

Summary
Genome-wide association studies (GWASs) have identified hundreds of risk loci for coronary artery disease (CAD). However, non-European populations are underrepresented in GWASs, and the causal gene-regulatory mechanisms of these risk loci during atherosclerosis remain unclear. We incorporated local ancestry and haplotypes to identify quantitative trait loci for expression (eQTLs) and splicing (sQTLs) in coronary arteries from 138 ancestrally diverse Americans. Of 2,132 eQTL-associated genes (eGenes), 47% were previously unreported in coronary artery; 19% exhibited cell-type-specific expression. Colocalization revealed subgroups of eGenes unique to CAD and blood pressure GWAS. Fine-mapping highlighted additional eGenes, including TBX20 and IL5. We also identified sQTLs for 1,690 genes, among which TOR1AIP1 and ULK3 sQTLs demonstrated the importance of evaluating splicing to accurately identify disease-relevant isoform expression. Our work provides a patient-derived coronary artery eQTL resource and exemplifies the need for diverse study populations and multifaceted approaches to characterize gene regulation in disease processes.
Keywords: eQTL, genetic diversity, coronary artery, RNA-seq, fine-mapping, gene regulation, genome-wide association studies
Graphical abstract
Highlights
-
•
Human coronary artery is a relevant tissue for profiling atherosclerosis gene expression
-
•
Multifaceted QTL mapping methodology improves discovery in diverse populations
-
•
Colocalization and epigenomic fine-mapping prioritize candidate risk variants and genes
-
•
Publicly available pipeline is generalizable across phenotypes and study designs
Hodonsky et al. perform gene expression profiling and a multifaceted genetic analysis of gene regulation in coronary artery tissues from an ancestrally diverse patient cohort. This new resource provides further insights into the heritable mechanisms of coronary artery disease risk and could serve as a template for multi-ancestry studies of other complex phenotypes.
Introduction
Coronary artery disease (CAD) is the leading cause of death worldwide, and it results from chronic inflammatory processes involving both genetic and environmental risk factors. CAD manifests as the development of atherosclerotic plaques in the coronary arteries of the heart, which can lead to erosion or plaque rupture and ultimately myocardial infarction. Genome-wide association studies (GWASs) have now reported more than 400 independent loci for CAD and related clinical outcomes.1,2,3,4,5,6,7,8,9,10,11 As with other common complex traits, the majority of lead CAD GWAS variants reside in non-coding genomic regions, implicating regulatory effects on gene expression.12 Previous studies have mapped CAD GWAS variants to specific cell types in the vessel wall (e.g., smooth muscle cells [SMCs],13 endothelial cells, and immune cells)14 and refined candidate cis-acting regulatory elements responsible for context-specific gene expression patterns.14,15 However, cultured vascular cells do not recapitulate the in vivo cell phenotype: for instance, high-passage SMCs reprogram toward a fibroblast-like state accompanied by rapid loss of differentiated marker gene expression.
Fine-mapping GWAS loci can help prioritize candidate causal variants within association signals, but identifying the causal variant or target gene within a locus can still be difficult. Overrepresentation of European- and East Asian-ancestry populations in most GWASs to date has also limited the capacity to identify independent associations within a locus and the generalizability of findings to global populations.16,17,18 Furthermore, genes within most CAD loci have not been associated with traditional risk factors (e.g., lipid levels or cholesterol metabolism), suggesting molecular mechanisms underlying physiological effects on the coronary artery vessel wall itself.
Molecular quantitative trait locus mapping in a disease-relevant tissue or cell line is a powerful approach to prioritize candidate causal genes and underlying mechanisms for complex GWAS loci.19 Prior studies have identified CAD-relevant expression quantitative trait loci (eQTLs) in bulk arterial tissues20 or vessel wall cell types, including human aortic endothelial cells,21 human coronary artery smooth muscle cells (HCASMCs),13,22 and monocytes.23 Similar to CAD GWASs and other eQTL studies, published summary statistics represent exclusively or primarily European-ancestry populations, often lack detailed phenotyping for the patients/participants.
To identify variants associated with coronary artery-specific gene expression and fine-map colocalized CAD GWAS associations, we performed a QTL mapping study in coronary artery tissues from an ancestrally diverse American patient-derived sample. We utilized multiple methods to identify eQTLs and splicing quantitative trait loci (sQTLs) in human coronary artery tissue, followed by bioinformatic characterization of potential eGene-phenotype associations. Our results not only highlight new coronary artery eQTLs at promising GWAS loci such as TBX20, but they also replicate and refine eQTLs previously reported in other arterial tissues, including ARHGAP42. Our approach represents an improved capacity to characterize gene regulation in coronary artery tissue through all stages of CAD progression. This dataset will therefore be a highly beneficial resource for better characterization of functional variants and molecular mechanisms driving CAD development.
Results
Study overview for transcriptomic profiling of human coronary artery
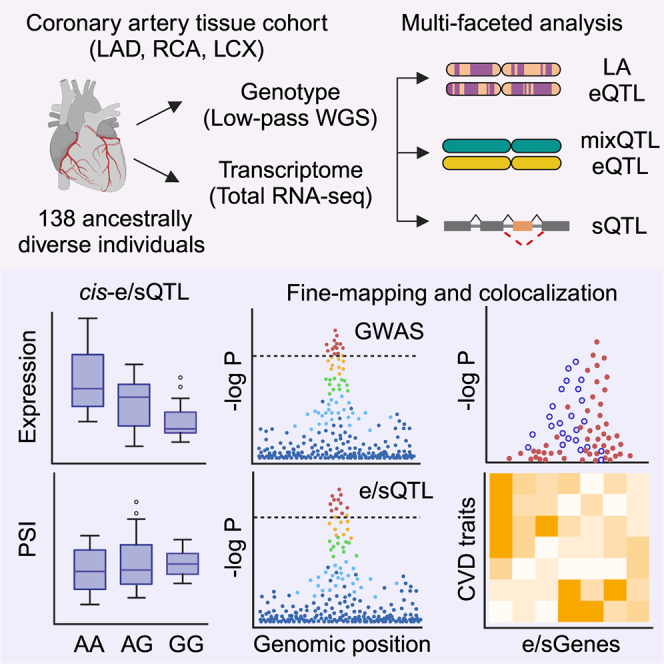
We conducted transcriptome-wide QTL mapping of autosomal gene expression in human coronary artery tissue samples from explanted transplant tissue as well as samples collected from rejected transplant donors (STAR Methods, Table S1). An overview of our sample characteristics and primary diagnoses are described in Figure 1 and Tables S1–S3. The study comprised 138 individuals from 19 to 72 years old, with 30% being female. While 57% of these individuals were of exclusively European ancestry, 15% were of majority South Asian ancestry, 5% East Asian, 5% Indigenous American, 7% African, and 10% had no majority ancestry, representing the broad genetic diversity of the American population (Figures 1A–1C and S1). Samples were derived from all three major coronary arteries (i.e., left anterior descending coronary artery [LAD], right coronary artery [RCA], and left circumflex artery [LCX], Figure 1D). Majority inferred ancestry groups were represented across diagnoses, but only majority-European and -South Asian genetic ancestries were represented in all primary diagnostic categories (Figure 1E; Table S1). Up to 5.83 million variants genotyped via low-pass whole-genome sequencing were included depending on method-specific allele frequency and annotation requirements (described in STAR Methods, Table S2).
Figure 1.

Overview of patient and sample characteristics
(A) Boxplot showing the age range of study participants (y axis) in females and males (x axis; orange and green, respectively). Lines represent median, boxes represent interquartile range (IQR), and upper and lower whiskers represent 1.5 × IQR.
(B) Genetic ancestry principal components 1 (x axis) and 2 (y axis) mapped onto the 1000 Genomes phase 3 reference population (bold color dots), with color corresponding to Gencove-assigned majority ancestry in our samples (lighter color dots with black outline).
(C) Local ancestry inference reveals a complex genetic substructure for individuals with ancestral admixture, with each row in the plot representing inferred local ancestry for one haplotype of one study participant and x axis representing position on respective chromosomes. Inset, zoomed in region on chr 20 showing genetic substructure for a subset of the individual haplotypes.
(D) Number (y axis) of coronary artery segments by type (x axis) used for RNA isolation in samples from explants (turquoise) and rejected donors (gold).
(E) Number (y axis) of coronary artery segments by primary ICD-10 code diagnosis for explanted hearts or donor hearts. Samples were grouped into main categories listed in Table S1.
We performed total RNA sequencing to a median depth of 102.6 million reads per sample (Table S3) to profile both protein-coding and non-coding RNAs (Figure S1). To determine the similarity of our expression profiles to bulk RNA profiles of other tissues in GTEx as well as cultured HCASMCs, we performed multidimensional scaling. Our samples form a cluster located near the left ventricle, muscle, pancreas, fibroblasts, and liver tissues (Figure S1). This distinct but proximal clustering aligns with expectations given differences in sample collection/storage methods and cold ischemia times (time lapsed after cessation of blood flow). Since eQTL studies have primarily been performed in genetically homogeneous populations, information on preferred methods for inclusive study populations is limited. We therefore applied two complementary approaches to attempt to maximize power for identifying associations that may not have globally consistent allele frequencies, i.e., by evaluating haplotype-specific associations (“mixQTL” for the total sample or “mixQTLEUR” for the 100% European-ancestry subset analysis) or ancestry-specific associations (local ancestry adjusted, henceforth referred to as “LA”).24,25 For mixQTL analyses, we incorporated three global genetic principal components; for LA, locus-specific genetic ancestry was statistically inferred on a continental scale using a reference panel of genotypes from 1000 Genomes (1000G) participants (see STAR Methods). After filtering for method-specific criteria, up to 20,100 autosomal protein-coding genes and lncRNAs met inclusion criteria for eQTL and sQTL analyses.
Coronary artery eQTL discovery
To identify genetic variants associated with gene expression in our diverse coronary artery tissue cohort, we performed eQTL analyses incorporating haplotype-specific (mixQTL)24 or LA information. Overall, we identified 2,132 and 793 coronary artery eGenes using mixQTL or LA, respectively (Tables 1, S4, and S5; Figure 2A). Between LA and mixQTL analyses, 457 shared eGenes were identified (Figure 2B); 45 lead SNPs were common to both approaches (Table S6). Of note, across all analyses, we report 735 total discovery eGenes (351 mixQTL, 163 LA, 210 mixQTLEUR, 395 mixQTLdownsample; 213 protein-coding genes, 514 lncRNAs) with no expression QTLs reported in any arterial tissue in GTEx or Stockholm-Tartu Atherosclerosis Reverse Networks Engineering Task (STARNET), including genes with established roles in vascular cell types (e.g., lipase G, endothelial type [LIPG] and AKT serine/threonine kinase 3 [AKT3]).26,27 40% of discovery eQTLs were >100 kb from the gene transcription start site, in line with long-standing evidence of both short- and long-range cis-acting regulatory mechanisms.28,29 We report mixQTL results from the entire study sample as our primary findings given the higher statistical power of this method.
Table 1.
Discovery expression and splicing quantitative trait loci in human coronary artery tissue
| Molecular phenotype | Method | Gene type | No. evaluated | No. genes (FDR <0.01) | No. genes (FDR <0.05) | No. genes (FDR <0.1) | No. discovery genes |
|---|---|---|---|---|---|---|---|
| Gene expression | mixQTL | protein-coding | 14,235 | 1,118 | 1,457 | 1,668 | 127 |
| lncRNA | 5,874 | 510 | 682 | 794 | 224 | ||
| local ancestry | protein-coding | 14,274 | 330 | 482 | 602 | 41 | |
| lncRNA | 7,395 | 234 | 311 | 378 | 122 | ||
| mixQTL (Euro) | protein-coding | 14,082 | 671 | 916 | 1,087 | 94 | |
| lncRNA | 5,219 | 285 | 395 | 467 | 116 | ||
| mixQTL (diverse) | protein-coding | 14,084 | 766 | 1,075 | 1,303 | 127 | |
| lncRNA | 5,222 | 323 | 477 | 569 | 294 | ||
| Splicing | QTLtools | protein-coding | 13,103 | 1,134 | 1,496 | 1,735 | 357 |
| lncRNA | 1,700 | 152 | 194 | 232 | 93 |
Figure 2.

Overview of eQTL analysis and generalization to published arterial eQTLs
(A) Miami plot of lead eQTLs for mixQTL (top) and local ancestry (LA) adjusted (bottom). Navy blue and orange dots represent reported and discovery eGenes Benjamini-Hochberg adjusted p-value (pBH) < 0.05, respectively; gray dots represent non-significant genes. A subset of top eGenes are labeled for clarity.
(B) Venn diagram showing the overlap of mixQTL and LA-based eGenes and LeafCutter sQTL sGenes.
(C) Circos plot portraying generalized (UVA pBH <0.05) published arterial eGenes from GTEx AOR (blue green), COR (orange), and TIB (purple), and STARNET AOR (pink) and MAM (light green) tissues, with significance increasing toward the outer edge of the circle.
(D) Direction of effect for genes in which the UVA lead eQTL was significant (pBH < 0.05) in any of the aforementioned tissues using the same color scheme for GTEx (top) and STARNET (bottom). Pearson’s r correlation coefficients shown for overlapping significant UVA coronary eQTL detected in GTEx or STARNET eQTL with tissue indicated in parentheses. AOR: aorta; COR: coronary artery; TIB: tibial artery; MAM: mammary artery.
Overview of mixQTL results
Of the 2,132 protein-coding or lncRNA mixQTL eGenes, 16% (n = 351) have not been previously reported in published arterial tissue QTL studies (Table S4). In concordance with published studies, most eQTLs were annotated as intergenic or intronic to their eGenes (2,859 of 3,952 available annotations for 1,122 eGenes, Figure S2). Only 2% of lead eQTLs (n = 59) were protein coding within their respective eGene, implicating the regulation of gene expression through transcriptional, splicing, or epigenetic mechanisms.20 Thirty-nine and 119 eGenes were identified based solely on allele-specific expression and total read count tests, respectively. Overall, 1,779 published arterial eGenes had significant eQTLs in our study sample (Figure 2C; Table S7). Fewer than 5% of shared associations had the same lead eQTL, but among shared lead eQTLs, we observed 98% directional consistency (64% of all replicated lead eQTLs, Figures 2D and S2). Nearly one in five eGenes exhibited cell-type-specific expression in a coronary artery single-cell RNA sequencing reference dataset (cell specificity expression score ≥0.7, Figure S2; Table S8),30 and functional enrichment analysis revealed several pathways for cell adhesion and inflammation (Table S9).31
Local-ancestry-adjusted and ancestry-specific eQTLs
With regard to LA analyses, 337 eGenes were identified that did not exceed a false discovery rate (FDR) of 5% in the overall mixQTL analysis, demonstrating the merit of incorporating multiple approaches in a diverse study sample with genetic admixture (Table S5). Among LA-specific eGenes was YY1-associated protein 1 (YY1AP1), which has no reported coronary artery eQTLs but has been associated with vascular diseases including Grange syndrome and sudden coronary artery dissection.32,33 Seventeen LA lead variants were monomorphic in the 1000G East Asian superpopulation (Table S10A). Despite high-confidence calling of 1000G continental ancestries in our study sample, the small numbers of shared haplotypes at any given locus likely limited our ability to identify ancestry-specific associations using this method. Using mixQTL, 54 eGenes with lead SNPs monomorphic in one or more 1000G superpopulations were identified; four overlapped with the LA results, and lead eQTLs were either shared or in high linkage disequilibrium (“LD,” Figure S2; Tables S10B and S11).
Among the genes with no eQTLs exceeding genome-wide significance in the mixQTL were several interesting genes with sub-significant associations, including VPS37B (vacuolar protein sorting-associated protein 37B). VPS37B is involved in endosomal protein binding activity, and the genomic region has been associated with CAD-relevant traits including adiponectin levels, BMI, and cholesterol traits.34,35,36,37,38 MixQTL and LA methods resulted in the same lead variant, rs897392, in the intron of neighboring gene HIP1R, which exhibits modest to strong LD with reported arterial eQTLs for VPS37B (Table S11) as well as different expression between CAD cases and controls in multiple tissues in the STARNET study population (Table S12).20,39 rs897392 had a mixQTL adjusted p value of 0.14, meaning VPS37B would not be considered an eGene using this method alone, despite evidence favoring genetic regulation of this gene in cardiac tissues. In combination, these results indicate the benefit of including genetically diverse individuals as well as multiple approaches to improve effective sample size across the lower end of the global allele frequency range.
Colocalization of eQTLs
We next evaluated the overlap between coronary artery gene expression and genetic associations with CAD and intermediate risk factors including blood pressure, cholesterol, and arterial calcification traits. Across all phenotypes, 108 GWAS association signals colocalized with eQTLs, including 25 discovery eGenes (Figure 3A; Table S13). Thirty-one eGenes exhibiting cell type specificity colocalized to one or more GWASs, including Rho GTPase-activating protein 42 (ARHGAP42) in pericytes and discovery eGenes LIPG and adhesion G protein-coupled receptor G6 (ADGRG6) in endothelial cells (Figure 3A; Table S8). We further assessed GWAS associations overlapping our eQTL associations using summarized Mendelian randomization (SMR).40 Given LD-dependent restrictions for both colocalization and SMR, as expected, we identified fewer associations but notable overlap between the two methods (25 overlapping signals and 18 unique to SMR, Table S15).
Figure 3.

Colocalization reveals trait- and cell-type-specific associations
(A) GWAS colocalization to eGene associations: each column represents the –log10(p value) of the study (first author last name) and relevant GWAS trait, with intensity of shading corresponding to a higher posterior probability of a shared association at that locus. Each row represents one protein-coding gene with a PPH4 ≥0.8 in at least one GWAS. The leftmost column (“Target cell type”) indicates cell-type specificity for SMC (dark blue), pericyte (light blue), endothelial (green), fibroblast (gold), or blood (pink) cells. Size of the circle represents the CELLEX combined gene score (range: 0.7–1.0).
(B and C) Top: normalized expression (y axis) by lead eQTL genotype (x axis) for TCF21 (B) and TBX20 (C); bottom: regional association plots showing overlap between our study (violet) and GWAS associations for CAD (red triangle) and blood pressure traits (gold diamonds).
(D and E) Human artery atherosclerosis single-cell RNA sequencing (left) and single-nuclear ATAC sequencing (right) showing TCF21 (D) and TBX20 (E) gene expression- and chromatin accessibility-based gene scores, respectively, with changes indicated by intensity of red (left) and pink/yellow (right).
We observed strong evidence of colocalization and generalization at the TCF21/TARID locus. TCF21, a known regulator of the SMC phenotype transition to fibromyocytes in plaque,41,42 exhibited a significant eQTL for the same variant, rs12190287, in our study as well as in STARNET aorta (AOR) and GTEx coronary artery (COR) samples (Figures 3B and S3). The TCF21 eQTL overlapped with the association for the adjacent lncRNA TARID (TCF21 antisense RNA inducing promoter demethylation). Our lead TARID eQTL, rs1535616, was also significant in GTEx COR, though not for other arterial tissues (Figure S3). Interestingly, GTEx COR exhibited similarly strong associations at both ends of the TARID coding region, while AOR showed a much stronger association at the 3′ end (Figure S3). TCF21 and TARID also colocalized with CAD and BP (blood pressure) trait associations but not coronary artery calcification or cholesterol traits (Figure 3B), suggesting the mechanism for this known CAD locus may be functioning via a causal blood pressure pathway.
TBX20, coding for transcription factor T-box 20, is another gene with established cardiac development and disease associations.43 However, the mechanism of its genetic regulation in CAD risk remains unclear. Our lead eQTL, rs11976145 (Figure 3C), is located in the fourth intron and colocalizes with a CAD GWAS signal but is independent from the DBP association at the same locus, suggesting a multifaceted approach may be required to ascertain disease-relevant mechanisms underlying transcriptional regulation. While TBX20 has no reported eQTLs for GTEx arterial tissues, we identify an overlapping association with the eQTL for STARNET (AOR), for which the lead eQTL rs10249005 is in high LD (r2 ≥ 0.94) with rs11976145 in all European-ancestry 1000G reference populations (Table S11). Coronary artery single-cell RNA and assay for transposase-accessible chromatin sequencing (scRNA-seq, snATAC-seq) reference datasets show that TARID and TCF21 are both expressed most highly and most accessible in fibroblasts and SMCs (including SMC-derived fibromyocytes resulting from SMC phenotypic switching during atherosclerosis, Figures 3D, 3E, and S3).30,44 TBX20 is most abundant in cardiac muscle and vascular tissues in GTEx, with coronary artery reference expression predominantly restricted to SMCs and fibroblasts (Figures 3D and 3E).
Among LA eGenes, colocalization was limited to 27 associations, but four of these were not eGenes in mixQTL, and a further three were mixQTL eGenes that did not colocalize to any GWAS trait (Table S15; Figure S4). Of particular interest is ANAPC13, a component of an anaphase-associated E3 ubiquitin ligase for which the LA eQTL (led by rs9809619, pLA = 2.3E−6) colocalized to the MVP CAD association signal but did not meet the threshold for colocalization (PPH4 ≥ 0.8) for any trait in micQTL. Rs9809619 is in close proximity to and exhibits near perfect LD globally with the lead mixQTL variant, rs4367113 (pmixQTL = 1.6E−5, Table S11), across 1000G populations. LA adjustment resulted in lower p values for ANAPC13-associated variants compared to mixQTL (Figure S4), showing the benefit of complementary approaches for a locus with similar associations but different significance between methods.
Fine-mapping significant coronary artery eQTLs
Next, we used a combination of methods to identify both credible sets (CSs) and independent associations within previously unreported coronary artery eGenes. First, we used the Bayesian mixFine function from the mixQTL package to identify independent CSs for significant associations (Table S16). Only 3% of eGenes with converging association signals fine-mapped to a single variant (n = 44 of 1,388). As expected, 91% of CSs contained the lead eQTL, which was the sole credible variant for 29 eGenes. Eighty-three eGenes exhibited multiple independent eQTL signals.
Including prior functional annotations in relevant tissues can refine association signals and prioritize variants and candidate cis-regulatory mechanisms.45,46 Therefore, we employed FastPaintor to fine-map associations with epigenomic annotations (ENCODE coronary artery H3K4me3 and H3K27Ac marks and activity-by-contact scores for human coronary artery SMCs as well as BP and CAD GWAS), which both exhibited strong evidence of colocalization. 1,964 eGenes had a sufficient number of eQTLs to converge for fine-mapping annotation for one or both GWAS traits (Tables S17A and S17B). Across both traits and three epigenetic marks, CSs for 106 eGenes were narrowed to a single likely causal variant. The lead eQTL was included in 11% of CSs. Regardless of epigenetic annotation, most CSs (>80%) were narrowed to five or fewer SNPs (Figure S5). While the majority of CSs contained the same variants regardless of epigenetic annotation, they often differed by GWAS annotation. Lead eQTLs in the 247 loci for which at least one variant was shared between BP trait and CAD annotations exhibited lower p values and nearer to the eGene transcription start site (TSS) on average (median p values 1E−08 vs. 5E−4, Figure S5).
We demonstrated the utility of combining multi-omic data with fine-mapped associations in ARHGAP42, a Rho-A GTPase-activating protein with functional evidence for disease relevance, and IL5, a discovery coronary eGene. Lead ARHGAP42 eQTL rs2455569 exhibited similar effect direction and magnitude in STARNET aorta tissue (Figure S6), in which decreased ARHGAP42 expression was significantly associated with case status (Table S12). The ARHGAP42 eQTL overlapped with GTEx AOR and COR associations, and it colocalized with both BP traits and CAD GWAS associations (Table S13; Figure S6). ARHGAP42 (also known as GRAF3) regulates vascular tone via expression predominantly in mural cells and fibroblasts (Figure S6),30,44,47 and insufficiency causes hypertension.48,49 Rs2455569 is located in an intronic pericyte-specific chromatin-accessibility peak, with fine-mapping highlighting proximal SNPs encompassing a region accessible in multiple vascular cell types (Figures 4A–4C).44 Interestingly, rs604723, 25 kb upstream of and in high LD with rs2455569 (padj = 4.6E−13, Table S11), has been shown to modulate ARHGAP42 expression in SMCs via SRF (serum response factor) binding.50 Rs604723 is also predicted to disrupt a binding motif for STAT6, which has been implicated in the proliferation of vascular SMCs in an injury-response murine model,51 suggesting multiple functional avenues for genetic regulation of ARHGAP42 and its downstream effects (Figure 4D).
Figure 4.

Fine-mapping identifies candidate causal variants for ARHGAP42 and IL5 eQTLs at known GWAS loci
(A) ARHGAP42 association driven by lead eQTL rs2455569, exhibiting differences in expression by genotype but not sex or majority continental ancestry. p values calculated from paired t test (t) or Kruskal-Wallis test (KW).
(B) Regional association plot depicting variants in PAINTOR credible sets specific to BP (violet) or CAD (golden) GWAS annotations, mixFine only (light blue), and variants not in any credible set (light gray).
(C) Variants of interest (rs604723 and rs2455569) are indicated by blue lines, and rs604723 is highlighted in gray box and corresponds to UCSC Genome Browser tracks indicating cell-type-specific chromatin accessibility.
(D) Location of rs604723 in critical nucleotides (in gray outlined boxes) of consensus transcription factor binding sequences for SRF and STAT6 as identified using the JASPAR 2022 database.
(E) IL5 association driven by lead eQTL rs7719499, exhibiting differences in expression by genotype but not sex or majority continental ancestry. p values calculated from paired t test (t) or Kruskal-Wallis test (KW).
(F) Regional association plot depicting variants in PAINTOR credible sets specific to BP (violet) or CAD (golden) GWAS annotations, mixFine only (light blue), mixFine and PAINTOR CS (green), and variants not in any credible set (light gray).
(G) Variants of interest (rs7719499 and rs10065633) are indicated by blue lines, and rs7719499 is highlighted in gray box and corresponds to UCSC Genome Browser tracks indicating cell-type-specific chromatin accessibility.
(H) Location of rs7719499 in critical nucleotides (in gray outlined boxes) of consensus transcription factor binding sequences for RUNX3 and nearby IRF4 (+10 bp), as identified using the JASPAR 2022 database.
IL5 encodes for interleukin 5, an inflammatory cytokine with no eQTLs reported in any GTEx tissue but highly significant eQTLs in both STARNET arterial tissues (Figure S7). Lead eQTL rs7719499 lies 75 kb downstream of IL5 adjacent to a plasma-specific chromatin accessible region for human coronary artery, proximal to IRF1 (lead eQTL rs72797327, pBH = 1, Figures 4E–4G). Rs7719499 is also predicted to alter a RUNX3 transcription factor binding motif and is proximal to an IRF4 motif, supporting putative cis-regulatory mechanisms in plasma cell types (Figure 4H). While IL5 is expressed at low levels across most tissue types and coronary cell types (Figure S7), it is significantly upregulated in aortic tissues from patients with CAD compared to controls (Figure S7; Table S12).39
Sensitivity analysis: European ancestry only compared to random downsampling
We also assessed whether our inclusive study design affected discovery, colocalization, and fine-mapping by restricting our sample to European-ancestry individuals (n = 80), as well as a random subset of 80 members approximating the representation of genetic ancestry of the total sample. In the European-ancestry-only subset, we identified 1,311 eGenes (16% discovery eGenes; 79% eGenes in the combined analysis), compared to 1,469 eGenes in the genetically diverse subset (27% discovery, 71% present in combined analysis, Figure S8; Tables S18A and S18B).
With regard to generalization of published arterial eQTLs, 983 and 1,074 eGenes in the European-only and representative subsets respectively also had eQTLs in a GTEx or STARNET arterial tissue, and directional consistency with GTEx coronary was over 90% (Figure S8; Tables S19A and S19B). Compared to the overall sample, less than half the number of eGenes from either subset colocalized to relevant GWAS traits—fewer than would be expected if colocalization were linearly correlated with sample size (Figure S4; Tables S20A and S20B). The reduction in associations across all analyses in the European-only subset compared to the genetically diverse subset reinforces the benefits of methodological approaches designed to maximize study sample size and diverse genetic ancestry representation.
Coronary artery splicing QTL discovery
Differential isoform expression affects a wide array of complex diseases, and genetic variants affecting splicing events have been shown to be a major and distinct source of regulation underlying disease phenotypes.52 However, tissue-specific transcript specificity and isoform switching are not detected through eQTL methods unless total expression is affected. To identify genetic contributions to isoform-specific expression, we therefore evaluated genetic associations with 132,373 splice junctions in 14,815 genes. We identified 3,590 sQTLs (pBH<0.05) in 1,690 sGenes (Figure 5A; Table S21). Only 296 sGenes (17.5%) were also identified as eGenes using mixQTL or LA approaches (Figure 2B), pointing to the importance of evaluating isoform-related events in addition to total gene expression.
Figure 5.

Coronary artery sQTL overview and characterization
(A) Manhattan plot of lead sQTLs. Navy blue and orange dots represent reported and discovery sGenes (pBH < 0.05), respectively; gray dots represent non-significant genes.
(B) Upset plot of generalization of GTEx arterial sQTLs. Black bars represent GTEx-specific sGenes; orange bars represent generalized sGenes, and the blue bar represents previously unreported sGenes.
(C) Upset plot of histone modifications in aorta, coronary, and liver from one individual in ENCODE as well as HCASMCs. Light blue bars represent lead sQTLs overlapping modifications specific to one cell or tissue type, gold bars for H3K27 acetylation in coronary and any other tissue or cell type, and magenta bars for both coronary artery H3K4 tri-methylation and H3K27 acetylation.
(D) GWAS colocalization to sQTL associations: each row represents the colocalization PPH4 of the study (first author last name) and relevant GWAS trait, with intensity of shading corresponding to a higher posterior probability of a shared association at that locus. Each column represents one protein-coding gene with a PPH4 ≥0.8 in at least one GWAS.
(E and F) Percent spliced in of ULK3 and TOR1AIP1 exons by genotype of their lead sQTLs (rs2290572 and rs2245425, respectively) in our study (left) and GTEx (right).
(G) Schematic of TOR1AIP1 gene and alternatively spliced isoforms, showing location and effect estimates for top splice acceptor variant (rs2245425) identified in our samples. rs2245425-G creates TAGCAG splice acceptor sequence at 3′ end of intron-exon 3 junction. Spliced CAG nucleotides (orange) encoding alanine amino acid distinguish LAP1B isoforms 1 and 2.
Lead sQTLs for 96 splice junctions (71 total sGenes) were annotated as splice region variants or splice acceptors in SnpEff (Table S22), and nine discovery sGenes (Table S21) had protein-coding variants as lead QTLs. While effect size was not correlated with broadly defined variant category (pANOVA = 0.62), there was a significant association with p value (Figure S9, pANOVA = 3.65E−11), suggesting small effects in isoform-specific differences may be functional. While fine-mapping sQTL associations is difficult without isoform-specific expression information, for sGenes with lead sQTLs within 10 kb of the overall TSS, approximately half overlapped enhancer histone modification marks in relevant tissues or cells (Figure 5B).
Generalization and colocalization of lead sQTLs
Tissue-specific differences in isoform proportion cannot necessarily be detected with bulk sequencing, implicating potentially distinct regulatory mechanisms for splicing compared to overall expression. Therefore, we first directly compared the gene sets represented by eQTLs and sQTLs. Only modest overlap was observed (297 eGenes were also sGenes, 23 of which shared the same lead variant, Table S23), suggesting that splicing analyses likely represent unique disease-relevant pathways compared to overall expression. Among 71 sGenes with lead sQTLs reported to have splicing functions in SnpEff, 59 have no eQTLs, with five having no expression variants exceeding even nominal significance (Table S23). This difference is further exemplified by differences in cell type specificity in our scRNA-seq reference dataset: while nearly all cell-type-specific (score >0.7 as described in STAR Methods) reference genes were eGenes (408 of 411 total), only 37 sGenes (of 83 total) exhibited cell-type specificity. Compared to published arterial sQTL data, 871 (52%) were also sGenes in GTEx coronary artery, while 875 sGenes had no reported sQTLs in GTEx AOR, COR, or tibial artery (TIB) tissues (Figure 5C; Table S21).
Colocalization of sQTL data exhibited a similar pattern to eQTLs, with a small subset of overlapping colocalized signals shared between CAD and BP trait GWAS and a higher number of quantitative trait associations (Figure 5D; Table S24). Despite a similar pattern of colocalization with CAD and BP traits being most well represented, none of the eQTLs for shared associations colocalized with tested GWAS traits, affirming the likely different causal mechanisms underlying genetic effects on gene expression compared to splicing activity.
Splicing contributes to CAD-relevant gene regulation
We highlight two plausible candidates for functional splicing effects in the coronary artery: ULK3 and TOR1AIP1 (Figures S9). ULK3 is a broadly expressed serine/threonine kinase exhibiting autophosphorylation activity.53,54 The ULK3 locus has also been associated with various CAD-relevant traits, including blood pressure, total cholesterol, and estimated glomerular filtration rate.18,55,56,57 Of thirteen junctions tested in our study, common missense variant rs2290572 was the lead sQTL for both significant associations: chr15:74837435:74837757 and chr15:74837435:74837751 (Figure 5E). Rs2290572 was also significantly associated with chr15:74837435:74837751 in GTEx tissues with directional consistency (Figure S9). The lead sQTL for GTEx arterial tissues, rs12898397, is in high LD with rs2290572 in reference populations (Table S11) and represents a two-codon difference in an MIT domain in the fourteenth exon for which the T allele is predicted by SpliceAI to cause loss of a splice donor (Δ score = 0.67, https://spliceailookup.broadinstitute.org).58 Rs12898397 is our lead eQTL (pBH = 1.3E−14), and its contribution to isoform specificity makes it a clear candidate for functional follow-up.
TOR1AIP1 (torsin 1A-interacting protein 1) is a broadly expressed lamin-binding protein that localizes to the inner nuclear membrane. Causal TOR1AIP1 variants have been found for several autosomal recessive disorders, including limb-girdle muscular dystrophy with cardiac failure.59,60 Located in the first intron, splice acceptor variant rs2245425 is the lead sQTL for both significant splice excision events (of six tested in our study; chr1:179884769:179889313 and chr1:179884769:179889310), exhibiting nearly binary effects on the PSI of the third exon in coronary artery (Figures 5F and 5G). Rs2245425 is also the lead sQTL of GTEx arterial tissues (Figure S9); TOR1AIP1 has no eQTLs in our study or any GTEx artery tissue, pointing to splicing rather than transcription levels as the main effect of genetic regulation for this gene.
Discussion
We report a coronary artery eQTL mapping study accounting for local ancestry and allele-specific expression. Our study sample’s representation of both ancestral diversity and phenotypic heterogeneity allowed us to capture eQTLs that likely affect vessel wall integrity and maintenance throughout the life course. The disruption of these transcriptional regulatory networks may be critical to plaque progression in coronary artery atherosclerosis. Our approach maximizes the likelihood of identifying eGenes in a rarely available tissue type, providing a roadmap for future QTL studies of diverse populations.
While tissues from patients with advanced CAD are useful for therapeutic development, profiling gene expression changes in subclinical CAD patients with multiple risk factors may point to avenues to prevent lesion progression. For example, molecular pathways for the strong epidemiologic association between blood pressure and CAD61 are incompletely described, while our work importantly begins to elucidate these mechanisms by highlighting a subset of eGenes colocalizing to GWAS findings of both traits. Furthermore, eGenes colocalizing to one or more GWAS traits can be prioritized for functional characterization, particularly those exhibiting cell-type specificity, e.g., heparanase 2 (HPSE2), which is enriched in SMCs in coronary artery and for which our eQTL colocalizes with SBP and DBP.30 HPSE2 would not be a strong candidate gene in a QTL study focused on disease outcomes rather than intermediate phenotypes, but recent characterization of this extracellular matrix protein in endothelial maintenance suggests a contribution to vascular function during inflammation.62,63
Statistical fine-mapping analyses may prioritize causal genes and mechanisms of CAD loci, and we highlight example eGenes TBX20 and TCF21. Here we identify the first genetic evidence of TBX20 regulation in the human coronary artery, filling in a key knowledge gap in characterizing this gene in atherosclerosis. TBX20 is required for normal cardiovascular development and was recently identified as regulating PROK2, a critical component of angiogenesis.64 TBX20 has been implicated in causal pathways for congenital heart defects and continuous traits involving the great vessels.65,66,67 However, the role of TBX20 in atherosclerosis has been minimally investigated, despite expression in arterial tissues and a well-replicated CAD GWAS signal.5,10,11 Conflicting evidence about cell-type-specific expression of TBX20 related to vascular function and neointimal hyperplasia68 demands functional characterization of regulatory elements in this locus that may modulate vessel wall pathways both during development and in a diseased state. Our TCF21/TARID findings also build on existing work by our lab and others characterizing an established CAD locus.13,69,70,71 This relationship is supported by research demonstrating regulation of TCF21 expression by long non-coding RNA TARID in the context of CAD via promoter demethylation.72,73 The mural cell-enriched expression and chromatin accessibility of both genes, as well as overlapping association signals, compel a deeper look into the potential co-regulation of TCF21 and TARID during SMC phenotypic transition.
It is now well appreciated that a large fraction of candidate regulatory variants are predicted to function via transcription factor binding-independent mechanisms, requiring comprehensive fine-mapping of candidate loci to prioritize likely causal variants.20 Our results expand the current focus on disease outcomes such as CAD by incorporating intermediate phenotype summary statistics into fine-mapping analyses. Using this approach, we narrowed colocalized GWAS signals to CSs of candidate variants, highlighting ARHGAP42 and IL5. The ARHGAP42 locus is associated with blood pressure and cIMT: carotid intima-media thickness,74 and gene expression changes affect smooth muscle cell contractility.75 Multi-omic fine-mapping revealed two candidate causal SNPs, of which the rs604723 risk allele has been shown to generate a cryptic SRF binding site to increase expression.50 We also highlight a putative STAT6 binding site created by the risk allele, suggesting a potential IL-4/IL-13-mediated activation of SMCs, both of which are normally lowly expressed in the coronary artery.
Another candidate gene identified through our eQTL discovery and fine-mapping analyses, IL5, resides in a gene-dense SBP GWAS locus.76 While IL5 is reported to function in Th2 cells or eosinophils associated with atherosclerosis progression,77,78,79 its precise role and regulation in the coronary artery remains unknown. Notably, we did not identify eQTLs for neighboring inflammatory genes IL4, IL13, and CSF2,80 despite proposed overlapping functions and regulatory mechanisms.81,82 Aorta-specific upregulation of IL5 in STARNET CAD cases supports a potential disease-specific effect for IL5 in multiple arterial tissues.39 Our lead eQTL, rs7719499, overlaps a plasma cell-specific chromatin-accessibility peak in the coronary artery. Given the known influence of inflammatory cytokines on endothelial and SMC activation, future studies are warranted to investigate the immune cell-derived IL5-mediated vascular wall injury in cell and animal models.
Finally, we demonstrated distinct genetic contributions for expression and splicing activity in the coronary artery. The relationship between isoform specificity and overall expression is complex.83,84 We observed a modest sharing of eGenes and sGenes (14%) and <10% overlap for GWAS colocalization, supporting orthogonal effects of genetic variation on total transcription compared to isoform regulation. Development in the areas of single-cell long-read sequencing will provide a more complete understanding of isoform-specific regulatory mechanisms in coronary artery.
We explored two sGenes with potential roles in coronary artery disease—ULK3 and TOR1AIP1. TOR1AIP1 mutations have been reported for monogenic dystrophic developmental disorders via dysregulation of necessary protein complex formation at the inner nuclear membrane.59,85 The TOR1AIP1 sQTL regulates the addition of an alanine residue to the third exon (position 185). While this variant has not been associated with disease, its high allele frequency suggests differential isoform expression may have an important functional role, particularly given the lack of TOR1AIP1 eQTLs. Regarding ULK3, though mainly described in the context of cancers,86 this gene may mediate vascular disease through autophagy dysregulation and interactions in the Shh signaling pathway.87,88 Although the ULK3 locus has also been reported in BP and cholesterol GWAS, the published associations are located downstream of the ULK3 coding region and are statistically independent of our sQTL and eQTL.9,55,76 Publicly available data do not implicate a particular cell type of interest for functional studies, pointing to the future benefits of long-read sequencing to characterize isoform-specific and cell-type-specific mechanisms relevant to disease pathways.89,90 Our findings for both transcriptional and isoform-specific regulation by variants with no GWAS signal point to the importance of considering multiple ‘omics datasets when evaluating candidate genes for complex diseases and traits.
Overall, our findings emphasize the importance of context in interpreting genetic associations with disease. Characterizing loci within a comprehensive genomic and physiologic setting helps prioritize top candidate genes uniquely relevant to atherosclerosis disease processes. While coronary artery tissue remains the most relevant single tissue type for prioritizing CAD candidate genes, the gene expression program changes during atherosclerosis likely involve multi-tissue and multi-cellular gene-regulatory networks. Multi-tissue network analyses may further resolve underlying paracrine signaling pathways and regulatory mechanisms for eGenes without an obvious role in predominant intimal or medial cell types.91 For instance, inflammatory processes driven by cytokine signaling may be difficult to detect in target cell types, but significantly different expression of IL5 specifically in the aorta in STARNET cases compared to controls provides both validation of our approach and options for identifying other genes functioning in the same molecular pathway.39
A major strength of our work is demonstrating the feasibility and promise of incorporating local genetic ancestry and allele-specific expression into eQTL analyses to discover disease-relevant genes/pathways. Our inclusive study design increased statistical power in both our diverse downsampled subset and overall study population compared to a genetically homogeneous European-ancestry-only subset. This is significant given the predominantly European genetic architecture of GTEx and published GWASs: while these resources have been crucial for genomics discovery to date, work highlighting the limitations of genetically restricted samples92 and technologies developed based on those samples93 points to the necessity of new, more expansive approaches.94,95,96,97 This also aligns with current appeals in basic science and public health to promote equitable research benefiting all populations, rather than studies that may extend the health disparity gap.17,98,99 Furthermore, incorporation of single-cell chromatin accessibility datasets in coronary artery complemented our eQTL-based gene prioritization approach to nominate cis-regulatory mechanisms underlying complex diseases associations.44 Finally, our findings benefited from comparing epigenomic and genetic annotations, providing a more tenable suite of candidate variants for future functional work. Our multiple-phenotype fine-mapping approach will be particularly relevant as the field moves toward functional characterization of disease-associated lncRNAs and splice isoforms, for which traditional metrics such as evolutionary conservation cannot be consistently applied.
In summary, we present a genetically diverse evaluation of coronary artery gene expression across the phenotypic spectrum of atherosclerosis. Our inclusive study design with respect to ancestry and robust pipeline facilitated the discovery of atherosclerosis-associated genes with plausible functional roles in the vascular wall. Molecular characterization of these genes in environments representing subclinical atherosclerosis will improve the identification of therapeutic targets for CAD patients.
Limitations of the study
It is worth noting limitations both common to eQTL studies and unique to our approach. First, restricting to 5% minor-allele frequency variants limited detection of ancestry-specific associations; previous work has shown that effectively capturing lower frequency variants increases discovery both across and within ancestral populations.100 A second limitation relates to interpretability of our colocalization and fine-mapping results based on genetically homogeneous public datasets.17 Individuals with genetic ancestry from Africa and the Americas in particular continue to be severely underrepresented in both GWASs and ‘omics reference datasets. Homogeneous study populations not only prevent identification of ancestry- or haplotype-specific associations, but they also limit generalizability of global associations when fine-mapping is restricted to genetic architecture from a single ancestry group.92,101 With the generation of new eQTL datasets from admixed populations, establishing best practices such as minimizing LD mismatch and using local ancestry estimates is needed to improve data standards, integration, and replication efforts. Additionally, we acknowledge that both sample quality and disease status may affect interindividual cell-type proportions and therefore eQTL detection.102 Adjusting for estimated cell type proportions using single-cell reference-based deconvolution may improve discovery across tissues103 and complement cell-type-specific QTL studies.104
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Genotypes from low-pass whole genome sequencing | This paper | dbGaP: phs002855.v1.p1 |
| Bulk RNA sequencing (raw files and TPMs) | This paper | GEO: GSE225650 |
| eQTL and sQTL summary statistics | This paper | Zenodo: https://doi.org/10.5281/zenodo.7581778 |
| snATAC sequencing (raw and processed data) | This paper | GEO: GSE175621 and GEO: GSE188422 |
| ATAC sequencing and H3K27 Hi-ChIP for human coronary artery smooth muscle cells (HCASMCs) | This paper | GEO: GSE113348 and GEO: GSE101498 |
| GWAS summary statistics for coronary artery disease | Tcheandjieu et al.11 | GWAS catalog: GCST90132305 |
| GWAS summary statistics for myocardial infarction | Hartiala et al.9 | GWAS catalog: GCST011365 |
| GWAS summary statistics for coronary artery disease | Matsunaga et al.10 | GWAS catalog: GCST010480 |
| GWAS summary statistics for coronary artery disease | Koyama et al.8 | Biobank Japan: https://biobankjp.org |
| GWAS summary statistics for coronary artery disease | van der Harst et al.5 | GWAS catalog: GCST005194 |
| GWAS summary statistics for coronary artery calcification | Kavousi et al.105 | GWAS catalog: GCST90278455 and GCST90278456 |
| GWAS summary statistics for lipid traits (HDL, LDL, TC, logTG) | Graham et al.18 | GWAS catalog: GCST90239649, GCST90239655, GCST90239661, and GCST90239673 |
| GWAS summary statistics for blood pressure traits (DBP, SBP, PP) | Evangelou et al.76 | GWAS catalog: GCST006624, GCST006629, and GCST006630 |
| GWAS summary statistics for carotid plaque and IMT | Franceschini et al.106 | GWAS catalog: GCST001231 |
| ENCODE CTCF, H3K27Ac, and H3K4me3 annotations for coronary artery | ENCODE15 | ENCODE Pproject: https://www.encodeproject.org/ |
| GTEx summary statistics for eQTLs in AOR, COR, and TIB tissues | GTEx20 | GTEx Portal: https://www.gtexportal.org/home/ |
| STARNET eQTL summary statistics, gene expression, and clinical trait enrichment for AOR & MAM tissues | Koplev et al.39 | dbGaP: phs001203.v2.p1 http://starnet.mssm.edu/ |
| scRNAseq data for coronary and carotid tissues (integrated single-cell reference) | Verdezoto et al.30; Wirka et al.41; Pan et al.107; Alsaigh et al.108; Hu et al.109 | GEO: GSE131778, GSE155512, and GSE159677. Zenodo: https://doi.org/10.5281/zenodo.6032099 |
| Software and algorithms | ||
| Custom R and shell scripts scripts (data processing; plot generation; coloc, Paintor, SnpEff, SMR, RFMix implementation) | This paper | GitHub: https://github.com/MillerLab-CPHG/CAD_QTL and zenodo: https://doi.org/10.5281/zenodo.10095581 |
| R 4.0.3 | R Core Team | https://www.r-project.org/ |
| R package SNPRelate v1.24.0 | Zheng et al.110 | https://github.com/zhengxwen/SNPRelate |
| Beagle v5.2 | Browning et al.111 | https://bioinformaticshome.com/tools/imputation/descriptions/BEAGLE.html |
| STAR | Dobin et al.112 | https://github.com/alexdobin/STAR/releases |
| samtools v1.10 | Li & Durbin113 | http://samtools.sourceforge.net/ |
| bedtools v2.29.2 | Quinlan et al.114 | https://github.com/arq5x/bedtools2 |
| VCFtools v0.1.16 | Danecek et al.115 | https://vcftools.github.io |
| BCFtools v1.9 | https://doi.org/10.1093/gigascience/giab008 | http://www.htslib.org/ |
| Tabix v0.6 | https://doi.org/10.1093/gigascience/giab008 | https://www.htslib.org/doc/tabix.html |
| Python library phaser (requires python 3.7) | Castel et al.116 | https://github.com/secastel/phaser |
| WASP v0.3.4 | van de Geijn et al.117 | https://github.com/bmvdgeijn/WASP |
| SnpEff (requires java 1.11.0) | https://pcingola.github.io/SnpEff/adds/SnpEff_paper.pdf | https://pcingola.github.io/SnpEff/ |
| Local ancestry inference R pipeline | Martin et al.118 | https://github.com/armartin/ancestry_pipeline |
| QTLtools v1.3.1 | Delaneau et al.119 | https://qtltools.github.io/qtltools/ |
| R package mixqtl v0.2 | Liang et al.24 | https://github.com/hakyimlab/mixqtl |
| R package coloc v5.1.1 | Wallace et al.120 | https://github.com/chr1swallace/coloc |
| SMR v1.03 | Zhu et al.40 | https://yanglab.westlake.edu.cn/software/smr/ |
| paintor v3.0 | Kichaev et al.121 | https://github.com/gkichaev/PAINTOR_V3.0 |
| R package tidyverse v1.3.1 | https://cran.r-project.org/ | |
| R package dplyr v1.0.7 | https://cran.r-project.org/ | |
| R package data.table v1.14.2 | https://cran.r-project.org/ | |
| R package ggplot2 v3.4.0 | https://cran.r-project.org/ | |
| R package susie v0.11.92 | https://cran.r-project.org/ | |
| R package BiocManager v1.30.6 | https://cran.r-project.org/ | |
| R package devtools v2.4.3 | https://cran.r-project.org/ | |
| R package reshape2 v1.4.4 | https://www.jstatsoft.org/article/view/v021i12 | https://cran.r-project.org/ |
| R package ggrepel v0.9.2 | https://cran.r-project.org/ | |
| R package VennDiagram v1.7.3 | https://cran.r-project.org/ | |
| R package UpSetR v1.4.0 | https://cran.r-project.org/ | |
| R package circlize v0.4.15 | Gu et al.122 | https://cran.r-project.org/ |
| R package GetoptLong v1.0.5 | https://cran.r-project.org/ | |
| R package RColorBrewer v1.1-3 | https://cran.r-project.org/ | |
| R package CMplot v4.2.0 | Yin et al.123 | https://cran.r-project.org/ |
| R package extrafont v0.19 | https://cran.r-project.org/ | |
| R package ComplexHeatmap v2.14.0 | Gu et al.124 | https://github.com/jokergoo/ComplexHeatmap |
| R package peer v1.0 | Stegle et al.125 | https://github.com/PMBio/peer |
| LeafCutter v1.0 | Yang et al.126 | https://github.com/davidaknowles/leafcutter |
| RFMix v2.0 | Maples et al.127 | https://github.com/slowkoni/rfmix |
| Plink v1.9 | Chang et al.128 | https://www.cog-genomics.org/plink/ |
| Custom QTL scripts | This paper | GitHub: https://github.com/MillerLab-CPHG/CAD_QTL and zenodo: https://doi.org/10.5281/zenodo.10095581 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Clint L. Miller (clintm@virginia.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data
All raw and processed bulk RNA-sequencing data are available in the Gene Expression Omnibus database (GEO: GSE225650). Low-pass whole-genome sequencing-based genotyping data are available on dbGaP: phs002855.v1.p1. The full summary statistics for the mixQTL eQTL analyses (full sample as well as subsamples for sensitivity analyses), as well as the local ancestry eQTL and the sQTL analyses are available on zenodo: https://doi.org/10.5281/zenodo.7992146
The single-cell RNA-seq datasets from coronary and carotid artery were re-analyzed and integrated from the original datasets available through GEO: GSE131778, GSE155512, GSE159677, and zenodo: https://doi.org/10.5281/zenodo.6032099.109 The raw and processed single-nucleus ATAC-seq datasets are available through GEO: GSE175621 and GEO: GSE188422. The reprocessed and analyzed human scRNA-seq datasets are also available on PlaqView (https://plaqview.com). GTEx gene expression and eQTL data were obtained from the v8 portal website (https://gtexportal.org). STARNET gene expression, eQTL, and clinical trait enrichment data were obtained from dbGaP: phs001203.v2.p1 and are also available at http://starnet.mssm.edu. The HCASMC ATAC-seq and H3K27ac HiChIP data used to calculate ABC scores are available through GEO: GSE113348 and GEO: GSE101498).
Code
All custom scripts used to generate the results are available on GitHub: https://github.com/MillerLab-CPHG/CAD_QTL and zenodo: https://doi.org/10.5281/zenodo.10095581. Specific parameters and versions for published software tools are also included in the key resources table and method details.
Experimental model and study participant details
Ethics statement
All research described herein complies with ethical guidelines for human subjects research under approved Institutional Review Board (IRB) protocols at Stanford University (#4237 and #11925) and the University of Virginia (#20008), for the procurement and use of human tissues and information, respectively.
Sample acquisition
Freshly explanted hearts from heart transplant recipients were obtained at Stanford University under approved Institutional Review Board protocols and written informed consent. Hearts were arrested in cardioplegic solution and rapidly transported from the operating room to the lab on ice. The proximal 5-6cm of three major coronary vessels (left anterior descending, left circumflex, and right coronary artery) were dissected from the epicardium on ice, trimmed of surrounding adipose and adventitia, rinsed in cold phosphate buffered saline, and rapidly snap frozen in liquid nitrogen. Similarly, aortic root and left ventricular free-wall tissues were also processed and stored at −80C until processing. Throughout the manuscript, these tissues will be referred to as "Explants.” Normal coronary artery, aorta, and left ventricle tissues were obtained by Stanford University (from Donor Network West and California Transplant Donor Network) from donor hearts rejected for transplantation, procured for research studies and were treated following the same protocol as the explanted hearts. The collected tissues will be referred to hereby as “Donors.” Tissues were de-identified and clinical information (e.g., ICD-10 codes) was used to classify normal, ischemic and non-ischemic hearts. Frozen tissues were transferred to the University of Virginia through a material transfer agreement and IRB-approved protocols.
Method details
DNA genotyping
Genomic DNA isolation and sequencing
Approximately 20-25mg of frozen left ventricle or coronary artery tissue was used to isolate genomic DNA for each donor sample following the manufacturer’s protocol (Qiagen DNeasy Blood and Tissue Kit, cat# 69504). Genomic DNA samples for all donors in the study were diluted using TE buffer to [5–15 ng/μL] in skirted 96-well PCR plates. Plates were sealed and shipped to Gencove (New York, USA) for 0.4X low-pass genomic DNA sequencing.
Genomic DNA sequence processing
Phasing and imputation
Unphased low-pass whole genome sequencing files were provided by Gencove in build b37. VCFs provided by Gencove included just over 38 million variants imputed to 1000G using a proprietary pipeline–imputation quality scores were not provided. Because phasing was necessary for downstream analyses, samples were phased and subsequently re-imputed to 1000G phase 3 b37 reference panel using Beagle with impute = true and gp = true options.111,129 No additional variants had been imputed at the conclusion of this process.
Liftover of genomic coordinates
Phased autosomal VCFs were lifted over from b37 to hg19 to hg38 using Picard (“Picard Toolkit", 2019, Broad Institute. GitHub Repository: http://broadinstitute.github.io/picard/). After excluding approximately 10,000 variants that could not be mapped, approximately 38 million total variants were available for consideration in analyses.
Principal component estimation
We calculated ancestral principal components using the R package SNPrelate.110 Briefly, SNPRelate uses LD pruning to restrict genotype data to ∼500,000 independent biallelic SNPs with a MAF >1% across the autosomal chromosomes. We used all participants from 1000 Genomes Phase 3 as a reference panel given the diversity of our sample.130 Eigenvectors (EVs) 1 through 3 demonstrated clustering and correlation with Gencove-assigned majority continental ancestry, but subsequent EVs were driven by one or a small number of individuals (Figure S1). Analyses adjusting for global ancestry were therefore restricted to the first three EVs.
Local ancestry estimation
Self-reported race/ethnicity and Gencove-reported regional ancestry estimates suggested that adjustment for local ancestry (LA) might improve discovery compared to global EVs. We adapted the local-ancestry pipeline developed by Alicia Martin (https://github.com/armartin/ancestry_pipeline), including incorporation of several Python scripts.118 We used RFmix2 (https://github.com/slowkoni/rfmix) to calculate ancestry from one of five continental reference populations127; ancestry was recorded as “missing” for a region if >90% probability of concordance with a reference was not attained. We randomly selected genotypes representing continental ancestries from each superpopulation for 1,200 total 1000 Genomes participants: 400 AFR (African), and 200 each of AMR (Indigenous to the Americas), EAS (East Asian), EUR (European), and SAS (South Asian) superpopulations.130 Because the most recent dataset with a 1000G genetic map is b37, we performed LA estimation and all downstream analyses using hg19 positions and gencode ‘v37lift37’ annotations.131
Bulk RNA sequencing and processing
Coronary artery tissue processing and RNA isolation
Coronary artery samples were selected for RNA sequencing based tissue availability (>50mg) and disease status (prioritizing capturing a range of phenotypes). Total RNA was extracted from using the QIAGEN miRNeasy Mini RNA Extraction kit (catalog #217004). Approximately 20 mg of frozen tissue pulverized using a pre-chilled mortar and pestle under liquid nitrogen was added to 1.5 mL RINO tubes (Next Advance, SKU TUBE1R5-S) which were stored on dry ice. As expected, ease of sample processing under liquid nitrogen varied depending on sample calcification. Tissue powder was then further homogenized in Qiazol lysis buffer using stainless steel beads in a Bullet Blender (Next Advance) homogenizer, followed by column-based purification according to the manufacturer’s instructions. RNA concentration was determined using Qubit 3.0; RNA quality was determined using Agilent 4200 TapeStation. Three to five samples were processed per day by one of three individuals. Samples with RNA Integrity Number (RIN) > 4.5 and Illumina DV200 values > 75 were included for library construction.
RNA library sequencing
Total RNA libraries were constructed using the Illumina TruSeq Stranded Total RNA Gold kit (catalog #20020599) and barcodes were added to RNA libraries using the IDT for Illumina-TruSeq RNA Unique Dual Indexes (96 indices, 96 samples) Kit (IDT, Illumina, catalog #20022371. This library preparation captured coding RNAs and some noncoding RNAs, while depleting ribosomal RNAs. After re-evaluating library quality using TapeStation, individually barcoded libraries were sent to Novogene for next generation sequencing. After passing additional QC, libraries were multiplexed and subjected to paired-end 150bp read sequencing on an Illumina NovaSeq S4 Flowcell to a median depth of 100 million total reads (>30G) per library.
RNA-seq mapping
Our RNA sequencing QC pipeline and scripts pertaining to all analyses described can be publicly accessed at: (https://github.com/MillerLab-CPHG/CAD_QTL)
RNA-seq read mapping and quality control
The raw passed filter sequencing reads obtained from Novogene were demultiplexed using the bcl2fastq script. Read quality was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, version 0.11.9) and the adapter sequences were trimmed using Trim Galore version 0.6.5 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore). Reads with Phred scores <20 were also removed, after which all samples passing the quality control analysis were considered for inclusion. Trimmed reads were mapped to the hg38 human reference genome using STAR v2.7.3a according to the GATK Best Practices for RNA-seq.132 To increase mapping efficiency and sensitivity, new splice junctions discovered in a first alignment pass with high stringency were used as annotation in a second pass to permit lower stringency alignment and therefore increase sensitivity. PCR duplicates were marked using Picard; WASP was used to filter reads prone to mapping bias. Total read counts and RPKM were calculated with RNA-SeQC v1.1.8 using default parameters and additional flags “-n 1000 -noDoC -strictMode” and GENCODE v32 reference annotation. Transcript and isoform expression levels were estimated using RSEM v1.3.3.133
Detection of RNA-seq sample swaps
Using known genotypes for an individual from our cohort, we used verifyBamID134 to check for contaminated reads or sample swaps, we identified four swapped samples. To crosscheck these swaps, we ran NGSCheckMate, which uses a depth-dependent correlation model of allele fractions of known SNPs to identify samples from the same individual.135 Consistent with VerifyBamID, we found four swapped samples, two of which we rematched and two of which were duplicates and therefore excluded from downstream analysis.
RNA sequencing data phasing for allele-specific expression
In order to capture allele-specific expression within RNAseq data, we phased all reads using RASQUAL.136 Haplotype phasing of RNA sequencing reads was performed using phASER v1.1.1.116 Known sites for allelic mapping bias and HLA genes were excluded because of their high mapping error rates and introduction of bias using the ENCODE Unified GRCh38 Blacklist (as of 5-5-2020).137 The phASER pipeline was performed using the guided tutorial with additional flags; “--paired_end 1 --mapq 255 --baseq 10”. Generation of the haplotype expression quantifications was performed with the companion tool called “phASER Gene AE” using the standard pipeline and the GENCODE v36 GRCh38 gene coordinates for haplotypic expression calculation.131
Multidimensional scaling
We performed nonparametric clustering in transcriptomic space to determine whether the transcriptomes of our coronary artery samples were unique from GTEx tissues as well as previously published HCASMC transcriptomes. We performed multidimensional scaling on log-transformed RPKM values (excluding genes in our study which did not have ≥0.1 RPKM in ≥10 individuals) of all GTEx tissues, our samples, and HCASMC cells using Kruskal’s non-parametric method. The 54 unique GTEx tissue sites were grouped into 27 broad tissue categories as previously described.13
Quantitative trait loci mapping
eQTL analysis
PEER factor calculation and optimization
Probabilistic Estimation of Expression Residuals (PEER) was used to account for unmeasured confounders related to RNA sequencing (e.g., batch effects).125 No covariates were included. Based on recommendations regarding sample size and statistical models, we expected to incorporate up to five PEER factors in all models. In order to determine how the number of PEER factors may affect our results, we selected a random subset of 400 genes on chr17 (∼30% of annotated lncRNAs or protein-coding genes on that chromosome). We then performed linear regression using QTLtools (described below but with an allele frequency cutoff of 1% rather than 5%) with no additional covariates and compared the number of eGenes with an FDR <0.05 adjusting for 1, 2, 3, 5, 8, 10, 15, 20, and 25 PEER factors (Figure S1). The number of eGenes was not meaningfully affected by the number of PEER factors, hence we continued with five as expected.
Regression analyses
To maximize true positive associations and minimize Type I error, we excluded variants that did not meet the following criteria: biallelic single-nucleotide variants with a minor allele frequency >5% and HWE p value >1E−6 (http://samtools.github.io/bcftools/bcftools.html). Approximately 6,100,000 variants were available for inclusion in each analysis. Bed files were generated using the criteria implemented by GTEx: all lncRNA and protein-coding genes exceeding 0.1 TPM for at least 20% of samples were included.29 Genes were annotated with name, genomic coordinates, and strand using Gencode v32 (https://www.gencodegenes.org/human/release_32.html) for hg38 analyses and gencode v37liftb37 for hg19 analyses.131 Due to strong selection and high likelihood of population stratification contributing to false-positive identification of genes in the MHC region which is under high selective pressure, HLA genes and other MHC components were not considered candidates for fine-mapping in downstream analyses.
Statistical significance reporting
For all regression analyses, the following standards will be used for reporting p values throughout the manuscript. For single variant association tests, pnom refers to the p value reported in the “pval_meta” column for mixQTL or the 12th output column for QTLtools run under the nominal pass model. For variants that were not lead QTLs, padj refers to the Bonferroni-corrected pnom, where the correction is adjusting for the number of SNPs tested for each gene or splice junction respectively. For lead QTLs, pBH refers to the value obtained by correcting for the total number of genes or splice junctions tested within each method using the Benjamini-Hochberg FDR correction applied to the padj for mixQTL analyses and the 19th output column for QTLtools analyses.
Local ancestry adjustment
Local-ancestry-adjusted cis-eQTL analyses were performed using a new pipeline which incorporates QTLtools (a computationally efficient implementation of MatrixQTL which allows adjustment for SNP-level covariates).119,138 Models adjusted for age, sex, local ancestry, and five PEER factors. Because local ancestry designations occur at the SNP level and cannot be included as traditional covariates in genome-wide eQTL regression analyses, proportions of estimated continental ancestry for each individual were calculated for each gene, and gene-specific covariate and bed files were used in QTLtools. Local ancestry interpolation scripts as well as scripts to run each gene individually and select lead SNPs with adjusted p values can be found on the lab GitHub repository referenced above.
Permutation pass mode was run in QTLtools using up to 100,000 iterations per gene based on the method described by Gay et al.,25 to generate a lead variant for each gene tested. “Nominal-pass” results were run for significant eGenes only to obtain a non-permuted p value for each variant within 500kb up- or downstream of the transcription start site (TSS). Isoforms were not evaluated separately, therefore the TSS used for each gene was the most upstream TSS for transcripts with multiple annotated isoforms.
Combined global-ancestry-adjusted and allele-specific expression analysis
We implemented the R package ‘mixQTL’ (https://github.com/hakyimlab/mixqtl) to identify eQTLs incorporating allele-specific expression in our data.24 All mixQTL analyses were performed using genome build hg38; RNA transcripts were mapped to Gencode v32 (https://www.gencodegenes.org/human/release_32.html). Briefly, mixQTL tests for an association between a genotype and total read count; an association between specific alleles and corresponding haplotype expression (allele-specific expression); and the meta-analysis of both scores when inclusion criteria are met for both methods. MixQTL inputs include phased genotypes, total read counts, allele-specific read counts, and covariate information. Because mixQTL has to be run separately for each gene and does not have a way to account for missing data, all data frames (covariate, haplotype, and expression) would need to be re-generated for each gene in the course of running the R script to account for “missing” values for any covariate, haplotype, or expression value for one or more individuals. We did not have missing data for any included covariates or genetic variants. Most genes with any missing expression values were lowly expressed, therefore we do not expect inflation that would not be accounted for by adjusting the resulting p values and set all missing values in the phased RNAseq data to zero. We adjusted the example script on the mixQTL Github (https://github.com/liangyy/mixqtl) to limit our window to variants within 500kb of the transcription start sites of each gene for a maximum window size of 1Mb, and analyzed associations all genes with ≥0.1 mean TPM. MixQTL models included three ancestry EVs, age, sex, and five PEER factors as covariates.
Sensitivity analyses in European-ancestry-specific and ancestrally proportional sample subsets
Most statistical fine-mapping methods and publicly available genomics references continue to over-represent ancestrally homogeneous European-ancestry populations. To evaluate whether discovery or replication could be improved in a genetically homogeneous study sample despite a meaningful decrease in sample size, we performed mixQTL and downstream analyses in subsets of our sample restricted either to individuals with 100% European ancestry (n = 80) or an 80-person downsample of the total study population randomly selected within each majority-assigned-ancestry group. Regression, generalization, and colocalization were performed within both subsets as described above for the combined sample.
Splicing analysis
Generation and processing of BAM and junction files for splice QTL analysis
We aligned to the human genome the FASTQ files from RNAseq data of 138 ancestrally diverse heart transplant patients using the aligner tool STAR (version 2.7.2b) in 2-pass mode.112 An STAR index was generated using Gencode v37 annotations and the UCSC hg38 reference genome. BAM files were generated using the --twopassmode basic and --outSAMtype BAM SortedByCoordinate options with a minimum overhang of 8 bp for spliced alignments. Additionally, the WASP correction option, --waspOutputMode SAMtag, was used to mitigate allelic mapping bias.117 The generated BAM files were further indexed using the option --index from the package Samtools.139 Next, junction files were generated using the package RegTools.140 The option “-junctions extract” was used with the following parameters: (1) minimum intron size of 50 bp, (2) maximum intron length (100 kbp), (3) minimum overlap between junctions of 8 bp, and (4) leaving the strand specificity as ‘unstranded’ to analyze both positive and negative strands. We removed unknown or unwanted chromosomes from junction files.
LeafCutter
QTL input files were generated using the modified LeafCutter-GTEX pipeline.126,141 Junction files were used as input to generate a count matrix of intron excision ratios. Clusters of alternatively spliced introns were identified using split reads that mapped with a minimum of 8 bp into each exon. Singleton introns that did not cluster with other introns were discarded. LeafCutter iteratively analyzed each intron cluster and removed introns in two ways: (1) introns with fewer than 50 reads across all samples, or (2) intron reads present in less than 0.1% of the total number of reads in the entire cluster. LeafCutter re-clustered introns and only included those with a maximum length of 100,000 bp. The count matrix was further processed to (1) generate a BED file and its index in a format compatible for QTL mappers, such as QTLtools; and (2) to calculate splice principal components. The LeafCutter protocol specifically outputs percent spliced in (PSI) for each splice junction, which adjusts for splicing events with overlapping start or stop positions to account for gene-level variation.
sQTL association
Genetic variants affecting the quantity of proximal splice junctions (cis-sQTLs) were estimated using QTLtools as described above, with age, sex, and four ancestry principal components included in the model. Nominal-pass was used to obtain non-permuted p values for each variant within 250kb up- or downstream of the LeafCutter-generated BED file of splice junction expression. 100,000 permutations were run with a permutation pass to generate an adjusted p value for each lead sQTL.
Characterization and fine-mapping of QTLs
Characterization of coronary artery QTLs
STARNET gene and protein expression analysis
In order to compare magnitude and direction of effect at eGenes of interest, we generated expression plots by genotype for normalized expression and applied the FDR of 0.05 in the published work as a threshold for significance. Stockholm-Tartu Atherosclerosis Reverse Networks Engineering Task study (STARNET) subject recruitment and tissue collection were performed as described previously.142 Briefly, patients with coronary artery disease (CAD) who were eligible for open-thorax surgery at the Department of Cardiac Surgery, Tartu University Hospital in Estonia were enrolled after informed consent. Venous blood was drawn, and DNA was isolated for genotyping using the Illumina Infinium assay. Tissue biopsies from aorta and mammary artery were obtained to study tissue-specific gene expression and the disease. RNA was extracted from tissues and sequenced for whole transcriptome as described.142 In addition, 250 subjects who were eligible for open-heart surgery for reasons other than coronary artery bypass graft surgery were consented and recruited as controls.39 For case control study cohort, whole transcriptome sequencing was performed on selected CAD cases and non-CAD controls with matched age, gender and BMI. Transcriptome data was processed and differential gene expression between cases and controls was estimated using DESeq2.143 Association of gene expression with clinical phenotypes of subjects was calculated by Pearson’s correlation coefficients. Matrix eQTL was used for the inference of cis-regulated eQTLs analysis on genotype and expression data from individuals with CAD cases.144 Association of gene expression with genotypes of SNPs were plotted using ggplot2 in R. For protein study, EDTA plasma was collected after blood drawn. In total, 304 cases and 217 controls were selected with matched age, sex, and BMI. Full-scale screening of Olink’s library of 11 panels targeting 1,012 proteins was performed by multiplex Proximity Extension Assay (Olink Proteomics, Uppsala, Sweden). R package DEqMS was used for quality control, data normalization, and statistical analysis for differential protein expression.145
Generalization of previously published eQTLs in our study
We compared LA and mixQTL eGenes to published eQTL datasets in relevant tissues. Specifically, we assessed whether eGenes in our study also had significant eQTLs–and whether our lead variants were correlated using European-ancestry LD–in GTExv8 coronary, thoracic aorta, and tibial artery tissues29; and STARNET internal mammary artery and aortic root tissues.39 Due to differences in annotation, overlap between each dataset and genes meeting our inclusion criteria was identified using the maximum set of gene names or primary ENSG identifiers (excluding post-decimal identifying numbers).
Annotation of eQTLs and sQTLs
To identify potential causal mechanisms of QTLs, we annotated lead eQTLs and sQTLs to their respective eGenes and sGenes using the “GRCh38.p13” database in SnpEff to correspond to the Gencode v32 annotation we aligned our RNAseq data to.146 SnpEff reports variant annotations by gene (i.e., specific variant-gene annotations are not provided in the main output). For this reason, we utilized the verbose (-v) option and the log file to identify gene-variant-specific annotations to ensure the eQTL-eGene association was maintained. Corresponding scripts are available on our Github. We further evaluated gene-based and region-based annotations using the annotate_variation.pl script with Annovar, also for build hg38.147
Co-localization of eQTLs and sQTLs with GWAS variants
We tested for colocalization of eQTLs with significant (p < 5x10−8) published genetic associations in GWAS of coronary artery disease and myocardial infarction.5,8,9,10,11 Due to the phenotypic heterogeneity within our study population, we additionally evaluated quantitative traits that are traditional risk factors for CAD including blood pressure traits (systolic [SBP], diastolic [DBP], and pulse [PP] pressures),76 cholesterol traits (total cholesterol [TC], high-density lipoprotein [HDL], low-density lipoprotein [LDL], and triglycerides (reported as a log concentration, logTG]),18 carotid artery calcification (CAC),105 and intima media thickness (IMT).106 The GWAS catalog (https://www.ebi.ac.uk/gwas) was most recently accessed on November 1, 2022. We considered PPH3 >0.8 to implicate two independent causal SNPs associated with eQTL and GWAS association signals, and PPH4 >0.8 to support evidence of a shared causal variant.120,148
Summary-data-based Mendelian randomization (SMR)
SMR evaluates evidence for statistical pleiotropy by comparing summary statistics from GWAS to eQTL results from expression data in relevant tissues; i.e., to test whether the association between a genetic variant and CAD-related phenotypes is mediated by gene expression in our study sample.40 Due to the complexity and low informativeness likely in LD generated from our genetically diverse but modestly sized sample, we generated bed files in Plink using 1000G EUR population (excluding Finnish samples due to genetic distinction of that population, which was not represented in our samples) as our LD ref. 128,130. We then performed SMR (https://yanglab.westlake.edu.cn/software/smr) using mixQTL results and GWAS summary statistics from CAD and BP traits included in colocalization analyses.5,9,11,76,106 We were not able to evaluate Japanese CAD GWAS8,10 due to the large number of variants with allele frequency differences >30% between the GWAS study populations and ours. Options included a minor allele frequency threshold of 0.01, a 1Mb window surrounding the most significant eQTL per locus, and a maximum mixQTL p value of 5E−06 required for variant inclusion.
Fine-mapping of eGene association signals
mixFine
To identify independent associations and prioritize credible sets for discovery coronary artery eGenes, we used the command line implementation of mixFine (https://github.com/hakyimlab/mixqtl).24 Mixfine runs SuSiE internally to accommodate the possibility of multiple causal variants in a dataset where many of the input variables (SNPs) are likely highly correlated.148 Input files utilized by mixFine are the same as those for mixQTL and were prepared as described above.
Fine-mapping with GWAS and variant annotations
To combine the effects of GWAS association signals, eQTLs identified in our coronary artery tissue samples, and tissue-relevant variant annotations, we fine-mapped mixQTL eGene associations using fast Paintor (v3.0, https://github.com/gkichaev/PAINTOR_V3.0).121 Due to FastPaintor input requirements and the ancestral representation in published GWAS of relevant traits, we restricted published datasets included in these analyses to European-only ancestry. MixQTL results were combined with overlapping SNPs from GWAS summary statistics for two groups of traits–CAD/MI and blood pressure traits.5,9,10 For each eGene (+/−500kb from TSS), VCFs and LD matrices were generated with Plink, using hg38 1000G EUR samples excluding FIN samples (which are minimally represented in our study or published GWAS).115,128,130 We evaluated four annotations based gene repression, activation, transcription, and chromatin-contact-based enhancer activity: CTCF only, H3K4me3 only, and H3K4me3/H3K27Ac combined. To do this, we included ENCODE binary SNP annotations for chromatin accessibility (CTCF, H3K27ac, and H3K4me3 in coronary artery tissue from one 53yo female).15 To apply enriched enhancer regulatory activity in coronary artery tissue, we also used the activity-by-contact model (ABC; https://github.com/broadinstitute/ABC-Enhancer-Gene-Prediction). ABC scores were obtained from previous H3K27ac HiChIP and ATAC data in HCASMC and coronary artery from our previous work are publicly available (see “Data and Code Availability”).13,149
Acknowledgments
This work was supported by grants from the following: the National Institutes of Health (grant numbers R01HL148239 and R01HL164577 to C.L.M.; T32HL007284 to C.J.H.; and R01HL125863 to J.L.M.B.; R01HL130423, R01HL135093, and R01HL148167 to J.C.K.), the American Heart Association (grant number 20POST35120545 to A.W.T.; AHA909150 to J.V.M.; and A14SFRN20840000 to J.L.M.B.), the Swedish Research Council and Heart Lung Foundation (grant numbers 2018-02529 and 20170265 to J.L.M.B.), the Fondation Leducq (grant number “PlaqOmics” 18CVD02 to C.L.M. and J.L.M.B.), and the Single-Cell Data Insights award from the Chan Zuckerberg Initiative, LLC, and Silicon Valley Community Foundation (to C.L.M.). This work was also supported by fellowship grants from the Bench to Bassinet Pediatric Cardiac Genomics Consortium (PCGC) and Cardiovascular Development Data Resource Center (CDDRC) (to C.J.H.), as well as the UVA MSTP training grant (NIH T32GM007267, to W.F.M.). S.W.v.d.L. is supported by EU H2020 TO_AITION (grant number: 848146). We are thankful for the support of the Netherlands CardioVascular Research Initiative of the Netherlands Heart Foundation (CVON 2011/B019 and CVON 2017-20: Generating the best evidence-based pharmaceutical targets for atherosclerosis [GENIUS I&II]) and the ERA-CVD program “druggable-MI-targets” (grant number: 01KL1802). The research for this contribution was made possible in part by the AI for Health working group of the EWUU alliance and the CZI Foundation. The authors would like to thank Yipei Song and Wesley Craig for assistance with scripting and troubleshooting; Catherine Robertson for assistance with fine-mapping study design; Katia Sol-Church and Yongde Bao for assistance with library preparation and sequencing; Peter Chiu, Paul Chang, A.J. Pedroza, Tiffany Koyano, Euan Ashley, Tom Quertermous, and all of the transplant recipients, heart donors, family members, study coordinators, and transplant procurement team at Stanford for coronary artery tissue procurement.
Author contributions
We used CRediT taxonomy to determine the authors’ contributions. C.J.H., A.W.T., J.L.M.B., and C.L.M. conceptualized the study. C.J.H., A.W.T., M.D.K., and N.G.L. curated the data. C.J.H., A.W.T., M.D.K., N.B.B., R.M., L.M., and J.V.M., performed formal data analysis. C.J.H., A.W.T., N.J.L., J.C.K., J.L.M.B., and C.L.M. acquired funding. C.J.H., A.W.T., and E.F. performed the experiments. M.D.K., N.B.B., R.M., E.F., D.W., and S.O.-G. contributed to methods development. G.A. and S.O.-G. performed project administration. A.W.T., L.M., N.G.L., J.V.M., W.F.M., M.K., P.A.P., S.W.v.d.L., N.J.L., J.C.K., J.L.M.B., and C.L.M. contributed biospecimens, datasets, scripts, or other resources. C.J.H. and W.F.M. contributed software tools. C.L.M. supervised the project. L.M., J.V.M., and J.L.M.B. contributed to validation of the results. C.J.H., A.W.T., M.D.K., N.B.B., J.V.M., and C.L.M. contributed to data visualization. C.J.H., A.W.T., and C.L.M. prepared the manuscript draft. All authors reviewed and edited the manuscript.
Declaration of interests
J.L.M.B. is a shareholder in Clinical Gene Network AB and has an invested interest in STARNET. J.C.K. is the recipient of an Agilent Thought Leader Award, which includes funding for research that is unrelated to the current manuscript. S.W.v.d.L. has received Roche funding for unrelated work. C.L.M. has received AstraZeneca funding for unrelated work.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 15, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100465.
Supplemental information
References
- 1.Khera A.V., Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat. Rev. Genet. 2017;18:331–344. doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watkins H., Farrall M. Genetic susceptibility to coronary artery disease: from promise to progress. Nat. Rev. Genet. 2006;7:163–173. doi: 10.1038/nrg1805. [DOI] [PubMed] [Google Scholar]
- 3.Schunkert H., König I.R., Kathiresan S., Reilly M.P., Assimes T.L., Holm H., Preuss M., Stewart A.F.R., Barbalic M., Gieger C., et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Webb T.R., Erdmann J., Stirrups K.E., Stitziel N.O., Masca N.G.D., Jansen H., Kanoni S., Nelson C.P., Ferrario P.G., König I.R., et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J. Am. Coll. Cardiol. 2017;69:823–836. doi: 10.1016/j.jacc.2016.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Harst P., Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ. Res. 2018;122:433–443. doi: 10.1161/CIRCRESAHA.117.312086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikpay M., Goel A., Won H.-H., Hall L.M., Willenborg C., Kanoni S., Saleheen D., Kyriakou T., Nelson C.P., Hopewell J.C., et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson C.P., Goel A., Butterworth A.S., Kanoni S., Webb T.R., Marouli E., Zeng L., Ntalla I., Lai F.Y., Hopewell J.C., et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017;49:1385–1391. doi: 10.1038/ng.3913. [DOI] [PubMed] [Google Scholar]
- 8.Koyama S., Ito K., Terao C., Akiyama M., Horikoshi M., Momozawa Y., Matsunaga H., Ieki H., Ozaki K., Onouchi Y., et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 2020;52:1169–1177. doi: 10.1038/s41588-020-0705-3. [DOI] [PubMed] [Google Scholar]
- 9.Hartiala J.A., Han Y., Jia Q., Hilser J.R., Huang P., Gukasyan J., Schwartzman W.S., Cai Z., Biswas S., Trégouët D.A., et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur. Heart J. 2021;42:919–933. doi: 10.1093/eurheartj/ehaa1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsunaga H., Ito K., Akiyama M., Takahashi A., Koyama S., Nomura S., Ieki H., Ozaki K., Onouchi Y., Sakaue S., et al. Transethnic Meta-Analysis of Genome-Wide Association Studies Identifies Three New Loci and Characterizes Population-Specific Differences for Coronary Artery Disease. Circ. Genom. Precis. Med. 2020;13 doi: 10.1161/CIRCGEN.119.002670. [DOI] [PubMed] [Google Scholar]
- 11.Tcheandjieu C., Zhu X., Hilliard A.T., Clarke S.L., Napolioni V., Ma S., Lee K.M., Fang H., Chen F., Lu Y., et al. Large-scale genome-wide association study of coronary artery disease in genetically diverse populations. Nat. Med. 2022;28:1679–1692. doi: 10.1038/s41591-022-01891-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kichaev G., Bhatia G., Loh P.-R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu B., Pjanic M., Wang T., Nguyen T., Gloudemans M., Rao A., Castano V.G., Nurnberg S., Rader D.J., Elwyn S., et al. Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am. J. Hum. Genet. 2018;103:377–388. doi: 10.1016/j.ajhg.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Örd T., Õunap K., Stolze L.K., Aherrahrou R., Nurminen V., Toropainen A., Selvarajan I., Lönnberg T., Aavik E., Ylä-Herttuala S., et al. Single-Cell Epigenomics and Functional Fine-Mapping of Atherosclerosis GWAS Loci. Circ. Res. 2021;129:240–258. doi: 10.1161/CIRCRESAHA.121.318971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van de Geijn B., Finucane H., Gazal S., Hormozdiari F., Amariuta T., Liu X., Gusev A., Loh P.-R., Reshef Y., Kichaev G., et al. Annotations capturing cell type-specific TF binding explain a large fraction of disease heritability. Hum. Mol. Genet. 2020;29:1057–1067. doi: 10.1093/hmg/ddz226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bien S.A., Wojcik G.L., Hodonsky C.J., Gignoux C.R., Cheng I., Matise T.C., Peters U., Kenny E.E., North K.E. The Future of Genomic Studies Must Be Globally Representative: Perspectives from PAGE. Annu. Rev. Genom. Hum. Genet. 2019;20:181–200. doi: 10.1146/annurev-genom-091416-035517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham S.E., Clarke S.L., Wu K.-H.H., Kanoni S., Zajac G.J.M., Ramdas S., Surakka I., Ntalla I., Vedantam S., Winkler T.W., et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600:675–679. doi: 10.1038/s41586-021-04064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong D., Turner A.W., Miller C.L. Genetic insights into smooth muscle cell contributions to coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2019;39:1006–1017. doi: 10.1161/ATVBAHA.119.312141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stolze L.K., Conklin A.C., Whalen M.B., López Rodríguez M., Õunap K., Selvarajan I., Toropainen A., Örd T., Li J., Eshghi A., et al. Systems Genetics in Human Endothelial Cells Identifies Non-coding Variants Modifying Enhancers, Expression, and Complex Disease Traits. Am. J. Hum. Genet. 2020;106:748–763. doi: 10.1016/j.ajhg.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slenders L., Landsmeer L.P.L., Cui K., Depuydt M.A.C., Verwer M., Mekke J., Timmerman N., van den Dungen N.A.M., Kuiper J., de Winther M.P.J., et al. Intersecting single-cell transcriptomics and genome-wide association studies identifies crucial cell populations and candidate genes for atherosclerosis. Eur. Heart J. Open. 2022;2:oeab043. doi: 10.1093/ehjopen/oeab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rotival M., Zeller T., Wild P.S., Maouche S., Szymczak S., Schillert A., Castagné R., Deiseroth A., Proust C., Brocheton J., et al. Integrating genome-wide genetic variations and monocyte expression data reveals trans-regulated gene modules in humans. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang Y., Aguet F., Barbeira A.N., Ardlie K., Im H.K. A scalable unified framework of total and allele-specific counts for cis-QTL, fine-mapping, and prediction. Nat. Commun. 2021;12:1424. doi: 10.1038/s41467-021-21592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gay N.R., Gloudemans M., Antonio M.L., Abell N.S., Balliu B., Park Y., Martin A.R., Musharoff S., Rao A.S., Aguet F., et al. Impact of admixture and ancestry on eQTL analysis and GWAS colocalization in GTEx. Genome Biol. 2020;21:233. doi: 10.1186/s13059-020-02113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robert J., Osto E., von Eckardstein A. The endothelium is both a target and a barrier of hdl’s protective functions. Cells. 2021;10 doi: 10.3390/cells10051041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu H., Littlewood T., Bennett M. Akt isoforms in vascular disease. Vasc. Pharmacol. 2015;71:57–64. doi: 10.1016/j.vph.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosquera J.V., Wong D., Auguste G., Turner A.W., Hodonsky C.J., Lino Cardenas C.L., Theofilatos K., Bos M., Kavousi M., Peyser P., et al. Integrative single-cell meta-analysis reveals disease-relevant vascular cell states and markers in human atherosclerosis. bioRxiv. 2022 doi: 10.1016/j.celrep.2023.113380. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carss K.J., Baranowska A.A., Armisen J., Webb T.R., Hamby S.E., Premawardhana D., Al-Hussaini A., Wood A., Wang Q., Deevi S.V.V., et al. Spontaneous coronary artery dissection: insights on rare genetic variation from genome sequencing. Circ. Genom. Precis. Med. 2020;13 doi: 10.1161/CIRCGEN.120.003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo D.-C., Duan X.-Y., Regalado E.S., Mellor-Crummey L., Kwartler C.S., Kim D., Lieberman K., de Vries B.B.A., Pfundt R., Schinzel A., et al. Loss-of-Function Mutations in YY1AP1 Lead to Grange Syndrome and a Fibromuscular Dysplasia-Like Vascular Disease. Am. J. Hum. Genet. 2017;100:21–30. doi: 10.1016/j.ajhg.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S.-B., Choi J.-E., Park B., Cha M.-Y., Hong K.-W., Jung D.-H. Dyslipidaemia-Genotype Interactions with Nutrient Intake and Cerebro-Cardiovascular Disease. Biomedicines. 2022;10 doi: 10.3390/biomedicines10071615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson T.G., Leyden G.M., Wang Q., Bell J.A., Elsworth B., Davey Smith G., Holmes M.V. Characterising metabolomic signatures of lipid-modifying therapies through drug target mendelian randomisation. PLoS Biol. 2022;20 doi: 10.1371/journal.pbio.3001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christakoudi S., Evangelou E., Riboli E., Tsilidis K.K. GWAS of allometric body-shape indices in UK Biobank identifies loci suggesting associations with morphogenesis, organogenesis, adrenal cell renewal and cancer. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-89176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y., Gao H., Li H., Tabara Y., Nakatochi M., Chiu Y.-F., Park E.J., Wen W., Adair L.S., Borja J.B., et al. A meta-analysis of genome-wide association studies for adiponectin levels in East Asians identifies a novel locus near WDR11-FGFR2. Hum. Mol. Genet. 2014;23:1108–1119. doi: 10.1093/hmg/ddt488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Z., Guo Y., Shi H., Liu C.-L., Panganiban R.A., Chung W., O’Connor L.J., Himes B.E., Gazal S., Hasegawa K., et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. J. Allergy Clin. Immunol. 2020;145:537–549. doi: 10.1016/j.jaci.2019.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koplev S., Seldin M., Sukhavasi K., Ermel R., Pang S., Zeng L., Bankier S., Di Narzo A., Cheng H., Meda V., et al. A mechanistic framework for cardiometabolic and coronary artery diseases. Nat. Cardiovasc. Res. 2022;1:85–100. doi: 10.1038/s44161-021-00009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M., Yang J. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 41.Wirka R.C., Wagh D., Paik D.T., Pjanic M., Nguyen T., Miller C.L., Kundu R., Nagao M., Coller J., Koyano T.K., et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019;25:1280–1289. doi: 10.1038/s41591-019-0512-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagao M., Lyu Q., Zhao Q., Wirka R.C., Bagga J., Nguyen T., Cheng P., Kim J.B., Pjanic M., Miano J.M., Quertermous T. Coronary Disease-Associated Gene TCF21 Inhibits Smooth Muscle Cell Differentiation by Blocking the Myocardin-Serum Response Factor Pathway. Circ. Res. 2020;126:517–529. doi: 10.1161/CIRCRESAHA.119.315968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olson E.N. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner A.W., Hu S.S., Mosquera J.V., Ma W.F., Hodonsky C.J., Wong D., Auguste G., Song Y., Sol-Church K., Farber E., et al. Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk. Nat. Genet. 2022;54:804–816. doi: 10.1038/s41588-022-01069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Çalışkan M., Manduchi E., Rao H.S., Segert J.A., Beltrame M.H., Trizzino M., Park Y., Baker S.W., Chesi A., Johnson M.E., et al. Genetic and epigenetic fine mapping of complex trait associated loci in the human liver. Am. J. Hum. Genet. 2019;105:89–107. doi: 10.1016/j.ajhg.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S., Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai X., Mangum K., Kakoki M., Smithies O., Mack C.P., Taylor J.M. GRAF3 serves as a blood volume-sensitive rheostat to control smooth muscle contractility and blood pressure. Small GTPases. 2020;11:194–203. doi: 10.1080/21541248.2017.1375602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fjorder A.S., Rasmussen M.B., Mehrjouy M.M., Nazaryan-Petersen L., Hansen C., Bak M., Grarup N., Nørremølle A., Larsen L.A., Vestergaard H., et al. Haploinsufficiency of ARHGAP42 is associated with hypertension. Eur. J. Hum. Genet. 2019;27:1296–1303. doi: 10.1038/s41431-019-0382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q., Dibus M., Casey A., Yee C.S.K., Vargas S.O., Luo S., Rosen S.M., Madden J.A., Genetti C.A., Brabek J., et al. A homozygous stop-gain variant in ARHGAP42 is associated with childhood interstitial lung disease, systemic hypertension, and immunological findings. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bai X., Mangum K.D., Dee R.A., Stouffer G.A., Lee C.R., Oni-Orisan A., Patterson C., Schisler J.C., Viera A.J., Taylor J.M., Mack C.P. Blood pressure-associated polymorphism controls ARHGAP42 expression via serum response factor DNA binding. J. Clin. Invest. 2017;127:670–680. doi: 10.1172/JCI88899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baetta R., Soma M., De-Fraja C., Comparato C., Teruzzi C., Magrassi L., Cattaneo E. Upregulation and activation of Stat6 precede vascular smooth muscle cell proliferation in carotid artery injury model. Arterioscler. Thromb. Vasc. Biol. 2000;20:931–939. doi: 10.1161/01.atv.20.4.931. [DOI] [PubMed] [Google Scholar]
- 52.Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K.C., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R., et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347 doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maloverjan A., Piirsoo M., Michelson P., Kogerman P., Osterlund T. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp. Cell Res. 2010;316:627–637. doi: 10.1016/j.yexcr.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 54.Caballe A., Wenzel D.M., Agromayor M., Alam S.L., Skalicky J.J., Kloc M., Carlton J.G., Labrador L., Sundquist W.I., Martin-Serrano J. ULK3 regulates cytokinetic abscission by phosphorylating ESCRT-III proteins. Elife. 2015;4 doi: 10.7554/eLife.06547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levy D., Ehret G.B., Rice K., Verwoert G.C., Launer L.J., Dehghan A., Glazer N.L., Morrison A.C., Johnson A.D., Aspelund T., et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinnott-Armstrong N., Tanigawa Y., Amar D., Mars N., Benner C., Aguirre M., Venkataraman G.R., Wainberg M., Ollila H.M., Kiiskinen T., et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 2021;53:185–194. doi: 10.1038/s41588-020-00757-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann T.J., Ehret G.B., Nandakumar P., Ranatunga D., Schaefer C., Kwok P.-Y., Iribarren C., Chakravarti A., Risch N. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat. Genet. 2017;49:54–64. doi: 10.1038/ng.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 59.Ghaoui R., Benavides T., Lek M., Waddell L.B., Kaur S., North K.N., MacArthur D.G., Clarke N.F., Cooper S.T. TOR1AIP1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. 2016;26:500–503. doi: 10.1016/j.nmd.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 60.Shin J.-Y., Méndez-López I., Wang Y., Hays A.P., Tanji K., Lefkowitch J.H., Schulze P.C., Worman H.J., Dauer W.T. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev. Cell. 2013;26:591–603. doi: 10.1016/j.devcel.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lewington S., Clarke R., Qizilbash N., Peto R., Collins R., Prospective Studies Collaboration Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. doi: 10.1016/s0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- 62.Pinhal M.A.S., Melo C.M., Nader H.B. The Good and Bad Sides of Heparanase-1 and Heparanase-2. Adv. Exp. Med. Biol. 2020;1221:821–845. doi: 10.1007/978-3-030-34521-1_36. [DOI] [PubMed] [Google Scholar]
- 63.Kiyan Y., Tkachuk S., Kurselis K., Shushakova N., Stahl K., Dawodu D., Kiyan R., Chichkov B., Haller H. Heparanase-2 protects from LPS-mediated endothelial injury by inhibiting TLR4 signalling. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-50068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meng S., Gu Q., Yang X., Lv J., Owusu I., Matrone G., Chen K., Cooke J.P., Fang L. TBX20 Regulates Angiogenesis Through the Prokineticin 2-Prokineticin Receptor 1 Pathway. Circulation. 2018;138:913–928. doi: 10.1161/CIRCULATIONAHA.118.033939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tcheandjieu C., Xiao K., Tejeda H., Lynch J.A., Ruotsalainen S., Bellomo T., Palnati M., Judy R., Klarin D., Kember R.L., et al. High heritability of ascending aortic diameter and trans-ancestry prediction of thoracic aortic disease. Nat. Genet. 2022;54:772–782. doi: 10.1038/s41588-022-01070-7. [DOI] [PubMed] [Google Scholar]
- 66.Škorić-Milosavljević D., Tadros R., Bosada F.M., Tessadori F., van Weerd J.H., Woudstra O.I., Tjong F.V.Y., Lahrouchi N., Bajolle F., Cordell H.J., et al. Common genetic variants contribute to risk of transposition of the great arteries. Circ. Res. 2022;130:166–180. doi: 10.1161/CIRCRESAHA.120.317107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luyckx I., Kumar A.A., Reyniers E., Dekeyser E., Vanderstraeten K., Vandeweyer G., Wünnemann F., Preuss C., Mazzella J.-M., Goudot G., et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur. J. Hum. Genet. 2019;27:1033–1043. doi: 10.1038/s41431-019-0364-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ji Y., Ma Y., Shen J., Ni H., Lu Y., Zhang Y., Ma H., Liu C., Zhao Y., Ding S., et al. TBX20 Contributes to Balancing the Differentiation of Perivascular Adipose-Derived Stem Cells to Vascular Lineages and Neointimal Hyperplasia. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.662704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller C.L., Haas U., Diaz R., Leeper N.J., Kundu R.K., Patlolla B., Assimes T.L., Kaiser F.J., Perisic L., Hedin U., et al. Coronary heart disease-associated variation in TCF21 disrupts a miR-224 binding site and miRNA-mediated regulation. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller C.L., Pjanic M., Wang T., Nguyen T., Cohain A., Lee J.D., Perisic L., Hedin U., Kundu R.K., Majmudar D., et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 2016;7 doi: 10.1038/ncomms12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller C.L., Anderson D.R., Kundu R.K., Raiesdana A., Nürnberg S.T., Diaz R., Cheng K., Leeper N.J., Chen C.-H., Chang I.-S., et al. Disease-related growth factor and embryonic signaling pathways modulate an enhancer of TCF21 expression at the 6q23.2 coronary heart disease locus. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arab K., Park Y.J., Lindroth A.M., Schäfer A., Oakes C., Weichenhan D., Lukanova A., Lundin E., Risch A., Meister M., et al. Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol. Cell. 2014;55:604–614. doi: 10.1016/j.molcel.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 73.Cheng Z., Zhang Y., Zhuo Y., Fan J., Xu Y., Li M., Chen H., Zhou L. LncRNA TARID induces cell proliferation through cell cycle pathway associated with coronary artery disease. Mol. Biol. Rep. 2022;49:4573–4581. doi: 10.1007/s11033-022-07304-5. [DOI] [PubMed] [Google Scholar]
- 74.Yeung M.W., Wang S., van de Vegte Y.J., Borisov O., van Setten J., Snieder H., Verweij N., Said M.A., van der Harst P. Twenty-Five Novel Loci for Carotid Intima-Media Thickness: A Genome-Wide Association Study in >45 000 Individuals and Meta-Analysis of >100 000 Individuals. Arterioscler. Thromb. Vasc. Biol. 2022;42:484–501. doi: 10.1161/ATVBAHA.121.317007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bai X., Lenhart K.C., Bird K.E., Suen A.A., Rojas M., Kakoki M., Li F., Smithies O., Mack C.P., Taylor J.M. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat. Commun. 2013;4:2910. doi: 10.1038/ncomms3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Evangelou E., Warren H.R., Mosen-Ansorena D., Mifsud B., Pazoki R., Gao H., Ntritsos G., Dimou N., Cabrera C.P., Karaman I., et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marx C., Novotny J., Salbeck D., Zellner K.R., Nicolai L., Pekayvaz K., Kilani B., Stockhausen S., Bürgener N., Kupka D., et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood. 2019;134:1859–1872. doi: 10.1182/blood.2019000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fernández-Gallego N., Castillo-González R., Méndez-Barbero N., López-Sanz C., Obeso D., Villaseñor A., Escribese M.M., López-Melgar B., Salamanca J., Benedicto-Buendía A., et al. The impact of type 2 immunity and allergic diseases in atherosclerosis. Allergy. 2022;77:3249–3266. doi: 10.1111/all.15426. [DOI] [PubMed] [Google Scholar]
- 79.Knutsson A., Björkbacka H., Dunér P., Engström G., Binder C.J., Nilsson A.H., Nilsson J. Associations of Interleukin-5 With Plaque Development and Cardiovascular Events. JACC. Basic Transl. Sci. 2019;4:891–902. doi: 10.1016/j.jacbts.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wenderfer S.E., Slack J.P., McCluskey T.S., Monaco J.J. Identification of 40 genes on a 1-Mb contig around the IL-4 cytokine family gene cluster on mouse chromosome 11. Genomics. 2000;63:354–373. doi: 10.1006/geno.1999.6100. [DOI] [PubMed] [Google Scholar]
- 81.Chang L., Yang H.-W., Lin T.-Y., Yang K.D. Perspective of immunopathogenesis and immunotherapies for kawasaki disease. Front. Pediatr. 2021;9 doi: 10.3389/fped.2021.697632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laporte J.C., Moore P.E., Baraldo S., Jouvin M.H., Church T.L., Schwartzman I.N., Panettieri R.A., Kinet J.P., Shore S.A. Direct effects of interleukin-13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am. J. Respir. Crit. Care Med. 2001;164:141–148. doi: 10.1164/ajrccm.164.1.2008060. [DOI] [PubMed] [Google Scholar]
- 83.Garrido-Martín D., Borsari B., Calvo M., Reverter F., Guigó R. Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun. 2021;12:727. doi: 10.1038/s41467-020-20578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li Y.I., van de Geijn B., Raj A., Knowles D.A., Petti A.A., Golan D., Gilad Y., Pritchard J.K. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–604. doi: 10.1126/science.aad9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rebelo S., da Cruz E Silva E.F., da Cruz E Silva O.A.B. Genetic mutations strengthen functional association of LAP1 with DYT1 dystonia and muscular dystrophy. Mutat. Res. Rev. Mutat. Res. 2015;766:42–47. doi: 10.1016/j.mrrev.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 86.Liu B., Gao W., Sun W., Li L., Wang C., Yang X., Liu J., Guo Y. Promoting roles of long non-coding RNA FAM83H-AS1 in bladder cancer growth, metastasis, and angiogenesis through the c-Myc-mediated ULK3 upregulation. Cell Cycle. 2020;19:3546–3562. doi: 10.1080/15384101.2020.1850971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Han Y., Wang B., Cho Y.S., Zhu J., Wu J., Chen Y., Jiang J. Phosphorylation of ci/gli by fused family kinases promotes hedgehog signaling. Dev. Cell. 2019;50:610–626.e4. doi: 10.1016/j.devcel.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goruppi S., Procopio M.-G., Jo S., Clocchiatti A., Neel V., Dotto G.P. The ULK3 Kinase Is Critical for Convergent Control of Cancer-Associated Fibroblast Activation by CSL and GLI. Cell Rep. 2017;20:2468–2479. doi: 10.1016/j.celrep.2017.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hardwick S.A., Hu W., Joglekar A., Fan L., Collier P.G., Foord C., Balacco J., Lanjewar S., Sampson M.M., Koopmans F., et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat. Biotechnol. 2022;40:1082–1092. doi: 10.1038/s41587-022-01231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gupta I., Collier P.G., Haase B., Mahfouz A., Joglekar A., Floyd T., Koopmans F., Barres B., Smit A.B., Sloan S.A., et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018;36:1197–1202. doi: 10.1038/nbt.4259. [DOI] [PubMed] [Google Scholar]
- 91.Lempiäinen H., Brænne I., Michoel T., Tragante V., Vilne B., Webb T.R., Kyriakou T., Eichner J., Zeng L., Willenborg C., et al. Network analysis of coronary artery disease risk genes elucidates disease mechanisms and druggable targets. Sci. Rep. 2018;8:3434. doi: 10.1038/s41598-018-20721-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wojcik G.L., Graff M., Nishimura K.K., Tao R., Haessler J., Gignoux C.R., Highland H.M., Patel Y.M., Sorokin E.P., Avery C.L., et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Joshi A., Mayr M. Aptamers They Trust: The Caveats of the SOMAscan Biomarker Discovery Platform from SomaLogic. Circulation. 2018;138:2482–2485. doi: 10.1161/CIRCULATIONAHA.118.036823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Katz D.H., Tahir U.A., Bick A.G., Pampana A., Ngo D., Benson M.D., Yu Z., Robbins J.M., Chen Z.-Z., Cruz D.E., et al. Whole genome sequence analysis of the plasma proteome in black adults provides novel insights into cardiovascular disease. Circulation. 2022;145:357–370. doi: 10.1161/CIRCULATIONAHA.121.055117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kachuri L., Chatterjee N., Hirbo J., Schaid D.J., Martin I., Kullo I.J., Kenny E.E., Pasaniuc B., Witte J.S., et al. Polygenic Risk Methods in Diverse Populations (PRIMED) Consortium Methods Working Group. Principles and methods for transferring polygenic risk scores across global populations. Nat. Rev. Genet. 2023;25:8–25. doi: 10.1038/s41576-023-00637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liao W.-W., Asri M., Ebler J., Doerr D., Haukness M., Hickey G., Lu S., Lucas J.K., Monlong J., Abel H.J., et al. A draft human pangenome reference. Nature. 2023;617:312–324. doi: 10.1038/s41586-023-05896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou W., Kanai M., Wu K.-H.H., Rasheed H., Tsuo K., Hirbo J.B., Wang Y., Bhattacharya A., Zhao H., Namba S., et al. Global Biobank Meta-analysis Initiative: Powering genetic discovery across human disease. Cell Genom. 2022;2 doi: 10.1016/j.xgen.2022.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Polygenic Risk Score Task Force of the International Common Disease Alliance Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps. Nat. Med. 2021;27:1876–1884. doi: 10.1038/s41591-021-01549-6. [DOI] [PubMed] [Google Scholar]
- 99.Browning S.R., Waples R.K., Browning B.L. Fast, accurate local ancestry inference with FLARE. Am. J. Hum. Genet. 2023;110:326–335. doi: 10.1016/j.ajhg.2022.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mohammadi P., Castel S.E., Brown A.A., Lappalainen T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 2017;27:1872–1884. doi: 10.1101/gr.216747.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Long E., García-Closas M., Chanock S.J., Camargo M.C., Banovich N.E., Choi J. The case for increasing diversity in tissue-based functional genomics datasets to understand human disease susceptibility. Nat. Commun. 2022;13:2907. doi: 10.1038/s41467-022-30650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim-Hellmuth S., Aguet F., Oliva M., Muñoz-Aguirre M., Wucher V., Kasela S., Castel S.E., Hamel A.R., Viñuela A., Roberts A.L., et al. Cell type specific genetic regulation of gene expression across human tissues. bioRxiv. 2019 doi: 10.1126/science.aaz8528. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maria M., Pouyanfar N., Örd T., Kaikkonen M.U. The Power of Single-Cell RNA Sequencing in eQTL Discovery. Genes. 2022;13 doi: 10.3390/genes13030502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kavousi M., Bos M.M., Barnes H.J., Lino Cardenas C.L., Wong D., Lu H., Hodonsky C.J., Landsmeer L.P.L., Turner A.W., Kho M., et al. Multi-ancestry genome-wide analysis identifies effector genes and druggable pathways for coronary artery calcification. Nat. Genet. 2023;55:1651–1664. doi: 10.1038/s41588-023-01518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Franceschini N., Giambartolomei C., de Vries P.S., Finan C., Bis J.C., Huntley R.P., Lovering R.C., Tajuddin S.M., Winkler T.W., Graff M., et al. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat. Commun. 2018;9:5141. doi: 10.1038/s41467-018-07340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pan H., Xue C., Auerbach B.J., Fan J., Bashore A.C., Cui J., Yang D.Y., Trignano S.B., Liu W., Shi J., et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation. 2020;142:2060–2075. doi: 10.1161/CIRCULATIONAHA.120.048378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alsaigh T., Evans D., Frankel D., Torkamani A. Decoding the transcriptome of atherosclerotic plaque at single-cell resolution. bioRxiv. 2020 doi: 10.1038/s42003-022-04056-7. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hu Z., Liu W., Hua X., Chen X., Chang Y., Hu Y., Xu Z., Song J. Single-Cell Transcriptomic Atlas of Different Human Cardiac Arteries Identifies Cell Types Associated With Vascular Physiology. Arterioscler. Thromb. Vasc. Biol. 2021;41:1408–1427. doi: 10.1161/ATVBAHA.120.315373. [DOI] [PubMed] [Google Scholar]
- 110.Zheng X., Levine D., Shen J., Gogarten S.M., Laurie C., Weir B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Browning B.L., Zhou Y., Browning S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018;103:338–348. doi: 10.1016/j.ajhg.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Castel S.E., Mohammadi P., Chung W.K., Shen Y., Lappalainen T. Rare variant phasing and haplotypic expression from RNA sequencing with phASER. Nat. Commun. 2016;7 doi: 10.1038/ncomms12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van de Geijn B., McVicker G., Gilad Y., Pritchard J.K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin A.R., Gignoux C.R., Walters R.K., Wojcik G.L., Neale B.M., Gravel S., Daly M.J., Bustamante C.D., Kenny E.E. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet. 2017;100:635–649. doi: 10.1016/j.ajhg.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Delaneau O., Ongen H., Brown A.A., Fort A., Panousis N.I., Dermitzakis E.T. A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 2017;8 doi: 10.1038/ncomms15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kichaev G., Roytman M., Johnson R., Eskin E., Lindström S., Kraft P., Pasaniuc B. Improved methods for multi-trait fine mapping of pleiotropic risk loci. Bioinformatics. 2017;33:248–255. doi: 10.1093/bioinformatics/btw615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 123.Yin L., Zhang H., Tang Z., Xu J., Yin D., Zhang Z., Yuan X., Zhu M., Zhao S., Li X., Liu X. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Dev. Reprod. Biol. 2021;19:619–628. doi: 10.1016/j.gpb.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 125.Stegle O., Parts L., Piipari M., Winn J., Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012;7:500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li Y.I., Knowles D.A., Humphrey J., Barbeira A.N., Dickinson S.P., Im H.K., Pritchard J.K. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018;50:151–158. doi: 10.1038/s41588-017-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maples B.K., Gravel S., Kenny E.E., Bustamante C.D. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 2013;93:278–288. doi: 10.1016/j.ajhg.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Delaneau O., Howie B., Cox A.J., Zagury J.-F., Marchini J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 2013;93:687–696. doi: 10.1016/j.ajhg.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Auwera G.A.V. der, O’Connor B.D. 1st ed. O’Reilly Media; 2020. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. [Google Scholar]
- 133.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jun G., Flickinger M., Hetrick K.N., Romm J.M., Doheny K.F., Abecasis G.R., Boehnke M., Kang H.M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 2012;91:839–848. doi: 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee S., Lee S., Ouellette S., Park W.-Y., Lee E.A., Park P.J. NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res. 2017;45:e103. doi: 10.1093/nar/gkx193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kumasaka N., Knights A.J., Gaffney D.J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 2016;48:206–213. doi: 10.1038/ng.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhong Y., Perera M.A., Gamazon E.R. On using local ancestry to characterize the genetic architecture of human traits: genetic regulation of gene expression in multiethnic or admixed populations. Am. J. Hum. Genet. 2019;104:1097–1115. doi: 10.1016/j.ajhg.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cotto K.C., Feng Y.-Y., Ramu A., Skidmore Z.L., Kunisaki J., Conrad D.F., Lin Y., Chapman W., Uppaulri R., Govindan R. RegTools: Integrated analysis of genomic and transcriptomic data for discovery of splicing variants in cancer. bioRxiv. 2023 doi: 10.1038/s41467-023-37266-6. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.GTEx Consortium. Laboratory, Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA. Biospecimen Collection Source Site—NDRI Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Franzén O., Ermel R., Cohain A., Akers N.K., Di Narzo A., Talukdar H.A., Foroughi-Asl H., Giambartolomei C., Fullard J.F., Sukhavasi K., et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016;353:827–830. doi: 10.1126/science.aad6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhu Y., Orre L.M., Zhou Tran Y., Mermelekas G., Johansson H.J., Malyutina A., Anders S., Lehtiö J. Deqms: A method for accurate variance estimation in differential protein expression analysis. Mol. Cell. Proteomics. 2020;19:1047–1057. doi: 10.1074/mcp.TIR119.001646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Series B Stat. Methodol. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nasser J., Bergman D.T., Fulco C.P., Guckelberger P., Doughty B.R., Patwardhan T.A., Jones T.R., Nguyen T.H., Ulirsch J.C., Lekschas F., et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data
All raw and processed bulk RNA-sequencing data are available in the Gene Expression Omnibus database (GEO: GSE225650). Low-pass whole-genome sequencing-based genotyping data are available on dbGaP: phs002855.v1.p1. The full summary statistics for the mixQTL eQTL analyses (full sample as well as subsamples for sensitivity analyses), as well as the local ancestry eQTL and the sQTL analyses are available on zenodo: https://doi.org/10.5281/zenodo.7992146
The single-cell RNA-seq datasets from coronary and carotid artery were re-analyzed and integrated from the original datasets available through GEO: GSE131778, GSE155512, GSE159677, and zenodo: https://doi.org/10.5281/zenodo.6032099.109 The raw and processed single-nucleus ATAC-seq datasets are available through GEO: GSE175621 and GEO: GSE188422. The reprocessed and analyzed human scRNA-seq datasets are also available on PlaqView (https://plaqview.com). GTEx gene expression and eQTL data were obtained from the v8 portal website (https://gtexportal.org). STARNET gene expression, eQTL, and clinical trait enrichment data were obtained from dbGaP: phs001203.v2.p1 and are also available at http://starnet.mssm.edu. The HCASMC ATAC-seq and H3K27ac HiChIP data used to calculate ABC scores are available through GEO: GSE113348 and GEO: GSE101498).
Code
All custom scripts used to generate the results are available on GitHub: https://github.com/MillerLab-CPHG/CAD_QTL and zenodo: https://doi.org/10.5281/zenodo.10095581. Specific parameters and versions for published software tools are also included in the key resources table and method details.