

Abstract
Amyloid-β, tau pathology, and biomarkers of neurodegeneration make up the core diagnostic biomarkers of Alzheimer disease (AD). However, these proteins represent only a fraction of the complex biological processes underlying AD, and individuals with other brain diseases in which AD pathology is a comorbidity also test positive for these diagnostic biomarkers. More AD-specific early diagnostic and disease staging biomarkers are needed. In this study, we performed tandem mass tag proteomic analysis of paired cerebrospinal fluid (CSF) and serum samples in a discovery cohort comprising 98 participants. Candidate biomarkers were validated by parallel reaction monitoring–based targeted proteomic assays in an independent multicenter cohort comprising 288 participants. We quantified 3,238 CSF and 1,702 serum proteins in the discovery cohort, identifying 171 and 860 CSF proteins and 37 and 323 serum proteins as potential early diagnostic and staging biomarkers, respectively. In the validation cohort, 58 and 21 CSF proteins, as well as 12 and 18 serum proteins, were verified as early diagnostic and staging biomarkers, respectively. Separate 19-protein CSF and an 8-protein serum biomarker panels were built by machine learning to accurately classify mild cognitive impairment (MCI) due to AD from normal cognition with areas under the curve of 0.984 and 0.881, respectively. The 19-protein CSF biomarker panel also effectively discriminated patients with MCI due to AD from patients with other neurodegenerative diseases. Moreover, we identified 21 CSF and 18 serum stage-associated proteins reflecting AD stages. Our findings provide a foundation for developing blood-based tests for AD screening and staging in clinical practice.
Graphical abstract
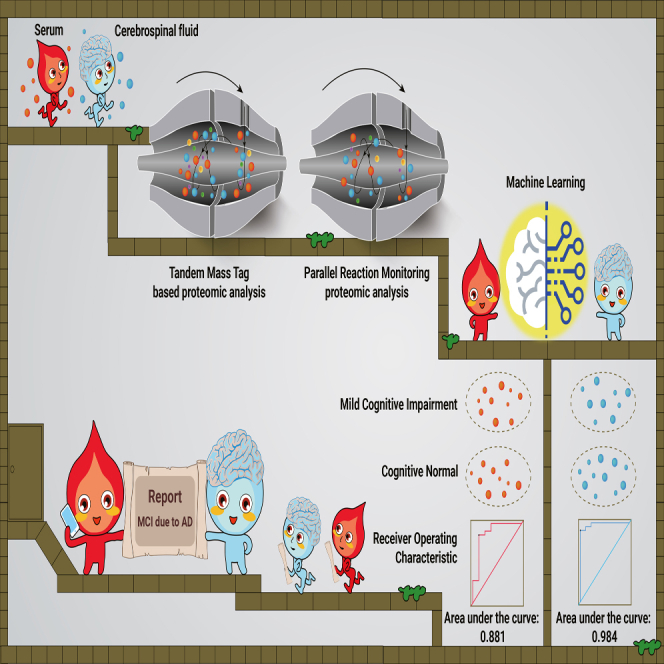
Public summary
-
•
In-depth proteomics workflow for profiling paired cerebrospinal fluid (CSF) and serum proteomes.
-
•
Independent multicenter set combined with multiple methods for validation.
-
•
Development of 19 CSF- and 8 serum-protein panels for early of Alzheimer disease (AD) diagnosis.
-
•
Twenty-one CSF and 18 serum proteins dysregulated in different AD stages.
-
•
Groundwork laid for AD blood tests in clinical screening and staging.
Introduction
Alzheimer disease (AD) is the most common type of neurodegenerative disease and is pathologically characterized by the deposition of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles.1 AD patients typically present with progressive cognitive decline. Extensive efforts have been made to determine the optimal strategy for diagnosing this devastating disease.2,3,4 A research framework defining AD on the basis of Aβ deposition, phosphorylated tau (p-tau), and neurodegeneration was proposed in 2018.5 However, positivity in these diagnostic biomarkers can also be observed in individuals with other brain diseases in which AD pathology has been recognized a comorbidity.6 Furthermore, several limitations, such as high cost, insufficient accessibility, and invasiveness, impede the use of cerebrospinal fluid (CSF) and positron emission tomography (PET) biomarkers as first-line diagnostic strategies.
Blood testing is advantageous due to its convenience, minimal invasiveness, and affordability. Recently, several blood-based AD biomarkers, such as the plasma Aβ42/40 ratio, p-tau, neurofilament light polypeptide (NEFL), and glial fibrillary acidic protein (GFAP), have been reported.7,8 Notably, plasma tau phosphorylated at threonine 217 (p-tau217) could be used to accurately determine AD and was correlated with Aβ and p-tau pathology in the brain.9 Nevertheless, plasma biomarkers for Aβ and tau pathology are not currently recommended for use in clinical practice because they remain to be further standardized and validated.6 In addition, these clinical manifestations lose their correlation with both Aβ and p-tau protein levels as the disease progresses, suggesting that Aβ and p-tau biomarkers are not suitable for staging.10 Converging findings indicate the presence of many additional pathological mechanisms underlying the pathogenesis of AD that are independent of Aβ and p-tau pathology.11 Comprehensive research is necessary to identify early diagnostic and staging biomarkers for AD. We previously used proteomic analyses to identify blood biomarkers for evaluating the severity of coronavirus disease 2019.12 In recent years, several mass spectrometry (MS)–based proteomics profiling studies of CSF and plasma have revealed protein biomarkers and insights into the biological processes underlying AD, with minimum sample amounts and high-throughput workflows.2,13,14,15,16 However, early diagnostic and staging blood-based biomarkers of AD remain incompletely defined.
To systemically identify early diagnostic and staging biomarkers of AD, we performed high-throughput MS-based proteomics analysis in a discovery cohort with paired CSF and serum samples and validated the results in an independent multicenter validation cohort.
Results
Study design
The study design is outlined in Figure 1. To explore early diagnostic and staging biomarkers of AD, we performed tandem mass tag (TMT)–based proteomic analysis of paired CSF and serum samples in the discovery cohort (Figure 1A; Table S1). We achieved a robust workflow with high proteome depth, quantifying 3,238 CSF (Figure S1) and 1,702 serum proteins (Figure S2; Table S2). The ranking of the median abundance of CSF proteins is shown in Figure S3A, and the identification depth of serum proteins is shown in Figure S3B. After filtering out proteins with a missing rate higher than 80%, 2,461 CSF proteins and 1,330 serum proteins remained for subsequent data analysis. The missing values of the two protein matrices were both 0.19. Potential early diagnosis and staging biomarkers in CSF and serum were determined. Mfuzz analysis was used to cluster the proteins that were dysregulated during the different stages of AD.17 Then, the identified specific dysregulated proteins that differed between the cognitive normal (CN) group and mild cognitive impairment (MCI) due to AD group, as well as stage-dependent dysregulated proteins, were further validated in an independent multicenter cohort (Figure 1B). Machine learning models were built to classify MCI due to AD patients from CN participants. In addition, a cluster of CSF and serum proteins that were dysregulated in an AD stage–dependent manner was validated.
Figure 1.

Overview of the study populations and schematic of the proteomic workflow
(A) The workflow of the TMT-based proteome for the discovery of potential early AD diagnostic and staging biomarkers. Paired CSF and serum samples were collected from the discovery cohort, comprising patients with AD, MCI due to AD, HD, and ALS and CN participants.
(B) The workflow of targeted proteomics for validating the early AD diagnostic and staging biomarkers. CSF and serum samples were collected from the validation cohort, comprising AD patients, patients with MCI due to AD, CN participants, and HD, ALS, and FTD patients.
TMT-based MS proteomics reveals proteins significantly altered in MCI due to AD
To identify the early CSF diagnostic biomarkers of AD, we compared the CSF proteomes of the MCI due to AD and CN participants in the discovery cohort. Compared with those of CN participants, the expression levels of 185 CSF proteins were upregulated (p < 0.05, log2 fold change [FC] > 0.25), and those of 5 CSF proteins were downregulated (p < 0.05, log2 FC <−0.25). To improve the specificity of the biomarkers, dysregulated proteins between CN and amyotrophic lateral sclerosis (ALS)/Huntington disease (HD) were excluded. The remaining 171 dysregulated proteins, including 167 with upregulated expression levels and 4 with downregulated expression levels, were CSF-specific biomarkers for MCI due to AD and were used for further validation (Figure 2A).
Figure 2.

Dysregulated CSF and serum proteins and pathways in the diagnosis of early AD
(A) Dysregulated (p < 0.05, |log2 FC| >0.25) CSF proteins between MCI due to AD, ALS, and HD patients and CN participants.
(B) The significantly dysregulated pathways (−log10 p > 1.3) enriched by IPA using 190 dysregulated CSF proteins.
(C) Heatmap of key dysregulated CSF proteins in various associated pathways in patients relative to CN participants.
(D) Dysregulated (p < 0.05, |log2 FC| >0.25) serum proteins between MCI due to AD, ALS, and HD patients and CN participants.
(E) The significantly dysregulated pathways (−log10 p > 1.3) enriched by IPA using 49 dysregulated CSF proteins.
(F) Heatmap of key dysregulated serum proteins in various associated pathways in patients relative to CN participants.
The 190 significantly dysregulated CSF proteins were analyzed using ingenuity pathway analysis (IPA) to identify enriched pathways. IPA showed molecular changes in the CSF of MCI due to AD patients relative to CN participants, implicating significant dysregulation (−log10 p > 1.3) of insulin growth factor 1 (IGF-1) signaling, dermatan sulfate biosynthesis, chondroitin sulfate biosynthesis, matrix metalloproteases, xenobiotic metabolism/pregnane X receptor (PXR), the constitutive androstane receptor (CAR) signaling pathway, heparan sulfate biosynthesis, and others, as shown in Figure 2B. Notably, we found that several identified pathways were closely related to the extracellular matrix (ECM; i.e., dermatan sulfate biosynthesis, chondroitin sulfate biosynthesis, and matrix metalloproteases). Our results imply the critical role of abnormal ECM metabolism in the pathogenesis of AD. In addition, several key dysregulated proteins in the CSF involved in these pathways are shown in Figure 2C.
To identify the blood-based early diagnostic biomarkers of AD, we analyzed the paired serum proteomes in the above participants. We identified 49 dysregulated proteins (p < 0.05, |log2 FC| > 0.25) between the MCI due to AD group and CN group. After excluding dysregulated serum proteins overlapping with the HD and ALS groups, 37 potential specific serum biomarkers for MCI due to AD were selected for further validation (Figure 2D).
In addition, IPA was used to analyze the 49 significantly dysregulated proteins to identify enriched pathways. The results showed molecular differences in the serum between the MCI due to AD patients and the CN group involving the following pathways: retinoate biosynthesis; the human leukocyte antigen-F adjacent transcript 10 cancer signaling pathway, natural killer cell signaling, the HOX transcript antisense RNA regulatory pathway, dendritic cell maturation, and others, as shown in Figure 2E. Several key dysregulated proteins involved in these pathways in MCI due to AD are shown in Figure 2F.
Verification of differentially expressed proteins in MCI due to AD in the independent validation cohort
To verify the results of the TMT-based proteomic analysis, a parallel reaction monitoring (PRM)-based targeted proteomic experiment was performed in an independent multicenter cohort. The validation cohort comprised 221 participants with CSF samples and 288 participants with serum samples (Figure 1B; Table S3). A total of 57 and 12 dysregulated CSF and serum proteins in the MCI due to AD group were verified, respectively (Table S4). In addition, we conducted real-time PCR to validate the transcription level of the differentially expressed proteins using the cortexes of 5×FAD mice. Two important candidates (GFAP and GM2A) were selected and verified (Figure S4).
We next explored the possibility of distinguishing between MCI due to AD and CN based on the dysregulated proteins. We formulated a random forest machine learning model using CSF proteomic data from 21 MCI due to AD and 52 CN participants. To ascertain the smallest number of proteins required to sufficiently differentiate MCI due to AD from CN, the prioritization of dysregulated proteins was explored by machine learning. A panel was constructed comprising the 19 proteins that were determined as CSF early diagnostic biomarkers (Figure 3A). The abundance ranking of the 19 proteins among the identified CSF proteins in this study can be seen in Figure S3A. We then tested the model on 9 MCI due to AD and 21 CN participants. This model demonstrated a high level of accuracy, with an area under the curve (AUC) of 0.984 in the test set (Figure 3B). The classifier model had similar accuracy when stratifying by sex, age, or APOE ε4 genotype. Moreover, the 19-protein CSF biomarker panel could effectively discriminate MCI due to AD patients from frontotemporal dementia (FTD), ALS, and HD patients (Figure S5A).
Figure 3.

Differentiation of MCI due to AD and CN subjects by machine learning of CSF proteomic features
(A) Top 19 proteins prioritized by random forest analysis ranked by the mean decrease in accuracy.
(B) ROC curve of the random forest model.
(C) Expression level change (Z scored original value) of 8 core proteins with significant differences between patients with MCI due to AD and CN participants in both TMT-based and PRM proteomic analyses. p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.005.
Most of the selected proteins showed the same expression tendency in the discovery cohort and validation cohort that were assayed by the TMT and PRM experiments, respectively (Figure 3C). Notably, four (MGAT2, GM2A, MAN1C1, and MAN2A2) of the 19 proteins were related to the Golgi and lysosome pathways (i.e., function as enzymes or participate in the transport process to the Golgi). It is well known that pathways related to the Golgi apparatus and lysosomes have critical roles in the pathogenesis of neurodegenerative diseases such as AD, Parkinson disease (PD), and ALS.18 The expression levels of all four proteins were upregulated in the MCI due to AD group relative to the CN group, suggesting that compensatory mechanisms may be initiated during the early stage of AD to protect against protein misfolding and aggregation. Among the 19 proteins in the panel, PCDHGC5 and IGHM are related to immune function (Figure 3C). IGHM has been reported to have an inverse correlation with Aβ burden in an AD mouse model.19 Our data further supported the underlying connections between Aβ pathobiology and immunoglobulins.
Concomitantly, a random forest machine learning model was constructed based on the serum proteomic data from 23 participants with MCI due to AD and 45 CN participants. A panel consisting of 8 core proteins was selected to distinguish MCI due to AD from CN (Figure 4A). The rankings of the concentrations of these 8 proteins in the blood can be seen in Figure S3B. A cohort of 8 MCI due to AD and 21 CN participants was used to test the model, achieving an AUC of 0.881 (Figure 4B). The expression levels of 5 representative proteins with high confidence in the panel are shown in boxplots (Figure 4C). However, the ability to discriminate MCI due to AD from FTD, ALS, and HD was relatively low using the 8-protein serum biomarker panel (Figure S5B), which indicates that this analysis needs to be optimized further.
Figure 4.

Differentiation of MCI due to AD and CN by machine learning of serum proteomic features
(A) Top 8 proteins prioritized by random forest analysis ranked by the mean decrease in accuracy.
(B) ROC curve of the random forest model.
(C) Expression level change (Z scored original value) of the 5 core proteins with significant differences between patients with MCI due to AD and CN participants in both TMT-based and PRM proteomic analyses. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.005.
Subsequently, we proceeded to analyze the diagnostic efficiency for MCI due to AD based on the levels of serum GFAP and NEFL detected by commercially available Single Molecular Immunity Detection kits in a subset of the validation cohort, comprising 41 MCI due to AD patients, 14 FTD patients, 14 ALS patients, and 31 CN controls (Table S5). Compared to those of the CN controls, elevated levels of GFAP and NEFL were found in the sera of patients with ALS and FTD. In addition, patients with MCI due to AD exhibited increased levels of GFAP (Figure S6A). Receiver operating characteristic (ROC) curve analysis was used to evaluate the efficacy of serum GFAP, NEFL, and their combination in distinguishing between MCI due to AD participants and CN controls. The findings revealed that serum GFAP achieved an AUC of 0.714, whereas NEFL achieved an AUC of 0.511 (Figure S6B). In contrast to the two established serum biomarkers, our biomarker panel demonstrated superior diagnostic efficacy, with an AUC of 0.881. In addition, the levels of GFAP and NEFL were insufficient in differentiating patients with MCI due to AD from those with FTD and ALS (Figure S6C).
Correlations between the levels of core CSF and serum dysregulated proteins and the CSF levels of Aβ42, t-tau, and p-tau181 in patients with MCI due to AD
We then explored whether the levels of the core dysregulated CSF and serum proteins in patients with MCI due to AD were correlated with the levels of CSF Aβ42, t-tau, and p-tau181. Interestingly, we found that among the 19 core CSF early diagnostic biomarkers, the levels of 2 (CHRD and COL18A1) were correlated with the levels of CSF Aβ42, the levels of 4 others (B3GALNT1, GM2A, SHBG, and IGHM) were correlated with the levels of CSF p-tau181, and the levels of yet 2 others (MAN2A2 and PCDHGC5) were correlated with the levels of both CSF Aβ42 and p-tau181 (Figure S7). Among the 8 core serum early diagnostic biomarkers, C4B levels were correlated with CSF Aβ42 levels, IGHV3-35 levels were correlated with CSF t-tau levels, and SERPINA11 levels were correlated with both CSF t-tau and p-tau181 levels (Figure S7). These results showed that only a fraction of the core dysregulated proteins we identified, 8/19 in CSF and 3/8 in serum, were associated with Aβ and p-tau pathology. This implies that the progression of AD is influenced by a variety of molecular pathways in addition to Aβ and p-tau.
Paired CSF and serum proteomes reveal proteins that are significantly altered during the progression of AD
To identify AD staging biomarkers, we compared the dysregulated proteins identified during the different stages of AD, including the MCI due to AD mild stage (ADM), AD moderate stage (ADMO), and AD severe stage (ADS). A total of 2,461 identified CSF proteins were clustered using Mfuzz into 8 discrete clusters (Figure S8). Among them, proteins in cluster 7 and cluster 8 showed the same regulatory trend with disease progression (Figure 5A). The levels of 18 selected significantly dysregulated proteins (ANOVA p < 0.05 in cluster 7, ANOVA p < 0.01 in cluster 8) are shown in the heatmap in Figure 5B. Network pathway analysis of the dysregulated proteins revealed several important molecular-related pathways, including pathways involving insulin, prolactin, calcium-dependent protein kinase II, microtubule-associated protein tau (MAPT), and others, as shown in Figure S9. Several previously recognized AD CSF biomarkers,20 including GFAP, MAPT, NEFL, and neurogranin (NRGN), were among them; the levels of these biomarkers are shown by the boxplots in Figure 5C.
Figure 5.

Dysregulated CSF and serum proteins as potential biomarkers for AD staging
(A) Two clusters of CSF proteins identified with the Mfuzz analysis showed the same regulatory trend with disease progression.
(B) Heatmap of the expression levels of the core dysregulated CSF proteins (ANOVA p < 0.05 in cluster 7, ANOVA p < 0.01 in cluster 8) in different stages of AD.
(C) Expression level change (Z scored original value) of 4 recognized AD biomarkers with significant changes in the different stages of AD.
(D) Two clusters of serum proteins identified with the Mfuzz analysis showed the same regulatory trend with disease progression.
(E) Heatmap of the expression levels of the core dysregulated serum proteins (ANOVA p < 0.05) in different stages of AD.
To identify blood-based AD staging biomarkers, we compared the dysregulated proteins in serum samples along the trajectory of AD. A total of 1,330 identified proteins were clustered using Mfuzz into 8 discrete clusters (Figure S10). Among them, proteins in cluster 5 and cluster 6 showed the same regulatory trend with disease progression (Figure 5D). The levels of 18 selected significantly dysregulated proteins (ANOVA p < 0.05 in cluster 5 and cluster 6) demonstrating stage-dependent dysregulation are shown by the heatmap in Figure 5E. Several important proteins, including insulin, IGHM, PRDX2, and others, were identified by network pathway analysis, as shown in Figure S11.
Verification of proteins with AD stage–dependent dysregulation in the validation cohort
The validation cohort comprised 221 participants with CSF samples and 288 participants with serum samples. Regarding the CSF samples, a total of 21 core proteins with stage-dependent alterations were validated. The expression levels of 9 of them were downregulated with disease progression, whereas those of 12 of them were upregulated (Figure 6A). Remarkably, the expression level of ATRN was upregulated as the disease progressed. This protein has both membrane-bound and secreted protein isoforms (Figure 6B). ATRN has been reported to be differentially regulated in the blood samples of asymptomatic familial AD patients carrying PSEN1 mutations.21 In addition, 2 proteins, 14-3-3 protein epsilon (YWHAE) and 14-3-3 protein gamma (YWHAG), were upregulated over the disease progression (Figure 6B). CSF 14-3-3 proteins have already been used as surrogate markers of neuronal damage for AD, PD, and ALS but lack specificity.22 Our results are consistent with several recent proteomic studies showing that YWHAE and YWHAG are promising AD staging biomarkers.13,23
Figure 6.

Verification of AD stage-dependent dysregulated proteins in the validation cohort
(A) Heatmap of the levels of the validated core dysregulated CSF proteins in different stages of AD.
(B) Expression level change (Z scored original value) of 4 selected CSF proteins dysregulated over the course of AD progression.
(C) Heatmap of the levels of the validated core dysregulated serum proteins in different stages of AD.
(D) Expression level change (Z scored original value) of 4 selected serum proteins dysregulated over the course of AD progression.
Regarding the serum samples, a total of 18 core proteins exhibited stage-dependent alterations. The levels of 5 of them were downregulated, whereas those of 13 of them were upregulated with disease progression (Figure 6C). Notably, CFI was upregulated over the course of AD progression (Figure 6D). CFI is essential for regulating the complement cascade. Complement pathway hyperactivation has been found in an AD mouse model.24 In addition, PRDX2 was found to be dysregulated in serum during the development of AD (Figure 6D). Interestingly, similar to the results found in CSF, another 14-3-3 protein, YWHAQ, was found to be dysregulated in serum in the different stages of AD (Figure 6D).
Discussion
In this study, we performed one of the most in-depth proteomic analyses using paired CSF and serum samples acquired from AD patients. Our proteomic data mirrored a variety of disease-associated changes in the early stage and developmental trajectory of AD. Furthermore, based on an independent multicenter validation cohort, we developed a 19-protein CSF panel and an 8-protein serum panel for the early diagnosis of AD by machine learning. We used these panels to establish a highly accurate model for discriminating participants with MCI due to AD from CN participants. In addition, we verified 21 CSF and 18 serum core AD stage-dependent dysregulated proteins.
The leading dysregulated CSF and serum molecules during the early stage of AD are involved in IGF-1 signaling, ECM dysfunction, immune response pathways, lipid metabolism, Golgi and lysosome pathways, and oxidative species metabolism. Our CSF discovery dataset showed partial overlap with the results of several recent AD CSF proteomic studies.13,23,25 Notably, one plasma proteomic study developed a 19-protein blood-based panel that could accurately distinguish AD patients from healthy controls (AUC = 0.9690–0.9816).26 We narrowed the number of proteins to 8 in our serum-based panel that could accurately discriminate MCI due to AD from CN (AUC = 0.881), making it more convenient for clinical application in identifying MCI due to AD.
Our study revealed the differential activation of various biological pathways throughout the trajectory of AD, with the inflammatory pathway, synapse impairment pathway, and ECM dysfunction pathways emerging as key players. Neuroinflammation and related immune changes in the AD brain have been discussed intensely.27 Our results identified several biomarkers involved in the inflammatory and immune response pathways that were significantly changed during the trajectory of the disease, including CXCL16, ATRN, CFI, and others. These findings, which are consistent with those from previous studies,28 supported the growing evidence that neuroinflammation is an early event in AD and dynamically changes with disease development. It is well known that the levels of proteins associated with neuron or synapse degeneration pathways are increased in the CSF of AD patients.29 Our data uncovered a series of known degeneration-related biomarkers (tau, NEFL, and NRGN), as well as several novel synapse-related biomarkers (YWHAE, YWHAG, YWHAQ, NRN1, NPTX2, and NPTXR).
In addition, our study found that the levels of proteins associated with ECM dysfunction, including COL15A1, PCOLCE, and PVR, were increased in the CSF and serum in AD. The role of ECM dysfunction in AD has attracted tremendous attention in recent years.30 It is closely related to blood‒brain barrier breakdown, which has been considered an early event of cognitive decline in AD patients.31,32 These results are consistent with several recent proteomic studies performed in AD patients.33,34
The use of paired CSF and serum proteomics is more conducive to investigating the link between protein alterations in the central and peripheral environments in AD. Remarkably, proteins involved in the immune response–related, ECM-related, insulin-related, and synapse impairment pathways were significantly altered in both the CSF and serum of AD patients.
This study has several limitations. First, owing to the cross-sectional design of the study and the relatively small scale of the discovery cohort, further studies are necessary to confirm these results in a larger, longitudinal cohort. Second, the validation study used 4 specifically selected cohorts, and it would be better to validate the results in unselected longitudinal populations. Finally, in this study, only representative neurodegenerative diseases were selected as disease controls.
In conclusion, our study identified an 8-protein serum biomarker panel that has comparable power in identifying the early stage of AD to a 19-protein CSF biomarker panel. Moreover, we identified 21 CSF and 18 serum proteins reflecting different AD stages. Our findings provide a foundation for developing blood-based tests for clinical AD screening and staging.
Materials and methods
Participants
In the discovery cohort, 98 individuals with paired CSF and serum samples were recruited from the Second Affiliated Hospital of Zhejiang University School of Medicine between August 2015 and January 2021 (Tables S1 and S3A). The diagnoses of AD and MCI due to AD were based on the National Institute on Aging-Alzheimer’s Association (NIA-AA) AT(N) criteria.5 ALS patients were diagnosed according to the El Escorial criteria for ALS.35 HD patients were diagnosed by the presence of typical clinical manifestations and a positive HTT gene genetic test.36 FTD patients were diagnosed according to previous criteria.37 Patients received neuropsychological assessments, including the Mini-Mental State Examination, the Clinical Dementia Rating (CDR), and the Montreal Cognitive Assessment. Measurements of CSF biomarkers (Aβ42, tau, and p-tau181) or Pittsburgh compound B (PET/PIB) PET imaging were also conducted. The AD group was further classified into the ADM, ADMO, and ADS groups according to the CDR. The CN group comprised patients with other noncentral nervous system diseases without dementia and evidence of an underlying AD pathophysiologic process.
The participants in the validation cohort were recruited from Xuanwu Hospital affiliated with Capital Medical University, the First and Second Affiliated Hospitals of Zhejiang University School of Medicine, and the First Affiliated Hospital of Xiamen University. The validation cohort comprised 221 participants with CSF samples and 288 participants with serum samples (Tables S2 and S4A). The demographic data are presented in Tables S1 and S2.
ELISAs for measurement of CSF Aβ42, t-tau, and p-tau181 levels
CSF Aβ42, t-tau, and p-tau181 levels were measured by ELISA kits (Fujirebio, Ghent, Belgium) according to the manufacturer’s instructions, as described in our previous study.38
Blood biomarker assessment
Serum GFAP and NEFL were quantified by commercially available Single Molecular Immunity Detection kits (Astrabio, R14060 and R14040). All of the measurements were conducted on an AST-Sc-Lite analyzer (Astrabio) and were performed according to the manufacturer’s instructions.
Proteome analysis
Immunodepleting was implemented before digestion to increase the depth of the CSF and serum proteomes. Briefly, 100 and 175 μL of depletion resin (Thermo Scientific) were used for 100 μL of CSF and 4 μL of serum, respectively.
TMT-based proteomics analysis was performed as previously described.12 The detailed procedures are described in the supplemental information.
Quality control of TMT-based proteome data
We divided the 98 samples into 7 batches according to disease type, age, and sex (Table S3A). We randomly selected one CSF and serum sample from each type of patient as a biological replicate to control the quality of the proteome discovery workflow. Our data exhibited a high degree of consistency and reproducibility, with median coefficients of variation (CVs) for 7 biological replicates below 0.16 (Figures S12A and S12B). Visualization of the data obtained from the pooled serum and CSF peptide samples showed minimal batch effects (Figures S12C and S12D).
Targeted proteomics analysis
Peptide samples were prepared in the same way as described in the previous proteomic section, except that no depletion or TMT labeling was performed. The detailed procedures are described in the supplemental information. A total of 120 peptides, including 15 common internal retention time (CiRT) peptides39 (Table S4B), were included in the CSF PRM experiment, whereas 52 peptides, including 13 CiRT peptides, were included in the serum PRM experiment (Table S4D). CiRT peptides were used for the prediction of retention time to improve the confidence in target peptide identification.40 The PRM data were manually analyzed with Skyline41 and ProteomeExpert.42
Quality control of PRM proteome data
We selected 11 CSF samples and 16 serum samples as technical replicates, and the median CVs were below 0.15 (Figures S13A and S13B). We also prepared pooled CSF and serum peptide samples to evaluate the reproducibility of the PRM workflow. The Pearson correlation coefficient (r) of the proteomics data for 8 CSF pooled samples and 10 serum pooled samples was calculated, and all of the median r values were 0.99 (Figures S13C and S13D). We also observed fewer batch effects using the pooled samples (Figures S13E and S13F).
Statistical analysis and machine learning
For each pair of compared groups, the two-sided unpaired Welch’s t test was conducted for statistical analysis. Pearson’s r was calculated using the “cor” function in R (version 4.0.2) with the “pairwise.complete.obs” parameter to handle missing values. Machine learning was carried out using the R package randomForest (version 4.6.14), with some modifications based on previous work,12 briefly described as follows. The key random forest parameters, including the mean decrease accuracy cutoff, number of iterations of cross-validation, and number of trees, were optimized. The input protein features were selected based on the mean decrease accuracy cutoff. Five-fold cross-validation was performed, and a total of 1,000 and 600 trees were built for the CSF model and the serum model, respectively. The minimum decreasing mean accuracy of protein features was set to 3 for the CSF model and 0 for serum features. The mtry values for the CSF and serum models were set to the square roots of 4 and 2, respectively.
Data and code availability
The raw MS data in this study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD039146.
Acknowledgments
This work was supported by grants from the Key Research and Development project of Zhejiang Province (2019C03039), the National Natural Science Foundation of China (81970998), the Science Innovation 2030-Brain Science and Brain-Inspired Intelligence Technology Major Projects (nos. 2021ZD0201103 and 2021ZD0201803), and the Integrative Traditional Chinese and Western Medicine Innovation Team for Neurodegenerative Diseases of Zhejiang Province.
Author contributions
Conception and design, Q.-Q.T., X.C., T.G., and Z.-Y.W.; clinical sample/data collection, Q.-Q.T., Y.-Y.X., X.-Y.L., R.-R.L., G.-P.P., K.-M.Z., B.J., J.-P.J., and Z.-Y.W.; development of methodology, Q.-Q.T., X.C., W.G., Y.-Y.X., L.Y., W.J., and S.L.; data management and statistical analyses, W.G.; manuscript drafting, Q.-Q.T. and X.C.; study supervision, T.G. and Z.-Y.W.
Declaration of interests
The authors declare no competing interests.
Published Online: January 2, 2024
Footnotes
It can be found online at https://doi.org/10.1016/j.xinn.2023.100544.
Contributor Information
Jian-Ping Jia, Email: jpjia@ccmu.edu.cn.
Tiannan Guo, Email: guotiannan@westlake.edu.cn.
Zhi-Ying Wu, Email: zhiyingwu@zju.edu.cn.
Lead contact website
Supplemental information
References
- 1.Scheltens P., De Strooper B., Kivipelto M., et al. Alzheimer's disease. Lancet. 2021;397:1577–1590. doi: 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang H., Dey K.K., Chen P.C., et al. Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer's disease. Mol. Neurodegener. 2020;15:43. doi: 10.1186/s13024-020-00384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmqvist S., Insel P.S., Stomrud E., et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer's disease. EMBO Mol. Med. 2019;11 doi: 10.15252/emmm.201911170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panyard D.J., McKetney J., Deming Y.K., et al. Large-scale proteome and metabolome analysis of CSF implicates altered glucose and carbon metabolism and succinylcarnitine in Alzheimer's disease. Alzheimers Dement. 2023 doi: 10.1002/alz.13130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jack C.R., Jr., Bennett D.A., Blennow K., et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubois B., Villain N., Frisoni G.B., et al. Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol. 2021;20:484–496. doi: 10.1016/S1474-4422(21)00066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teunissen C.E., Verberk I.M.W., Thijssen E.H., et al. Blood-based biomarkers for Alzheimer's disease: towards clinical implementation. Lancet Neurol. 2022;21:66–77. doi: 10.1016/S1474-4422(21)00361-6. [DOI] [PubMed] [Google Scholar]
- 8.Oeckl P., Anderl-Straub S., Von Arnim C.A.F., et al. Serum GFAP differentiates Alzheimer's disease from frontotemporal dementia and predicts MCI-to-dementia conversion. J. Neurol. Neurosurg. Psychiatry. 2022;93:659–667. doi: 10.1136/jnnp-2021-328547. [DOI] [PubMed] [Google Scholar]
- 9.Palmqvist S., Janelidze S., Quiroz Y.T., et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs Other Neurodegenerative Disorders. JAMA. 2020;324:772–781. doi: 10.1001/jama.2020.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egan M.F., Kost J., Voss T., et al. Randomized Trial of Verubecestat for Prodromal Alzheimer's Disease. N. Engl. J. Med. 2019;380:1408–1420. doi: 10.1056/NEJMoa1812840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long J.M., Holtzman D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. 2019;179:312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen B., Yi X., Sun Y., et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai B., Vanderwall D., Li Y., et al. Proteomic landscape of Alzheimer's Disease: novel insights into pathogenesis and biomarker discovery. Mol. Neurodegener. 2021;16:55. doi: 10.1186/s13024-021-00474-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson E.C.B., Dammer E.B., Duong D.M., et al. Large-scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020;26:769–780. doi: 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen M., Xia W. Proteomic Profiling of Plasma and Brain Tissue from Alzheimer's Disease Patients Reveals Candidate Network of Plasma Biomarkers. J. Alzheimers Dis. 2020;76:349–368. doi: 10.3233/JAD-200110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehtewish H., Mesleh A., Ponirakis G., et al. Blood-Based Proteomic Profiling Identifies Potential Biomarker Candidates and Pathogenic Pathways in Dementia. Int. J. Mol. Sci. 2023;24:8117. doi: 10.3390/ijms24098117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar L., E Futschik M. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J., Huang Y., Li T., et al. The role of the Golgi apparatus in disease (Review) Int. J. Mol. Med. 2021;47:38. doi: 10.3892/ijmm.2021.4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H., Williams D., Griffin J., et al. Time-course global proteome analyses reveal an inverse correlation between Abeta burden and immunoglobulin M levels in the APPNL-F mouse model of Alzheimer disease. PLoS One. 2017;12 doi: 10.1371/journal.pone.0182844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leuzy A., Cullen N.C., Mattsson-Carlgren N., et al. Current advances in plasma and cerebrospinal fluid biomarkers in Alzheimer's disease. Curr. Opin. Neurol. 2021;34:266–274. doi: 10.1097/WCO.0000000000000904. [DOI] [PubMed] [Google Scholar]
- 21.Muenchhoff J., Poljak A., Thalamuthu A., et al. Changes in the plasma proteome at asymptomatic and symptomatic stages of autosomal dominant Alzheimer's disease. Sci. Rep. 2016;6 doi: 10.1038/srep29078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinacker P., Aitken A., Otto M. 14-3-3 proteins in neurodegeneration. Semin. Cell Dev. Biol. 2011;22:696–704. doi: 10.1016/j.semcdb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Bader J.M., Geyer P.E., Müller J.B., et al. Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer's disease. Mol. Syst. Biol. 2020;16 doi: 10.15252/msb.20199356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan B.P. Complement in the pathogenesis of Alzheimer's disease. Semin. Immunopathol. 2018;40:113–124. doi: 10.1007/s00281-017-0662-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whelan C.D., Mattsson N., Nagle M.W., et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer's disease. Acta Neuropathol. Commun. 2019;7:169. doi: 10.1186/s40478-019-0795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y., Zhou X., Ip F.C., et al. Large-scale plasma proteomic profiling identifies a high-performance biomarker panel for Alzheimer's disease screening and staging. Alzheimers Dement. 2022;18:88–102. doi: 10.1002/alz.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinney J.W., Bemiller S.M., Murtishaw A.S., et al. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. (N Y) 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ismail R., Parbo P., Madsen L.S., et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer's disease: a longitudinal PET study. J. Neuroinflammation. 2020;17:151. doi: 10.1186/s12974-020-01820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramachandran A.K., Das S., Joseph A., et al. Neurodegenerative Pathways in Alzheimer's Disease: A Review. Curr. Neuropharmacol. 2021;19:679–692. doi: 10.2174/1570159X18666200807130637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y., Xu S., Jiang M., et al. Role of the Extracellular Matrix in Alzheimer's Disease. Front. Aging Neurosci. 2021;13 doi: 10.3389/fnagi.2021.707466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montagne A., Nation D.A., Sagare A.P., et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581:71–76. doi: 10.1038/s41586-020-2247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nation D.A., Sweeney M.D., Montagne A., et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019;25:270–276. doi: 10.1038/s41591-018-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai B., Wang X., Li Y., et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer's Disease Progression. Neuron. 2020;106:700. doi: 10.1016/j.neuron.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higginbotham L., Ping L., Dammer E.B., et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer's disease. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aaz9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks B.R., Miller R.G., Swash M., et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 36.Li H.L., Li X.Y., Dong Y., et al. Clinical and Genetic Profiles in Chinese Patients with Huntington's Disease: A Ten-year Multicenter Study in China. Aging Dis. 2019;10:1003–1011. doi: 10.14336/AD.2018.0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boeve B.F., Boxer A.L., Kumfor F., et al. Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022;21:258–272. doi: 10.1016/S1474-4422(21)00341-0. [DOI] [PubMed] [Google Scholar]
- 38.Ye L.Q., Gao P.R., Zhang Y.B., et al. Application of Cerebrospinal Fluid AT(N) Framework on the Diagnosis of AD and Related Cognitive Disorders in Chinese Han Population. Clin. Interv. Aging. 2021;16:311–323. doi: 10.2147/CIA.S294756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker S.J., Rost H., Rosenberger G., et al. Identification of a Set of Conserved Eukaryotic Internal Retention Time Standards for Data-independent Acquisition Mass Spectrometry. Mol. Cell. Proteomics. 2015;14:2800–2813. doi: 10.1074/mcp.O114.042267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu T., Zhu Y., Xuan Y., et al. DPHL: A DIA Pan-human Protein Mass Spectrometry Library for Robust Biomarker Discovery. Dev. Reprod. Biol. 2020;18:104–119. doi: 10.1016/j.gpb.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacLean B., Tomazela D.M., Shulman N., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu T., Chen H., Yan X., et al. ProteomeExpert: a Docker image-based web server for exploring, modeling, visualizing and mining quantitative proteomic datasets. Bioinformatics. 2021;37:273–275. doi: 10.1093/bioinformatics/btaa1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw MS data in this study have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD039146.