

Abstract
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) functions as a key regulator in inflammation and cell death and is involved in mediating a variety of inflammatory or degenerative diseases. A number of allosteric RIPK1 inhibitors (RIPK1i) have been developed, and some of them have already advanced into clinical evaluation. Recently, selective RIPK1i that interact with both the allosteric pocket and the ATP-binding site of RIPK1 have started to emerge. Here, we report the rational development of a new series of type-II RIPK1i based on the rediscovery of a reported but mechanistically atypical RIPK3i. We also describe the structure-guided lead optimization of a potent, selective, and orally bioavailable RIPK1i, 62, which exhibits extraordinary efficacies in mouse models of acute or chronic inflammatory diseases. Collectively, 62 provides a useful tool for evaluating RIPK1 in animal disease models and a promising lead for further drug development.
Key words: RIPK1, Necroptosis, Type-II kinase inhibitors, Rational design, Lead optimization, Structure‒activity relationship, Anti-inflammation, Preclinical drug discovery
Graphical abstract
Based on the rediscovery of a reported RIPK3 inhibitor (GSK’840) and structure-guided lead optimization, a potent, selective, and orally bioavailable type-II kinase inhibitor of RIPK1 (62) has been developed.

1. Introduction
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) plays a key role in inflammatory response and programmed cell death. It is well known as a master regulator in the TNFR1-mediated activations of the pro-survival NF-κB, the pro-apoptotic FADD-caspase-8, and the pro-necroptotic RIPK3-MLKL signal pathways1. The kinase activity of RIPK1 has been demonstrated to mediate or aggravate the pathological conditions in a broad set of inflammatory or degenerative diseases, such as sepsis, ischemia, rheumatoid arthritis (RA), inflammatory bowel disease (IBD), psoriasis, multiple sclerosis, amyotrophic lateral sclerosis, and severe COVID-192,3. Thus, RIPK1 is considered a therapeutic target with great potential, and plenty of RIPK1 inhibitors (RIPK1i) have been developed in the past decade2,4,5.
Facilitated by the robust necroptotic phenotypes induced by TNFα, a large part of RIPK1i was developed based on cell-based high-throughput screening (HTS). Examples of them include Nec-1s (1)6,7, the first RIPK1i widely used to interrogate RIPK1 in diverse disease models, GSK2982772 (2)8, the first RIPK1i to enter clinical trials, RIPA-56 (3), GSK’547, and Nec-34 (Fig. 1)9, 10, 11. To obtain more optimized RIPK1i, various analogs of 2 had been generated, such as the Takeda compound 22 (4), GNE684, R552, and ZB-R-552,4,12, 13, 14, 15. Structural studies discovered that most selective RIPK1i bind to an allosteric and hydrophobic pocket adjacent to the ATP-binding pocket of RIPK1, rending RIPK1 in an uncommon DLG-out/Glu-out inactive conformation16,17. Additional interactions with the ATP-binding pocket could significantly improve the potency of 2, especially against mouse RIPK1 (mRIPK1)14,15. These findings indicated that RIPK1i could be developed as bona fide type-II kinase inhibitors (KI) with further improved potency and retained selectivity by concomitantly occupying both binding pockets of the targeted kinases, which could provide novel pharmacophores and potentially advantageous candidates for current clinical drug discovery18. Early-stage type-II RIPK1i had already been discovered based on HTS (5) or structure-guided hybridization (6), despite notable flaws17,19,20. Recently, more optimized RIPK1i, such as Zharp1-211 (7) and RI-962, had been successfully developed21, 22, 23, further supporting the potentials of type-II RIPK1i.
Figure 1.

Chemical structures of representative RIPK1i (A) or RIPK3i (B).
Herein, we report the discovery of a series of potent and selective type-II RIPK1i based on a known RIPK3i and structure-guided rational design. RIPK3 is a major downstream effector and close homolog of RIPK1, but it does not possess the structurally unique hydrophobic pocket as RIPK124. Two selective RIPK3i, GSK’843 (8) and GSK’872 (9), were reported to induce apoptosis, with 8 confirmed as a type-I KI based on co-crystal structure determination (Fig. 1)25. In our preliminary studies, 9 could be readily docked into the co-crystal structure of 8, whereas another RIPK3i, GSK’840 (10), was incompatible with the binding mode of 8, and did not induce apoptosis. These findings led us to speculate that 10 could be a type-II KI and encouraged us to optimize 10 using classical strategies to develop type-II KI. However, initial modification efforts on 10 serendipitously resulted in activity against RIPK1 other than improved potency against RIPK3, eventually leading to the development of bona fide type-II RIPK1i in this work. We then discuss the structure‒activity relationship (SAR) of these inhibitors and describe the lead optimization of 62, an orally bioavailable and tolerable type-II RIPK1i, which exhibited extraordinary efficacies in RIPK1-dependent acute and chronic inflammation animal models.
2. Results and discussion
Both RIPK3i 9 and 10 were tested for their protective effects against the cell death of L929 mouse fibroblasts induced by TNFα treatments (Fig. 2A). Consistent with the literature25, treatments with 9 led to aggravated cell death reportedly via induction of RIPK3-dependent apoptosis. In contrast, treatments with 10 remarkably protected the L929 cells from cell death caused by TNFα, indicating that 10 might inhibit RIPK3 through a different mechanism compared to 9. Based on the inconsistent docking results and phenotypic differences in the context of cell death between 9 and 10, we speculated that 10 might be a type-II KI of RIPK3, with its tert-butyl ester moiety interacting with the hydrophobic pocket of RIPK3 near its DFG-motif18. Then, a meta-trifluoromethylaniline-based amide moiety commonly adopted by type-II KI was introduced to replace the tert-butyl ester moiety of 10, yielding a more prototypical type-II inhibitor 11 (Fig. 2B). Replacing the phenylacetamide linkage of 11 with phenylurea or benzamide provided two analogs, 12 and 13.
Figure 2.

Identification of RIPK1/3 dual inhibitors. (A) Bar graph showing the protective effects of RIPK3i and derivatives in L929 cells. Cells were treated with TNFα at 40 ng/mL in the presence of DMSO or compounds for 15 h before the cell viability test. (B) The chemical structure of the derivatives of 10. (C) Dose–response curves showing the protective effects of each compound compared to DMSO in HT-29 cells stimulated by TNFα (10 ng/mL), SM164 (25 nmol/L) and zVAD (25 μmol/L) for 24 h. (D) HT-29 cells stimulated by TNFα, SM164, and zVAD in the presence of DMSO or compounds at 5 μmol/L for 4 h, then analyzed by Western blotting. (E) Bar graph showing the anti-necroptotic effects of each compound at 10 μmol/L in FKBP-RIPK3-expressing NIH-3T3 cells. Cells were treated with the FKBP-dimerizing agent, AP20187 (100 nmol/L), in the presence of DMSO or compounds for 15 h before the viability test. All data represent mean ± SD (n = 3); ∗∗P < 0.01, ∗∗∗P < 0.001, unpaired Student's t-test.
Both 11 and 12 showed protective effects similar to 10 in L929 cells, while 13 was relatively weaker (Fig. 2A). These compounds also protected HT-29 human colon cancer cells against necroptosis induced by a cocktail of TNFα, SM164 (cIAP1/2 inhibitor) and zVAD (caspase inhibitor) (Fig. 2C). The Western blot results showed that, in the stimulated HT-29 cells, 11 and 12 at 5 μmol/L comparably diminished the phosphorylation of RIPK3 and the downstream MLKL as 9 and 10 did, confirming their inhibition of the necroptosis pathway (Fig. 2D). Surprisingly, 11 and 12 also diminished the phosphorylation of the upstream effector RIPK1, whereas 9 or 10 did not at all. On the other hand, 9 and 10 fully protected FKBP-RIPK3-expressing NIH-3T3 cells from necroptosis caused by AP20187, a compound capable of inducing FKBP dimerization and the consequent RIPK3 activation26, whereas 11 and 12 only exerted partial protection (Fig. 2E). These results indicated that 11 and analogs are RIPK1/3 dual inhibitors.
11 was found to be comparably potent as 1 against mRIPK1 with sub-micromolar EC50 values to protect L929 cells from necroptosis. However, it was almost inactive in the FADD-deficient (FADD−/−) Jurkat human T lymphocyte cells, indicating that it is rather weak against human RIPK1 (hRIPK) (Table 1). To optimize the potency against hRIPK1, an N-benzylhydroxylamine group was adopted from a reported RIPK1i, 3, to replace the general type-II aniline tail, which only slightly improved the potency. Then, the amide bond of 11 was reversed and the triazole moiety of 2 was adopted, which unexpectedly led to a complete loss of potency (15). Nevertheless, replacing the 1H-1,2,4-triazole ring of 15 with a 1,2,4-oxadiazole ring yielded an inhibitor more selective for hRIPK1, 16, whereas 17 with a 1,3,4-oxadiazole ring was less potent. 18 with an oxazole ring was better than 17, while 19 with an isoxazole ring was even better than 16. Replacing the isoxazole with a N-methyl pyrazole (20) slightly increased the potency of 19. On the contrary, 21, the isomer of 20, was completely inactive. Notably, increases in the size of the N-substituents on the pyrazole were less favored (22, 23). On the other hand, 24 with a 1,2,3-triazole ring showed moderate potency, indicating the naked pyrazole NH of 15 to be disfavored. 25 with a tetrazole ring was even more potent than 24. However, these analogs of 14 by far, all with a benzyl group located at the meta-position on the 5-membered heterocycles, were all weak against mRIPK1. In contrast, 26 with a 5-fluoropyrrole ring and an ortho-N-benzyl exhibited outstanding potency against both hRIPK1 and mRIPK1. Consistently, the bioisosteric 27 with an N-methyl pyrazole and an ortho-benzyl showed even better potency.
Table 1.
Optimization of the tail subunits (R)a.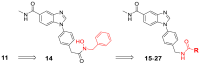
| Compd. | R | EC50 (nmol/L)b | |||
| FADD−/− Jurkat | L929 | ||||
| TNFα | T + S | TNFα | T + S | ||
| 1 | 94 | 590 | 112 | 689 | |
| 11 | >10,000 | >10,000 | 190 | 713 | |
| 14 | 3805 | >10,000 | 50 | 98 | |
| 15 | 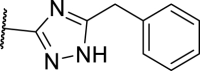 |
>10,000 | >10,000 | >10,000 | >10,000 |
| 16 |  |
24 | 176 | 962 | 4735 |
| 17 | 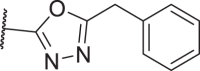 |
185 | 1516 | >10,000 | >10,000 |
| 18 | 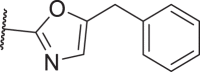 |
70 | 380 | 831 | 3975 |
| 19 |  |
8.1 | 113 | 2138 | 3348 |
| 20 |  |
5.1 | 44 | 312 | 2022 |
| 21 | 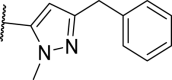 |
>10,000 | >10,000 | >10,000 | >10,000 |
| 22 | 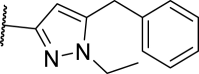 |
5.9 | 43 | n.d. | >10,000 |
| 23 | 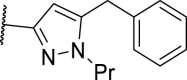 |
109 | 738 | 3007 | >10,000 |
| 24 |  |
233 | 1594 | >10,000 | >10,000 |
| 25 | 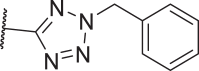 |
24 | 150 | 1106 | 4310 |
| 26 |  |
1.7 | 17 | 6 | 34 |
| 27 |  |
0.5 | 5.2 | 20 | 77 |
Data represent means of three replicates.
Cell viability assays, T = TNFα, S = SM164.
To better understand the pharmacophore of our RIPK1i, we determined the co-crystal structure of the RIPK1‒20 complex at 2.3 Å resolution (PDB 8I2N) (Fig. 3 and Supporting Information Table S1). Unlike 1 and 2 which interact mainly with the hydrophobic pocket adjacent to the DLG-motif of RIPK1 (Fig. 3A), 20 interacts with both the hydrophobic pocket and the ATP-binding pocket (Fig. 3B). Its benzylpyrazole ‘tail’ occupies the allosteric pocket, forcing RIPK1 into a DLG-out/inactive conformation18. Notably, the α-helix of RIPK1 adopts a “Glu-out” conformation upon binding to 20, just as it does upon binding to 1 or 2. In comparison, a previously reported type-II RIPK1i, 5, binds to RIPK1 in a DLG-out but Glu-in conformation (Fig. 3C), just as a promiscuous type-II KI, ponatinib, binds to RIPK227, or a reported type-II RIPK3i binds to RIPK3 (Supporting Information Fig. S1)28. On the other hand, the phenylacetamide linker of 20 helps position its benzimidazole ‘head’ toward the hinge region of RIPK1. Two hydrogen bonds (H-bonds) are formed between 20 and RIPK1: one between the linker carbonyl of 20 and the amide NH of Arg156 in the DLG-motif; another between a nitrogen atom of 20's benzimidazole head and the amide NH of Met95 in the hinge (Fig. 3D). Additionally, each subunit of 20 interacts with several surrounding residues through van der Waals forces (Fig. 3E). An intramolecular H-bond may also form a pseudo-5-membered ring within 20 between the linker amide NH and the 2-nitrogen atom in the pyrazole ring.
Figure 3.

Determination of the co-crystal structure of RIPK1‒20 complex. (A) Superimposition of the co-crystal structures of RIPK1 (green ribbons) in complex with 1 (purple sticks, PDB 4ITH) or 2 (grey sticks, PDB 5TX5). (B) The co-crystal structure of RIPK1 (green ribbons) in complex with 20 (gold sticks, PDB 8I2N). (C) Superimposition of the co-crystal structures of RIPK1‒20 complex (green ribbons and gold sticks) and RIPK1–5 complex (gray ribbons and sticks, PDB 4NEU). (D) Cartoon showing interactions between 20 and the ATP-binding pocket of RIPK1, key residues were shown as sticks, and H-bonds were indicated as red dashes. (E) The residue side chains of RIPK1 potentially interacting with 20 through van der Waals forces.
Consistent with the binding mode, replacing the benzimidazole of 20 with an indole (28) or indazole (29) weakened the hinge H-bond and led to the loss of activity (Table 2). 27 was found to be more potent than 20, especially against mRIPK1. Therefore, optimization efforts were then focused on the scaffold of 27. As the N-methyl amide group of 27 appeared to protrude toward the solvent phase, more hydrophilic groups such as 2-(dimethylamino)ethyl (30), 2-(4-methylpiperazin-1-yl)ethyl (31) and 2-morpholinoethyl (32) were introduced into 27. However, these analogs were less effective against mRIPK1. In contrast, introducing a hydrophobic methoxy group (33–35) considerably increased the potency against both hRIPK1 and mRIPK1 but caused cytotoxicity at high concentrations. Weakening the hinge H-bond of 34 resulted in a dramatic decrease in potency (36), and the quinazoline head (37) was also less potent than the benzamide head. We hypothesized that adding an H-bond donor in the head subunit of 27 could increase its potency. However, introducing a pyrrolopyrimidine head resulted in loss of activity (38). Inspired by the structure of ATP, we introduced an adenine head to 27 and discovered 39, which had single-digit nanomolar or subnanomolar IC50 values in both human and mouse cell models. Further modifications to the adenine head of 39 by changing the pyrimidine ring into a pyridine ring (40) or removing the H-bond acceptor (41) only decreased its potency. Consistently, introducing an adenine head to 21 provided a more potent inhibitor (43), and removing nitrogen atoms from the adenine of 43 (44–46) decreased its potency.
Table 2.
Optimization of the head subunits (R) based on 20 and 27a.
| Compd. | R |
EC50 (nmol/L)b |
Compd. |
R |
EC50 (nmol/L)b |
||||||
| FADD–/– Jurkat |
L929 |
FADD–/– Jurkat |
L929 |
||||||||
| TNFα |
T+S |
TNFα | T+S |
TNFα |
T+S |
TNFα | T+S |
||||
| 20 |  |
5.1 | 44 | 312 | 2022 | 37 |  |
233 | 1503 | n.d. | n.d. |
| 27 |  |
0.5 | 5.2 | 20 | 77 | 38 | 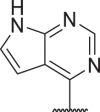 |
152 | 747 | >10,000 | >10,000 |
| 28 |  |
>10,000 | >10,000 | >10,000 | >10,000 | 39 |  |
0.04 | 1 | 0.9 | 3.6 |
| 29 |  |
>10,000 | >10,000 | >10,000 | >10,000 | 40 | 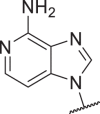 |
0.3 | 2.3 | 1.2 | 6.3 |
| 30 |  |
53 | 368 | >10,000 | >10,000 | 41 | 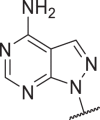 |
0.4 | 6.9 | 9.9 | 23 |
| 31 | 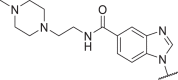 |
0.01 | 0.6 | 5632 | >10,000 | 42 | 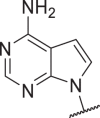 |
4.2 | 8 | 0.1 | 0.9 |
| 32 | 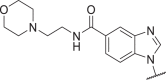 |
0.5 | 3 | 32 | 190 | 43 | 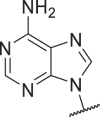 |
0.06 | 0.3 | 25 | 384 |
| 33c |  |
0.02 | 0.1 | 0.06 | 0.4 | 44 | 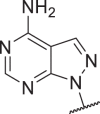 |
71 | 451 | 6548 | >10,000 |
| 34c | 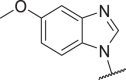 |
0.002 | 0.1 | 0.005 | 0.08 | 45 | 137 | 980 | 2887 | >10,000 | |
| 35c | 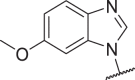 |
0.01 | 0.6 | 0.03 | 0.4 | 46 | 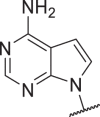 |
361 | >10,000 | 2316 | >10,000 |
| 36 | 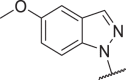 |
18 | 128 | n.d. | n.d. | 1 |  |
94 | 590 | 112 | 689 |
| 2 | 0.5 | 3.9 | 1195 | 8279 | |||||||
Data represent means of three replicates.
Cell viability assays, T = TNFα, S = SM164.
With cytotoxicity at high concentrations.
In this round of optimization, 39 exhibited extraordinary potency against both hRIPK1 and mRIPK1. To elucidate the SAR, we conducted a molecular docking study using the co-crystal structure of hRIPK1-20 (Fig. 4). The results predicted a reliable binding mode for 39 that was highly similar to that of 20, with a docking score of −13.6. In this binding mode, the adenine head of 39 forms two H-bonds with Met95 in the hinge region of RIPK1 (Fig. 4A). The phenyl ring in the tail subunit of 39 is positioned exactly where that of 20 is located, resulting in distinct orientation for the benzyl methylene (Fig. 4B). While the N-methyl of 39 occupies the same position as the methylene of 20 did, the methylene of 39 could interact with Ala155 of RIPK1 and contribute to stronger affinity. Compared to 20, 43 shares the same head subunit as 39 but was less effective against mRIPK1. This indicated the sensitivity of mRIPK1 to be more dependent on the tail subunit of 39. To investigate why this occurs, we compared the amino acid residue sequences of mouse and human RIPK1 (Fig. 4B). Most residues around the tail subunit of inhibitors are identical between both homologs; however, mRIPK1 has a DLGV-motif instead of hRIPK1's DLGL-motif, which could result in differences in sensitivity.
Figure 4.

The predicted binding mode of 39 (azure sticks) based on the RIPK1-20 (yellow sticks) co-crystal structure. The docking study predicted an additional H-bond (red dash) for 39 with the hinge region of RIPK1 (A), and similar but distinct interactions for the tail subunit of 39 compared to that of 20 (B). In panel B, the structure of mRIPK1 (gray ribbons and sticks) was aligned with that of hRIPK1 (green ribbons and sticks).
Based on 39, we further studied the SAR of the tail subunit (Table 3). Consistent with those of 20, larger N-substitution in the pyrazole ring led to a decrease in potency (47). To stabilize conformations favored by RIPK1 through an intramolecular H-bond similar to that of 20, we introduced a 3-chloro to the scaffold of 39 and obtained 48 with comparable potency. Moreover, replacing the pyrazole ring with a 1,2,3-triazole ring led to a significant increase in potency (49). Docking results predicted a binding mode for 49 similar to that of 39 with an intramolecular H-bond and a docking score of −13.8 (Fig. 5A). Notably, removing the N-methyl from 49 resulted in a serious loss of potency. On the other hand, 51 with a bioisosteric 1-methyl-5-fluoro-1H-pyrrole ring showed comparable potency to 39. However, analogs with larger substituents at 5-position (52, 53) showed decreased potency.
Table 3.
Optimization of the subunit R based on 39a.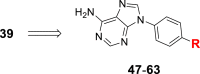
| Compd. | R | EC50 (nmol/L)b | Compd. | R | EC50 (nmol/L)b | ||||||
| FADD−/− Jurkat | L929 | FADD−/− Jurkat | L929 | ||||||||
| TNFα | T + S | TNFα | T + S | TNFα | T + S | TNFα | T + S | ||||
| 39 |  |
0.04 | 1 | 0.9 | 3.6 | 56 | 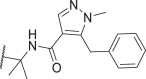 |
7.5 | 28 | 550 | 6964 |
| 47 | 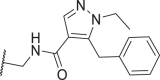 |
1.2 | 35 | 22 | 74 | 57c | 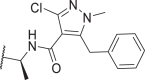 |
0.09 | 0.3 | 0.3 | 0.4 |
| 48 | 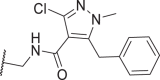 |
0.2 | 0.7 | 0.5 | 4.6 | 58c | 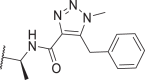 |
<0.01 | <0.01 | <0.01 | <0.01 |
| 49 |  |
0.06 | 0.5 | 0.03 | 0.06 | 59 |  |
0.2 | 2.3 | 8.4 | 39 |
| 50 | 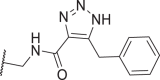 |
2.8 | 273 | n.d. | n.d. | 60 | 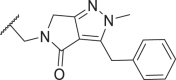 |
0.05 | 2.9 | 4.8 | 116 |
| 51 |  |
0.1 | 1.6 | 0.9 | 74 | 61 |  |
0.3 | 0.4 | 1390 | 2729 |
| 52 | 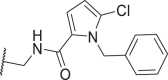 |
6.6 | 80.6 | n.d. | n.d. | 62 |  |
0.02 | 0.1 | 0.2 | 1.3 |
| 53 |  |
214 | 1860 | 875 | 3770 | 63 |  |
0.03 | 0.4 | 0.3 | 1.6 |
| 54 | 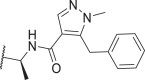 |
0.08 | 0.3 | 0.4 | 0.6 | 1 | 94 | 590 | 112 | 689 | |
| 55 |  |
36 | 132 | 52 | 219 | 2 | 0.5 | 3.9 | 1195 | 8279 | |
Data represent means of three replicates.
Cell viability assays, T = TNFα, S = SM164.
With cytotoxicity at high concentrations.
Figure 5.

The predicted binding mode of 49 (orange sticks) and 62 (magenta sticks) based on the RIPK1-20 co-crystal structure, which indicates an intramolecular H-bond (red dash) in 49 (A) and a highly similar conformation for 62 (B).
In addition to optimizing the pyrazole tail of 39, we also attempted to optimize its linker (Table 3). Our results showed that (S)-methylation of the linker methylene (54) further improved the potency of 39, whereas (R)-methylation (55) or di-methylation (56) led to opposite effects. Combining the intramolecular H-bond with the (S)-methyl feature resulted in even stronger potency but also increased cytotoxicity at higher concentrations (57, 58). Inspired by the bicyclic tail of 4 with more favorable pharmacokinetic (PK) properties12, we introduced a fused 6-membered ring to replace the pseudo-5-membered ring of 49. The resulting compound, 59, showed slightly weaker potency against mRIPK1 than its predecessor. Its 5,5-bicyclic analog, 60, also exhibited weaker potency. In contrast, 61 with a bicyclic tail more similar to that of 4 had a much weaker effect against mRIPK1. After adding a (S)-methyl to 59 and 60, we obtained 62 and 63 with cellular potencies between those of 39 and 49. Docking results predicted 62 binds similarly to 49, with a docking score of −14.587 (Fig. 5B). In this binding mode, the 6,5-bicyclic tail of 62 adopts a similar orientation to the pseudo-5,5-bicyclic tail of 49. Additionally, the (S)-methyl of 62 potentially interacts with Leu159 of RIPK1 via hydrophobic effects, which could explain its increased potency. So far in the cellular assays, several of our inhibitors have shown significantly better potency against RIPK1‒especially mRIPK1‒compared to many reported RIPK1i.
The RIPK1i with outstanding cellular potency were further assessed for their stability in liver microsomes (Fig. 6A). 20 and 27, which share the same benzamide head, showed good stability in both mouse or human liver microsomes (MLM/HLM), with half-lives (t1/2) around 40 min. In contrast, 33 and 34 with more hydrophobic heads showed moderate stability in HLM and rather short t1/2s in MLM. Meanwhile, 39 with an adenine head showed good stability in both HLM and MLM. However, its close analogs with fewer nitrogen atoms, 40 and 42, were significantly less stable. For the tail subunit, adding a chlorine (48) or one more nitrogen atom (49) to 39 remarkably improved the MLM stabilities, whereas the pyrrole tail (51) was much less stable. Nevertheless, (S)-methylation (54) and cyclization (62, 63) barely affected the stability of 39.
Figure 6.

In vitro characterization of lead compounds. (A) Metabolic stability of selected RIPK1i in MLM and HLM. (B) Representative inhibition curves and IC50 values for lead compounds against hRIPK1 or hRIPK3 based on enzymatic assays. (C) Kinase selectivity profiles for 62 at 100 nmol/L against a diverse panel of 468 kinases. Scores for primary screen hits are reported as a percent of the DMSO control (% control), and the lower score indicated a higher probability of being a hit. (D) IC50 values for lead compounds against a panel of CYPs and hERG. Data represent means of three replicates; n.d., not detected.
Next, inhibitors that were stable in microsomes were selected for evaluation in an in vitro hRIPK1 kinase activity assay (Fig. 6B). In this assay, 1 and 2 were used as controls with IC50 values of 445 and 10.2 nmol/L, respectively. 39 was found to be comparably active to 2 with an IC50 of 9.5 nmol/L, while 49 and 54 were slightly more potent with IC50 values around 6 nmol/L 62 was found to be threefold more potent than 2 against hRIPK in vitro, with an IC50 of 3.5 nmol/L. Meanwhile, 62 was at least 200-fold less potent against hRIPK3 with an IC50 of 730 nmol/L, while 39, 49 and 54 were even weaker with IC50 values over 1 μmol/L (Fig. 6B).
62 was then profiled against a diverse panel of 468 kinases using an in vitro ATP-site competition binding assay29,30 at a concentration of 100 nmol/L, and exhibited good overall selectivity among the kinome (Fig. 6C and Supporting Information Fig. S2). The profiling results suggested that RIPK1 was the top hit and that TRKA and DDR1/2 could potentially be off-targets for 62. To assess the inhibitory effects of our RIPK1i against TRKA in cellular contexts, 62 and 63 were tested for their antiproliferative abilities in engineered Ba/F3 cells whose proliferation is dependent on a TEL-TRKA fusion (Fig. S2A). The results showed that 62 was relatively weak against TRKA with an IC50 of 634 nmol/L, while 63 was almost inactive against TRKA (IC50 > 1000 nmol/L). On the other hand, reported siRNA screening results indicated that knockdown of neither TRKA nor DDR1/2 could affect the necroptotic pathway31, consistently, inhibition of DDR1/2 with a reported inhibitor32 showed no effect on necroptosis in cells (Fig. S2B).
These inhibitors were also preliminarily evaluated for potential drug–drug interactions and hepatoxicity. In a panel of selected cytochrome P450 (CYP) enzymatic assays, they were inactive against most CYPs with IC50 values over 10 μmol/L, except that 39 and 49 weakly inhibited CYP2C8 (Fig. 6D). At a concentration of 50 μmol/L, these inhibitors showed negligible cytotoxic effects in the HepG2 human hepatocellular carcinoma cells compared to 1 and 2 (Fig. S2C). Additionally, these inhibitors were assessed for potential cardiotoxicity in a hERG safety assay, and all were found to be inactive with IC50 values over 30 μmol/L (Fig. 6D). Overall, 62 demonstrated satisfactory selectivity for RIPK1 both in vitro and in cells.
To evaluate its inhibitory effects on the necroptotic signaling downstream of TNFα, 62 was titrated in FADD−/− Jurkat and L929 cells stimulated with TNFα and SM164 (Fig. 7A). Western blot results showed that in the FADD−/− Jurkat cells, 62 at 8 nmol/L was able to abolish the stimulated phosphorylation and activation of RIPK1 and MLKL, and eliminated most phosphorylation of RIPK3. In comparison, 2 was slightly weaker than 62, and 1 was the least potent with effective concentrations at a submicromolar level. In L929 cells, 62 almost completely blocked the phosphorylation cascade at 40 nmol/L. In comparison, 1 at 1 μmol/L showed similar effects whereas 2 became inactive even at 1 μmol/L 62 was also tested in the RIPK3-dimerization-induced necroptosis assay and showed no protective effects at all, confirming that RIPK1 is its sole target in the necroptotic pathway (Fig. 7B). On the other hand, 62 at 5 μmol/L completely abolished the activation of RIPK1 in the human lung organoids infected with SARS-CoV-2, and exerted very strong antiviral effects compared to reported RIPK1i (Fig. 7C). Collectively, 62 potently and selectively inhibited the kinase activity of RIPK1 in various cell types.
Figure 7.

Interrogating RIPK1 in cellular models with 62. (A) Human FADD−/− Jurkat or mouse L929 cells were treated with TNFα (40 ng/mL) and SM164 (25 nmol/L) for 15 h in the presence of 1, 2, or 62 at indicated concentrations, then lysed and followed by Western blotting analysis using species-specific antibodies. (B) Bar graph showing the anti-necroptotic effects of each compound at 10 μmol/L in mouse 3T3 cells engineered with an FKBP-RIPK3-fusion. Cells were treated with the FKBP-dimerizing agent, AP20187 (100 nmol/L), in the presence of DMSO or compounds for 15 h before the viability test. Data represent means ± SD (n = 3); ∗∗P < 0.01, ∗∗∗P < 0.001, unpaired Student's t-test. (C) Human lung organoids were infected with SARS-CoV-2 (0.3 MOI) for 1 h, then washed out with PBS and cultured with normal medium and indicated compounds (5 μmol/L) for an additional 48 h, then lysed and followed by Western blotting analysis. The viral loads of SARS-CoV-2 were indicated by its N protein levels.
To select a suitable inhibitor for animal studies, we evaluated the PK properties of selected inhibitors in mice following intravenous (iv) and oral (po) delivery (Table 4). 39, 49, 54, and 63 showed overall comparable PK properties following an oral dose at 10 mg/kg (mpk). Their t1/2s ranged from 0.90 to 1.65 h, maximum concentrations (Cmax) ranged from 714 to 1338 μg/L, area under the curve (AUC) values ranged from 1695 to 4110 μg h/L, and bioavailabilities ranged from 25.4% to 37.0%. In comparison, 48 at 10 mpk (po) showed more optimized PK properties with remarkably higher Cmax, AUC, and bioavailability parameters. 62 at 5 mpk (po) was even better with the longest t1/2 (3.86 h) and the highest bioavailability (82.2%). Additionally, 62 showed a favorable PK profile in rats as well with a t1/2 of 4.19 h, a Cmax of 505 μg/L, an AUC of 2202 μg·h/L, and a bioavailability of 34.1% following an oral dose at 5 mpk (Table 4). Taken together, these results suggested that 62 is a more optimal choice for further animal studies.
Table 4.
PK parameters of optimized RIPK1ia.
| Compd. | Route |
t1/2 |
Tmax |
Cmax |
AUC0–t |
AUC0–inf |
CL |
MRT0–inf |
Vss |
F |
|---|---|---|---|---|---|---|---|---|---|---|
| (h) | (h) | (μg/L) | (h·μg/L) | (h·μg/L) | (L/h/kg) | (h) | (L/kg) | (%) | ||
| 39b | po | 1.34 | 0.33 | 1338 | 2190 | 2210 | 2.55 | 25.4 | ||
| iv | 0.81 | 862 | 882 | 1.1 | 2.08 | 2.35 | ||||
| 48b | po | 1.39 | 0.42 | 3053 | 8276 | 8395 | 4.05 | 63.9 | ||
| iv | 0.81 | 1295 | 1314 | 0.8 | 1.97 | 1.55 | ||||
| 49b | po | 1.65 | 0.67 | 1218 | 4110 | 4334 | 5.75 | 33.3 | ||
| iv | 1.03 | 1235 | 1415 | 0.7 | 10.50 | 6.98 | ||||
| 54b | po | 0.90 | 0.50 | 1200 | 2437 | 2485 | 3.05 | 37.0 | ||
| iv | 0.44 | 659 | 672 | 1.5 | 1.98 | 2.98 | ||||
| 62c | po | 3.86 | 0.50 | 1977 | 6501 | 6517 | 2.81 | 82.2 | ||
| iv | 1.29 | 471 | 476 | 0.6 | 1.49 | 0.94 | ||||
| 63b | po | 1.05 | 0.67 | 714 | 1695 | 1726 | 2.75 | 26.4 | ||
| iv | 0.61 | 643 | 644 | 1.6 | 0.97 | 1.60 | ||||
| 62d | po | 4.19 | 0.50 | 505 | 2202 | 2253 | 6.82 | 34.1 | ||
| iv | 0.91 | 390 | 397 | 0.8 | 1.03 | 0.77 |
Data represent means of three replicates.
Mice administered at 10 mpk (po) or 1 mpk (iv).
Mice or.
rats administered at 5 mpk (po) or 0.5 mpk (iv).
In the end, we evaluated the efficacy and tolerability of 62 in mouse models of RIPK1-dependent acute or chronic inflammatory disease (Fig. 8). In a TNFα-induced systemic inflammatory response syndrome (SIRS) mouse model, 62 at 2 mpk (po) completely mitigated the acute hypothermia in mice and almost eliminated the symptoms even at the dose of 1 mpk (Fig. 8A). Furthermore, 62 at 1 mpk (po) protected all mice from the fatality while 6 out of 7 mice from the control group died of SIRS within 36 h. The pharmacodynamic study results showed that treatments with 62 at 1 mpk almost eliminated TNFα-induced activation of RIPK1 in mouse organs (Supporting Information Fig. S3). Meanwhile, the phosphorylation levels of the downstream effector, MLKL, were also significantly suppressed by treatments with 62 at 2 mpk (Supporting Information Fig. S4).
Figure 8.

The anti-inflammatory effects of 62in vivo. (A) Evaluation of 62 (po) in TNFα (10 μg) induced SIRS mouse model measuring reduction in body temperature and survival rates (n = 7). (B) Quantification of the bodyweight for each group of Ripk1K612R/K612R mice treated with vehicle, 1 or 62 (po) (n = 4). (C) Quantification of the RA symptoms induced by collagen antibodies and LPS for each group of mice treated with vehicle or 62 (po) (n = 3). Data represent means ± SD; ∗∗P < 0.01, ∗∗∗P < 0.001, paired Student's t-test.
On the other hand, in a chronic IBD mouse model with adult-onset intestinal inflammation and splenomegaly based on a Ripk1K612R/K612R knockin33, we observed significantly reduced diarrhea and weight loss symptoms in mice orally dosed with 62 at 10 or 20 mpk per day (QD) for a continuous period of 40 days, compared to the control group (Fig. 8B). Notably, the colorectal stricture, splenomegaly, and thymus hyperplasia symptoms had been significantly alleviated in the Ripk1K612R/K612R mice dosed with 62, and no adverse effects had been observed in these mice during this long-term treatment (Supporting Information Fig. S5). Finally, in a RA mouse model induced by collagen antibodies and LPS13, daily oral administration of 62 at 15 or 30 mpk for 8 days could effectively eliminate most RA symptoms, including paw edema, erythema, and joint stiffness, without affecting the bodyweight in mice (Fig. 8C and Supporting Information Fig. S6). These findings suggested that 62 is an orally bioavailable and tolerable RIPK1i that can effectively suppress RIPK1-dependent acute or chronic inflammations in vivo.
3. Conclusions
Despite being homologs and that both can be inhibited by type-II KI, RIPK1 and RIPK3 have very limited structural similarities in their hydrophobic pockets. So far only the pocket of RIPK1 has been proved utterly exploitable for selective targeting20,24. The RIPK3i, 10, was reportedly to be ineffective against mRIPK325, but in our assays, it effectively protected mouse L929 cells from necroptosis, without inducing apoptosis like other RIPK3i. This indicates that its inhibitory mechanism is distinct from that of type-I RIPK3i. Nevertheless, modifications of 10 did generate moderately active dual inhibitors of RIPK1/3 with a typical type-II KI pharmacophore.
By utilizing ligands that are more specific to the hydrophobic pocket of RIPK1, we were able to further modify these moderate RIPK1/3 dual inhibitors and successfully obtained potent and selective type-II RIPK1 inhibitors. The co-crystal structure and molecular docking results showed clear interactions between these inhibitors and both the hinge region and the hydrophobic pocket of RIPK1, which facilitated our lead optimization process. Notably, our inhibitors rendered RIPK1 in DLG-out/Glu-out conformations as the selective type-III RIPK1i did, whereas typical type-II KIs usually adopt DLG-out/Glu-in binding modes, which could explain the good selectivity of our inhibitors. In addition, minor optimization of the tail subunit resulted in significant improvement in potency, especially against mRIPK1. The optimized lead, 62, exhibited excellent potency against both human and mouse RIPK1 in cells with subnanomolar EC50 values and hundredfold selectivity windows. It also showed stronger effects in suppressing the viral replication of SARS-CoV-2 in human lung organoids, compared to reported RIPK1i. With a favorable PK profile, low oral doses of 62 exerted incredible anti-inflammatory effects in various mouse models of acute or chronic inflammation mediated by RIPK1. In summary, 62 and its analogs provide useful probes for the pharmacological perturbation of RIPK1 in animal models and promising leads for clinical drug developments.
4. Experimental
4.1. Chemistry
The synthesis of 20 and 62 is outlined in Scheme 1. Other final compounds were prepared using similar procedures. Starting from the commercially available protected 4-iodobenzylamines (64) and benzimidazoles (65) or adenine (66), the intermediates 67 and 68 can be obtained by Ullmann coupling. After deprotection of 67 or 68, the resulting benzylamines can be coupled with carboxylic acids (69) to form amides, directly producing 20 and its analogs. On the other hand, the arylamine 70 can be prepared with reported procedures and converted into aryl iodide 71 through diazotization34. Suzuki–Miyaura coupling of 71 with an alkene boronic ester yields the enol ether 72, which can be hydrolyzed to the aldehyde 73 under acidic conditions. The benzylamine resulting from deprotection of 68 can then be coupled with 73 through reductive amination, to provide the secondary amine intermediate 74. Finally, intramolecular cyclization of 74 under Lewis acid catalysis produces 62 and its analogs with bicyclic tails.
Scheme 1.

Reagents and conditions: (a) CuI, Cs2CO3, 4,7-dimethoxy-1,10-phenanthroline, DMSO, 100 °C, under N2; (b) (1) TFA, CH2Cl2, (2) HATU, DIEA, CH2Cl2/DMF; (c) H2SO4, NaNO2, CH3CN/H2O, 0 °C, then KI, 0 °C; (d) (E)-1-ethoxyethene-2-ylboronic acid pinacol ester, Pd(dppf)Cl2, Cs2CO3, DME/H2O, 90 °C, under N2; (e) 6 N HCl, THF, 0 °C to rt; (f) NaBH(OAc)3, AcOH, DCE/DMF, rt; (g) AlMe3, xylene, 120 °C, under N2.
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. Reactions were run in round bottom flasks or glass vials and stirred with Teflon-coated magnetic stir bars. Solvent evaporation was performed on rotary evaporators under reduced pressure. Reactions were monitored by thin layer chromatography, LC‒MS or UPLC/MS using water +0.05% formic acid (solvent A) and acetonitrile + 0.05% formic acid (solvent B). Preparative HPLC was performed on a C18 column (19 × 100 mm, 5 μmol/L) using a gradient of 10%–95% acetonitrile in water containing 0.05% trifluoroacetic acid (TFA) over 10 min (15 min run time) at a flow rate of 15 mL/min. All final compounds are >95% pure as determined by HPLC analysis. 1H NMR (400 MHz) and 13C NMR (101 MHz) spectra were recorded at ambient temperature in the specified deuterated solvents. Observed proton absorptions are reported as δ units of parts per million (ppm) relative to tetramethylsilane (δ 0.0). Multiplicities are reported: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), and m (multiplet). Coupling constants are reported as a J value in Hertz (Hz).
tert-Butyl(4-(5-(methylcarbamoyl)-1H-benzo[d]imidazole-1-yl)benzyl)carbamate (67).64A (200 mg, 0.60 mmol), 65 (126 mg, 0.72 mmol), Cs2CO3 (390 mg, 1.20 mmol), cuprous iodide (57 mg, 0.30 mmol), 4,7-dimethoxy-1,10-phenanthroline (43 mg, 0.18 mmol) were stirred in DMSO (3.0 mL) and heated to 100 °C and stirred overnight under nitrogen protection. The reaction mixture was filtered and purified by reverse phase C18 column (CH3CN:H2O = 0–80%) to afford compound 67 in 42% yield as a brown oil. 1H NMR (400 MHz, chloroform-d) δ 8.22 (d, J = 1.6 Hz, 1H), 8.16 (s, 1H), 7.90–7.79 (m, 1H), 7.56–7.46 (m, 5H), 6.27 (s, 1H), 5.01 (s, 1H), 4.43 (d, J = 6.2 Hz, 2H), 3.08 (d, J = 4.8 Hz, 3H), 1.49 (s, 9H). UPLC‒MS (ESI) calculated for C21H24N4O3 [M + H]+: 380.2, found: 381.1.
1-(4-((5-Benzyl-1-methyl-1H-pyrazole-3-carboxamido)methyl)phenyl)-N-methyl-1H-benzo[d]imidazole-5-carboxamide (20). To a solution of 67 (203 mg, 0.53 mmol) in CH2Cl2 (5.0 mL) was added TFA (1.0 mL). The reaction mixture was stirred at room temperature for 3 h, then the solution was diluted with CH2Cl2 and evaporated in vacuo several times to remove TFA. The crude material was used for the next step without further purification.
To a stirred suspension of 69 (15 mg, 0.07 mmol), HATU (32 mg, 0.09 mmol), and DIEA (23 mg, 0.18 mmol) in CH2Cl2 (2.0 mL) were added the above-mentioned product (20 mg, 0.07 mmol). The reaction mixture was stirred at room temperature for 4 h. The solution was diluted with CH2Cl2 and partitioned between CH2Cl2 (10 mL) and water (5 mL). The organic layer was washed by brine and dried over Na2SO4, then evaporated in vacuo and the residue was purified by silica gel column chromatography (CH3OH:CH2Cl2 = 0–20%) to afford compound 20 in 59% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 8.77 (t, J = 6.4 Hz, 1H), 8.55 (d, J = 4.5 Hz, 1H), 8.31 (d, J = 1.1 Hz, 1H), 7.91–7.86 (m, 1H), 7.72–7.63 (m, 3H), 7.56 (t, J = 9.3 Hz, 2H), 7.34 (t, J = 7.3 Hz, 2H), 7.28–7.22 (m, 3H), 6.37 (d, J = 1.7 Hz, 1H), 4.49 (t, J = 5.7 Hz, 2H), 4.07 (s, 2H), 3.78 (s, 3H), 2.80 (dd, J = 14.9, 4.5 Hz, 3H). HRMS (ESI) calculated for C28H26N6O2 [M + H]+: 478.2117, found: 479.2182.
Ethyl 5-benzyl-3-iodo-1-methyl-1H-pyrazole-4-carboxylate (71). Con. H2SO4 (265 mg, 2.7 mmol) was added dropwise to the solution of 70 (350 mg, 1.35 mmol) in MeCN/H2O (3 mL/6 mL), stirred at 0 °C for 10 min. Sodium nitrite (102 mg, 1.48 mmol) in H2O (3.0 mL) was added dropwise to the solution and stirred at 0 °C for 1 h, then potassium iodide (672 mg, 4.05 mmol) was added slowly to the mixture and stirred at 0 °C for another 1 h before quenched by sodium thiosulfate solution. The solution was diluted with ethyl acetate and partitioned between ethyl acetate (60 mL) and water (10 mL). The organic layer was washed by H2O and brine, dried over Na2SO4, then evaporated in vacuo and the residue was purified by silica gel column chromatography (EA:PE = 0–50%) to afford compound 71 in 30% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.29 (dd, J = 8.2, 6.5 Hz, 2H), 7.25–7.19 (m, 1H), 7.14–7.03 (m, 2H), 4.42 (s, 2H), 4.32 (q, J = 7.1 Hz, 2H), 3.71 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H). LC‒MS (ESI) calculated for C14H15IN2O2 [M + H]+: 370.0, found: 370.1.
Ethyl (E)-5-benzyl-3-(2-ethoxyvinyl)-1-methyl-1H-pyrazole-4-carboxylate (72).71 (150 mg, 0.41 mmol), (E)-1-ethoxyethene-2-ylboronic acid pinacol ester (161 mg, 0.81 mmol, Pd(dppf)Cl2 (30 mg, 0.04 mmol), Cs2CO3 (294 mg, 0.90 mmol), were stirred in DME/H2O (2.0 mL/0.4 mL) and heated at 90 °C, stirred overnight under nitrogen protection. The reaction mixture was filtered and purified by reverse phase C18 column (CH3CN: H2O = 10%–100%) to afford compound 72 in 87% yield as a brown solid. 1H NMR (400 MHz, chloroform-d) δ 7.37 (s, 0.5H), 7.34 (s, 0.5H), 7.28 (dd, J = 9.7, 2.6 Hz, 2H), 7.20 (t, J = 7.2 Hz, 1H), 7.13–7.07 (m, 2H), 6.33 (d, J = 13.0 Hz, 1H), 4.38 (s, 2H), 4.27 (q, J = 7.1 Hz, 2H), 3.94 (q, J = 7.0 Hz, 2H), 3.64 (s, 3H), 1.35 (t, J = 7.1 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H). UPLC‒MS (ESI) calculated for C18H22N2O3 [M + H]+: 314.2, found: 315.0.
Ethyl 5-benzyl-1-methyl-3-(2-oxoethyl)-1H-pyrazole-4-carboxylate (73). 6 N HCl (1.5 mL) was added to 72 (112 mg, 0.36 mmol) in THF (2.0 mL) at 0 °C, then the reaction was warmed to room temperature and stirred overnight. The solution was diluted with ethyl acetate and partitioned between ethyl acetate (15 mL) and water (5 mL). The organic layer was washed by NaHCO3 saturated solution and brine, dried over Na2SO4, then evaporated in vacuo, and the crude product was used for the next step without further purification.
tert-Butyl (S)-(1-(4-(6-amino-9H-purin-9-yl)phenyl)ethyl)carbamate (68).64B (1.62 g, 4.66 mmol), Cs2CO3 (2.39 g, 7.34 mmol), cuprous iodide (444 mg, 2.33 mmol), 4,7-dimethoxy-1,10-phenanthroline (112 mg, 0.47 mmol) were stirred in DMSO (25.0 mL) and heated at 100 °C and stirred overnight under nitrogen protection. The reaction mixture was filtered and purified by reverse phase C18 column (CH3CN:H2O = 0–80%) to afford compound 68 in 39% yield as a yellow-white solid. 1H NMR (400 MHz, chloroform-d) δ 8.42 (s, 1H), 8.06 (s, 1H), 7.70–7.59 (m, 2H), 7.51 (d, J = 8.1 Hz, 2H), 5.87 (s, 2H), 4.96–4.86 (m, 1H), 1.96 (s, 1H), 1.48 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). UPLC‒MS (ESI) calculated for C18H22N6O2 [M + H]+: 354.2, found: 355.1.
Ethyl(S)-3-(2-((1-(4-(6-amino-9H-purin-9-yl)phenyl)ethyl)amino)ethyl)-5-benzyl-1-methyl-1H-pyrazole-4-carboxylate (74). To a solution of 68 (1.45 g, 4.08 mmol) in CH2Cl2 (20.0 mL) was added TFA (2.0 mL). The reaction mixture was stirred at room temperature for 4 h, then the reaction solution was diluted with CH2Cl2 and evaporated in vacuo for several times to remove TFA. The crude material was used for the next step without further purification.
To a stirred suspension of the above-mentioned product (1.1 g, 4.2 mmol), 73 (1.7 mmol) in DCE/DMF (10.0 mL/2.0 mL) were added acetic acid (306 mg, 5.1 mmol). The reaction mixture was stirred at room temperature for 30 min then added sodium triacetoxyborohydride (899 mg, 4.2 mmol) to the mixture, and stirred overnight at room temperature. DCE was evaporated in vacuo, the residue was filtered and purified by reverse phase C18 column (CH3CN:H2O = 10%–100%) to afford compound 74 in 66% yield as a yellow foam. 1H NMR (400 MHz, chloroform-d) δ 8.43 (s, 1H), 8.06 (s, 1H), 7.65–7.50 (m, 4H), 7.27 (t, J = 3.7 Hz, 3H), 7.20 (t, J = 7.3 Hz, 1H), 7.14–7.04 (m, 2H), 5.74 (s, 2H), 4.37 (s, 2H), 4.24 (q, J = 7.1 Hz, 2H), 3.93 (q, J = 6.5 Hz, 1H), 3.65 (s, 3H), 3.07 (t, J = 6.8 Hz, 2H), 2.96–2.75 (m, 2H), 1.37 (d, J = 6.4 Hz, 3H), 1.26 (t, J = 7.0 Hz, 3H). UPLC‒MS (ESI) calculated for C29H32N8O2 [M + H]+: 524.3, found: 525.2.
(S)-5-(1-(4-(6-Amino-9H-purin-9-yl)phenyl)ethyl)-3-benzyl-2-methyl-2,5,6,7-tetrahydro-4H-pyrazolo[4,3-c]pyridin-4-one (62). Trimethylaluminium (1.5 mmol) was added dropwise to the solution of 74 (262 mg, 0.5 mmol) in dry xylene (20.0 mL), then heated to 120 °C and stirred overnight under nitrogen protection. The reaction was quenched by potassium sodium tartrate solution, then partitioned between ethyl acetate (60 mL) and water (20 mL). The organic layer was washed by brine, dried over Na2SO4, then evaporated in vacuo and the residue was purified by silica gel column chromatography (CH3OH:CH2Cl2 = 0–20%) to afford compound 62 in 44% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 1H), 8.21 (s, 1H), 7.98–7.76 (m, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.43 (s, 2H), 7.37–7.27 (m, 4H), 7.26–7.16 (m, 1H), 6.01 (q, J = 7.0 Hz, 1H), 4.48–4.32 (m, 2H), 3.64 (s, 3H), 3.55–3.46 (m, 1H), 3.18–3.09 (m, 1H), 2.82–2.66 (m, 2H), 1.58 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.65, 150.54, 149.66, 148.13, 145.28, 143.15, 142.97, 142.33, 137.56, 132.47, 128.65, 128.39, 128.16, 126.50, 124.12, 118.94, 110.02, 48.44, 41.42, 36.14, 29.00, 22.32, 16.45. HRMS (ESI) calculated for C27H26N8O [M + H]+: 478.2230, found: 479.2303.
N-(4-(6-Amino-9H-purin-9-yl)benzyl)-5-benzyl-1-methyl-1H-pyrazole-4-carboxamide (39). The title compound was obtained as described in the general procedure (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.76–8.66 (m, 2H), 8.35 (s, 1H), 7.99 (s, 1H), 7.82–7.77 (m, 2H), 7.52 (d, J = 8.6 Hz, 2H), 7.31–7.25 (m, 2H), 7.21–7.17 (m, 3H), 4.51 (d, J = 5.8 Hz, 2H), 4.46 (s, 2H), 3.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.65, 150.54, 149.66, 148.13, 145.28, 143.15, 142.97, 142.33, 137.56, 132.47, 128.65, 128.39, 128.16, 126.50, 124.12, 118.94, 110.02, 48.44, 41.42, 36.14, 29.00, 22.32, 16.45. HRMS (ESI) calculated for C24H22N8O [M + H]+: 438.1917, found: 439.1993.
N-(4-(6-Amino-9H-purin-9-yl)benzyl)-5-benzyl-3-chloro-1-methyl-1H-pyrazole-4-carboxamide (48). The title compound was obtained as described in the general procedure (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.50 (s, 1H), 8.45 (t, J = 6.0 Hz, 1H), 8.14 (s, 1H), 7.83–7.69 (m, 2H), 7.49–7.38 (m, 2H), 7.35 (s, 2H), 7.23 (dd, J = 8.0, 6.6 Hz, 2H), 7.18–7.07 (m, 3H), 4.44 (d, J = 6.0 Hz, 2H), 4.22 (s, 2H), 3.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.69, 152.55, 148.50, 148.10, 144.96, 141.95, 139.85, 136.97, 134.68, 132.77, 128.69, 128.27, 126.63, 123.59, 119.12, 113.10, 42.05, 36.78, 29.37. HRMS (ESI) calculated for C24H21ClN8O [M + H]+: 472.1527, found: 473.1600.
N-(4-(6-Amino-9H-purin-9-yl)benzyl)-5-benzyl-1-methyl-1H-1,2,3-triazole-4-carboxamide (49). The title compound was obtained as described in the general procedure (white solid). 1H NMR (400 MHz, DMSO-d6) δ 9.30 (t, J = 6.3 Hz, 1H), 8.71 (s, 1H), 8.35 (s, 1H), 7.83–7.70 (m, 2H), 7.55 (d, J = 8.3 Hz, 2H), 7.37–7.26 (m, 2H), 7.27–7.14 (m, 3H), 4.54 (d, J = 6.3 Hz, 2H), 4.47 (s, 2H), 3.90 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 161.07, 153.14, 148.91, 148.61, 141.61, 139.99, 138.41, 138.24, 136.65, 132.90, 128.77, 128.44, 128.32, 126.78, 123.58, 119.12, 41.53, 34.67, 27.40. HRMS (ESI) calculated for C23H21N9O [M + H]+: 439.1869, found: 440.1944.
(S)-N-(1-(4-(6-Amino-9H-purin-9-yl)phenyl)ethyl)-5-benzyl-1-methyl-1H-pyrazole-4-carboxamide (54). The title compound was obtained as described in the general procedure (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.55 (s, 1H), 8.48 (d, J = 7.9 Hz, 1H), 8.19 (s, 1H), 8.08 (s, 1H), 7.86–7.74 (m, 2H), 7.60–7.51 (m, 2H), 7.41 (s, 2H), 7.27 (dd, J = 8.0, 6.8 Hz, 2H), 7.20–7.12 (m, 3H), 5.20 (p, J = 7.2 Hz, 1H), 4.56–4.22 (m, 2H), 3.65 (s, 3H), 1.50 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.31, 156.33, 153.12, 149.19, 144.68, 143.36, 139.67, 137.81, 137.53, 133.49, 128.51, 128.21, 127.02, 126.28, 123.03, 119.22, 114.46, 47.46, 36.31, 28.82, 22.33. HRMS (ESI) calculated for C25H24N8O [M + H]+: 452.2073, found: 453.2147.
(S)-5-(1-(4-(6-Amino-9H-purin-9-yl)phenyl)ethyl)-3-benzyl-2-methyl-5,6-dihydropyrrolo[3,4-c]pyrazol-4(2H)-one (63). The title compound was obtained as described in the general procedure (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.35 (s, 1H), 7.85–7.79 (m, 2H), 7.54 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 5.7 Hz, 4H), 7.26–7.19 (m, 1H), 5.51 (q, J = 7.1 Hz, 1H), 4.44 (d, J = 16.3 Hz, 1H), 4.19 (s, 2H), 4.06 (d, J = 16.3 Hz, 1H), 3.79 (s, 3H), 1.65 (d, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.38, 157.19, 152.94, 148.65, 148.56, 142.19, 141.68, 137.93, 137.25, 133.14, 128.77, 128.42, 127.79, 126.70, 123.81, 119.11, 115.48, 48.88, 41.58, 36.75, 29.51, 18.07. HRMS (ESI) calculated for C26H24N8O [M + H]+: 464.2073, found: 465.2148.
4.2. Reagents
Cell lines were cultured as follows: L929 and NIH-3T3 cells were cultured in DMEM supplemented with 10% (v/v) FBS; Jurkat and Ba/F3 cells were cultured in RPMI-1640 with 10% FBS; HT-29 was cultured in McCoy's 5A with 10% FBS; HepG2 was cultured in MEM with 10% FBS; All the cells were maintained at 37 °C and 5% CO2. All culture media and FBS were GIBCO™ and purchased from ThermoFisher (Waltham, MA, USA), common reagents were purchased from Sigma–Aldrich (Burlington, MA, USA) unless otherwise specified. The following commercial reagents were used in this study: TNFα (Novoprotein, Shanghai, China), mTNFα (Cell Sciences, Newburyport, MA, USA), monoclonal anti-collagen antibodies cocktail (Chondrex, Woodinville, USA). The following antibodies were used in this study: p-S345-MLKL (ab196436, mouse-specific), p-S358-MLKL (ab187091, human-specific), MLKL (ab172868, mouse-specific), MLKL (ab189612, human-specific) and RIPK3 (ab72106, human-specific) were purchased from Abcam (Cambridge, UK); RIPK1 (3493), p-S166-RIPK1 (31122, human-specific), p-T231/S232-RIPK3 (57220, mouse-specific) and p-S227-RIPK3 (93654, human-specific) were purchased from Cell Signaling Technology (Danvers, MA, USA); RIPK3 (AHP1797, mouse-specific) was purchased from BIO-RAD (Hercules, CA, USA); β-actin (I10813) and tubulin (I11107) were purchased from TransGen (Beijing, China); SARS-CoV-2 N protein (40588-T62) was purchased from Sino Biological (Beijing, China); p-S166-RIPK1 (mouse specific) was purchased from Biolynx (Hangzhou, China). Chemical probes were purchased from Selleck (Shanghai, China) or prepared according to the literature.
4.3. Co-crystal structure determination and molecular docking
The expression, purification, and co-crystallization of recombinant RIPK1 protein (1-294, C34A, C127A, C233A, C240A) were following the reported procedures and conditions16. Diffraction datasets were collected at the beamline BL17U1 of the Shanghai Synchrotron Radiation Facility (Shanghai, China). The diffraction data sets were processed with the XDS program and the autoPROC suite35, 36, 37. The initial phase for the complex structure was solved by molecular replacement using the PDB entry 4ITH as the search model with the program Phaser38. The structural model was further manually re-built in Coot and refined with Phenix39,40. The qualities of the final models were validated by MolProbity41 and listed in Supporting Information Table S1. The docking studies were performed based on coordinates of RIPK1-20 (PDB 8I2N) using Schrödinger 2021 software suites (Schrödinger, New York, NY, USA)42. The best ligand poses were chosen based on the docking score, and scores of −10 or lower usually represent very good binding. All the structural figures were prepared in the program PyMol (Schrödinger).
4.4. In vitro profiling assays
The RIPK1 kinase activities were tested using a commercially available assay kit from Promega (Madison, Wisconsin, USA), and the RIPK3 kinase activities were tested similarly except that recombinant RIPK3 other than RIPK1 protein was used (SignalChem, Richmond, BC, Canada). The KINOMEscan assays were done by DiscoverX (Fremont, CA, USA), and the scores were reported as a percent of DMSO control, with the lower score usually indicating a higher probability of being a hit29,30. The assays for microsomal stability, CYP450 inhibition and hERG inhibition were performed according to the literature43, 44, 45.
4.5. Cell viability assays
The cells were plated at a density of 2000 (adherent) or 10,000 (suspended) cells per well in a 384-well white plate and incubated with DMSO or inhibitors. For necroptosis cell models, the cells were treated with inhibitors for 30 min before TNFα stimulation (10 ng/mL with 25 nmol/L SM164, or 40 ng/mL without SM164), then analyzed for viabilities 16 h after the stimulation. For cytotoxicity assessment, cells were treated with inhibitors for 72 h before viability analysis. The relative cell viabilities were determined using the Cell Titer Glo assay (Promega) and reported as percentages of the DMSO controls.
4.6. Immunoblotting analyses
HT29 or L929 cells were seeded in 6-well plates at a density of 4 × 105 cells per well 24 h before experiment. FADD−/− Jurkat cells were seeded in 6-well plates at a density of 2 × 106 cells per well. Cells were pretreated with inhibitors for 1 h before applying stimulations. The human lung organoids infected with SARS-CoV-2 were prepared according to literature3, and treated with inhibitors for 48 h before being lysed. The whole-cell lysates were collected with 1% SDS lysis buffer (1% SDS, 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.4)), and then boiled at 95 °C for 10 min. Total protein concentration was analyzed by BCA kit (ThermoFisher) and aligned with lysis buffer. Lysates were mixed with an equal volume of 2X loading buffer (100 mmol/L Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, bromophenol blue, and 5% β-mercaptoethanol) and boiled at 95 °C for 10 min, then separated in 10% polyacrylamide gel electrophoresis. Protein was transferred onto 0.2 μm nitrocellulose membrane and blotted with specific antibodies as indicated.
4.7. In vivo studies
For PK studies, male ICR mice or SD rats were dosed via tail vein (iv, 10% v/v DMA, 10% w/v Solutol HS 15 in normal saline at a dose of 0.5 or 1 mpk) or oral gavage (po, 3% v/v DMSO, 97% v/v aqueous HP-β-CD solution (30% w/v) at a dose of 10 or 5 mpk). Blood samples were collected at 5, 15, 30, 60, 120, 240, 360, 480, 720 and 1440 min (iv) and 15, 30, 60, 120, 240, 360, 480, 720 and 1440 min (po). The animal was restrained manually at the designated time points, and approximately 30–50 μL of blood sample was collected via the retro-orbital plexus into tubes with EDTA2K. Plasma samples were separated by centrifugation of whole blood and stored below −80 °C until bioanalysis using LC‒MS/MS.
For the efficacy study in the SIRS model, six-week-old C57BL/6J male mice received 62 at 1, 2, or 5 mpk (po) 30 min before injection with mouse TNFα (iv, 10 μg), then monitored for survival and rectal temperature every hour. The vehicle- or 1-treated groups were used as controls. For the pharmacodynamic study, mice were anesthetized, perfused, and dissected 8 h after the injection of TNFα (20 μg) on a cryostat, then the organs were collected and sectioned. For immunostaining, tissue sections were mounted and blocked with 10% normal goat serum and 1% BSA, and then incubated with primary antibodies at 4 °C overnight.
For the efficacy study in the IBD model, eight-week-old Ripk1K612R/K612R-knockin male mice were administered with 62 at a dose of 10 or 20 mpk (po, QD) for continuous 40 days. Mice were monitored every other day for their weights and stool scales. Ripk1WT background mice, and the vehicle- or 1-treated groups were used as controls.
For the efficacy study in the RA model, eight-week-old BALB/c male mice received 1.5 mg (iv) of the anti-collagen antibodies cocktail on Day 0, followed by intraperitoneal injection with LPS (10 μg) on day 3. After 24 h, 62 was administered (po) at a dose of 15 or 30 mpk, using the vehicle as a negative control. Mice were weighed and scored every day according to a qualitative scoring system to assess the severity of paw inflammation46.
The animal protocols were approved by the Institutional Animal Care and Use Committee of IRCBC, SIOC, and all experiments were performed in accordance with IRCBC, SIOC policies on the care, welfare, and treatment of laboratory animals.
4.8. Statistics
All statistical analyses and curves were performed using the GraphPad software package (La Jolla, CA, USA). Statistical significance between conditions was calculated using a t-test (two-tailed) when comparing two groups. All the enzymatic activity assays were performed in triplicate and quantified data were presented as mean ± SD. IC50 values were calculated using non-linear regression and shown as data points representing mean ± SD.
4.9. Accession codes
Coordinates and structure factors for the co-crystal structure of RIPK1 kinase domain (1-294, C34A, C127A, C233A, C240A) and 20 have been deposited in the Protein Data Bank with the accession number 8I2N.
Author contributions
Conceptualization: Li Tan and Ying Li; Methodology: Li Tan, Ying Li, Ying Qin, Lifeng Pan, Zheng Zhang and, Yechun Xu; Investigation and Formal Analysis: Ying Qin, Dekang Li, Chunting Qi, Huaijiang Xiang, Huyan Meng, Jingli Liu, Shaoqing Zhou, Xinyu Gong, Ying Li, Guifang Xu, Rui Zu, Hang Xie, and Gang Xu; Software: Huaijiang Xiang, Xinyu Gong, and Lifeng Pan; Resources: Shi Chen; Writing-Original Draft: Li Tan, Ying Li, Ying Qin, and Dekang Li; Writing-Review & Editing: All the authors; Supervision: Li Tan, Ying Li, Lifeng Pan, Shi Chen, Zheng Zhang, and Yechun Xu; Funding acquisition: Li Tan, Ying Li, and Zheng Zhang.
Conflicts of interest
Li Tan, Ying Li, Ying Qin, Chunting Qi, and Huaijiang Xiang are inventors on patent applications relating to this work, owned by SIOC, CAS.
Acknowledgments
We thank Prof. Junying Yuan (IRCBC of CAS, Shanghai, China) and Dr. Jidong Zhu (Etern Therapeutics, Shanghai, China) for their generous help on this work, Dr. Sudan He (ISM of CAMS, Suzhou, China) for providing RIPK3-FKBP NIH/3T3 cells, and National Facility for Protein Science in Shanghai (China) for the help in animal studies. This work was supported by grants from the National Natural Science Foundation of China (Grants Nos. 21837004, 82151212, and 32170755), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39050500, China), Shanghai Municipal Science and Technology Major Project (Grant No.2019SHZDZX02, China).
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.10.021.
Contributor Information
Ying Li, Email: liying@sioc.ac.cn.
Li Tan, Email: tanli@sioc.ac.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Ofengeim D., Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14:727–736. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 2.Mifflin L., Ofengeim D., Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov. 2020;19:553–571. doi: 10.1038/s41573-020-0071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu G., Li Y., Zhang S., Peng H., Wang Y., Li D., et al. SARS-CoV-2 promotes RIPK1 activation to facilitate viral propagation. Cell Res. 2021;31:1230–1243. doi: 10.1038/s41422-021-00578-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi K., Zhang J., Zhou E., Wang J., Wang Y. Small-molecule receptor-interacting protein 1 (RIP1) inhibitors as therapeutic agents for multifaceted diseases: current medicinal chemistry insights and emerging opportunities. J Med Chem. 2022;65:14971–14999. doi: 10.1021/acs.jmedchem.2c01518. [DOI] [PubMed] [Google Scholar]
- 5.Wu Y., Dong G., Sheng C. Targeting necroptosis in anticancer therapy: mechanisms and modulators. Acta Pharm Sin B. 2020;10:1601–1618. doi: 10.1016/j.apsb.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degterev A., Hitomi J., Germscheid M., Ch'en I.L., Korkina O., Teng X., et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 8.Harris P.A., Berger S.B., Jeong J.U., Nagilla R., Bandyopadhyay D., Campobasso N., et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem. 2017;60:1247–1261. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 9.Ren Y., Su Y., Sun L., He S., Meng L., Liao D., et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of receptor-interacting protein 1 (RIP1) for the treatment of systemic inflammatory response syndrome. J Med Chem. 2017;60:972–986. doi: 10.1021/acs.jmedchem.6b01196. [DOI] [PubMed] [Google Scholar]
- 10.Wang W., Marinis J.M., Beal A.M., Savadkar S., Wu Y., Khan M., et al. RIP1 Kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell. 2018;34:757–774. doi: 10.1016/j.ccell.2018.10.006. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng H., Wu G., Zhao X., Wang A., Li D., Tong Y., et al. Discovery of a cooperative mode of inhibiting RIPK1 kinase. Cell Discov. 2021;7:41. doi: 10.1038/s41421-021-00278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshikawa M., Saitoh M., Katoh T., Seki T., Bigi S.V., Shimizu Y., et al. Discovery of 7-oxo-2,4,5,7-tetrahydro-6H-pyrazolo[3,4-c]pyridine derivatives as potent, orally available, and brain-penetrating receptor interacting protein 1 (RIP1) kinase inhibitors: analysis of structure-kinetic relationships. J Med Chem. 2018;61:2384–2409. doi: 10.1021/acs.jmedchem.7b01647. [DOI] [PubMed] [Google Scholar]
- 13.Patel S., Webster J.D., Varfolomeev E., Kwon Y.C., Cheng J.H., Zhang J., et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2020;27:161–175. doi: 10.1038/s41418-019-0347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris P.A. Inhibitors of RIP1 kinase: a patent review (2016-present) Expert Opin Ther Pat. 2021;31:137–151. doi: 10.1080/13543776.2021.1854729. [DOI] [PubMed] [Google Scholar]
- 15.Yang X., Lu H., Xie H., Zhang B., Nie T., Fan C., et al. Potent and selective RIPK1 inhibitors targeting dual-pockets for the treatment of systemic inflammatory response syndrome and sepsis. Angew Chem Int Ed Engl. 2022;61 doi: 10.1002/anie.202114922. [DOI] [PubMed] [Google Scholar]
- 16.Xie T., Peng W., Liu Y., Yan C., Maki J., Degterev A., et al. Structural basis of RIP1 inhibition by necrostatins. Structure. 2013;21:493–499. doi: 10.1016/j.str.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Najjar M., Suebsuwong C., Ray S.S., Thapa R.J., Maki J.L., Nogusa S., et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 2015;10:1850–1860. doi: 10.1016/j.celrep.2015.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y., Gray N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 19.Harris P.A., Bandyopadhyay D., Berger S.B., Campobasso N., Capriotti C.A., Cox J.A., et al. Discovery of small molecule RIP1 kinase inhibitors for the treatment of pathologies associated with necroptosis. ACS Med Chem Lett. 2013;4:1238–1243. doi: 10.1021/ml400382p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martens S., Hofmans S., Declercq W., Augustyns K., Vandenabeele P. Inhibitors targeting RIPK1/RIPK3: old and new drugs. Trends Pharmacol Sci. 2020;41:209–224. doi: 10.1016/j.tips.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Yu X., Ma H., Li B., Ji Y., Du Y., Liu S., et al. A novel RIPK1 inhibitor reduces GVHD in mice via a nonimmunosuppressive mechanism that restores intestinal homeostasis. Blood. 2023;141:1070–1086. doi: 10.1182/blood.2022017262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y., Zhang L., Wang Y., Zou J., Yang R., Luo X., et al. Generative deep learning enables the discovery of a potent and selective RIPK1 inhibitor. Nat Commun. 2022;13:6891. doi: 10.1038/s41467-022-34692-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J., Xin M., Xu C., He Y., Zhang W., Wang Z., et al. Ligand-based substituent-anchoring design of selective receptor-interacting protein kinase 1 necroptosis inhibitors for ulcerative colitis therapy. Acta Pharm Sin B. 2021;11:3193–3205. doi: 10.1016/j.apsb.2021.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuny G.D., Degterev A. RIPK protein kinase family: atypical lives of typical kinases. Semin Cell Dev Biol. 2021;109:96–105. doi: 10.1016/j.semcdb.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandal P., Berger S.B., Pillay S., Moriwaki K., Huang C., Guo H., et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X.N., Yang Z.H., Wang X.K., Zhang Y., Wan H., Song Y., et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21:1709–1720. doi: 10.1038/cdd.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canning P., Ruan Q., Schwerd T., Hrdinka M., Maki J.L., Saleh D., et al. Inflammatory signaling by NOD-RIPK2 is inhibited by clinically relevant type II kinase inhibitors. Chem Biol. 2015;22:1174–1184. doi: 10.1016/j.chembiol.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hart A.C., Abell L., Guo J., Mertzman M.E., Padmanabha R., Macor J.E., et al. Identification of RIPK3 type II inhibitors using high-throughput mechanistic studies in hit triage. ACS Med Chem Lett. 2020;11:266–271. doi: 10.1021/acsmedchemlett.9b00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein D.M., Gray N.S., Zarrinkar P.P. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- 30.Miduturu C.V., Deng X., Kwiatkowski N., Yang W., Brault L., Filippakopoulos P., et al. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem Biol. 2011;18:868–879. doi: 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hitomi J., Christofferson D.E., Ng A., Yao J., Degterev A., Xavier R.J., et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terai H., Tan L., Beauchamp E.M., Hatcher J.M., Liu Q., Meyerson M., et al. Characterization of DDR2 inhibitors for the treatment of DDR2 mutated nonsmall cell lung cancer. ACS Chem Biol. 2015;10:2687–2696. doi: 10.1021/acschembio.5b00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., Zhang M., Huang X., Liang W., Li G., Lu X., et al. Ubiquitination of RIPK1 regulates its activation mediated by TNFR1 and TLRs signaling in distinct manners. Nat Commun. 2020;11:6364. doi: 10.1038/s41467-020-19935-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Xia S.C., Czarniecki Michael, Tsai Hsingan, Henry Vaccaro, Cleven Renee, Cook John, et al. Synthesis and evaluation of polycyclic pyrazolo[3,4-d]pyrimidines as PDE1 and PDE5 cGMP phosphodiesterase inhibitors. J Med Chem. 1997;40:4372–4377. doi: 10.1021/jm970495b. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tickle I.J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., et al. Global Phasing Ltd; Cambridge, United Kingdom: 2018. STARANISO. [Google Scholar]
- 38.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liebschner D., Afonine P.V., Baker M.L., Bunkoczi G., Chen V.B., Croll T.I., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein‒ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 43.Li X., He Y., Ruiz C.H., Koenig M., Cameron M.D., Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37:1242–1250. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J.J., Guo J.J., Zhan J., Bu H.Z., Lin J.H. An in-vitro cocktail assay for assessing compound-mediated inhibition of six major cytochrome P450 enzymes. J Pharm Anal. 2014;4:270–278. doi: 10.1016/j.jpha.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamiya K., Niwa R., Morishima M., Honjo H., Sanguinetti M.C. Molecular determinants of hERG channel block by terfenadine and cisapride. J Pharmacol Sci. 2008;108:301–307. doi: 10.1254/jphs.08102fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caplazi P., Baca M., Barck K., Carano R.A., DeVoss J., Lee W.P., et al. Mouse models of rheumatoid arthritis. Vet Pathol. 2015;52:819–826. doi: 10.1177/0300985815588612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.