

SUMMARY
Osteoarthritis is a degenerative joint disease that causes pain, degradation, and dysfunction. Excessive canonical Wnt signaling in osteoarthritis contributes to chondrocyte phenotypic instability and loss of cartilage homeostasis, however, the regulatory is unknown. Using multiple models of temporomandibular joint osteoarthritis, we identify Lgr5-expressing secretory cells as forming a Wnt inhibitory niche that instruct Wnt-inactive chondroprogenitors to form nascent synovial joint and regulate chondrocyte lineage and identity. Lgr5 ablation or suppression during joint development, aging, or osteoarthritis results in depletion of Wnt-inactive chondroprogenitors and a surge of Wnt-activated, phenotypically unstable chondrocytes with osteoblast-like properties. We recapitulate the Wnt inhibitory niche critical for cartilage maintenance and create StemJEL™, an injectable hydrogel formula combining hyaluronic acid and sclerostin. Local delivery of StemJEL™ to post-traumatic osteoarthritic joints in rabbit, rat and mini-pig models restores cartilage homeostasis, chondrocyte identity and joint function. We provide proof-of-principal that preserving chondrocyte niche and identity mitigates osteoarthritis.
Keywords: chondroprogenitor cells, cartilage, temporomandibular joint, skeletal development, stem cell niche, WNT signaling, Lgr5, osteoarthritis, biomaterials, hydrogels, regeneration
INTRODUCTION
Skeletal mobility is reliant upon synovial joints comprised of articular cartilage connecting adjacent bones. Osteoarthritis (OA) causes progressive synovial joint deterioration, pain, and dysfunction1. Age and trauma are major risk factors for OA, which is the primary cause of disability in aging adults2. OA impacts 500 million people, however current treatments are limited to palliative care or invasive surgery 2. Minimally invasive, disease modifying OA drugs (DMOADs) 3 remains an unmet clinical need.
Adult articular cartilage is avascular, harbors a restricted number of cells, lacks a source of circulating progenitors, and consequently has limited regenerative capacity 4. Chondrocytes are mature cartilage cells that reside in a dense extracellular matrix (ECM), undergo low anaerobic metabolism 5, and maintain minimal ECM turnover 6. OA is marked by a loss of cartilage tissue homeostasis, where the balance is tipped toward cartilage catabolism7. Chondrocyte phenotypic instability significantly contributes to the failure of cartilage tissue maintenance 8,9 Phenotypically unstable chondrocytes pathologically express genes related to cartilage development 10 and osteoblast differentiation 11–13, such as RUNX2, a transcription factor critical for chondrocyte hypertrophy14, transdifferentiation of chondrocytes into osteoblasts15, and osteoblast differentiation16, and osteocalcin (OCN), a protein expressed by osteoblasts and in osteoarthritic chondrocytes 12,17. Osteoarthritic chondrocytes exhibit high anaerobic glycolysis 5, a process critical for osteoblast differentiation and bone formation 18, and acquire multiple, aberrant cell fates that collectively contribute to cartilage demise, such as hypertrophy, apoptosis, osteoblast differentiation, senescence, and production of degradative enzymes/cytokines 8,13,19. While the maintenance of chondrocyte identity is crucial for cartilage homeostasis, OA therapies that restore chondrocyte identity are not available clinically.
In skeletal progenitor cells the β-catenin-dependent canonical Wnt (cWnt) pathway controls a dynamic switch in determining a chondrocyte versus osteoblast fate and is critical for chondrocyte identity 20. During limb development 21,22 and bone fracture healing 23, conditional β-catenin ablation drives skeletal progenitors toward a chondrocyte fate, whereas β-catenin is required for osteoblast differentiation 21,22. A cWnt inhibitory environment is crucial for maintenance of resting zone chondrocytes in growth plate cartilage 24,25; while excessive cWnt in chondrocytes contributes to OA by promoting inflammation, senescence, hypertrophy, osteoblast differentiation and osteophytes 20,26. Therefore, sustaining low cWnt levels is crucial for chondrocyte identity and deters hypertrophy and/or an osteoblast-like fate. However, the niche governing low cWnt in cartilage remains elusive.
To define the cWnt inhibitory niche critical for chondrocyte identity, we interrogated cWnt signaling during skeletal joint morphogenesis. Here we used mammalian lower jaw joint or temporomandibular joint (TMJ) as model. The TMJ developed as a secondary cartilage during evolution when several ancestral lower jaw bones were transformed into the middle ear bones to adapt for greater bite force and hearing in mammals27,28. Unlike axial skeletal articular cartilage, the mandibular condylar cartilage is lined with a fibrous, perichondrium-like tissue that persists in adults and dually operates as an articular cartilage and a growth cartilage 29,30. Postnatal mandibular condylar cartilage is stratified into cellular zones consisting of a perichondrium-like tissue lining the surface of the condyle containing heterogenous COL1A1+/COL2A1− perichondrial cells (PCs), including an outer superficial zone (SZ) and an inner polymorphic zone (PZ) harboring heterogeneous chondroprogenitor cells (CPCs) that express SOX9 and RUNX229,30. Directly underlying the perichodrium reside the condylar cartilage consisting of the flattened chondrocyte and hypertrophic chondrocyte zones comprised of COL2A1+/RUNX2− chondrocytes and COL10A1+/RUNX2+ hypertrophic chondrocytes, respectively 29,30. Here we exploit the mandibular condylar cartilage as a model system for elucidating the cWnt inhibitory niche critical for chondrocyte identity.
The Wnt target gene Lgr5 is a well-known marker for multiple adult stem cells 31–33 and also labels secretory cells critical for tissue maintenance 34. Recently Lgr5-expressing cells have been identified in developing joints within interzone cells and SZ, but their function is unknown 35. Here we define Lgr5-expressing cells in localized in the outer SZ of the perichondrium (PC) as secretory niche cells crucial for tempering cWnt signaling in chondroprogenitors and chondrocytes. Lgr5-expressing cells are required to maintain an appropriate pool of cWnt-inactive chondroprogenitors and maintain chondrocyte phenotypic identity. Upon Lgr5 deletion or suppression during TMJ development, aging, or OA, chondrocytes become phenotypically unstable and abnormally express RUNX2 and/or OCN. Based on our model system, we developed StemJEL™, a minimally invasive OA therapy that blends high molecular weight hyaluronic acid and the Wnt inhibitor sclerostin to provide a cWnt inhibitory niche, restore chondrocyte identity, and promote cartilage tissue maintenance. We demonstrate in post-traumatic osteoarthritic rat knee joints and rabbit and pig jaw joints that local delivery of StemJEL™ reinstates chondrocyte identity, cartilage homeostasis and joint function.
RESULTS
Osteoarthritic chondrocytes are phenotypically unstable and have high canonical Wnt activity.
To test the hypothesis that osteoarthritic chondrocytes lose their phenotypic identity, we surgically induced secondary, post-traumatic OA by perforating the disc in the jaw joints of Yucatan miniature pigs 36–38 and analyzed differential gene expression using bulk RNAseq (Figure 1a). Clustering analysis showed that biological quadruplicates of cartilages from the injury and naive healthy groups each clustered together (Figure 1b), indicating that injured cartilages possessed a biologically unique pattern of transcriptomes. Analyses of differentially expressed genes (DEGs) revealed 993 genes were different between the groups (fold change≥2; FDR-adjusted p value ≤0.05), with 596 upregulated genes and 397 downregulated genes in the cartilage injury group. Gene ontology (GO) enrichment analyses of DEGs at the level of biological processes (Figure 1c) and molecular functions (Figure S1a) revealed that the cartilage injury group had down-regulation of functions/processes related to mitochondrial energy consumption and oxidative phosphorylation (Table S1), suggesting chondrocytes shift to anaerobic metabolism, consistent with osteoarthritic chondrocytes 5. Evaluation of DEGs confirmed significant increase in genes related to ECM degradation, inflammation, and OA in the cartilage injury group (MMP13, COMP, MMP9, MMP25, ADAM15, IL-16, ILRL1) (Figures 1d, S1b) 39. GO analyses showed the cartilage injury group had upregulated cartilage development process with increased expression of joint development genes (SOX5, SOX6, SOX9, SNORC, PTCH1) (Figures 1d, S1c, Table S1), consistent with osteoarthritic chondrocytes 39. GO analyses showed upregulation of osteoblast differentiation and bone mineralization processes and Frizzled binding in the injury group (Figures 1c, S1a, Table S1). Gene expression levels confirmed a significant upregulation osteogenesis genes (Figures 1d, S2a) and differentially expressed Wnt-related genes (Figures 1d, S2b) in the injury group. These data suggest that the cartilage injury induces OA chondrocyte phenotype and dysregulated Wnt signaling.
Figure 1. Osteoarthritic chondrocytes are phenotypically unstable and have high Wnt/β–CATENIN.
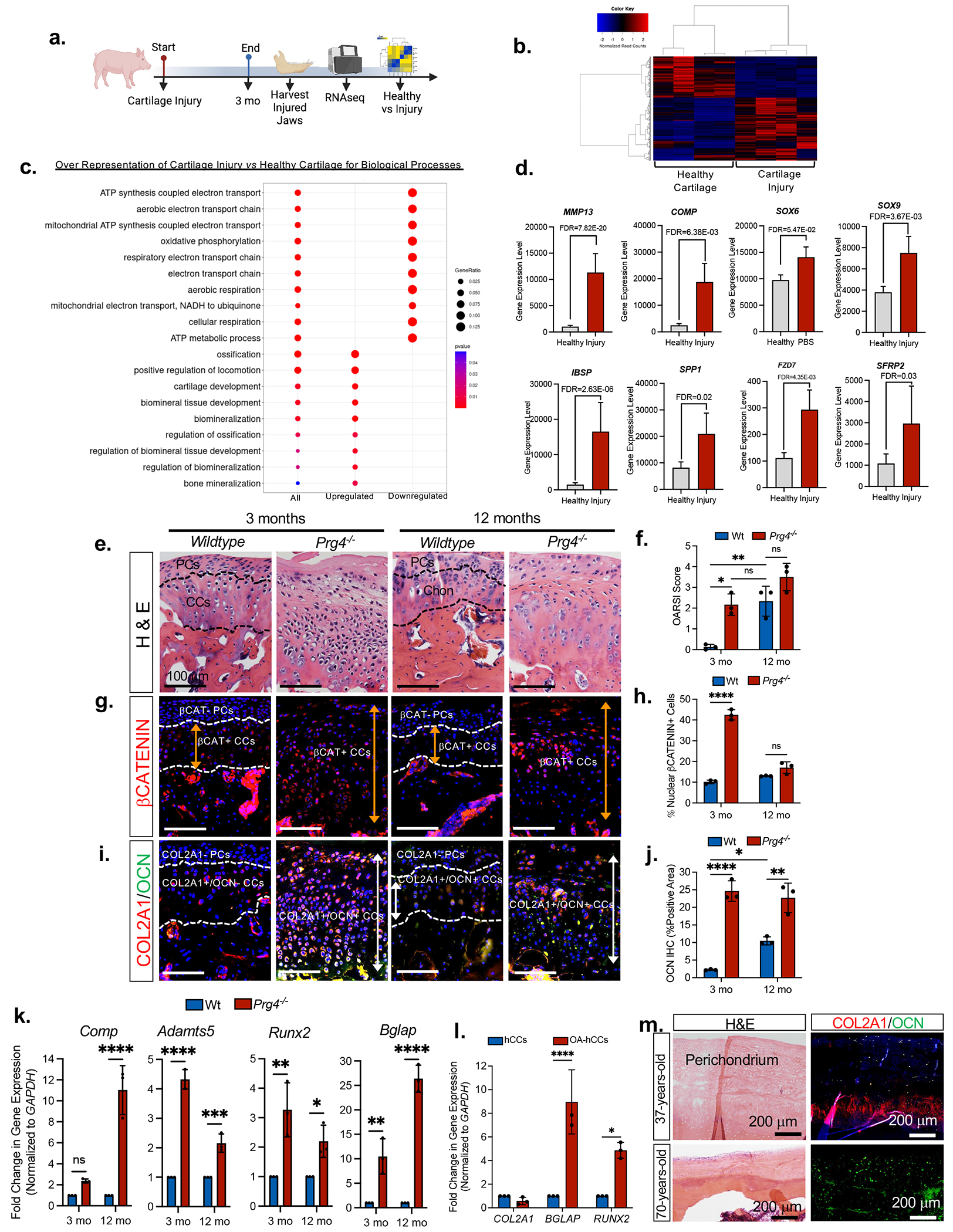
(a) Schematic depicting miniature pig cartilage injury model. (b) Gene expression heat map and hierarchical clustering for 993 genes significantly up- or downregulated (FC > ±2, FDR-adjusted p value=0.05) in injured cartilage (versus healthy). (c) Dot plot of GO terms of biological processes from B. (d) Relative gene expression levels from GO enrichment analyses. (e) H&E staining and (f) OARSI scores of Wildtype and Prg4−/− mice. Data presented are mean score from 3 reviewers ± SD. *p≤0.05, **p≤0.01; Two-way ANOVA followed by Tukey’s post hoc; n=3 mice. (g) Immunohistochemistry of βCatenin expression in Wildtype and Prg4−/− mice. PCs=perichondrial cells, CCs=condylar chondrocytes (h) Percentage of nuclear β-CATENIN-expressing cells in Wildtype and Prg4−/− mice. Data are mean percentage ± SD normalized to total cell number; ***p≤0.001; Two-way ANOVA followed by Tukey’s post hoc; n=3 mice. (i) Representative image of immunohistochemistry of type II collagen (COL2A1) and osteocalcin (OCN) in Wildtype and Prg4−/− mice. (j) Area of OCN expression from immunohistochemistry in Wildtype and Prg4−/− mice. Data are mean % area ± SD normalized to total area; *p≤0.05; **p≤0.01; ****p≤0.0001; Two-way ANOVA followed by Tukey’s post hoc; n=3 mice. (k) qRT-PCR using Wildtype and Prg4−/− mouse condyles. Data presented are mean fold change ± SD normalized to GAPDH. *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two-way ANOVA followed by Tukey’s post hoc; n=3 mice. (l) qRT-PCR using healthy human mandibular condylar chondrocytes (hMCCs) relative to osteoarthritic human mandibular condylar chondrocytes (OA-hMCCs). Data are mean fold change ± SD normalized to GAPDH. *p≤0.05, ***p≤0.001; two-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (m) H&E and immunohistochemistry of type II collage (COL2A1) and osteocalcin (OCN) in the mandibular condylar cartilages of a healthy 37-year-old and a 70-year-old OA patient .
Phenotypically unstable OA chondrocytes pathologically express genes/proteins found in hypertrophic chondrocytes and/or osteoblasts 11–13, including Runx2/RUNX2 13,40 and Bglap/OSTEOCALCIN 12. We tested whether excessive cWnt promotes phenotypically unstable chondrocytes in vitro. Porcine condylar chondrocytes (CCs) were cultured in pellet cultures with WNT3A or the porcupine inhibitor WNT-C59 (Figures S3a–S3b). Immunohistochemistry (Figure S3a) and qRT-PCR (Figure S3b) showed WNT3A promoted the expression of chondrocyte genes/proteins (ACAN, CO2A1) and hypertrophic chondrocyte and osteoblast genes (RUNX2/BGLAP), suggesting that WNT3A induces chondrocyte instability. However, there were no changes in ACAN/COL2A1 or RUNX2/BGLAP expression in CC pellets upon WNT-C59 treatment (Figures S3a–S3b). To test whether WNT3A-mediated phenotypic instability is chondrocyte-specific, CCs were first induced to undergo osteogenesis followed by treatment with WNT3A or WNT-C59 (Figures S3c–S3d). CCs underwent mineralization and osteoblast differentiation (alizarin red+, RUNX2); however, the addition of WNT3a inhibited mineralization and RUNX2 expression, suggesting that WNT3A-induced phenotypic instability is chondrocyte-specific (Figures S3c–S3d).
We next evaluated whether porcine perichondial cells (PCs) form phenotypically unstable chondrocytes in vitro 36 (Figures S3e–S3f). In PC pellets WNT3A significantly induced the expression of RUNX2 and BGLAP, but not ACAN or COL2A1 (Figures S3e–S3f), suggesting that Wnt suppresses chondrogenesis of PCs. On the contrary treatment with WNT-C59 promoted ACAN and COL2A1 expression in PC pellets, suggesting that Wnt inhibition promotes chondrogenesis of PCs. In osteogenic assays WNT3A enhanced mineralization (+alizarin red) in PCs (Figures S3g–S3h). These data are consistent with our previous findings demonstrating the presence of multipotent stem/progenitor cells in the mandibular condyle PC, where Wnt signaling promotes osteogenesis and Wnt inhibition promotes chondrogenesis 37,41. Unlike mandibular CCs, PCs do not form phenotypically unstable chondrocytes upon WNT3A treatment.
To test whether excessive cWnt corresponds with chondrocyte instability in OA, we evaluated intracellular βCatenin and the loss of chondrocyte identity in the Prg4−/− OA mouse model 42,43. Relative to Wildtype mice, 3-month-old Prg4−/− mice (Figure 1e) had significantly increased OARSI structure score (Figure 1f). In 3-month-old Wildtype mice nuclear β–CATENIN expression was present in COL2A1+ CCs and absent in COL2A1− PCs (Figures 1g–1i). However, 3-month-old Prg4−/− mice showed a significant expansion of nuclear β–CATENIN+ CCs (Figure 1g orange arrow, Figure 1h) and phenotypically unstable COL2A1+/OCN+ CCs (Figure 1i white arrow, Figure 1j). The expansion of nuclear β–CATENIN+ CCs and phenotypically unstable COL2A1+/OCN+ CCs in Prg4−/− mouse condyles was concomitant with a loss of COL2A1− PCs. These data suggest that excessive cWnt signaling and chondrocyte phenotypic instability may contribute to OA in the Prg4−/− mice. Interestingly, aged 12 months-old Wildtype mice acquired phenotypically unstable COL2A1+/OCN+ CCs (Figure 1i white arrow, Figure 1j) concomitantly with increased OARSI scores (Figure 1f), suggesting that chondrocyte phenotypic instability and OA develops when aging. qRT-PCR analysis confirmed increased expression of genes related to OA (Comp, Adamts5) and phenotypically unstable chondrocytes (Runx2, Bglap) in the Prg4−/− mice relative to Wildtype (Figure 1k). We next evaluated phenotypically unstable chondrocytes in humans. Relative to young human condylar chondrocytes (hCCs), aged osteoarthritic human condylar chondrocytes (OA hCCs) had significantly increased RUNX2 and BGLAP expression (Figure 1I). Immunohistochemistry showed that, unlike healthy young condylar cartilage, aged and osteoarthritic human condylar cartilage showed marked OCN expression (Figure 1m). These data suggest that aged and osteoarthritic human chondrocytes lose their phenotypic identity as evidenced by upregulation of RUNX2 and OCN expression in chondrocytes. Our studies across multiple species indicate that during OA high intracellular βCatenin is correlated with phenotypically unstable chondrocytes that express hypertrophic chondrocyte and/or osteoblast gene/proteins and depletion of COL2A1−/βCatenin− PCs.
Lgr5-expressing cells are enriched in juvenile mandibular condylar cartilage perichondrium but decrease in aging and osteoarthritis.
Lgr5 is a Wnt target gene that has recently emerged as marking secretory precursors and cells in the stomach and gut 34,44,45. To test the hypothesis that Lgr5-expressing cells represent a secretory cell in cartilage, we first examined Lgr5 expression during jaw joint morphogenesis in Wildtype and Lgr5EFP-IRES-creERT2+/− reporter mice (Figures 2a, S4, S5a). At E14.5 Lgr5-expressing cells reside on the surface of the condylar blastema (CB) (Figures 2a, S4a), surrounding Meckel’s cartilage (orange arrows, MK, Figure S5a), and surrounding the middle ear bones (blue arrows, Figure S5a). At E16.5 after superior joint cavity (SJC) formation, Lgr5 is expressed in disc progenitor cells (DPCs) (Figures 2a, S4b). When the disc separates from the condyle and the inferior joint cavity (IJC) is formed, Lgr5 is expressed in disc cells at E18.5 and P0 (Figures 2a, S4c, S4d), but is restricted to the outer SZ of the PC during embryonic and post-natal timepoints (black triangles, Figures 2a, white triangles, S4b–S4e).
Figure 2. Lgr5 expression is enriched in the superficial zone of articular cartilage perichondrium, but decreases in aging and osteoarthritis.
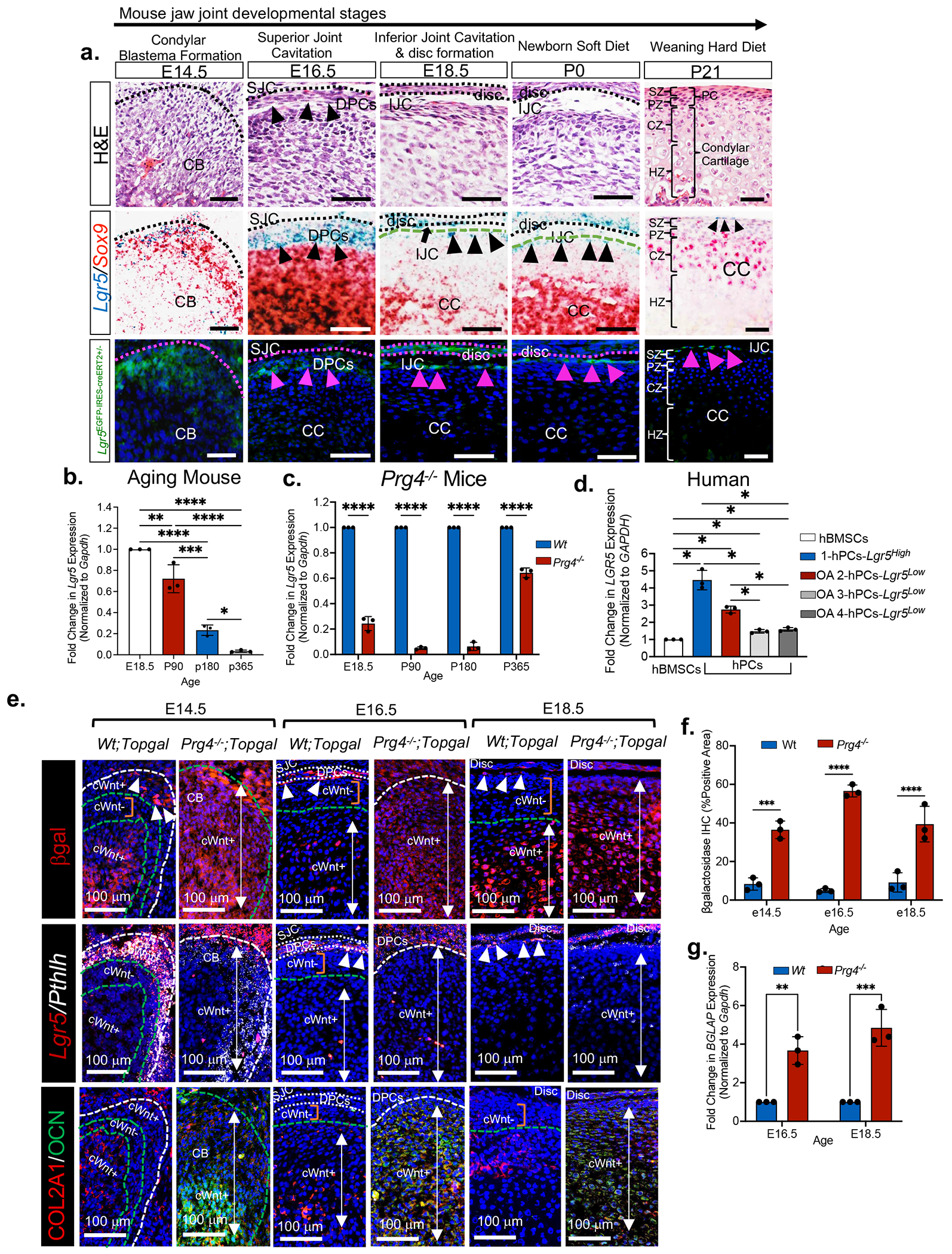
(a) H&E images (top row) of key mouse jaw joint developmental stages. in situ hybridization (middle row) of Lgr5 (black triangles) and Sox9 genes in mice. Lgr5-GFP cells (bottow row, pink triangles) in jaw joint superficial zone of Lgr5EFPcre+/− mice. CB=condylar blastema; DPCs=disc progenitor cells; SJC=superior joint cavity; CC=mandibular condylar cartilage; IJC=inferior joint cavity (black arrow), SZ=superficial zone, PZ=polymorphic zone, PC= perichondrium, CZ=chondrocyte zone, HZ=hypertrophic zone. (b) qRT-PCR of Lgr5 in aging mice. (c) qRT-PCR of Lgr5 in Wildtype and Prg4−/− mouse condyles. Data are mean fold change ± SD normalized to GAPDH. ****p≤0.0001; two-way ANOVA followed by Tukey’s post hoc; n=3 mice. (d) qRT-PCR using human bone marrow stromal cells (hBMSCs), healthy human perichondral cells (hPCCs LGR5High) and osteoarthritic human perichondrial cells (OA-2-4-hPCCs LGR5Low). Data are mean fold change ± SD normalized to GAPDH. *p≤0.05; two-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (e) in situ hybridization of Lgr5 and Pthlh and immunohistochemistry of βgalactosidase (βgal), type II collagen (COL2A1) and osteocalcin (OCN) Wt;Topgal and Prg4−/−;Topgal mouse condyles. CB=condylar blastema, SJC=superior joint cavity, DPCs=disc progenitor cells, cWnt+=canonical WNT activity (white arrows), cWnt− = cWnt inactivity (orange bar), white triangles=Lgr5-expressing cells. (f) Quantification of βgal immunostainings from 2e. Data presented are mean % area ± SD normalized to total area; ***p≤0.001; ****p≤0.0001; Two-way ANOVA followed by Tukey’s post hoc; n=3 mice. ***p≤0.01 (g) qRT-PCR of Wildtype and Prg4−/− condyles. Data are mean fold change in gene expression ± SD normalized to GAPDH. **p≤0.01, ***p≤0.001; two-way ANOVA followed by Tukey’s post hoc; n=3 mice.
We next characterized Lgr5-expressing cells relative to other cell types in the mandibular condyle, including Sox9+/Runx2+ chondroprogenitor cells (CPCs)46 and ACAN+/COL2A1+ chondrocytes. Lgr5-expressing cells do not express Sox9 or Runx2 and differ from neighboring Sox9+/Runx2+ CPCs localized in the inner polymorphic zone of the PC (Figure 2a, Figure S4). Lgr5-expressing cells do not express aggrecan (ACAN) or type II collagen (COL2A1) unlike ACAN+/COL2A1+ chondrocytes (Figure S5b). Indian hedgehog (IHH) produced by prehypertrophic/hypertrophic chondrocytes regulates parathyroid-related protein-expressing (PTHRP/Pthlh) skeletal stem/progenitor cells and Ptch1+ chondrocytes in long bone and jaw growth plate 47–49. In the mandibular condyle, we observed Lgr5+/Pthlh+ superficial zone cells (SZCs), Lgr5+/Pthlh− SZCs, (white triangles, Figure S4) and Lgr5−/Pthlh− SZCs that differ from Ptch1+ chondrocytes and Ihh+ prehypertrophic/hypertrophic chondrocytes (Figure S4). To test the proliferation of Lgr5-expression cells, we evaluated EDU uptake in Lgr5EFP-IRES-creERT2+/− mouse condyles. After a 4-hour pulse Lgr5-expressing cells did not retain EDU, unlike EDU+ CPCs localized in the inner polymorphic zone (Figure S5c). Together these data show that Lgr5-expressing cells are heterogeneous and different from CPCs and chondrocytes.
We subsequently evaluated Lgr5 expression during aging and OA. Lgr5 expression persists until 6 months in mouse mandibular condyles, but there was age-dependent loss of expression (Figure 2b). In the Prg4−/− OA mouse model 42,43 there was a significant decrease in Lgr5 expression relative to aged-match Wildtype mice (Figure 2c). We evaluated LGR5 expression in humans using primary human periochondrial cells (hPCs) derived from the mandibular condyle of a healthy 37-year-old cadaver jaw joint relative to hPCs derived from the mandibular condyles of older patients undergoing jaw joint replacement surgery for OA treatment. Lgr5 expression in human PCs derived from a healthy 37-year-old cadaver was significantly higher (hPCs Lgr5High) than in OA hPCCs derived from older patients with OA (OA 2-4 hPCs Lgr5Low) (Figure 2d), suggesting a loss in Lgr5-expressing PCs in aging and in disease. These data in aged and osteoarthritic condyles from mice and humans suggest that loss of Lgr5-expressing PCs may contribute to OA progression.
To test the hypothesis that loss of Lgr5-expressing cells is concomitant with high cWnt activity during OA, we crossed Prg4−/− mice with Wnt reporter mice TOPGAL (Figures 2e–2g). In Wildtype mice, βGalactosidase expression (βGal, red) labeled cWnt activity (cWnt+) in SZCs on the surface of the condylar blastema and condyle (white triangles. Figure 2e) and in DPCs at E16.5 (Figure 2e white dashed lines). βGal showed cWnt activity in COL2A1+ chondrocytes (cWnt+, white arrows), but was absent in COL2A1β CPCs (cWntβ, orange bar) (Figure 2e), suggesting that CPCs are maintained in a low cWnt environment. In Prg4−/−/TOPGAL mice, the loss of Lgr5 expression was concurrent surge of cWnt-activated cells (βGal, white arrows Figures 2e–2f), phenotypically unstable COL2A1+/OCN+/Bglap chondrocytes (Figures 2e, 2g), and a loss of cWnt-inactive COL2A1− CPCs (Figure 2e). These data suggest that Lgr5− expressing cells are crucial for regulating low cWnt activity required to maintain a pool of cWNT inactive CPCs and chondrocyte phenotypic identity.
We next evaluated Lgr5 progeny using Lgr5-EGFP-IRES-creERT2+/−;R26R-tdTomato mice (Figures 3, S5c–S5f). Dams were administered tamoxifen daily at E12.5-E13.5 and chased over 1 year (Figure 3a). Lgr5-progeny (red) were localized in the disc and SZ (white triangles) at E19.5 and P21 (Figures 3b, S3d). At P365 Lgr5-progeny (red) are localized in the periosteum (+Periostin, POSTN) and osteocytes (white arrows), but do not become chondrocytes (Figure 3b). Tamoxifen-induced recombination at E15.5 and E16.5 (Figure S5e), prior to meniscus/disc formation, resulted in the Lgr5-progeny comprising the disc, SZ and PC (Figure S5f). Postnatal tamoxifen-induced recombination at P1-P3 (Figure 3c) also resulted in Lgr5-progeny encompassing the disc, articular cartilage SZ/PC, and periosteum of the ramus (Figure 3d). These data support a model where Lgr5-expressing cells localized around the condylar blastema give rise to cells localized in the SZ of the PC, disc, periosteum and bone (Figure 3e). However, Lgr5-expressiong cells do not become CPCs or chondrocytes during TMJ development, favoring our hypothesis that Lgr5-expressing cells provide a supportive role to CPCs and chondrocytes.
Figure 3. Lor5-expressing cells supply progeny to synovial joint meniscus/disc, articular cartilage superficial zone, perichondrium, periosteum and bone, but do not become chondroprogenitor cells or chondrocytes.
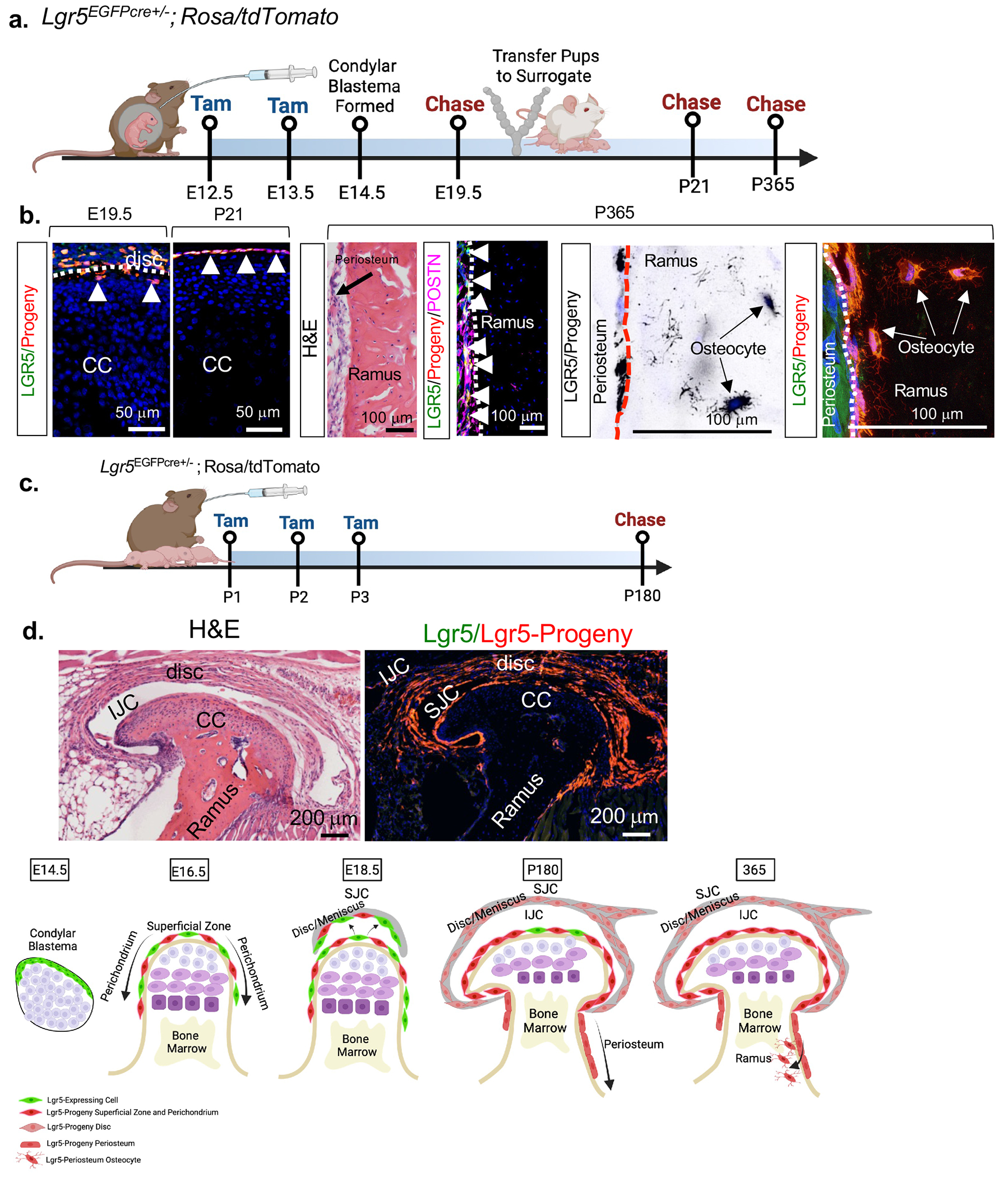
(a) Schematic of lineage tracing experiment in b. (b) Fluorescent imaging and immunostainings for lineage tracing in A. CC=mandibular condylar cartilage. (c) Schematic of lineage tracing experiment in d. (d) Fluorescent imaging for lineage tracing in C. CC=mandibular condylar cartilage, IJC=inferior joint cavity, SJC=superior joint cavity. (e) Proposed model.
Ablation of Lgr5-expressing cells impairs TMJ development, diminishes the chondroprogenitor cell pool, and generates a surge of Wnt-activated, phenotypically unstable chondrocytes.
To determine the function of Lgr5-expressing cells in articular cartilage, we used the Lgr5DTR-EGFP mouse model 33 to systematically ablate Lgr5-expressing cells in the presence of diphtheria toxin (DT) during TMJ development (Figures 4a–4c, S6). DT was applied once at E13.5, E15.5 and E17.5 and the TMJ was analyzed after 24 hours (Figures S6a–S6f). Ablation Lgr5-expressing cells for 24 hours at E13.5 (Figures S6a–S6b) and E15.5 (Figures S6c–S6d) disrupted the formation of the condyle blastema with a reduction in Sox9+ CPCs and Ihh+ prehypertrophic/hypertrophic chondrocytes (Figures. S6b, S6d). Ablation of Lgr5-expressing cells at E17.5 for 24 hours (Figures. S6e–S6f) was less disruptive to TMJ morphogenesis (Figure S6f), suggesting that Lgr5-expressing cells are more critical for early stages of TMJ development. We tested the hypothesis that Lgr5-expressing cells modulate cWnt activity in CPCs and chondrocytes through regulation of Wnt inhibitors by crossing Lgr5DTR-EGFP mice with Wnt reporter mice TOPGAL and evaluating the expression of the Wnt inhibitor dickkopf Wnt signaling pathway 3 (Dkk3) (Figures 4a–4c, S6g–S6h). Lgr5DTR-EGFP; TOPGAL and Wildtype mice were treated with DT at E13.5 and E15.5 and pups were examined at E16.5 and P0 (Figures 4a, S6g), βgal immunostaining in Wt/TOPGAL mice at P0 showed cWnt-activity in cells localized in the outer SZ of the PC (green triangles), cWnt-inactivity (cWnt−) in CPCs localized in the inner polymorphic zone of the PC and a gradient of cWnt activity in COL2A1+ chondrocytes (cWnt+, white triangle) (Figure 4b, first and second rows). Dkk3 expression was co-localized with Lgr5-expressing cells (orange triangles), within the inner perichondrium harboring cWnt− CPCs, and within the upper portion of chondrocytes, suggesting that Dkk3 may regulate cWnt activity in Lgr5-expressing cells and support low cWnt− activity in CPCs and chondrocytes (Figure 4b). Ablation of Lgr5-expressing cells in Lgr5DTR-EGFP;TOPGAL + DT mice showed a delay in CB formation with a loss Ptch1+ CPCs/chondrocytes and Ihh+ prehypertrophic/hypertrophic chondrocytes at E16.5 (Figure S6h). At P0 βGal immunohistochemistry showed that ablation of Lgr5-expressing cells in Lgr5DTR-EGFP;TOPGAL + DT mice resulted in a reduction of cWnt− CPCs in the inner PC and a surge of cWnt activated COL2A1+/OCN+ phenotypically unstable chondrocytes (Figure 4b, last row, white and yellow arrows, Figure 4c). In Lgr5DTR-EGFP;TOPGAL mice and Lgr5DTR-EGFP;TOPGAL + DT mice, diminished Lgr5-expressing cells also resulted in reduced Dkk3 expression (orange arrow) suggesting that Lgr5-expressing cells may support a cWnt inhibitory niche for cWnt− CPCs (Fig. 4b, third and fourth row). Relative to the wildtype mice, untreated Lgr5DTR-EGFP mice also demonstrated a significant loss of Lgr5 and Dkk3 expression in their condyles, indicating that the Lgr5DTR-EGFP transgenic mouse model has an abnormal TMJ phenotype (Figures 4b–4c). However, the expression of Lgr5 and Dkk3 is significantly lower in the Lgr5DTR-EGFP + DT mice relative to untreated Lgr5DTR-EGFP mice, suggesting that the introduction of DT induces a greater loss of Lgr5 expression and phenotype (Figures 4b–4c). Together these data demonstrate the loss of Lgr5-expressing cells is correlated with high cWnt activity, loss of cWnt-inactive CPCs, dysregulated Dkk3 expression, and marked chondrocyte phenotypic instability.
Figure 4. Lar5-expressing cells provide a Wnt inhibitory niche critical for maintaining chondroprogenitor cell pool and chondrocyte phenotypic identity.
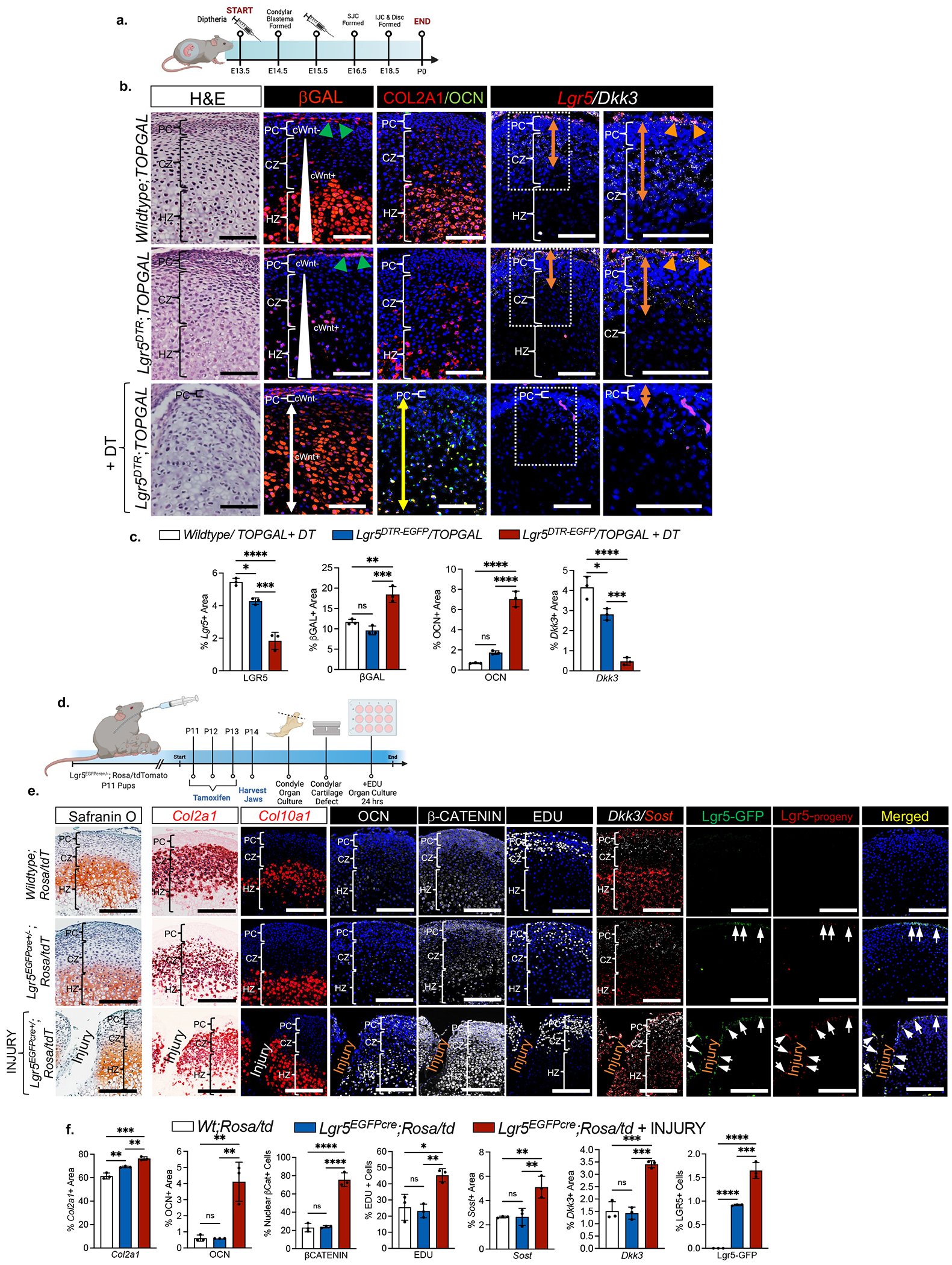
(a) Experimental timeline of Lgr5 ablation. (b) H&E staining, immunohistochemistry of βgalactosidase (βgal), type II collagen (COL2A1) and osteocalcin (OCN), and in situ hybridization of Lgr5 and Dkk3 in P0 mice from experiment in 4a. PC=perichondrium, CZ=chondrocyte zone, HZ=hypertrophic zone. Green triangles=cWnt-activated superficial zone cells in perichondrium, white triangle = cWnt gradient, white arrow=cWnt activated cells, yellow arrow=area of OCN expression, orange arrow = area of Dkk3 expression, orange triangles= Lgr5/Dkk3-expressing cells. (c) Quantification of immunohistochemistry and in situ hybridization from 4b. Data are mean % area expression ± SD normalized to total area; *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3 mice. (d) Schematic of cartilage injury model using organ cultures. (e) Safranin O staining, in situ hybridization of Col2a1, Col10a1, Dkk3, and Sost, immunohistochemistry of osteocalcin (OCN) and βCatenin, EDU uptake, and Lgr5-GFP+ cells (green) and Lgr5-progeny (red) in mice from experiment in 4d. Quantification of immunohistochemistry, in situ hybridization, EDU+ cells, and Lgr5-GFP+ cells from experiment in 4d. Data are mean % area expression ± SD normalized to total area; **p≤0.01, ***p≤0.001, ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3 mice.
During cartilage injury Lgr5-expressing cells modulate cWnt signaling and self-renew, but do not differentiate into chondrocytes.
To exclude Lgr5-expressing cells as the origin of phenotypically unstable chondrocytes and to confirm Lgr5-expressing cells as secretory niche cells, we performed lineage tracing studies using injured Lgr5-EGFP-IRES-creERT2+/−;R26R-tdTomato mice (Figures 4d–fj). We induced tamoxifen recombination in dams, harvested condyles and created a cartilage defect in P14 pups, and cultured the injured cartilage explants with EDU for 24 hours (Figure 4d). Safranin O staining confirmed the injury in organ cultures (Figure 4e). Injured organ cultures had significant expansion of OCN expression localized within Col2a1-expressing chondrocytes and Col10a1-expressing hypertrophic chondrocytes (Figures 4e–4f), suggesting that cartilage injury induces chondrocyte instability. Cartilage injury also caused a significant increase in the percentage of intracellular β-CATENIN-expressing cells, suggesting that increased cWnt-activity is correlated with loss of chondrocyte identity (Figures 4e–4f). There was a significant increase in EDU uptake and Dkk3 expression within the PC upon cartilage injury, suggesting that the injury may activate PCs to modulate cWnt activity (Figure 4e–4f). Relative to the uninjured Lgr5-EGFP-IRES-creERT2+/−;R26R-tdTomato and Wildtype;/R26R-tdTomato organ cultures, the injury group showed that GFP-Lgr5+ cells significantly increased, implicating that Lgr5-expressing cells self-renew during joint injury. GFP-Lgr5+ cells (green) and Lgr5-progeny (red, arrows) were localized to the periphery of the cartilage injury in the outer SZ of the PC, but did not populate the inner polymorphic zone of the PC, the chondrocyte zone, or the hypertrophic zone (Figure 4e– 4f). These data demonstrate that during cartilage injury Lgr5-expressing cells do not differentiate into CPCs or chondrocytes and may function as a potent cWnt modulator for neighboring cells.
Lgr5-expressing secretory cells provide a Wnt inhibitory niche for chondrocytes.
We hypothesized that Lgr5-expressing cells are secretory niche cells that produce cWnt inihbitors to modulate cWnt activity in CPCs and chondrocytes (Figure 5a). To interrogate this concept in vitro, we isolated mini-pig bone marrow stromal cells (BMSCs), heterogeneous primary mini-pig perichondrial cells (PCs, Lgr5+, Prg4+, Col1a1+) and mini-pig condylar chondrocytes (CCs, Lgr5−, Prg4−, Col1a−, Col2a1+) (Figure S7a) and used an ELISA to measure DKK3 and SOST produced in conditioned media. DKK3 was significantly increased in PCs, relative to CCs and BMSCs, whereas SOST was significantly increased in BMSCs and CCs relative to PCs (Figure 5b). These data support our model (Figure 5a), whereby PC production of DKK3 and chondrocyte production of SOST cooperatively regulate cWnt signaling in cartilage.
Figure 5. StemJEL™ recapitulates the Wnt inhibitory niche in cartilage and rescues chondrocyte phenotypic identity.
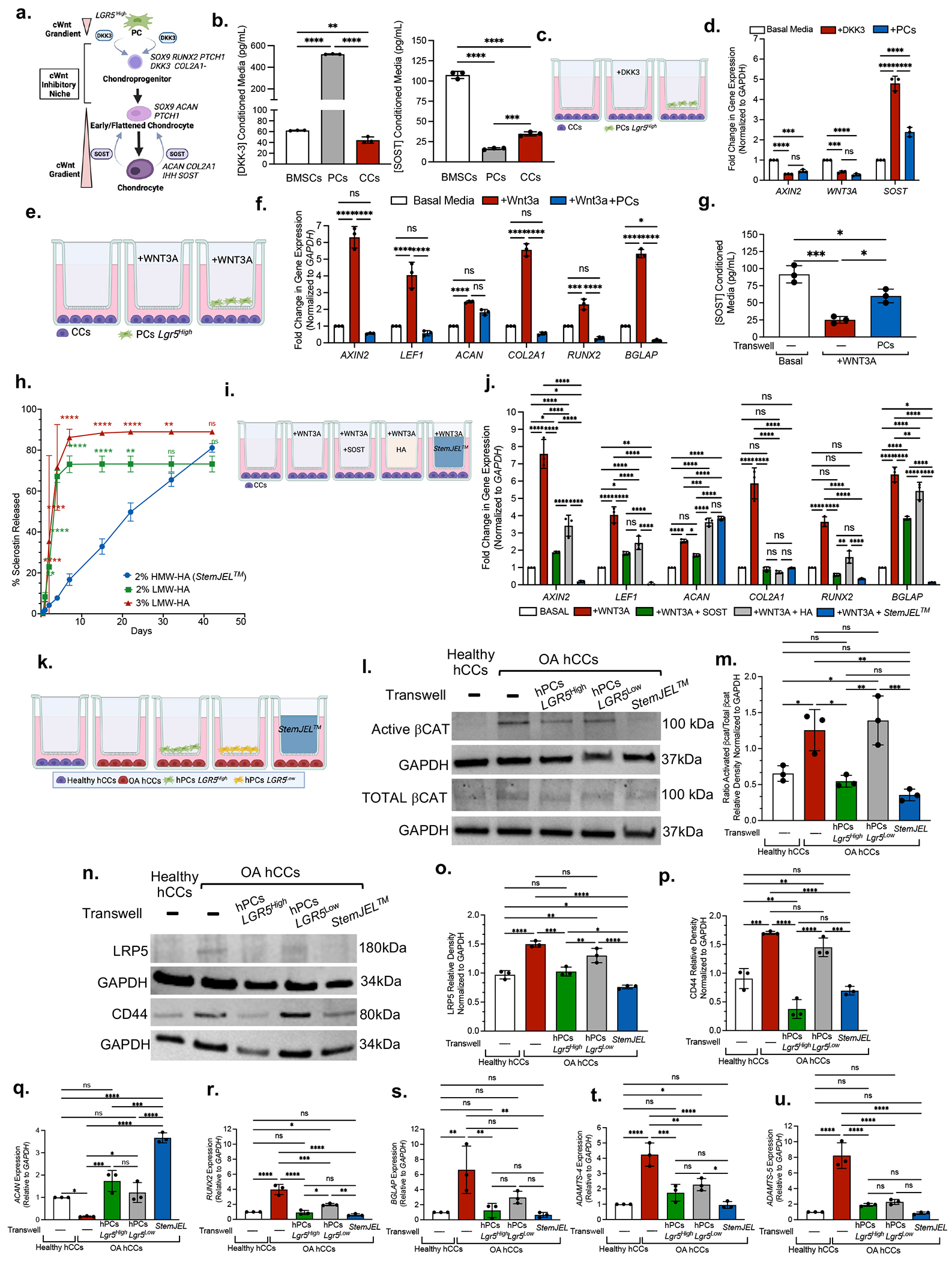
(a) Schematic model. (b) ELISA using conditioned media from mini-pig-derived cells. Data are mean protein concentration ± SD. **p≤0.01, ***p≤0.001, ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (c) Experimental schematic in d. (d) qRT-PCR of CCs from c. Data presented are mean fold change ± SD normalized to GAPDH. ***p≤0.001, ****p≤0.0001; two-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (e) Experimental schematic in f-g. (f) qRT-PCR of CCs in e. Data are mean fold change ± SD normalized to GAPDH; *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (g) ELISA using conditioned media in e. Data presented are mean ± SD. *p≤0.05, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (h) SOST release curve. Data are mean ± SD; *p≤0.05 **p≤0.01; ***p≤0.001; ****p≤0.0001 relative to Day 0, two-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (i) Experimental schematic in j. (j) qRT-PCR of CCs in i. Data are mean fold change ± SD normalized to GAPDH; *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (k) Experimental schematic of l-u (I) Western blot analyses in k. (m) Quantification of westerns in k-l. Data are mean ± SD normalized to GAPDH; *p≤0.05, **p≤0.01, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (n) Western blot in k. (o,p) Quantification of western blots n. Data presented are mean ± SD normalized to GAPDH; *p≤0.05, **p≤0.01, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=3 experiments. (q-u) qRT-PCR of OA hCCs in k. Data are mean fold change ± SD normalized to GAPDH; *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3 experiments.
To corroborate the functional role of Lgr5-expressing cells as forming a cWnt inhibitory niche and to evaluate the crosstalk between Lgr5+ PCs and CCs, we employed transwell co-culture experiements (Figures 5c–5g). To test the hypothesis that Lgr5+ PCs support cWnt inhibition in CCs through cWnt inhibitor secretion, we compared CCs co-cultured with Lgr5+ PCs to CCs treated with DKK3 (Figures 5c–5d). qRT-PCR showed that CCs treated with DKK3 and CCs co-cultured with Lgr5+ PCs both showed a significant decrease in AXIN2 and WNT3A expression and an increase SOST expression, suggesting that secreted factors derived from PCs behave similiarly to exogenous DKK3 (Figure 5d). We next tested in a high Wnt environment the ability of Lgr5+ PCs to reduce the expression of downstream Wnt targets and rescue chondrocyte phenotype (Figure 5e–5g). Relative to CCs in basal media, CCs treated with WNT3A induced the expression of downstream Wnt targets (AXIN2, LEF1) and phenotypically unstable chondrocytes (ACAN+, COL2A1+, RUNX2+, BGLAP+) (Figure 5f). In the presence of WNT3A, CCs co-cultured with PCs caused a downregulation of Wnt target genes (AXIN2, LEF1) and rescued chondrocyte phenotype (ACAN+, COL2A1+, RUNX2−, BGLAP−) (Figure 5f, blue bar). These data suggest that in a high Wnt environment, Lgr5-expressing cells may function to temper cWnt activity in CCs and restore their phenotypic identity. Given that Wnt inhibitor sclerostin (SOST) has been implicated as chondro-protective during osteoarthritis50, we evaluated the effect of Lgr5+ PCs on CC production of SOST. WNT3A treatment in CCs inhibited SOST production, but co-cultures of PCs and CCs enhanced SOST secretion (Figure 5g). These data suggest that Lgr5+ PCs may support a cWnt inhibitory environment in part by promoting SOST secretion.
StemJEL™ provides a Wnt inhibitory niche and rescues chondrocyte identity.
We hypothesized that restoring the cWnt inhibitory niche would rescue chondrocyte phenotype and establish cartilage homeostasis during OA. We and others have previously demonstrated that weekly, intra-articular injections of the Wnt inhibitor SOST ameliorates OA37,50. Given weekly injections are impractical for clinical use, we developed StemJEL™, an injectable hydrogel therapy that combines 2% high molecular weight hyaluron (HMW-HA) and recombinant SOST protein to provide sustained SOST release and reduce cWnt activity. We found HMW-HA in our StemJEL™ formula (2% HMW-HA) provides sustained release of SOST protein relative to SOST combined with 2% or 3% lower molecular weight hyaluronic acid (Figure 5h). To evaluate key rheological properties for StemJEL™, we performed a shear rate sweep of viscosity test. Strain and frequency sweep tests were first used to determine the linear viscoelastic limit of 2M Da HA hydrogel, including a strain of 1% and frequency limit of 10 Hz (Figures S7b–S7c). Within these parameters, our data show that as the shear stress increases in response to an increase in shear rate (Figure S7d–S7e), the viscosities of the high molecular weight 2 M Da HA and 2 M Da HA + SOST (StemJEL™) hydrogels decreased relative to low molecular weight HA (1 M Da HA or 500 KDa HA). These data suggest that StemJEL™ possesses a shear-thinning behavior that is indicative of good performance injectability and is also able to maintain its hydrogel structural integrity following injection 51. We next performed time sweep and temperature sweep tests to determine whether StemJEL™ maintained gel-like properties. We found that unlike 1M Da or 500 KDa HA hydrogels (with and without SOST) but similar to 2M Da HA, StemJEL™ possessed distinctively higher storage modulus (G’) over loss modulus (G”) during time and temperature sweep tests (Figures S7f–S7g). These data suggest that StemJEL™ peformed better than other formulas consisting of low molecular weight HA in mechanical strength by maintaining gel-like properties at all timepoints and temperatures that were tested. Together our data demonstrate that StemJEL™ possesses key structural hydrogel properties crucial for local delivery to articular joint cartilages via injection and its clinical use as a drug delivery system.
To test the hypothesis that StemJEL™ formula inhibits cWnt in CCs and maintains their phenotypic identity, we treated mini-pig CCs with WNT3A in transwell cultures with StemJEL™ in comparison to HMW-HA and exogenous SOST protein (Figure 5i). In the presence of WNT3A, StemJEL™ significantly downregulated Wnt target genes (AXIN2, LEF1) in CCs relative to CCs alone or treated with HA and SOST, suggesting that StemJEL™ combination formula synergistically downregulates cWnt activity (Figures 5i–5j). StemJEL™ treatment also significantly decreased RUNX2 expression in CCs relative to the HA group and downregulatured BGLAP expression in CCs relative to the HA and SOST groups (Figure 5j), corroborating that StemJEL™ rescues chondrocyte identity in a high Wnt environment.
We next tested the therapuetic efficacy of StemJEL™ to inhibit cWnt signaling and maintain chondrocyte identity in humans using primary healthy human CCs (hCCs) and OA human CCs (OA hCCs) (Figures 5k–5u). To test the hypothesis that StemJEL™ functions similary to human LGR5-expressing perichondrial cells (hPCs LGR5) in providing a cWnt inhibitory niche, we compared healthy hCCs to OA hCCs cultured alone or OA hCCs co-cultured StemJEL™, hPCs LGR5High and hPCs LGR5Low in transwell experiments (Figure 5k). Western blot analyses showed that OA hCCs had significantly higher ratio of activated βCatenin levels relative to healthy hCCs (Figures 5I, 5m), suggesting that Wnt activity is higher in OA hCCs. Relative to OA hCCs cultured alone, OA hCCs co-cultured with hPCs LGR5High or treatment with StemJEL™ caused a significant reduction in the ratio of activated β–CATENIN protein levels in OA hCCs, suggesting that StemJEL™ downregulates cWnt activity in OA hCCs (Figures 5I, 5m). We next tested the hypothesis that StemJEL™ inhibits Wnt signaling by blocking the action of the Wnt receptor LRP5/LRP6 and the hyaluron receptor CD4452. Western blot analyses showed that there was no difference in LRP6 expression among the groups (Figures S7h–S7i), suggesting that LRP6 does not play a major role in our model system. However, LRP5 expression (Figures 5n, 5o) and CD44 expression (Figures 5n, 5p) was significantly increased in OA hCCs relative to healthy hCCs. Co-cultures of OA hCCs with either hPCs LGR5Low, hPCs LGR5High or StemJEL™ significantly reduced LRP5 expression (Figures 5n, 5o) and co-cultures of OA hCCs with either hPCs LGR5High or StemJEL™ significantly reduced CD44 expression (Figures 5n, 5p). There was no significant difference in CD44 levels between healthy hCCs and OA hCCs co-cultured with StemJEL™ (Figs. 5n, 5p). Thus StemJEL™ may reduce high Wnt signaling in OA hCCs by blocking the action of LRP5 and CD44 receptors. We tested the ability of StemJEL™ to rescue chondrocyte identity in OA hCCs. qRT-PCR showed that, relative to OA hCCs cultured alone, OA hCCs cultured with hPCs LGR5Low, hPCs LGR5High, or StemJEL™ signficantly increase ACAN expression (Figure 5q). Additonally, relative to OA hCCs cultured alone, OA hCCs treated with StemJEL™ significantly reduced the expression of RUNX2 (Figure 5r), BGLAP (Figure 5s), ADAMTS-4 (Figure 5t), and ADAMTS-5 (Figure 5u). Together these data suggest StemJEL™ supports a Wnt inhibitory niche and rescues chondrocyte identity during OA.
StemJEL™ restores chondrocyte identity and articular cartilage homeostasis in post-traumatic osteoarthritis.
We tested the therapeutic efficacy of StemJEL™ to ameliorate OA using a surgically induced rabbit TMJ OA model 36–38 (Figures 6a–6m). Following TMJ injury, StemJEL™, SOST, high molecular weight-hyaluronic acid (HA) or PBS were injected once a month for 2 months (Figure 6a). Unlike the SOST or HA groups, relative to the PBS group StemJEL™ significantly improved equilibrium contact modulus, a biomechanical property critical for jaw force 53 (Figure 6b), and reduced OARSI macroscopic score (Figure 6c). H&E staining (Figure 6d) and OARSI structure score (Figure 6e) demonstrated that, unlike SOST and HA treatment groups, StemJEL™ treatment improved the cartilage histological structural integrity relative to the PBS group and was similar to OARSI structure score for healthy control group. These data suggest that StemJEL™ treatment supports the maintenance of cartilage mechanical and histological integrity. In corroboration of this concept, safranin O staining showed that unlike the PBS, HA or, SOST treatment groups, StemJEL™ treatment maintained the organization of cellular zones (Figure 6f). There was no signficant change in the area of Safranin O staining in the StemJEL™ treatment group relative to healthy control group (Figures 6g), suggesting that StemJEL™ treatment maintained cartilage homeostasis upon injury.
Figure 6. StemJEL™ ameliorates post-traumatic osteoarthritis and restores chondrocyte identity in rabbit temporomandibular joints and rat knee joints.
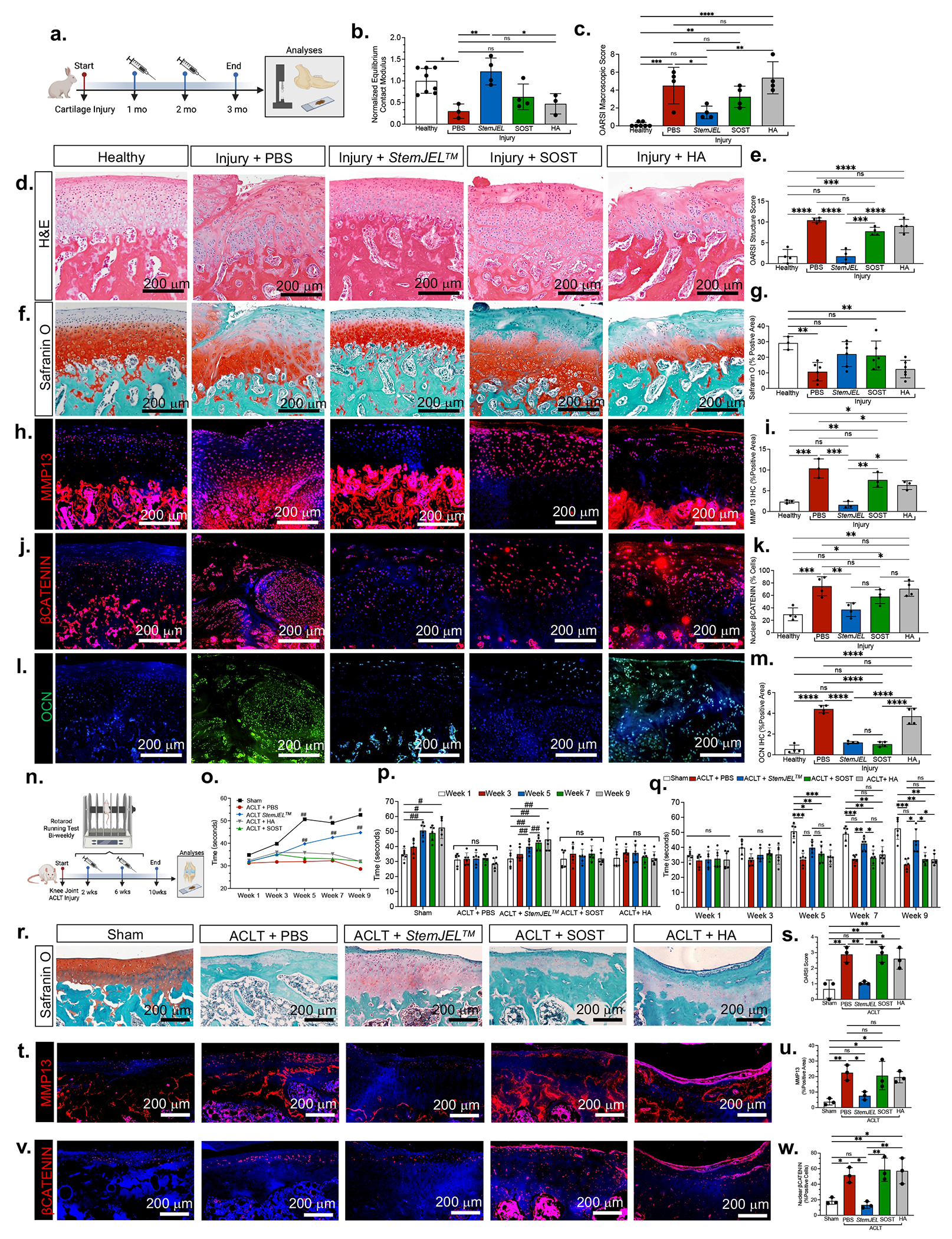
(a) Schematic of rabbit TMJ injury model. (b) Normalized equilibrium contact modulus in rabbit condyles in a. Data are normalized mean ± SD; *p≤0.05, **p≤0.01; one-way ANOVA followed by Tukey’s post hoc; n=3-8 rabbits. (c) OARSI macroscopic score of rabbits a. Data are mean ± SD; **p≤0.01, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=3-8 rabbits. (d) H&E of rabbits in a. (e) OARSI structure score of rabbit condyles in a. Data are mean ± SD; ***p≤0.001, ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=4 rabbits. (f) Safranin O of rabbit condyles in a. (g). Quantification of safranin O staining in f. Data are mean ± SD; **p≤0.01; one-way ANOVA followed by Tukey’s post hoc; n=3-6 rabbits. (h) Immunostaining of MMP13 in rabbits from a. (i) Area of MMP13 immunostaining in h. Data are mean ± SD; *p≤0.05, **p≤0.01, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=3 rabbits. (j) Immunostaining of βCATENIN in rabbits from a. (k) Percentage of nuclear βCatenin+ cells j. Data are mean percent nuclear βCATENIN+ cells ± SD; *p≤0.05, **p≤0.01, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=4 rabbits. (l) Immunohistochemistry of osteocalcin (OCN) in rabbits from a. (m) The area of OCN immunostaining in I. Data are mean area ± SD; ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=4 rabbits. (n) Schematic of rat anterior cruciate ligament transection (ACLT) injury model. (o) Running time on rotarod of rats in n. Data are mean ± SD; *p≤0.05, **p≤0.01; significance is relative to time at week 1 within same group; repeated measures one-way ANOVA; n=6 rats. (p) Running time of rats depicted in n. Data presented are mean running time ± SD; #p≤0.05, ##p≤0.01; repeated measures one-way ANOVA; n=6 rats. (q) Running time on rotarod of rats depicted in n. Data presented are mean running time ± SD; *p≤0.01; **p≤0.01, ***p≤0.001; two-way ANOVA followed by Tukey’s post hoc; n=6 rats. (r) Safranin O staining of rat knees in n. (s) OARSI score of knee joints in n. Data are mean score ± SD; *p≤0.05, **p≤0.01; one-way ANOVA followed by Tukey’s post hoc; n=3 rats. (t) Immunohistochemistry of MMP13 in rat knee joints from n. (u) The area of MMP13 expression in t. Data are mean ± SD; *p≤0.05, **p≤0.01; one-way ANOVA followed by Tukey’s post hoc; n=3 rats. (v) Immunohistochemistry of βCATENIN in knee joints from rats in n. (w) The percentage of nuclear βCatenin+ cells from immunostaining in v. Data are mean percent ± SD; *p≤0.05, **p≤0.01; one-way ANOVA followed by Tukey’s post hoc; n=3 rats.
Immunohistochemistry showed that MMP13, a degradative enzyme cruicial for OA pathogenesis54, was signficantly reduced in StemJEL™ treatment group relative to PBS, HA, and SOST treatment groups, confirming the therapuetic efficacy of StemJEL™ for OA (Figure 6h–6i). Immunohistochemisry also showed that relative to the PBS and HA treatment groups StemJEL™ treatment significantly reduced the percentage of nuclear βCATENIN-expressing cells (Figures 6j–6k) and osteocalcin (OCN) expression (Figures 6l–6m), suggesting that StemJEL™ reduces Wnt signaling and promotes maintenance of chondrocyte phenotypic identity.
We next tested whether StemJEL™ ameliorates knee OA using an anterior cruciate ligament transection (ACLT) model in rats (Figure 6n–6w). StemJEL™, PBS, SOST or HA was injected intra-articularly in rat knees at 2 and 6 weeks following ACLT surgery and knee joint function was measured using a rotarod performance test55 (Figure 6n–6q). Relative to week 1, the sham group showed a significant increase in running time by weeks 5, 7 and 9 (Figs. 6o–6p), suggesting that the rats learned how to run on the rod over time. Upon ACLT injury, treatment with PBS, SOST, or HA did not improve running time, suggesting that the ACLT injury impaired their knee joint function and treatment with SOST and HA does not improve knee joint function (Figs. 6o–6p). On the contrary, the ACLT injury + StemJEL™ group demonstrated a significant increase in running time by weeks 5, 7 and 9 compared to their run time at 1 week (Figs. 6o–6p). Furthermore at 7 weeks the StemJEL™ treatment group had significantly increased running time compared to the PBS and SOST groups (Fig. 6q). At week 9 the StemJEL™ group had significantly increased running time compared to the PBS, SOST, and HA treatment groups and there was no difference in running time between the StemJEL™ and sham group (Fig. 6q). These data suggest that, unlike HA or SOST, StemJEL™ treatment rescues knee joint function upon ACLT injury. Histological analyses further confirmed that the StemJEL™ treatmnt group had significantly lower OARSI score compared to the PBS, SOST, and HA groups (Figs. 6r–6s), suggesting that StemJEL™ improved OA histopathology relative to the other groups. Relative to the PBS group, StemJEL™ treatment significantly reduced MMP13 expression (Figs. 6t–6u), suggesting that StemJEL™ can ameliorate OA pathogenesis. The StemJEL™ treatment group also had significantly lower nuclear βCatenin-expressing cells relative to the SOST, HA or PBS groups (Figs. 6v–6w). These data support the idea that StemJEL™ improves knee joint function and ameliorates OA by suppressing Wnt activity.
We scaled up our translational studies and tested the therapeutic efficacy of StemJEL™ in a surgically-induced TMJ OA 36–38 mini-pig model (Figure 7). At 1 and 2 months following injury, mini-pigs 36 were treated intra-articularly with either StemJEL™, SOST, HA or PBS (Figure 7a). MRI analyses was used to define the location of the injury site on the posterior/lateral portion of the mandibular condyle (top panel Figure 7b). Cross-section of MRI analyses showed that the StemJEL™ group had thicker cartilage relative to PBS, SOST or HA groups (bottom panel Figure 7b). To evaluate joint biomechanical fuction, we determined equilibrium contact modulus in injured mini-pig condyles within the injury site and normalized the injury site to non-injury site within the same condyle (Figure 7c). Relative to the PBS group, the normalized equilibrium contact modulus in the StemJEL™ treated group was significantly increased (Figure 7c), suggesting that StemJEL™ promotes biomechanical recovery critical for jaw function. qRT-PCR showed that relative to the PBS, SOST or HA treatment groups, StemJEL™ treatment significantly reduced the expression degradative enzymes and markers in mini-pig condyles critical for OA pathogenesis (MMP13, ADAMTS-4, ADAMTS-5, COMP) (Figures 7c–7g). Consistent with our murine injury model (Figure 4e), LGR5 expression was upregulated in the PBS group upon jaw joint injury in mini-pigs (Figure 7h), suggesting that Lgr5-expressing cells self-renew during joint injury. Histological evaluation (Figure 7i) confirmed induction of OA upon cartilage injury in PBS treated groups, where, relative to uninjured healthy controls, PBS groups showed a significant increase in OARSI structure score (Figure 7j), abnormal expression of osteocalcin (OCN) (Figures 7k–7l) and RUNX2 in condylar cartilage (Figures 7m–7n), and expansion of nuclear βCATENIN+ cells (Figure 7o–7p).
Figure 7. StemJEL™ ameliorates post-traumatic osteoarthritis and restores chondrocyte identity in pre-clinical mini-pig jaw joints.
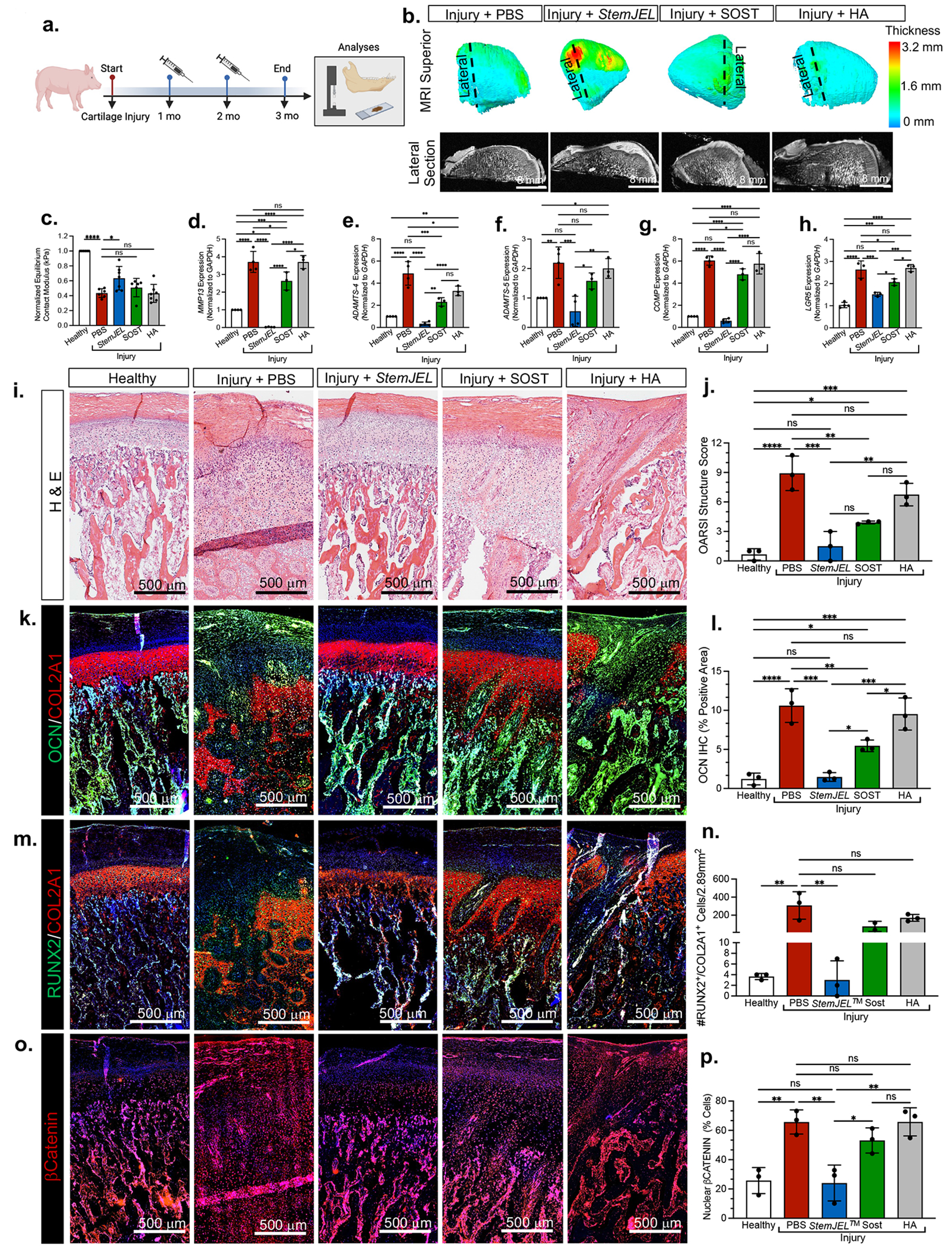
(a) Schematic of mini-pig jaw joint injury model. (b) MRI of superior view of mini-pig condyles (top). Cross section of MRI (bottom). (c) Normalized equilibrium contact modulus in injured mini-pig condyles normalized to uninjured site within the same condyle. Data are normalized mean ± SD; *p≤0.05, ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=6-8 mini-pigs. (d-h) qRT-PCR using mini-pig mandibular condyles from a. Data are mean normalized to GAPDH ± SD; *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3-4 mini-pigs. (i) H&E staining of mini-pigs from a. (j) OARSI macroscopic score of mini-pig condyles from a. Data are mean score ± SD; *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3 mini-pigs. (k) Immunohistochemistry of osteocalcin (OCN, green) and type II collagen (COL2A1, red) in mini-pig condyles from a. (I) Area of OCN immunostaining in k. Data are mean area ± SD; *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤0.0001; one-way ANOVA followed by Tukey’s post hoc; n=3 mini-pigs. (m) Immunohistochemistry of RUNX2 (green) and type II collagen (COL2A1, red) in mini-pig condyles from a. (n). The percentage of RUNX2+/COL2A1+ cells from immunostaining in m. Data are mean percent ± SD; **p≤0.01; ***p≤0.001; one-way ANOVA followed by Tukey’s post hoc; n=3 mini-pigs. (o) Immunohistochemistry of βCATENIN from mini-pigs in a. (p) The percentage of βCatenin+ cells from o. Data are mean ± SD; **p≤0.01; one-way ANOVA followed by Tukey’s post hoc; n=3 mini-pigs.
Relative to the PBS group, StemJEL™ significantly reduced OARSI structure score (Figures 7i–7j), suggesting that StemJEL™ maintains strucutural integrity. The expression of OCN was significantly reduced in StemJEL™ group relative to the PBS, SOST or HA groups (Figures 7k–7l), Unlike the HA or SOST treatment groups, compared to the PBS group, StemJEL™ treatment significantly reduced the expression of RUNX2 in chondrocytes (Figures 7m–7n) and nuclear βCatenin cells (7o–7p). Together these data corroborate the hypothesis that StemJEL™ ameliorates OA by restoring the cWnt inhibitory niche and chondrocyte phenotypic identity.
DISCUSSION
Despite the critical role of low cWnt in chondrocytes and the recent intensive focus on Wnt inhibitors for OA treatment 56–58, the niche modulating a low cWnt environment in cartilage remains unknown 25. We used the TMJ as a model to elucidate adult Lgr5-expressing cells as forming the crucial cWnt inhibitory niche in cartilage. Based our model system, we designed StemJEL™ to restore the chondrocyte niche for OA treatment. Our model reveals invaluable insight into the sophisticated network of cells and cues supporting the cWnt inhibitory niche required for cartilage homeostasis. Our therapy emulates this system and can be broadly applied to other articular cartilages, given cWnt signaling is highly conserved and plays a vital role in cartilage homeostasis in other joints, such as hips and knees 20,56. Here, we provide proof of principle and demonstrate that application of StemJEL™ to rat injured knee joints diminishes OA pathogenesis.
Our work substantiates the jaw joint articular cartilage is lined with a fibrous tissue that essentially functions like a perichondrium 59 by secreting instructive cues to regulate the underlying cartilage and providing progeny to perichondrium, disc/meniscus, periosteum and bone 59. Lgr5-expressing cells in the mandibular condyle do not constitute chondroprogenitor or chondrocyte progeny, and therefore distinct from skeletal stem cells in growth plate 24, knee articular cartilage 60 and bone marrow 61. However, Lgr5 is a well-known marker for adult stem cells 31–33 and we speculate that TMJ Lgr5-expressing cells are derived from neural crest 31. Given that we discovered Lgr5-expressing cells surrounding the middle ear and condyle blastema and the TMJ evolved when jaw bones were integrated into the middle ear 27,28, we suspect that both TMJ and middle ear Lgr5-expressing cells arise from a common precursor. Thus Lgr5-expressing cells possibly adapted to provide cWnt inhibitory cues to existing skeletal stem/progenitor cells necessary to form secondary cartilage and a new craniofacial synovial joint during mammalian lower jaw joint evolution 27,28. We corroborate this idea and show that ablation of Lgr5-expressing cells during early stages of TMJ morphogenesis obliterates the formation of the condylar blastema.
Our data also support the concept that chondroprogenitors and chondrocytes could be derived from Lgr5− PCs that represent a currently unidentified local subset of skeletal stem cells 24,61. We favor this hypothesis given that we have previously identified multi-potent stem/progenitor cells in the mandibular condyle that regenerate cartilage, bone and bone marrow in serial transplantation experiments 37,41. Similar to growth plate cartilage harboring PTHRP skeletal stem cells 24, we observed neighboring Pthlh+Lgr5− PCs. We surmise Lgr5− expressing cells may provide the cWnt inhibitory niche critical for controlling Pthlh+Lgr5− PC differentiation into chondrocytes 25.
In healthy adult cartilage, a low cWnt environment is required for homeostasis, where chondrocytes typically endure low activity levels and nominal ECM turnover. Our data confirm that during OA a high cWnt niche significantly contributes to chondrocyte pathological activities, including inflammation, degradation, hypertrophy, and osteoblast differentiation. We demonstrate that OA chondrocytes abnormally express RUNX2 and OCN, indicating a change in their phenotypic identity. Given that our data is limited to expression analyses, the cell of origin and the fate of these pathological chondrocytes remains unclear. Our utilization of RUNX2 to denote a phenotypically unstable chondrocyte indicates several plausible pathological fates in our model system, including hypertrophy14, transdifferentiation into osteoblasts15, and osteoblast differentiation16. Our use of osteocalcin (OCN) to mark unstable chondrocytes, may indicate chondrocytes undergo hypertrophy and/or osteoblast differention12,17.
Another possibility is that phenotypically unstable chondrocytes may represent a hybrid chondrocyte-osteoblast cell that is transiently present during development, absent in adults, and pathologically reappears during injury/OA. Here we show Sox9+/Runx2+ cells emerge within the condylar blastema and diminish in adults 46. In post-traumatic joints and during Lgr5 suppression/deletion, we discovered eruption of putative hybrid chondrocytes concomitant with pervasive βCatenin and a loss of CPCs. In post-traumatic mini-pig joints putative hybrid chondrocytes pathologically produce bone-like ECM enriched with OCN. In support of this idea, recent studies have identified a skeletal progenitor population with a hybrid chondrocyte-osteoblast phenotype within rib fracture cartilage callous, growth plate cartilage, and osteophytes that mature to bone-producing osteoblasts 62–64. These studies corroborate our hypothesis where a hybrid chondrocyte-osteoblast cell may be selectively employed during repair. The resurgence and persistence of hybrid chondrocytes during articular joint injury and OA may be the tissue’s botched attempt to regenerate cartilage, but instead produces a bonelike ECM. We surmise that hybrid chondrocytes may be derived from a unknown skeletal progenitor, CPC and/or transdifferentiated chondrocytes 65,66.
We show several lines of evidence to demonstrate that Lgr5-expressing cells form the critical cWnt inhibitory niche for neighboring CPCs and chondrocytes to maintain their phenotypic identity. During aging in mice and humans, Prg4 deficiency in mice, and ablation/suppression of Lgr5-expressing cells, we show depletion of Wnt-inactive CPCs and an explosion of cWnt-activated unstable chondrocytes. These data suggest that Lgr5-expressing cells function to temper cWnt activity to maintain an appropriate reservoir of CPCs and chondrocyte identity. To confirm our hypothesis, we found that upon cartilage injury in mice and pigs, Lgr5-expressing cells self-renew to meet the demands of cWnt hyperactivity in pathological chondrocytes. Our expression analyses and transwell experiments, suggest that Lgr5+ PCs produce DKK3 to cue adjacent CPCs to sustain low βCatenin levels and an undifferentiated state. We suspect that as CPCs mature into chondrocytes a low cWnt/βCat gradient is reinforced by mature chondrocytes through SOST production. Our transwell experiments also suggest that Lgr5+ PCs production of DKK3 may support chondrocyte production of SOST. We speculate that DKK3 producing PCs and SOST producing chondrocytes fortifies two borders ensuring a low cWnt niche is maintained. However to conclusively determine whether PCs and chondrocytes secrete DKK3 and SOST to support a Wnt inhibitory niche, genetic knock-outs or mutations of SOST and DKK3 are required.
By the year 2040, approximately 78.4 million adults in the US alone will be diagnosed with arthritis with OA among the most common 2. Disease modifying osteoarthritis drugs (DMOADs) 3 remains an unmet clinical need. While current DMOADs in clinical trials target cartilage anabolism, we take a different approach and focus on stabilizing chondrocyte identity. Our hydrogel consists of high molecular weight HA (HMW-HA), which is critical for joint health by providing structure, lubrication, pain relief 67,68 and reducing inflammation 67,68. Through blocking CD44 action, HMW-HA also inhibits membrane localization of the Wnt receptor LRP6 52. While we found no changes in LRP6, we found significant increase in LRP5 and CD44 expression during OA that was reversed with StemJEL™. These data suggest that StemJEL™ may be acting through LRP5 and CD44 to reduce Wnt signaling; however, future mechanistic studies are warranted. We surmise our therapy is more suited for early to mid-stages of OA and has the potential to minimize the risk of surgery. Human clinical trials testing the therapeutic efficacy of StemJEL™ to attenuate pain is essential. We speculate that given HMW-HA is FDA approved as an anti-nociceptive agent 69 and SOST has been shown to reduce inflammation, that our combination therapy has the potential to serve as a pain-reducing agent. Our data indicate a minimally-invasive OA therapy that preserves chondrocyte identity and supports cartilage tissue maintenance.
Limitations of Study
We demonstrate the therapeutic efficacy of StemJEL™ in post-traumatic knee and jaw joints, which is limited to a surgically-induced OA model. The therapeutic efficacy of StemJEL™ may be joint or model dependent and requires further testing in other model systems. Our in vitro studies using primary cells from mini-pigs and humans are restricted, given that we use heterogenous PCs that contain a mixture of SZCs/CPCs and heterogeneous CCs harboring chondrocytes and hypertrophic chondrocytes. Further characterization of primary cell populations is necessary. We were unsuccessful in finding an antibody that recognizes active βCatenin in pig, so confirmation of Wnt signaling in our primary mini-pig cell lines are limited. Identification of phenotypically unstable chondrocytes is limited to OCN and RUNX2 expression, and lineage studies are necessary to determine their origin and fate. To definitively confirm that PCs and chondrocytes secrete DKK3 and SOST, the generation of cell-specific knock-outs is required.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Dr. Mildred C Embree (mce2123@cumc.columbia.edu).
Materials Availability
The primary human cell lines used in this paper are not available due to IRB restrictions. There are no new additional materials generated from this paper.
Data and Code Availability
RNA-seq data have been deposited at Gene Expression Omnibus and are publicly available as of the date of publication. The accession number is listed in the Key Resources Table. Additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Aggrecan | Thermofisher | MA3-16888 |
| Rabbit ant-βcatenin | Abcam | ab6302 |
| Rabbit ant-βgalactosidase | MBL Life Science | PM049 |
| Mouse anti-Collagen 2A1 | Millipore | MAB8887 |
| Rabbit anti-Osteocalcin | EMD Millipore | ab10911 |
| Goat anti-Periostin | R&D Systems | af2955 |
| Rabbit anti-Runx2 | Abcam | ab23981 |
| Rabbit anti-total βcatenin | Invitrogen | 71-2700 |
| Rabbit anti-non-phospho (active)-βcatein | Cell Signaling | 8814 |
| Rabbit anti-LRP5 | Proteintech | 24899-1-AP |
| Rabbit anti-LRP6 | Cell Signaling | 3395 |
| Mouse anti-CD44 | Proteintech | 60224-1 |
| Mouse anti-GAPH | Invitrogen | MA116757 |
| Goat anti-mouse HRP | Invitrogen | G21040 |
| Goat anti-rabbit HRP | Invitrogen | G21234 |
| Goat anti-rabbit Alexa Fluor 546 | Invitrogen | A11010 |
| Goat anti-rabbit Alexa Fluor 488 | Invitrogen | A11008 |
| Goat anti-mouse Alexa Fluor 546 | Invitrogen | A11003 |
| Goat anti-mouse Alexa Fluor 488 | Invitrogen | A11001 |
| Donkey anti- rabbit Alexa Fluor 647 | Invitrogen | A32795 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Protein Sclerostin Protein | R&D Systems | 1406-ST-025/CF |
| Sodium Hyaluronate | Lifecore Biomedical | HA2M-5 |
| Recombinant Human Wnt3A Protein | R&D Systems | 5036-WN-010/CF |
| Recombinant Human Dickkopf Related 3 Protein | R&D Systems | 1118-DK-050/CF |
| Wnt-C59 | Peprotech | 1248913 |
| Critical commercial assays | ||
| Porcine Dickkopf Related Protein 3 ELISA Kit | Mybioscience | MBS099882 |
| Sclerostin Human ELISA Kit | Invitrogen | EHSOST |
| Invitrogen™ Click-iT™ Plus EdU Cell Proliferation Kit for Imaging | Invitrogen | C10639 |
| SYBR™ Green PCR Master Mix | ThermoFisher | 4309155 |
| RNAscope™ Multiplex Fluorescent Reagent Kit v2 | Advanced Cell Diagnostics | 323100 |
| RNAscope™ 2.5 HD Duplex Detection Reagents | Advanced Cell Diagnostics | 322500 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE236909 |
| Experimental models: Primary Cell lines | ||
| 1-Human Perichondrial Cells Lgr5High | Embree Lab, this paper | N/A |
| OA 2-Human Perichondrial Cells Lgr5Low | Embree Lab, this paper | N/A |
| OA 3-Human Perichondrial Cells Lgr5Low | Embree Lab, this paper | N/A |
| OA 4-Human Perichondrial Cells Lgr5Low | Embree Lab, this paper | N/A |
| Healthy Human Mandibular Condylar Chondrocytes | Embree Lab, this paper | N/A |
| Osteoarthrithic Human Mandibular Condylar Chondrocytes | Embree Lab, this paper | N/A |
| Mini-pig mandibular condylar chondrocytes | Embree Lab, this paper | N/A |
| Mini-pig perichondrial cells | Embree Lab, this paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: prg4tm1Mawa/J | Jackson Laboratory | 025737 |
| Mouse: Tg (TCF/Lef1-lacZ)34Efu/J | Jackson Laboratory | 004623 |
| Mouse: Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | 007909 |
| Mouse: Lgr5tm1(cre/ERT2)Cle/J | Jackson Laboratory | 008874 |
| Mouse: CD1-IGS | Charles River | 022 |
| Mouse: Lgr5DTR-EGFP | Genetech | N/A |
| Rat: Sprague Dawley | Charles River | 400 |
| Rabbit: New Zealand White | Charles River | 052 |
| Pig: Yucatan Miniature Swine | Sinclair Bioresources | N/A |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Table S5 | This paper | N/A |
| Primers for genotyping, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| Prism Graphpad | N/A | https://www.graphpad.com |
| ImageJ | Schneider et al. 2012 | https://imagej.nih.gov/ij/ |
| Biorender | N/A | https://www.biorender.com |
| StringTie software | Kovaka et al. 2019 | https://ccb.jhu.edu/software/stringtie/ |
| RUVSeq software | Risso et al. 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| SQANTI3 software | Tardgauila et al. 2018 | https://github.com/ConesaLab/SQANTI3 |
| IsoformSwitchAnalyzeR software | Vitting-Seerup et al. 2019 | https://bioconductor.org/packages/release/bioc/html/IsoformSwitchAnalyzeR.html |
| DESeq2 software | Love et al. 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| clusterProfiler 4.0 software | Wu et al. 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Salmon aligner | Patro et al. 2017 | https://combinelab.github.io/salmon/ |
| STAR aligner | Dobin et al. 2013 | https://github.com/alexdobin/STAR/tree/master |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
ANIMALS
Yucatan Miniature Pigs Post-traumatic TMJ OA Model.
Six-month-old Yucatan miniature pigs (n=44, equal number of males and females, Sinclair Bioresources) were used with approval from the Institution of Animal Care and Use Committee at the at the Medical University of South Carolina (2017-00047). Post-traumatic TMJ OA was surgically induced using a disc perforation model as previously described36–38. An oblique incision was created superior to the zygomatic process. A periosteal elevator was placed under the disc to protect the condyle from secondary injury. A punch biopsy was used to create a 5.0 mm perforation in the posterior-lateral portion of the joint disc and the perforated disc tissue was excised. The disc was allowed to reduce to its normal anatomical location. One-month post-surgery either 1mL of SOST (150 ng/ml in PBS, R&D 1406-ST-025/CF), PBS, 2% Hyaluronic acid (HA) (Lifecore Biomedical HA2M-5), or StemJEL™ (2% HA with SOST) was injected into the intra-articular space unilaterally once a month for two months. All animals were euthanized 3 months post-surgery. Aged-matched non-operated Yucatan miniature pigs were used as controls (n=18, equal number of males and females).
Mouse Genotyping.
All mouse strains (Key Resources Table), Supplemental Table 2) ages E14.5 to 1 years-old were used with approval from the Institution of Animal Care and Use Committee at Columbia University Medical Center (AC-AAAU6480, AC-AAAO4651 AABL5583 and AC-AABP1553). To collect embryonic timepoints, breeding pairs were placed together in the evening and females were checked for plugs after 12 hours followed by separation. Mice were genotyped using tail clip or toe clip in pups P7 or younger. DNA was isolated from mouse tissues and PCR was performed with VeritiPro Thermal Cycler (Thermo Fisher, A48141) using primers listed in Supplemental Table 2.
Mouse Lineage Tracing, Cesarian Section and Surrogacy Preparation.
For lineage tracing Lgr5tm1(cre/ERT2)Cle/J male mice were bred with with Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J female mice (Jackson Labs, #007909). Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J dams were treated with 25mg/mL tamoxifen (Sigma, T5648) in corn oil per mass at different timepoints. Dams were administered tamoxifen vial oral gavage at gestational and post-partum timepoints including: 1) E12.5 & E13.5 and pups were euthanized at P0, P21, and 1 year; 2) E15.5 & E16.5 and pups were euthanized at P0; 3) P1-P3 and pups were euthanized at 6 months. Given tamoxifen administration during pregnancy inhibits estrogen and parturition, to ensure pup survival in dams that were administered tamoxifen at E12.5 & E13.5, a cesarian surgery was performed to harvest E18.5 pups. The dam was euthanized and the surviving litter was transferred to a CD1 surrogate dam that was timed to give birth on the same day. For all lineage tracing experiments, the Lgr5tm1(cre/ERT2)Cle/J x Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J positive untreated mice and Lgr5tm1(cre/ERT2)Cle/J x Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J negative treated mice were evaluated as negative controls. At least 4 pups were collected per time point for analyses (equal number males and females).
Mouse Diphtheria Toxin Injections.
To ablate Lgr5-expressing cells, diphtheria toxin (50ug/kg, IP, Sigma D0564) was administrated to Lgr5DTR/+ dams (Genentech) intraperitoneally, at e13.5 and e16.5 and euthanized at P0. Single injections were also done at e13.5, e15.5, e17.5, and e18.5 and euthanized e14.5, e16.5, e 18.5, and P0 respectively. For all timed DT injections in the Lgr5-DTR-EGFP mice, 3-4 pregnant females were prepared. Litter-matched Wildtype + DT pups were utilized as a control for Lgr5DTR-EGFP+/− DT pups, along with untreated Lgr5DTR-EGFP+/− pups from a separate litter. For the DT injections on day E13.5 and analyzed at E15.5, 3 pregnant females were utilized, which yielded 5-6 Lgr5DTR-EGFP+/− pups for histological evaluation.
Sprague Dawley Rat Knee Joint Anterior Cruciate Ligament Transection (ACLT) Surgery and Rotarod Performance Test.
All rat surgeries and procedures were approved by Columbia Institute of Comparative Medicine (AC-AABG8555). Sprague Dawley rats at 3 months-old underwent unilateral anterior cruciate ligament transaction (ACLT) surgery to induce knee joint post-traumatic OA (n=24, equal number of males and females, Charles River), while age-matched sham-operated rats with no transection injury served as controls (n=6, equal number of males and females). To create the injury an incision was made over the medial aspect of the femoral-tibial joint and the anterior cruciate ligament was exposed and transected. The joint space was returned to normal and the skin was sutured. At 2- and 6-weeks postsurgery either 50 μl of SOST (150 ng/ml in PBS, R&D 1406-ST-025/CF), PBS, 2% Hyaluronic acid (HA) (Lifecore Biomedical HA2M-5), or StemJEL (2% HA with SOST) was injected into the intra-articular space unilaterally. All rats underwent rotarod running performance test55 (IITC Series 8 Roto-Rod) at 1, 3, 5, 7, 9 weeks. To train the rats to run, all rats were placed on the accelerating rotarod 2-3 trials prior to recording their time. All rats were euthanized 10 weeks post-surgery for analyses.
New Zealand White Rabbits Post-traumatic TMJ OA Model.
Six-month-old New Zealand white rabbits (n=36, equal number of males and females, Charles River) were used with approval from the by Columbia University Institute of Comparative Medicine (ACAAAM8955). Post-traumatic TMJ OA was surgically induced using a disc perforation model as previously described36–38. An oblique incision was created superior to the zygomatic process. A periosteal elevator was placed under the disc to protect the condyle from secondary injury. A punch biopsy was used to create a 2.5 mm perforation in the posterior-lateral portion of the joint disc and the perforated disc tissue was excised. The disc was allowed to reduce to its normal anatomical location. One-month post-surgery 100 μL of SOST (150 ng/ml in PBS, R&D 1406-ST-025/CF), or PBS, or 2% Hyaluronic acid (HA) (Lifecore Biomedical HA2M-5), or StemJEL (2% HA with SOST) was injected into the intra-articular space unilaterally once a month for two months. All animals were euthanized 3 months post-surgery. Aged-matched non-operated rabbits were used as controls (n=16, equal number of males and females).
Human Samples.
Human joint tissue samples were collected and analyzed from cadavers or patients undergoing joint replacement surgery. The study was approved by the Institutional Review Board of Columbia University Irving Medical Center (AAAQ8195) and the Institutional Review Board of Weill Cornell New York Presbyterian (1608017486-A002). All subjects were provided with written informed consent prior to study enrollment. All experiments were performed in accordance with relevant guidelines and regulations.
PRIMARY CELL CULTURES
Mini-Pig Primary Cell Lines.
Mandibular condyles were dissected from 6-month-old Yucatan miniature pig TMJ (n=4, equal number of males and females). Perichondrium tissue was physically separated from the surface of the mandibular condyle. Heterogeneous perichondrial cells (PCs) were isolated from the perichondrium and heterogeneous condylar chondrocytes (CCs) were isolated from the mandibular condyle as previously described37. The tissues from multiple pigs (n=4) were pooled, digested in an enzyme mixture of dispase II (4 mg/mL) and collagenase I (3 mg/mL) in a waterbath shaker for 45 minutes at 37°C. The cell supernatant was collected in lot-selected fetal bovine serum on ice. The tissue digestion and cell supernatant collection was repeated 3 times or until the tissue was completely digested. Single-cell suspensions were cultured (5% CO2, 37 C) in basal medium consisting of DMEM (Invitrogen 11885-092) supplemented with 20% lot-selected fetal bovine serum (FBS, Gibco ES Cell FBS), glutamax (Invitrogen 35050-061), penicillin-streptomycin (Invitrogen 15140-163) and 55 mM 2-mercaptoethanol (Gibco) for 4–6 days. Cells were detached with trypsin-EDTA (Gibco) and either frozen in 100% FBS or plated at P1 for in vitro experiments.
Human Primary Cell Lines.
The generation of primary human cell lines was approved by the Institutional Review Board of Columbia University Irving Medical Center (AAAQ8195) and the Institutional Review Board of Weill Cornell New York Presbyterian (1608017486-A002). Human TMJ tissues were harvested from two sources including: 1) cadaver specimens and 2) patients with osteoarthritis undergoing a TMJ procedure or surgery that generates specimens that would otherwise be discarded as medical waste. Healthy human perichondrial cells (hPCs Lgr5High) and healthy human mandibular condylar chondrocytes (hCCs) were isolated from a 37-year female cadaver with no TMJ OA. To isolated primary OA human cell lines, some specimens from OA patients with severely degenerated TMJs were eliminated due to our inability to identify perichondrium and condylar cartilage tissues. Primary OA perichondrial cells were isolated from 3 females with TMJ OA (54-70 years-old, OA 2-4 hPCs Lgr5Low) and primary OA condylar chondrocytes (OA hCCs) were isolated from 1 female with TMJ OA (70 years-old). Perichondrium tissue was physically separated from the surface of the mandibular condyle. Heterogeneous hPCs were isolated from the perichondrium and heterogeneous hCCs were isolated from the mandibular condyle as previously described37. Briefly, the tissues were digested in an enzyme mixture of dispase II (4 mg/mL) and collagenase I (3 mg/mL) in a waterbath shaker for 45 minutes at 37°C. The cell supernatant was collected in lot-selected fetal bovine serum on ice. The tissue digestion and cell supernatant collection was repeated 2-3 times or until the tissue was completely digested. Single-cell suspensions were cultured (5% CO2, 37 C) in basal medium consisting of DMEM (Invitrogen 11885-092) supplemented with 20% lot-selected fetal bovine serum (FBS, Gibco ES Cell FBS), glutamax (Invitrogen 35050-061), penicillin-streptomycin (Invitrogen 15140-163) and 55 mM 2-mercaptoethanol (Gibco) for 4–6 days. Cells were detached with trypsin-EDTA (Gibco) and either frozen in 100% FBS and stored in liquid nitrogen or plated at P1 for in vitro experiments.
METHOD DETAILS
Condyle Organ Culture and Injury Model.
Lgr5tm1(cre/ERT2)Cle/J male mice were bred with with Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J female mice. The female dams were administered tamoxifen (25mg/mL, Sigma, T5648) via oral gavage at post-partum timepoints daily (P11-P13). At P14 the mandibular condyles were harvested from male and female Lgr5tm1(cre/ERT2)Cle/J x Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J pups under sterile conditions. The articular condyle and approximately 3 mm of the ramus was dissected from the mandible. A razor blade was utilized to create a cartilage defect in the posterior portion of the condyle and the explants were cultured in a 24-well plate with DMEM media supplemented with 1% penicillin/streptomycin (Invitrogen 15140–163), 100 mmol/L ascorbic acid, and EdU (10μM, Sigma A10044) . The contralateral side was not injured. The condyle explants (n=3 explants) were incubated for 24 hrs. (5% CO2, 37°C) and were fixed in 4% PFA for histology.
RNA Amplification and Deep Sequencing.
Total RNA was extracted from tissue samples using RNeasy Micro kit (Qiagen, 74004). To evaluate RNA quality (RIN number), Bio-Analyzer 2100 (Agilent Biotechnologies) was used. Total RNA samples were first subjected to DNase treatment using 2.2x beads cleanup. After treatment, QIAseq FastSelect rRNA HMR Kit (Qiagen) was used for rRNA depletion, followed by library preparation using NEBNext Ultra II Directional RNA Library Prep Kit (NEB). The samples were then sequenced using the lllumina HiSeq sequencer.
RNA-seq Analysis.
Following quality trimming, reads were mapped to the reference genome Sscrofa11.1 with the STAR aligner70. The de novo transcriptome assembly was performed using StringTie software71 and was guided by existing annotation from ENSEMBL database. The assembled transcripts from individual StringTie runs were merged into one reference transcriptome using StringTie software, which generated a GTF format file to store the combined de novo transcriptome assembly. The resulting de novo transcripts in the merged GTF file were further annotated with SQANTI3 software72. To ensure that the analysis of the individual samples were comparable, all samples were re-quantified in transcript level using the merged GTF file with a quasi-mapping aligner called Salmon. An additional quality filtering was performed using IsoformSwitchAnalyzeR software73 based on the transcript level mapping results from Salmon software74. The median read number of all transcripts of a gene was used to estimate the expression level of this gene. The correction of batch effect was performed on the gene expression level using RUVSeq software75 and the significant differentially expressed genes (DEGs) were detected by DESeq2 software76.
Gene Ontology Analysis.
Over Representation Analysis was conducted for functional annotation by using the Gene Ontology (GO) (http://geneontology.org) databases based on the GO biological processes (BPs) and molecular functions (MFs) through the clusterProfiler 4.0 software77. The analyses were performed based on DEGs identified in each group. The BPs and MFs significantly associated with the gene lists were determined based on their p value (p≤0.05).
Histology and Immunohistochemistry.
Tissue samples were fixed in 4% paraformaldehyde, decalcified in EDTA or Morse Solution, and prepared for either paraffin or frozen embedded sections. To section the mandibular condyle, sagittal sections were collected beginning from the lateral side of the jaw and every 5th slide was stained with H&E. The H&E sections were used to ensure similar depths of the sections were compared for each speciment. For the rabbit and mini-pig injury models, the posterior lateral regions were easily accessed for joint injury, therefore we selected the posterior lateral areas for histological comparison. For mouse histological comparisons, we also choose the middle to posterior regions, given that the majority of mechanical forces and pathology occur within the middle/posterior regions of the condyle. Serial tissue sections were also stained with Safranin O. For immunohistochemistry sections were enzymatically treated with DAKO Target Retrieval Solution (DAKO, S1700), blocked with 3% BSA for 1 hour, and immunolabelled with primary antibodies at 4°C overnight followed by secondary antibody (1 hour at room temperature) to detect immunoactivity (Supplemental Tables 3 & 4). Isotype-matched negative control antibodies were used under the same conditions. ProLong Gold antifade reagent with DAPI (Invitrogen, P36931) was used for the nuclear counterstain. The area of antibody expression or chemical stain was captured using Olympus cellSens Dimension imaging software. The percent area of antibody expression and the number of immunopositive cells was determined using ImageJ software using at least 3 biological replicates. Immunohistochemistry and quantification are performed on comparable tissue sections from at least 3 different mice and the mean representative image is presented.
OARSI Scoring.
We used the Osteoarthritis Research Society International (OARSI) osteoarthritis cartilage histopathology assessment system (OARSI system) to demonstrate the progression of osteoarthritis in our samples. H&E and/or safranin O staining were utilized to determine the Structure score and Macroscopic score. Observers (n=4) blindly assessed OARSI histopathological osteoarthritic degenerative scores in our rabbit and mini-pig jaw joint injury models 78 , the rat ACLT knee injury model 79, and in the Prg4−/− mice 80.
RNA Isolation and qRT-PCR.
Total RNA was purified from whole tissues or cells (Qiagen, 74004) and treated with DNAse I (Ambion AM2222) to remove genomic DNA. RNA quantity and purity were determined using Nanodrop. RNA samples (260/280≥1.8) were used to obtain cDNA (Biorad AM2222). Quantitative RT-PCR was performed using SYBER Green PCR Master Mix (Applied Biosystems, 4309155) and pre-designed primers (Supplemental Table 5) (Integrated DNA Technologies). Gene expression levels were normalized to housekeeping gene GAPDH.
Western Blot Analysis.
Protein was isolated using RIPA buffer (Thermofisher, 89900) with the addition of Protease (Thermofisher, 1862209) and Phosphatase (Thermofisher, 1862495) inhibitors. The cell suspension was agitated for 30 mins at 4C, sonicated 3 times for 15 seconds, and spun at 16000g for 20 min. at 4C. Bolt LDS Sample Buffer (4x) (Invitrogen, B0007) and Bolt/NuPAGE Reducing Agent (Invitrogen, B0009) were added to the protein lysate and heated for 10 minutes at 70C and then 85C for 2 minutes. 40 uL of sample was then loaded on Bolt 4-12% Bis-Tris Plus Gels (Invitrogen, NW0422BOX) and run using MES SDS Running Buffer (Invitrogen, B0002) in Bolt Mini Gel Tank (Thermo Fisher. A25977) for 35 minutes at 200V. iBlot2 NC Mini Stacks (nitrocellulose) (Invitrogen, IB23001) were used for transfer in the iBlot2 (Thermo Fisher, IB21001) for 7minutes at 20V. The membrane was blocked for 3 hours with 5% non-fat milk/TBST at room temperature. Primary antibodies were diluted in 5% non-fat milk/TBST overnight at 4C (Supplemental Table 6) and secondary antibodies were diluted in 5% non-fat milk/TBST rocking at room temperature for an hour (Supplemental Table 4). The membranes were washed 5x’s for 5 minutes each with TBST and used SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher, 34577) for development. The membrane was imaged using the iBright™ CL 1500 imaging system (Invitrogen, A44114) and density was measured using the iBright analysis software using Thermo Fisher Connect. Normalization, using GAPDH, was used to calculate the density of the bands. The protocol used followed the manufacturer’s guidelines from Life Technologies for the Mini Gel Tank and Blot Module Set (Thermofisher, NW2000) (MAN0006968).
Multi-lineage Differentiation.
Multi-lineage differentiation was tested in vitro using chemically defined media for chondrogenesis and osteogenesis. For chondrogenesis, cells (1 x106 per pellet) were pelleted in 15 ml polypropylene tubes by centrifugation and cultured (5% CO2, 37° C) for 3 weeks in Dulbecco’s Modified Eagle medium supplemented with 10−7 mol 1−1 dexamethasone, 100 mmol I −1 ascorbic acid, 1% insulin, transferrin, selenium (ITS), 1 mmol I −1 pyruvate, and 10 ng/ mL TGF-β1 The pellets were analyzed via immunohistochemistry. To induce osteogenesis, cells (5 x 104) were culture in 12-well plate for 4–5 weeks in media containing αMEM supplemented with 20% FBS, dexamethasone (10−8 M), 100 mM L-ascorbic acid, and 2 mM β-glycerophosphate. Calcium nodules were visualized by staining with alizarin. Wnt overexpression was done by adding Wnt3a (500ng/ml, R&D 5036-WN-010/CF) and inhibition was done using Wnt-C59 (10uM, Peprotech, 1248913).
In situ hybridization.
RNAscope in situ hybridization (ISH, Advanced Cell Diagnostics, #323100, #322500) was performed with 5μm sections of fixed frozen mouse tissues. Tissue samples were pre-treated by boiling in Target Retrieval solution for 5 minutes followed by Protease III treatment for 30 minutes at 40°C. Subsequently the tissue samples were processed with the RNAscope Multiplex Fluorescent/Colormetric Assay according to manufacturer protocols using probes listed in Supplemental Table 7.
EDU label-retaining cells.
EDU labeled cells were detected in explant frozen sections (Invitrogen, C10639) and quantified (ImageJ).
Hydrogel Preparation.
In a 15mL conical tube, sclerostin (SOST, 150ng ml−1 in PBS, R&D 1406-ST-025/CF), hyaluronic acid (HA, 2%, Lifecore Biomedical HA2M-5), and tissue grade Dulbecco’s Phosphate Buffered Saline ( DPBS, Corning 21-031-CV), which was filter sterilized (.22μM, Millex-GP, SLGPR33RS) were added in that order. Then it was placed at 4C for 2hrs. to allow the HA to solidify to a gel.
Rheology Analysis
HAAKE MARS Rheometer was used to perform rheological analysis of the hydrogels. The hydrogel was placed between the two plates of the rheometer heated to 37°C with a gap size of 1mm. Shear viscosity measurements were obtained through shear rate increase from 1 to 10 1/s with constant displacement at 1% strain. The oscillation time sweep was performed to record the storage modulus (G’) and loss modulus (G”) at 37°C as strain and angular frequency were set up at 1% and 10.0 Hz, respectively. The oscillation temperature sweep was performed as a temperature ramp study to record G’ and G” over a constant strain of 1% applied at frequency of 10 Hz. Strain and frequency logarithmic sweep measurements were conducted with strain sweep from 0.01 to 10 % and frequency sweep from 0.1 to 100 Hz, prior to time and temperature sweep tests. Each hydrogel sample was used for only one test. Each test was performed in triplicate and the data represents the average of the three tests.
Transwell Experiments.
Mini-pig or human condylar cartilage cells (CCs) (5x104) were seeded in a 12 well transwell plates (Corning 3460) in DMEM (Invitrogen 11885-092) supplemented with 20% lot-selected fetal bovine serum (FBS, Gibco ES Cell FBS), glutamax (Invitrogen 35050-061), penicillin-streptomycin (Invitrogen 15140-163) and 55 mM 2-mercaptoethanol (Gibco). Upon 60-70% confluency after 2 days, the transwells seeded with mini-pig or human perichondrial cells (5x104). Either DKK3 (60ng/ml, R&D 1118-DK-050/CF), Wnt3a (500ng/ml, R&D 5036-WN-010/CF), SOST (150ng ml−1 in PBS, R&D 1406-ST-025/CF), StemJEL, or HA (2%, Lifecore Biomedical HA2M-5) was added to the transwell. When the CCs reached 100% confluency after an additional 3 days, CCs were removed using a cell scraper for downstream protein and RNA isolation. Conditioned media was collected for ELISA.
ELISA.
The DKK3 (Mybiosource, MBS099882) and SOST (Invitrogen, EHSOST) in pig condyle cells were measured using the quantitative sandwich enzyme immunoassay technique following the manufacturer’s instructions. Briefly, standard or sample (100 μL) was added to each well and incubated for 2 hr. at 37°C, followed by treatment with primary antibody using either biotinylated antibody for 1 hr. at 37°C. For antibody detection, 100 μL of HRP-Avidin was for 1 hr. at 37°C. After washing, TMB substrate was added to each well for 15-30 min at 37°C, followed by the addition of 50 μL of Stop Solution. The optical density (OD) of each well was determined using a microplate reader at 450 nm. The DKK3 & SOST concentrations were calculated by comparing the OD of each sample to the standard curve.
MRI Analysis.
Fresh pig condyles were harvested and imaged with a 7T MRI scanner (BioSpec 70/30 USR, Bruker Corp, Germany) with a spin echo sequence. The settings for the MRI scanning were: repetition time/echo time = 4000 ms/12 ms, field of view = 25.6 mm × 12.8 mm, matrix size = 256 × 128, in plane resolution = 0.1 mm × 0.1 mm, slide thickness = 0.3 mm with zero gap, total scanning time = 22 minutes. Following the MRI imaging, the condyle cartilage was manually segmented out from the MRI images using the Amira software (Amira 6.4, FEI Co., Hillsboro, OR) to generate the 3D cartilage geometry. The cartilage thickness was then calculated using the build-in surface thickness function in Amira and presented as a thickness color map over the surface of the condyle cartilage.
Mechanical Testing.
Fresh mini-pig and rabbit jaw joint condyles were harvested and tested with a micro-indentation system (UNHT3 Bio, Anton Paar, Switzerland). The system resolution for the force and displacement measurement were down to 0.001 μN and 0.006 nm, respectively. Samples were carefully mounted on a sample holder using a small amount of cyanoacrylate and immersed in the PBS solution during the whole testing period. The sample holder was on a 2-axis leveling ball and can be adjusted to ensure that the indenter was normal to the condyle cartilage surface at the testing site. For pig condyles, a spherical ruby ball indenter with 1 mm in diameter was used. A 10 μN tare load were applied and held for 400 s, followed by a 300 μN step load for 1800 s to obtain the creep data. For the rabbit condyles, a spherical ruby ball indenter with 0.2 mm in diameter was used owing to the thin condyle cartilage thickness in rabbit condyles. A 2 μN tare load were applied and held for 300 s, followed by a 40 μN step load for 1500 s to generate the creep data. The force and displacement data were acquired at 20 Hz for both tests. Each condyle was indented in at least seven different sites, including anterior, posterior, central, lateral, medial, posterior-lateral, and injury sites (injury site were identified by referencing the MRI images and the morphological appearance of the condyle). Condyle cartilage thickness were determined through the MRI images and the condyle histology slides. The creep data were then analyzed with the Hertzian biophysical theory to calculate the equilibrium contact modulus. The mechanical data from the injury site of the pig condyle were normalized to the values from the corresponding sites of control healthy pig condyles to reveal the changes of the condyle cartilage mechanical properties relative to the healthy one.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics.
All statistics were calculated using Prism 9 GraphPad Software. The statistical significance between two groups was determined using Student’s t test assuming Gaussian distribution. The normality of distribution was confirmed using the Kolmogorov–Smirnov test and the resulting two-tailed P value ≤ .05 was regarded as a statistically significant difference. Among the two groups, one-way ANOVA followed by Tukey’s post hoc test was used for statistical comparisons. For multiple comparisons, a two-way ANOVA followed by Tukey’s post hoc or a multiple Student’s t-test assuming Gaussian distribution and a two-stage linear step-up procedure was used for statistical comparisons.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Gerard Karsenty (Columbia University Irving Medical Center) for critical feedback and advice. We thank Dr. Fred de Sauvage (Genentech) for kindly providing the Lgr5DTR-EGFP mouse model. We thank Thomas Gallien, Dr. Roxanna Swagel and the vet staff for their support on mini-pig surgeries at the Medical University of South Carolina. We thank Dr. Alex Romanov and the vet staff at Columbia University Irving Medical Center for their support on animal surgeries and procedures.
Funding Sources:
National Institutes of Health/NIDCR R00DE022060 (MCE)
National Institutes of Health/NIDCR 1R01DE029068 (MCE and HY)
National Institutes of Health/NIDCR 1R41DE028215 (MCE and MC)
National Institutes of Health/NIDCR 1R42 DE028215 (MCE and MC)
Columbia University Clinical and Translational Sciences Award: Translational Accelerator Program Grant (MCE)
Columbia University Faculty Diversity Award (MCE)
National Institutes of Health R01DE021134 (HY)
National Institutes of Health P20GM121342 (HY)
National Institutes of Dental and Craniofacial Research T32DE017551 (HY)
National Institutes of Health F31AR076917 (DB)
Footnotes
DECLARATION OF INTERESTS
M.C.E. and M.C. are inventors of a patent application on an osteoarthritis therapy related to this work (US patent 17/253993), which has been licensed to Wnt Scientific for commercialization. M.C. is a founder and equity holder of Wnt Scientific. H.Y. is an advisor and equity holder of Wnt Scientific. All other authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- 1.Berenbaum F, Wallace IJ, Lieberman DE, and Felson DT (2018). Modern-day environmental factors in the pathogenesis of osteoarthritis. Nat Rev Rheumatol 14, 674–681. 10.1038/s41584-018-0073-x. [DOI] [PubMed] [Google Scholar]
- 2.Hunter DJ, and Bierma-Zeinstra S (2019). Osteoarthritis. Lancet 393, 1745–1759. 10.1016/S0140-6736(19)30417-9. [DOI] [PubMed] [Google Scholar]
- 3.Latourte A, Kloppenburg M, and Richette P (2020). Emerging pharmaceutical therapies for osteoarthritis. Nat Rev Rheumatol 16, 673–688. 10.1038/s41584-020-00518-6. [DOI] [PubMed] [Google Scholar]
- 4.Huey DJ, Hu JC, and Athanasiou KA (2012). Unlike bone, cartilage regeneration remains elusive. Science 338, 917–921. 10.1126/science.1222454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, and Fearon U (2017). The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol 13, 302–311. 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
- 6.Heinemeier KM, Schjerling P, Heinemeier J, Moller MB, Krogsgaard MR, Grum-Schwensen T, Petersen MM, and Kjaer M (2016). Radiocarbon dating reveals minimal collagen turnover in both healthy and osteoarthritic human cartilage. Sci Transl Med 8, 346ra390. 10.1126/scitranslmed.aad8335. [DOI] [PubMed] [Google Scholar]
- 7.Hunter DJ, March L, and Chew M (2020). Osteoarthritis in 2020 and beyond: a Lancet Commission. Lancet 396, 1711–1712. 10.1016/S0140-6736(20)32230-3. [DOI] [PubMed] [Google Scholar]
- 8.Aigner T, Soder S, Gebhard PM, McAlinden A, and Haag J (2007). Mechanisms of disease: role of chondrocytes in the pathogenesis of osteoarthritis--structure, chaos and senescence. Nat Clin Pract Rheumatol 3, 391–399. 10.1038/ncprheum0534. [DOI] [PubMed] [Google Scholar]
- 9.Haseeb A, Kc R, Angelozzi M, de Charleroy C, Rux D, Tower RJ, Yao L, Pellegrino da Silva R, Pacifici M, Qin L, and Lefebvre V (2021). SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2019152118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pitsillides AA, and Beier F (2011). Cartilage biology in osteoarthritis--lessons from developmental biology. Nat Rev Rheumatol 7, 654–663. 10.1038/nrrheum.2011.129. [DOI] [PubMed] [Google Scholar]
- 11.Pesesse L, Sanchez C, Walsh DA, Delcour JP, Baudouin C, Msika P, and Henrotin Y (2014). Bone sialoprotein as a potential key factor implicated in the pathophysiology of osteoarthritis. Osteoarthritis Cartilage 22, 547–556. 10.1016/j.joca.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Pullig O, Weseloh G, Ronneberger D, Kakonen S, and Swoboda B (2000). Chondrocyte differentiation in human osteoarthritis: expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcif Tissue Int 67, 230–240. 10.1007/s002230001108. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Kim DJ, Shen J, Zou Z, and O’Keefe RJ (2020). Runx2 plays a central role in Osteoarthritis development. J Orthop Translat 23, 132–139. 10.1016/j.jot.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo J, Chung UI, Yang D, Karsenty G, Bringhurst FR, and Kronenberg HM (2006). PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol 292, 116–128. 10.1016/j.ydbio.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 15.Qin X, Jiang Q, Nagano K, Moriishi T, Miyazaki T, Komori H, Ito K, Mark KV, Sakane C, Kaneko H, and Komori T (2020). Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLoS Genet 16, e1009169. 10.1371/journal.pgen.1009169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komori T (2010). Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 339, 189–195. 10.1007/s00441-009-0832-8. [DOI] [PubMed] [Google Scholar]
- 17.Kumm J, Tamm A, Lintrop M, and Tamm A (2013). Diagnostic and prognostic value of bone biomarkers in progressive knee osteoarthritis: a 6-year follow-up study in middle-aged subjects. Osteoarthritis Cartilage 21, 815–822. 10.1016/j.joca.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, Takarada T, Lezaki T, Pessin JE, Hinoi E, and Karsenty G (2015). Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 161, 1576–1591. 10.1016/j.cell.2015.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen CG, Thuillier D, Chin EN, and Alliston T (2012). Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum 64, 3278–3289. 10.1002/art.34566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y, Wang T, Hamilton JL, and Chen D (2017). Wnt/beta-catenin Signaling in Osteoarthritis and in Other Forms of Arthritis. Curr Rheumatol Rep 19, 53. 10.1007/s11926-017-0679-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day TF, Guo X, Garrett-Beal L, and Yang Y (2005). Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 8, 739–750. 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Hill TP, Spater D, Taketo MM, Birchmeier W, and Hartmann C (2005). Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 8, 727–738. 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Whetstone HC, Lin AC, Nadesan P, Wei Q, Poon R, and Alman BA (2007). Beta-catenin signaling plays a disparate role in different phases of fracture repair: implications for therapy to improve bone healing. PLoS Med 4, e249. 10.1371/journal.pmed.0040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizuhashi K, Ono W, Matsushita Y, Sakagami N, Takahashi A, Saunders TL, Nagasawa T, Kronenberg HM, and Ono N (2018). Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 563, 254–258. 10.1038/s41586-018-0662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hallett SA, Matsushita Y, Ono W, Sakagami N, Mizuhashi K, Tokavanich N, Nagata M, Zhou A, Hirai T, Kronenberg HM, and Ono N (2021). Chondrocytes in the resting zone of the growth plate are maintained in a Wnt-inhibitory environment. Elife 10. 10.7554/eLife.64513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castano Betancourt MC, Cailotto F, Kerkhof HJ, Cornelis FM, Doherty SA, Hart DJ, Hofman A, Luyten FP, Maciewicz RA, Mangino M, et al. (2012). Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc Natl Acad Sci U S A 109, 8218–8223. 10.1073/pnas.1119899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lautenschlager S, Gill PG, Luo ZX, Fagan MJ, and Rayfield EJ (2018). The role of miniaturization in the evolution of the mammalian jaw and middle ear. Nature 561, 533–537. 10.1038/s41586-018-0521-4. [DOI] [PubMed] [Google Scholar]
- 28.Ji Q, Luo ZX, Zhang X, Yuan CX, and Xu L (2009). Evolutionary development of the middle ear in Mesozoic therian mammals. Science 326, 278–281. 10.1126/science.1178501. [DOI] [PubMed] [Google Scholar]
- 29.Ochiai T, Shibukawa Y, Nagayama M, Mundy C, Yasuda T, Okabe T, Shimono K, Kanyama M, Hasegawa H, Maeda Y, et al. (2010). Indian hedgehog roles in postnatal TMJ development and organization. J Dent Res 89, 349–354. 10.1177/0022034510363078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kubiak M, and Ditzel M (2016). A Joint Less Ordinary: Intriguing Roles for Hedgehog Signalling in the Development of the Temporomandibular Synovial Joint. J Dev Biol 4. 10.3390/jdb4030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boddupally K, Wang G, Chen Y, and Kobielak A (2016). Lgr5 Marks Neural Crest Derived Multipotent Oral Stromal Stem Cells. Stem Cells 34, 720–731. 10.1002/stem.2314. [DOI] [PubMed] [Google Scholar]
- 32.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 33.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, and de Sauvage FJ (2011). A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478, 255–259. 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sigal M, Reines MDM, Mullerke S, Fischer C, Kapalczynska M, Berger H, Bakker ERM, Mollenkopf HJ, Rothenberg ME, Wiedenmann B, et al. (2019). R-spondin-3 induces secretory, antimicrobial Lgr5(+) cells in the stomach. Nat Cell Biol 21, 812–823. 10.1038/s41556-019-0339-9. [DOI] [PubMed] [Google Scholar]
- 35.Feng C, Chan WCW, Lam Y, Wang X, Chen P, Niu B, Ng VCW, Yeo JC, Stricker S, Cheah KSE, et al. (2019). Lgr5 and Col22a1 Mark Progenitor Cells in the Lineage toward Juvenile Articular Chondrocytes. Stem Cell Reports 13, 713–729. 10.1016/j.stemcr.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruscitto A, Morel MM, Shawber CJ, Reeve G, Lecholop MK, Bonthius D, Yao H, and Embree MC (2020). Evidence of vasculature and chondrocyte to osteoblast transdifferentiation in craniofacial synovial joints: Implications for osteoarthritis diagnosis and therapy. FASEB J 34, 4445–4461. 10.1096/fj.201902287R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Embree MC, Chen M, Pylawka S, Kong D, Iwaoka GM, Kalajzic I, Yao H, Shi C, Sun D, Sheu TJ, et al. (2016). Exploiting endogenous fibrocartilage stem cells to regenerate cartilage and repair joint injury. Nat Commun 7, 13073. 10.1038/ncomms13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Embree MC, Iwaoka GM, Kong D, Martin BN, Patel RK, Lee AH, Nathan JM, Eisig SB, Safarov A, Koslovsky DA, et al. (2015). Soft tissue ossification and condylar cartilage degeneration following TMJ disc perforation in a rabbit pilot study. Osteoarthritis Cartilage 23, 629–639. 10.1016/j.joca.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldring MB (2012). Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis 4, 269–285. 10.1177/1759720X12448454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagata K, Hojo H, Chang SH, Okada H, Yano F, Chijimatsu R, Omata Y, Mori D , Makii Y, Kawata M, et al. (2022). Runx2 and Runx3 differentially regulate articular chondrocytes during surgically induced osteoarthritis development. Nat Commun 13, 6187. 10.1038/s41467-022-33744-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nathan J, Ruscitto A, Pylawka S, Sohraby A, Shawber CJ, and Embree MC (2018). Fibrocartilage Stem Cells Engraft and Self-Organize into Vascularized Bone. J Dent Res 97, 329–337. 10.1177/0022034517735094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coles JM, Zhang L, Blum JJ, Warman ML, Jay GD, Guilak F, and Zauscher S (2010). Loss of cartilage structure, stiffness, and frictional properties in mice lacking PRG4. Arthritis Rheum 62, 1666–1674. 10.1002/art.27436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koyama E, Saunders C, Salhab I, Decker RS, Chen I, Um H, Pacifici M, and Nah HD (2014). Lubricin is Required for the Structural Integrity and Post-natal Maintenance of TMJ. J Dent Res 93, 663–670. 10.1177/0022034514535807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, and Winton DJ (2013). Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495, 65–69. 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 45.van Es JH, Sato T, van de Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, et al. (2012). Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 14, 1099–1104. 10.1038/ncb2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibata S, and Yokohama-Tamaki T (2008). An in situ hybridization study of Runx2, Osterix, and Sox9 in the anlagen of mouse mandibular condylar cartilage in the early stages of embryogenesis. J Anat 213, 274–283. 10.1111/j.1469-7580.2008.00934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, and Tabin CJ (1996). Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273, 613–622. 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 48.Purcell P, Joo BW, Hu JK, Tran PV, Calicchio ML, O’Connell DJ, Maas RL, and Tabin CJ (2009). Temporomandibular joint formation requires two distinct hedgehog-dependent steps. Proc Natl Acad Sci U S A 106, 18297–18302. 10.1073/pnas.0908836106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kronenberg HM (2006). PTHrP and skeletal development. Ann N Y Acad Sci 1068, 113. 10.1196/annals.1346.002. [DOI] [PubMed] [Google Scholar]
- 50.Chang JC, Christiansen BA, Murugesh DK, Sebastian A, Hum NR, Collette NM, Hatsell S, Economides AN, Blanchette CD, and Loots GG (2018). SOST/Sclerostin Improves Posttraumatic Osteoarthritis and Inhibits MMP2/3 Expression After Injury. J Bone Miner Res 33, 1105–1113. 10.1002/jbmr.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thakur A, Jaiswal MK, Peak CW, Carrow JK, Gentry J, Dolatshahi-Pirouz A, and Gaharwar AK (2016). Injectable shear-thinning nanoengineered hydrogels for stem cell delivery. Nanoscale 8, 12362–12372. 10.1039/C6NR02299E. [DOI] [PubMed] [Google Scholar]
- 52.Schmitt M, Metzger M, Gradl D, Davidson G, and Orian-Rousseau V (2015). CD44 functions in Wnt signaling by regulating LRP6 localization and activation. Cell Death Differ 22, 677–689. 10.1038/cdd.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuroda S, Tanimoto K, Izawa T, Fujihara S, Koolstra JH, and Tanaka E (2009). Biomechanical and biochemical characteristics of the mandibular condylar cartilage. Osteoarthritis Cartilage 17, 1408–1415. 10.1016/j.joca.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 54.Phillips R (2021). Targeting articular Mmp13 in OA. Nat Rev Rheumatol 17, 645. 10.1038/s41584-021-00696-x. [DOI] [PubMed] [Google Scholar]
- 55.Ruan MZ, Patel RM, Dawson BC, Jiang MM, and Lee BH (2013). Pain, motor and gait assessment of murine osteoarthritis in a cruciate ligament transection model. Osteoarthritis Cartilage 21, 1355–1364. 10.1016/j.joca.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Fan X, Xing L, and Tian F (2019). Wnt signaling: a promising target for osteoarthritis therapy. Cell Commun Signal 17, 97. 10.1186/s12964-019-0411-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monteagudo S, Cornelis FMF, Aznar-Lopez C, Yibmantasiri P, Guns LA, Carmeliet P, Cailotto F, and Lories RJ (2017). DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat Commun 8, 15889. 10.1038/ncomms15889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yazici Y, McAlindon TE, Gibofsky A, Lane NE, Lattermann C, Skrepnik N, Swearingen CJ, Simsek I, Ghandehari H, DiFrancesco A, et al. (2021). A Phase 2b randomized trial of lorecivivint, a novel intra-articular CLK2/DYRK1A inhibitor and Wnt pathway modulator for knee osteoarthritis. Osteoarthritis Cartilage 29, 654–666. 10.1016/j.joca.2021.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Kronenberg HM (2007). The role of the perichondrium in fetal bone development. Ann N Y Acad Sci 1116, 59–64. 10.1196/annals.1402.059. [DOI] [PubMed] [Google Scholar]
- 60.Murphy MP, Koepke LS, Lopez MT, Tong X, Ambrosi TH, Gulati GS, Marecic O, Wang Y, Ransom RC, Hoover MY, et al. (2020). Articular cartilage regeneration by activated skeletal stem cells. Nat Med 26, 1583–1592. 10.1038/s41591-020-1013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y, et al. (2015). Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269–284. 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuwahara ST, Serowoky MA, Vakhshori V, Tripuraneni N, Hegde NV, Lieberman JR, Crump JG, and Mariani FV (2019). Sox9+ messenger cells orchestrate large-scale skeletal regeneration in the mammalian rib. Elife 8. 10.7554/eLife.40715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paul S, Schindler S, Giovannone D, de Millo Terrazzani A, Mariani FV, and Crump JG (2016). Ihha induces hybrid cartilage-bone cells during zebrafish jawbone regeneration. Development 143, 2066–2076. 10.1242/dev.131292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muruganandan S, Pierce R, Teguh DA, Perez RF, Bell N, Nguyen B, Hohl K, Snyder BD, Grinstaff MW, Alberico H, et al. (2022). A FoxA2+ long-term stem cell population is necessary for growth plate cartilage regeneration after injury. Nat Commun 13, 2515. 10.1038/s41467-022-30247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X, von der Mark K, Henry S, Norton W, Adams H, and de Crombrugghe B (2014). Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet 10, e1004820. 10.1371/journal.pgen.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang L, Tsang KY, Tang HC, Chan D, and Cheah KS (2014). Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A 111, 12097–12102. 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang CT, Lin YT, Chiang BL, Lin YH, and Hou SM (2006). High molecular weight hyaluronic acid down-regulates the gene expression of osteoarthritis-associated cytokines and enzymes in fibroblast-like synoviocytes from patients with early osteoarthritis. Osteoarthritis Cartilage 14, 1237–1247. 10.1016/j.joca.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 68.Takahashi K, Goomer RS, Harwood F, Kubo T, Hirasawa Y, and Amiel D (1999). The effects of hyaluronan on matrix metalloproteinase-3 (MMP-3), interleukin-1beta(IL-1beta), and tissue inhibitor of metalloproteinase-1 (TIMP-1) gene expression during the development of osteoarthritis. Osteoarthritis Cartilage 7, 182–190. 10.1053/joca.1998.0207. [DOI] [PubMed] [Google Scholar]
- 69.Boettger MK, Kummel D, Harrison A, and Schaible HG (2011). Evaluation of long-term antinociceptive properties of stabilized hyaluronic acid preparation (NASHA) in an animal model of repetitive joint pain. Arthritis Res Ther 13, R110. 10.1186/ar3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kovaka S, Zimin AV, Pertea GM, Razaghi R, Salzberg SL, and Pertea M (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 278. 10.1186/s13059-019-1910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tardaguila M, de la Fuente L, Marti C, Pereira C, Pardo-Palacios FJ, Del Risco H, Ferrell M, Mellado M, Macchietto M, Verheggen K, et al. (2018). SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res 28, 396–411. 10.1101/gr.222976.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vitting-Seerup K, and Sandelin A (2019). IsoformSwitchAnalyzeR: analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinformatics 35, 4469–4471. 10.1093/bioinformatics/btz247. [DOI] [PubMed] [Google Scholar]
- 74.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902. 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Laverty S, Girard CA, Williams JM, Hunziker EB, and Pritzker KP (2010). The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rabbit. Osteoarthritis Cartilage 18 Suppl 3, S53–65. 10.1016/j.joca.2010.05.029. [DOI] [PubMed] [Google Scholar]
- 79.Gerwin N, Bendele AM, Glasson S, and Carlson CS (2010). The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the rat. Osteoarthritis Cartilage 18 Suppl 3, S24–34. 10.1016/j.joca.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 80.Glasson SS, Chambers MG, Van Den Berg WB, and Little CB (2010). The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18 Suppl 3, S17–23. 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at Gene Expression Omnibus and are publicly available as of the date of publication. The accession number is listed in the Key Resources Table. Additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Aggrecan | Thermofisher | MA3-16888 |
| Rabbit ant-βcatenin | Abcam | ab6302 |
| Rabbit ant-βgalactosidase | MBL Life Science | PM049 |
| Mouse anti-Collagen 2A1 | Millipore | MAB8887 |
| Rabbit anti-Osteocalcin | EMD Millipore | ab10911 |
| Goat anti-Periostin | R&D Systems | af2955 |
| Rabbit anti-Runx2 | Abcam | ab23981 |
| Rabbit anti-total βcatenin | Invitrogen | 71-2700 |
| Rabbit anti-non-phospho (active)-βcatein | Cell Signaling | 8814 |
| Rabbit anti-LRP5 | Proteintech | 24899-1-AP |
| Rabbit anti-LRP6 | Cell Signaling | 3395 |
| Mouse anti-CD44 | Proteintech | 60224-1 |
| Mouse anti-GAPH | Invitrogen | MA116757 |
| Goat anti-mouse HRP | Invitrogen | G21040 |
| Goat anti-rabbit HRP | Invitrogen | G21234 |
| Goat anti-rabbit Alexa Fluor 546 | Invitrogen | A11010 |
| Goat anti-rabbit Alexa Fluor 488 | Invitrogen | A11008 |
| Goat anti-mouse Alexa Fluor 546 | Invitrogen | A11003 |
| Goat anti-mouse Alexa Fluor 488 | Invitrogen | A11001 |
| Donkey anti- rabbit Alexa Fluor 647 | Invitrogen | A32795 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Protein Sclerostin Protein | R&D Systems | 1406-ST-025/CF |
| Sodium Hyaluronate | Lifecore Biomedical | HA2M-5 |
| Recombinant Human Wnt3A Protein | R&D Systems | 5036-WN-010/CF |
| Recombinant Human Dickkopf Related 3 Protein | R&D Systems | 1118-DK-050/CF |
| Wnt-C59 | Peprotech | 1248913 |
| Critical commercial assays | ||
| Porcine Dickkopf Related Protein 3 ELISA Kit | Mybioscience | MBS099882 |
| Sclerostin Human ELISA Kit | Invitrogen | EHSOST |
| Invitrogen™ Click-iT™ Plus EdU Cell Proliferation Kit for Imaging | Invitrogen | C10639 |
| SYBR™ Green PCR Master Mix | ThermoFisher | 4309155 |
| RNAscope™ Multiplex Fluorescent Reagent Kit v2 | Advanced Cell Diagnostics | 323100 |
| RNAscope™ 2.5 HD Duplex Detection Reagents | Advanced Cell Diagnostics | 322500 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE236909 |
| Experimental models: Primary Cell lines | ||
| 1-Human Perichondrial Cells Lgr5High | Embree Lab, this paper | N/A |
| OA 2-Human Perichondrial Cells Lgr5Low | Embree Lab, this paper | N/A |
| OA 3-Human Perichondrial Cells Lgr5Low | Embree Lab, this paper | N/A |
| OA 4-Human Perichondrial Cells Lgr5Low | Embree Lab, this paper | N/A |
| Healthy Human Mandibular Condylar Chondrocytes | Embree Lab, this paper | N/A |
| Osteoarthrithic Human Mandibular Condylar Chondrocytes | Embree Lab, this paper | N/A |
| Mini-pig mandibular condylar chondrocytes | Embree Lab, this paper | N/A |
| Mini-pig perichondrial cells | Embree Lab, this paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: prg4tm1Mawa/J | Jackson Laboratory | 025737 |
| Mouse: Tg (TCF/Lef1-lacZ)34Efu/J | Jackson Laboratory | 004623 |
| Mouse: Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | 007909 |
| Mouse: Lgr5tm1(cre/ERT2)Cle/J | Jackson Laboratory | 008874 |
| Mouse: CD1-IGS | Charles River | 022 |
| Mouse: Lgr5DTR-EGFP | Genetech | N/A |
| Rat: Sprague Dawley | Charles River | 400 |
| Rabbit: New Zealand White | Charles River | 052 |
| Pig: Yucatan Miniature Swine | Sinclair Bioresources | N/A |
| Oligonucleotides | ||
| Primers for qRT-PCR, see Table S5 | This paper | N/A |
| Primers for genotyping, see Table S2 | This paper | N/A |
| Software and algorithms | ||
| Prism Graphpad | N/A | https://www.graphpad.com |
| ImageJ | Schneider et al. 2012 | https://imagej.nih.gov/ij/ |
| Biorender | N/A | https://www.biorender.com |
| StringTie software | Kovaka et al. 2019 | https://ccb.jhu.edu/software/stringtie/ |
| RUVSeq software | Risso et al. 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| SQANTI3 software | Tardgauila et al. 2018 | https://github.com/ConesaLab/SQANTI3 |
| IsoformSwitchAnalyzeR software | Vitting-Seerup et al. 2019 | https://bioconductor.org/packages/release/bioc/html/IsoformSwitchAnalyzeR.html |
| DESeq2 software | Love et al. 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| clusterProfiler 4.0 software | Wu et al. 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Salmon aligner | Patro et al. 2017 | https://combinelab.github.io/salmon/ |
| STAR aligner | Dobin et al. 2013 | https://github.com/alexdobin/STAR/tree/master |