Of note, “No authentication” needs to be selected from the drop-down list “Authentication type”. We provide further information at the Neo4COVID19 website (https://neo4covid19.ncats.io) under “ACCESS” tab regarding how the Neo4COVID19 graph database can be accessed programmatically via the Neo4j Bolt protocol using API. Moreover, we provide detailed instruction how the database can be imported into Cytoscape in a user-friendly manner, see: section “Reproducing the Use Cases” in SI. The source code repository of the workflow utilized to construct the Neo4COVID19 database is publicly available at https://github.com/ncats/neo4covid19. In the same repository we provide detailed instructions on how the workflow can be replicated to build a replica of the Neo4COVID19 graph database.
REFERENCES
- [1].Houlihan CF and Whitworth JA, “Outbreak Science: Recent Progress in the Detection and Response to Outbreaks of Infectious Diseases,” Clin. Med. (Northfield. Il), vol. 19, no. 2, pp. 140–144, Mar. 2019, doi: 10.7861/clinmedicine.19-2-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ravi SJ, Meyer D, Cameron E, Nalabandian M, Pervaiz B, and Nuzzo JB, “Establishing a Theoretical Foundation for Measuring Global Health Security: A Scoping Review,” BMC Public Health, vol. 19, no. 1, p. 954, Dec. 2019, doi: 10.1186/s12889-0197216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Berger K. et al. , “Policy and Science for Global Health Security: Shaping the Course of International Health,” Trop. Med. Infect. Dis, vol. 4, no. 2, p. 60, Apr. 2019, doi: 10.3390/tropicalmed4020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gaulton A. et al. , “ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery,” Nucleic Acids Res., vol. 40, no. D1, pp. D1100–D1107, Jan. 2012, doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].A. P. Bento et al. , “The ChEMBL Bioactivity Database: an Update.,” Nucleic Acids Res., vol. 42, no. Database issue, pp. D1083–90, Jan. 2014, doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Croft D. et al. , “Reactome: a Database of Reactions, Pathways and Biological Processes,” Nucleic Acids Res., vol. 39, no. Database, pp. D691–D697, Jan. 2011, doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nguyen D-T et al. , “Pharos: Collating Protein Information to Shed Light on the Druggable Genome,” Nucleic Acids Res., vol. 45, no. D1, pp. D995–D1002, Nov. 2016, doi: 10.1093/nar/gkw1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cerami EG et al. , “Pathway Commons, a Web Resource for Biological Pathway Data.,” Nucleic Acids Res., vol. 39, no. Database issue, pp. D685–90, Jan. 2011, doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang R. et al. , “The NCATS BioPlanet – An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics,” Front. Pharmacol, vol. 10, Apr. 2019, doi: 10.3389/fphar.2019.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Avram S. et al. , “DrugCentral 2021 Supports Drug Discovery and Repositioning,” Nucleic Acids Res., 2020, doi: 10.1093/nar/gkaa997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Holshue ML et al. , “First Case of 2019 Novel Coronavirus in the United States,” N. Engl. J. Med, vol. 382, no. 10, pp. 929–936, Mar. 2020, doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li R. et al. , “Substantial Undocumented Infection Facilitates the Rapid Dissemination of Novel Coronavirus (SARS-CoV-2),” Science (80-. )., vol. 368, no. 6490, pp. 489–493, May 2020, doi: 10.1126/science.abb3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pei S, Kandula S, and Shaman J, “Differential Effects of Intervention Timing on COVID-19 Spread in the United States,” medRxiv, 2020, doi: 10.1101/2020.05.15.20103655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jones D, Neal RD, Duffy SRG, Scott SE, Whitaker KL, and Brain K, “Impact of the COVID-19 Pandemic on the Symptomatic Diagnosis of Cancer: The View From Primary Care,” Lancet Oncol., vol. 21, no. 6, pp. 748–750, Jun. 2020, doi: 10.1016/S1470-2045(20)30242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang Y, Shen C, and Hu C, “Effect of COVID-19 Epidemic on Delay of Diagnosis and Treatment Path for Patients With Nasopharyngeal Carcinoma,” Cancer Manag. Res, vol. Volume 12, pp. 3859–3864, May 2020, doi: 10.2147/CMAR.S254093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Madhusoodanan J, “News Feature: to Counter the Pandemic, Clinicians Bank on Repurposed Drugs,” Proc. Natl. Acad. Sci, vol. 117, no. 20, pp. 10616–10620, May 2020, doi: 10.1073/pnas.2007346117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gil C. et al. , “COVID-19: Drug Targets and Potential Treatments,” J. Med. Chem, p. acs.jmedchem.0c00606, Jun. 2020, doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- [18].“Knowledge Graph on COVID-19,” 2020. http://www.odbms.org/2020/03/we-build-a-knowledge-graph-on-covid-19/.
- [19].Smith M. and Smith JC, “Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface,” 2020, doi: 10.26434/chemrxiv.11871402.v4. [DOI] [Google Scholar]
- [20].Batra R, Chan H, Kamath G, Ramprasad R, Cherukara MJ, and Sankaranarayanan S, “Screening of Therapeutic Agents for COVID-19 using Machine Learning and Ensemble Docking Simulations,” Apr. 2020, [Online]. Available: http://arxiv.org/abs/2004.03766. [DOI] [PubMed] [Google Scholar]
- [21].Brimacombe KR et al. , “An OpenData Portal to Share COVID-19 Drug Repurposing Data in Real Time,” bioRxiv, 2020, doi: 10.1101/2020.06.04.135046. [DOI] [Google Scholar]
- [22].“COVID-19 Disease Portal.” rgd.mcw.edu/rgdweb/portal/home.jsp?p=14.
- [23].Kuleshov MV et al. , “The COVID-19 Drug and Gene Set Library,” Patterns, vol. 1, no. 6, p. 100090, Sep. 2020, doi: 10.1016/j.patter.2020.100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Q. et al. , “COVID-19 Literature Knowledge Graph Construction and Drug Repurposing Report Generation,” Jul. 2020, [Online]. Available: http://arxiv.org/abs/2007.00576. [Google Scholar]
- [25].Ioannidis VN et al. , “DRKG - Drug Repurposing Knowledge Graph for Covid-19,” 2020. https://github.com/gnn4dr/DRKG/. [Google Scholar]
- [26].Haendel MA, Chute CG, and Gersing K, “The National COVID Cohort Collaborative (N3C): Rationale, Design, Infrastructure, and Deployment,” J. Am. Med. Informatics Assoc, Aug. 2020, doi: 10.1093/jamia/ocaa196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hopkins AL, “Network Pharmacology.,” Nat. Biotechnol, vol. 25, no. 10, pp. 1110–1, Oct. 2007, doi: 10.1038/nbt1007-1110. [DOI] [PubMed] [Google Scholar]
- [28].Maron BA et al. , “A Global Network for Network Medicine,” npj Syst. Biol. Appl, vol. 6, no. 1, p. 29, Dec. 2020, doi: 10.1038/s41540-020-00143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barabási A-L, Gulbahce N, and Loscalzo J, “Network Medicine: A Network-Based Approach to Human Disease,” Nat. Rev. Genet, vol. 12, no. 1, pp. 56–68, Jan. 2011, doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Womack F, McClelland J, and Koslicki D, “Leveraging Distributed Biomedical Knowledge Sources to Discover Novel Uses for Known Drugs,” bioRxiv, 2019, doi: 10.1101/765305. [DOI] [Google Scholar]
- [31].Chen H, Cheng F, and Li J, “iDrug: Integration of Drug Repositioning and Drug-Target Prediction via Cross-Network Embedding,” PLOS Comput. Biol, vol. 16, no. 7, p. e1008040, Jul. 2020, doi: 10.1371/journal.pcbi.1008040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Krogan, “A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing,” [Online]. Available: https://www.biorxiv.org/content/ 10.1101/2020.03.22.002386v3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gordon DE et al. , “A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing,” Nature, vol. 583, no. 7816, pp. 459–468, Jul. 2020, doi: 10.1038/s41586020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].“Archived: WHO Timeline - COVID-19.” https://www.who.int/news-room/detail/27-042020-who-timeline---covid-19.
- [35].“Update - Timeline of WHO’s Response to COVID-19.” https://www.who.int/newsroom/detail/29-06-2020-covidtimeline.
- [36].Zahoránszky-Kőhalmi G, Sheils T, and Oprea TI, “SmartGraph: A Network Pharmacology Investigation Platform,” J. Cheminform, vol. 12, no. 1, p. 5, Dec. 2020, doi: 10.1186/s13321-020-0409-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Himmelstein DS et al. , “Systematic Integration of Biomedical Knowledge Prioritizes Drugs for Repurposing,” Elife, vol. 6, Sep. 2017, doi: 10.7554/eLife.26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mering C. v., “STRING: a Database of Predicted Functional Associations Between Proteins,” Nucleic Acids Res., vol. 31, no. 1, pp. 258–261, Jan. 2003, doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Szklarczyk D. et al. , “STRING v11: Protein–Protein Association Networks With Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets,” Nucleic Acids Res., vol. 47, no. D1, pp. D607–D613, Jan. 2019, doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Doncheva NT, Morris JH, Gorodkin J, and Jensen LJ, “Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data,” J. Proteome Res, vol. 18, no. 2, pp. 623–632, Feb. 2019, doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Apweiler R, “UniProt: The Universal Protein Knowledgebase,” Nucleic Acids Res., vol. 32, no. 90001, pp. 115D–119, Jan. 2004, doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].“UniProt: A Worldwide Hub of Protein Knowledge,” Nucleic Acids Res., vol. 47, no. D1, pp. D506–D515, Jan. 2019, doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Patient S, Wieser D, Kleen M, Kretschmann E, Jesus Martin M, and Apweiler R, “UniProtJAPI: A remote API for Accessing UniProt Data,” Bioinformatics, vol. 24, no. 10, pp. 1321–1322, May 2008, doi: 10.1093/bioinformatics/btn122. [DOI] [PubMed] [Google Scholar]
- [44].Consortium TU, “UniProt: The Universal Protein Knowledgebase,” Nucleic Acids Res., vol. 45, no. D1, pp. D158–D169, 2016, doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].“UniProt Proggrammatic Service for ID Mapping.” https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/idmapping/by_organism/HUMAN_9606_idmapping.dat.gz.
- [46].Ursu O. et al. , “DrugCentral 2018: An Update,” Nucleic Acids Res., vol. 47, no. D1, pp. D963–D970, Jan. 2019, doi: 10.1093/nar/gky963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].“Neo4j Graph Database.” https://neo4j.com/.
- [48].“Python Core Team. Python: A Dynamic, Open Source Programming Language. Python Software Foundation.” https://www.python.org/.
- [49].“Code Repository ‘neo4covid19.’” https://github.com/ncats/neo4covid19.git.
- [50].“Python Library ‘py2neo’ v4.”.
- [51].Colvis CM and Austin CP, “Innovation in Therapeutics Development at the NCATS,” Neuropsychopharmacology, vol. 39, no. 1, pp. 230–232, Jan. 2014, doi: 10.1038/npp.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shannon P. et al. , “Cytoscape: a Software Environment for Integrated Models of Biomolecular Interaction Networks.,” Genome Res., vol. 13, no. 11, pp. 2498–504, Nov. 2003, doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Warris S, Dijkxhoorn S, van Sloten T, and van de Vossenberg B, “Mining Functional Annotations Across Species,” bioRxiv, 2018, doi: 10.1101/369785. [DOI] [Google Scholar]
- [54].“CDC - Coronavirus Disease 2019 (COVID-19).” https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html#diabetes.
- [55].Crouse A, Grimes T, Li P, Might M, Ovalle F, and Shalev A, “Metformin Use Is Associated With Reduced Mortality in a Diverse Population With Covid-19 and Diabetes,” medRxiv, 2020, doi: 10.1101/2020.07.29.20164020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].“ChemAxon Ltd., Marvin Suite. Molecules Were Depicted with ChemAxon’s MarvinSketch 17.15.0.” https://chemaxon.com/products/marvin.
- [57].Shannon P, “Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks,” Genome Res., vol. 13, no. 11, pp. 2498–2504, Nov. 2003, doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wiese R KM, Eiglsperger M, “yFiles: Visualization and Automatic Layout of Graphs,” in Proceedings of the 9th International Symposium on Graph Drawing (GD 2001), 2001, p. 453 ff. [Google Scholar]
- [59].Daniloski Z. et al. , “Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells.,” Cell, vol. 184, no. 1, pp. 92–105.e16, 2021, doi: 10.1016/j.cell.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Abbott TR et al. , “Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza.,” Cell, vol. 181, no. 4, pp. 865–876.e12, 2020, doi: 10.1016/j.cell.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wei J. et al. , “Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection.,” Cell, vol. 184, no. 1, pp. 76–91.e13, 2021, doi: 10.1016/j.cell.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schneider WM et al. , “Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks.,” Cell, vol. 184, no. 1, pp. 120–132.e14, 2021, doi: 10.1016/j.cell.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhou Y, Hou Y, Shen J, Huang Y, Martin W, and Cheng F, “Network-Based Drug Repurposing for Novel Coronavirus 2019-nCoV/SARS-CoV-2,” Cell Discov., vol. 6, no. 1, p. 14, Dec. 2020, doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhou Y. et al. , “A Network Medicine Approach to Investigation and Population-Based Validation of Disease Manifestations and Drug Repurposing for COVID-19,” PLOS Biol., vol. 18, no. 11, p. e3000970, Nov. 2020, doi: 10.1371/journal.pbio.3000970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhou Y, Wang F, Tang J, Nussinov R, and Cheng F, “Artificial Intelligence in COVID-19 Drug Repurposing,” Lancet Digit. Heal, vol. 2, no. 12, pp. e667–e676, Dec. 2020, doi: 10.1016/S2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zeng X. et al. , “Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning,” J. Proteome Res, vol. 19, no. 11, pp. 4624–4636, Nov. 2020, doi: 10.1021/acs.jproteome.0c00316. [DOI] [PubMed] [Google Scholar]
- [67].Mehra MR, Ruschitzka F, and Patel AN, “Retraction—Hydroxychloroquine or Chloroquine with or without a Macrolide for Treatment of COVID-19: A Multinational Registry Analysis,” Lancet, vol. 395, no. 10240, p. 1820, Jun. 2020, doi: 10.1016/S01406736(20)31324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mehra MR, Desai SS, Kuy S, Henry TD, and Patel AN, “Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19,” N. Engl. J. Med, vol. 382, no. 25, p. e102, Jun. 2020, doi: 10.1056/NEJMoa2007621. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [69].Heath LS and Sioson AA, “Multimodal Networks: Structure and Operations.,” IEEE/ACM Trans. Comput. Biol. Bioinforma, vol. 6, no. 2, pp. 321–32, doi: 10.1109/TCBB.2007.70243. [DOI] [PubMed] [Google Scholar]
- [70].Bojkova D. et al. , “Proteomics of SARS-CoV-2-Infected Host Cells Reveals Therapy Targets,” Nature, vol. 583, no. 7816, pp. 469–472, Jul. 2020, doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Klann K, Tascher G, and Münch C, “Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2α,” Mol. Cell, vol. 77, no. 4, pp. 913–925.e4, Feb. 2020, doi: 10.1016/j.molcel.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hsu PD, Lander ES, and Zhang F, “Development and Applications of CRISPR-Cas9 for Genome Engineering,” Cell, vol. 157, no. 6, pp. 1262–1278, Jun. 2014, doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wei J. et al. , “Genome-wide CRISPR Screen Reveals Host Genes that Regulate SARS-CoV-2 Infection,” bioRxiv, 2020, doi: 10.1101/2020.06.16.155101. [DOI] [Google Scholar]
- [74].Stern M, Jensen R, and Herskowitz I, “Five SWI Genes are Required for Expression of the HO Gene in Yeast,” J. Mol. Biol, vol. 178, no. 4, pp. 853–868, Oct. 1984, doi: 10.1016/0022-2836(84)90315-2. [DOI] [PubMed] [Google Scholar]
- [75].“Reactome, RUNX3 Regulates CDKN1A Transcription.” https://reactome.org/content/detail/R-HSA-8941855.
- [76].“The GTEx Consortium Atlas of Genetic Regulatory Effects Across Human Tissues,” Science (80-. )., vol. 369, no. 6509, pp. 1318–1330, Sep. 2020, doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Stathias V. et al. , “LINCS Data Portal 2.0: Next Generation Access Point for Perturbation-Response Signatures,” Nucleic Acids Res., vol. 48, no. D1, pp. D431–D439, Jan. 2020, doi: 10.1093/nar/gkz1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Fabregat A. et al. , “The Reactome Pathway Knowledgebase,” Nucleic Acids Res., vol. 44, no. D1, pp. D481–D487, Jan. 2016, doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Oprea TI, Yang JJ, Byrd DR, and Deretic V, “Autophagy Dark Genes: Can We Find Them With Machine Learning?,” bioRxiv, 2019, doi: 10.1101/715037. [DOI] [Google Scholar]
- [80].Chen T. and Guestrin C, “XGBoost,” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Aug. 2016, pp. 785–794, doi: 10.1145/2939672.2939785. [DOI] [Google Scholar]
- [81].Himmelstein DS and Baranzini SE, “Heterogeneous Network Edge Prediction: A Data Integration Approach to Prioritize Disease-Associated Genes.,” PLoS Comput. Biol, vol. 11, no. 7, p. e1004259, Jul. 2015, doi: 10.1371/journal.pcbi.1004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].“Code Repository ‘ProteinGraphML.’” https://github.com/unmtransinfo/ProteinGraphML.
- [83].Lasso G. et al. , “A Structure-Informed Atlas of Human-Virus Interactions,” Cell, vol. 178, no. 6, pp. 1526–1541.e16, Sep. 2019, doi: 10.1016/j.cell.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Dunham WH, Mullin M, and Gingras A-C, “Affinity-Purification Coupled to Mass Spectrometry: Basic Principles and Strategies,” Proteomics, vol. 12, no. 10, pp. 1576–1590, May 2012, doi: 10.1002/pmic.201100523. [DOI] [PubMed] [Google Scholar]
- [85].“P-HIPSTer.” http://phipster.org/.
- [86].“Anatomical Therapeutic Chemical (ATC) Classification (WHO).” https://www.who.int/medicines/regulation/medicines-safety/toolkit_atc/en/.
- [87].“International Nonproprietary Names (WHO).”
- [88].Armstrong JF et al. , “The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending Immunopharmacology Content and Introducing the IUPHAR/MMV Guide to Malaria Pharmacology,” Nucleic Acids Res., Nov. 2019, doi: 10.1093/nar/gkz951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Olah M OT, Rad R, Ostopovici L, Bora A, Hadaruga N, Hadaruga D, Moldovan R, Fulias A, Mracec M, “WOMBAT and WOMBAT-PK: Bioactivity Databases for Lead and Drug Discovery.,” in Chemical Biology: From Small Molecules to Systems Biology and Drug Design., Wiley-VCH, New York, 2007. [Google Scholar]
- [90].Wishart DS et al. , “DrugBank 5.0: A Major Update to the DrugBank Database for 2018,” Nucleic Acids Res., vol. 46, no. D1, pp. D1074–D1082, Jan. 2018, doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, and Tanabe M, “Data, Information, Knowledge and Principle: Back to Metabolism in KEGG.,” Nucleic Acids Res., vol. 42, no. Database issue, pp. D199–205, Jan. 2014, doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sheahan TP et al. , “An Orally Bioavailable Broad-Spectrum Antiviral Inhibits SARS-CoV-2 and Multiple Endemic, Epidemic and Bat Coronavirus,” bioRxiv, 2020, doi: 10.1101/2020.03.19.997890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hoffmann M. et al. , “SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor,” Cell, vol. 181, no. 2, pp. 271–280.e8, Apr. 2020, doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mestres J, “The Target Landscape of N4-Hydroxycytidine Based on its Chemical Neighborhood,” bioRxiv, 2020, doi: 10.1101/2020.03.30.016485. [DOI] [Google Scholar]
- [95].“CLARITY v4 (2019). Chemotargets S.L., Barcelona.” https://www.chemotargets.com/PRODUCTS/CLARITY-v4.
- [96].Garcia-Serna R, Vidal D, Remez N, and Mestres J, “Large-Scale Predictive Drug Safety: From Structural Alerts to Biological Mechanisms,” Chem. Res. Toxicol, vol. 28, no. 10, pp. 1875–1887, Oct. 2015, doi: 10.1021/acs.chemrestox.5b00260. [DOI] [PubMed] [Google Scholar]
- [97].Oprea TI et al. , “Unexplored Therapeutic Opportunities in the Human Genome,” Nat. Rev. Drug Discov, vol. 17, p. 317, Mar. 2018, [Online]. Available: 10.1038/nrd.2018.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1 – “Importing Neo4COVID19 graph database into Cytoscape – part 1.”
Fig S2 – “Importing Neo4COVID19 graph database into Cytoscape – part 2.”
Fig S3 – “Customizing network visualization.”
Table S1 – “Node and edge attributes of the Neo4COVID19 graph database.”
Table S2 – “Structure of data registry file.”
Table S3 – “Definition of mandatory and optional fields of internal data structure.”
Data Availability Statement
The Neo4COVID19 database is publicly available at https://aspire.covid19.ncats.io:7473. Of note, “No authentication” needs to be selected from the drop-down list “Authentication type”. We provide further information at the Neo4COVID19 website (https://neo4covid19.ncats.io) under “ACCESS” tab regarding how the Neo4COVID19 graph database can be accessed programmatically via the Neo4j Bolt protocol using API. Moreover, we provide detailed instruction how the database can be imported into Cytoscape in a user-friendly manner, see: section “Reproducing the Use Cases” in SI. The source code repository of the workflow utilized to construct the Neo4COVID19 database is publicly available at https://github.com/ncats/neo4covid19. In the same repository we provide detailed instructions on how the workflow can be replicated to build a replica of the Neo4COVID19 graph database.