

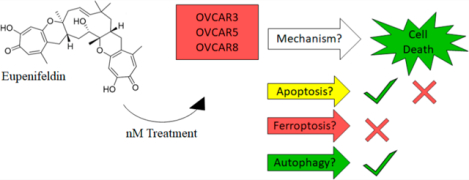
Abstract
High-grade serous ovarian cancer (HGSOC) is the most common and lethal ovarian cancer histotype. Lack of early detection methods, limited therapeutic agents, and low 5-year survival rate reflect the urgent need to develop new therapies. Eupenifeldin, a bistropolone, originally isolated from Eupenicillium brefeldianum, is a cytotoxic fungal metabolite. In three HSGOC cell lines (OVCAR3, OVCAR5, OVCAR8), eupenifeldin was found to have an IC50 value less than 10 nM, while 10 times higher concentrations were required for cytotoxicity in nontumorigenic fallopian tube secretory epithelial cell lines (FTSEC). An in vivo hollow fiber assay showed significant cytotoxicity in OVCAR3. Eupenifeldin significantly increased Annexin V staining in OVCAR3 and -8, but not OVCAR5. Eupenifeldin activated caspases 3/7 in OVCAR3, OVCAR5, and OVCAR8; however, cleaved PARP was only detected in OVCAR3. Quantitative proteomics performed on OVCAR3 implicated ferroptosis as the most enriched cell death pathway. However, validation experiments did not support ferroptosis as part of the cytotoxic mechanism of eupenifeldin. Autophagic flux and LC3B puncta assays found that eupenifeldin displayed weak autophagic induction in OVCAR3. Inhibition of autophagy by cotreatment with bafilomycin reduced the toxicity of eupenifeldin, supporting the idea that induction of autophagy contributes to the cytotoxic mechanism of eupenifeldin.
Graphical Abstract
High-grade serous ovarian cancer is the most lethal gynecologic malignancy in the U.S. and the fifth leading cause of cancer-related death in women.1 Standard treatment for epithelial ovarian cancer is surgical debulking followed by combination chemotherapy.2 Targeted therapies such as bevacizumab have been incorporated into maintenance therapy.3 While the approvals of PARP inhibitors as frontline therapy for patients with mutations in the BRCA1 or BRCA2 genes have led to an increase in progression-free and overall survival, these agents are only effective in a subpopulation with homologous recombination deficiencies (HRD).2 Additionally even in the HRD subpopulations, many patients ultimately relapse and succumb to the disease. Thus, there still exists an unmet need for drugs that modulate multiple pathways to combat mutation and recurrence.
Natural products serve as an inspiration for novel chemotherapeutics.4 Eupenifeldin was isolated as a fungal secondary metabolite in abundant quantities from Neosetophoma sp. [strain MSX50044], and it displayed nanomolar cytotoxic activity.5 Previous studies found that eupenifeldin was cytotoxic in a panel of carcinoma cell lines, including MDA-MB-231 (triple negative breast cancer), OVCAR3 and OVCAR8 (high-grade serous ovarian cancer), MSTO-211H (mesothelioma), LLC (murine lung cancer), and A549 (human lung cancer).5 Eupenifeldin induced submicromolar cytotoxicity in the HCT-116 colon carcinoma line as well as in the HCT-VM46 model, which is resistant to paclitaxel, suggesting a mechanism that is distinct.6 Additionally, three fungal meroterpenoids with some structural similarity to eupenifeldin were tested for the ability to cause mitochondrial toxicity and were not toxic up to 12.5 μM.5
Eupenifeldin has also shown efficacy in two in vivo murine cancer models. First, it was able to increase overall survival in a P388 leukemia model when dosed i.p. once daily for 5 days.6 Eupenifeldin was also encapsulated into a biofilm formulation and tested in vivo as a local-delivery system in a recurrent nonsmall cell lung carcinoma model.7 Eupenifeldin’s inherent low solubility was improved by slow release from the biofilm formulation. Eupenifeldin-loaded biopolymer films improved progression-free, disease-specific, and overall survival compared to the blank film.7 However, the mechanism of action for this compound has not been studied.
Eupenifeldin was evaluated for cytotoxic and cytostatic activity in addition to an in vivo hollow fiber assay. Eupenifeldin was tested for the ability to induce annexin V expression, caspase 3/7 activation, and PARP cleavage. A TMT-labeled proteomic analysis of eupenifeldin-treated OVCAR3 cells (25 nM, 72 h) was performed. When analyzed, the most enriched cell death pathway was ferroptosis which was prioritized for testing using known activators and inhibitors combined with eupenifeldin to determine if ferroptosis is required for inducing cytotoxicity. Finally, the role of autophagy induction was studied as a contributing factor in the cytotoxicity activity.
RESULTS AND DISCUSSION
Eupenifeldin Is Cytotoxic in Multiple Ovarian Cancer Cell Lines.
Previously, eupenifeldin (Figure 1A) was reported to exhibit cytotoxicity in breast, ovarian, colon, and lung cancer cell lines.5 To further evaluate the cell killing in high-grade serous ovarian cancer tumor models, OVCAR3, OVCAR5, and OVCAR8 were selected and compared to the nontumorigenic fallopian tube line, FT33-TAg. Cell lines were treated in a dose-dependent manner with either eupenifeldin, paclitaxel as a positive control, or DMSO. IC50 values were determined to be 9.7 ± 6.2 nM, 11 ± 2 nM, 12 ± 7 nM, and 170 ± 40 nM in OVCAR3, OVCAR5, OVCAR8, and FT33 cells, respectively (Figure 1B,C). These IC50 values demonstrate eupenifeldin to be more cytotoxic in the three ovarian cancer lines compared to FT33. The impact of eupenifeldin on cell migration was measured in a wound healing assay since compounds that block cell migration would be useful in metastatic disease such as ovarian cancer. OVCAR5 and OVCAR8 have previously been demonstrated to be good representative models for migration and were therefore selected for this study, while OVCAR3 was not selected.8 Cells were treated with either eupenifeldin (10 nM or 25 nM) or paclitaxel (10 nM), and wound closure was measured after 48 h. Significant inhibition of wound closure was observed with both eupenifeldin concentrations in both cell lines. (Figure 1D,E).
Figure 1.
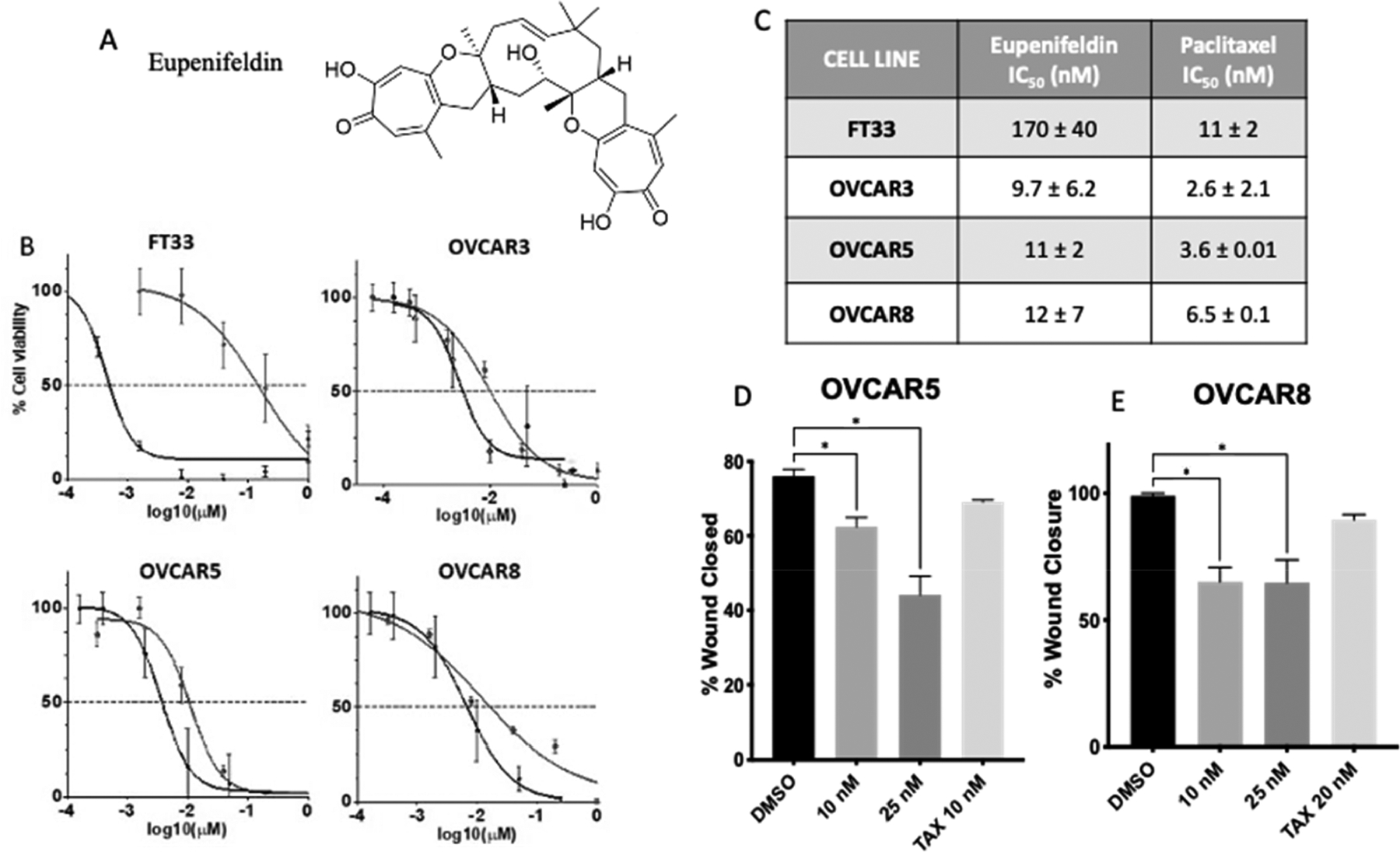
(A) The structure of eupenifeldin. (B, C) Eupenifeldin is cytotoxic in high-grade serous ovarian cancer cell lines at 72 h. Black line indicates paclitaxel control, and gray line indicates eupenifeldin treatment. (D) OVCAR5 and (E) OVCAR8 HGSOC cells have significantly decreased wound closure when treated with eupenifeldin. All data are mean ± SEM from three biological replicates, and statistical analysis was conducted via one-way ANOVA with Dunnett’s multiple comparison. *P < 0.05.
To determine if these effects were indicative of cytotoxic and not cytostatic growth arrest, a 2D foci assay was performed. OVCAR3, OVCAR5, and OVCAR8 cell lines were treated with either eupenifeldin (10 nM), paclitaxel (10 nM), or the solvent control DMSO (0.1%) for 8 h and then grown in untreated media for 15 days to allow for foci formation (Figure 2A,B). In the eupenifeldin-treated samples, there was a significant reduction in colony formation compared to the vehicle. In contrast, paclitaxel behaved as a cytostatic agent, in which fewer colonies grew over 2 weeks. Next, a hollow fiber assay was performed to determine if eupenifeldin was effective against ovarian cancer in vivo. OVCAR3 and OVCAR8 cells were embedded into hollow fibers and implanted intraperitoneally in female mice. Mice were treated with vehicle control (DMSO/PEG/H2O) (n = 8), chemotherapeutic control (paclitaxel, 5 mg/kg) (n = 6), or eupenifeldin (0.5 mg/kg) (n = 8) once per day for 4 days. Upon retrieval of the fibers on day 7, OVCAR3 cell survival was significantly lower in the eupenifeldin-treated group compared to the vehicle (Figure 2C). OVCAR8 cells were not significantly growth inhibited in the fibers in vivo (Supporting Information, S2.2).
Figure 2.
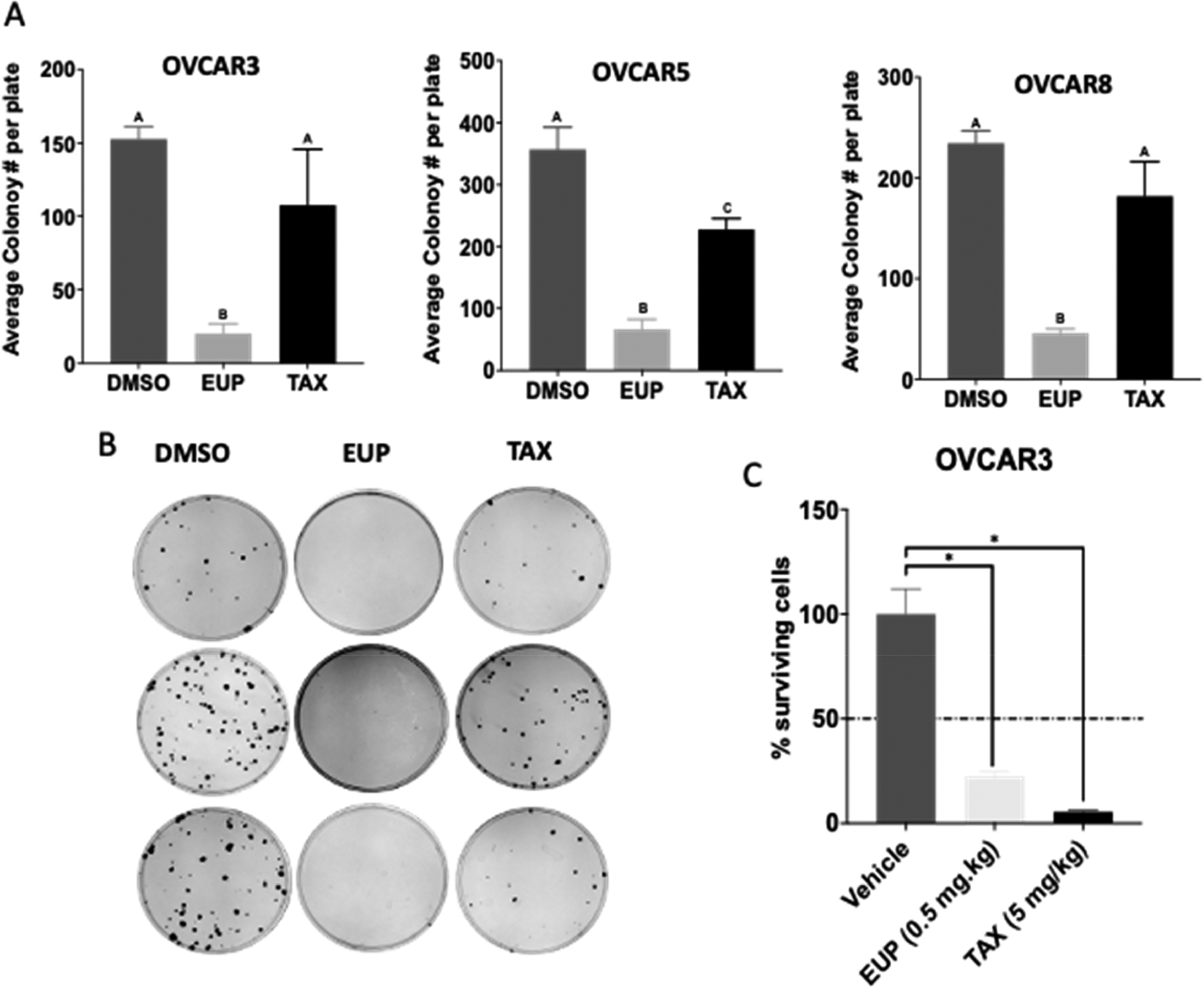
(A) Eupenifeldin is cytotoxic in 2D foci assay. Cell lines were treated for 8 h with either eupenifeldin (10 nM), paclitaxel (10 nM), or solvent control. Letters represent statistically significantly different groups with p < 0.05. (B) Representative 2D foci assay images. (C) A hollow fiber assay demonstrated decreased OVCAR3 cell survival after eupenifeldin treatment. Mice were treated with vehicle control (0.2% DMSO/PEG/H2O) (n = 8), chemotherapeutic control (paclitaxel, 5 mg/kg) (n = 6), or eupenifeldin (0.5 mg/kg)(n = 8) once per day for 4 days. Data represented as mean ± SEM and analyzed with one-way ANOVA using PRISM and statistical analysis conducted using Dunnett’s multiple comparison (* P = < 0.05).
In the in vivo hollow fiber assay eupenifeldin was generally well tolerated by the mice. The body weights of the eupenifeldin-treated mice did not change more than 12% over the course of the study and did not appear to suffer from systemic toxicity (Supporting Information, S2.1). However, previous work using surgical buttresses on lung tissue demonstrated systemic toxicity.7 As there is a potential for toxicity when delivery is improved, further testing on systemic effects may be required and a delivery platform may be required.
Eupenifeldin Increases Annexin V Staining and Caspase 3/7 Activity.
Apoptosis is a common cell death mechanism known to be activated by many chemotherapeutics. To investigate if eupenifeldin’s cytotoxic activity is due to apoptosis, single cell annexin V-FITC (AV) and propidium iodide (PI) staining were performed. In OVCAR3 and OVCAR8, 25 nM eupenifeldin treatment significantly increased both apoptosis and necrosis (AV+,PI− and AV+,PI+, respectively), but there was no significant induction in OVCAR5 (Figure 3A). A representative scatterplot of the DMSO control is included in Supporting Information, S3.1. To further test apoptotic induction, the activity of caspase 3/7 was measured using a fluorescent reporter assay after treatment with eupenifeldin. Staurosporine, a compound known to induce apoptosis through mitochondrial dysfunction and caspase activation, was used as the positive control. All three cell lines showed significant increases in caspase 3/7 activity after 48 h of eupenifeldin (50 nM) treatment. This activity was significantly attenuated in all three cell lines by cotreatment with the caspase inhibitor, ZVAD-FMK (Figure 3B).
Figure 3.
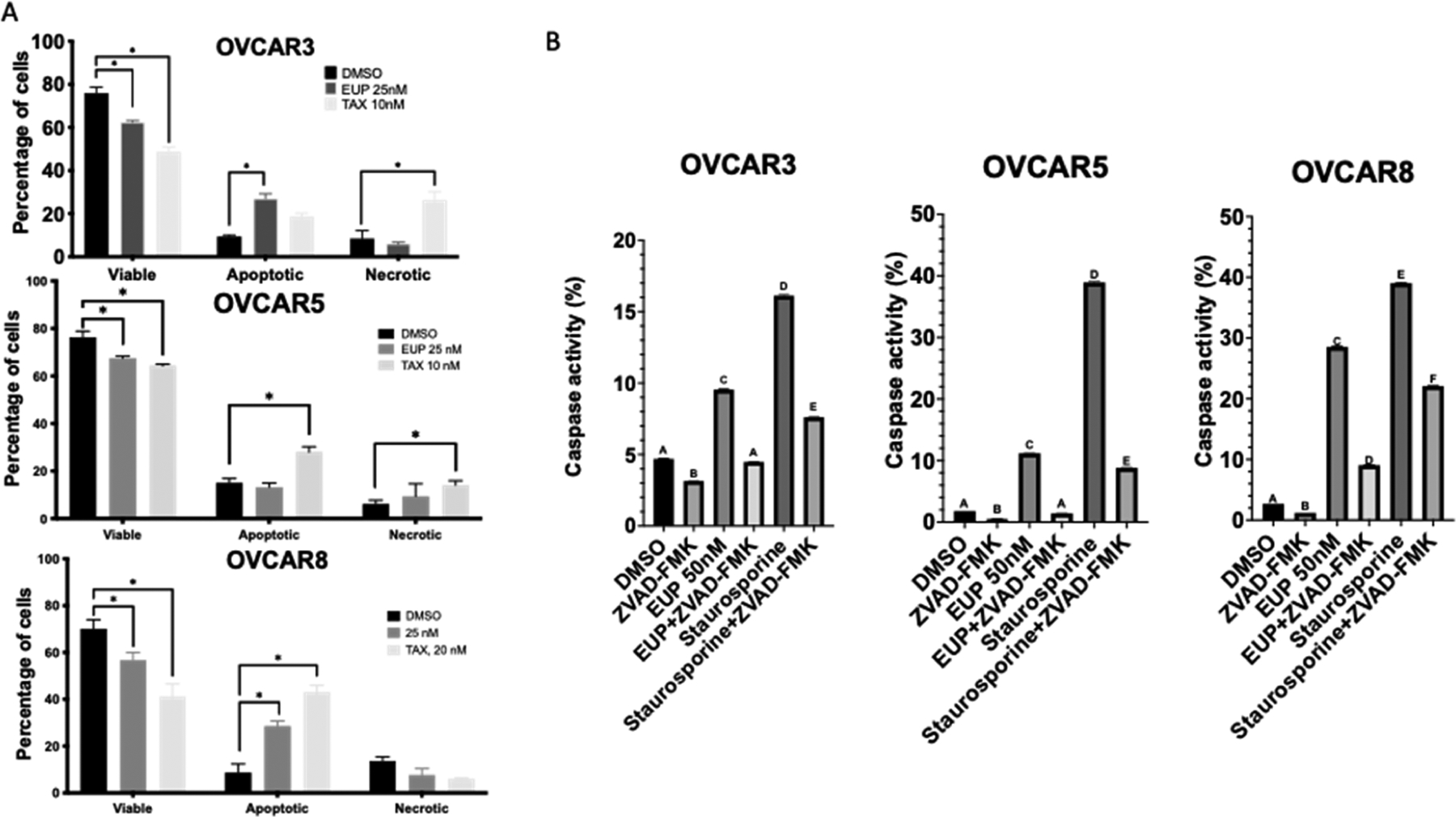
(A) Eupenifeldin treatment increases apoptosis in OVCAR3 and OVCAR8. Cells were treated with eupenifeldin (25 nM), paclitaxel (10 nM), or vehicle control (DMSO) for 48 h and stained with annexin-V/PI. Each experiment is represented by three biological replicates, and data are represented using mean ± SEM using one-way ANOVA and Dunnett’s posthoc with p < 0.05. (B) Eupenifeldin activates caspase 3/7 in all three cell lines. Data represented using mean ± SEM using two-way ANOVA and Tukey’s posthoc. Letters represent statistically different groups with a p < 0.05.
As further confirmation of apoptotic activity, especially given the lack of AV+,PI− staining in OVCAR5, we investigated whether eupenifeldin induced PARP cleavage via Western blot. Cleaved PARP was only observed in OVCAR3 cells treated with ≥10 nM eupenifeldin (Figure 4A,B). This suggested that apoptosis was not the primary mechanism of cytotoxicity of eupenifeldin in OVCAR5 and OVCAR8 or that partial apoptosis is occurring. The cleavage of PARP may explain why the OVCAR3 cells responded to eupenifeldin in the in vivo hollow fiber assay when OVCAR8 cells did not.
Figure 4.
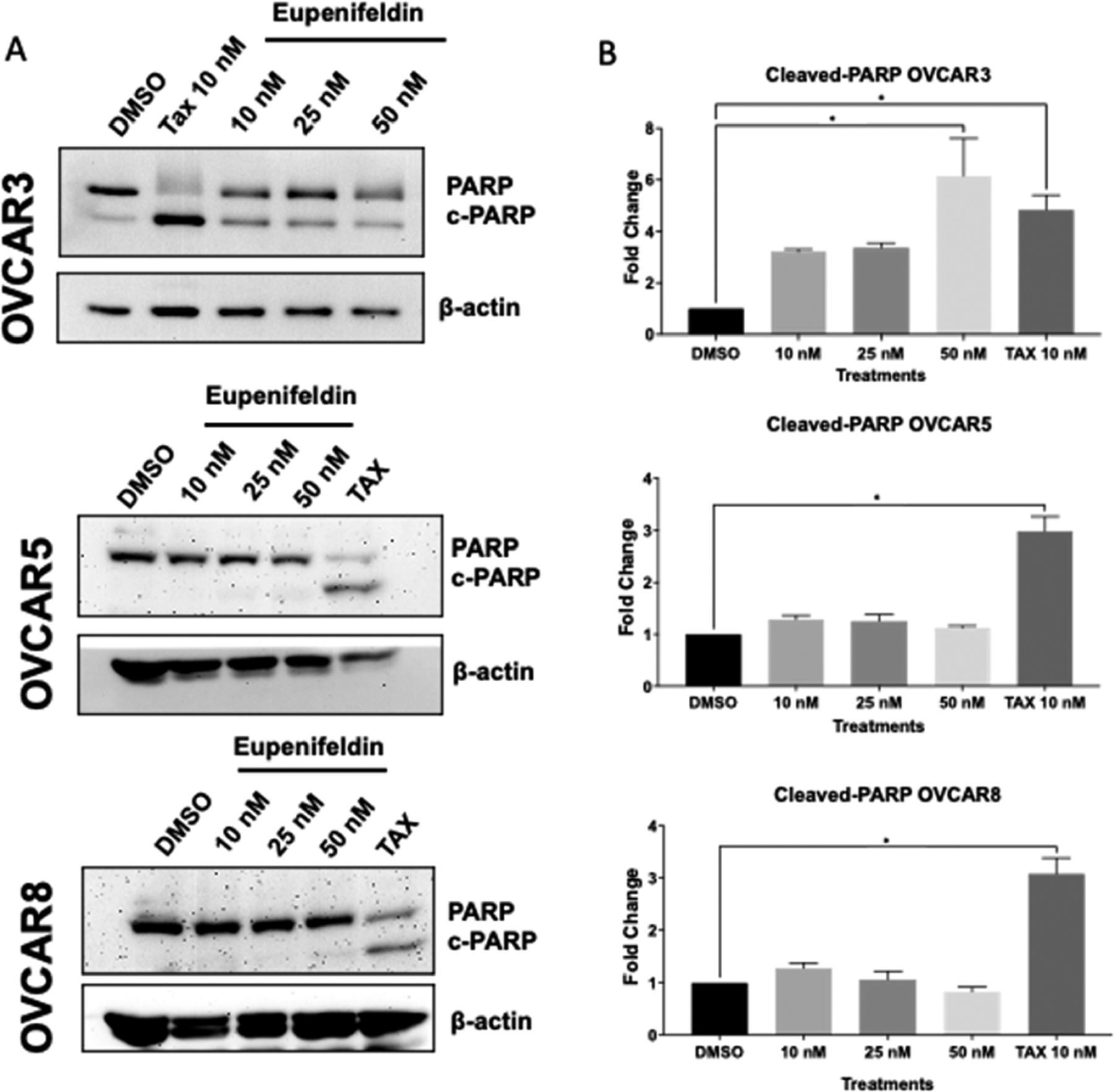
(A) Western blots and (B) densiometric analysis show an increase in cleaved PARP in eupenifeldin-treated OVCAR3 cells (72 h) but not OVCAR5 or OVCAR8 (48 h). Data represents mean ± SEM and three biological replicates. Data were quantified using ImageJ of cleaved PARP normalized to β-actin, and data points generated using one-way ANOVA with Dunnett’s multiple comparison test were used to produce P-values. (*P < 0.05).
The activation of caspases could be evidence of partial or incomplete apoptosis. Nonlethal caspase activation leading to incomplete apoptosis has been shown to increase cancer cell adhesion and invasiveness in metastatic melanoma.9 Low levels of caspase activation can trigger noncanonical responses.10 Stronger caspase activation results in apoptosis, suggesting that caspase activation below a certain threshold triggers a different cellular response.11 Further, there is a growing body of evidence suggesting nonapoptotic roles for caspases including cell differentiation.11,12 These nonlethal caspase-dependent pathways are still tightly regulated and frequently rely on temporal, spatial, or substrate controls to achieve the desired nonapoptotic result. By limiting the substrate, the effects of caspase 3/7 activation can be more precisely applied leading to nonapoptotic outcomes.12 In the case of eupenifeldin it is possible that caspase activation is below the level needed to induce apoptosis. Alternatively, sequestration or blocking of some caspase substrates could be responsible for the observed activation of caspases and incomplete apoptosis.
Proteomic Analysis Suggests Apoptosis and Ferroptosis as Potential Cytotoxic Pathways.
In order to gain a more complete understanding of the downstream changes that occur in response to eupenifeldin treatment, a proteomic analysis of eupenifeldin-treated OVCAR3 cells was conducted. Tandem mass tag (TMT) labeled proteomics identified 75 differentially expressed proteins (S2.1). Differentially expressed proteins were submitted to the Search Tool for the Retrieval of Interacting Genes/Proteins Database (STRING DB) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (Figure 5A), which identified ferroptosis as the most enriched cell death pathway with a strength of 1.28 and a false discovery rate of 0.0152. As eupenifeldin was previously found not to be toxic to mitochondria, the cell death specific pathway was prioritized over the higher strength mitochondrial associated pathways. Within the ferroptotic pathway, multiple proteins, including transferrin receptor protein 1(TRFC) and 4F2 cell-surface antigen light chain (TFL), were altered. TRFC was upregulated, and TFL was downregulated both with fold changes of 0.4. Autophagic degradation of FTL is required for ferroptosis, suggesting that ferroptosis activity may play a role in eupenifeldin’s cytotoxic mechanism.13 A heatmap of ferroptotic protein expression was generated (Figure 5B). The alterations in these proteins suggested ferroptosis as a potential mechanism of eupenifeldin cytotoxicity, and given the high strength and low false discovery rate, and number of identified proteins, ferroptosis was chosen for follow-up.
Figure 5.
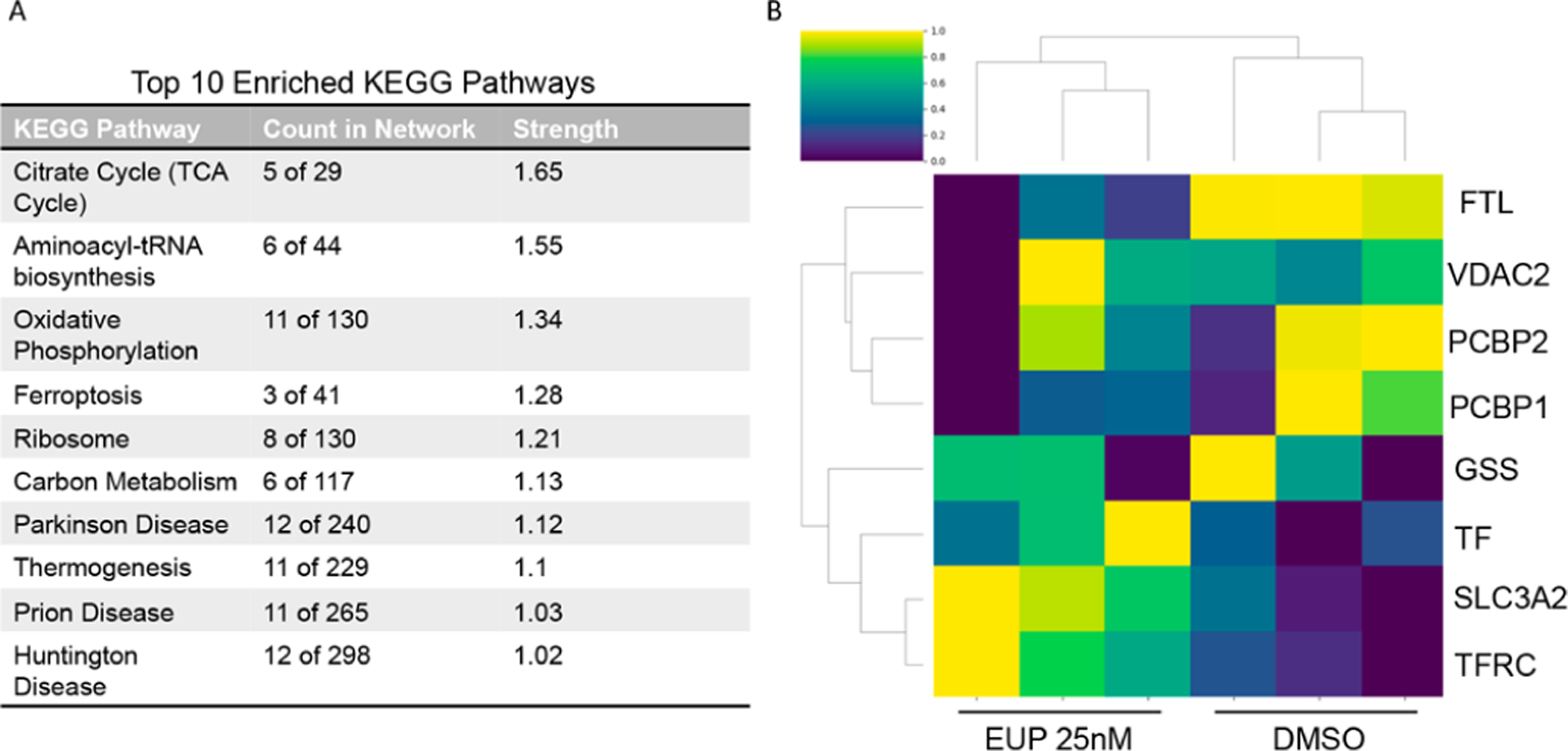
(A) The significant proteins were submitted to String DB for pathway analysis. Of the top 10 pathways, ferroptosis was identified as one of the most enriched cell death pathways. (B) Changes in ferroptotic protein expression with eupenifeldin treatment compared to DMSO. Normalized protein abundance levels are denoted by color, from bright yellow to green and to dark blue for high, medium, and low abundance.
Ferroptosis Inhibition Does Not Impact Toxicity of Eupenifeldin.
Ferroptosis is an iron-dependent nonapoptotic form of cell death caused by lipid peroxide accumulation by ROS.14 In order to understand if ferroptosis was involved eupenifeldin-induced cytotoxicity, eupenifeldin was combined with activators and inhibitors of the pathway. Ferroptosisinducing compounds, RAS-selective lethal 3 (RSL3) and imidazole ketone erastin (IKE), caused substantial cell death in OVCAR3 cells that was reversed by the addition of 2 μM ferroptosis inhibitor ferrostatin-1 (Fer-1), indicating that this cell line is sensitive to ferroptosis and that it can be blocked by inhibitors (Figure 6A,B). When OVCAR3 cells were treated with increasing concentrations of eupenifeldin (100 nM to 10 μM) and 2 μM Fer-1, there was no rescue of cell death (Figure 6C). This suggests that ferroptosis induction is not responsible for the cytotoxic effects of eupenifeldin. As further confirmation that ferroptosis is not involved, lipid peroxidation, a common hallmark of ferroptosis, was measured by the alterations in fluorescence of C11-BODIPY on the green channel by flow cytometry. OVCAR3 cells were treated with 50 nM eupenifeldin for 24 h in the presence or absence of Fer-1 (Figure 6D). The lack of rightward shift indicative of increased lipid peroxidation further supports that eupenifeldin does not induce ferroptosis as a mechanism of cytotoxicity. These experiments were also carried out in HT1080 cells with the same results (S3.1). While ferroptosis was concluded not to be the primary mechanism of action, autophagy has been shown to promote ferroptosis through ferritin degradation, which may explain why it appeared as an enriched pathway.15
Figure 6.
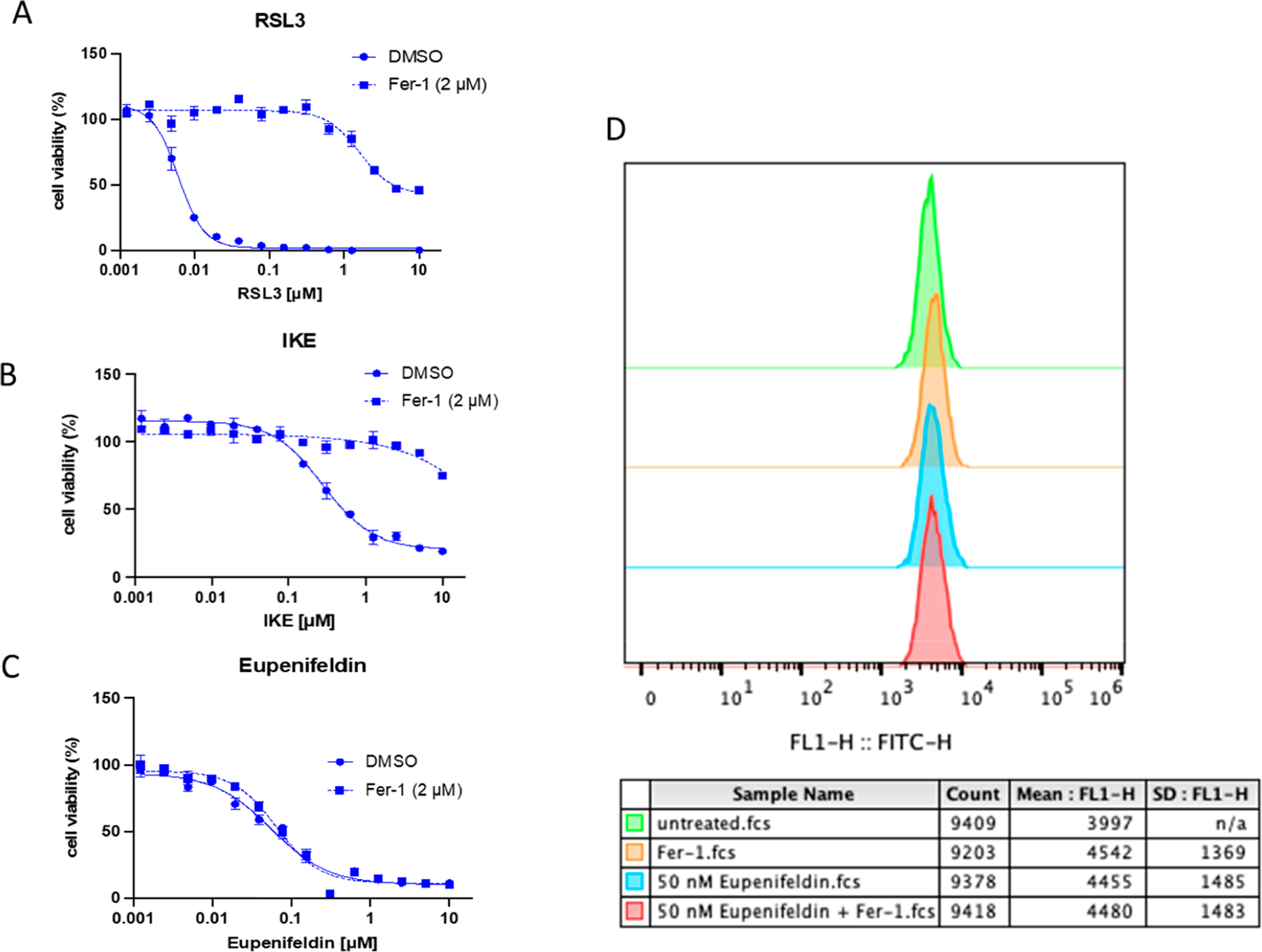
(A, B) The sensitivity of OVCAR3 cells to ferroptosis induction with RSL3 and IKE was validated. (C) The cytotoxicity induced by eupenifeldin treatment was not reversed by ferrostatin-1 addition, implicating that a nonferroptotic death pathway is involved. (D) The ability of eupenifeldin to induce lipid peroxidation was tested, and no increase in lipid peroxidation was observed.
Eupenifeldin Causes Weak Autophagic Induction.
In the absence of ferroptosis induction, the autophagy pathway was explored. Ferroptosis has been called an autophagic form of cell death, which suggested that perhaps the reason ferroptotic proteins were enriched with eupenifeldin treatment while ferroptosis was not required for cytotoxic activity was due to overlap with the process of autophagy. Autophagic induction has been previously demonstrated by our lab to be the mechanism of the cytotoxic effects of other natural products, including silvestrol.16 Autophagy was examined through tracking of LC3 puncta and an autophagic flux assay.17 HeLa cells (eGFP-LC3) were treated with serially diluted eupenifeldin (starting at 250 nM) for 4 h and were found to accumulate LC3 puncta indicative of modulation of autophagy (Figure 7A). In the autophagic flux assay, 24 h eupenifeldin treatment increased the number of autolysosomes and slightly increased the number of autophagosomes. Chloroquine (CQ) acts as a control, as it is known to inhibit autophagy. As formation of autolysosomes is required for autophagy, this is indicative of a weak induction of autophagy (Figure 7B,C). It is interesting to note that the autophagosomes localized to the tips of the cells in the autophagic flux assay, as this is atypical. To determine if induction of autophagy contributed to the cytotoxicity, OVCAR3, OVCAR5, and OVCAR8 cells were treated with eupenifeldin (25 nM) alone or in combination with bafilomycin (5 nM) (BAF), an autophagic inhibitor. Cotreatment with BAF blocked the cytotoxic effects of eupenifeldin in OVCAR3 and OVCAR5 (Figure 7D). BAF alone was too toxic in OVCAR8 to combine with eupenifeldin, but 1 nM BAF cotreatment also demonstrated reduction in the cytotoxic effect of eupenifeldin (Figure 7D).
Figure 7.
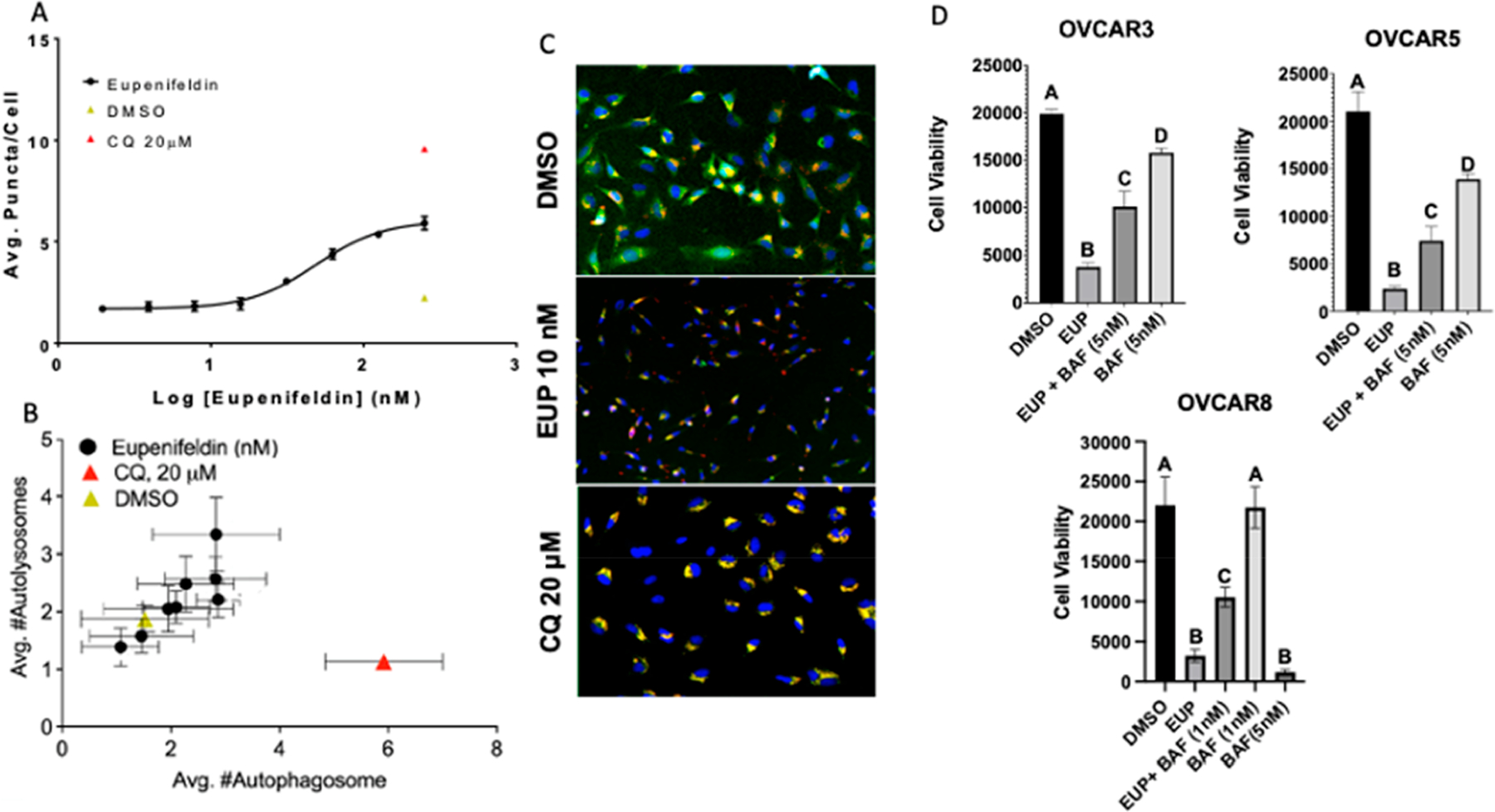
(A) An increasing number of LC3 puncta per HeLa cell were observed with increasing eupenifeldin treatment. All treatments were normalized to the vehicle (DMSO). (B) Autophagic flux assay demonstrates weak induction of autophagy by eupenifeldin. All treatments were normalized to vehicle (DMSO). Autophagy data represent mean ± SEM and three biological replicates, each in duplicate. (C) Representative images of B taken at 10× showing puncta formation indicative of autolysosome formation compared to the late-stage autophagy inhibitor CQ, which induces an accumulation of autophagosomes. (D) Bafilomycin cotreatment shows partial rescue of eupenifeldin-induced cytotoxicity. Data represent mean ± SEM and three biological replicates. Significance was determined with two-way ANOVA with Tukey’s post hoc. Letters represent significantly different treatments, p < 0.05.
The attenuation of eupenifeldin cytotoxicity with bafilomycin cotreatment highlights the role of autophagy in the cytotoxic mechanism of eupenifeldin. Induction of autophagy can serve as either a survival or death signal.18 In this case we hypothesize the induction of autophagy by eupenifeldin to be part of the cytotoxic mechanism, as inhibition by bafilomycin A1 reduces the cytotoxicity. However, the incomplete rescue compared to BAF treatment alone does suggest that the weak induction of autophagy may not fully explain eupenifeldin’s potent cytotoxicity. Further autophagy studies in additional ovarian cancer cell lines such as OVCAR5 and OVCAR8 may be warranted to determine if different cell models of high-grade serous cancer undergo specific cytotoxic mechanisms in response to eupenifeldin.
In conclusion, eupenifeldin is a natural product isolated from fungi with cytotoxic activity in the nanomolar range. In agreement with previous research, we have demonstrated eupenifeldin to be effective in killing several ovarian cancer cell lines in vitro. Eupenifeldin treatment was also cytotoxic to OVCAR3 cells in vivo in a hollow fiber study. Treatment with eupenifeldin causes caspase 3/7 activation in OVCAR3, OVCAR5, and OVCAR8 in addition to an induction of apoptosis in OVCAR3 and OVCAR8. Cleavage of PARP was only detected in OVCAR3, suggesting another cytotoxic mechanism. Proteomics performed in OVCAR3 cells identified 75 differentially regulated proteins, and ferroptosis was the strongest predicted potential cell death pathway based on KEGG clustering. However, ferroptosis was found not to be induced by eupenifeldin treatment. Eupenifeldin was found to modulate autophagy as a weak inducer, and inhibition of autophagy reduced the toxicity of eupenifeldin.
EXPERIMENTAL SECTION
General Experimental Procedures.
Paclitaxel (Taxol), bafilomycin A1, and FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) were obtained from Sigma-Aldrich Corp. The irreversible pan-caspase inhibitor, Z-VAD-FMK, was obtained from Abcam. All compounds were resuspended in DMSO. Primary and secondary antibodies were commercially available. Antibodies were used according to the supplier recommendations. Secondary antirabbit and antimouse antibodies coupled to horseradish peroxidase were obtained from Cell Signaling Technology, Inc. Eupenifeldin (verified >97% pure by HPLC Supporting Information S1.1 and S1.2) was isolated from Neostophoma sp. (strain MSX50044) as described previously.5
Biological Material.
OVCAR3, OVCAR5, and OVCAR8 cells were purchased from the American Type Culture Collection. As previously described, OVCAR3 were grown in RPMI1640 supplemented with 10% FBS and penicillin/streptomycin (final concentration: 100 IU/mL and 100 μg/mL, respectively).19 OVCAR5 and OVCAR8, cells were grown in DMEM with 10% FBS and penicillin/streptomycin.18 Normal immortalized human fallopian tube secretory epithelial cells (hFT33) were generously gifted by Dr. Ronny Drapkin from University of Pennsylvania, Pearlman School of Medicine, Department of Obstetrics and Gynecology. hFT33 cells were grown in DMEM-Ham’s F12 50/50 supplemented with fetal bovine serum (10%).
All cultured cells were mycoplasma free and validated by short tandem repeat analysis in 2021. Cells were passaged a maximum of 20 times and maintained in a humidified incubator at 37 °C in a 5% CO2 environment.
Cytotoxicity Assay.
A total of 5000 cells were seeded in triplicates in 96-well clear, flat-bottom tissue culture-treated plates and allowed to attach overnight. Compounds were dissolved in DMSO (0.05%), diluted to final concentrations, and added to the appropriate wells. Cells were incubated for 72 h, and cell viability was evaluated with a commercially available fluorometric assay (Promega CellTiter-Blue Cell Viability Assay; Promega). IC50 values were expressed in nanomolar relative to vehicle (DMSO) control. Dose–response curves were generated using GraphPad Prism software.
2D Foci Assay.
A total of 200 cells per 600 mm dish were seeded and allowed to attach overnight. Cells were treated with either vehicle (DMSO) control, a known chemotherapeutic agent (paclitaxel, 10 nM), or eupenifeldin (10 nM) for 8 h. Following a 15-day incubation in growth media, cells were fixed with 4% (w/v) paraformaldehyde and stained with 0.05% crystal violet and washed with distilled water. Images were taken with a FluoroChemE system (ProteinSimple, Bio-Techne), and colony formations were quantified and analyzed by ImageJ software (NIH) and normalized to vehicle control.
Wound Healing Assay.
Cells were seeded into a defined cell-free gap monolayer in 24-well clear, flat-bottom plates and allowed to reach 100% confluence overnight. A single, uniform scratch was made down the center of each monolayer with a 1000 μL sterile pipet tip, and cells were gently washed with 1X PBS to remove any detached cells. Treatments occurred with either vehicle (DMSO) control, paclitaxel (10 nM), or eupenifeldin (10 nM or 25 nM) and allowed to incubate for 48 h. Images were captured at 4× magnification at 0 and 48 h. The area of each scratch was analyzed at the different time points with ImageJ software using the Freehand selection tool and normalized to vehicle control using Excel for each treatment.
Annexin V-FITC/Propidium Iodide.
OVCAR5 and OVCAR8 cells were seeded at 175 000 cells per 60 mm dish, and OVCAR3 cells were seeded at 190 000 cells per 60 mm dish and allowed to attach overnight. Cells were treated with either vehicle (DMSO) control, paclitaxel (10 nM), or eupenifeldin (10 nM, 25 nM, or 50 nM) and allowed to incubate for 48 h. Media and cells were collected, washed with 1X PBS, and subjected to annexin V-FITC/propidium iodide assay (Nexcelom Biosciences) according to manufacturer’s protocol, and fluorescence was measured on a K2 Cellometer (Nexcelom Biosciences). Data were analyzed using FCS Express Cytometry Software (De Novo Software, LLC). Gating channels occurred according to the manufacturer’s protocol.
Immunoblot Analysis.
Whole cell lysates were prepared with RIPA lysis buffer (50 mM Tris pH 7.6, 150 mM NaCl, 1% Triton-X-100, and 0.1% SDS) supplemented with phosphatase inhibitors (Sigma-Aldrich) and protease inhibitors (Roche Applied Sciences). Protein concentrations were measured using Bradford assay (Bio-Rad) and resolved on SDS-PAGE gels using a 30 μg protein concentration. Proteins were immobilized onto a nitrocellulose membrane and saturated in either 5% nonfat milk in TBST or 5% BSA in TBST for 30 min. Membranes were probed with the appropriate primary antibody at 4 °C overnight. Membranes were washed with TBST 3X for each and incubated overnight in secondary antibody (horseradish peroxidase-conjugated IgG). Proteins were visualized via a FluoroChemE system (ProteinSimple, Bio-Techne).
Autophagy and Cytotoxicity Assays.
Cells (OVCAR3, OVCAR5, and OVCAR8) were seeded at 5000 cells per well in a 96-well clear, flat-bottom plate and allowed to attach overnight. Cells were treated with either vehicle (DMSO) control, positive control (bafilomycin A1 5 nM), or eupenifeldin (25 nM) ± bafilomycin A1 (5 nM). OVCAR8 cells were also tested with 1 nM bafilomycin A1. Incubation was done over 72 h, and then cell viability was measured using Promega CellTiter-Blue Cell Viability Assay (Promega), as previously described.
Caspase-Activity Assay.
Cultured OVCAR3, OVCAR5, and OVCAR8 cells were seeded at 5000 cells per well in a 96-well, flat-bottom plate and allowed to attach overnight. Cells were treated with either vehicle (DMSO) control, positive control (staurosporine, 20 μM) or eupenifeldin (50 nM) ± Z-VAD-FMK (1 μM). All treatments included 2 μM of ViaStain Live Caspase 3/7 Detection for 2D/3D Culture. Cells were incubated with ViaStain Live Caspase 3/7 Detection for 2D/3D Culture for 30 min, and then fluorometric reading occurred on a Celigo iPaas Platform for day 0. Cells were then incubated additionally for 48 h, a fluorometric reading was conducted on the Celigo iPaas Platform, and data were normalized to vehicle control using Excel.
In Vivo Hollow Fiber Assay.
All animals were treated in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and the established Animal Care and Use Committees at the University of Illinois at Chicago (UIC, protocol 19-011). Immunodeficient NCr nu/nu mice purchased from Taconic Biosciences were allowed to grow for 6 to 8 weeks in age. Mice were separated into three groups of eight and housed in temperature- and light-controlled environments under 12:12 h light:dark cycle and provided food and water ad libitum. OVCAR3 and OVCAR8 cells were grown and on day 0 were embedded into sterile conditioned, biocompatible, polyvinylidene fluoride hollow fibers and heat sealed. Paclitaxel and eupenifeldin were both dissolved in a [2% DMSO:48% PEG300:50% H2O] solution and diluted to final concentrations with H2O. Mice were dosed intraperitoneally once per day for 4 days (days 3–6). Treatment group sizes were as follows: six mice received paclitaxel, six received eupenifeldin, and six received vehicle. On day 7, the remaining mice were sacrificed via CO2 inhalation followed by cervical dislocation, and the hollow fibers retrieved. To evaluate the cell viability, a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (Promega) was used.
Autophagic Flux and LC3B Puncta Assay.
HeLa cells with stably expressing mCherry-eGFP (autophagic flux) or stably expressing eGFP-LC3 (LC3 puncta assay) were seeded in 384-well plates and allowed to attach overnight. Test compounds and controls were incubated for 4 h (eGFP-LC3) or 24 h (mCherry-eGFP-LC3) followed by fixation with 4% formaldehyde in PBS for 12 min. Hoechst 33342 (Molecular Probes) or DAPI (4′,6-diamidino-2-phenylindole) was used for nuclear staining, and plates were sealed using a PlateMax Semi-Automatic Plate Sealer (Corning Axygen, Inc.) and FITC filters (autophagic flux and LC3B puncta assay) or Texas Red filter (autophagic flux assay) and imaged at 10× by ImageXpress Micro XLS (Molecular Devices LLC). The assay was quantified by MetaXpress High-Content Image Analysis Software with Transfluor Application Module (Molecular Devices LLC) and CellProfiler Software (Broad Institute of MIT and Harvard). All treatments were normalized to vehicle (DMSO) solvent, and dose–response curves and EC50 values were generated using GraphPad Prism Software.
Ferroptosis Potentiation Assays.
OVCAR3 or HT1080 cells were seeded at 1000 cells per well in 384-well plates. Cells were treated with increasing concentrations of RSL3 or IKE in combination with 2 μM Fer-1, in triplicates. Viability was measured with CellTiterGlo (Promega). Alterations in lipid peroxidation were measured via fluorescence of C11-BODIPY on the green channel by flow cytometry.
TMT-Labeled Proteomic Sample Preparation.
OVCAR3 cells were treated with 25 nM eupenifeldin or vehicle control for 72 h. Cells were then resuspended in TNI buffer (50 mM Tris (pH 7.5), 250 mM NaCl, 0.5% Igepal CA-630, 1 mM EDTA, and protease inhibitor (ThermoFisher Pierce Protease Inhibitor Mini Tablets). Cells were sonicated 3× at 55 kHz 30 s each, with 15 s intervals on ice in between rounds. Supernatant was separated by centrifugation at 18 000g, 4 °C, 20 min. Protein was precipitated with methanol and chloroform and then resuspended in 8 M urea, 100 mM Tris. Protein was quantified using Pierce BCA assay. Protein was digested with trypsin, 1:50 (trypsin to protein) ratio, overnight (~18 h) at 37 °C. Protein was reduced with TCEP (5 mM) and alkylated with 2-chloroacetamide (50 mM). Peptides were desalted using Pierce peptide desalting spin columns. Peptides were speed-vacuumed dried before being redissolved in 0.1% formic acid (FA). Peptides were labeled with TMTsixplex Isobaric Label Reagent (ThermoFisher Scientific) following the manufacturer’s instruction. For each sample, 2 μg of peptide was labeled in each channel. Samples were pooled, dried with speed-vacuum, and then resuspended in 0.1% FA for LC-MS/MS analysis.
Proteomic LC-MS/MS Analysis.
Peptide (500 ng) was injected for LC-MS/MS analysis with a Q-Exactive HF Orbitrap and Dionex Ultimate 3000. Electrospray ionization was applied with a voltage of 1.8 kV. Peptides were separated with an Acclaim PepMap RSLC C18 column (2 μm, 100 Å, 75 μm i.d. × 15 cm). Solvent A (0.1% FA) and solvent B (80% MeCN/0.1% FA) were used as mobile phases for HPLC separation. Peptides were eluted during a 90 min gradient at a flow rate of 0.300 μL/min. The LC gradient was as follows: 1–55% solvent B within 70 min, 55–90% solvent B over 10 min, 1% solvent B for 10 min. MS1 resolution was set to 120 000, and MS2 resolution was set to 60 000 with a scan range of 375–1400 m/z and a normalized collision energy (NCE) of 32.
Proteomic Data Analysis.
Raw data were analyzed using FragPipe (v15.0) software to search MS/MS data against the UniProt Homo sapiens reversed FASTA database (release date March 2018). Trypsin cleavage rule was selected with miss cleavage equal to 2. Methionine oxidation, N-terminal acetylation, and lysine TMT-6plex modification were selected as variable modifications. 2-Chloroacetamide alkylation and N-terminal TMT-6plex modification were selected as fixed modifications. Peptide and protein false discovery rates (FDRs) were set to 1%, and peptide length was set to 7–50 amino acids with a peptide mass range of 500–5000 Da. For isobaric quantification, the TMT-integrator was run with TMT-6plex default settings. Data were normalized using both median centering and variance scaling.
Statistical Analysis.
All data presented are provided as the mean ± the standard error mean with three biological replicates. Biostatistics and graphs were generated using GraphPad PRISM Software (GraphPad Software Inc.). One-Way Analysis of Variance (ANOVA) with Dunnett’s multiple comparison test, compared to vehicle (DMSO) control, was used on 2D-Foci Formation, Wound Healing Assay, annexin-V/propidium iodide stains, immunoblot densitometry, and hollow fiber assay, to generate P-values (*,P < 0.05). P-values of 0.05 or less were considered statistically significant. One-way analysis of variance (ANOVA) with Tukey’s multiple comparison test, compared to vehicle (DMSO) control, was used on the caspase activity assay and cytotoxicity rescue assay to generate P-values (*,P < 0.05). P-values of 0.05 or less were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grant P01 CA125066 from the National Cancer Institute and T32 AT007533 from the National Center for Complementary and Integrated Health of the NIH (Bethesda, MD), and NIH/NCI grant R35CA209896. IVIS imaging was performed using an Xenogen IVIS Spectrum Imager at University of Illinois, Chicago Research Resource. eGFP-LC3, and mCherry-eGFP-LC3 HeLa cells were a gift from Ramnik Xavier from Massachusetts General Hospital.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.3c00186.
Eupenifeldin isolation purity data, hollow fiber assay data, representative annexin V vs PI scatterplot, PARP Western blots, proteomic analysis, and additional ferroptosis potentiation assays (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jnatprod.3c00186
The authors declare the following competing financial interest(s): N.H.O. and C.J.P. are members of the Scientific Advisory Board of Clue Genetics, Inc. N.H.O. is also a member of the Scientific Advisory Boards of Mycosynthetix, Inc. and Ionic Pharmaceuticals, Inc. B.R.S. is an inventor on patents and patent applications involving ferroptosis, cofounded and serves as a consultant to ProJenX, Inc. and Exarta Therapeutics, holds equity in Sonata Therapeutics, serves as a consultant to Weatherwax Biotechnologies Corporation and Akin Gump Strauss Hauer and Feld LLP, and receives sponsored research support from Sumitomo Dainippon Pharma Oncology.
Contributor Information
Amanda C. Maldonado, Chicago Biomedical Consortium, Northwestern University, Evanston, Illinois 60208, United States.
Monica A. Haughan, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States;.
Manead Khin, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States;.
Julia Ekiert, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
Ziwei Zhang, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
Daniel Lantvit, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
Zeinab Y. Al Subeh, Department of Medicinal Chemistry and Pharmacognosy, Jordan University of Science and Technology, Irbid 22110, Jordan;
Herma C. Pierre, Department of Chemistry and Biochemistry, University of North Carolina at Greensboro, Greensboro, North Carolina 27402, United States
Maryna Salkovski, Department of Chemistry, College of Liberal Arts and Sciences, University of Illinois at Chicago, Chicago, Illinois 60607, United States.
Tal Hirschhorn, Department of Biological Sciences, Columbia University, New York, New York 10027, United States.
Yu Gao, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States;.
Cedric J. Pearce, Mycosynthetix Inc., Hillsborough, North Carolina 27278, United States
Brent R. Stockwell, Department of Biological Sciences and Department of Chemistry, Columbia University, New York, New York 10027, United States
Leslie N. Aldrich, Department of Chemistry, College of Liberal Arts and Sciences, University of Illinois at Chicago, Chicago, Illinois 60607, United States;
Nicholas H. Oberlies, Department of Chemistry and Biochemistry, University of North Carolina at Greensboro, Greensboro, North Carolina 27402, United States
Joanna E. Burdette, Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois 60607, United States;
REFERENCES
- (1).Kuroki L; Guntupalli SR BMJ. (Clinical research ed.). 2020, 371, m3773. [DOI] [PubMed] [Google Scholar]
- (2).Mittica G; Ghisoni E; Giannone G; Genta S; Aglietta M; Sapino A; Valabrega G Recent patents on anti-cancer drug discovery. 2018, 13 (4), 392–410. [DOI] [PubMed] [Google Scholar]
- (3).Kurnit KC; Fleming GF; Lengyel E Obstetrics and Gynecology (New York. 1953). 2021, 137 (1), 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Newman DJ; Cragg GM Journal of Natural Products (Washington, D.C.). 2020, 83 (3), 770–803. [DOI] [PubMed] [Google Scholar]
- (5).El-Elimat T; Raja HA; Ayers S; Kurina SJ; Burdette JE; Mattes Z; Sabatelle R; Bacon JW; Colby AH; Grinstaff MW; Pearce CJ; Oberlies NH Org. Lett 2019, 21 (2), 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mayerl F; Gao Q; Huang S; Klohr SE; Matson JA; Gustavson DR; Pirnik DM; Berry RL; Fairchild C; Rose WC Journal of antibiotics. 1993, 46 (7), 1082–1088. [DOI] [PubMed] [Google Scholar]
- (7).Al Subeh ZY; Chu N-Q; Korunes-Miller JT; Tsai LL; Graf TN; Hung YP; Pearce CJ; Grinstaff MW; Colby AH; Colson YL; Oberlies NH J. Controlled Release 2021, 331, 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Haley J; Tomar S; Pulliam N; Xiong S; Perkins SM; Karpf AR; Mitra S; Nephew KP; Mitra AK Oncotarget. 2016, 7 (22), 32810–32820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Berthenet K; Castillo Ferrer C; Fanfone D; Popgeorgiev N; Neves D; Bertolino P; Gibert B; Hernandez-Vargas H; Ichim G Cell Reports (Cambridge) 2020, 31 (10), 107731–107731. [DOI] [PubMed] [Google Scholar]
- (10).Nakajima Y; Kuranaga E Cell Death and Differentiation. 2017, 24 (8), 1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lamkanfi M; Festjens N; Declercq W; Vanden Berghe T; Vandenabeele P Cell Death and Differentiation. 2007, 14 (1), 44–55. [DOI] [PubMed] [Google Scholar]
- (12).Conde-Rubio M; Mylonas R; Widmann C Cell Death Discovery. 2021, 7 (1), 164–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shi ZZ; Fan ZW; Chen YX; Xie XF; Jiang W; Wang WJ; Qiu YT; Bai J Onco Targets Ther. 2019, 12, 11291–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yang WS; Stockwell BR Trends in Cell Biology. 2016, 26 (3), 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hou W; Xie Y; Song X; Sun X; Lotze MT; Zeh HJ; Kang R; Tang D Autophagy. 2016, 12, 1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chen Q; Pan L; Kinghorn AD; Swanson SM; Burdette JE BMC Cancer. 2016, 16 (17), 17–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Aldrich LN; Kuo SY; Castoreno AB; Goel G; Kuballa P; Rees MG J. Am. Chem. Soc 2015, 137 (16), 5563–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Singh SS; Vats S; Chia AY; Tan TZ; Deng S; Ong MS; Arfuso F; Yap CT; Goh BC; Sethi G; Huang RY; Shen HM; Manjithaya R; Kumar AP Oncogene. 2018, 37 (9), 1142–1158. [DOI] [PubMed] [Google Scholar]
- (19).Mitra AK; Davis DA; Tomar S; Roy L; Gurler H; Xie J; Lantvit DD; Cardenas H; Fang F; Liu Y; Loughran E; Yang J; Sharon Stack M; Emerson RE; Cowden Dahl KD; Barbolina MV; Nephew KP; Matei D; Burdette JE Gynecologic Oncology. 2015, 138 (2), 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.