

Abstract
A current bottleneck in the development of proteolysis targeting chimeras (PROTACs) is the empirical nature of linker length structure-activity relationships (SARs). A multidisciplinary approach to alleviate the bottleneck is detailed here. Firstly, we examine four published synthetic approaches that have been developed to increase synthetic throughput. We then discuss advances in structural biology and computational chemistry that have led to successful rational PROTAC design efforts and give promise to de novo linker design in silico. Lastly, we present a model generated from a curated list of linker SARs studies normalized to reflect how linear linker length affects the observed degradation potency (DC50).
Keywords: Proteolysis targeting chimera, PROTAC, linker, structure activity relationships, ternary complex, rational design, heterobifunctional degrader, chimeric small molecule, degrader, chimera
Graphical Abstract
INTRODUCTION
Since the first proof of concept in 2001,1 small molecule–induced targeted protein degradation has become an exciting modality for pharmacological intervention. The two main strategies used to induce the degradation are the use of molecular glues which are small molecules that alter the substrate recognition domain of an E3 ligase thereby allowing the recruitment of neosubstrates for proteolysis, and the use of chimeric small molecules that consist of an E3 ligase binding moiety linked to a motif that binds desired target protein.2 The later will be the focus of this perspective. Coined proteolysis targeting chimeras (PROTACs, also known as SNIPERs, uSMITEs or degraders), bifunctional small molecule degraders have become known for their catalytic activity (potency), ability to induce isozyme selectivity through interprotein interaction, and their deviation from the ‘rule–of–five’.3–6 The utility of six unique chimeric degrader molecules in humans are currently being investigated in clinical trials.7 Of note, the indications of metastatic castration–resistant prostate cancer (ARV–110)8 and metastatic breast cancer (ARV–471),9 through the targeted degradation of the androgen receptors and estrogen receptors, respectively, and the non-oncogenic target IRAK4 degrader (KT-474) which could demonstrate their use in chronic dosing.7 Their advance into the clinic along with their unique ability to degrade challenging protein targets highlights the importance of understanding the salient features that guide successful PROTAC discovery. In this mini–perspective, we explore the strategies involved in enhancing throughput, examine successful rational design efforts, and provide a synopsis of the molecular trends within the current linker designs.
The unique metabolic activity of PROTACs comes from their ability to appropriate RING ubiquitin ligases from the ubiquitin–proteasome system (UPS), which regulates protein homeostasis by tagging proteins with poly–ubiquitin chains, thereby marking them for proteolysis.10 The UPS signal cascade (Figure 1a) begins with the E1 activating enzyme, which adenylates the C–terminus of ubiquitin, followed by a transfer of the ubiquitin to an active site cysteine. The E1 activating enzyme then engages an E2 conjugating enzyme and transfers the ubiquitin to the E2 active site cysteine through a trans–thioesterification. The ubiquitin–loaded E2 conjugating enzyme then binds a multisubunit E3 ligase complex, which transfers the ubiquitin to a peripheral lysine residue on the substrate protein, labeling the substrate for degradation by the proteasome.11 With each step of the cascade (Figure 1a), the UPS confers specificity for its substrate protein. Efforts have been made at therapeutic intervention at each step in the UPS cascade.12–14
Figure 1.
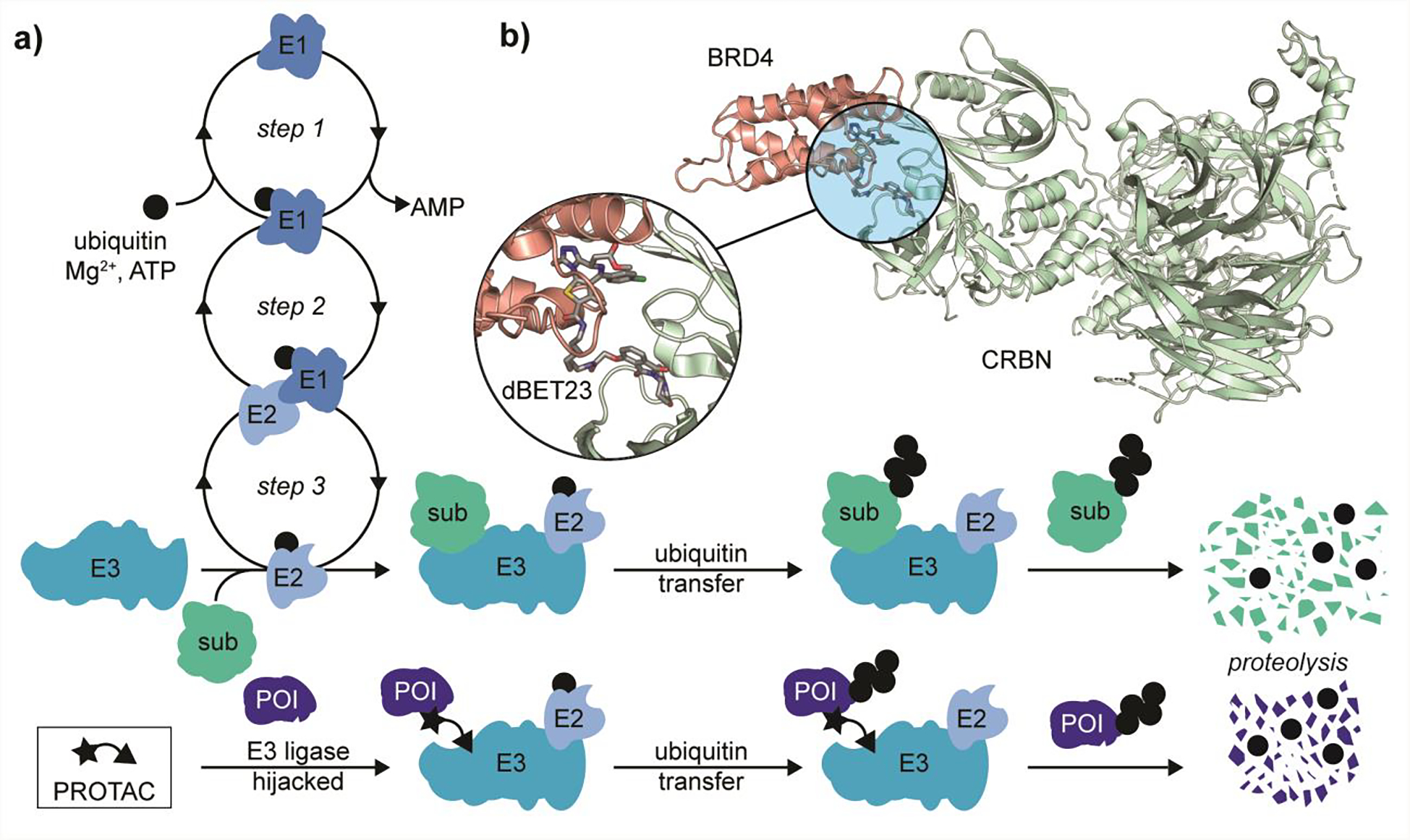
Metabolic activity of a PROTAC. a) Representative comparison of UPS pathway for cullin–RING E3 ligases in both the presence and absence of PROTAC molecules. Step 1: ubiquitin activation by E1. Step 2: Engagement of ubiquitin loaded E1 with E2 conjugating enzyme. Step 3: transfer of ubiquitin to E2 via trans–thioesterification and subsequent formation of E3 ligase complex. Introduction of PROTAC molecules can redirect the E3 ligase from its cognate substrate (sub) to a non–cognate protein of interest (POI). b) Representative example of a non–cognate ternary complex formed by the PROTAC dBET23, bromodomain BRD4, and E3 ligase substrate binding domain CRBN (PDB 6BN7). Note: E3 cartoon represents a multi-component complex in which the PROTAC is able to bind the substrate adapter protein in order to redirect its metabolic activity.
PROTACs are chimeric small molecules that are able to engage the substrate recognition domain of an E3 ligase and a protein of interest (POI) simultaneously, thereby inducing non–cognate ubiquitination and subsequent degradation of the POI (Figure 1a).15 A number of E3 ligases have been targeted for PROTAC development,16 however here we focus on PROTACs that hijack the substrate adapter domains cereblon (CRL4CRBN)17 and Von–Hippel Lindau (CRL2VHL)18 RING E3 ligases. Interestingly, PROTACs can exhibit an alluring ‘plug–and–play’ architecture,19 where in some cases the same POI can be degraded by recruiting different E3 ligases. For example, the bromodomain (BRD4) degraders MZ1 and dBET–23 (Figure 1b) recruit Von–Hippel Lindau (CRL2VHL) and cereblon (CRL4CRBN) E3 ligases, respectively.20,21 In other cases swapping E3 ligase recruitment can show exquisite target selectivity through tertiary interaction as demonstrated by the development of tyrosine kinase (ABL/BCR-ABL) degraders developed with promiscuous kinase inhibitor warheads.19
The potency and isozyme selectivity of PROTACs can be optimized through structure–activity relationships (SARs) within the linker (Figure 1b). Here, the length and chemical composition has been shown to influence, among others, a PROTAC’s structural rigidity, hydrophobicity, and solubility.22 To date, linker length SAR studies are largely empirical and have proven to be time and labor intensive. While great strides have been made towards rational PROTAC design through structural biological and computational studies, linker design still presents a significant synthetic burden.
APPROACHES TO ENHANCE THROUGHPUT
The current approaches to streamline linker variant SAR studies involve increasing synthetic throughput via use of orthogonally protected bifunctional linkers23 solid phase synthesis,24 copper–catalyzed click chemistry,25 activated esters,26 and Staudinger ligation chemistry27 (Figure 2). PROTAC synthesis involves an asymmetric three–part diversification between the two binding motifs and a linker region (Figure 2b). By exploiting simplified purification procedures and parallel synthetic strategies, these approaches decrease material lead-time without altering the empirical nature of the study.
Figure 2.
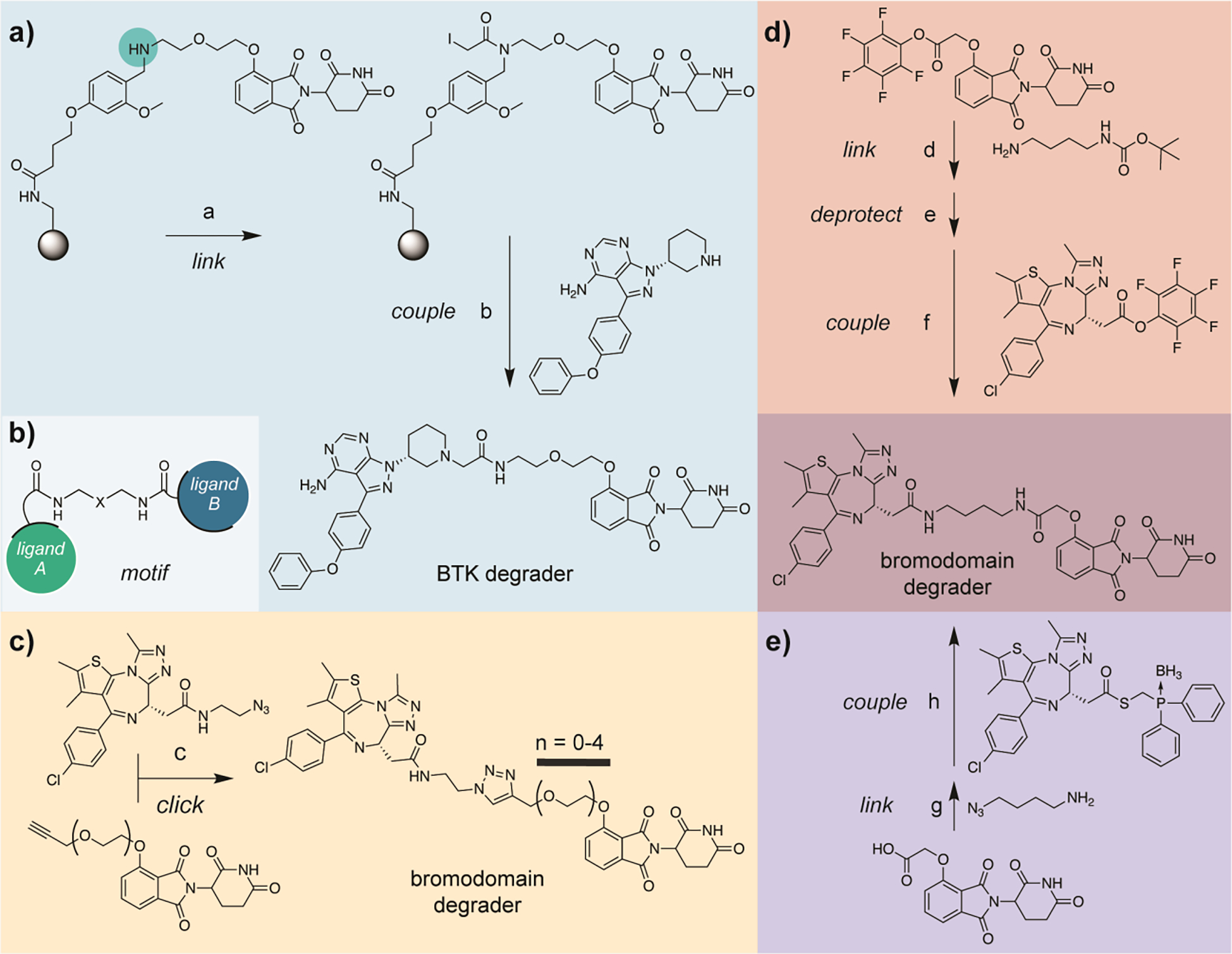
Published synthetic methods for accessing PROTACs. a) Synthesis of a BTK degrader using thalidomide preloaded resin (TPR). Secondary amine shown highlighted in COLOR is used as a linker diversification handle. b) Representative structure of a PROTAC bearing amide linker chemistry. c) Synthesis of bromodomain degraders using copper–catalyzed click chemistry. d) Synthesis of dBET1 utilizing pentafluorophenyl activated esters. e) Synthesis of dBET1 via Staudinger ligation chemistry. Reagents: a, iodoacetic acid, DIC, DCM, rt; b, despropenoyl ibrutinib, DIPEA, DMSO, rt then TFA/DCM (1:1), rt, 49% overall for a and b; c, CuSO4 (20 mol%), sodium ascorbate (20 mol%), THF/H2O, rt, 67–90%; d, N–Boc–1,4–butanediamine, DIPEA, DMF, rt, 81%; e, TFA/DCM (1:5), rt, 99%; f, DIPEA, DMF, rt, 81%; g, 4–azido–1–butanamine, HATU, DABCO, DMF, rt, 50%; h, DMF, 40 deg., 54%. Note: steps g and h can be accomplished in a one–pot fashion.
Krajcovicova et. al.24 was able to prepare a suite of PROTACs consisting of five different kinase inhibitors using a thalidomide preloaded resin (TPR) (Figure 2a). The aminomethyl polystyrene–divinylbenzene resin (PS–DVB) was acylated with 4–(4–formyl–3–methoxyphenoxy)–butanoic acid yielding a terminal aldehyde, which after amidation and subsequent reduction to a secondary amine, provided a synthetic handle for linker diversification. The resulting resin was then treated with iodoacetic acid (with and without additional polyethylene glycol (PEG) spacer), producing electrophilic TPR.
Subsequent reactions of this electrophilic TPR with a family of nucleophilic kinase inhibitors yielded a family of PROTACs. The fully elaborated PROTACs were then cleaved from the PS–DVB resin with trifluoroacetic acid (TFA) yielding the crude PROTACs in 24–85% yield and 78–98% purity and were then further purified by reverse–phase high pressure liquid chromatography (RP–HPLC) on a 200 mg scale. Though in this study, the authors prepared a PROTAC library using an electrophilic TPR and nucleophilic kinase inhibitors, they noted that this methodology is generalizable and other synthetic strategies to append POI–targeting inhibitors to the TPR can be accommodated, including use of electrophilic inhibitors with a nucleophilic TPR. If TPR resin is stable upon storage, this strategy will undoubtedly buoy efforts to produce ‘user–friendly’ tool kits for the diversification of CRBN–recruiting PROTACs.
The copper–catalyzed click chemistry platform demonstrated by Wurz et. al.25 (Figure 2c) relied upon the preparation of a library of alkyne–terminal PEG–linked CRBN ligands and VHL ligands (not shown), as well as an azide–linked derivative of known bromodomain inhibitor JQ1. The two ligands were then united through triazole formation using copper–catalyzed click chemistry.28 A panel of ten bromodomain targeting PROTACs (five CRBN recruiting, five VHL recruiting) were prepared on 100 mg scale in 55–90% yields, and purity was demonstrated via liquid chromatography-mass spectrometry (LCMS). Purification was accomplished using reverse–phase chromatography (ISCO Combiflash). This is the only published synthetic platform that directly synthesized both VHL recruiting PROTACs in addition to CRBN recruiting PROTACs.
An approach developed out by Papatzimas et. al.26 utilized pentafluorophenyl (Pfp) esters as synthons to access previously published PROTAC dBET117 (Figure 2d) in 81% yield on 40 mg scale and could be readily purified on normal–phase silica gel. This strategy entailed isolating both the E3 ligase ligand and bromodomain inhibitor JQ1 as Pfp–esters. In a three–step sequence, a Boc–protected amine linker was then reacted with the thalidomide–Pfp–ester to yield the first amide bond formation, the Boc–protecting group was removed with TFA, and finally the JQ1 Pfp–ester was added to yield dBET117 (Figure 2d).
We recently developed27 a synthetic method that leverages the chemoselectivity of the Staudinger ligation in order to assemble PROTACs in an asymmetric, one–pot fashion (Figure 2e). We also targeted dBET117 (BRD4 degrader) and two analogs as a model system to demonstrate that the entire PROTAC assembly could be choreographed in a single reaction flask. However, the resulting reaction mixture was quite complex and would require HPLC purification. We then chose to isolate azido–terminal linked thalidomide intermediates and showed that all of the linker variants could be synthesized in parallel in 39–85% yields from a stock solution of JQ1 borane–protected phosphine thioester. This only required silica microcolumn purification to obtain highly pure compound on 10 mg scale.
Taken together, there are a number of strategies that have been demonstrated to achieve modular synthesis of PROTAC linker variant suites for IMiD–based (CRBN recruiting) systems, although none of them appear to have widespread application in the field as of yet. Although it is conceivable that most of the strategies presented could be translatable to VHL or other E3 ligase recruiting systems, only the copper–catalyzed click platform25 specifically demonstrated this, however introduction of the triazole moiety to the linker may not yield desirable physiochemical properties due to its high topological total polar surface area (TPSA).29 All of these strategies would greatly benefit from the availability of libraries of functionalized (alkyne, Boc–protected amine terminal, or azide terminal) linked E3 ligase ligands prepared and aliquoted for posterity. Indeed, many are commercially available. Key considerations for choosing a platform include stability upon long term storage of the linked E3 ligase ligand libraries, purification capabilities of the laboratory, and desired linker chemistry (linear vs. triazole containing). Collectively, these strategies increase the synthetic throughput of linker length SAR studies, and we see them as a first step (potentially coupled with computational techniques) in PROTAC development to determine POI•E3 ligase compatibility. Once an optimal linker length hit is detected further linker SAR could be implemented to optimize rigidity, solubility, cell permeability, and pharmacological (PK/PD) profile to produce a lead degrader compound. This is commonly done by replacing PEG and linear aliphatic linkers with piperine and piperazine based linkers which reduce the degrees of freedom in the degrader and provide more favorable pharmacological properties, such as in the development of mutant BRAF kinase degrader SJF-062830 and recently released structures of ARV-110 and ARV-471, two of the degraders currently in the clinic. However, we view the linker rigidifying SAR as a next step in lead degrader development after the optimal linker length has been determined either by biological evaluation or computational prediction.
RATIONAL PROTAC DESIGN
Alternatively, efforts have been made towards rational PROTAC design utilizing both X–ray crystallographic data and computational modeling.31 The first X-ray structure of a degrader (MZ1) in ternary complex was solved by Gadd et. al.32 and using these data they were able to rationally design a more selective bromodomain degrader (AT1). Farnaby and coworkers33 were able to identify crucial stabilizing interactions between the PEG linker of PROTAC 1 (Figure 3a) and VHL from co–crystal structures of SMARCA2:PROTAC 1:VHL (PDB 6HAY). Based on these data, they introduced an additional T–shaped–stacking interaction and increased rigidity via insertion of a phenyl moiety in the linker region without sacrificing the key PEG interactions yielding ACBI1 (Figure 3a); an improved SMARCA2/4 degrader.
Figure 3:
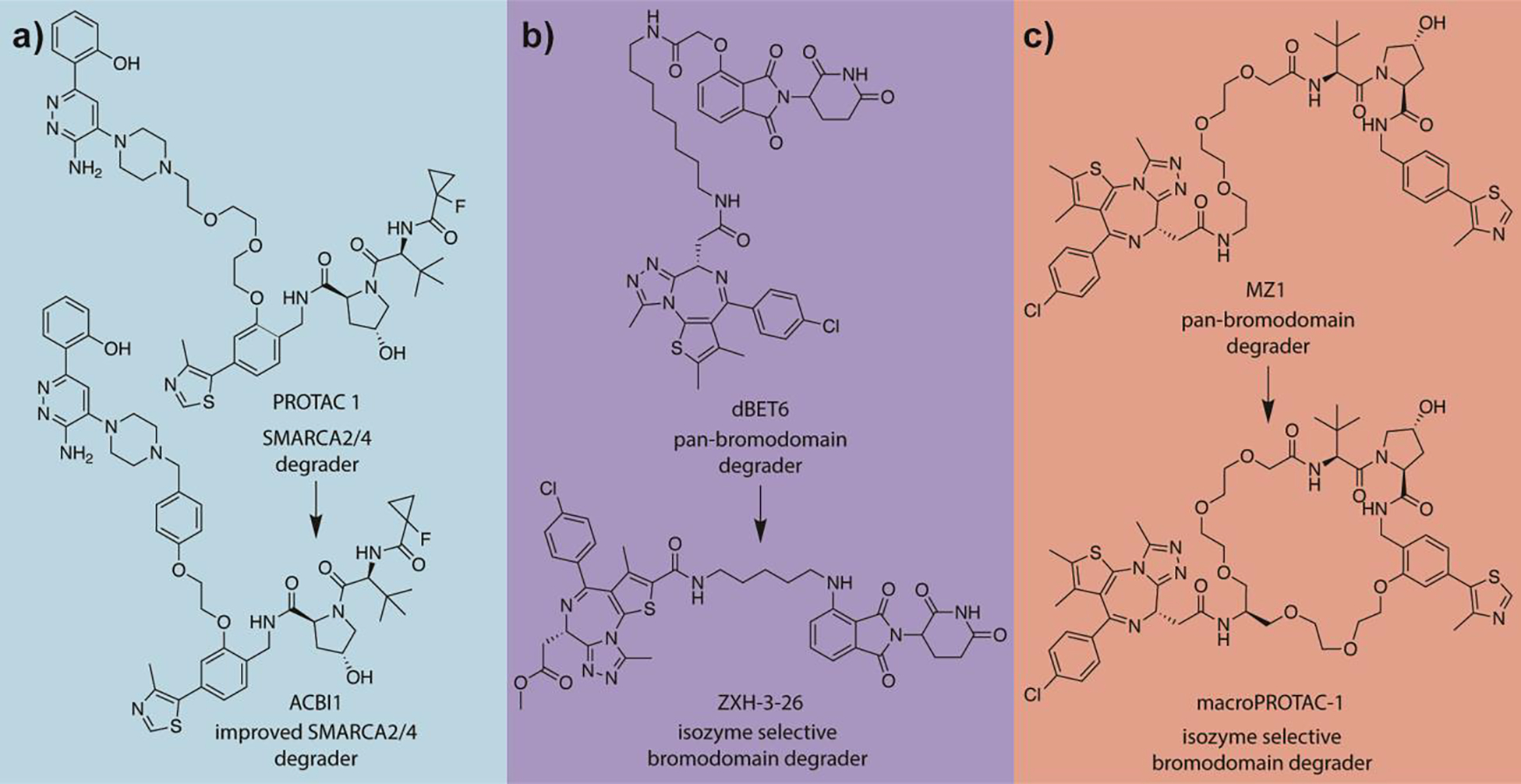
Exemplary rational PROTAC development campaigns. a) Development of improved SMARCA2/4 degrader ACBI1 through identification of key interactions with the PEG linker. b) Design of isozyme selective bromodomain degrader ZXH-3–26 through limitation of the degrees of freedom via computationally-aided linker length reduction. c) Development of isozyme selective bromodomain degrader macroPROTAC-1 through limitation of the degrees of freedom via introduction of a macrocyclic linker.
Notably, Nowak et al.21 were able to develop a novel BRD4 selective degrader ZXH–3–26 based upon a known pan–bromodomain degrader, dBET6, (Figure 3b) by performing RosettaDock simulations that utilized the X–ray structures of a set of related, PROTAC-bound ternary complexes.34 Stochastic sampling of low energy conformations using Rosetta34 was able to recapitulate experimental structures. Structural predictions also suggested that minimization of PROTAC linker length would enhance degrader selectivity by reducing the number of favorable binding modes for the POI•PROTAC•ligase ternary complex.
Testa and coworkers35 have also demonstrated the utility of computer-aided PROTAC design. They identified a potent and isoform selective (BRD4BD2 selective) bromodomain degrader using molecular dynamics simulations of the complex of BRD4BD2 and VHL bound with known bromodomain degrader MZ1 (PDB 5T35) (Figure 3c). Analysis of the simulations suggested that a macrocyclic linker could improve MZ1 effectiveness by reducing the degrees of freedom of the PROTAC, pre-organizing its POI and ligase moiety for ternary complex formation. Synthesis of a macrocyclic analog of MZ1 was realized using a ‘bespoke’ PEG-based linker. The resulting macrocyclic PROTAC demonstrated a lower binding affinity for both BRD4 and VHL in binary complex compared to MZ1, while maintaining a comparable degradation potency. Taken together, this suggests an increase in ternary complex formation efficiency.
More rigorous computational methods have been developed in efforts towards rationally designed de novo PROTAC development using both Molecular Operating Environment (MOE) and the open source Rosetta software suites. These methods have not only successfully reproduced, in silico, PROTAC binding modes identified from X-ray crystallography, but are also consistent with trends in potency and selectivity based upon prior biological evaluation. Herein, we describe select examples to illustrate the value of such efforts. Further reading on computer-aided PROTAC development may be found here.36–41
Using the MOE software suite, Drummond et. al.42,43 developed a series of protocols for generating and analyzing PROTAC ternary complexes (Figure 4). Of these protocols, the most effective sampled the conformational space of the degrader in the absence of both POI and ligase in order to identify degrader conformations that in a subsequent step would be subjected to docking simulations. These methods used co-crystal structures of the binding moieties of the PROTAC (no linker) bound to their respective targets. This team has also shown that computationally docked structures may also be used as input. Next global protein•protein docking simulations performed using MOE’s docking protocol in order to generate an ensemble of states and determine how the proteins might interact proximal to their ligated pockets.
Figure 4.
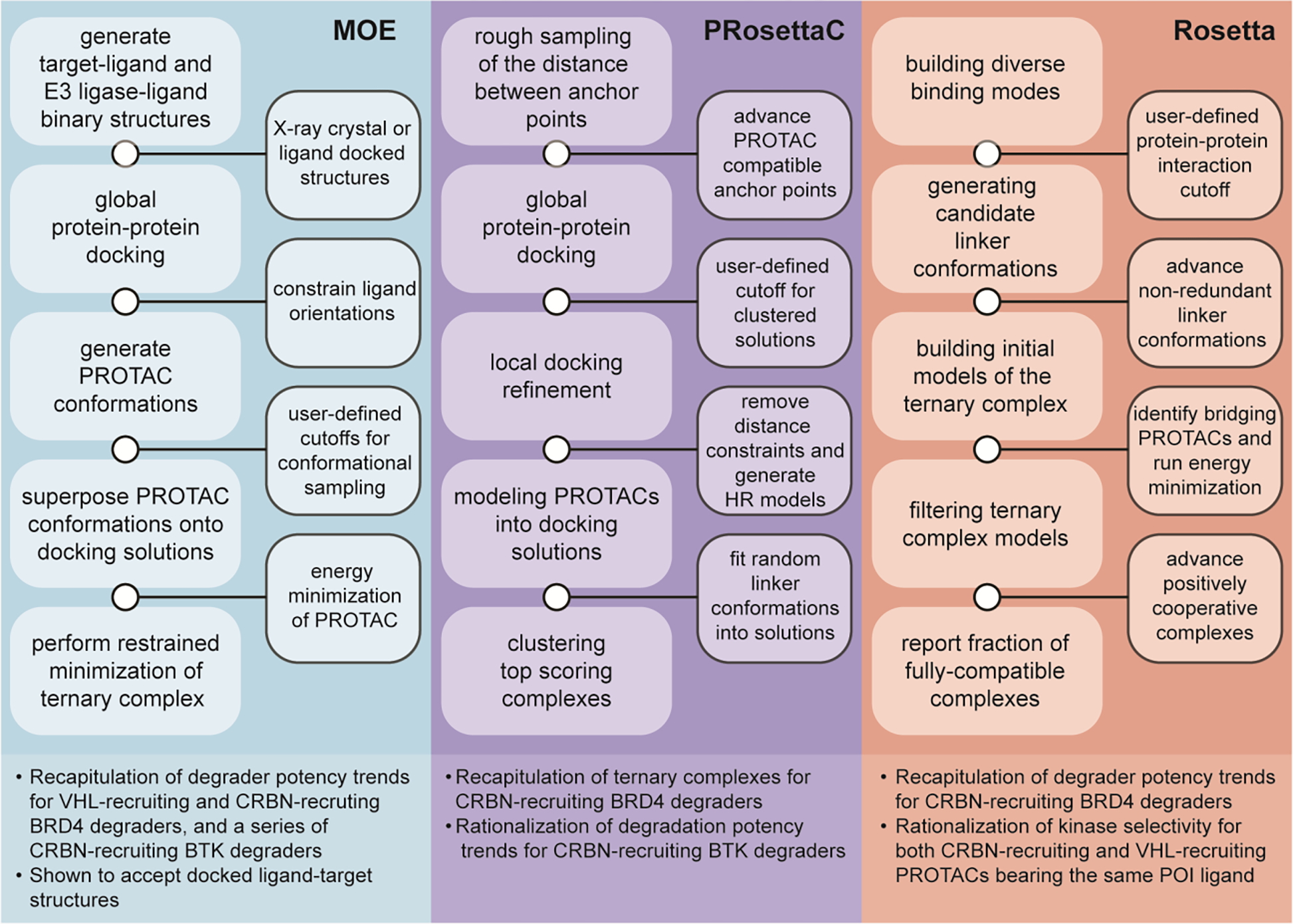
Recent advances in computational approaches to PROTAC development. MOE: General approach and highlights of the protocol used by Drummond et. al.36 on the Molecular Operating Environment (MOE) platform. PRosettaC: General approach and highlights of the protocol used by Zaidman et al.42 on the Rosetta platform. Rosetta: General approach and highlights of the protocol used by Bai et al.46 on the Rosetta platform.
Next, the ligands were then constrained to their bound conformation used in the global protein•protein docking operation, and a series of PROTAC conformers were generated using LowModeMD.44 The resulting PROTAC conformers were then superimposed onto the docked POI•ligase structures, multiple energy minimizations were performed on the PROTAC, and the coordinates of the bound relaxed PROTACs were compared to the unlinked ligand-protein structures. Finally, a restrained minimization protocol was performed on the ternary complex as a whole to remove steric clashes between the proteins and the PROTAC. The method described was able to recapitulate degrader binding modes and potency trends for both VHL-recruiting and CRBN-recruiting degraders,21,32,33 rationalize the degradation potency trends of macrocyclic PROTAC-1 (Figure 3c),35 a series of CRBN-recruiting Bruton’s tyrosine kinase (BTK) degraders,45 PROTACs bearing a more rigid linker design,46 and predict with reasonable accuracy the degradation selectivity among kinases of a degrader bearing a pan-kinase inhibitor.47
Alternatively, Zaidman et. al.48 used the open source Rosetta software suite in order to develop PRosettaC (Figure 4). PRosettaC is similar to that of Drummond and coworkers42,43 insofar as that the structures of the binary complexes of both ligands with their respective targets are requisite inputs. The PRosettaC protocol requires the definition of two anchor points corresponding to the binding epitopes on the degrader. The anchor points are then used to perform a rough sampling of the distances and ligand positions between the two anchor points that could accommodate a full length PROTAC, yielding an ensemble of degrader conformations that are binned based on the distance between the anchor atoms. These simulations provide information about the distance constraints for global protein•protein docking simulations using PatchDock.49 This approach facilitates the rapid sampling of protein•protein interaction space. Restraint-free local protein•protein docking using RosettaDock50 are subsequently performed to refine the structures of modeled POI•E3 ligase complexes.
Lastly, each of the solutions had their ligand positions fixed and were superposed with 100 full PROTAC conformations generated using RDkit, and the optimal conformation was chosen using Rosetta Packer.51 The resulting ternary complexes were then filtered via Rosetta energy score, and clustered with the assumption that the near native solutions would be sampled many times. This method was able to recapitulate experimentally determined ternary complexes of CRBN-recruiting and VHL-recruiting degraders,21,32,33,35 as well as rationalize the degradation potencies of a series of CRBN-recruiting BTK degraders.45
Bai et. al.52 described an alternative Rosetta-based protocol (Figure 4). The protocol was still reliant on ligand-bound binary X-ray crystal structures as initial inputs, however it used a global docking protocol initially developed for antibody-antigen docking53 to generate the initial ensemble of diverse poses of the POI and ligase about a fixed ligand position. Poses were evaluated based upon the stabilizing effect of protein•protein interactions formed between the POI and the ligase with the most highly scored decile of poses chosen for further refinement. Independently, the OMEGA software was used to generate a series of low energy linker conformations that were assembled from various linkers examined attached to small chemical moieties representing the functional group used at the binding moiety attachment site.54 Up to 1,000 conformers were generated for each linker and aligned with the ligand bound global docking solutions to determine if the linker adequately bridged the gap between proteins. The selected linkers were then merged with the bound ligands to create the full PROTAC and an energy minimization was performed to eliminate steric clash and bond angle distortions. The resulting ternary complexes were then filtered based on the median interaction energy of the initial protein•protein interaction and normalized by the number of initially docked models and linker conformations. The protocol was able to recapitulate degrader potency data for CRBN-recruiting BRD4 degraders,21,46 as well as rationalize kinase selectivity between both CRBN-recruiting and VHL-recruiting bearing the same kinase inhibitor.37
Both MOE and Rosetta were able to successfully recapitulate ternary complex binding modes elucidated from prior X-ray crystallographic studies, although these binding modes could represent thermodynamic artifacts from the process of crystallization.52 For this reason, we find the ability of the protocols to rationalize biological trends in potency and target selectivity is, perhaps, a more apt benchmark of success as it pertains to de novo PROTAC development.
Due to the non-cognate nature of the induced protein•protein interactions, a common theme between global docking strategies was the biasing towards hydrophobic interactions, which is consistent with the plastic protein•protein interface model21 where multiple binding modes may be populated with direction-dependent polar interactions playing a secondary role in binding. A hurdle for the field to overcome will be the reliance on input of X-ray crystallographic data of ligand-bound POI targets, however Drummond et. al.42,43 have demonstrated moderate success with computationally docked structures, attributing the protocols shortcomings to the deviation of the position of the ligand anchor point atom in the docked structures from that of the X-ray crystal structure. Improvement in ligand docking protocols and strategies with reduced reliance on anchor points in global docking could overcome this issue.
Overall, these studies demonstrate that PROTAC development has benefitted from computer-aided optimization (Figure 3). Recent advances in computational protocols for small molecule design presage an era of rational de novo PROTAC design (Figure 4) as recently demonstrated in the design of the enhancer lysine acetyltransferase (CBP/p300) degrader dCBP-1 where both the ligand and linkage point were successfully determined prior to synthesis using Rosetta.55 With available tools, we envision a workflow that entails the computational protocols first being implemented as a method to determine POI•E3 ligase compatibility through examination of the energy landscapes of the protein•protein interfaces. Potent degrader hits could then be accessed through a mixture of in silico linker length prediction combined with the synthetic strategies outlined previously (Figure 2). Translating degrader leads that are selective between highly conserved protein targets would be carried out more rigorously, such that degrader potency is maintained, while degrees of freedom of the ternary complex are minimized, as previously shown (Figure 3). Ultimately, the goal of in silico PROTAC development aims to increase throughput by minimizing the number of synthesized and biologically evaluated molecules in order to satisfactorily degrade a target protein. To this end, these protocols have already achieved their goal, and future advancements bode well for this hybrid approach.
TABULATING THE EMPERICAL SAR OF LINKER LENGTH
One of the most critical features observed early in PROTAC discovery, was the role of linker length.22 Early studies21,45 showed that there is complex interplay between having a too short or too long of a linker. In an effort to further reduce the synthetic burden of linker length SAR studies, we explored the PROTAC literature and systematically tabulated potency data as a function of linker length in order to search for trends. Our goal was to explore a possible model with predictive properties that would be agnostic to the PROTAC system under study and could suggest optimal linker length from minimal empirical inputs. This strategy would not eliminate the empirical nature of an SAR study but could potentially reduce the number of molecules needed to determine the linker length that produces the most potent degrader.
PROTACs are primarily characterized through binary and ternary complex formation dissociation constants (Kd), half maximal inhibitory concentrations (IC50 values), maximal degradation percentage (Dmax), and half maximal degradation concentration (DC50 values).56 For this study we chose to examine the reported DC50 values, defined as the concentration at which half-maximal degradation is observed, of the PROTACs in comparison to their relative linker lengths based on the fact that they encapsulate in a single measured value the abundance of metabolic processes at play. Using DC50 values one can assume that the effects of target protein degradation and resynthesis kinetics, cellular permeability and efflux, native protein expression levels, rate of ubiquitination and subsequent degradation, and other metabolic considerations between different protein targets and cell lines would be included within the dataset.
Similar efforts have been made to correlate heterogeneous IC50 values with chemical structure, and it has been shown57 that this strategy does not work due to variability in the assays and conditions used to measure the values, as well as errors in data tabulation on large databases. To avoid errors in data tabulation we chose to normalize the degradation potency data by hand, with careful consideration to compare PROTACs across different studies. Maple et. al.29 developed a degrader scoring measurement and compared 422 degraders from 73 different articles and were able to show correlations between degrader efficacy and increasing clogP, decreasing total polar surface area, and decreasing hydrogen bond donor count. Here we take a narrow view and attempt to add to these findings as it pertains to linker length specifically.
In terms of PROTAC structure, comparison of degradation values were only made when they utilized the same E3 ligase ligand, POI ligand, linkage point on each ligand, linkage functionality (such as amide bond or analine linkages) and linker chemical composition (exemplified by aliphatics or PEGs). Also, only linear linkers were considered for the study due to a lack of extensive published SAR series for more rigid (piperazine-type) linkers,58 as it pertains directly to linear length versus DC50 values. We also excluded studies of covalent degraders and homo-degraders due to their implicit differences in mechanism. In terms of biological evaluation, comparison was only made when they shared the same target, were evaluated in the same cell line, DC50 values were collected at the same time points via the same methodology. These normalization restrictions effectively allow only for direct comparison of linker variants across an SAR series within the same study; however, it also allows comparison of all the various compound series for which there is a sufficiently large (> 2 compounds) data set.
Due to the conservative normalization strategy pursued, the number of eligible SAR campaigns was greatly narrowed. An exhaustive review of the literature (as of July 12, 2020) revealed 26 series of compounds across 12 SAR studies25,45,59–68 that were normalized and compared for this analysis. There are likely more unpublished PROTAC DC50 values that were omitted from publication due to lackluster performance, and we urge the future publication of such negative data. Even though some compounds may not make useful preclinical candidates, there is great value in sharing all data sets for the gestalt of PROTAC development. To this end, we have provided the raw data from this analysis for others to access and elaborate upon the model (see Supporting Information).
To begin our analysis we normalized our entire data set to the single most potent degrader across all 26 compound series and compared how linker length affected the degradation potency (ΔDC50 value). As might be expected, no coherent correlation was found when directly comparing the eligible literature degradation data as a whole (Figure 5a). In order to overcome the issues ingrained with direct comparison, we applied the normalization strategy described above. The most optimal degrader from each SAR compound series, as chosen by the authors, had both coordinates set to zero (ΔDC50 value) and zero linker length in order to observe how a change in the number of linear atoms affects potency. Other PROTACs from within the same series would then be normalized in reference to the most potent compound for linker length (number of linear atoms)(Figure 5b). A final consideration had to be made pertaining to compounds reported as having a DC50 > 3000 nM.25,60 We have reported these values as the maximum (3000 nM) for graphical representation only, however, the overall trend is apparent without their inclusion. We also expect evaluation of the less potent degraders to be complicated by the hook-effect,69 as increasing concentrations of PROTAC are required.
Figure 5.
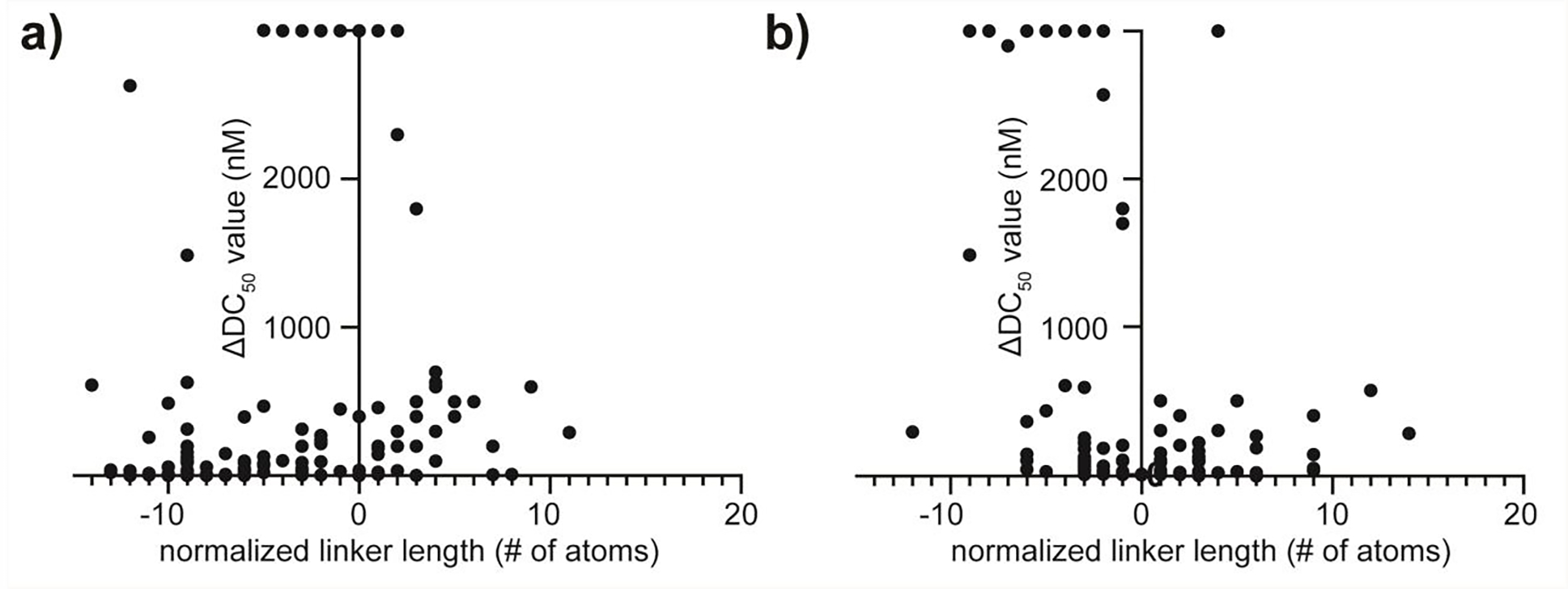
Comparison of aggregate and filtered linker length SAR studies after normalization. a) The selected SAR studies normalized to the most potent reported compound as an aggregate. b) The selected SAR studies normalized to other compounds that meet the more rigorous filtering criteria described.
The results of normalization (Figure 6) show an apparent ‘boot-shaped’ pattern across all of the compound series examined, and although the data is not coherent enough to provide useful quantitative (non-linear regression based) interpretation, useful qualitative inferences can be made. We qualitatively break this data into three sections relative to linker length. The section left of Δ linker length = 0 yields a steep drop-off in PROTAC potency, which we attribute to increased contributions of steric clash due to shorter linker lengths, which likely diminishes ternary complex formation efficiency (Figure 6). The section centered around 0 yields the most potent PROTACs (due to normalization procedures), but when compared to the section on the left suggests this zone is likely where protein•protein interactions are most favorable for the given system, regardless of whether they are cooperative,32,33,70 non-cooperative,45 or negatively cooperative.21,45 The zone to the right of +6 yields effective PROTACs with diminishing potency, which we attribute to entropic effects intrinsic to longer linker lengths and more degrees of freedom in the ternary complex. Ongoing debate has focused on the importance of protein•protein interaction between the E3 ligase and POI as it pertains to PROTAC development, and although leveraging this interaction is likely paramount to considerations of selectivity, this analysis suggests the proximity model could also coexist with regard to degradation potency.
Figure 6.
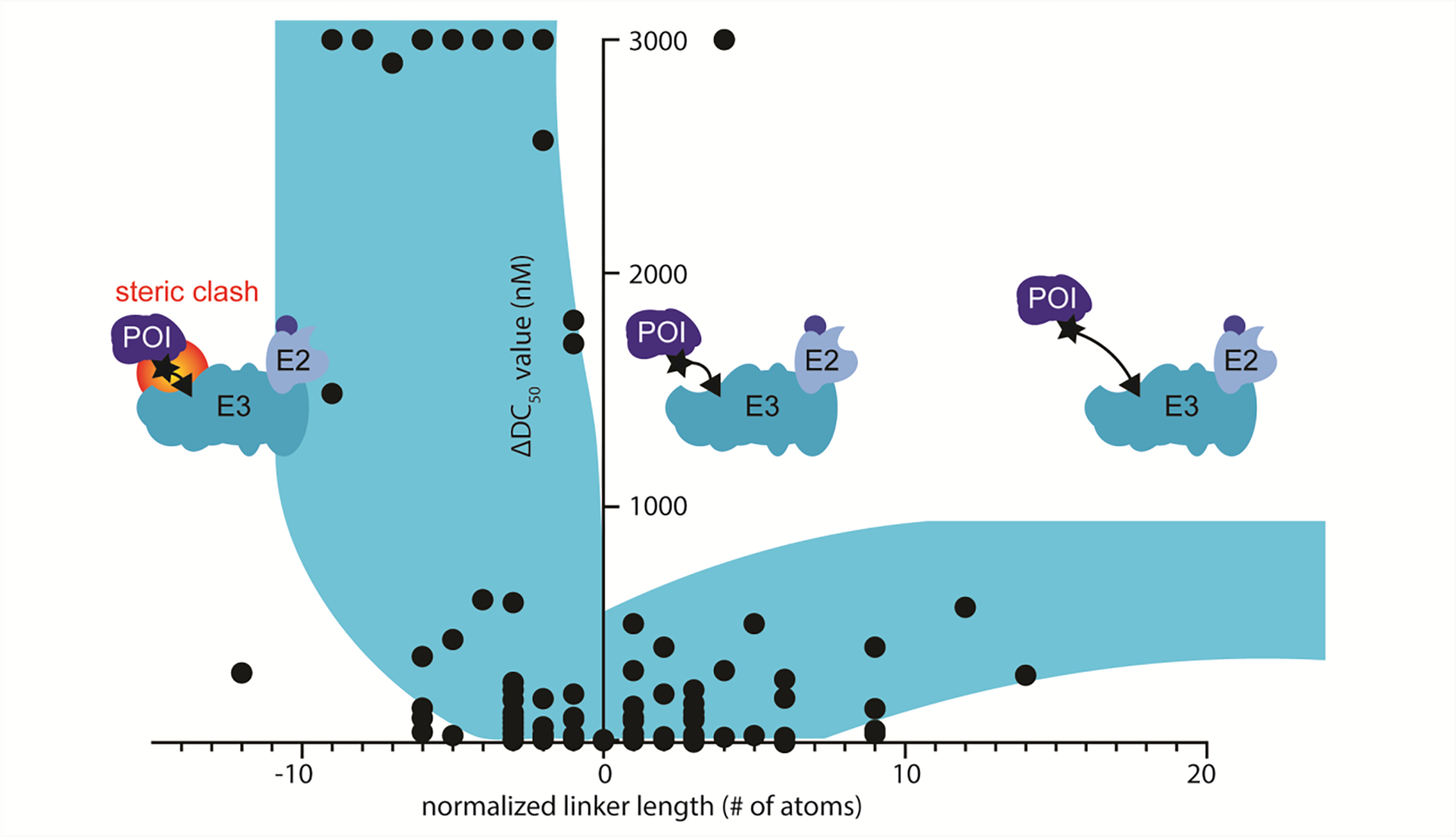
Representation of the qualitative inferences made. Qualitatively we describe this trend in three zones. Left of center represents a region where steric clash can hinder successful ternary complex formation leading to a sharp drop off in degradation potency. Center represents the most potent degraders. Right of center represents the diminishing degradation potency due to increased entropy associated with longer linkers.
This analysis suggests that an empirical linker length SAR study might be best initiated with longer linker lengths as a means to test for compatibility between the E3 ligase and POI. However, the data does not necessarily suggest that any linker length larger than the optimum will produce a successful degrader; only that, in general, the chances of finding a degrader are increased with longer linkers due to the absence of steric clash. If a compatible E3 ligase•POI system is identified, systematically shortening the length of the linker will identify the sharp cutoff point in potency and locate the linker length that induces potency abolishing steric clash. In general, this analysis suggests that the most potent PROTACs were found within a few atoms of that linker length. In addition, this region may provide the best chances of selectivity by minimizing degrees of freedom in the ternary complex while maintaining potency.
The model presented was comprised of limited published data and can only be used to make qualitative inferences about how linker length can affect degrader potency. We would expect that no such model could be generated for degrader selectivity, as each system will have its own unique interprotein contacts, and normalization for direct comparison would be difficult between systems. However, we can imagine that, were enough data reported and added to the normalization, a quantitative description of the trend may become relevant through non-linear regression. This would likely take the form of a weakly fitted curve due to the intrinsic biological and pharmacological complexities inherent in DC50 measurements, yet it could wield suggestive power by fitting a novel limited linker SAR dataset to the determined curve to examine which zones (Figure 6) have been identified for a compound series in a novel system. For this reason, we intend to update and evaluate the publicly available dataset into the future. This could offer another tool to reduce the synthetic bottleneck caused by linker length SAR studies.
SUMMARY
In conclusion, the empirical nature of PROTAC development campaigns is being eroded by the harmonious development in synthetic methodologies of chimeric small molecules, structural characterization of ternary complexes, advances in computational protocols, and systematic analysis of prior studies. PROTAC development presents a dramatic detour for drug design, in that the focus has shifted to small molecules capable of stabilizing non-cognate protein interactions long enough for poly-ubiquitination. This event–driven pharmacology allows for repurposing of previously abandoned ligands that had been shelved due to lack of potency or lack of selectivity. It also offers a method to effectively target non–catalytic protein targets, a significant expansion of possible leads. Through continued cooperation between the fields of expertise outlined here, we expect the coupled reduction of lead-time for development of PROTACs through the development of a solution to the empirical nature of the linker domain. We await further validation of this exciting pharmacological modality in the clinic.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by UC San Diego and NIH grant GM095970. We thank Ashay Patel (UC San Diego) for his critical reading end comments on this manuscript.
ABBREVIATIONS
- Boc
tert-butoxycarbonyl
- BRD4
bromodomain-containing protein 4
- BTK
Bruton’s tyrosine kinase
- CRBN
cereblon
- CRL2VHL
E3 ligase Cul2 with Von Hippel-Lindau substrate binding domain
- CRL4CRBN
E3 ligase Cul4 with cereblon substrate binding domain
- DABCO
1,4–diazabicyclo[2.2.2]octane
- DC50 value
half-maximal degradation concentration
- DIC
N,N’–diisopropylcarbodiimide
- DIPEA
diisopropylethylamine
- Dmax
maximal degradation percentage
- HATU
O–(7–azabenzotriazol–l–yl)–N,N,N’,N’–tetramethyluronium hexafluorophosphate
- HPLC
high pressure liquid chromatography
- IC50 value
half-maximal inhibitory concentration
- IMiD
immunomodulatory imide drugs
- Kd
dissociation constant
- LCMS
liquid chromatography-mass spectrometry
- MOE
Molecular Operating Environment
- PD
pharmacodynamic
- PEG
polyethylene glycol
- Pfp
pentafluorophenyl
- PK
pharmacokinetic
- POI
protein of interest
- PROTAC
proteolysis targeting chimera
- PS-DVB
polystyrene-divinyl benzene
- RP-HPLC
reverse phase high pressure liquid chromatography
- SAR
structure activity relationship
- SMARCA2
probable global transcription activator SNF2L2
- SMARCA4
transcription activator BRG1
- TFA
trifluoroacetic acid
- TPR
thalidomide preloaded resin
- UPS
ubiquitin-proteasome system
- VHL
Von Hippel-Lindau
Biographies
Biographies
Troy A. Bemis received his BSc in Chemistry from the University of Wisconsin – Madison where he worked with Prof. Sandro Mecozzi developing theranostic nanoparticles. In 2015 he began his Ph.D studies at the University of California – San Diego with Prof. Michael D. Burkart where he is currently a Ph.D candidate with a focus on the development of chimeric small molecules for the purposes of drug development and characterization of natural product biosynthetic pathways.
James J. La Clair received his Ph.D. degree in Chemistry from SUNY Buffalo under the tutelage of Prof. Peter T. Lansbury. His studies continued through a postdoctoral fellowship with Prof. Gilbert Stork at Columbia University. He then undertook a position at the Scripps Research Institute. In 1999, he shifted his focus to work on a project–by–project basis. This has included research positions at UC San Diego and the Salk Institute, among other collaborative endeavors
Michael D. Burkart is the Teddy Traylor Faculty Scholar of Chemistry & Biochemistry at the University of California, San Diego. He received a BS in Chemistry from Rice University and a Ph.D. in Chemistry from The Scripps Research Institute, followed by a post-doctoral fellowship at Harvard University. The Burkart Laboratory pursues interdisciplinary research in the fields of organic and biological chemistry, with a focus on natural product synthesis and biosynthesis.
Footnotes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Supporting Information containing unprocessed linker SAR data.
The following files are available free of charge.
Tabulated linker SAR data of studies discussed herein (.xlsx)
REFERENCES
- (1).Sakamoto KM.; Kim KB.; Kumagai A.; Mercurio F; Crews CM.; Deshaies RJ. Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Faust TB; Donovan KA; Yue H; Chamberlain PP; Fischer ESP Small-Molecule Approaches to Targeted Protein Degradation. Annual Review of Cancer Biology 2020, 181–201. [Google Scholar]
- (3).António JPM; Gonçalves LM; Guedes RC; Moreira R; Gois PM. Diazaborines as New Inhibitors of Human Neutrophil Elastase. ACS Omega 2018, 3, 7418–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dang CV; Reddy EP; Shokat KM; Soucek L Drugging the “undruggable” Cancer Targets. Nat. Rev. Cancer 2017, 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Crews CM Targeting the Undruggable Proteome: The Small Molecules of My Dreams. Chem. Biol. 2010, 17, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (7).Mullard A Targeted Protein Degraders Crowd into the Clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [DOI] [PubMed] [Google Scholar]
- (8).ClinicalTrials.gov Identifier: NCT03888612
- (9).ClinicalTrials.gov Identifier: NCT04072952
- (10).Pohl C; Dikic I Cellular Quality Control by the Ubiquitin–Proteasome System and Autophagy. Science 2019, 366, 818–822. [DOI] [PubMed] [Google Scholar]
- (11).Kleiger G; Mayor T Perilous Journey: A Tour of the Ubiquitin–Proteasome System. Trends Cell Biol. 2014, 24, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wertz IE; Wang X From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [DOI] [PubMed] [Google Scholar]
- (13).Huang X; Dixit VM Drugging the Undruggables: Exploring the Ubiquitin System for Drug Development. Cell Res. 2016, 26, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nalepa G; Rolfe M; Harper JW Drug Discovery in the Ubiquitin–Proteasome System. Nat. Rev. Drug Discov. 2006, 5, 596–613. [DOI] [PubMed] [Google Scholar]
- (15).Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y Protacs: Great Opportunities for Academia and Industry. Signal Transduct. Target. Ther. 2019, 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Konstantinidou M; Li J; Zhang B; Wang Z; Shaabani S; Ter Brake F; Essa K; Dömling A PROTACs a Game–Changing Technology. Expert Opin. Drug Disc. 2019, 14, 1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe–Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Buckley DL; Van Molle I; Gareiss PC; Tae HS; Michel J; Noblin DJ; Jorgensen WL; Ciulli A; Crews CM Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF–1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lai AC; Toure M; Hellerschmied D; Salami J; Jaime–Figueroa S; Ko E; Hines J; Crews CM Modular PROTAC Design for the Degradation of Oncogenic BCR–ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zengerle M; Chan KH; Ciulli A Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nowak RP; Deangelo SL; Buckley D; He Z; Donovan KA; An J; Safaee N; Jedrychowski MP; Ponthier CM; Ishoey M; Zhang T; Mancias JD; Gray NS; Bradner JE; Fischer ES Plasticity in Binding Confers Selectivity in Ligand–Induced Protein Degradation Article. Nat. Chem. Biol. 2018, 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cyrus K; Wehenkel M; Choi EY; Han HJ; Lee H; Swanson H; Kim KB Impact of Linker Length on the Activity of PROTACs. Mol. Biosyst. 2011, 7, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Steinebach C; Kehm H; Lindner S; Vu LP; Köpff S; López Mármol Á; Weiler C; Wagner KG; Reichenzeller M; Krönke J; Gütschow M PROTAC-Mediated Crosstalk between E3 Ligases. Chem. Commun. 2019, 55, 1821–1824. [DOI] [PubMed] [Google Scholar]
- (24).Krajcovicova S; Jorda R; Hendrychova D; Krystof V; Soural M Solid–Phase Synthesis for Thalidomide–Based Proteolysis–Targeting Chimeras (PROTAC). Chem. Commun. 2019, 55, 929–932. [DOI] [PubMed] [Google Scholar]
- (25).Wurz RP; Dellamaggiore K; Dou H; Javier N; Lo MC; McCarter JD; Mohl D; Sastri C; Lipford JR; Cee VJA “Click Chemistry Platform” for the Rapid Synthesis of Bispecific Molecules for Inducing Protein Degradation. J. Med. Chem. 2018, 61, 453–461. [DOI] [PubMed] [Google Scholar]
- (26).Papatzimas JW; Gorobets E; Brownsey DK; Maity R; Bahlis NJ; Derksen DJ A General Strategy for the Preparation of Thalidomide–Conjugate Linkers. Synlett 2017, 28, 2881–2885. [Google Scholar]
- (27).Bemis TA; La Clair JJ; Burkart MD Traceless Staudinger Ligation Enabled Parallel Synthesis of Proteolysis Targeting Chimera Linker Variants. Chem. Commun. 2021, 57, 1026–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Himo F; Lovell T; Hilgraf R; Rostovtsev VV; Noodleman L; Sharpless KB; Fokin VV Copper(I)–Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [DOI] [PubMed] [Google Scholar]
- (29).Maple HJ; Clayden N; Baron A; Stacey C; Felix R Developing Degraders: Principles and Perspectives on Design and Chemical Space. Medchemcomm 2019, 10, 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Alabi S; Jaime-Figueroa S; Yao Z; Gao Y; Hines J; Samarasinghe KTG; Vogt L; Rosen N; Crews CM Mutant-Selective Degradation by BRAF-Targeting PROTACs. Nat. Commun. 2021, 12, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Leissing TM; Luh LM; Cromm PM Structure Driven Compound Optimization in Targeted Protein Degradation. Drug Discovery Today: Technologies. Elsevier Ltd; December 10, 2020. [DOI] [PubMed] [Google Scholar]
- (32).Gadd MS; Testa A; Lucas X; Chan KH; Chen W; Lamont DJ; Zengerle M; Ciulli A Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol. 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Farnaby W; Koegl M; Roy MJ; Whitworth C; Diers E; Trainor N; Zollman D; Steurer S; Karolyi–Oezguer J; Riedmueller C; Gmaschitz T; Wachter J; Dank C; Galant M; Sharps B; Rumpel K; Traxler E; Gerstberger T; Schnitzer R; Petermann O; Greb P; Weinstabl H; Bader G; Zoephel A; Weiss–Puxbaum A; Ehrenhöfer–Wölfer K; Wöhrle S; Boehmelt G; Rinnenthal J; Arnhof H; Wiechens N; Wu MY; Owen–Hughes T; Ettmayer P; Pearson M; McConnell DB; Ciulli A BAF Complex Vulnerabilities in Cancer Demonstrated via Structure–Based PROTAC Design. Nat. Chem. Biol. 2019, 15, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sircar A; Chaudhury S; Kilambi KP; Berrondo M; Gray JJ A Generalized Approach to Sampling Backbone Conformations with RosettaDock for CAPRI Rounds 13–19. Protein. Struct. Funct. Bioinforma. 2010, 78, 3115–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Testa A; Hughes SJ; Lucas X; Wright JE; Ciulli A Structure–Based Design of a Macrocyclic PROTAC. Angew. Chem. Int. Ed. 2020, 59, 1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Smith BE; Wang SL; Jaime–Figueroa S; Harbin A; Wang J; Hamman BD; Crews CM Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime–Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lebraud H; Wright DJ; Johnson CN; Heightman TD Protein Degradation by In–Cell Self–Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schiedel M; Herp D; Hammelmann S; Swyter S; Lehotzky A; Robaa D; Oláh J; Ovádi J; Sippl W; Jung M Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [DOI] [PubMed] [Google Scholar]
- (40).Imrie F; Bradley AR; van der Schaar M; Deane CM Deep Generative Models for 3D Linker Design. J. Chem. Inf. Model. 2020, 60, 1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Pérez–Benito L; Henry A; Matsoukas MT; Lopez L; Pulido D; Royo M; Cordomí A; Tresadern G; Pardo L The Size Matters? A Computational Tool to Design Bivalent Ligands. Bioinformatics 2018, 34, 3857–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Drummond ML; Williams CI In Silico Modeling of PROTAC–Mediated Ternary Complexes: Validation and Application. J. Chem. Inf. Model. 2019, 59, 1634–1644. [DOI] [PubMed] [Google Scholar]
- (43).Drummond ML; Henry A; Li H; Williams CI Improved Accuracy for Modeling ProTAC–Mediated Ternary Complex Formation and Targeted Protein Degradation via New in Silico Methodologies. J. Chem. Inf. Model. 2020, 60, 5234–5254. [DOI] [PubMed] [Google Scholar]
- (44).Labute P LowModeMD–Implicit Low–Mode Velocity Filtering Applied to Conformational Search of Macrocycles and Protein Loops. J. Chem. Inf. Model. 2010, 50, 792–800. [DOI] [PubMed] [Google Scholar]
- (45).Zorba A; Nguyen C; Xu Y; Starr J; Borzilleri K; Smith J; Zhu H; Farley KA; Ding WD; Schiemer J; Feng X; Chang JS; Uccello DP; Young JA; Garcia–Irrizary CN; Czabaniuk L; Schuff B; Oliver R; Montgomery J; Hayward MM; Coe J; Chen J; Niosi M; Luthra S; Shah JC; El–Kattan A; Qiu X; West GM; Noe MC; Shanmugasundaram V; Gilbert AM; Brown MF; Calabrese MF Delineating the Role of Cooperativity in the Design of Potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 7285E7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Qin C; Hu Y; Zhou B; Fernandez–Salas E; Yang CY; Liu L; McEachern D; Przybranowski S; Wang M; Stuckey J; Meagher J; Bai L; Chen Z; Lin M; Yang J; Ziazadeh DN; Xu F; Hu J; Xiang W; Huang L; Li S; Wen B; Sun D; Wang S Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra–Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61, 6685–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Huang HT; Dobrovolsky D; Paulk J; Yang G; Weisberg EL; Doctor ZM; Buckley DL; Cho JH; Ko E; Jang J; Shi K; Choi HG; Griffin JD; Li Y; Treon SP; Fischer ES; Bradner JE; Tan L; Gray NS A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi–Kinase Degrader. Cell Chem. Biol. 2018, 25, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zaidman D; Prilusky J; London N PRosettaC: Rosetta Based Modeling of PROTAC Mediated Ternary Complexes. J. Chem. Inf. Model. 2020, 60, 4894–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Duhovny D; Nussinov R; Wolfson HJ Efficient Unbound Docking of Rigid Molecules. In Algorithms in Bioinformatics; Guigó R., Gusfield D., Eds.; Springer Verlag, 2002; 2452, 185–200. [Google Scholar]
- (50).Lyskov S; Gray JJ The RosettaDock Server for Local Protein•protein Docking. Nucleic Acids Res. 2008, 36, W233–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kuhlman B; Baker D Native Protein Sequences Are Close to Optimal for Their Structures. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bai N; Kirubakaran P; Karanicolas J Rationalizing PROTAC–Mediated Ternary Complex Formation Using Rosetta. bioRxiv. 2020. 10.1101/2020.05.27.119347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Weitzner BD; Jeliazkov JR; Lyskov S; Marze N; Kuroda D; Frick R; Adolf–Bryfogle J; Biswas N; Dunbrack RL; Gray JJ Modeling and Docking of Antibody Structures with Rosetta. Nat. Protoc. 2017, 12, 401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hawkins PCD; Nicholls A Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [DOI] [PubMed] [Google Scholar]
- (55).Vannam R; Sayilgan J; Ojeda S; Karakyriakou B; Hu E; Kreuzer J; Morris R; Herrera Lopez XI; Rai S; Haas W; Lawrence M; Ott CJ Targeted Degradation of the Enhancer Lysine Acetyltransferases CBP and P300. Cell Chem. Biol. 2021, 28, 503–514. [DOI] [PubMed] [Google Scholar]
- (56).Hughes SJ; Ciulli A Molecular Recognition of Ternary Complexes: A New Dimension in the Structure–Guided Design of Chemical Degraders. Essays Biochem. 2017, 61, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kalliokoski T; Kramer C; Vulpetti A; Gedeck P Comparability of Mixed IC50 Data–A Statistical Analysis. PLoS One 2013, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Krönke J; Steinebach C; Ng YLD; Sosič I; Lee CS; Chen S; Lindner S; Vu LP; Bricelj A; Haschemi R; Monschke M; Steinwarz E; Wagner KG; Bendas G; Luo J; Gütschow M Systematic Exploration of Different E3 Ubiquitin Ligases: An Approach towards Potent and Selective CDK6 Degraders. Chem. Sci. 2020, 11, 3474–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Yang K; Wu H; Zhang Z; Leisten ED; Nie X; Liu B; Wen Z; Zhang J; Cunningham MD; Tang W Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Li MX; Yang Y; Zhao Q; Wu Y; Song L; Yang H; He M; Gao H; Song BL; Luo J; Rao Y Degradation versus Inhibition: Development of Proteolysis–Targeting Chimeras for Overcoming Statin–Induced Compensatory Upregulation of 3–Hydroxy–3–Methylglutaryl Coenzyme a Reductase. J. Med. Chem. 2020, 63, 4908–4928. [DOI] [PubMed] [Google Scholar]
- (61).Zhou H; Bai L; Xu R; Zhao Y; Chen J; McEachern D; Chinnaswamy K; Wen B; Dai L; Kumar P; Yang CY; Liu Z; Wang M; Liu L; Meagher JL; Yi H; Sun D; Stuckey JA; Wang S Structure–Based Discovery of SD–36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J. Med. Chem. 2019, 62, 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhao Q; Ren C; Liu L; Chen J; Shao Y; Sun N; Sun R; Kong Y; Ding X; Zhang X; Xu Y; Yang B; Yin Q; Yang X; Jiang B Discovery of SIAIS178 as an Effective BCR–ABL Degrader by Recruiting von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. J. Med. Chem. 2019, 62, 9281–9298. [DOI] [PubMed] [Google Scholar]
- (63).Su S; Yang Z; Gao H; Yang H; Zhu S; An Z; Wang J; Li Q; Chandarlapaty S; Deng H; Wu W; Rao Y Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wu H; Yang K; Zhang Z; Leisten ED; Li Z; Xie H; Liu J; Smith KA; Novakova Z; Barinka C; Tang W Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62, 7042–7057. [DOI] [PubMed] [Google Scholar]
- (65).Cromm PM; Samarasinghe KTG; Hines J; Crews CM Addressing Kinase–Independent Functions of Fak via PROTAC–Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [DOI] [PubMed] [Google Scholar]
- (66).Crew AP; Raina K; Dong H; Qian Y; Wang J; Vigil D; Serebrenik YV; Hamman BD; Morgan A; Ferraro C; Siu K; Neklesa TK; Winkler JD; Coleman KG; Crews CM Identification and Characterization of von Hippel–Lindau–Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK–Binding Kinase 1. J. Med. Chem. 2018, 61, 583–598. [DOI] [PubMed] [Google Scholar]
- (67).Chan KH; Zengerle M; Testa A; Ciulli A Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra–Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I–BET726) BET Inhibitor Scaffolds. J. Med. Chem. 2018, 61, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sun N; Ren C; Kong Y; Zhong H; Chen J; Li Y; Zhang J; Zhou Y; Qiu X; Lin H; Song X; Yang X; Jiang B Development of a Brigatinib Degrader (SIAIS117) as a Potential Treatment for ALK Positive Cancer Resistance. Eur. J. Med. Chem. 2020, 193, 112190. [DOI] [PubMed] [Google Scholar]
- (69).Pettersson M; Crews CM PROteolysis TArgeting Chimeras (PROTACs)–Past, Present and Future. Drug Discov. Today Technol. 2019, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Roy MJ; Winkler S; Hughes SJ; Whitworth C; Galant M; Farnaby W; Rumpel K; Ciulli A SPR–Measured Dissociation Kinetics of PROTAC Ternary Complexes Influence Target Degradation Rate. ACS Chem. Biol. 2019, 14, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.