

ABSTRACT
There is a need in clinical microbiology and public health for assays that enable sensitive and rapid detection of infectious agents. Next Generation Sequencing (NGS) is increasingly used in the fields of oncology and personalized genome medicine but has not gained a wider acceptance for clinical microbiology due to operational and bioinformatics complexity, as well as lower sensitivity compared to agent-specific quantitative polymerase chain reaction (qPCR) assays. VirCapSeq-VERT is a positive selection system for detection, typing, and strain differentiation of both RNA and DNA viruses with sensitivity comparable to qPCR. Here we report the analytical and clinical validation of the VirCapSeq-VERT system for detection of viruses in plasma and nasal secretions that cause systemic and/or respiratory infections.
IMPORTANCE
Broad range assay for accurate and sensitive diagnostics.
KEYWORDS: VirCapSeq, diagnostics, analytical validation, clinical validation, sequencing, clinical microbiology
INTRODUCTION
A cluster of severe respiratory infections emerged in December 2019, linked to a wet market in Wuhan, China. By the spring of 2020, the world was in the grips of a pandemic with the near collapse of healthcare systems, leading to unprecedented global lockdowns (1, 2). Subsequent reports of Mpox, polio, enterovirus D68 (EV-D68), respiratory syncytial virus (RSV), and measles outbreaks confirmed our vulnerability to the emergence and reemergence of viral diseases (3–12). Despite the advances made in healthcare and medicine, microbial infections are still one of the leading causes of death in both developing as well as developed countries (13). The impact of infectious diseases includes not only morbidity and mortality but also a substantive economic burden (14, 15).
Polymerase chain reaction (PCR) has transformed clinical microbiology by providing methods for the detection of viruses and quantitation of viral load. It is nonetheless applicable only for the detection of known and closely related pathogens and has limited potential for multiplexing. Unbiased Next Generation Sequencing (NGS) is not constrained in multiplex capacity, but has other limitations that include higher cost, longer time to deliver results, greater complexity of workflow and data analysis, and lower sensitivity (1,000–10,000 copies/mL versus 10–100 copies/mL) (16, 17).
VirCapSeq-VERT is a positive selection system for detection, typing, and strain differentiation of both RNA and DNA viruses. It has sensitivity similar to qPCR (5–50 copies/mL), and enables high throughput detailed genomic analyses in less than 36 hours. Total nucleic acid is extracted and subjected to first and second-strand cDNA synthesis. Products are sheared prior to library construction. After pooling of bar-coded libraries, viral targets are enriched by VirCapSeq-VERT capture oligonucleotides, followed by washing to remove host products prior to sequencing (schematic in Fig. 1). VirCapSeq-VERT has been used with multiple specimen types including saliva, nasopharyngeal swabs, serum, plasma, urine, feces, cerebrospinal fluid, environmental samples, and organ tissues (16, 18–31). The enabling assay component is a library of oligonucleotide probes designed to bind and capture sequences of all known vertebrate viruses. The capture probes cover all relevant sequences in Genbank, RefSeq, EMBL, or GISAID (a listing of viral targets included in VirCapSeq-VERT capture library construction can be found in Table S1 of reference 16; the capture library was updated in May 2021 by adding probes for newly reported sequence entries, including the novel SARS-CoV-2 virus). With the goal of extending this method to clinical microbiology, we undertook a rigorous assessment of the performance characteristics of VirCapSeq-VERT including its limit of detection (LoD), repeatability and reproducibility, differential diagnosis in mixed infections, clinical accuracy, and precision. This assessment was undertaken to obtain formal approval for the use of VirCapSeq-VERT as a certified diagnostic test by the New York State Department of Health (NYSDOH) under the auspices of the Clinical Laboratory Evaluation Program (CLEP), a stringent state licensure program run by the NYSDOH (32).
Fig 1.

Schematic of VirCapSeq-VERT workflow. (A) sample collection, total nucleic acid (TNA) extraction, and library preparation; (B) Capture enrichment and sequencing.
MATERIALS AND METHODS
Participating institutions and ethical clearance
The validation was a collaborative effort between the Center for Infection and Immunity in the Mailman School of Public Health and the Personalized Genomic Medicine laboratory in the Department of Pathology and Cell Biology in the Vagelos College of Physicians and Surgeons, at Columbia University New York, NY. All work was pursued with Columbia University IRB approval.
Viruses
A representative set of viruses with RNA and DNA genomes that vary in polarity, size, and structure, and are relevant to the practice of clinical microbiology were selected for validation (Table 1).
TABLE 1.
Genome structures and sizes for 22 representative viruses used for validation study in plasma and nasal swab samples
| Plasma | Nasal swab | ||||
|---|---|---|---|---|---|
| Virus | Genome Type | Genome Size (nt) | Virus | Genome Type | Genome Size (nt) |
| Adenovirus 1 | dsDNA linear | 40,000 | Adenovirus 1 | dsDNA linear | 40,000 |
| BK Polyomavirus | dsDNA linear | 5,000 | Coronavirus 229E | ss(+) RNA | 30,000 |
| Coxsackievirus A16 | ss(+) RNA | 7,400 | Coronavirus OC43 | ss(+) RNA | 30,000 |
| Coxsackievirus B4 | ss(+) RNA | 7,300 | SARS-CoV-2 | ss(+) RNA | 30,000 |
| Enterovirus A71 | ss(+) RNA | 7,000 | Enterovirus D68 | ss(+) RNA | 7,000 |
| Hepatitis B | dsDNA circular | 3,200 | Human metapneumovirus | ss(-) RNA | 14,000 |
| Human herpesvirus 1 | dsDNA linear | 152,000 | Respiratory syncytial virus | ss(-) RNA | 15,000 |
| Human herpesvirus 2 | dsDNA linear | 155,000 | Human parainfluenza virus 1 | ss(-) RNA | 16,000 |
| Human herpesvirus 5 | dsDNA linear | 240,000 | Human parainfluenza virus 2 | ss(-) RNA | 16,000 |
| Parvovirus B19 | ssDNA | 5,600 | Human parainfluenza virus 3 | ss(-) RNA | 16,000 |
| Zika virus | ss(+) RNA | 11,000 | Influenza A virus | ss(-) RNA | 13,500 |
| Influenza B virus | ss(-) RNA | 14,500 | |||
Validation was performed using a mix of contrived samples, controls, and clinical specimens. Virus culture supernatants were procured either pre-quantified or were quantified in copies per milliliter using Conformitè Europëenne (CE) marked commercial qPCR kits from Siemens Healthineers (Resp21 FTD Plus and Neuro9 FTD) with the exceptions of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and Zika virus (ZIKV) which were quantified using FDA Emergency Use Authorization (EUA) approved assays (33, 34). To replicate conditions found in clinical materials, we spiked live viruses into appropriate negative matrices that were confirmed to be negative for the viruses included in the validation. The only exception being SARS-CoV-2, where, for safety reasons, pre-extracted total nucleic acid (TNA) from both the virus and pooled nasal swabs were mixed. Viruses or TNA were spiked into the background at desired concentrations for all performance characteristics studies.
Controls
Negative controls
Negative controls included either single donor plasma samples or a pool of anterior nasal swabs in viral transport media (VTM) (Becton Dickinson, USA; Copan, Italy) that were confirmed in specific PCR assays to be free of nucleic acids of viruses used in validation studies.
Positive control
The External RNA Controls Consortium (ERCC, ThermoFisher Scientific, USA) has created a commercial kit for use as a common set of RNA controls for various molecular platforms including NGS (35). We used ERCC spike-in mix 1 in a salmon sperm DNA background as a technical post-extraction control to monitor the performance of the library and capture hybridization workflow as well as spillover/contamination events of sample sequences (viral or human) into ERCC/salmon sperm or vice versa. The capture probe set was designed to include probes matching only half of the ERCC RNA control set that represented the various template sizes and concentrations included in the set to specifically measure the efficacy of the capture enrichment.
Extractions
TNA was extracted either from 300 µL of plasma or 250 µL of anterior nasal swabs collected in VTM on the NucliSENS easyMAG platform (bioMérieux, France). A no-template control (nuclease-free water) was included in each extraction run.
Library preparation, capture hybridization, and sequencing
For the plasma samples, libraries were prepared as per our published protocol (16). For analyses of respiratory samples, we employed Twist library preparation kits and Twist Fast Hybridization Reagents (Twist Biosciences, USA). Amplified and purified barcoded libraries were pooled and set up for the capture using VirCapSeq-VERT probes and using either Roche Hybridization Kits (Roche, USA) or Twist Fast Hybridization Reagents (Twist Biosciences, USA). The enriched pools were sequenced on Illumina NextSeq instruments. Our target range was 10 million raw reads per sample.
Creating contrived samples and analytical validation studies designs
Analytical validation was done with contrived samples, comprised of quantified live cultured viral isolates or viral TNA from SARS-CoV-2, spiked into negative background matrices. Single donor plasma from 5 individuals sourced from the New York Blood Center and nasal swabs collected from 10 anonymized volunteers in 2 mL VTM were screened by VirCapSeq-VERT. A sample was defined as negative if it did not contain sequences of any of the viruses used in the validation. The acceptable negative background matrices used were one single donor plasma and a pool of nasal swabs in VTM. The viral isolates were sourced from our own archived repository and from commercial sources (ATCC, USA; Zeptometrix, USA). Viruses were quantified and serially diluted in 10-fold dilutions, ranging from 500,000 copies/mL to 0.5 copies/mL for nasal swabs and from 500,000 copies/mL to five copies/mL for plasma, due to the limited single donor sample volume.
Using the LoD data, we proceeded to determine repeatability and reproducibility, and accuracy in differential diagnosis of mixed infections. Repeatability and reproducibility assays were done at 5x LoD for each virus. Following guidance from CLEP, three independent runs, with each run containing three replicates of all viruses, were conducted by three different operators, starting from distinct extractions, on three separate starting dates. For mixed infections, samples were generated that contained one agent at LoD and another at a higher concentration.
Clinical concordance
We examined diagnostic concordance between VirCapSeq-VERT and approved qPCR assays. Clinical samples (plasma and nasal swabs in VTM) were sourced through the New York Presbyterian Hospital (NYP) network and from Associated Regional and University Pathologists, Inc. (ARUP Labs, USA). ARUP samples included 30 plasma specimens that had been tested using qPCR for the following viruses: BK Polyomavirus (BKPyV), human herpesvirus 5 (HHV-5), hepatitis B virus (HBV), human parvovirus B19 (B19V), human herpesvirus 4 (HHV-4), human herpes virus 6 (HHV-6), and hepatitis C virus (HCV) based on availability. Additionally, to test assay performance for viruses for which we had no relevant clinical specimens, live virus at concentrations ranging from 10,000 to 100 copies/mL were seeded in the negative plasma diluent to create contrived specimens [10 adenovirus C (ADV-C), 5 coxsackievirus B4 (CV-B4), 6 enterovirus A71 (EV-A71), 10 human herpesvirus 1 (HHV-1), 11 ZIKV, 2 BKPyV, 10 (HBV), and 4 B19V]. For respiratory infections, we tested 50 banked and de-identified clinical nasal swab specimens that had been previously tested for a range of respiratory viruses and were made available to us through NYP.
Data analysis, interpretation, and reporting
Sequencing data sets were analyzed using an automated bioinformatics pipeline and a custom viral database. Demultiplexed fastq files had adapters trimmed using Cutadapt program (36). The reads were then quality filtered and end-trimmed with PRINSEQ software (37). The host reads were removed by mapping quality-filtered reads against a custom host reference database using Bowtie2 mapper (38). Then reads that match any of these criteria were removed: shorter than 50 bases after adapter trimming; reads with Q scores below 25; low complexity reads with an entropy value ≤70; and those with 20 or more Ns. The first 9 bases of the remaining reads were trimmed as the quality tends to be inherently poor. The final host-subtracted reads were subjected to homology search using GenBank MegaBLAST against a curated custom viral database that was created by downloading viral sequences from GenBank and removing faulty and misannotated sequences. All reads were subsequently remapped to the reference viral sequences identified by the initial homology search to validate the MegaBLAST results. Finally, stringency filters including e-value of ≤1.00E-35, identity of ≥95%, alignment lengths of ≥100 nucleotides (nt), minimum number of unique reads aligned, minimum number of regions ≥ 100 nt, and cumulative minimum length of ≥500 nt were applied. All viral sequences that were identified at this stage were included in the final results report.
RESULTS
Limits of detection and linearity
LoDs and performance of VirCapSeq-VERT in the context of virus quantity were determined using qPCR quantitated virus stocks or TNA (SARS-CoV-2) serially 10-fold diluted in the respective background matrix (plasma or nasal swab in VTM), starting at a concentration of 500,000 copies/mL. A LoD of 5 copies/mL was determined in plasma for ADV-C, coxsackieviruses (CV) A16 and B4, EV-A71 and HHV-1 and HHV-5, and in nasal swabs for EV-D68 and human parainfluenza viruses (HPIV) 1 and 2. An LoD of 50 copies/mL was determined in plasma for BKPyV, HBV, HHV-2, B19V, ZIKV, and in nasal swabs for ADV-C, coronaviruses 229E, OC43, and SARS-CoV-2, human metapneumovirus (HMPV), RSV, HPIV-3, and influenza viruses (FLUV) A and B. Linearity data for plasma and respiratory swab samples are shown in Fig. 2 and LoDs are listed in Table 2; Tables S1 and S2 shows raw and normalized read counts and the fraction of viral reads per sample.
Fig 2.
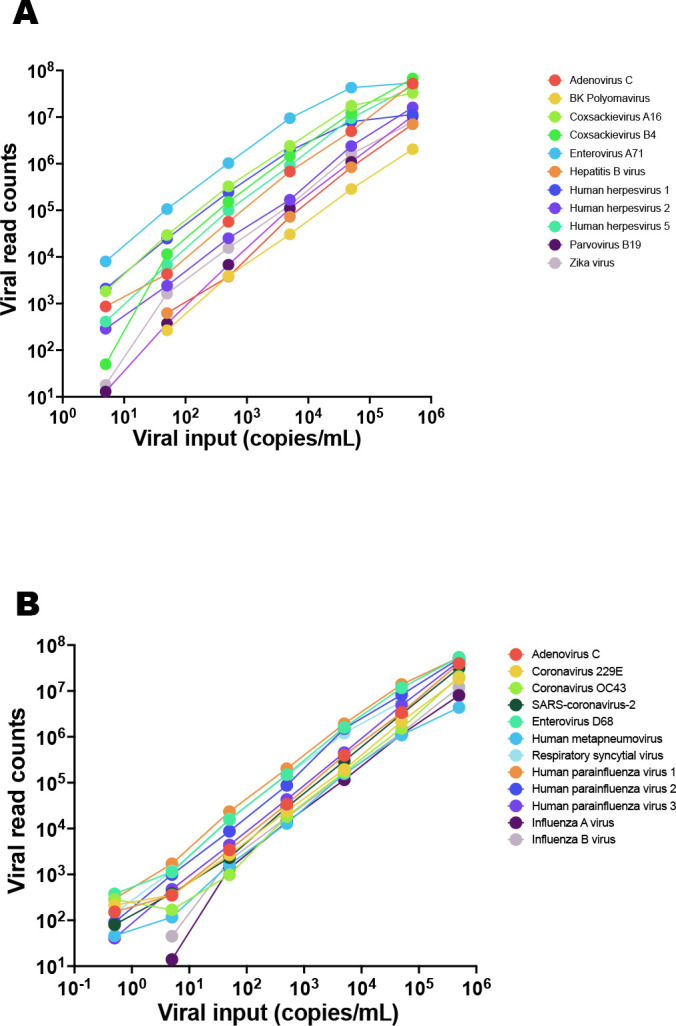
Linearity data for (A) systemic and (B) respiratory infection test validation. Virus stock was serially diluted in sample matrix (plasma or nasal swabs in VTM, respectively) that had been tested negative for the viruses used for validation, prior to processing for VirCapSeq-VERT.
TABLE 2.
Limits of detection (LoD) for 22 representative viruses used for validation study in plasma and nasal swab samples
| Plasma | Nasal swab | ||
|---|---|---|---|
| Virus | LoD (copies/ml) | Virus | LoD (copies/ml) |
| Adenovirus 1 | 5 | Adenovirus 1 | 50 |
| BK Polyomavirus | 50 | Coronavirus 229E | 50 |
| Coxsackievirus A16 | 5 | Coronavirus OC43 | 50 |
| Coxsackievirus B4 | 5 | SARS-CoV-2 | 50 |
| Enterovirus A71 | 5 | Enterovirus D68 | 5 |
| Hepatitis B | 50 | Human metapneumovirus | 50 |
| Human herpesvirus 1 | 5 | Respiratory syncytial virus | 50 |
| Human herpesvirus 2 | 50 | Human parainfluenza virus 1 | 5 |
| Human herpesvirus 5 | 5 | Human parainfluenza virus 2 | 5 |
| Parvovirus B19 | 50 | Human parainfluenza virus 3 | 50 |
| Zika virus | 50 | Influenza A virus | 50 |
| Influenza B virus | 50 | ||
Repeatability and reproducibility
Based on the LoD information, contrived specimens were created at 5x LoD for each virus to measure intra-run repeatability and inter-run reproducibility. Three independent runs, each containing triple replicates of every virus, were conducted by three different operators, starting from independent extractions, on three separate starting dates. VirCapSeq-VERT achieved a high degree of precision for all viruses tested with an overall coefficient of variance of 18% for plasma and 10% for respiratory swab samples. Data for both sample types are plotted in Fig. 3.
Fig 3.
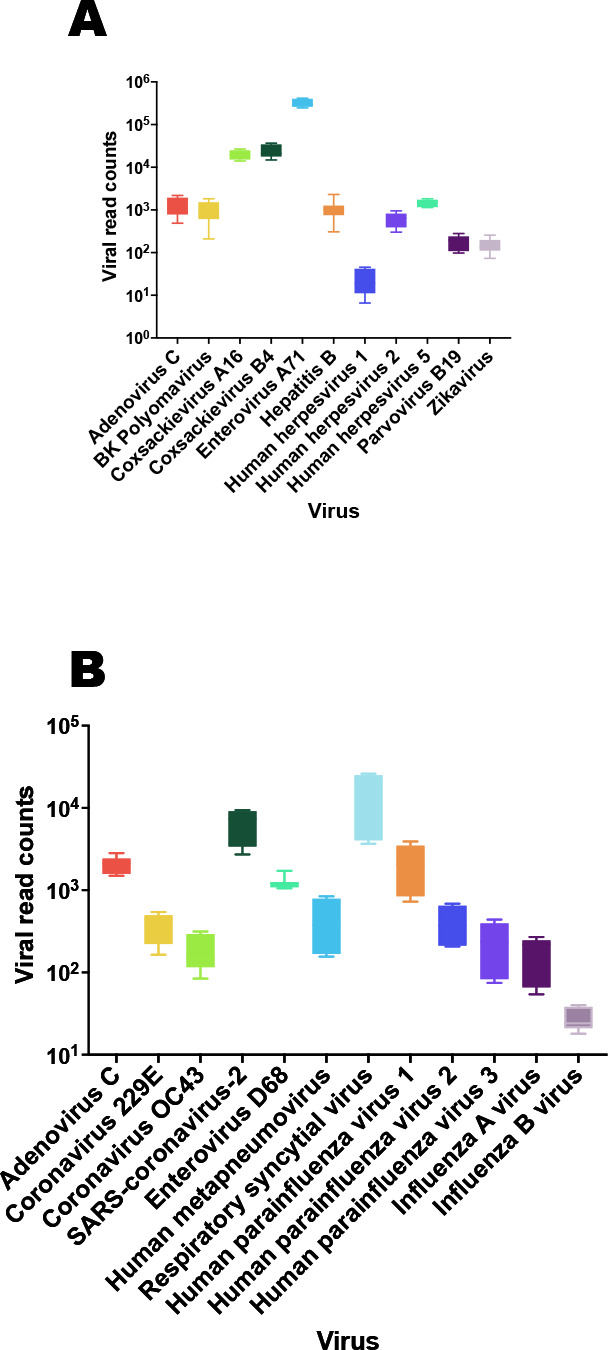
Repeatability and reproducibility for the (A) systemic and (B) respiratory test validation experiments. Negative specimen matrices (plasma or nasal swabs in VTM) were spiked with viruses at 5x their respective LoD (see Table 2). The boxplots represent values for the 10–90% of the normalized viral read counts after host subtraction.
Coinfections/mixed infections
We tested for the detection of coinfections using samples that contained one agent at LoD and another at 1000x LoD. VirCapSeq-VERT accurately identified coinfection in 25 samples (Tables 3 and 4). We did not detect HHV-5 at the LoD in one sample in the presence of EV-A71 at 50,000 copies/mL (Table 3). On recommendations from CLEP, we validated the LoD of 5 copies/mL for HHV-5 by generating another 10 replicates of HHV-5 in plasma at LoD and all were correctly detected (Table S3).
TABLE 3.
Detection of co-infections in plasma samples
| Co-infections | |||||||
|---|---|---|---|---|---|---|---|
| Mix | Virus | Viral Load | Conc./mL | Detected | Total reads | Viral Reads | Viral Reads/M |
| M1 | Adenovirus 1 | High | 5.00E + 04 | + | 3,048,365 | 795,972 | 261,114 |
| Coxsackievirus A16 | LoD | 5.00E + 00 | + | 1,404 | 461 | ||
| M2 | Adenovirus 1 | LoD | 5.00E + 00 | + | 2,924,053 | 85 | 29 |
| Coxsackievirus A16 | High | 5.00E + 04 | + | 438,814 | 150,070 | ||
| M3 | Human herpesvirus 5 | High | 5.00E + 04 | + | 2,186,442 | 2,329 | 1,066 |
| Enterovirus A71 | LoD | 5.00E + 00 | + | 16,663 | 7,621 | ||
| M4 | Human herpesvirus 5 | LoD | 5.00E + 00 | - | 37,343,993 | 10 | 1 |
| Enterovirus A71 | High | 5.00E + 04 | + | 30,033,914 | 804,250 | ||
| M5 | BK polyomavirus | LoD | 5.00E + 01 | + | 2,310,452 | 53 | 28,372 |
| Hepatitis B virus | High | 5.00E + 04 | + | 65,553 | 24 | ||
| M6 | Human herpesvirus 1 | High | 5.00E + 04 | + | 4,824,399 | 1,418,998 | 294,130 |
| Human parvovirus B19 | LoD | 5.00E + 01 | + | 126 | 26 | ||
| M7 | Human herpesvirus 1 | LoD | 5.00E-01 | + | 2,115,787 | 224 | 106 |
| Human parvovirus B19 | High | 5.00E + 04 | + | 222,780 | 105,294 | ||
| M8 | Human herpesvirus 2 | High | 5.00E + 04 | + | 3,056,605 | 474,699 | 325 |
| Zika virus | LoD | 5.00E + 01 | + | 994 | 155,303 | ||
| M9 | Human herpesvirus 2 | LoD | 5.00E-01 | + | 2,386,911 | 606 | 4,328 |
| Zika virus | High | 5.00E + 04 | + | 10,331 | 254 | ||
| M10 | Coxsackievirus B4 | High | 5.00E + 04 | + | 3,479,489 | 948,246 | 272,525 |
| Enterovirus A71 | LoD | 5.00E + 01 | + | 19,391 | 5,573 | ||
| M11 | Coxsackievirus B4 | LoD | 5.00E-01 | + | 97,052,301 | 3,361 | 35 |
| Enterovirus A71 | High | 5.00E + 04 | + | 79,274,785 | 816,825 | ||
| M12 | BK polyomavirus | High | 5.00E + 05 | + | 24,259,301 | 4,462 | 184 |
| Hepatitis B virus | High | 5.00E + 05 | + | 8,042 | 332 | ||
| Human herpesvirus 1 | High | 5.00E + 05 | + | 37,386 | 1,542 | ||
| Human parvovirus B19 | High | 5.00E + 05 | + | 41,967 | 1,730 | ||
TABLE 4.
Detection of coinfections in nasal swab samples
| Coinfections | |||||||
|---|---|---|---|---|---|---|---|
| Mix | Virus | Viral Load | Concentration Copies/mL | Detected | Total reads | Viral Reads | Viral Reads/M |
| M1 | Influenza A virus | LoD | 5.00E + 01 | + | 41,026,700 | 1,050 | 26 |
| Coronavirus OC43 | High | 5.00E + 04 | + | 35,158,111 | 856,957 | ||
| M2 | Influenza A virus | High | 5.00E + 04 | + | 3,517,029 | 110,729 | 31,484 |
| Coronavirus OC43 | LoD | 5.00E + 01 | + | 1,524 | 434 | ||
| M3 | Adenovirus C | LoD | 5.00E + 01 | + | 4,486,231 | 17,335 | 3,864 |
| Human metapneumovirus | High | 5.00E + 04 | + | 1,112,083 | 247,888 | ||
| M4 | Adenovirus C | High | 5.00E + 04 | + | 7,830,480 | 3,934,006 | 502,397 |
| Human metapneumovirus | LoD | 5.00E + 01 | + | 1,253 | 161 | ||
| M5 | Human parainfluenza virus 2 | LoD | 5.00E + 00 | + | 18,636,157 | 1,117 | 60 |
| Respiratory syncytial virus | High | 5.00E + 03 | + | 12,985,557 | 696,794 | ||
| M6 | Human parainfluenza virus 2 | High | 5.00E + 03 | + | 4,563,732 | 1,327,348 | 290,847 |
| Respiratory syncytial virus | LoD | 5.00E + 00 | + | 105,840 | 23,192 | ||
| M7 | Influenza B virus | LoD | 5.00E + 01 | + | 2,994,401 | 493 | 165 |
| Human parainfluenza virus 3 | High | 5.00E + 04 | + | 123,490 | 41,240 | ||
| M8 | Influenza B virus | High | 5.00E + 04 | + | 2,698,984 | 122,712 | 45,466 |
| Human parainfluenza virus 3 | LoD | 5.00E + 01 | + | 170 | 63 | ||
| M9 | Coronavirus 229E | LoD | 5.00E + 01 | + | 22,887,286 | 4,578 | 200 |
| Enterovirus D68 | High | 5.00E + 04 | + | 18,213,661 | 795,799 | ||
| M10 | Coronavirus 229E | High | 5.00E + 03 | + | 5,676,372 | 2,873,451 | 506,213 |
| Enterovirus D68 | LoD | 5.00E + 00 | + | 5,660 | 997 | ||
| M11 | Human parainfluenza virus 1 | LoD | 5.00E + 00 | + | 2,687,212 | 2,394 | 891 |
| SARS-CoV-2 | High | 5.00E + 03 | + | 2,530,082 | 941,527 | ||
| M12 | Human parainfluenza virus 1 | High | 5.00E + 04 | + | 21,726,230 | 7,880,254 | 362,707 |
| SARS-CoV-2 | LoD | 5.00E + 01 | + | 31,929 | 1,470 | ||
| M13 | Influenza A virus | High | 5.00E + 04 | + | 25,261,588 | 72,567 | 2,873 |
| Respiratory syncytial virus | High | 5.00E + 04 | + | 6,748,813 | 267,157 | ||
| Influenza B virus | High | 5.00E + 04 | + | 143,347 | 5,675 | ||
| SARS-CoV-2 | High | 5.00E + 04 | + | 8,634,842 | 341,817 | ||
Diagnostic concordance
For systemic infections, we tested clinical specimens sourced commercially. Clinical specimens positive for additional viruses to those used for analytical validation (Table 1) were included based on availability. These were HHV-4, HHV-6, and HCV-positive specimens. Findings with these specimens provided additional support for the utility of the platform in detecting and differentiating between related members within a virus family, subfamily, or genus. VirCapSeq-VERT results matched previous diagnoses with the exception of three HHV-4 samples and two HBV samples. The provided qPCR Ct values for the negative HHV-4 samples were 42.7, 42.1, and 40.6. The negative HBV samples had a load <10 and 20 IU. We conducted independent qPCR with CE-cleared tests, confirmed our VirCapSeq-VERT results, and concluded that discordance may reflect sample degradation from the date of the initial clinical testing and receipt for VirCapSeq-VERT analysis. CLEP advised us to create additional contrived material for viruses not adequately represented in the available clinical samples. Table 5 lists data for all plasma samples examined during clinical validation. VirCapSeq-VERT demonstrated a 100% clinical sensitivity and specificity on the valid samples. For respiratory infections, we tested 50 banked clinical nasal swab specimens that were diagnosed for respiratory pathogens using the BioFire Diagnostics FilmArray Respiratory Panel. We were blinded at the time of sample processing and data analysis but were provided results of the initial molecular characterization. Some of these samples had coinfections at the time of initial testing (see Table S4). There were instances where we did not detect the purported virus by VirCapSeq-VERT analysis. Accordingly, we conducted additional qPCR tests to examine whether discordance represented the failure of VirCapSeq-VERT. Ten of eleven discordant samples were found to be negative in subsequent CE or FDA EUA qPCR assays. These results suggest that discordance was due to sample degradation between the initial tests and later VirCapSeq-VERT and qPCR analyses. One of the discordant samples was positive for adenovirus sequence in qPCR after 36 cycles, demonstrating a low viral load in the specimen. Table 6 and S4 summarize the results of the clinical specimen testing and concordance between the BioFire assay and VirCapSeq-VERT, and results of CE or FDA EUA confirmation qPCR assays for negative discordant specimens. Viral genome coverages across a range of viral reads for a few clinical specimens are displayed in Fig. S1.
TABLE 5.
Data demonstrating diagnostic concordance with qPCR assay for plasma specimens
| Virus | # of specimen tested (true clinical +contrived) | VirCapSeq results (positive/tested) | In-house qPCR results on discordant samples (positive/tested) |
|---|---|---|---|
| Adenovirus 1 | 10 (0 + 10) | 10/10 | |
| BK polyomavirus | 10 (8 + 2) | 10/10 | |
| Coxsackievirus B4 | 5 (0 + 5) | 5/5 | |
| Enterovirus A71 | 6 (0 + 6) | 6/6 | |
| Human herpesvirus 4a | 4 (4 + 0) | 1/4 | 0/3 |
| Human herpesvirus 5 | 4 (4 + 0) | 4/4 | |
| Human herpesvirus 1 | 10 (0 + 10) | 10/10 | |
| Human herpesvirus 6a | 4 (4 + 0) | 4/4 | |
| Hepatitis B virus | 14 (4 + 10) | 12/14 | 0/2 |
| Hepatitis C virusa | 2 (2 + 0) | 2/2 | |
| Human parvovirus B19 | 8 (4 + 4) | 8/8 | |
| Zika virus | 11 (0 + 11) | 11/11 |
Included based on clinical specimen availability but not part of the validation test.
TABLE 6.
Data demonstrating diagnostic concordance with BioFire FilmArrays for the respiratory specimens
| Virus in specimen | BioFire results (positive/tested) | VirCapSeq-VERT results (positive/tested) | In-house qPCR results on discordant samples (positive/tested) |
|---|---|---|---|
| Adenovirus | 12/12 | 9/12 | 1/3 |
| Coronavirus OC43 | 3/3 | 3/3 | |
| SARS-CoV-2 | 1/1 | 0/1 | 0/1 |
| Enterovirus/rhinovirus | 14/14 | 13/14 | 0/1 |
| Influenza A virus | 5/5 | 4/5 | 0/1 |
| Human metapneumovirus | 4/4 | 4/4 | |
| Human parainfluenza virus 3 | 5/5 | 4/5 | 0/1 |
| Respiratory syncytial virus | 15/15 | 12/15 | 0/3 |
VirCapSeq-VERT provided insights that were not achieved with PCR assays and in two instances detected coinfections (RSV in one sample and influenza C in another, Table S4) that were not indicated by the initial tests. For nasal swab testing VirCapSeq-VERT demonstrated significant concordance for valid specimens with a clinical sensitivity of 98% and specificity of 100%.
DISCUSSION
NGS has transformed the field of pathogen discovery and surveillance (39–42). It has nonetheless not been widely applied in clinical microbiology laboratories due to cost, complexity, insensitivity, and lengthy turnaround times. Capture sequencing addresses each of these limitations. Through positive selection, relevant targets are enriched, while nonrelevant nucleic acids from the host and environment are substantially reduced. This approach results in two to three orders of magnitude enhancement in sensitivity at a reduced cost. An additional advantage of capture sequencing is that it mitigates Health Insurance Portability and Accountability Act (HIPAA) concerns because host sequence data are not collected.
In previous studies, VirCapSeq-VERT has enabled differential diagnosis of unexplained febrile illnesses in Tanzania, clusters of severe respiratory infections in Uganda, as well as meningitis in the UK, and myocarditis in Canada and Switzerland (19, 23, 25, 29). These studies were conducted under the research use only designation because, at the time the work was conducted, NGS-based infectious diseases assays, including VirCapSeq-VERT, were not certified as diagnostic tests. A critical step in the transition of NGS from research facilities to clinical microbiology laboratories is validation by regulatory agencies based on the demonstration of assay performance. We report here the validation of the VirCapSeq-VERT platform for detecting viral nucleic acid in plasma and nasal swabs. With LoDs established at ≤50 copies/mL it has sensitivity comparable to qPCR, is highly reproducible, can detect multiple viruses in a single sample, and matches the results obtained with accepted gold standard assays such qPCRs and FilmArrays.
A key challenge we encountered in our validation efforts was accurate quantitation of the viral cultures. Other groups have reported sensitivity in terms of plaque forming units or 50% tissue culture infectious dose (43–46). Both of these methods are cumbersome and subjective, and at times yield conflicting results for identical starting cultures in a direct comparison (47, 48). Other methods used for virus quantitation include qPCR and western blots (47). Though faster and easier to use, qPCRs nevertheless pose a similar challenge where two independent assays targeting the same virus can yield differing titers (46, 49). We observed this discrepancy firsthand when we used multiple PCR assays in parallel. For consistency, we used titers quantified by regulatory approved assays. One limitation of our validation is that we did not have the resources to conduct additional 2-fold dilutions or Probit analyses to determine LoDs with better accuracy and established them based on our current data for the 10-fold dilutions.
VirCapSeq-VERT achieved results that are highly concordant with other molecular platforms currently used in clinical microbiology laboratories. We do not have information about the nature of the PCR assays that were used for the initial tests on plasma samples procured from ARUP Labs. The respiratory specimens were previously tested using BioFire FilmArrays. It has been reported that the BioFire FilmArray is unable to differentiate between adenovirus species and between entero- and rhinoviruses (43). As anticipated, VirCapSeq-VERT detected as well as enabled species identification for these viruses in clinical specimens. Additionally, VirCapSeq-VERT identified coinfections that were not indicated in the clinical tests.
Clinical samples also included sequences of anelloviruses, herpesviruses HHV-6, HHV-7, HHV-8, papillomaviruses, and endogenous retroviruses. Their significance is uncertain as they are not known to be associated with the acute febrile illness that led to clinical analysis.
VirCapSeq-VERT is designed to detect all vertebrate viruses. It is not feasible to test all known viruses; however, the representative viruses we did test varied in genome structure and length and included both RNA and DNA viruses. The strength of this platform is that it is not limited to pathogens with DNA genomes (50), a limited repertoire of pathogens (51), and that it has sensitivity comparable to qPCR and has obtained regulatory approval for clinical testing from the NYSDOH. Additionally, although we have not yet obtained regulatory approval for expedited processing, we have preliminary data to indicate that the time required from sample receipt to agent identification can be reduced from the current 24–36 hours to around 12 hours. As the NGS field continues to evolve, we anticipate that clinicians will have access to actionable data in a time frame that reduces mortality, morbidity, and healthcare costs.
ACKNOWLEDGMENTS
We are grateful to Kelly Magnus for assistance in manuscript preparation and to the Department of Health and Human Services; Administration for Strategic Preparedness and Response; Biomedical Advanced Research and Development Authority (75A50122C00012), NIH/NIAID (Center for Research in Diagnostics and Discovery, U19AI109761), and the Skoll Foundation (Global Alliance for Preventing Pandemics, 20–45017) for federal and private financial support.
Contributor Information
W. Ian Lipkin, Email: wil2001@cumc.columbia.edu.
Alexander Mellmann, Westfalische Wilhelms-Universitat Munster, Münster, Germany.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jcm.00612-23.
Supplemental figure and tables.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Girum T, Lentiro K, Geremew M, Migora B, Shewamare S, Shimbre MS. 2021. Optimal strategies for COVID-19 prevention from global evidence achieved through social distancing, stay at home, travel restriction and lockdown: a systematic review. Arch Public Health 79:150. doi: 10.1186/s13690-021-00663-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guan W-J, Chen R-C, Zhong N-S. 2020. Strategies for the prevention and management of Coronavirus disease 2019. Eur Respir J 55:2000597. doi: 10.1183/13993003.00597-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hakim MS, Widyaningsih SA. 2023. The recent re-emergence of human monkeypox: would it become endemic beyond Africa? J Infect Public Health 16:332–340. doi: 10.1016/j.jiph.2023.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guan H, Gul I, Xiao C, Ma S, Liang Y, Yu D, Liu Y, Liu H, Zhang CY, Li J, Qin P. 2023. Emergence, phylogeography, and adaptive evolution of mpox virus. New Microbes New Infect 52:101102. doi: 10.1016/j.nmni.2023.101102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pallansch MA. 2022. Circulating Poliovirus in New York - new instance of an old problem. N Engl J Med 387:1725–1728. doi: 10.1056/NEJMp2212115 [DOI] [PubMed] [Google Scholar]
- 6. Rai A, Uwishema O, Uweis L, El Saleh R, Arab S, Abbass M, Wellington J, Musabirema F, Adanur I, Patrick Onyeaka CV. 2022. Polio returns to the USA: an epidemiological alert. Ann Med Surg (Lond) 82:104563. doi: 10.1016/j.amsu.2022.104563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benschop KS, Albert J, Anton A, Andrés C, Aranzamendi M, Armannsdóttir B, Bailly J-L, Baldanti F, Baldvinsdóttir GE, Beard S, Berginc N, Böttcher S, et al. 2021. Re-emergence of enterovirus D68 in Europe after easing the COVID-19 lockdown, September 2021. Euro Surveill 26:2100998. doi: 10.2807/1560-7917.ES.2021.26.45.2100998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shah MM, Perez A, Lively JY, Avadhanula V, Boom JA, Chappell J, Englund JA, Fregoe W, Halasa NB, Harrison CJ, Hickey RW, et al. 2021. Enterovirus D68-associated acute respiratory illness ─ new vaccine surveillance network, United States, July-November 2018-2020. MMWR Morb Mortal Wkly Rep 70:1623–1628. doi: 10.15585/mmwr.mm7047a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pruccoli G, Castagno E, Raffaldi I, Denina M, Barisone E, Baroero L, Timeus F, Rabbone I, Monzani A, Terragni GM, Lovera C, Brach Del Prever A, Garazzino S. 2023. The importance of RSV epidemiological surveillance: multicenter observational study of RSV infection during the COVID-19 pandemic. Viruses 15:280. doi: 10.3390/v15020280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghosh A, Annigeri S, Hemram SK, Dey PK, Mazumder S. 2022. Clinico-demographic profile and predictors of intensive care need in children with respiratory syncytial virus-associated acute lower respiratory illness during its recent outbreak alongside ongoing COVID-19 pandemic: an Eastern Indian perspective. Indian J Crit Care Med 26:1210–1217. doi: 10.5005/jp-journals-10071-24350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rana MS, Alam MM, Ikram A, Salman M, Mere MO, Usman M, Umair M, Zaidi SSZ, Arshad Y. 2021. Emergence of measles during the COVID-19 pandemic threatens Pakistan's children and the wider region. Nat Med 27:1127–1128. doi: 10.1038/s41591-021-01430-6 [DOI] [PubMed] [Google Scholar]
- 12. Abbasi J. 2023. Amid Ohio measles outbreak, new global report warns of decreased vaccination during COVID-19 pandemic. JAMA 329:9–11. doi: 10.1001/jama.2022.23241 [DOI] [PubMed] [Google Scholar]
- 13. WHO . 1996. Infectious diseases kill over 17 million people a year: WHO warns of global crisis. World Health Organization News Room; [PubMed] [Google Scholar]
- 14. Fonkwo PN. 2008. Pricing infectious disease. the economic and health implications of infectious diseases. EMBO Rep 9 Suppl 1:S13–7. doi: 10.1038/embor.2008.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schlaberg R, Chiu CY, Miller S, Procop GW, Weinstock G, Professional Practice Committee and Committee on Laboratory Practices of the American Society for Microbiology, Microbiology Resource Committee of the College of American Pathologists . 2017. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch Pathol Lab Med 141:776–786. doi: 10.5858/arpa.2016-0539-RA [DOI] [PubMed] [Google Scholar]
- 16. Briese T, Kapoor A, Mishra N, Jain K, Kumar A, Jabado OJ, Lipkin WI. 2015. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 6:e01491–15. doi: 10.1128/mBio.01491-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jarvie T. 2005. Next generation sequencing technologies. Drug Discov Today Technol 2:255–260. doi: 10.1016/j.ddtec.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 18. Anderson ME, Nagy-Szakal D, Jain K, Patrone CC, Frattini MG, Lipkin WI, Geskin LJ. 2018. Highly sensitive Virome capture sequencing technique VirCapSeq-VERT identifies partial noncoding sequences but no active viral infection in cutaneous T-cell lymphoma. J Invest Dermatol 138:1671–1673. doi: 10.1016/j.jid.2018.01.024 [DOI] [PubMed] [Google Scholar]
- 19. Cummings MJ, Tokarz R, Bakamutumaho B, Kayiwa J, Byaruhanga T, Owor N, Namagambo B, Wolf A, Mathema B, Lutwama JJ, Schluger NW, Lipkin WI, O’Donnell MR. 2019. Precision surveillance for viral respiratory pathogens: virome capture sequencing for the detection and genomic characterization of severe acute respiratory infection in Uganda. Clin Infect Dis 68:1118–1125. doi: 10.1093/cid/ciy656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dugue R, Cay-Martínez KC, Thakur KT, Garcia JA, Chauhan LV, Williams SH, Briese T, Jain K, Foca M, McBrian DK, Bain JM, Lipkin WI, Mishra N. 2020. Neurologic manifestations in an infant with COVID-19. Neurology 94:1100–1102. doi: 10.1212/WNL.0000000000009653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Franke N, Bette M, Marquardt A, Briese T, Lipkin WI, Kurz C, Ehrenreich J, Mack E, Baying B, Beneš V, Rodepeter FR, Neff A, Teymoortash A, Eivazi B, Geisthoff U, Stuck BA, Bakowsky U, Mandic R. 2018. Virome analysis reveals no association of head and neck vascular anomalies with an active viral infection. In Vivo 32:1323–1331. doi: 10.21873/invivo.11382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldstein T, Anthony SJ, Gbakima A, Bird BH, Bangura J, Tremeau-Bravard A, Belaganahalli MN, Wells HL, Dhanota JK, Liang E, et al. 2018. Author correction: the discovery of bombali virus adds further support for bats as hosts of ebolaviruses. Nat Microbiol 3:1486. doi: 10.1038/s41564-018-0315-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heidecker B, Williams SH, Jain K, Oleynik A, Patriki D, Kottwitz J, Berg J, Garcia JA, Baltensperger N, Lovrinovic M, Baltensweiler A, Mishra N, Briese T, Hanson PJ, Lauten A, Poller W, Leistner DM, Landmesser U, Enseleit F, McManus B, Lüscher TF, Lipkin WI. 2020. Virome sequencing in patients with myocarditis. Circ Heart Fail 13:e007103. doi: 10.1161/CIRCHEARTFAILURE.120.007103 [DOI] [PubMed] [Google Scholar]
- 24. Kim KW, Horton JL, Pang CNI, Jain K, Leung P, Isaacs SR, Bull RA, Luciani F, Wilkins MR, Catteau J, Lipkin WI, Rawlinson WD, Briese T, Craig ME. 2019. Higher abundance of enterovirus a species in the gut of children with islet autoimmunity. Sci Rep 9:1749. doi: 10.1038/s41598-018-38368-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGill F, Tokarz R, Thomson EC, Filipe A, Sameroff S, Jain K, Bhuva N, Ashraf S, Lipkin WI, Corless C, Pattabiraman C, Gibney B, Griffiths MJ, Geretti AM, Michael BD, Beeching NJ, McKee D, Hart IJ, Mutton K, Jung A, Miller A, Solomon T. 2022. Viral capture sequencing detects unexpected viruses in the cerebrospinal fluid of adults with meningitis. J Infect 84:499–510. doi: 10.1016/j.jinf.2021.12.042 [DOI] [PubMed] [Google Scholar]
- 26. Mishra N, Ng TFF, Marine RL, Jain K, Ng J, Thakkar R, Caciula A, Price A, Garcia JA, Burns JC, Thakur KT, Hetzler KL, Routh JA, Konopka-Anstadt JL, Nix WA, Tokarz R, Briese T, Oberste MS, Lipkin WI, Biron CA, Whitley R, Jacobson S. 2019. Antibodies to enteroviruses in cerebrospinal fluid of patients with acute flaccid Myelitis. mBio 10:e01903-19. doi: 10.1128/mBio.01903-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Souza TML, Vieira YR, Delatorre E, Barbosa-Lima G, Luiz RLF, Vizzoni A, Jain K, Miranda MM, Bhuva N, Gogarten JF, Ng J, Thakkar R. 2019. Emergence of the East-central-South-African genotype of Chikungunya virus in Brazil and the city of Rio de Janeiro may have occurred years before surveillance detection. Sci Rep 9:2760. doi: 10.1038/s41598-019-39406-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tokarz R, Hyams JS, Mack DR, Boyle B, Griffiths AM, LeLeiko NS, Sauer CG, Shah S, Markowitz J, Baker SS, Rosh J, Baldassano RN, Kugathasan S, Walters T, Tagliafierro T, Sameroff S, Lee B, Che X, Oleynik A, Denson LA, Lipkin WI. 2019. Characterization of stool virome in children newly diagnosed with moderate to severe ulcerative colitis. Inflamm Bowel Dis 25:1656–1662. doi: 10.1093/ibd/izz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams SH, Cordey S, Bhuva N, Laubscher F, Hartley M-A, Boillat-Blanco N, Mbarack Z, Samaka J, Mlaganile T, Jain K, d’Acremont V, Kaiser L, Lipkin WI. 2018. Investigation of the plasma virome from cases of unexplained febrile illness in tanzania from 2013 to 2014: a comparative analysis between unbiased and VirCapSeq-VERT high-throughput sequencing approaches. mSphere 3:e00311-18. doi: 10.1128/mSphere.00311-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gogarten JF, Ulrich M, Bhuva N, Garcia J, Jain K, Lee B, Löhrich T, Oleynik A, Couacy-Hymann E, Fuh Neba T, Mishra N, Briese T, Calvignac-Spencer S, Lipkin WI, Leendertz FH. 2019. A novel orthohepadnavirus identified in a dead Maxwell’s Duiker (Philantomba maxwellii) in tai national park, cote D’ivoire. Viruses 11:279. doi: 10.3390/v11030279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paulson JN, Williams BL, Hehnly C, Mishra N, Sinnar SA, Zhang L, Ssentongo P, Mbabazi-Kabachelor E, Wijetunge DSS, von Bredow B, Mulondo R, Schiff SJ, et al. 2020. Paenibacillus infection with frequent viral coinfection contributes to postinfectious hydrocephalus in Ugandan infants. Sci Transl Med 12. doi: 10.1126/scitranslmed.aba0565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goverment US . 2015. Medicare, Medicaid, and CLIA programs; clinical laboratory improvement amendments of 1988 exemption of permit-holding Laboratories in the state of New York. Available from: https://www.federalregister.gov/documents/2015/03/27/2015-07113/medicare-medicaid-and-clia-programs-clinical-laboratory-improvement-amendments-of-1988-exemption-of. Retrieved 15 Apr 2023.
- 33. Mishra N, Ng J, Rakeman JL, Perry MJ, Centurioni DA, Dean AB, Price A, Thakkar R, Angus AG, Williamson P, Delwart E, Carrington C, Sahadeo N, Che X, Briese T, Tokarz R, Lipkin WI. 2019. One-step pentaplex real-time polymerase chain reaction assay for detection of zika, dengue, chikungunya, West Nile viruses and a human housekeeping gene. J Clin Virol 120:44–50. doi: 10.1016/j.jcv.2019.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. PGM . 2020. Accelerated emergency use authorization (EUA) summary the triplex CII-SARS-Cov-2 rRT-PCR test
- 35. Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B. 2011. Synthetic spike-in standards for RNA-seq experiments. Genome Res 21:1543–1551. doi: 10.1101/gr.121095.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin MJEj. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j 17:10. doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 37. Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi: 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rusňáková D, Sedláčková T, Radvák P, Böhmer M, Mišenko P, Budiš J, Bokorová S, Lipková N, Forgáčová-Jakúbková M, Sládeček T, et al. 2022. Systematic genomic surveillance of SARS-CoV-2 virus on Illumina sequencing platforms in the slovak republic-one year experience. Viruses 14:2432. doi: 10.3390/v14112432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wallace ZS, Davis J, Niewiadomska AM, Olson RD, Shukla M, Stevens R, Zhang Y, Zmasek CM, Scheuermann RH. 2022. Early detection of emerging SARS-CoV-2 variants of interest for experimental evaluation. Front Bioinform 2:1020189. doi: 10.3389/fbinf.2022.1020189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hofman P, Bordone O, Chamorey E, Benzaquen J, Schiappa R, Lespinet-Fabre V, Lanteri E, Brest P, Mograbi B, Maniel C, Tanga V, Allegra M, Salah M, Fayada J, Boutros J, Leroy S, Heeke S, Hofman V, Marquette C-H, Ilié M. 2021. Setting-up a rapid SARS-CoV-2 genome assessment by next-generation sequencing in an academic hospital center (LPCE, Louis Pasteur Hospital, Nice, France).. Front Med (Lausanne) 8:730577. doi: 10.3389/fmed.2021.730577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robles-Escajeda E, Mohl JE, Contreras L, Betancourt AP, Mancera BM, Kirken RA, Rodriguez G. 2023. Rapid shift from SARS-CoV-2 delta to Omicron sub-variants within a dynamic southern U.S. Borderplex. Viruses 15:658. doi: 10.3390/v15030658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Babady NE, England MR, Jurcic Smith KL, He T, Wijetunge DS, Tang Y-W, Chamberland RR, Menegus M, Swierkosz EM, Jerris RC, Greene W. 2018. Multicenter evaluation of the ePlex respiratory pathogen panel for the detection of viral and bacterial respiratory tract pathogens in nasopharyngeal swabs. J Clin Microbiol 56:e01658-17. doi: 10.1128/JCM.01658-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen JHK, Lam H-Y, Yip CCY, Wong SCY, Chan JFW, Ma ESK, Cheng VCC, Tang BSF, Yuen K-Y. 2016. Clinical evaluation of the new high-throughput luminex NxTAG respiratory pathogen panel assay for multiplex respiratory pathogen detection. J Clin Microbiol 54:1820–1825. doi: 10.1128/JCM.00517-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang Y-W, Gonsalves S, Sun JY, Stiles J, Gilhuley KA, Mikhlina A, Dunbar SA, Babady NE, Zhang H. 2016. Clinical evaluation of the luminex NxTAG respiratory pathogen panel. J Clin Microbiol 54:1912–1914. doi: 10.1128/JCM.00482-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jullien S, Fitzgerald F, Keddie S, Baerenbold O, Bassat Q, Bradley J, Falconer J, Fink C, Keogh R, Hopkins H, Voice M. 2022. Diagnostic accuracy of multiplex respiratory pathogen panels for influenza or respiratory syncytial virus infections: systematic review and meta-analysis. BMC Infect Dis 22:785. doi: 10.1186/s12879-022-07766-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Masci AL, Menesale EB, Chen W-C, Co C, Lu X, Bergelson S. 2019. Integration of fluorescence detection and image-based automated counting increases speed, sensitivity, and robustness of plaque assays. Mol Ther Methods Clin Dev 14:270–274. doi: 10.1016/j.omtm.2019.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smither SJ, Lear-Rooney C, Biggins J, Pettitt J, Lever MS, Olinger GG. 2013. Comparison of the plaque assay and 50% tissue culture infectious dose assay as methods for measuring filovirus infectivity. J Virol Methods 193:565–571. doi: 10.1016/j.jviromet.2013.05.015 [DOI] [PubMed] [Google Scholar]
- 49. Baylis SA, Wallace P, McCulloch E, Niesters HGM, Nübling CM. 2019. Standardization of nucleic acid tests: the approach of the world health organization. J Clin Microbiol 57:e01056-18. doi: 10.1128/JCM.01056-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blauwkamp TA, Thair S, Rosen MJ, Blair L, Lindner MS, Vilfan ID, Kawli T, Christians FC, Venkatasubrahmanyam S, Wall GD, Cheung A, Rogers ZN, Meshulam-Simon G, Huijse L, Balakrishnan S, Quinn JV, Hollemon D, Hong DK, Vaughn ML, Kertesz M, Bercovici S, Wilber JC, Yang S. 2019. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol 4:663–674. doi: 10.1038/s41564-018-0349-6 [DOI] [PubMed] [Google Scholar]
- 51. Illumina . 2020. Illumina respiratory pathogen ID/AMR panel.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure and tables.