

Abstract
Background
Although the constitutively activated Wnt/β‐catenin signaling pathway plays vital roles in gastric cancer (GC) progression, few Wnt inhibitors are approved for clinical use. Additionally, the clinical significance of long non‐coding RNAs (lncRNAs) in GC intraperitoneal dissemination (IPD) remains elusive. Here, we investigated the function and therapeutic potential of Wnt‐transactivated lncRNA, colon cancer‐associated transcript 5 (CCAT5), in GC metastasis.
Methods
LncRNA‐sequencing assay was performed to document abundance changes of lncRNAs induced by Wnt family member 3A (Wnt3a) and degradation‐resistant β‐catenin (S33Y mutated) in ascites‐derived GC cells with low Wnt activity. Luciferase reporter, Chromatin immunoprecipitation (ChIP)‐re‐ChIP assays were performed to determine how CCAT5 was transcribed. The clinical significance of CCAT5 was examined in 2 cohorts of GC patients. The biological function of CCAT5 was investigated through gain‐ and loss‐of‐function studies. The molecular mechanism was explored through RNA‐sequencing, mass spectrometry, and CRISPR/Cas9‐knocknout system. The therapeutic potential of CCAT5 was examined through RNAi‐based cell xenograft model and patient‐derived xenograft (PDX) model of IPD.
Results
We identified a novel Wnt‐regulated lncRNA, CCAT5, which was transactivated by the β‐catenin/transcription factor 3 (TCF3) complex. CCAT5 was significantly upregulated in GC and predicted poor prognosis. Functional studies confirmed the promotive role of CCAT5 in GC growth and metastasis. Mechanistically, CCAT5 bound to the C‐end domain of signal transducer and activator of transcription 3 (STAT3) and blocks Src homology 2 domain‐containing protein tyrosine phosphatase 1 (SHP‐1)‐mediated STAT3Y705 dephosphorylation, leading to STAT3 nuclear entry and transactivation, thus accelerating GC progression. Furthermore, we demonstrated that both Wnt3a and β‐catenin acted as activator of STAT3 signaling pathway, and the interplay between CCAT5 and STAT3 was functionally essential for Wnt‐drived STAT3 signaling and tumor evolution. Finally, we revealed in vivo si‐CCAT5 selectively attenuated growth and metastasis of Wnthigh GC, but not Wntlow GC. The combination of si‐CCAT5 and oxaliplatin displayed obvious synergistic therapeutic effects on Wnthigh PDX mice.
Conclusions
We identified a novel Wnt‐transactivated lncRNA, CCAT5. Our study revealed a mechanism of STAT3 signaling regulation via canonical Wnt signaling and the functional significance of CCAT5 as critical mediator. We provided conceptual advance that lncRNAs serve as therapeutic targets reversing GC progression.
Keywords: CCAT5, gastric cancer, intraperitoneal dissemination, STAT3, Wnt/β‐catenin
Abbreviations
- GC
gastric cancer
- lncRNAs
long non‐coding RNAs
- IPD
intraperitoneal dissemination
- Wnt3a
Wnt family member
- PDX
patient‐derived xenograft
- TCF3
transcription factor 3
- STAT3
signal transducer and activator of transcription 3
- FBS
fetal bovine serum
- GC‐AL
ascites‐derived GC cell line
- sgRNAs
short guide RNAs
- FISH
fluorescence in situ hybridization
- ChIP
chromatin immunopreciptation
- IHC
immunohistochemistry
- EdU
Ethynyldeoxyuridine
- RNA‐seq
RNA‐sequencing
- RIP
RNA immunoprecipitation
- Co‐IP
co‐immunoprecipitation
- PBS
phosphate buffered saline
- MNX1
motor neuron and pancreatic homeobox 1
- PTPs
protein tyrosine phosphatases
- PTKs
protein tyrosine kinases
- qRT‐PCR
quantitative real‐time PCR
1. BACKGROUND
Gastric cancer (GC) is the third most leading cause of cancer‐related death globally, due to its frequently advanced stage at diagnosis [1, 2]. Despite advances in systemic chemotherapy and targeted therapy, metastases remain the main cause for poor prognosis of GC [3]. Intraperitoneal dissemination (IPD) is one of the most common types of GC metastasis, which usually causes ascites accumulation [3, 4]. The poor peritoneal penetration of current chemotherapeutic drugs greatly limits the treatment options for metastatic GC [3]. Therefore, it is urgent to deepen our understanding of the molecular mechanism of IPD, and various alternative treatment strategies for metastatic GC are to be developed.
Genomic analysis identified aberrant canonical Wnt signaling among 46% (range 43%‐48%) of GC and the top driver pathway highly relevant to gastric carcinogenesis [5, 6, 7]. Deregulated Wnt signaling favors cancer stem cell survival and weaken cadherin‐mediated cell adhesion, and cooperatively promote epithelial‐mesenchymal transition, invasion and metastasis [7, 8]. Hence, blocking Wnt signaling is an attractive therapeutic strategy for GC treatment [7]. The key switch of this pathway is the versatile and tightly regulated protein β‐catenin. In the absence of Wnt signals, cytoplasmic β‐catenin is rapidly phosphorylated by the adenomatous polyposis coli protein (APC)/Axin/glycogen synthase kinase‐3 (GSK‐3)/casein kinase 1 alpha (CK1α) destruction complex and then degraded by the proteasome. Wnt ligands initiate signaling through frizzled class receptor (FZD) receptor and low‐density lipoprotein receptor related protein (LRP) co‐receptor, thereby disrupting the destruction complex, resulting in β‐catenin accumulation and nuclear entry. In the nucleus, β‐catenin binds to the transcription factor 1(TCF1), lymphoid enhancer‐binding factor 1 (LEF1), TCF3, and TCF4 to trans‐activate target genes implicated in carcinogenesis and metastasis [9]. The identification of various novel components and cross‐talk mechanisms has made the Wnt signaling network increasingly complex [10, 11]. Notably, cross‐talk between Wnt signaling and other pathways, such as the IL‐6/STAT3, Hippo‐YAP, PI3K/Akt signaling pathway, etc., contributes greatly to the dysregulation of Wnt signaling and subsequent tumor evolution [12, 13, 14, 15].
Accumulating evidence has extended the functional significance of long non‐coding RNAs (lncRNAs) in driving malignant phenotype and regulation of cellular signaling transduction [16, 17, 18]. The high specificity and ease of detection in tissues and body fluids have spurred the applications of lncRNAs in GC diagnosis and prognosis [19]. However, the therapeutic potential of lncRNAs remains largely unknown [20]. Although numerous inhibitors have been developed to control Wnt activity, none of them has been succeeded to clinical use, and only a few are under clinical trials [21, 22]. Thus, it is of great interest to identify lncRNAs involved in Wnt signaling cascade and explore their clinical feasibility in reversing Wnthigh metastatic GC.
This study identified a novel Wnt‐transactivated lncRNA‐CCAT5, and explored its role in promoting GC progression along with its specific mechanisms of action. Furthermore, preliminary investigations into the therapeutic potential of targeting CCAT5 offer new avenues for GC treatment.
2. METHODS AND MATERIALS
2.1. Patients, tissue samples, and ascites
This study consisted of 15 GC cases with malignant ascites, 3 GC cases with IPD and 2 independent cohorts of primary GC patients, cohort 1 (112 patients) and cohort 2 (186 patients). Written informed consents were received from all patients. After pathologically diagnosed with metastatic adenocarcinoma, ascites was obtained by abdominocentesis, and IPD tissues were collected by intraoperative biopsy. For primary GC cohorts followed until March 2016, inclusion criteria were as follows: (1) treatment with surgical resection; (2) pathologically confirmed by two independent experts; (3) no local or systemic treatment prior to the surgery. Exclusion criteria were as follows: (1) patients with simultaneous diagnoses of other cancers. (2) refusal by patients to allow the utilization of their surgical specimens for research purposes. The study protocol was approved by the Clinical Research Ethics Committee of Fudan University Shanghai Cancer Center (FUSCC, No. 050432‐4‐1911D). The clinical features are summarized in Supplementary Table S1.
2.2. Cell lines and cell culture
Human GC cell lines (AGS, BGC‐823, HGC‐27, MGC‐803, MKN‐28, MKN‐45, and SGC‐7901), normal gastric epithelial cell line GES‐1, and HEK293T were obtained from the Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China) and maintained in the RPMI 1640 (BasalMedia, Shanghai, China) medium. All medium contained 10% fetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA) and 1% penicillin‐streptomycin (BasalMedia). All cell lines were cultured in a humidified incubator of 5% CO2 at 37°C. Wnt3a‐conditioned (100 ng/mL, R&D Systems, Minneapolis, Minnesota, USA) and control medium were prepared as previously described [12].
2.3. Establishment of ascites‐derived GC cell line
Approximately 200‐1000 mL fresh ascites of GC patient with peritoneal metastasis was collected by centrifuge tube supplemented with heparin. The ascites was centrifuged at 560 ×g for 5 min. The cell precipitation was resuscitated with DMEM/F12 medium (BasalMedia, Shanghai, China) containing 5% FBS (supplemented with insulin, hydrocortisone, epidermal growth factor, fibroblast growth factor; Sigma, St. Louis, Missouri, USA), and then cultured in a humidified atmosphere of 5% CO2 at 37°C. The cells were purified through a combination of differential adhesion method and low concentration trypsin digestion methods [23]. Briefly, cells with good growth status were digested with low concentration pancreatic enzyme. When most fibroblasts were shed but tumor cell were not shed, digestion was immediately terminated using culture medium containing FBS. The supernatant was then discarded, and cells were continued to be cultured in conditioned medium. If necessary, this operation can be repeated multiple times to purify primary GC cells derived from ascites. After primary culture, the ascites‐derived GC cell line (named GC‐AL) that could passage steadily was obtained. The purification effect and epithelial source property of the GC‐AL cell line were confirmed by immunofluorescence assay showing EpCAM and Cytokeratin expression [24, 25].
2.4. Plasmid construction
For human CCAT5 and CCAT5‐△612‐781 (tagged with EGFP and puromycin) overexpression, the coding sequence was PCR‐amplified and subcloned into the pLenti‐CMV‐MCS vector (Biolink, Shanghai, China) for lentivirus production. Human β‐cateninS33Y (tagged with RFP and hygromycin B) plasmid, in which the serine 33 was substituted with tyrosine, was obtained from Oobio (Shanghai, China). For human TCF3 overexpression, the pcDNA3.1‐CMV‐Flag vector was obtained from Biolink. For CCAT5 knockdown, pLenti‐hU6‐MCS‐CMV (Biolink) was used for short hairpin RNA (shRNA) plasmid construction, and the target sequence was 5’‐GCTCTGCAGGTCGAACCTTAT‐3’ for shCCAT5‐1 and 5’‐GGGCCCTCTGCAGACTGTTGG‐3’ for shCCAT5‐2. The STAT3 and Jak2 expression plasmids were obtained from Oobio. All constructs were confirmed by Sanger sequencing.
CRISPR‐Cas9 genomic editing system was used to generate CCAT5 knock‐out GC cells. Four short guide RNAs (sgRNAs) targeting the CCAT5 genomic region were respectively cloned into the pLenti‐CRISPR v2 vector (Biolink) to enable lentiviral delivery of both Cas9 and sgRNA.
For luciferase assays, indicated fragments of CCAT5 promoter or mutation fragments were cloned into the pGL3‐basic luciferase reporter vector (Yeasen, Shanghai, China). The small interfering RNA (siRNA) transfection was performed using lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, MA, USA). Briefly, after co‐incubation of 5 μL siRNA and 5 μL transfection reagent for 30 minutes, the mixture was added to the 6‐well plates covered with cells, and the culture medium was replaced after 6 hours. The target sequences are described in Supplementary Table S2.
2.5. Luciferase reporter assay
Luciferase activity was detected using the dual‐luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer's instructions. Briefly, the expression or control vectors were co‐transfected with the reporter plasmids into cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). After 48 hours post‐transfection, the cells were lysed and the luciferase substrate was added to the cell lysate. The luciferase activity was measured using a Synergy H1 microplate reader (Biotek, Vermont, USA). TOP/FOP flash assay was performed to detect the Wnt luciferase activity. For the detection of the binding site of TCF3 on the CCAT5 promoter, indicated fragments of CCAT5 promoter or mutation fragments were cloned into the pGL3‐basic vector and transfected into HEK‐293T cells. The STAT3 luciferase reporter plasmids were obtained from Yeasen (Shanghai, China). The final constructs were confirmed by Sanger sequencing. Firefly luciferase activity was normalized to that of Renilla luciferase.
2.6. Fluorescence in situ hybridization (FISH) analysis
RNA‐FISH assay was performed to determine the localization of CCAT5. Fluorescence‐conjugated CCAT5 probes were generated according to protocols of Biosearch Technologies (Novato, CA, USA). For the detection of co‐localization of CCAT5 and STAT3, cells were treated in a non‐denaturing condition, followed by hybridization with probe sets. Subsequently, the cells were incubated with anti‐STAT3 antibody (Supplementary Table S3) overnight at 4°C, followed by incubation of fluorescein‐conjugated secondary antibody. Then the cells were counterstained with DAPI (Servicebio, Wuhan, Hubei, China), and the images were acquired through a confocal microscope (Leica, Wetzlar, Hesse, Germany).
2.7. Cytoplasmic/nuclear RNA purification
Cytoplasmic and nuclear RNAs were purified using the Nuclear/Cytoplasmic Isolation kit (Thermo Fisher Scientific). Briefly, 1×107 GC‐AL cells were harvested and incubated with lysis solution for 10 min. After centrifugation, cytoplasmic RNA was isolated from the supernatants, and nuclear RNA was isolated from the pellets according to the manufacturer's instructions.
2.8. Chromatin immunoprecipitation (ChIP)‐re‐ChIP assay
The AGS cells were transfected with a Flag‐TCF3 plasmid (GeneChem, Shanghai, China) and then subjected to ChIP‐re‐ChIP assay. ChIP assay was conducted using the EZ‐ChIP kit (Millipore, Belmont, MA, USA) according to the manufacturer's instructions. The antibodies used for ChIP are summarized in Supplementary Table S3. The primers were synthesized according to the β‐catenin/TCF3 binding site of the CCAT5 promoter. The primers used in ChIP assay are summarized in Supplementary Table S2. ChIP‐re‐ChIP was performed as described previously [26, 27]. Briefly, cross‐linked chromatin was sonicated into fragments, which were then immunoprecipitated using Flag or β‐catenin antibodies (Supplementary Table S3). The immunoprecipitated complexes were eluted with re‐ChIP buffer.
2.9. Quantitative real‐time PCR (qRT‐PCR) analysis
Total RNA of tissues or cells was extracted with Trizol reagent (Invitrogen). The cDNA was synthesized using Prime‐Script RT Reagent Kit (TaKaRa, Tokyo, Japan). The expression levels of lncRNAs and genes were determined using SYBR Premix (TaKaRa) and calculated using the 2−△△Ct method. The primer sequences for CCAT5, U6, GADPH, MMP2, COX2 and BCL2, along with the primer sequences for the luciferase reporter experiments, are detailed in Supplementary Table S2.
2.10. Western blot analysis
The total or nuclear protein was prepared using protein extraction kit (Beyotime, Shanghai, China). After protein quantification using a BCA assay, equal amounts of protein were resolved by sodium dodecyl‐sulfate polyacrylamide gel electrophoresis gels and transferred onto polyvinylidene fluoride membrane (Millipore). After blocking with 5% nonfat milk (Servicebio, Wuhan, Hubei, China), membranes were then incubated overnight at 4°C with primary antibody. After washing with phosphate buffered saline (PBS), the membrane was probed with HRP‐conjugated secondary antibody (Cell Signaling Technology, Danvers, Massachusetts, USA), and the signals were detected with ECL detection reagents (Millipore). The antibodies used in this study are listed in Supplementary Table S3.
2.11. Immunohistochemistry (IHC) analysis
IHC analysis was conducted with a tissue microarray containing 154 GC samples from cohort 2. Briefly, after baking at 60°C, deparaffinization, antigen retrieval, blocking endogenous peroxidase with 3% H2O2, the samples were then incubated with anti‐β‐catenin antibody or anti‐p‐STAT3 antibody (Supplementary Table S3) overnight at 4°C and HRP‐conjugated secondary antibody (Cell Signaling Technology). The IHC scoring was conducted according to the staining intensity and positive cell ratio with Aipathwell automatic analysis system (Servicebio, Wuhan, China). The quantitative analysis was refined by pathologists based on the nuclear staining intensity of β‐catenin. The total score was obtained by multiplying staining intensity (0, no staining; 1, mild staining; 2, moderate staining; 3, intense staining) and positive cell ratio (0, 0%∼5%; 1, 6%∼25%; 2, 26%∼50%; 3, more than 50%). A total score ≥ 4 was considered as high expression to simplify data analysis [28, 29].
2.12. Immunofluorescence assay
Cells were seeded on sterilized coverslips with appropriate density, and fixed in 4% paraformaldehyde at room temperature for 20 min, permeabilized with 0.5% Triton X‐100 (Beyotime) at room temperature for 5 min, and blocked with 1% BSA for 1 hour. The coverslips were subsequently incubated overnight with primary antibody (Supplementary Table S3), followed by 1‐hour incubation with fluorescent secondary antibody (Cell Signaling Technology). Finally, cells were counterstained with DAPI and imaged through a confocal microscope (Leica).
2.13. Cell proliferation assay
Indicated cells were seeded into 96‐well plates at a density of 1.5 × 103/well (MKN‐28 and AGS cells) or 2.0 × 103/well (MGC‐803 cells) for culture. In each sample, the original medium was replaced by medium with 10% CCK‐8 reagents (Dojondo Laboratories, Kumamoto, Japan) at different time points (24, 48, 72, and 96 hours), followed by 2 hours incubation at 37°C. The absorbance of each sample was then measured at 450‐nm. Each experiment was repeated 3 times with 5 technical repeats for each time point per group.
2.14. Colony formation assay
Cells were plated in 6‐well plates (1 × 103/well) and cultured for 14 days. The culture medium was replaced every 3 days. The colonies were fixed with 4% paraformaldehyde for 15 min, stained with Giemsa solution for 20 min, and then photographed.
2.15. Ethynyldeoxyuridine (EdU) incorporation assay
Cells at 50% confluency were cultured with EdU labeling medium (RiboBio, Guangzhou, China) for 2 hours, fixed with 4% paraformaldehyde for 30 min and treated with 1% Triton X‐100 for 30 min at room temperature. After 3 washes with PBS, the cells were dyed with Apollo (RiboBio) for 30 min. Then, Hoechst (RiboBio) was used to stain the DNA in the cells, and subsequently visualized with a confocal microscope (Leica).
2.16. Transwell migration and invasion assays
Indicated cells (2 × 104 for MKN‐28 cells, 5 × 104 for MGC‐803 and AGS cells) with FBS‐free medium were seeded into the upper chamber of 8.0 μm pore transwells (Millipore) precoated with or without Matrigel (BD Bioscience, Franklin Lakes, NJ, USA). The medium supplemented with 10% FBS was added into the lower compartment as chemokine. After 36 hours incubation, the cells attached to the lower surface of the chamber were counted in 5 random fields after crystal violet staining.
2.17. In vivo tumor growth assay and lung metastasis model
Four‐ to six‐week‐old male BALB/C nude mice were obtained from the Institute of Zoology, Chinese Academy of Sciences, Shanghai, China. Mice were kept in a controlled, pathogen‐free environment at 24°C, with 50% humidity, and a 12‐hour light‐dark cycle. All the mouse experiments were approved by the Animal Care and Use Committee of FUSCC (No. 2020JS‐139). For tumor xenograft model, 2 × 106 cells were injected subcutaneously into the groin of nude mice (n = 5 for each group). After one month of inoculation, all mice were sacrificed, and the tumors were removed and weighed. The tumor volume was calculated using the following formula: Volume (mm3) = 4π/3 × (width/2)2 × (length/2). The mice were euthanized using carbon dioxide anesthesia suffocation method at the end of the experiment.
Lung metastasis model was established to explore the effect of CCAT5 on GC metastatic ability in vivo. Briefly, 5 × 106 cells were injected into the tail vein of mice. All mice were sacrificed 6 weeks later, and the lungs were then removed for pathologic examination, H&E staining, and count of the lung metastatic nodules.
For luciferase imaging, mice were injected intraperitoneally with D‐luciferin, potassium salt (150 μg/g, Sciencelight, Shanghai, China). Eight minutes after injection, animals were anesthetized and imaged using an IVIS‐100 system (Caliper Life Sciences, Hopkinton, MA, USA). Signal was displayed as photons/s/cm2/sr.
2.18. RNA‐sequencing (RNA‐seq) and computational analysis
Approximate 1 μg of total RNA was used for RNA‐seq libraries preparation according to the manufacture's instruction using the TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, California, USA). High‐throughput sequencing was performed on the Illumina HiSeq 2500 platform. The raw RNA sequencing reads were qualified by FastQC software (Bioinformatics Group, Cambridge, Cambridgeshire, UK). The reads were aligned and mapped on reference Ensemble human genome assembly (GRCh38) by the spliced read aligner HISAT2. Gene expression levels were calculated as transcripts per kilobase million (TPM) quantification with GENCODE v29 annotation. The differentially expressed genes were analyzed by moderate Student's t‐test by limma package. The differently regulated targets were annotated by official gene ID and used for enrichment annotation of KEGG pathway and gene set enrichment analysis (GSEA) using the clusterProfiler package.
2.19. RNA pull‐down assay
Biotin‐labeled RNAs were transcribed in vitro with the Biotin RNA Labeling Mix (Roche, Mannheim, Germany) and T7 RNA polymerase (Roche), treated with RNase‐free DNase I (Roche), and purified with RNeasy Mini Kit (Qiagen, Hilden, North Rhine‐Westphalia, Germany). Protein‐RNA interactions were carried out using 1 mg whole‐cell lysates and 3 μg of purified biotinylated transcripts for 1 hour at 25°C. Complexes were isolated with streptavidin agarose beads (Invitrogen). The beads were washed briefly thrice and boiled in SDS buffer, and the retrieved protein was detected by mass spectrometry or standard western blotting procedures. Briefly, proteins were extracted from cells using RIPA lysis buffer (Yeason), digested into peptides, and then protein identification was performed using the mass spectrometer (Thermo Fisher Scientific).
2.20. RNA immunoprecipitation (RIP) assay
Cells were lysed to obtain cross‐linked proteins and RNA using the Magna RIP RNA‐binding protein immunoprecipitation kit (Millipore). The supernatants were then incubated with Protein G Dynabeads (Thermo Fisher Scientific) and indicated antibodies (Supplementary Table S3). The RNA was extracted using the Total RNA isolation kit (Thermo Fisher Scientific), and the co‐precipitated RNAs were detected by qRT‐PCR.
2.21. Co‐immunoprecipitation (Co‐IP) assay
Cells were lysed with lysis buffer supplemented with protease inhibitor cocktail (Roche). Total protein in cell lysates were incubated with 2 μg anti‐STAT3 or anti‐SHP‐1 antibody (Supplementary Table S3) at 4°C for 2 hours, and with 20 μL A/G PLUS‐Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. After incubation, the beads were separated from the lysis buffer, washed 5 times in cold NP‐40 buffer (Beyotime), and then subjected to western blotting.
2.22. In vitro phosphatase assay
Human p‐STAT3 were immunoprecipitated with anti‐Flag gel beads (Sigma) from HEK293T lysates after 24 hours co‐transfection with STAT3 and Jak2 expression plasmids. After stringent washing, immunoprecipitated STAT3 was incubated with recombinant human SHP1 (Supplementary Table S3) according to the manufacturer's instructions in the presence or absence of indicated RNA.
2.23. RNAi‐based therapeutics for cell‐based model and human PDX model
For in vivo treatment of cell‐based model, AGS, MKN‐28, and MKN‐45 cells stably labeled with luciferase were used. A total of 2 × 106 AGS‐luc (Wnthigh), MKN‐28‐luc (Wntlow) or MKN‐45‐luc (Wntlow) cells were injected subcutaneously into the groin of nude mice. After 11 days, when the tumor had grown to an appropriate volume (50‐100 mm3), the tumor‐bearing mice were randomly assigned to two groups (n = 5) to receive intratumoral injection of CCAT5 siRNA (si‐CCAT5) or scrambled siRNA‐Ctrl (200 μg/kg) with high‐purity and high‐stability (RiboBio) every 3 days [30]. In order to reduce the impact of the physical pressure generated by intratumoral injection on the tumor, the volume of each injection is fixed at 50 μL. The tumor volume was measured every 4 days to depict growth curve. After 1 month of inoculation, the tumors were luciferasely imaged, removed and weighed.
For endogastric xenograft tumor model, 2 × 106 AGS‐luc (Wnthigh) cells was firstly injected subcutaneously into nude mice to develop transplanted tumor. When the tumor mass grew to a diameter of 1 cm, 2∼3 mm3 tumor pieces were cut and orthotopically overlapping sutured into lesser gastric curvature of nude mice to establish endogastric xenograft model. Tumor growth was monitored using in vivo luciferase imaging system every 4 days. Five weeks after endogastric xenograft, all mice developed abdominal metastasis, and were randomly assigned to 2 groups (n = 4) to receive intraperitoneal injection of si‐CCAT5 or siRNA‐Ctrl (200 μg/kg, 200 μL/injection) every 3 days. After 4 weeks of treatment, the mice were luciferasely imaged and pathologically examined.
For human PDX model, 2 GC cases of malignant ascites and 3 cases with intraperitoneal dissemination were enrolled, and the strong β‐cateninnuc expression was confirmed by immunofluorescence. The purified ascites cells (5 × 105) and fresh IPD tumor tissues (30∼50 mm3) were subcutaneously transplanted into the NOD/SCID mice to construct the P0 model. Then 50∼100 mm3 tumors were transplanted into the nude mice to establish the P1 and P2 model. Finally, tumor‐bearing P2 mice were assigned to 5 groups: (1) the control group, PBS 200 μL/injection, intravenous, every 3 days; (2) the XAV939 group, 20 mg/kg, 200 μL/injection, intraperitoneal, every 3 days; (3) the si‐CCAT5 group, 200 μg/kg, 50 μL/injection, intratumoral, every 3 days; (4) the oxaliplatin group, 10 nmol/kg, 200 μL/injection, intraperitoneal, every 3 days; or (5) the oxaliplatin (10 nmol/kg, 200 μL/injection, intraperitoneal) + si‐CCAT5 (200 μg/kg, 50 μL/injection, intratumoral) group, every 3 days. Tumor volume was examined every 4 days. After 1 month of inoculation, all mice were sacrificed, and the tumors were removed and weighed.
2.24. Bioinformation analysis
Gene expression profiles in the form of fragments per kilobase million (FPKM) and corresponding clinical information of GC in the Cancer Genome Atlas (TCGA) were collected from the UCSC XENA (https://xenabrowser.net/datapages/) website. The gene expression profiles and corresponding clinical information of GSE62254 was acquired from the Gene Expression Omnibus (GEO) database. Kaplan‐Meier Plotter (http://kmplot.com/analysis/) was used to analysis the correlation between TCF3 and prognosis in GC patients. The software programs JASPAR (http://jaspar.genereg.net) was used to analyze promoter region of CCAT5.
2.25. Statistical analysis
All data were represented as mean ± standard deviation (SD). Kaplan‐Meier analysis and the log‐rank test were used to compare survival between different groups. Shapiro‐Wilk Normality test was first used to test the data distribution for continuous variables. When variables had a normal distribution, statistical significance was determined using t‐test between two groups and two‐way ANOVA analysis among multiple groups. If the measurements did not have a normal distribution, two‐group comparisons were conducted using the Mann‐Whitney U test, while multiple‐group comparisons were performed using the Kruskal‐Wallis test. Two‐tailed P value < 0.05 was considered statistically different.
3. RESULTS
3.1. Identification of lncRNAs induced by canonical Wnt signaling in GC cells
To screen for functional lncRNAs involved in Wnt signaling‐induced GC abdominal dissemination, we utilized the cellular model of Wnt3a and β‐catenin‐driven Wnt activation (Figure 1A). We firstly isolated, purified and primarily cultured ascites cells from 13 GC patients with abdominal metastasis. The ascites cells with the lowest nuclear β‐catenin expression were chosen to establish novel cell line capable of stable passage (named GC‐AL; Supplementary Figure S1A‐B). We confirmed the purification effect and epithelial source properties of GC‐AL cells by immunofluorescence assay of EpCAM and Cytokeratin (Figure 1B). To model the Wnt signaling activation, the GC‐AL cells were harvested after stimulation with Wnt3a (100 ng/mL) or overexpression of degradation‐resistant β‐catenin (S33Y mutated) protein for the indicated times. As expected, both Wnt3a and β‐cateninS33Y treatment resulted in substantial nuclear accumulation of β‐catenin (Figure 1C), increased TCF/LEF transcriptional activity (Figure 1D) and expression of Wnt target genes (Supplementary Figure S1C), thus confirming the validity of Wnt‐activated GC cell model.
FIGURE 1.
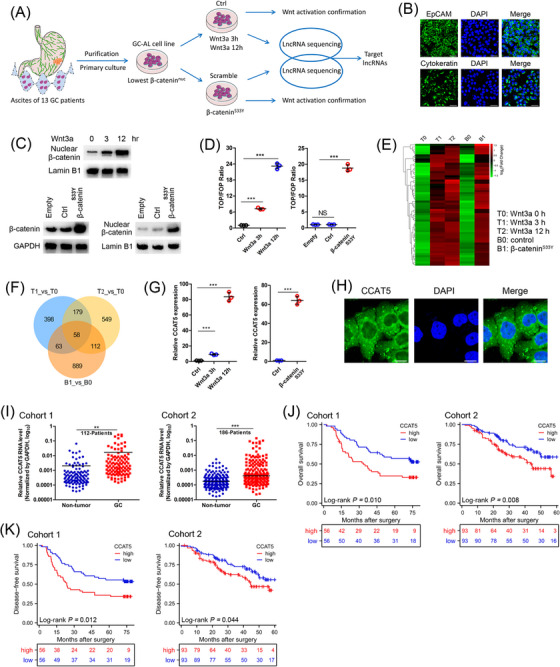
Identification of lncRNAs induced by Wnt/β‐catenin signaling and the clinical significance of lncRNA‐CCAT5 in GC. (A) Schematic diagram depicting the screening of lncRNAs induced by Wnt signaling in ascites‐derived GC cells. The cellular model of Wnt3a‐driven Wnt activation: ascites‐derived GC cell line GC‐AL was serum‐starved for 16 h and then treated with Wnt3a (100 ng/mL) for 0 (T0), 3 (T1), or 12 (T2) h. The cellular model of β‐cateninS33Y‐driven Wnt activation: GC‐AL cells were transfected with β‐cateninS33Y (B1) or control (Ctrl; B0) plasmids. Wnt activation was defined as nuclear accumulation of β‐catenin, increased TCF/LEF transcriptional activity, and enhanced expression of Wnt target genes.(B) Immunofluorescence assay of EpCAM and Cytokeratin confirmed the purification effect and epithelial source properties of GC‐AL cells.(C) Western Blot confirmed both Wnt3a (0, 3, 12 h) and β‐cateninS33Y treatment resulted in substantial nuclear accumulation of β‐catenin.(D) TOP/FOP flash luciferase assay indicated the upregulation of the Wnt transcriptional activity in GC‐AL cells treated with Wnt3a or β‐cateninS33Y. (n = 3)(E) Differential gene heatmap shows the gene expression patterns of T0 (Wnt3a treatment for 0 h), T1 (Wnt3a treatment for 3 h), T2 (Wnt3a treatment for 12 h), B0 (control plasmid), and B1 (β‐catenin S33Y mutation) groups.(F) Hierarchical clustering analysis of 58 up‐regulated lncRNAs correlated with the Wnt signaling activation induced by Wnt3a and β‐cateninS33Y treatment.(G) qRT‐PCR analysis elucidated the upregulation of CCAT5 in GC‐AL cells treated with Wnt3a or β‐cateninS33Y. (n = 3)(H) FISH assay showed that CCAT5 was mainly located in the cytoplasm in GC‐AL cells. Scale bars, 50 μm.(I) qRT‐PCR analysis using cancer tissues and paired normal mucosae from two independent cohorts (cohort 1, 112 patients and cohort 2, 186 patients) confirmed the significantly upregulated CCAT5 expression in GC.(J) Kaplan‐Meier analysis showed that patients with higher CCAT5 levels exhibited worse overall survival in two independent cohorts (cohort 1, 112 patients and cohort 2, 186 patients).(K) Kaplan‐Meier analysis showed that patients with higher CCAT5 levels exhibited worse disease‐free survival in 2 independent cohorts (cohort 1, 112 patients and cohort 2, 186 patients).All values are presented as the mean ± SD. ** P < 0.01, *** P < 0.001.Abbreviations: GC, gastric cancer; lncRNAs, long non‐coding RNAs; DAPI, 4',6‐diamidino‐2‐phenylindole; GC‐AL, ascites‐derived GC cell line; qRT‐PCR, quantitative real‐time PCR; Wnt3a, Wnt family member 3A; FISH, fluorescence in situ hybridization; SD, standard deviation.
Next, time‐course samples of Wnt3a/β‐cateninS33Y treated and control cells were subjected to lncRNA sequencing analysis. Hierarchical clustering showed systematic variations in lncRNA expression between these samples (Figure 1E, Supplementary Figure S1D). By intersecting the differentially expressed lncRNAs (FDR < 0.01, fold‐change > 2) of these datasets, we identified 58 up‐regulated and 60 down‐regulated lncRNAs (Figure 1F, Supplementary Figure S1E). Further screening showed that CCAT5 was induced most significantly by Wnt3a and β‐cateninS33Y treatment (Supplementary Tables S4‐S6). We also confirmed this induction effect using qRT‐PCR analysis (Figure 1G). CCAT5, also known as MNX1‐AS1, is a recently discovered long noncoding RNA located on chromosome 7q36.3. It generates antisense transcripts in close proximity to the 5’ end of the motor neuron and pancreatic homeobox 1 (MNX1) gene and originates from the bidirectional promoter of MNX1. RNA‐FISH assay showed that CCAT5 was mainly localized in the cytoplasm (Figure 1H), which was also confirmed by subcellular fractionation analysis (Supplementary Figure S1F).
3.2. Elevated expression of CCAT5 in GC patients is associated with poor prognosis
Next, the clinical significance of CCAT5 in GC was investigated. Bioinformatics analysis of GSE54129 dataset indicated that the CCAT5 expression was markedly higher in GC tissues than in normal tissues (Supplementary Figure S2A). Subsequently, the significantly upregulated CCAT5 expression in GC was confirmed by qRT‐PCR analysis using 298 cancer tissues and paired normal mucosae from 2 independent cohorts (Figure 1I, Supplementary Figure S2B). Notably, CCAT5 expression was gradually decreased with the improvement of tumor differentiation (Supplementary Figure S2C), while increasing with tumor TNM stage progression (Supplementary Figure S2D), prompting a putative role of CCAT5 in GC evolution. Subsequently, all patients were divided into high and low CCAT5 expression groups according to the median value. Kaplan‐Meier analysis showed that patients with higher CCAT5 levels exhibited worse overall survival and disease‐free survival (Figure 1J‐K). Multivariate analysis demonstrated that high CCAT5 level was an independent predictor for poor prognosis in GC patients (Supplementary Figure S2E‐F). Taken together, these data indicated that CCAT5 expression is upregulated in GC patients and associated with poor prognosis.
3.3. CCAT5 is regulated by Wnt/β‐catenin signaling cascade
Wnt ligand activates signaling through the binding of β‐catenin to the TCF/LEF transcription factors [9]. The potential influence of TCF/LEF on CCAT5 expression was examined to gain insight into how CCAT5 was transcribed in GC cells. According to the endogenous CCAT5 level, MKN‐28 and AGS cells were selected for further study (Figure 2A). Subsequent qRT‐PCR results suggested that knockdown of TCF3 (Figure 2B), but not the other TCF factors (Supplementary Figure S3A), significantly inhibited the CCAT5 expression in AGS cells. Meanwhile, TCF3 overexpression dramatically increased CCAT5 expression in MKN‐28 cells (Figure 2B). In addition, bioinformatics analyses of GSE62254 dataset indicated that TCF3 was significantly correlated with CCAT5 (Supplementary Figure S3B‐C). Further analyses of TCGA and Kaplan‐Meier Plotter (http://kmplot.com/analysis/) found that TCF3 was markedly upregulated in GC and predicted poor overall survival (Supplementary Figure S3D‐F). Meanwhile, TCF3 overexpression significantly promoted the TCF/LEF activity in GC cells (Supplementary Figure S3G). Subsequently, luciferase reporter assay was performed by cloning CCAT5 promoter into pGL3‐basic vector. The results suggested that the CCAT5 promoter activity was profoundly attenuated by TCF3 depletion, and stimulated by TCF3 overexpression (Figure 2C). Thus, CCAT5 transcription might be modulated by TCF3.
FIGURE 2.
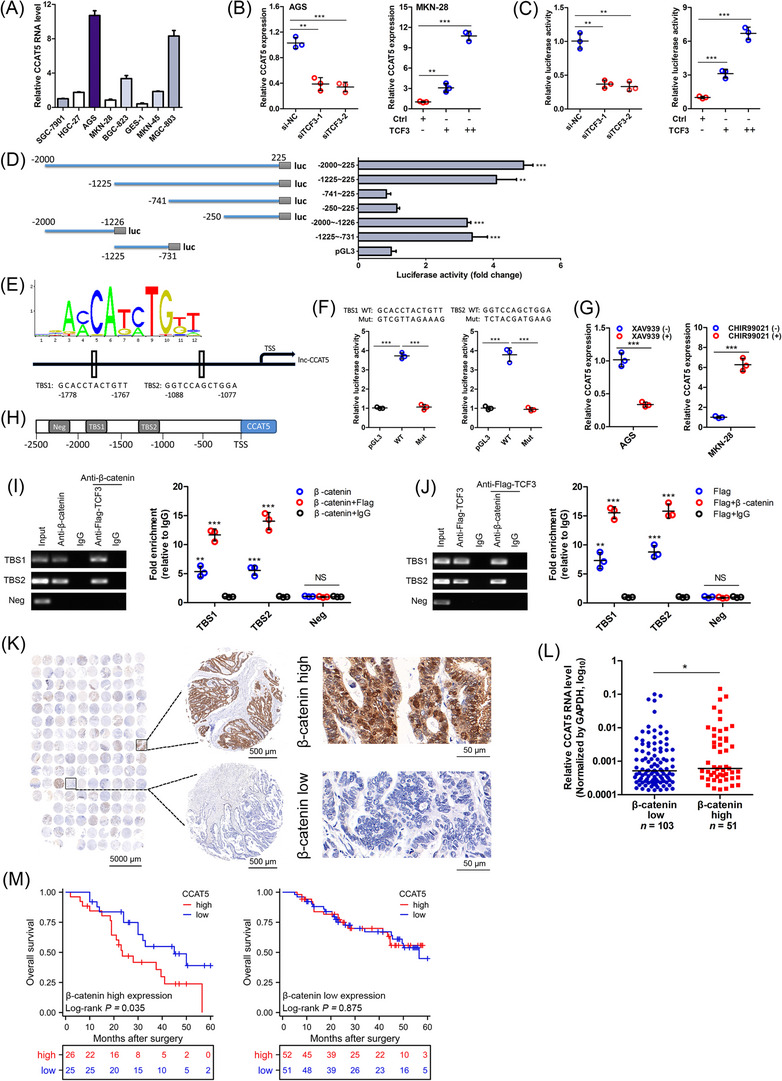
β‐catenin/TCF3 complex transactivates CCAT5.(A) The expression levels of CCAT5 in normal gastric epithelial cell line GES‐1 and 7 GC cell lines were examined by qRT‐PCR analysis. (n = 3).(B) qRT‐PCR analysis was conducted in AGS cells transfected with 2 siRNA targeting TCF3 or negative control (si‐NC), and MKN‐28 cells transfected with TCF3 overexpression or control (Ctrl) plasmids. (n = 3).(C) The CCAT5 promoter was subcloned into the pGL3‐basic vector to construct luciferase reporter plasmids. The relative luciferase activity was examined using HEK‐293T cells transfected with siRNA targeting TCF3 or si‐NC, TCF3 or Ctrl plasmids. (n = 3).(D) Scheme for the fragment mutant of CCAT5 promoter used in luciferase assay (left panel). The relative luciferase activities of 6 truncated constructs in HEK‐293T cells (right panel).(E) The sequence logo of a potential TCF3 binding site (TBS) by JASPAR (upper panel). Schematic diagram showing the CCAT5 upstream promoter region (lower panel), including the predicted TBS1 (‐1778 nt ∼ ‐1767 nt) and TBS2 (‐1088 nt ∼ ‐1077 nt).(F) The relative luciferase activity was examined in HEK‐293T cells. The predicted TCF3 binding sequence TBS1, TBS2, and indicated mutated variant (both sequences indicated) were respectively inserted into the pGL3‐basic luciferase reporter and analyzed for their responses to TCF3. (n = 3).(G) AGS or MKN‐28 cells were treated with XAV939 (15 μmol/L) or CHIR99021 (8 μmol/L) for 24 h. Then the CCAT5 expression was detected by qRT‐PCR analysis. (n = 3).(H) Putative TBSs on the promoter region of CCAT5. A random region without TBS acted as a negative control (Neg).(I) ChIP–re‐ChIP assay was performed using anti‐β‐catenin antibody first (β‐catenin) in AGS cells. The eluents were then subjected to a second ChIP assay using anti‐Flag‐TCF3 antibody (β‐catenin + Flag‐TCF3) or control IgG antibody (β‐catenin + IgG). (n = 3).(J) ChIP‐re‐ChIP assay was performed using anti‐Flag‐TCF3 antibody first (Flag‐TCF3) in AGS cells. The eluents were then subjected to a second ChIP assay using β‐catenin antibody (Flag‐TCF3 + β‐catenin) or control IgG antibody (Flag‐TCF3 + IgG). (n = 3).(K) Immunohistochemistry analysis was conducted to examine β‐catenin expression using a tissue microarray containing 154 GC samples from cohort 2. Scale bars were shown in each panel.(L) According to the immunohistochemistry score of β‐catenin, patients were stratified into high (n = 51) and low expression (n = 103) group. The CCAT5 expression of these 154 GC samples was also examined with qRT‐PCR analysis.(M) Kaplan‐Meier analysis of overall survival for 154 GC patients with low and high CCAT5 expression stratified by the median value. Patients were stratified according to β‐catenin high and low staining.All values are presented as the mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001.Abbreviations: GC, gastric cancer; lncRNA, long non‐coding RNA; siRNA, small interfering RNA; TCF3, Transcription factor 3; TBS, TCF3 binding site; ChIP, Chromatin immunoprecipitation; SD, standard deviation; qRT‐PCR, quantitative real‐time PCR; NS, not significant.
To test whether CCAT5 was a transcriptional target of TCF3, serial truncations of CCAT5 promoter were constructed and transfected into HEK‐293T cells. The highest activities were found in ‐2000 nt ∼ ‐1226 nt and ‐1225 nt ∼ ‐731 nt fragment (Figure 2D), indicating these 2 regions may be responsible for TCF3‐mediated transactivation of CCAT5. Then the software program JASPAR (http://jaspar.genereg.net) was used to analyze promoter region of CCAT5. Sequence analysis of ‐2000 nt ∼ ‐731 nt region identified 2 putative TCF3‐binding sites with the highest scores, located at ‐1088 nt ∼ ‐1077 nt and ‐1778 nt ∼ ‐1767 nt (Figure 2E). Next, we generated mutant constructs for these two sites, and both of these mutations exhibited significantly lower promoter activity than wild‐type promoter (Figure 2F). Thus, CCAT5 may be trans‐activated by TCF3.
TCF3 is characterized as the transcriptional factor of the canonical Wnt/β‐catenin pathway [31]. Hence, the potential relationship between Wnt/β‐catenin signaling and CCAT5 was investigated. Interestingly, CCAT5 expression was markedly repressed by Wnt specific inhibitor XAV939, and increased by Wnt specific activator CHIR99021 (Figure 2G). Finally, ChIP‐re‐ChIP assay was performed to test the co‐occupancy of β‐catenin and TCF3 on CCAT5 promoter. The results demonstrated that the promoter region of CCAT5 was amplifiable from the DNA recovered from the immunoprecipitation complex using specific antibodies for β‐catenin and TCF3 but not the negative control IgG (Figure 2H‐J), thus confirming that β‐catenin/TCF3 complex trans‐activates CCAT5 expression in GC cells.
To further clarify the clinical relevance between CCAT5 and Wnt/β‐catenin signaling, immunohistochemistry analysis of β‐catenin was conducted using a tissue microarray containing 154 GC samples from cohort 2 (Figure 2K). The results suggested that GC patients with high β‐catenin staining showed significantly higher CCAT5 expression compared to those with low β‐catenin staining (Figure 2L). Interestingly, subsequent subgroup analysis indicated that higher CCAT5 level was associated with worse overall survival in high β‐catenin group, while no survival difference between CCAT5‐high and low group was observed in low β‐catenin expression patients (Figure 2M), further confirming the close interaction between CCAT5 and Wnt/β‐catenin signaling.
3.4. CCAT5 promotes the growth and metastasis of GC cells
Functional assays were performed to further explore the role of CCAT5 in GC progression. We first overexpressed CCAT5 in MKN‐28 cells and knocked down CCAT5 in AGS and MGC‐803 cells based on endogenous CCAT5 levels (Figure 2A, Figure 3A). As determined by CCK‐8 and EdU assays, CCAT5 overexpression significantly increased, while CCAT5 knockdown decreased GC cell proliferation in vitro (Figure 3B‐D). Xenograft tumor grafting was performed in nude mice to examine the effect of CCAT5 on GC cell growth in vivo. Tumors from CCAT5‐overexpressed MKN‐28 cells exhibited more active growth than control group (Figure 3E). Moreover, the tumor volume and weight were considerably increased in MKN‐28‐CCAT5 group compared to control group (Figure 3F‐G). Collectively, these data showed a potential role of CCAT5 in promoting growth of GC cells.
FIGURE 3.
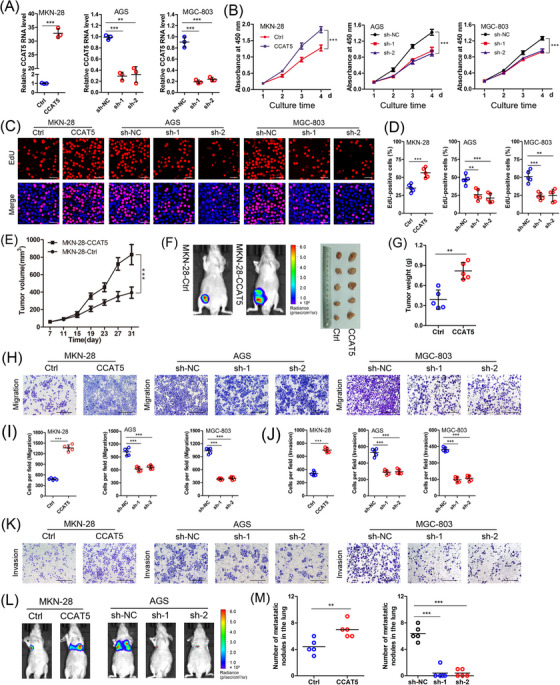
CCAT5 promotes the growth and metastasis of GC cells.(A) MKN‐28 cells were infected with lentiviral vectors containing CCAT5. AGS and MGC‐803 cells were infected with lentiviral vectors containing CCAT5‐targeting shRNAs. The CCAT5 overexpression and knockdown effects were confirmed by qRT‐PCR analysis. (n = 3).(B) The effect of modified CCAT5 expression on GC cell proliferation was monitored by the CCK8 assay. The absorbance at 450‐nm was measured at a different time point. (n = 3).(C‐D) EdU incorporation assay was performed in indicated cells with modified CCAT5 expression. Representative micrographs (C) and quantification (D) of EdU labeling were shown. Scale bars, 50 μm. (n = 5).(E) Tumor‐volume curves for xenograft models constructed by injecting CCAT5‐overexpressed and control MKN‐28 cells into the groin of nude mice (n = 5 for either group).(F‐G) Representative xenograft tumors formed by CCAT5‐overexpressed and control MKN‐28 cells were luciferasely imaged and shown (F), and the tumor weight was measured (G).(H‐K) Transwell assays detecting the influence of modified CCAT5 expression on in vitro migration and invasion ability of GC cells. Representative micrographs (H and K) and quantification (I and J) of cells attached to the lower surface of chamber were shown. Scale bars, 400 μm. (n = 5).(L) Lung metastasis models constructed by injecting GC cells with modified CCAT5 expression into the tail vein of nude mice (n = 5 for either group). Representative images of luciferase imaging were shown 6 weeks after injection.(M) The numbers of lung metastatic nodules were pathologically examined and counted.All values are presented as the mean ± SD. ** P < 0.01, *** P < 0.001.Abbreviations: GC, gastric cancer; shRNA, short hairpin RNA; qRT‐PCR, quantitative real‐time PCR; EdU, Ethynyldeoxyuridine; SD, Standard deviation.
Subsequently, the effect of CCAT5 on GC metastasis was examined. As determined by transwell assays, CCAT5 overexpression remarkably increased, while CCAT5 knockdown decreased the migration and invasion of GC cells (Figure 3H‐K). Furthermore, lung metastasis model was constructed to investigate the in vivo effect of CCAT5 on metastasis. Consistent with the in vitro data, significantly increased lung metastatic nodules were observed in CCAT5‐overexpressed mice compared with control group. Meanwhile, CCAT5 knockdown markedly decreased both the lung metastasis formation rate and numbers of metastatic nodules (Figure 3L‐M). Taken together, these results demonstrated a potential role of CCAT5 in promoting GC metastasis.
3.5. CCAT5 facilitated GC progression in a STAT3‐mediated manner
The mechanism underlying the promotive role of CCAT5 in GC development was investigated via RNA‐seq transcriptional profiles of CCAT5‐depleted AGS and control cells (Figure 4A, Supplementary Table S7). KEGG and GSEA enrichment analyses identified the Jak‐STAT as the top‐ranked signaling pathway (Figure 4B‐C, Supplementary Table S8). Subsequent luciferase reporter assay revealed that STAT3 transcriptional activity was significantly activated by CCAT5 overexpression, and inhibited by CCAT5 knockdown (Figure 4D). Furthermore, CCAT5 overexpression led to significantly increased phosphorylated STAT3 (p‐STAT3Y705) and substantial nuclear accumulation of p‐STAT3Y705. Conversely, CCAT5 knockdown resulted in markedly decreased p‐STAT3Y705 expression and redistribution of nuclear p‐STAT3 to cytoplasm (Figure 4E‐G). Moreover, CCAT5 overexpression significantly increased the protein and mRNA expression of STAT3 target genes (MMP2, COX2 and Bcl‐2) [32], whereas CCAT5 knockdown reduced their expression (Figure 4H‐I). Thus, CCAT5 was able to enhance STAT3 nuclear translocation, promote the transcriptional activity of STAT3, and consequently activate the STAT3 signaling pathway.
FIGURE 4.
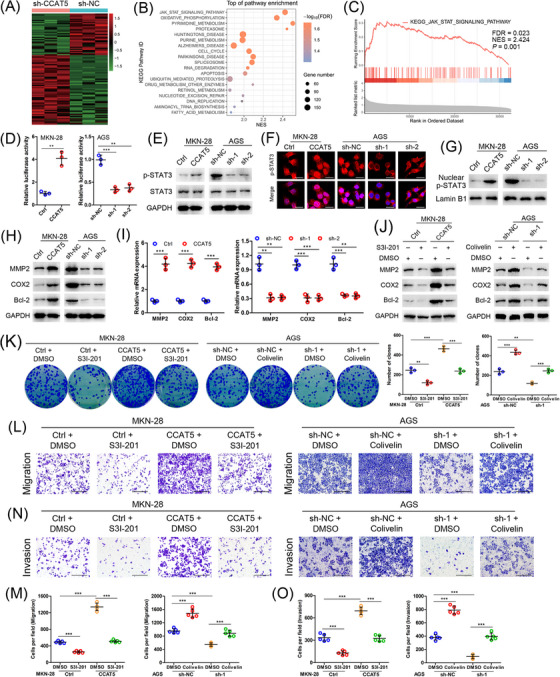
CCAT5 facilitated GC progression in a STAT3‐mediated manner.(A) Hierarchical clustering showing differentially expressed genes (P value <0.05, fold‐change >4) in the CCAT5‐depleted AGS and control cells. The genes are shaded with green, black, or red in the heat map to indicate low, intermediate or high expression, respectively.(B) Functional categories for differently expression genes of CCAT5‐depleted and control AGS cells through KEGG analysis.(C) JAK‐STAT signaling pathway was enriched by GSEA.(D) STAT3 luciferase reporter assay was performed in MKN‐28 and AGS cells with modified CCAT5 expression. Data were normalized against renilla luciferase activity. (n = 3).(E) Western blotting analysis of p‐STAT3Y705 and STAT3 expression in indicated cells with modified CCAT5 expression. GAPDH was used as internal control.(F) Subcellular p‐STAT3Y705 localization in indicated cells was assessed by immunofluorescence assay. The p‐STAT3Y705 was stained in red color, and the nuclei were stained with DAPI in blue color. Scale bars, 25 μm.(G) Western blotting analysis of the nuclear p‐STAT3Y705 expression in indicated GC cells with modified CCAT5 expression. Lamin B1 was used as an internal control.(H‐I) Western blotting (H) and qRT‐PCR (I) analyses of the expression of STAT3 signaling target genes (MMP2, COX2, and Bcl‐2) in MKN‐28 and AGS cells with modified CCAT5 expression. (n = 3).(J) CCAT5‐overexpressed MKN‐28 or control cells were treated with STAT3 inhibitor S3I‐201 (100 μmol/L) or DMSO for 2 h. CCAT5‐depleted AGS or control cells were treated with STAT3 activator Colivelin (1 μmol/L) or DMSO for 2 h. Then the expression of STAT3 signaling target genes was determined by western blotting analysis.(K) Colony formation assay in indicated cells with modified CCAT5 expression and/or S3I‐201 (100 μmol/L for 2 h every 3 days) or Colivelin (1 μmol/L for 2 h every 3 days) treatment. (n = 3).(L‐O) Transwell migration (L and M) and invasion (N and O) assays in indicated cells with modified CCAT5 expression and/or S3I‐201 (100 μmol/L for 2 h) or Colivelin (1 μmol/L for 2 h) treatment. Representative micrographs (L and N) and quantification (M and O) of cells attached to the lower surface of chamber were shown. Scale bars, 400 μm. (n = 5).All values are presented as the mean ± SD. ** P < 0.01, *** P < 0.001.Abbreviations: GC, gastric cancer; shRNA, short hairpin RNA; GSEA, gene set enrichment analysis; STAT3, signal transducer and activator of transcription 3; qRT‐PCR, quantitative real‐time PCR; GADPH, glyceraldehyde‐3‐phosphate dehydrogenase; DMSO, dimethyl sulfoxide; SD, standard deviation.
To further explore whether STAT3 activation is functionally required for CCAT5‐induced GC progression, the STAT3 inhibitor S3I‐201 and activator Colivelin were introduced. As expected, S3I‐201 neutralized the activation of STAT3 target genes resulted from CCAT5 overexpression, whereas Colivelin reversed the inhibition of STAT3 target genes caused by CCAT5 knockdown (Figure 4J), again confirming the crucial role of CCAT5 in the activation of the STAT3 pathway. Furthermore, administration of S3I‐201 and STAT3 knockdown markedly interfered with the enhanced proliferative capacity of GC cells caused by CCAT5 overexpression as determined by colony formation assay. Conversely, Colivelin treatment and STAT3 overexpression partially reversed the reduced proliferation of GC cells induced by CCAT5 knockdown (Figure 4K, Supplementary Figure S4A). In addition, transwell assays suggested that S3I‐201 treatment and STAT3 knockdown successfully blocked the increased migration and invasion of GC cells resulting from CCAT5 overexpression, while Colivelin and STAT3 overexpression impeded the inhibited metastasis of GC cells induced by CCAT5 knockdown (Figure 4L‐O, Supplementary Figure S4B). Furthermore, it was observed that GC patients with elevated phosphorylated STAT3 level exhibited higher expression level of CCAT5 (Supplementary Figure S4C). Taken together, the promotive role of CCAT5 in GC progression is mediated by STAT3.
3.6. CCAT5 enhances STAT3 signaling activity by directly interacting with STAT3 and blocking SHP‐1‐mediated STAT3Y705 dephosphorylation
LncRNAs interact with proteins to modulate protein function, regulate protein‐protein interactions, or direct localization within cellular compartments [33]. Hence, RNA pull‐down assay was performed to identify CCAT5‐interacting proteins (Figure 5A). The protein band specifically pulled down by CCAT5 at approximately 90 kDa was subjected to mass spectrometry analysis, and 15 potential interacting proteins were obtained (Supplementary Table S9). Of note, STAT3, the key switch of the JAK‐STAT3 signaling pathway, was identified as a CCAT5‐binding protein (Figure 5B). Furthermore, this specific interaction between CCAT5 and STAT3 in GC cells was confirmed by RNA pull‐down and RIP analyses (Figure 5C‐D). In addition, RNA FISH showed that CCAT5 co‐localized with STAT3 in the cytoplasm but not the nucleus of GC cells (Figure 5E). Collectively, these data demonstrated that CCAT5 physically associates with STAT3.
FIGURE 5.
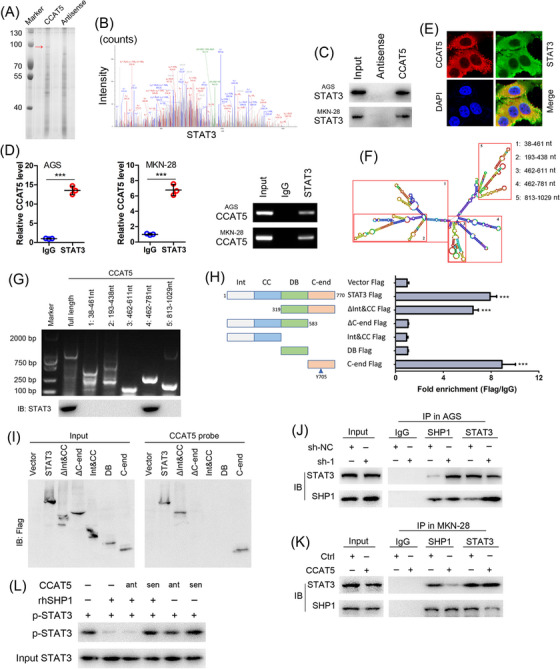
CCAT5 enhances STAT3 signaling activity by directly interacting with STAT3 and blocking SHP‐1‐mediated STAT3Y705 dephosphorylation.(A) RNA pull‐down assay was performed with lysates of AGS cells using full‐length CCAT5 and antisense RNA probes, followed by SDS‐PAGE separation, silver staining and MS. Red arrows indicate target band.(B) MS/MS profiles of target band (corresponding peptide sequences of STAT3) retrieved by CCAT5.(C) Western blot analysis of the proteins retrieved from the biotinylated CCAT5 pull‐down assay using anti‐STAT3 antibody in AGS and MKN‐28 cells.(D) RIP experiments were performed in AGS and MKN‐28 cells and the coprecipitated RNA was subjected to qRT‐PCR for CCAT5. Expression levels of CCAT5 are as fold enrichment in STAT3 relative to IgG immunoprecipitants. (n = 3).(E) Confocal RNA‐FISH and immunofluorescence images showing the co‐localization of CCAT5 and STAT3. Scale bars, 50 μm.(F) Graphic illustration of predicted CCAT5 secondary structure (LNCipedia, http://www.lncipedia.org), and the truncation diagram of CCAT5 according to the stem‐loop structure.(G) Deletion mapping of the STAT3‐binding domain in CCAT5. The in vitro transcribed full‐length CCAT5 and deletion fragments showing the correct sizes (upper panel). Immunoblotting analysis of STAT3 in protein samples pulled down by the different CCAT5 constructs (lower panel).(H) Deletion mapping of the CCAT5‐binding domain in STAT3. Diagrams of full‐length STAT3 and the domain truncated fragments (left panel). qPCR detection of CCAT5 retrieved by full‐length or domain truncated STAT3‐Flag (right panel). RIP assays were performed using AGS cells transfected with the indicated plasmids. Int, protein interaction domain; CC, coiled‐coil domain; DB, DNA‐binding domain; C‐end, C‐terminus.(I) Immunoblotting of the full‐length or domain truncated STAT3‐Flag in the lysates from AGS cells transfected with the indicated plasmids, or in the retrieved proteins by the biotinylated CCAT5 probe from the lysates of AGS cells transfected with the indicated plasmids.(J) Co‐IP experiments of STAT3 with SHP1 in CCAT5‐depleted AGS and control cells.(K) Co‐IP experiments of STAT3 with SHP1 in CCAT5‐overexpressed MKN‐28 or control cells.(L) Immunoblot of STAT3Y705 phosphorylation after incubation of phosphorylated STAT3 with rhSHP1 in the presence or absence of CCAT5, sense (sen) or antisense (ant).All values are presented as the mean ± SD. *** P < 0.001.Abbreviations: GC, gastric cancer; MS, mass spectrometry; RIP, RNA immunoprecipitation; FISH, fluorescence in situ hybridization; Co‐IP, co‐immunoprecipitation; SD, Standard deviation.
To further determine how CCAT5 regulates STAT3 activity, a series of deletion mapping analyses revealed that the truncated CCAT5 fragment 612‐781 nt was responsible for the interaction of CCAT5 with STAT3 (Figure 5F‐G). Next, truncated STAT3 mutants were constructed to unravel its binding sites with CCAT5. RIP and RNA pull‐down assays demonstrated that the C‐end domain of STAT3 (residues 583‐770 nt) was required for its interaction with CCAT5 (Figure 5H‐I). Considering that the Y705 residue of C‐end domain, whose phosphorylation is critical for STAT3 activation and tightly controlled by protein tyrosine phosphatases (PTPs) and tyrosine kinases (PTKs), such as SHP‐1, SHP‐2, and JAK [34, 35, 36], was enhanced by CCAT5 (Figure 4E), we speculated that CCAT5 might regulate PTPs‐STAT3 or PTKs‐STAT3 interaction to activate STAT3. Indeed, Co‐IP assay showed that CCAT5 knockdown promoted the association of SHP1 with STAT3 in AGS cells, while CCAT5 overexpression attenuated SHP1‐STAT3 interaction in MKN‐28 cells (Figure 5J‐K). Furthermore, in vitro kinase assay demonstrated that CCAT5 could protect STAT3 from Y705 dephosphorylation by SHP1 (Figure 5L). Taken together, CCAT5 activates STAT3 signaling by directly interacting with STAT3 and blocking SHP‐1‐mediated STAT3Y705 dephosphorylation.
3.7. The interaction between CCAT5 and STAT3 is required for Wnt‐induced activation of STAT3 signaling and GC development
Previous work showed that Wnt3a and Wnt/β‐catenin signaling function via activation of STAT3 pathway in kidney cells [37, 38]. Our results indicated that both XAV939 and si‐CCAT5 treatment markedly interfered with IL‐6‐induced nuclear accumulation of p‐STAT3 in GC cells (Supplementary Figure S5A), suggesting the critical role of Wnt and CCAT5 in STAT3 signaling activation. Furthermore, both β‐cateninS33Y overexpression and Wnt3a treatment significantly enhanced STAT3 luciferase activity (Figure 6A, Supplementary Figure S5B‐C), promoted the phosphorylation and nuclear translocation of STAT3 (Figure 6B, Supplementary Figure S5D‐E), and increased the expression of STAT3 target genes (Supplementary Figure S5F‐I) in both MKN28 (intestinal‐type, Wnt‐mutated) and MKN‐45 (diffuse‐type, Wnt‐wild) cells. Thus, aberrant Wnt signaling acts as driver of oncogenic STAT3 signaling in both intestinal‐type and diffuse‐type GC cells.
FIGURE 6.
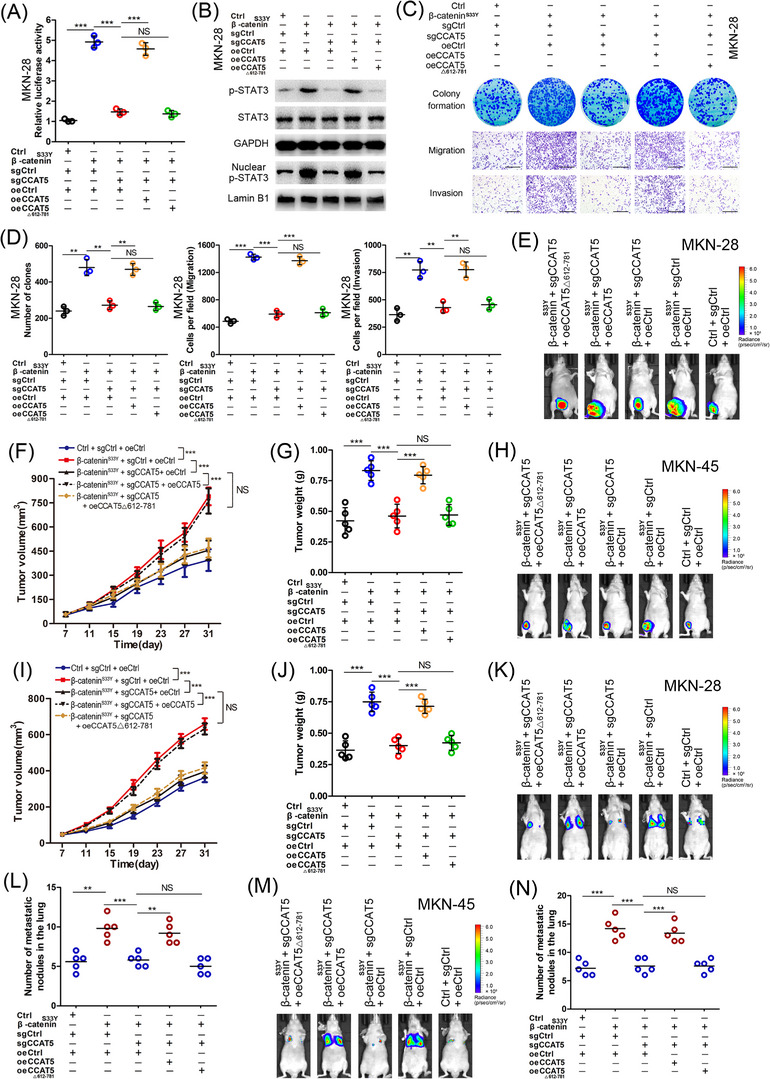
The interaction between CCAT5 and STAT3 is required for Wnt‐induced activation of STAT3 signaling and GC development.Stable MKN‐28 and MKN‐45 cells carrying control (Ctrl) or β‐cateninS33Y‐containing lentiviruses were subjected to the CRISPR/Cas9 system to generate CCAT5 knockout (sgCCAT5) or control (sgCtrl) cell lines. Then the MKN‐28 and MKN‐45 cells with manipulated β‐cateninS33Y and/or CCAT5 expression were infected with control (oeCtrl), full‐length CCAT5 (oeCCAT5), or 612‐781nt depleted CCAT5 (oeCCAT5△612‐781) lentiviruses.(A‐D) Stable MKN‐28 cells with modified β‐cateninS33Y or/and CCAT5 expression (Ctrl + sgCtrl + oeCtrl, β‐cateninS33Y + sgCtrl + oeCtrl, β‐cateninS33Y + sgCCAT5 + oeCtrl, β‐cateninS33Y + sgCCAT5 + oeCCAT5, β‐cateninS33Y + sgCCAT5 + oeCCAT5△612‐781) were subjected to STAT3 luciferase activity assay (A) (n = 3), western blotting of STAT3 and p‐STAT3Y705 protein (B), colony formation, transwell migration, invasion assays (C) and quantitation (D). Scale bars, 400 μm. (n = 3).(E‐G) Stable MKN‐28 cells with modified β‐cateninS33Y or/and CCAT5 expression (2 × 106 cells) were injected subcutaneously into the groin of nude mice (n = 5 for either group). One month after tumor cell injection, mice were euthanized and luciferasely imaged (E). Tumor volumes were measured on the indicated days (F) and the average tumor weight was measured (G).(H‐J) Stable MKN‐45 cells with modified β‐cateninS33Y or/and CCAT5 expression (2 × 106 cells) were injected subcutaneously into the groin of nude mice (n = 5 for either group). One month after tumor cell injection, mice were euthanized and luciferasely imaged (H). Tumor volumes were measured on the indicated days (I) and the average tumor weight was measured (J).(K‐L) Lung metastasis models were constructed by injecting MKN‐28 cells with modified β‐cateninS33Y or/and CCAT5 expression (5 × 106 cells) into the tail vein of nude mice (n = 5 for either group). Representative luciferase images were shown 6 weeks after injection (K). The numbers of lung metastatic nodules were pathologically examined and counted (L).(M‐N) Lung metastasis models were constructed by injecting MKN‐45 cells with modified β‐cateninS33Y or/and CCAT5 expression (5 × 106 cells) into the tail vein of nude mice (n = 5 for either group). Representative luciferase images were shown 6 weeks after injection (M). The numbers of lung metastatic nodules were pathologically examined and counted (N).All values are presented as the mean ± SD. ** P < 0.01, *** P < 0.001. NS, not significant.Abbreviations: GC, gastric cancer; SD, Standard deviation.
Given that CCAT5 is a direct transcriptional target of β‐catenin/TCF3 complex and facilitated GC progression in a STAT3‐mediated manner, it raised possibility that CCAT5 may be involved in Wnt‐induced activation of STAT3 signaling and GC development. Thus, CCAT5 knockout (KO) GC cell lines were generated using the CRISPR/Cas9 system (Supplementary Figure S6). Depletion of CCAT5 obviously suppressed the enhanced STAT3 luciferase activity, STAT3 activation and nuclear entry, and STAT3 target genes expression resulting from β‐cateninS33Y overexpression and Wnt3a stimulation in both MKN‐28 and MKN‐45 cells (Figure 6A‐B, Supplementary Figure S5B‐I). This suggested that Wnt‐induced STAT3 activation is indeed mediated by CCAT5. Furthermore, to test whether the CCAT5 fragment 612‐781 nt responsible for the interaction between CCAT5 and STAT3 is required for Wnt‐induced STAT3 activation, full length CCAT5 or 612‐781 nt depleted CCAT5△612‐781 was ectopically expressed in CCAT5‐KO cells. As a result, the full length CCAT5, but not the CCAT5△612‐781, restored the inhibited Wnt/β‐catenin‐induced STAT3 signaling activity resulting from CCAT5 depletion (Figure 6A‐B, Supplementary Figure S5B‐I). Thus, the interaction between CCAT5 and STAT3 is essential for Wnt‐induced STAT3 activation.
Subsequently, we investigated whether CCAT5 is functionally required for constitutively activated Wnt‐induced promotion of GC development. As shown in Figure 6C‐D and Supplementary Figure S5J‐K, β‐cateninS33Y overexpression markedly promoted the proliferative, migrative, and invasive abilities of GC cells in vitro, whereas depletion of CCAT5 diminished the distinct difference of malignant phenotype between β‐cateninS33Y‐overexpressed and control cells. Collectively, these data indicated a critical role for CCAT5 in mediating Wnt‐induced GC progression. Strikingly, re‐expression of full length CCAT5 but not CCAT5△612‐781 reversed the enhanced GC cell growth and metastasis induced by β‐cateninS33Y overexpression, demonstrating the importance of the interplay between CCAT5 and STAT3 in mediating Wnt‐induced GC development.
Finally, xenograft tumor model (Figure 6E‐J, Supplementary Figure S7A‐B) and lung metastasis model (Figure 6K‐N, Supplementary Figure S7C‐D) were established using stable MKN‐28 and MKN‐45 cells with modified β‐cateninS33Y and/or CCAT5 expression. The results suggested that β‐cateninS33Y overexpression dramatically enhanced the tumor growth and metastasis compared to control cells. More importantly, consistent with the in vitro data, CCAT5 depletion significantly inhibited β‐cateninS33Y‐induced tumorigenesis and lung metastasis. In addition, re‐expression of full length CCAT5 successfully rescued the growth inhibition and metastasis suppression resulting from CCAT5 depletion in β‐cateninS33Y‐overexpressed GC cells. However, overexpression of CCAT5△612‐781 failed to deliver this rescue phenotype (Figure 6E‐N). Taken together, the interaction between CCAT5 and STAT3 is required for the development of both intestinal‐type and diffuse‐type GC induced by Wnt signaling activation.
3.8. RNAi‐based therapeutics targeting CCAT5 selectively decreased the growth and metastasis of Wnthigh GC cells
We then synthesized high‐purity and high‐stability siRNA against CCAT5 to study its therapeutic efficacy. The endogenous Wnt activity was firstly examined in AGS (intestinal‐type, Wnt‐mutated), MKN‐28 (intestinal‐type, Wnt‐mutated), and MKN‐45 (diffuse‐type, Wnt‐wild) cells. The results indicated that AGS cells showed the highest Wnt activity, while MKN‐28 and MKN‐45 cells showed similar low Wnt activity (Figure 7A). These cells were then stably labeled with luciferase to construct RNAi‐based in vivo models (Figure 7B). The results suggested that intratumoral injection of si‐CCAT5 led to a significant reduction in the growth and weight of tumors formed by AGS‐luc (Wnthigh) cells (Figure 7C‐E, Supplementary Figure S8A), but no phenotype difference was observed in MKN‐28‐luc (Wntlow) and MKN‐45‐luc (Wntlow) tumor models (Figure 7F‐H, Supplementary Figure S8B‐F). Thus si‐CCAT5 selectively decreases tumor growth of GC with high Wnt signaling activity, and this inhibitory effect is independent of intestinal‐type or diffuse‐type GC.
FIGURE 7.
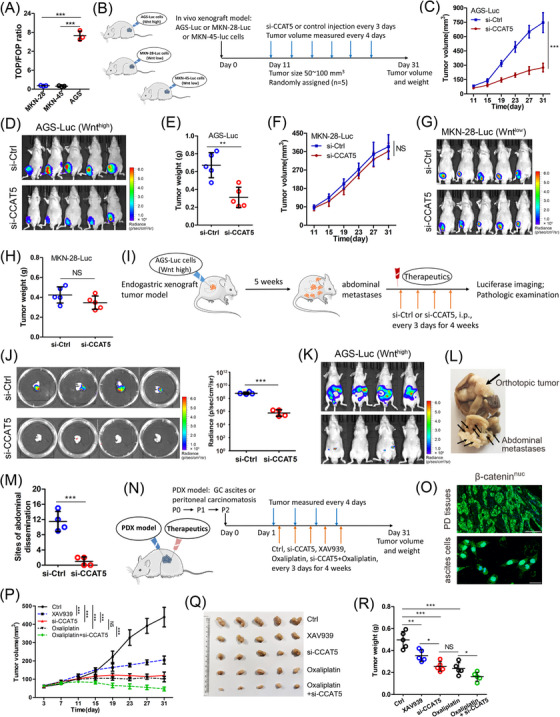
RNAi‐based therapeutics targeting CCAT5 selectively decreased the growth and metastasis of Wnthigh GC. (A) TOP/FOP flash luciferase assay was performed to examine the Wnt signaling activity of MKN‐28, MKN‐45 and AGS cells. (n = 3).(B) AGS, MKN‐28 and MKN‐45 cells were stably labeled with luciferase to establish AGS‐luc, MKN‐28‐luc and MKN‐45‐luc cells (left panel). The experimental workflow for RNAi‐based tumor xenograft model (right panel).(C‐H) RNAi‐based tumor xenograft models were established (n = 5 for either group), and tumor volumes were measured on the indicated days (C and F). One month after injection of AGS‐luc or MKN‐28‐luc cells, tumor‐bearing mice treated with si‐CCAT5 or si‐Ctrl were luciferasely imaged (D and G), and the average tumor weight was measured (E and H).(I) The experimental workflow for the establishment of RNAi‐based endogastric xenograft tumor model (n = 4 for either group).(J) After 5 weeks of model construction and 4 weeks of siRNA treatment, the endogastric xenograft tumors were pathologically examined, and the luciferase intensity was quantified.(K) Before mice were sacrificed, the abdominal dissemination status of tumor‐bearing mice was luciferasely detected.(L‐M) The representative specimen of si‐Ctrl group mice was shown (L), and the number of abdominal macro‐metastases was counted (M).(N) The experimental workflow for the establishment of PDX models with strong β‐catenin nuclear expression (β‐cateninnuc) using malignant ascites or abdominal dissemination tumor and subsequent therapeutic strategy.(O) The strong β‐cateninnuc expression of peritoneal dissemination (PD) tissues (n = 3) and ascites cells (n = 2) was confirmed using immunofluorescence assay, and representative images are displayed. The β‐catenin was stained in green color, and the nuclei were stained with DAPI in blue color. Scale bars, 50 μm.(P) Tumor growth curves of PDX model mice upon treatment with vehicle control, XAV939, si‐CCAT5, oxaliplatin, or oxaliplatin + si‐CCAT5.(Q‐R) One month after the tumor xenograft, the PDX mice were sacrificed (Q), the tumor weight was measured (R).Error bars represent mean ± SD. Statistical significance was determined using Student's two‐tailed t‐test between two groups (A, C, E, F, H, J, and M), and two‐way ANOVA analysis among multiple groups (P and R). * P < 0.05; ** P < 0.01; *** P < 0.001.Abbreviations: GC, gastric cancer; PDX, patient‐derived xenograft; SD, standard deviation; NS, not significant.
To better simulate the effect of CCAT5 blockade, we further constructed endogastric xenograft tumor model with AGS‐luc (Wnthigh) cells, and monitored metastases formation by luciferase imaging and pathologic examination. After 5 weeks, all mice developed IPD, and the mice were then intraperitoneally injected with si‐CCAT5 or control siRNA every 3 days (Figure 7I). Four weeks after injection, we observed that si‐CCAT5 treatment not only significantly decreased the volume and luciferase intensity of endogastric xenograft tumor (Figure 7J, Supplementary Figure S8G), but also reduced the number of abdominal macro‐metastases (Figure 7K‐M). Overall, our data support the therapeutic efficacy of targeting CCAT5 in combating growth and metastasis of GC with high Wnt activity.
Subsequently, to determine the clinical benefits of targeting CCAT5 in metastatic GC, we successfully established five patient‐derived IPD xenograft (PDX) models with strong β‐catenin nuclear expression (β‐cateninnuc) using malignant ascites or IPD tumor (Figure 7N‐O). We found that the xenograft tumor growth was effectively inhibited by intratumoral addition of si‐CCAT5. Moreover, the tumor suppression effect of si‐CCAT5 was much better than that of Wnt specific inhibitor XAV939, and equivalent to first‐line chemotherapeutic agent oxaliplatin. Of note, the combination of si‐CCAT5 and oxaliplatin displayed obvious synergistic therapeutic effects on β‐cateninnuc PDX mice (Figure 7P‐R). Collectively, RNAi‐based therapeutics targeting CCAT5 selectively decreased the progression of Wnthigh metastatic GC.
4. DISCUSSION
Genetic and epigenetic deregulation of Wnt/β‐catenin signaling contributes greatly to tumor evolution [21]. It is well‐established that Wnt signaling pathway acts as the downstream effector of lncRNAs in cancer metastasis [39, 40, 41]. LncRNAs drive malignant transformation through interactions with other cellular macromolecules to activate or suppress the Wnt/β‐catenin signaling pathway [41, 42, 43, 44]. However, few studies of whether Wnt‐induced lncRNAs exist and their potential role in tumor development (if these lncRNAs exist) have been reported. In this study, we identified CCAT5 as the direct transcriptional target of Wnt signaling cascade and confirmed that CCAT5 transcription is modulated by TCF3, but not the other TCF transcriptional factors. In addition, we also confirmed the co‐occupancy of β‐catenin and TCF3 on CCAT5 promoter. Thus, this study provides structural insight into β‐catenin/TCF3‐mediated CCAT5 transcription.
LncRNAs attracted widespread attention as biomarkers for diagnosis, prognosis, and regulator of chemoresistance [16]. Thus far, a series of lncRNAs have been reported to have a role in GC initiation and progression. We reported previously a novel onclncRNA‐626 which promoted malignancy of GC via inactivated the p53 pathway through interacting with SRSF1 [45]. Studies have also revealed that CCAT5 was aberrantly overexpressed in various types of cancer and exerts oncogenic functions [46, 47]. Consistent with literature, this study identified CCAT5 as a key promoter of GC growth and metastasis, and independent prognostic predictor.
Accumulating studies have indicated the involvement of lncRNAs in STAT3‐induced tumor evolution. For example, HOXA11‐AS drove cisplatin resistance of lung cancer via modulating miR‐454‐3p/STAT3 axis [48]. SNHG16 induces hemangioma progression by modulating miR‐520d‐3p/STAT3 axis [49]. By a series of mechanistic studies, this study demonstrated STAT3 as the top‐ranked signaling pathway regulated by CCAT5. Notably, our data also indicated CCAT5 facilitated GC progression in a STAT3‐mediated manner. CCAT5 was demonstrated to regulate miR‐34a/SIRT1, miR‐218‐5p/COMMD8, miR‐6785‐5p/BCL2, miR‐370/FoxM1, and miR‐744‐5p/BCL9/β‐catenin axis to promote aggressive characteristics of a series of tumors [50, 51, 52, 53, 54]. Various mechanisms have been implicated in the lncRNA‐mediated gene regulation and cancer phenotypes, which can be attributed to their ability to interact with DNA, RNA or protein [17]. CCAT5 was also reported to bind to YB1 protein and prevented its ubiquitination and degradation in colorectal cancer [55]. This study disclosed CCAT5 as a new STAT3 binding molecule and CCAT5 fragment 612‐781nt bound specifically to the C‐end domain of STAT3. Another new point of this study is that CCAT5 could attenuate SHP1‐STAT3 interaction and protect STAT3 from Y705 dephosphorylation by SHP1. These data characterized CCAT5 as a direct signal transducer through acting on the functional domains of STAT3.
The depth of research underscores the complex interplay between Wnt/β‐catenin and STAT3 signaling. Previous studies have reported Wnt3a as activator of STAT3 signaling in kidney disease models [37, 38]. Van Andel et al. [15] revealed that aberrant LGR4 expression driven by IL‐6/STAT3 signaling allows multiple myeloma cells to hijack R‐spondins produced by (pre)osteoblasts in the bone marrow niche, resulting in empowered Wnt signaling. However, this field lacks a unified model regarding the promotive mechanism of Wnt in STAT3 signaling. This study provides further evidence to support that both Wnt3a and β‐catenin act as promoter of STAT3 phosphorylation and transcription activity in both intestinal‐type and diffuse‐type GC cells. Notably, depletion of CCAT5 using CRISPR/Cas9‐based strategy obviously suppressed Wnt3a/β‐catenin‐induced STAT3 activation, thus revealing STAT3 signaling regulation via canonical Wnt signaling and the functional significance of CCAT5 as critical mediator. Most importantly, ectopic expression of full length CCAT5, but not CCAT5△612‐781, restored the decreased Wnt‐induced STAT3 activity and malignant phenotype resulting from CCAT5 depletion, further revealing the interplay between CCAT5 and STAT3 is necessary for Wnt‐drived STAT3 signaling and tumor evolution (Figure 8). While these findings highlight CCAT5's capability to drive JAK‐STAT3 pathway activation, it remains paramount to recognize the intricate intertwining of CCAT5 with various other signaling pathways, notably the MYC and PI3K‐AKT pathway. For forthcoming studies, a deeper dive into understanding CCAT5's influence on these signaling cascades becomes paramount. Furthermore, it is imperative to clarify the role of CCAT5 in the tumor microenvironment. This comprehensive pursuit is poised to enrich our grasp of the multifaceted role that CCAT5 plays in the intricate realm of cancer progression.
FIGURE 8.
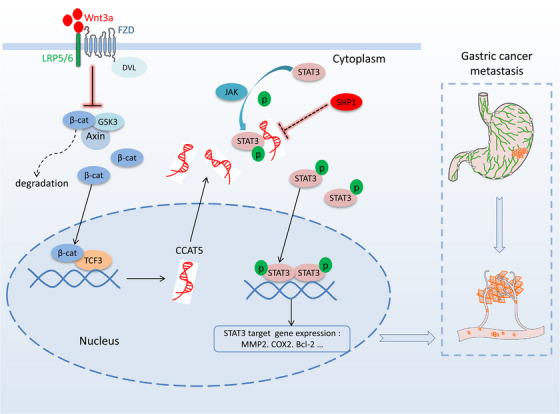
The graphic illustration of Wnt/β‐catenin‐CCAT5‐STAT3 signaling axis in GC metastasis. Abbreviations: Wnt3a, Wnt family member 3A; β‐cat, β‐catenin; TCF3, Transcription factor 3; STAT3, signal transducer and activator of transcription 3; FZD, frizzled class receptor; SHP1, SH2‐containing protein tyrosine phos‐phatase 1; GSK3, glycogen synthase kinase 3; LRP5/6, LDL receptor related protein 5/6; DVL, dishevelled segment polarity protein.
As the clinical significance of lncRNAs is increasingly appreciated, multiple therapeutics targeting cancer‐related lncRNAs, including siRNAs, ASO‐based strategies, and small molecular inhibitors, are under active investigation [20]. Although several lncRNAs have shown in vivo benefits in gastrointestinal cancers [56, 57], effective targets remain largely unknown. Through RNAi‐based cell xenograft model and human PDX model, this study showed that siRNA against CCAT5 can selectively reduce tumor growth and metastasis of Wnthigh GC, but not GC with low Wnt activity, independent of intestinal‐type or diffuse‐type GC. Moreover, combining in vivo si‐CCAT5 with Oxaliplatin chemotherapy conferred remarkable tumor regression in Wnthigh metastatic GC. Although no significant adverse reactions were observed in mice models during the siRNA treatment, further clinical trials are needed to explore the feasibility of siRNA against CCAT5 on human gastric tumors.
5. CONCLUSIONS
This study identified and systematically characterized the transcription mechanism of a novel Wnt‐transactivated lncRNA‐CCAT5, providing compelling association of high CCAT5 expression with aggressive malignancy and poor prognosis. Mechanistically, CCAT5 activates STAT3 signaling pathway to promote GC progression by directly binding to STAT3 and blocking SHP‐1‐mediated STAT3Y705 dephosphorylation. This study demonstrated the critical role of CCAT5 in Wnt‐drived STAT3 activation and GC metastasis and highlighted the therapeutic potential of targeting CCAT5 by high purity siRNA (alone or in combination with Oxaliplatin) for Wnthigh metastatic GC.
AUTHOR CONTRIBUTIONS
Yanong Wang and Xiaowen Liu designed this study. Chenchen Liu, Aiwen Shen, Junquan Song, Lei Cheng, and Meng Zhang performed the experiments. Yanong Wang and Xiaowen Liu supervised the direction of project and interpreted the data. Chenchen Liu, Xiaowen Liu, and Yanong Wang wrote the manuscript. All authors discussed the results and commented on the manuscript.
CONFLICT OF INTERESTS STATEMENT
The authors declare that they have no competing interests.
FUNDING INFORMATION
This work was sponsored by Shanghai Sailing Program (20YF1409200), Shanghai Pujiang Program (2019PJD007), and National Natural Science Foundation of China (82002545, 81902875, 81972213, 82372874 and 81902430).
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Clinical Research Ethics Committee of Fudan University Shanghai Cancer Center (No. 050432‐4‐1911D). Written informed consents were received from all patients. All animal studies were conducted in accordance with the Animal Care Guidelines of FUSCC (No. 2020JS‐139).
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
We thank Dr. Xiaoling Wang (Shanghai Institute of Hematology, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine) and anonymous reviewers for reading and commenting on the manuscript.
Liu C, Shen A, Song J, Cheng L, Zhang M, Wang Y, et al. LncRNA‐CCAT5‐mediated crosstalk between Wnt/β‐Catenin and STAT3 signaling suggests novel therapeutic approaches for metastatic gastric cancer with high Wnt activity. Cancer Commun. 2024;44:76–100. 10.1002/cac2.12507
Chenchen Liu and Aiwen Shen contributed equally to this work.
Contributor Information
Yanong Wang, Email: drwangyanong@126.com.
Xiaowen Liu, Email: liuxw1129@hotmail.com.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396(10251):635‐648. [DOI] [PubMed] [Google Scholar]
- 2. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guner A, Yildirim R. Surgical management of metastatic gastric cancer: moving beyond the guidelines. Transl Gastroenterol Hepatol. 2019;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chia DKA, So JBY. Recent Advances in Intra‐peritoneal Chemotherapy for Gastric Cancer. J Gastric Cancer. 2020;20(2):115‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ooi CH, Ivanova T, Wu J, Lee M, Tan IB, Tao J, et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 2009;5(10):e1000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, et al. Whole‐genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46(6):573‐582. [DOI] [PubMed] [Google Scholar]
- 7. Koushyar S, Powell AG, Vincan E, Phesse TJ. Targeting Wnt Signaling for the Treatment of Gastric Cancer. Int J Mol Sci. 2020;21(11):3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heuberger J, Birchmeier W. Interplay of cadherin‐mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2(2):a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang X, Cong F. Novel Regulation of Wnt Signaling at the Proximal Membrane Level. Trends Biochem Sci. 2016;41(9):773‐783. [DOI] [PubMed] [Google Scholar]
- 11.Ter Steege EJ, Bakker ERM. The role of R‐spondin proteins in cancer biology. Oncogene. 2021;40(47):6469‐6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell. 2015;162(4):780‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. β‐Catenin‐driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee G, Goretsky T, Managlia E, Dirisina R, Singh AP, Brown JB, et al. Phosphoinositide 3‐kinase signaling mediates beta‐catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology. 2010;139(3):869‐881, 81.e1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Andel H, Ren Z, Koopmans I, Joosten SP, Kocemba KA, de Lau W, et al. Aberrantly expressed LGR4 empowers Wnt signaling in multiple myeloma by hijacking osteoblast‐derived R‐spondins. Proc Natl Acad Sci U S A. 2017;114(2):376‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan L, Xu ZY, Ruan SM, Mo S, Qin JJ, Cheng XD. Long non‐coding RNAs towards precision medicine in gastric cancer: early diagnosis, treatment, and drug resistance. Mol Cancer. 2020;19(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng WX, Koirala P, Mo YY. LncRNA‐mediated regulation of cell signaling in cancer. Oncogene. 2017;36(41):5661‐5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan YT, Lin JF, Li T, Li JJ, Xu RH, Ju HQ. LncRNA‐mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun (Lond). 2021;41(2):109‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao Y, Wang JW, Ren JY, Guo M, Guo CW, Ning SW, et al. Long noncoding RNAs in gastric cancer: From molecular dissection to clinical application. World J Gastroenterol. 2020;26(24):3401‐3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin C, Yang L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018;28(4):287‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jung YS, Park JI. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond β‐catenin and the destruction complex. Exp Mol Med. 2020;52(2):183‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Wang L, Qu Y. Targeting the β‐catenin signaling for cancer therapy. Pharmacol Res. 2020;160:104794. [DOI] [PubMed] [Google Scholar]
- 23. Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, Krohn M, et al. Characterization of twenty‐five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun. 2015;6:7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smoot DT, Sewchand J, Young K, Desbordes BC, Allen CR, Naab T. A method for establishing primary cultures of human gastric epithelial cells. Methods Cell Sci. 2000;22(2‐3):133‐136. [DOI] [PubMed] [Google Scholar]
- 25. Ajani JA, Xu Y, Huo L, Wang R, Li Y, Wang Y, et al. YAP1 mediates gastric adenocarcinoma peritoneal metastases that are attenuated by YAP1 inhibition. Gut. 2020;70(1):55‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Furlan‐Magaril M, Rincon‐Arano H, Recillas‐Targa F. Sequential chromatin immunoprecipitation protocol: ChIP‐reChIP. Methods Mol Biol. 2009;543:253‐266. [DOI] [PubMed] [Google Scholar]
- 27. Yue B, Liu C, Sun H, Liu M, Song C, Cui R, et al. A Positive Feed‐Forward Loop between LncRNA‐CYTOR and Wnt/beta‐Catenin Signaling Promotes Metastasis of Colon Cancer. Mol Ther. 2018;26(5):1287‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luan H, Yan L, Zhao Y, Ding X, Cao L. Fucoxanthin induces apoptosis and reverses epithelial‐mesenchymal transition via inhibiting Wnt/β‐catenin pathway in lung adenocarcinoma. Discov Oncol. 2022;13(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature. 2015;523(7559):231‐235. [DOI] [PubMed] [Google Scholar]
- 30. Zhang H, Xu R, Li B, Xin Z, Ling Z, Zhu W, et al. LncRNA NEAT1 controls the lineage fates of BMSCs during skeletal aging by impairing mitochondrial function and pluripotency maintenance. Cell Death Differ. 2022;29(2):351‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 2008;22(6):746‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736‐746. [DOI] [PubMed] [Google Scholar]
- 33. Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29(4):452‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene. 2016;35(8):939‐951. [DOI] [PubMed] [Google Scholar]
- 35. Yuan J, Zhang F, Niu R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci Rep. 2015;5:17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang TT, Su JC, Liu CY, Shiau CW, Chen KF. Alteration of SHP‐1/p‐STAT3 Signaling: A Potential Target for Anticancer Therapy. Int J Mol Sci. 2017;18(6):1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng Y, Ren J, Gui Y, Wei W, Shu B, Lu Q, et al. Wnt/beta‐Catenin‐Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J Am Soc Nephrol. 2018;29(1):182‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fragoso MA, Patel AK, Nakamura RE, Yi H, Surapaneni K, Hackam AS. The Wnt/beta‐catenin pathway cross‐talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLoS One. 2012;7(10):e46892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang G, Shen T, Yi X, Zhang Z, Tang C, Wang L, et al. Crosstalk between long non‐coding RNAs and Wnt/β‐catenin signalling in cancer. J Cell Mol Med. 2018;22(4):2062‐2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zarkou V, Galaras A, Giakountis A, Hatzis P. Crosstalk mechanisms between the WNT signaling pathway and long non‐coding RNAs. Noncoding RNA Res. 2018;3(2):42‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu XY, Hou PF, Li TT, Quan HY, Li ML, Lin T, et al. The roles of Wnt/β‐catenin signaling pathway related lncRNAs in cancer. Int J Biol Sci. 2018;14(14):2003‐2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ma Y, Yang Y, Wang F, Moyer MP, Wei Q, Zhang P, et al. Long non‐coding RNA CCAL regulates colorectal cancer progression by activating Wnt/β‐catenin signalling pathway via suppression of activator protein 2α. Gut. 2016;65(9):1494‐1504. [DOI] [PubMed] [Google Scholar]
- 43. Lu Y, Zhao X, Liu Q, Li C, Graves‐Deal R, Cao Z, et al. lncRNA MIR100HG‐derived miR‐100 and miR‐125b mediate cetuximab resistance via Wnt/β‐catenin signaling. Nat Med. 2017;23(11):1331‐1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, He L, Du Y, Zhu P, Huang G, Luo J, et al. The long noncoding RNA lncTCF7 promotes self‐renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16(4):413‐425. [DOI] [PubMed] [Google Scholar]
- 45. Wu ZH, Liu CC, Zhou YQ, Hu LN, Guo WJ. OnclncRNA‐626 promotes malignancy of gastric cancer via inactivated the p53 pathway through interacting with SRSF1. Am J Cancer Res. 2019;9(10):2249‐2263. [PMC free article] [PubMed] [Google Scholar]
- 46. Li C, Wang S, Xing Z, Lin A, Liang K, Song J, et al. A ROR1‐HER3‐lncRNA signalling axis modulates the Hippo‐YAP pathway to regulate bone metastasis. Mol Cell Biol. 2017;19(2):106‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lv Y, Li H, Li F, Liu P, Zhao X. Long Noncoding RNA MNX1‐AS1 Knockdown Inhibits Cell Proliferation and Migration in Ovarian Cancer. Cancer Biother Radiopharm. 2017;32(3):91‐99. [DOI] [PubMed] [Google Scholar]
- 48. Zhao X, Li X, Zhou L, Ni J, Yan W, Ma R, et al. LncRNA HOXA11‐AS drives cisplatin resistance of human LUAD cells via modulating miR‐454‐3p/Stat3. Cancer Science. 2018;109(10):3068‐3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao W, Fu H, Zhang S, Sun S, Liu Y. LncRNA SNHG16 drives proliferation, migration, and invasion of hemangioma endothelial cell through modulation of miR‐520d‐3p/STAT3 axis. Cancer Med. 2018; 7(7):3311‐3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chu J, Li H, Xing Y, Jia J, Sheng J, Yang L, et al. LncRNA MNX1‐AS1 promotes progression of esophageal squamous cell carcinoma by regulating miR‐34a/SIRT1 axis. Biomed Pharmacother. 2019;116:109029. [DOI] [PubMed] [Google Scholar]
- 51. Ji D, Wang Y, Sun B, Yang J, Luo X. Long non‐coding RNA MNX1‐AS1 promotes hepatocellular carcinoma proliferation and invasion through targeting miR‐218‐5p/COMMD8 axis. Biochem Biophys Res Commun. 2019;513(3):669‐674. [DOI] [PubMed] [Google Scholar]
- 52. Shuai Y, Ma Z, Liu W, Yu T, Yan C, Jiang H, et al. TEAD4 modulated LncRNA MNX1‐AS1 contributes to gastric cancer progression partly through suppressing BTG2 and activating BCL2. Mol Cancer. 2020;19(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cui X, Yu H, Yu T, Xiao D, Wang X. LncRNA MNX1‐AS1 drives aggressive laryngeal squamous cell carcinoma progression and serves as a ceRNA to target FoxM1 by sponging microRNA‐370. Aging (Albany NY). 2021;13(7):9900‐9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma B, Ren G, Xu J, Yin C, Shi Y. LncRNA MNX1‐AS1 Contributes to Laryngeal Squamous Cell Carcinoma Growth and Migration by Regulating mir‐744‐5p/bcl9/β‐Catenin Axis. Cell Transplant. 2021;30:9636897211005682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu QN, Luo XJ, Liu J, Lu YX, Wang Y, Qi J, et al. MYC‐Activated LncRNA MNX1‐AS1 Promotes the Progression of Colorectal Cancer by Stabilizing YB1. Cancer Res. 2021;81(10):2636‐2650. [DOI] [PubMed] [Google Scholar]
- 56. Pichler M, Rodriguez‐Aguayo C, SY Nam, Dragomir MP, Bayraktar R, Anfossi S, et al. Therapeutic potential of FLANC, a novel primate‐specific long non‐coding RNA in colorectal cancer. Gut. 2020;69(10):1818‐1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhuo W, Liu Y, Li S, Guo D, Sun Q, Jin J, et al. Long Noncoding RNA GMAN, Up‐regulated in Gastric Cancer Tissues, Is Associated With Metastasis in Patients and Promotes Translation of Ephrin A1 by Competitively Binding GMAN‐AS. Gastroenterology. 2019;156(3):676‐691.e11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.