

Abstract
Introduction
Chronic hepatitis C infection can result in insulin resistance (IR). We have previously shown that it occurs through the interaction of pathways for glucose homeostasis, insulin signaling, and autophagy. But it is not known how soon the pathways are activated and how IR is related to the signals generated by catabolic and anabolic conditions occurring in infected cells. We have extended our studies to a cell culture system mimicking acute infection and to downstream pathways involving energy-sensor AMPK and nutrient-sensor mTOR that are active in catabolic and anabolic processes within the infected cells.
Methods
Huh7 liver cells in culture were infected with hepatitis C virus (HCV). We performed proteomics analysis of key proteins in infected cells by Western blotting and IP experiments, with or without IFNα exposure as a component of conventional therapeutic strategy.
Results
We present evidence that (a) IRS-1 Ser312, Beclin-1, protein conjugate Atg12-Atg5 or GS Ser641 are up-regulated early in infection presumably by activating the same pathways as utilized for persistent infection; (b) Bcl-XL, an inhibitor of both autophagy and apoptosis, is present in a core complex with IRS-1 Ser312 and Beclin-1 during progression of IR; (c) AMPK level remains about the same in infected cells where it is activated by phosphorylation at Thr172 concomitant with increased autophagy, a hallmark of catabolic conditions; (d) an mTOR level that promotes anabolism is increased rather than decreased under an expanded autophagy; (e) hypophosphorylation of translational repressor 4E-BP1 downstream of mTOR is suggestive of reduced protein synthesis; and (f) β-catenin, is up-regulated but not phosphorylated suggesting indirectly our previous contention that its kinase, GSK-3β, is mostly in an inactive state.
Conclusion
We report that in the development of IR following chronic infection, anabolic and catabolic pathways are activated early, and the metabolic interaction occurs possibly in a core complex with IRS-1 Ser312, Beclin-1, and autophagy inhibitor Bcl-XL. Induction of autophagy is usually controlled by a two-edged mechanism acting in opposition under anabolic and catabolic conditions by AMPK/mTOR/4E-BP1 pathway with GSK-3β-mediated feedback loops. However, we have observed an up-regulation of mTOR along with an up-regulation of AMPK caused by HCV infection is a deviation from the normal scenario described above which might be of therapeutic interest.
Keywords: Hepatitis C virus, Insulin resistance, Glucose homeostasis, Autophagy, Akt/GSK-3/mTOR pathway
Introduction
Hepatitis C virus (HCV) infection is a global health problem affecting over 170 million individuals worldwide [1]. An emerging concept considers HCV a factor in the occurrence of metabolic associated disease and its complications [2]. It induces insulin resistance (IR) that is the pathologic foundation underlying the metabolic syndrome, and is associated with steatohepatitis, cirrhosis, and possibly hepatocellular carcinoma (HCC). Epidemiological studies have established a 3-fold higher incidence of type 2 diabetes mellitus in patients chronically infected with HCV [2, 3]. Since insulin signaling is important both in HCC and in type 2 diabetes mellitus, it is of much interest to determine how this pathway is altered in HCV-infected cells.
Insulin transmits its signal through a cell surface tyrosine kinase receptor to induce tyrosine phosphorylation of two adapter proteins, insulin receptor substrates, IRS-1 and IRS-2, which then recruit and activate downstream effector molecules [4]. The immediate effect is the downstream activation of phosphatidylinositol 3-kinase (PI-3K) which is central to insulin’s regulatory action through Akt on glucose metabolism, such as acceleration of glucose transport and glycogen synthesis. This pathway is mediated by inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) [5, 6]. GSK-3 inhibits glycogen synthesis by phosphorylation of glycogen synthase (GS) at one or more of a string of serine residues at Ser652, 648, 644, and particularly at 641 [7]. Two isoforms of GSK-3, GSK-3α and GSK-3β, are broadly expressed and constitutively active in cells with distinct but overlapping tissue-specific functions, GSK-3α being more active in the liver and GSK-3β in the muscle cells [8, 9]. The GSK-3 isoforms are transiently inhibited by the protein kinase B/Akt signaling pathway through phosphorylation of Ser21 and Ser9 of GSK-3α and GSK-3β, respectively. Also, Akt is activated by phosphorylation at two different sites: Ser308 by PDK-1 and Ser473 by PDK2/mTORC2. Thus, the IRS-1/PI-3K-Akt/GSK-3 pathway plays a vital role in insulin-induced glycogen synthesis. When phosphorylation at Ser9 and Ser21 is blocked by PI-3K inhibitor (Wortmannin), insulin-induced activation of GS also is inhibited. Activation of GS by insulin is mediated by the stimulation of protein phosphatase 1 (PP-1), a Ser/Thr protein phosphatase, followed by dephosphorylation and activation of GS [10]. Along with dephosphorylation of GS, GSK-3 also dephosphorylates eukaryotic translation initiation factor 2B (elF-2B), thus stimulating both glycogen and protein synthesis [5]. Furthermore, hepatic autophagy is suppressed in the presence of IR and hyperinsulinemia by the inhibition of FOXO1-dependent expression of autophagy genes by insulin. A dysfunction of the insulin signaling pathway is expected to enhance autophagy in these cells [11]. In chronic HCV infection, we have shown that the pathways for insulin signaling and glucose homeostasis are dysregulated [12]. This is brought about by key events, such as phosphorylation of IRS-1 at Serine 312 (IRS-1 Ser312), dysregulation of the Akt pathway, inhibitory phosphorylation of glycogen synthase kinase-3β (GSK-3β Ser9), and regulation of GS phosphorylation. Although the roles of the two GSK-3 isoforms are not understood, GSK-3α is active in both HCV-infected and HCV-uninfected liver cells, whereas GSK-3β is overly phosphorylated at Ser9 and inactivated in persistently HCV-infected Huh7 cells but not in Huh7 uninfected control cells [12]. Also, active autophagy was detected in infected cells by the formation of LC3 puncta or by the increased level of Beclin-1 or the Atg5-Atg12 protein conjugate. Clearly, GSK-3β regulates and is regulated by proteins involved in IRS-1/PI-3K-Akt/GSK-3/GS or IRS-1/PI-3K-Akt/GSK-3/autophagy axes. This suggests that GSK-3 is at the center of multiple feedback loops leading to glycogen synthesis or autophagy [12, 13]. Thus, an understanding of the complex mechanistic details of these pathways is important for more affordable therapeutic development. To advance our knowledge, we want to know how early in infection the IR pathways are activated.
While the induction of autophagy during HCV infection is established, its biological role and the pathway leading to IR development is not clearly understood [12–15]. We previously observed that autophagy proteins, Beclin-1 and Atg12-Atg5, are up-regulated in HCV-infected cells and that the inhibition of autophagy by inhibitor 3-methyl adenine leads to suppression of IRS-1 Ser312 and GS Ser641 phosphorylation. These observations suggest a role of autophagy in the development of IR [12]. Along the same line, it is reported that Beclin-1 is inhibited by the anti-apoptotic protein Bcl-2 or Bcl-XL by direct binding to the BH3 domain of Beclin-1, stabilizing Beclin-1 homodimerization and inactivity due to its inability to bind other proteins while Bcl-XL maintains its anti-apoptotic function [15, 16]. Beclin-1 is phosphorylated by a kinase DAPK or by Akt [17, 18]. While this may connect the two cell death pathways, information is missing whether inhibitors of Beclin-1, such as Bcl-XL or Bcl-2, mediates IRS-1 Ser312- Beclin-1 interaction in IR development.
Two cellular proteins, AMP-activated protein kinase K (AMPK) and mammalian target of rapamycin (mTOR), are associated with autophagy [19–23]. But their interdependence and relationship with autophagy are complex because AMPK is energy-sensitive and mTOR is nutrient sensitive, therefore being controlled by catabolic and anabolic conditions, respectively [24]. AMPK is a heterotrimeric complex with one catalytic α and two regulatory β/γ subunits activated under a catabolic condition of decreasing energy status with increasing AMP/ATP ratios. GSK-3β forms a complex with the β regulatory subunit to phosphorylate AMPK α on threonine 172 (Thr172) which is required for its full activation. Phosphorylation is also mediated by several other kinases including the ubiquitously expressed liver kinase B1 (LKB1) [25]. Under energy crisis by glucose starvation, AMPK promotes autophagy by directly activating ULK-1 through phosphorylation at Ser317 and Ser777. Under nutrient sufficiency, high mTOR activity prevents ULK-1 activation directly by phosphorylating ULK-1 at Ser757 and disrupting interaction between ULK-1 and AMPK, thus inhibiting autophagy [19, 20]. mTOR forms two distinct complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 phosphorylates S6 kinase 1 (S6K1) and eukaryotic translation initiation factor 4E (eIF-4E)-binding protein (4E-BP1) to stimulate protein synthesis [26, 27], while mTORC2 phosphorylates Akt to promote cell survival and proliferation [21]. At the downstream of mTOR, 4E-BP1 inhibits cap-dependent translation by binding to eLF-4E [22, 23]. Phosphorylation of 4E-BP1 by mTOR results in the dissociation of elongation initiation factor (eIF)-4E, followed by an interaction with eIF-4G which is crucial for the assembly of translation initiation complex. There are limited data on AMPK associated with mTOR or 4E-BP1 in HCV infection except that it generates oxidative stress and can provoke ATP depletion, leading to an increase in the amount of AMP bound to AMPK [21, 27]. The activated AMPK also indirectly inhibits mTORC1 through phosphorylation of tuberous sclerosis 2 (TSC2) and of raptor thus inhibiting autophagy [23, 24]. Knowledge of the AMPK/mTOR/4E-BP1 pathway will elucidate their complex biochemical functions in association of protein synthesis and autophagy during transition of catabolic and anabolic conditions in IR development.
GSK-3β, being a master protein regulating many feedback pathways, is of special interest. Besides insulin signaling, another signaling pathway of Wnt/β-catenin is known to impinge on GSK-3β. This pathway is important in intracellular adhesion, embryonic development, and in promoting transcription of many oncogenes leading to tumor progression of several cancers, such as HCC [28]. There is a crosstalk between insulin and Wnt signaling dependent on insulin receptors/LRP5 co-receptor which is of significance in IR [29]. GSK-3β is present in a protein complex that also includes β-catenin, APC, and Axin, regulating the stability of β-catenin. In the absence of Wnt, β-catenin is constantly degraded by the ubiquitin-proteasome pathway [30]. This degradation depends on the phosphorylation of β-catenin in the amino terminus by GSK-3β [31]; when the site is mutated, the level of β-catenin is elevated [32]. Wnt signaling inhibits β-catenin phosphorylation to increase cytosolic β-catenin, followed by its transportation to the nucleus to associate with TCF/LEF transcription factors and activation of Wnt/β-catenin responsive genes [32, 33]. As mentioned earlier, our previous study shows that in HCV-IR, GSK-3β is overly phosphorylated at Ser9, leaving this kinase mostly inactive. Understanding the level of β-catenin may provide additional support of the phosphorylation status of GSK-3β, the master protein interacting with many other proteins, in HCV infection.
Using a cell culture system that mimics the early stage of HCV infection, we extend our previous study to determine whether the metabolic pathways of glucose homeostasis, insulin signaling, and autophagy are activated early in acute infection. Autophagy being clearly a hallmark of the catabolic condition, our major goal was to understand the basis and regulation of autophagy in IR development by the interplay of two oppositely acting anabolic and catabolic conditions through the IRS-1/PI-3K/Akt/GSK-3/AMPK/mTOR/4E-BP1 axis. Also, we wish to address the status of autophagy and apoptosis inhibitors (Bcl-XL, Bcl-2) as well as GSK-3β in the development of HCV-IR.
Materials and Methods
Cell Lines and Plasmids
The human liver cell line Huh7 was a gift from Dr. Charles Rice and was grown in Dulbecco modified Eagle Medium with 10% fetal bovine serum and 5,000 units of penicillin and streptomycin/mL The persistently infected cellular model for long-term infection, Huh.HCV2a, has been described earlier [12]. It contains an integrated cDNA copy of the HCV genotype 2a genome in an Huh7 background that releases mature infectious virions in the media [34]. To prepare infectious virions, Huh.HCV2a cells were cultured for 4–5 days. Cell culture media was supplemented with 25 mm HEPES buffer, pH 7.5. The media was collected and centrifuged at 2,000 g at 4°C to remove cellular debris. The supernatant was concentrated using Amicon Ultra 100K-Da (Millicon, UFC 9100096). RNA was prepared following a standard procedure, and genome equivalents (GE)/mL were determined by using a quantitative real-time reverse transcription polymerase chain reaction (RT-qPCR). To convert GE/mL to international units (IU)/mL, we obtained purified, intact, noninfectious human HCV particles of known concentration (IU/mL) from ZeptoMetrix® (NATtrols, Corporate Headquarters, Buffalo, NY, USA) to prepare RNA that was diluted sequentially by 10-fold increments and used for RT-qPCR for a calibration curve to estimate IU/mL of the RNA stock used for infection. For acute infection, Huh7 cells were infected with the HCV 2a virion in cell culture media at approximately 5 × 106 GE/mL (MOI 2.5). Cells were harvested generally on day 5 for analysis. Chronic infection was carried out as described earlier [12]. When required, interferon-α treatment was carried out at 100 pg/mL for 48 h. The analysis of viral RNA in the media and its fractionation when required are the same as described earlier [12].
Antibody, Chemicals, and Reagents
Antibodies to AMPK, phosphorylated (p)-AMPK, β-catenin, p-β-catenin, mTOR and p-mTOR, Akt, p-Akt Ser473, GSK-3β Ser9, GSK-3α Ser21, IRS-1, and GS Ser641 were obtained from Cell Signaling Technology (Danvers, MA, USA). IRS-1 Ser312 antibody was obtained from Abcam (Cambridge, MA, USA). One single secondary antibody (Cat # 7074) was used against all anti-rabbit primary antibody from Cell Signaling Technology.
Western Blotting and Immunoprecipitation Experiments
The Western blotting experiment was described in detail in our earlier publication [12]. Experiments were carried out at two different protein concentrations, as indicated. We used AMPK control cell extracts (cat # 9158, Cell Signaling Technology, Danvers, MA, USA) from C2C12 cells grown in the presence of serum (S+) or serum starved (S−) as controls. S− and S+ controls expressed low and elevated levels of both AMPK and pAMPK, respectively.
For IP experiments, 100–150 μg of total protein in the lysate was incubated with the primary antibody. After 2 h of incubation, 20 μL of agarose beads were added and mixed overnight in the cold room. The pellets were washed three times with PBS, fractionated on a 10% gel, and transferred to nitrocellulose membranes. The rest of the procedure was identical to that for Western blotting.
Results
IR Pathways Are Activated in the Early Stages of HCV Infection (Acute Phase) Along with Markers of Autophagy
Previously, we showed that the pathways for insulin signaling and glucose homeostasis were dysregulated in a liver cell line persistently infected with HCV that mimics chronic infection [12]. This was associated with an increased level of autophagy, where the autophagy regulatory protein Beclin-1 forms a complex with IRS-1 Ser312 in the Huh7 cells chronically infected with HCV. To investigate how soon the pathway is activated, we developed an acute infection system by infecting Huh7 cells with infectious HCV obtained from persistently infected Huh.HCV2a cells at about 107 copies of genomic RNA/mL. The level of viral RNA secreted in the media was assayed or fractionated as described earlier [12]. In parallel, the level of gene expression was evaluated by Western blot analysis.
Our Western blot analysis (Fig. 1a) shows that in control Huh7 cells, a very faint band both for IRS-1 and IRS-1 Ser312 is present, and those are overexpressed by about 200% in HCV-infected cells. The ratio of IRS-1 Ser312 to IRS-1 increased by about 40% in HCV-infected cells. In the downstream PI-3K/Akt pathway (Fig. 1b), we measured the phosphorylation of Akt Ser473 that is crucial for activation of many pathways in cell survival and proliferation [35]. As shown, phosphorylation of Akt Ser473 is increased by about 300% in the HCV-infected cells compared to uninfected cells. This increase is more impressive than was observed in the persistently infected cells [12]. We also determined the phosphorylation of GSK-3 isoforms (GSK-3α Ser21 and GSK-3β Ser9). GSK-3α is not phosphorylated in acutely infected cells, like what was observed in persistently infected cells [12]. This was further confirmed in an additional control experiment by using lysate from Jurkat cells in which Akt Ser473 and subsequently GSK-3α Ser21 phosphorylation is induced by curcumin as a positive control (online suppl. Fig. S1; for all online suppl. material, see https://doi.org/10.1159/000535787). In contrast, GSK-3β is present predominantly in an inactivated state. This isoform is also inactivated in Huh7 control cells in varying degrees, depending on the time in culture. This favors a modulation of the GSK-3 mediated processes. Analysis of our results (Fig. 1b) shows that in acutely infected cells, normalized band intensities for Akt Ser473 with respect to uninfected control cells was 3.8, whereas the same ratio for GSK-3β Ser9 was 0.9. The remarkable difference in the GSK-3β Ser9 ratio is due to high-level expression of this protein in both acutely infected and uninfected Huh7 cells. We also sought the status of GS that is phosphorylated at Ser641 as an important landmark of glucose metabolism and found that this phosphorylation is increased in HCV-infected cells by about 10–20-fold in different experiments, whereas in uninfected Huh7 cells, it is barely detectable (Fig. 1c). Earlier reports suggest that GSK-3β is responsible for phosphorylation of GS at Ser641 [36, 37], but it is not clear how increased inactivation of GSK-3β is correlated with increased phosphorylation of GS Ser641 (Fig. 1b, c).
Fig. 1.

Western blot analyses of the key proteins of insulin signaling, glucose homeostasis, and autophagy pathways in acute infection. Huh7 cells were infected with infectious HCV secreted in the media from persistently infected cells (Huh.HCV.2a). Infection was carried out at an HCV genome equivalence (GE) of 5 × 106 and at a GE/cell ratio of 100 (MOI approximately 2.5). Cell lysate was prepared at 48 h for Western blot analyses. Two different amounts of lysate (50 and 100 μg) indicated above each lane were fractionated on a 10% PAGE IRS-1 and IRS-1 Ser312 (a); Akt Ser473, GSK-3α Ser21, GSK-3β Ser9 (b); GS Ser641 (c); autophagy proteins Atg12-Atg5 conjugates and Beclin-1 (d). Actin is the loading control in Western blot. Band intensity was quantified and normalized with respect to this actin control as indicated beneath each lane. When appropriate, further normalization was carried out with respect to uninfected cells.
We also determined whether the autophagy marker Beclin-1 and the Atg12-Atg5 conjugate are up-regulated as observed in persistently infected cells and found that cell lysates prepared from Huh7 cells acutely infected with HCV showed an increase of about 200% in these markers (Fig. 1d). This suggests that the pathway to autophagy also is activated early in infection in our system.
Anti-Apoptotic Bcl-XL Is Present in a Multiprotein Immunocomplex with IRS-1 Ser312 and Beclin-1
Two anti-apoptotic proteins of the BH3-only family, Bcl-2 and Bcl-XL, are inhibitors of autophagy by complex formation with Beclin-1. Our Western blot analyses show that in acutely HCV-infected cells, Bcl-XL is up-regulated about 2-fold. In contrast, Bcl-2 protein is barely detectable, if at all (online suppl. Fig. S2). Since our previous study shows that Beclin-1 forms a complex with IRS-1 Ser312, we sought to determine whether the autophagy regulator Bcl-XL participates in this interaction by examining its presence in the immunoprecipitated immune complex. Lysates from both infected and uninfected cells were immunoprecipitated with Bcl-XL, and the blot was probed with antibodies against Bcl-XL, IRS-1 Ser312, and Beclin-1. The results show a Bcl-XL band in the IP complex and in the input control (Fig. 2a). The bands for Beclin-1 and IRS-1 Ser312 also are clearly present when probed with individual antibodies. In parallel experiments, when the lysates were immunoprecipitated with IRS-1 Ser312 or Beclin-1 antibodies individually, bands for all three proteins were present (Fig. 2b, c). When the lysates were immunoprecipitated with Bcl-2 or with IRS-1 antibody alone, no specific bands were visualized in the autoradiogram (not shown). These results suggest that the autophagy inhibitor Bcl-XL is present in the immunocomplex with Beclin-1 and IRS-1 Ser312, implicating a direct or indirect interaction of Bcl-XL with the other two proteins.
Fig. 2.

Co-immunoprecipitation of Bcl-XL, IRS-1 Ser312, and Beclin-1. Two different amounts (100 and 150 μg) of protein lysate (indicated above each lane) from infected and uninfected cells were immunoprecipitated with antibody against Bcl-XL (a), IRS-1 Ser312 (b), and Beclin-1 (c). The lysates were fractionated on a 10% PAGE and probed individually with all three antibodies. Respective cell lysates were included in each gel as a control for a direct Western blot. Specific protein bands, as well as those for IgG, are marked. The IRS-1 Ser12 and Beclin-1 bands in (b, c) were published previously but presented again for comparison [12]. The reduced intensity of the band for Bcl-XL in b is probably due to the loss of precipitated material in one lane during the course of processing of the sample.
Regulation of AMPK-mTOR Pathway
It is known that in response to nutrient stimuli, the nutrient-sensitive kinase, mTOR, integrates a signaling network to promote anabolism by blocking catabolic processes, thus regulating autophagy negatively. Since our previous study suggested that induction of autophagy is a contributory factor in IR, we wanted to elucidate how the AMPK-mTOR pathway is regulated in HCV infection in Huh7 cells. Considering that mTOR is a downstream target of AMPK under energy shortage conditions, we determined the status of AMPK phosphorylation and mTOR expression and their effect on downstream targets. To accomplish this, we used both acutely (HCVa) and persistently (HCVp) infected HCV cell models side by side when appropriate. Both types of cells were treated with interferon-α (IFNα) to understand its therapeutic significance. Lysates from serum-starved C2C12 muscle cells (S−), where AMPK is overexpressed, served as positive controls. AMPK levels were similar in infected and uninfected cells, but phosphorylation at Thr172 (p-AMPK) led to a dramatic 2–4-fold increase in both acute and persistent infection (lanes marked as HCVa and HCVp) (Fig. 3a). This increase is consistent with the AMPK-induced activation of catabolism through ULK-1 phosphorylation with increased autophagic induction. While unphosphorylated AMPK level in acute or persistently infected cells remains about the same in the presence of IFNα, Thr172 phosphorylation of AMPK (pAMPK) is inhibited by this treatment, implicating the significance of AMPK phosphorylation (lane marked +IFNα, Fig. 3a). Our results show that the expression of mTOR is negligible in uninfected cells but increases 2–4-fold in cells infected both acutely and persistently (Fig. 3b). mTOR also is phosphorylated at Ser2448 and is inhibited by IFNα, emphasizing the therapeutic significance of this antiviral compound (Fig. 3c).
Fig. 3.

Role of AMPK, mTOR, and 4E-BP1 in cellular energy homeostasis. Western blot analysis for expression and phosphorylation of AMPK (a), mTOR (b, c), 4E-BP1 (d). Two different amounts of lysate (50 and 75 μg) from infected (Huh.HCV) and uninfected (Huh) cells were analyzed. In a, lysates from acutely and persistently infected cells were marked as Huh.HCVa and Huh.HCVp, respectively, labeled as (A) and (P) in (b), and loaded side by side for a comparison. Interferon-treated cell lysate was identified as +IFNα (a, c). A control cell lysate from serum-starved (S−) C2C12 muscle cells that show an elevated level of AMPK expression is included as control (Cell Signaling Technology, Danvers, MA). p-AMPK is phosphorylated AMPK at Thr172. The blots in (d) were probed with 4E-BP1 and p-4E-BP1 antibody. Normalized band intensity with respect to actin is indicated beneath each lane except in (a, c), where inhibition of p-AMPK and p-mTOR by IFNα was demonstrated. The lower intensity of the bands in the first control lane may result from a technical problem of protein transfer and processing of the blot.
It is well known that mTOR regulates 4E-BP1 expression involved in translational regulation, where mTOR phosphorylates 4E-BP1 to stimulate protein synthesis [26]. Our Western blot analysis exhibits a modest decrease in the level of expression of this protein in acutely infected cells while the level of phosphorylation is decreased by about 3–5-fold (Fig. 3d). This suggests that the mTOR pathway could reduce the overall protein synthesis via 4E-BP1. While the induction of autophagy is accompanied by negative regulation by mTOR in a cellular context [19, 20], our data suggest that during suppression of hepatic autophagy in HCV-infected cells, IR may be independent of mTOR.
Wnt/β-Catenin Signaling Pathway in HCV Infection
Whereas GSK-3β phosphorylation appears to be critical in GS regulation and glucose homeostasis, we have shown previously that it is overly inactivated by phosphorylation at Ser9 in the presence of HCV IR. For additional support, we used another well-known substrate, β-catenin, and the Wnt/β-catenin pathway, that also controls cell growth and proliferation. Figure 4 shows Western blot analyses from Huh7 uninfected and HCV-infected cells at two different protein concentrations. Lysates from supplemented serum-plus (S+) and serum-minus (S−) C2C12 muscle cells were used as controls since under S− conditions AMPK is overexpressed, regulating other downstream targets, whereas S+ cells will have opposite effects. An increased level of β-catenin is shown in S− cells compared to S+ cells showing much lower level of β-catenin expression. This serves as positive and negative controls. When Huh7 cells were infected with HCV, the β-catenin level increased approximately 1.30–2.0-fold, normalized with respect to the control Huh7 cells. With antibody specific for phosphorylated β-catenin, no band was detected in Huh7 infected or uninfected cells indicating that β-catenin is either not phosphorylated or it is degraded following phosphorylation. The increase of β-catenin level, however, suggests that β-catenin is not phosphorylated by GSK-3β, rather it is transported to the nucleus for regulating other genes (upper panel). The presence of an inactive form of GSK-3β thus further supports our earlier conclusion.
Fig. 4.

Western blot analysis for Wnt/β-catenin (β-cat) using specific antibodies for phosphorylated or nonphosphorylated β-catenin. Lysates from acutely infected cells were prepared and analyzed. Two different amounts of lysates were fractionated (75 μg and 100 μg) as marked above each lane. Lysates from C2C12 muscle cells were induced by serum (S+) or were serum-starved (S−) and used as markers for low- and high-level expression for AMPK and included as controls. Band intensity is indicated beneath each lane. A weak and a strong band were present in the control lanes with an antibody specific for β-catenin and p-β-catenin.
Discussion
Previously, we showed that the normal pathways for glucose homeostasis and insulin signaling were dysregulated in liver cells chronically infected with HCV contributing to the development of IR [12]. The overlapping pathways in this process were activated under anabolic conditions characterized by reduced glucose uptake and the expression and phosphorylation of IRS-1, Akt Ser473, Akt Thr308, GSK-3 Ser9, and GS Ser641. Autophagy, a catabolic process inside the cells, plays a significant role as exemplified by the overexpression of autophagy initiator proteins such as Beclin-1, Atg12-Atg5, and LC3-PE conjugates. Additionally, Beclin-1 is complexed with IRS-1 Ser312 and apoptosis regulator Bcl-XL leading to the formation of a core complex during IR progression. However, it was not determined how early in infection the pathways are activated and what biochemical mechanisms are essential. Using a cell culture system that mimics early infection, we report that many of the changes observed in chronically infected cells are initiated early in infection with a notable difference observed in the phosphorylation status of several key proteins (Fig. 3, 4). Additionally, we extend our study along the AMPK-mTOR-4E-BP-1 axis, encompassing both catabolic and anabolic conditions in HCV infection. We also examined the involvement of GSK-3β in the Wnt/β-catenin pathway that is also known to be connected to IR. The absence of β-catenin phosphorylation provides additional support indirectly for our previous contention that GSK-3β remains predominantly in an inactive form during IR development [12].
Although acute and chronic HCV infection is better defined by a time-dependent manifestation of clinical criteria, it is defensible to set up an approximate comparison in a cell culture system. At about day 5 postinfection in Huh7 cells, the level of IRS-1 and IRS-1 Ser312 is increased about 2-fold like what we observed in persistent or chronic HCV infection. Although IRS-1 overexpression and phosphorylation have been reported in many cellular contexts including cancer [38–41], several studies have also reported down-regulation and/or degradation of IRS-1 as an alternate mechanism for the disruption of insulin signaling in IR [42–46]. This could be mediated by diverse mechanisms including targeted phosphorylation at Ser312 or other serine residues [44, 46, 47], down-regulation of SOCS-1 signaling [48], inhibition of IRS-1 by suppressing PTEN [49], ubiquitination of hyperphosphorylated IRS-1 through Cullin-RING E3 ubiquitin ligase 7 (CRL7] [50], or by an inappropriate activation of the TSC/Rheb/mTOR/S6K cassette [51]. Three stress kinases (JNK-1, p38, and MAPK) are elevated in HCV-infected cells to varying levels (15–217%) when compared to uninfected cells (unpublished observation), of which JNK is known to phosphorylate IRS-1 at Ser312 [52]. Since many target proteins, such as GSK-3, GS, Akt, or mTOR, are phosphorylated by multiple kinases, the significance of the up-regulation of p38 and MAPK is a matter of separate study outside the scope of the present work.
Phosphorylation of Akt Ser473 is central to the activation of the IRS-1/PI-3K/Akt/GSK-3/GS pathway. Previously, we reported a slight increase in Akt Ser473 phosphorylation in persistently infected cells (about 2-fold), but in acute infection, significantly higher levels of Akt phosphorylation (3.8-fold) occurred with respect to uninfected control cells. GSK-3β Ser9 is phosphorylated in both acute (Fig. 1b) and chronic infection in our previous study [12]. In chronic infection, an exceptionally low level of GSK-3β Ser9 was visually observed in control Huh7 cells, in contrast to more than a 5-fold increase of the same protein in control cells in acute infection. The GSK-3β Ser9 ratio in acute infection thus remains at a low level of 0.9 due to a comparable level of expression in the control cells. This increase in the uninfected control cells results from the activation of the anabolic pathway (PI-3K/Akt) in rapidly replicating cells due to the release of cellular genes under the negative control of GSK-3β [5, 6, 12]. In the absence of phosphorylation, the GSK-3α Ser21 isoform is active both in acute and in persistent infection (online suppl. Fig. S1). Both GSK-3 isoforms are substrates for Akt which, in turn, can phosphorylate GS [6]. In those instances where glycogen synthesis is considered an end point of glucose metabolism, regulation of GS by Ser641 phosphorylation may not only result from GSK-3β phosphorylation at this site but from alternate pathways for kinases and phosphatases as well. For example, AMPK inhibits glycogen synthesis through inhibitory phosphorylation of GS [53, 54]. It is conceivable that the IR pathway is regulated by several feedback loops under the control of GSK-3, as GSK-3 regulates and is regulated by IRS-1, Akt and AMPK or mTOR as illustrated in Figure 5.
Fig. 5.

Schematic diagrams show potential interaction between the pathways for glucose homeostasis (IRS-1-PI-3K-GSK-3-GS), insulin signaling (IRS-1-PI-3K-GSK-3), and autophagy (IRS-1-PI-3K-GSK-3-mTOR) under anabolic and catabolic conditions. Catabolic and anabolic pathways are shown in different colors. Arrow indicates activation and a bar repression.
Also, GSK-3β is known to be involved in the Wnt/β-catenin pathway, as GSK-3β-mediated phosphorylation of β-catenin triggers proteasomal degradation of β-catenin [29–32]. Our Western blotting did not detect a band specific for phosphorylated β-catenin in the infected (Huh.HCV) or uninfected (Huh7) cells (Fig. 4). But a band was present in the control lane with serum-starved (S−) C2C12 lysate which induces β-cat and p-β-cat as opposed to S+ cells. It suggests that β-cat is either not phosphorylated or it is totally degraded following phosphorylation. But an increase of total β-cat level by 3–5-fold (upper panel) based on band intensity in the same lysate favors the absence of phosphorylation but not for degradation. We are aware that another way to confirm this finding is to examine the status of the level of the various isoforms of the Wnt proteins along with their receptors and co-receptors, but this approach was outside the scope of the present study. Although in early infection, the difference in GSK-3β Ser9 level in infected and uninfected cells is not significant (Fig. 1b), absence of β-catenin phosphorylation supports our contention that GSK-3β is predominantly present in an inactive form during HCV-IR development [12]. Since it is known that AMPK is down-regulated by GSK-3β [24], an increase in AMPK level in our study may, at least partially, come from inactivation of GSK-3β. It is intriguing to imagine that the inactive fraction of GSK-3β is progressively increased during development of IR. This is supported by a finding that in HCC cells, HCV core protein increases and stabilizes β-catenin through inactivation of GSK-3β [55].
Induction of the catabolic autophagy pathway appears to be the central biochemical mechanism for developing IR in HCV infection [12, 14]. It has been elucidated by finding elevated levels of Beclin-1 and the Atg12-Atg5 conjugate, or by the increased presence of the lipidated form of LC3 (LC3-PE) [12, 15]. Also, we previously reported that the autophagy inhibitor 3 MA suppresses phosphorylation of IRS-1 Ser312 and GS Ser641 in the insulin signaling pathway further strengthening a connection between autophagic cell death and IR development [12]. In this paper (Fig. 1d), an increase of the Atg12-Atg5 conjugate and Beclin-1 as early as 5 days postinfection is consistent with the observation from other laboratories that lipidation of LC3 occurs at day 5 or even after 4 h of postinfection [56, 57]. Recently, the structures of Beclin-1 and Bcl-XL have been studied by nuclear magnetic resonance (NMR) spectroscopy, and it is reported that Bcl-XL binds to the BH3 domain of Beclin-1 at its N-terminal and forms a heterooligomer [15, 16]. Our results now extend this observation to an interaction of the Beclin-1/Bcl-XL complex directly or indirectly with IRS-1 when phosphorylated at Ser312 (Fig. 2). A complex formation between the anti-apoptotic protein Bcl-XL and an autophagy protein Beclin-1 is a clear indication of the convergence of cell death pathways. In this complex, Beclin-1 still maintains the characteristic of BH3 protein, and Bcl-XL retains its own anti-apoptotic function [16]. The other autophagy inhibitor, Bcl-2, is present at a low level, if at all, in HCV-infected Huh7 cells under our experimental conditions (online suppl. Fig. S2). Any complex formed with Beclin-1 is undetectable by IP experiments, in contrast to a report of Bcl-2 involvement in an immortalized human hepatoma cell line [58]. It is not understood whether the autophagy inhibitor Bcl-XL remains associated with Beclin-1 or if it is dissociated by phosphorylation while inducing autophagy. Depending on the IP running condition, occasionally we observed in preliminary experiments a slow-moving band as a doublet in Western blots from chronically infected cell lysates (online suppl. Fig. S3). This could be a hyperphosphorylated fraction of Beclin-1, and this phosphorylation event in the multiprotein core complex may thus offer a clue for the dissociation of Bcl-XL to trigger autophagy. Beclin-1 is known to be phosphorylated by both DAPK and Akt [17, 18], and the former is present at an elevated level in infected cell lysates (results not shown). An interplay of Beclin-1 and Bcl-XL may alter the dynamics of cell death involving both autophagy and apoptosis in IR.
As mentioned earlier, we found an overexpression and activation of AMPK by phosphorylation at Thr172 [59] associated with an increased autophagy induction, a hallmark of catabolic conditions. But up-regulation of mTOR along with an up-regulation of AMPK caused by HCV infection is a deviation from the normal scenario in a cellular system where autophagy is negatively regulated by mTOR to promote anabolism [19, 20]. This apparent dysfunction of the normal mTOR autophagy pathway agrees with another report [58] and appears to be a characteristic of HCV infection. HCV was shown to down-regulate both the AMPK and Akt-TSC2-mTOR C1 pathway inducing autophagy by ER stress [57]. While the inhibition of the Akt-TSC-mTORC1 pathway up-regulates autophagy, inhibition of AMPK down-regulated it. The net positive regulation of autophagy in HCV infection was assigned to Akt, to override the negative effect of AMPK. There are reports that mTOR-independent autophagy inhibition by Akt is operative through Beclin-1 phosphorylation [18]. Also, HCV-induced oxidative stress activates AMPK that depletes the ATP level, activating AMPK by binding AMP [60–62]. In another study, we find an up-regulation of AMPK and autophagy markers Beclin-1 and Atg12-Atg5 conjugate in both chronic [12] and acute infection (Fig. 1d) at about day 5 which is in good agreement with another report [57], except that we observe an overexpression of AMPK. Conversely, down-regulation of AMPK is reported to facilitate lipid accumulation for enhanced HCV genome replication [63]. The basis of this discrepancy is not currently understood, but it may result from the different stages of cell growth and culture conditions.
It is intriguing to imagine that IR is broadly under the control of two interactive but opposite programs inside the cells, designated the anabolic and catabolic pathways (Fig. 5), both regulating autophagy through coordinated communication between AMPK and mTOR. The role of mTOR is exactly opposite to the effect of AMPK, but there is always a coordinated phosphorylation of ULK-1 by mTORC1 and AMPK at different sites that provides a mechanism by which cells can properly respond to a wide range of stimuli and achieve a balance between their anabolic and catabolic pathways [19, 20, 24]. Interestingly, AMPK may not be coupled directly to mTOR phosphorylation at Ser2448 since it has been questioned that Ser2448 phosphorylation is not a measure of mTOR kinase activity. Instead, the inhibitory effect on kinase is targeted by the downstream effector, such as p70S6K or 4E-BP1, in a negative feedback mechanism [64]. As mentioned earlier, AMPK can maintain the energy balance by inhibiting the effect of mTOR in two opposing pathways: (1) by activating autophagy under a catabolic condition directly by phosphorylating ULK-1 which was otherwise under negative regulation of mTOR and (2) by inhibiting autophagy by mTOR through the activation of TSC-2/Rheb or by phosphorylation of raptor, an essential component of mTORC1 that results in the reduction of mTOR activity [20, 28]. Interestingly, we observe that 4E-BP1 is hypophosphorylated resulting in a reduced protein synthesis [65, 66]. This may be associated with caspase-cleavage-dependent cell death as reported by another laboratory [67]. It may be questioned how an increase in mTOR phosphorylation results in a decrease of 4E-BP1 phosphorylation. As mentioned earlier, mTOR forms two complexes, mTOR C1 and mTOR C2, of which mTOR C1 is associated with 4E-BP1 phosphorylation. mTOR also binds a regulatory protein, raptor along with other factors, to form mTORC1, and this binding may be reduced by phosphorylation of mTOR at Ser2448, thus reducing 4E-BP1 phosphorylation. The observed under-phosphorylation of 4E-BP1 is in good agreement with that reported by Su et al. [68] but contradicts the report by another laboratory probably due to the difference in the cell line used and/or different experimental conditions [58].
GSK-3α is not phosphorylated in our system. Thus, GSK-3β seems to play a key role as a coordinator in catabolic to anabolic transition by directly binding to the β subunit of AMPK to open a serine-threonine (ST)-stretch in the α subunit for phosphorylation and dephosphorylation. It continues to phosphorylate the ST-stretch to inhibit AMPK activity in a timely manner [24, 69]. Inhibition of GSK-3β also triggers a profound autophagic response, resulting in a significant decline in cellular ATP production leading to an increase of AMP/ATP ratio [25]. Since AMPK activity is down-regulated by GSK-3β, our observation of an increase of AMPK (Fig. 3a) may result, at least partially, from inactivation of GSK-3β, in agreement with our previous observation [12]. While mTOR stimulates anabolism by activating protein synthesis through the PI-3K/Akt pathway, under-phosphorylation of 4E-BP1 leads to reduced protein synthesis, favoring catabolism [70–72]. The association of autophagy with HCV infection is highly significant in IR development. It may be induced by one or more of the viral nonstructural proteins (NS3-5B) [56, 73]. Progression of the autophagic process may increase viral replication by providing a double-membrane structure of autophagic vacuoles [12, 74]. Autophagy may inhibit innate immunity [58, 75] and impede the alteration of lipid metabolism [73, 75, 76]. The role of GSK-3β in promoting cell survival by modulating the autophagic response overexpressing micro-RNA (miR)-122 has been shown recently [77–79]. In HCV-IR, we observe that the anti-apoptotic protein Bcl-XL is present in a core complex with IRS-1 Ser312 and Beclin-1, possibly disturbing the balance between pro- and anti-apoptotic protein. In summary, HCV-IR may present a two-sided mechanism by controlling the AMPK-mTOR-4E-BP1 pathway under opposite catabolic and anabolic conditions. GSK-3 may serve as a switch which becomes defective by Ser9 mutations under the control of the Akt pathway. A similar insight into the relationship between catabolic and anabolic IR has been proposed for a circadian control of metabolic disorders leading to diabetes [80].
As suggested by us and others, IRS-1 phosphorylation down-regulates, at least partially, the IFNα response through negative regulation of the PI-3K/Akt pathway by down-regulating GSK-3β Ser9 and GS Ser641 [12]. It is worth noting that IFNα treatment of HCV-infected cells also inhibited phosphorylation of mTOR, AMPK, and GSK-3β in early infection. An analysis of protein phosphorylation pathway(s) of Akt, GSK-3β, and AMPK may lead to potential affordable therapeutic targets. Thus, multiple pathways are involved in developing IR, and their combined inactivation will lead to a superior therapeutic outcome with better understood mechanisms.
Acknowledgment
The authors would like to thank Professor Charles M. Rice, Ph.D., Laboratory of Virology and Infectious Disease, The Rockefeller University, New York, NY, for providing the Huh7 cell line.
Statement of Ethics
The study protocol #25128 was reviewed and approved by the Institutional Biosafety Committee, Baylor College of Medicine, Houston, TX, USA. Ethics approval and written informed consents were not required.
Conflict of Interest Statement
The authors have declared that no financial or competing interest exists.
Funding Sources
We gratefully acknowledge the financial support from the Eugene B. Casey Foundation and William and Sonia Carpenter fund for carrying out this research.
Author Contributions
Conceived idea and designed experiment: G.C.D.; execution and interpretation of data and preparation of the manuscript and editing: G.C.D. and F.B.H.; and the authors agreed about the contents of the manuscript and its submission.
Funding Statement
We gratefully acknowledge the financial support from the Eugene B. Casey Foundation and William and Sonia Carpenter fund for carrying out this research.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.
Supplementary Material
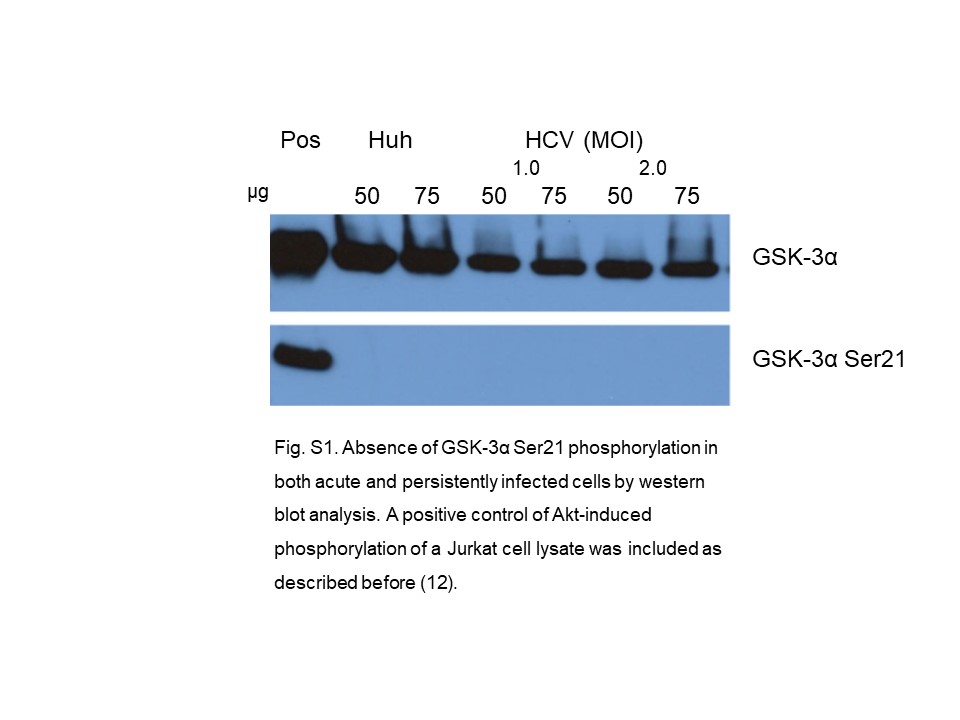{kind=link}
Supplementary Material
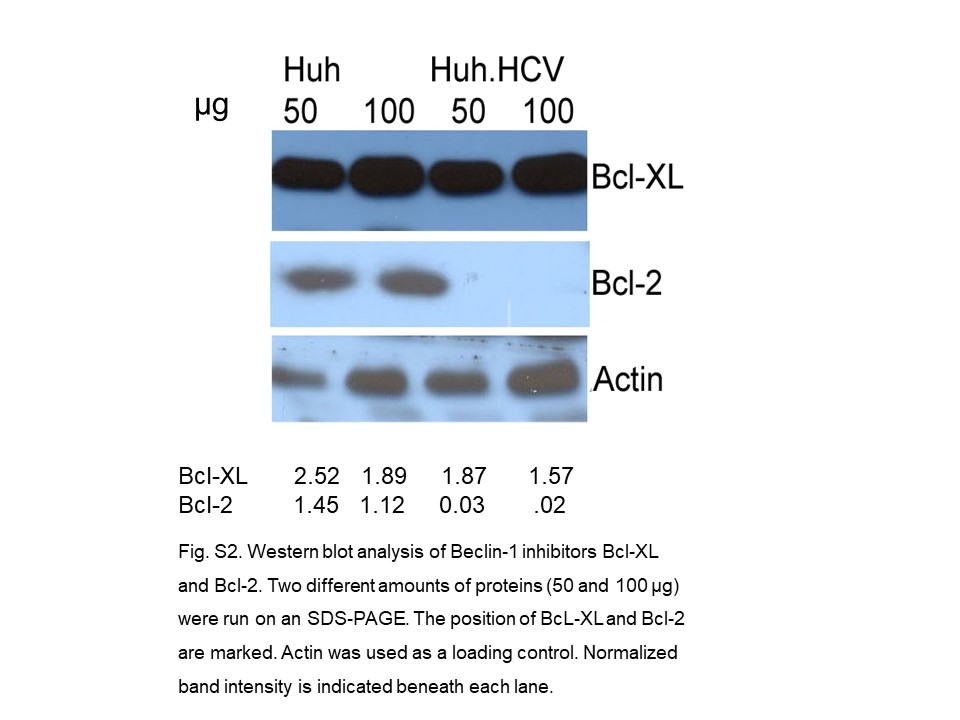{kind=link}
Supplementary Material
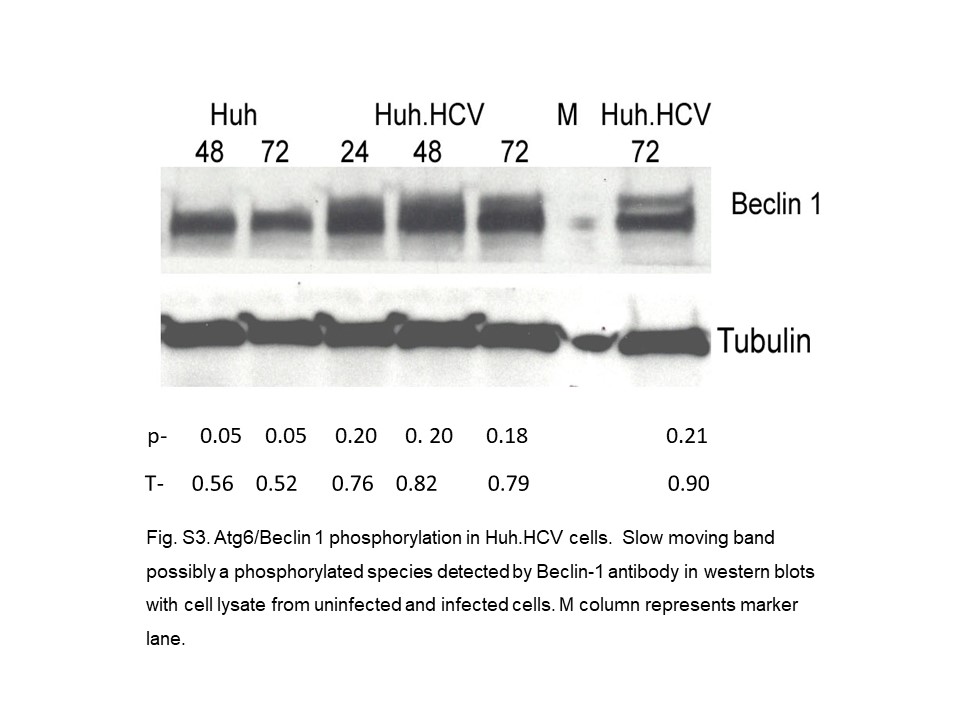{kind=link}
References
- 1. Petruzziello A, Marigliano S, Loquercio G, Cozzolino A, Cacciapuoti C. Global epidemiology of hepatitis C virus infection: an update of the distribution and circulation of hepatitis C virus genotypes. World J Gastro. 2016;22(34):7824–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta SH, Brancati FL, Sulkowski MS, Strathdee SA, Szklo M, Thomas DL. Prevalence of type-2 diabetes mellitus among persons with hepatitis C virus infection in the United States. Ann Intern Med. 2000;133(8):592–9. [DOI] [PubMed] [Google Scholar]
- 3. Allison M, Wreghitt T, Palmer CR, Alexander GJ. Evidence for a link between hepatitis C virus infection and diabetes mellitus in a cirrhotic population. J Hepatol. 1994;21(6):1135–9. [DOI] [PubMed] [Google Scholar]
- 4 . Summers SA, Yin VP, Whiteman EL, Garza LA, Cho H, Tuttle RL, et al. Signaling pathways mediating insulin-stimulated glucose transport. NY Acad Sci. 1999;892:169–86. [DOI] [PubMed] [Google Scholar]
- 5. Cohen P, Frame S. The renaissance of GSK-3. Nat Rev Mol Cell Biol. 2001;2(10):769–76. [DOI] [PubMed] [Google Scholar]
- 6. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK-3): regulation, actions and diseases. Pharmacol Ther. 2015;148:114–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–20. [DOI] [PubMed] [Google Scholar]
- 8. MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, et al. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007;6(4):329–37. [DOI] [PubMed] [Google Scholar]
- 9. Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28(20):6314–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ragolia L, Begum N. Protein phosphatase 1 and insulin action. Mol Cell Biochem. 1998;182(1–2):49–58. [PubMed] [Google Scholar]
- 11. Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284(45):31484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das GC, Hollinger FB. Molecular pathways for glucose homeostasis, insulin signaling and autophagy in hepatitis C virus induced insulin resistance in a cellular model. Virology. 2012;434(1):5–17. [DOI] [PubMed] [Google Scholar]
- 13. Hermida MA, Dinesh Kumar J, Leslie NR. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul. 2017;65:5–15. [DOI] [PubMed] [Google Scholar]
- 14. Lee Y-R, Lei H-Y, Liu M-T, Wang J-R, Chen S-H, Jiang-Shieh Y-F, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374(2):240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-XL’s target recognition versatility revealed by the structure of Bcl-XL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372(1):223–35. [DOI] [PubMed] [Google Scholar]
- 16. Wen J, Mai Z, Zhao M, Wang X, Chen T. Full anti-apoptotic function of Bcl-XL complexed with Beclin-1 verified by live-cell FRET assay. Biochem Biophys Res Comm. 2019;511:700–14. [DOI] [PubMed] [Google Scholar]
- 17. Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase mediated phosphorylation on the BH3 domain of Beclin-1 promotes dissociation of Beclin-1 from BCL-XL and induction of autophagy. EMBO Rep. 2009;10(3):285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin-1 phosphorylation. Science. 2012;338(6109):956–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of ULK1. Nat Cell Biol. 2011;13(2):132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Egan DF, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK-1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89(3):1025–78. [DOI] [PubMed] [Google Scholar]
- 22. Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR) conducting the cellular signaling symphony. J Biol Chem. 2010;285(19):14071–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Showkat M, Beigh MA, Andrabi KI. mTOR signaling in protein translation regulation: implications in cancer genesis and therapeutic interventions. Mol Biol Int. 2014;2014:686984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin J, et al. Inhibition of AMPK catabolic action by GSK 3. Mol Cell. 2013;50:407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun A, Li C, Chen R, Huang Y, Chen Q, Cui X, et al. GSK-3β controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate. 2016;76(2):172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma XM, Blenis J. Molecular mechanism of mTOR-mediated translational control. J Nat Rev Mol Cell Biol. 2009;l10:307–18. [DOI] [PubMed] [Google Scholar]
- 27. Qin XQ, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multi-functional protein. Cell Cycle. 2016;15(6):781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shang S, Hua F, Hu ZW. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8(20):33972–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Palsgaard J, Emanuelli B, Winnay JN, Sumara G, Karsenty G, Kahn CR. Cross-talk between insulin and Wnt signaling in preadipocytes: role of Wnt co-receptor low density lipoprotein receptor-related protein-5 (LRP5). J Biol Chem. 2012;287(15):12016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16(13):3797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peifer M. Cell adhesion and signal transduction: the Armadillo connection. Trends Cell Biol. 1995;5(6):224–9. [DOI] [PubMed] [Google Scholar]
- 32. Kim WK, Kwon Y, Jang M, Park M, Kim J, Cho S, et al. β-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci Rep. 2019;9(1):18440–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022;7(1):3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cai Z, Zhang C, Chang KS, Jiang J, Ahn BC, Wakita T, et al. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J Virol. 2005;79(22):13963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skurat AV, Roach PJ. Multiple mechanisms for the phosphorylation of c-terminal regulatory sites in rabbit muscle glycogen synthase expressed in cos cells. Biochem J. 1996;313 ( Pt 1)(Pt 1):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase -3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278(35):33067–77. [DOI] [PubMed] [Google Scholar]
- 38. Ito T, Sasaki Y, Wands JR. Overexpression of human insulin receptor substrate 1 induces cellular transformation with activation of mitogen-activated protein kinases. Mol Cell Biol. 1996;16(3):943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Surmacz E, Burgaud JL. Overexpression of insulin receptor substrate 1 (IRS-1) in the human breast cancer cell line MCF-7 induces loss of estrogen requirements for growth and transformation. Clin Cancer Res. 1995;1(11):1429–36. [PubMed] [Google Scholar]
- 40. Li L, Qi X, Williams M, Shi Y, Keegan AD. Overexpression of insulin receptor substrate-1, but not insulin receptor substrate-2, protects a T cell hybridoma from activation-induced cell death. J Immun. 2002;168(12):6215–23. [DOI] [PubMed] [Google Scholar]
- 41. Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon M, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor B kinase complex. J Biol Chem. 2002;277(50):48115–21. [DOI] [PubMed] [Google Scholar]
- 42. Yoneyama Y, Inamitsu T, Chida K, Iemura SI, Natsume T, Maeda T, et al. Serine phosphorylation by mTORC1 promotes IRS-1 degradation through scfβ-TRCP E3 Ubiquitin ligase. iScience. 2018;5:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J Virol. 2012;86(11):6315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taniyama Y, Hitomi H, Shah A, Alexander RW, Griendling KK. Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25(6):1142–7. [DOI] [PubMed] [Google Scholar]
- 45. Wang Y, Nishina PM, Naggert JK. Degradation of IRS-1 leads to impaired glucose uptake in adipose tissue of the type 2 diabetes mouse model TALLYHO/Jng. J Endocrinol. 2009;203(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greene MW, Sakaue H, Wang LW, Alessi DR, Roth RA. Modulation of insulin stimulated degradation of human insulin receptor substrate-1 by serine 312 phosphorylation. J Biol Chem. 2003;278(10):8199–211. [DOI] [PubMed] [Google Scholar]
- 47. Pederson TM, Kramer DL, Rondinone CM. Serine threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes. 2001;50(1):24–31. [DOI] [PubMed] [Google Scholar]
- 48. Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165(5):1499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gao TT, Qin ZL, Ren H, Zhao P, Qi ZT. Inhibition of IRS-1 by hepatitis C virus infection leads to insulin resistance in a PTEN-dependent manner. Virol J. 2015;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu X, Keshwani M, Meyer K, Sarikas A, Taylor S, Pan ZQ. Identification of the degradation determinants of insulin receptor substrate 1 for signaling cullin-RING E3 ubiquitin ligase 7-mediated ubiquitination. J Biol Chem. 2012;287(48):40758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6k cassette induces IRS-1/2 depletion, insulin resistance and cell survival deficiencies. Curr Biol. 2004;14(18):1650–6. [DOI] [PubMed] [Google Scholar]
- 52. Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. 2003;278(5):2896–902. [DOI] [PubMed] [Google Scholar]
- 53. Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3β-mediated phosphorylation of rictor. Sci Signal. 2011;4(161):ra10. [DOI] [PubMed] [Google Scholar]
- 54. Jeon SM. Regulation and function of AMPK in Physiology and diseases. Exp Mol Med. 2016;48(7):e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu J, Ding X, Tang J, Cao Y, Hu P, Zhou F, et al. Enhancement of canonical Wnt/β-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. Plos One. 2011;6(11):e27496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mohl BP, Tedbury PR, Griffin S, Harris M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J Virol. 2012;86(19):10724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang H, Kang R, Wang J, Luo G, Yang W, Zhao Z. Hepatitis C virus inhibits AKT-tuberous sclerosis complex (TSC), the mechanistic target of rapamycin (mTOR) pathway, through endoplasmic reticulum stress to induce autophagy. Autophagy. 2013;9(2):175–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol. 2012;86(16):8705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345(3):437–43. [PMC free article] [PubMed] [Google Scholar]
- 60. Douglas DN, Pu CH, Lewis JT, Bhat R, Anwar-Mohamed A, Logan M, et al. Oxidative stress attenuates lipid synthesis and increases mitochondrial fatty acid oxidation in hepatoma cells infected with hepatitis C virus. J Biol Chem. 2016;291(4):1974–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Pros Natl Acad Sci. 2006;103(14):5379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ando T, Imamura H, Suzuki R, Aizaki H, Watanabe T, Wakita T, et al. Visualization and measurement of ATP levels in living cells replicating hepatitis C Virus genome RNA. PLoS Pathog. 2012;8(3):e1002561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mankouri J, Tedbury PR, Gretton S, Hughes ME, Griffin SDC, Dallas ML, et al. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc Natl Acad Sci. 2010;107(25):11549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Figueiredo VC, Markworth JF, Cameron-Smith D. Considerations on mTOR regulation at serine 2,448: implications for muscle metabolism studies. Cell Mol Life Sci. 2017;74(14):2537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37(Pt 1):217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev. 1998;12(4):502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tee AR, Proud CG. Caspase cleavage of initiation factor 4E-binding protein 1 yields a dominant inhibitor of cap-dependent translation and reveals a novel regulatory motif. Mol Cell Biol. 2002;22(6):1674–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MMC. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011;85(20):10561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2(84):pe51. [DOI] [PubMed] [Google Scholar]
- 71. Saxton RA, Sabatani DM. mTOR signaling in growth, metabolism and disease. Cell. 2017;168:960–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yoon MS. mTOR as a key regulator in maintaining skeletal muscle mass. Front Physiol. 2017;8:788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chu JYK, Ou JHJ. Autophagy in HCV replication and protein trafficking. Int J Mol Sci. 2021;22(3):1089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, et al. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem. 2012;287(22):18036–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chan ST, Lee J, Narula M, Ou J-HJ. Suppression of host innate immune response by hepatitis C virus via induction of autophagic degradation of TRAF6. J Virol. 2016;90(23):10928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vescovo T, Romagnoli A, Perdomo AB, Corazzari M, Ciccosanti F, Alonzi T, et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gasteroenterol. 2012;142(3):644–53.e3. [DOI] [PubMed] [Google Scholar]
- 77. Yang J, Takahashi Y, Cheng E, Liu J, Terranova PF, Zhao B, et al. GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death. J Cell Sci. 2010;123(Pt 6):861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang L. Autophagy in hepatitis B or C virus infection: an incubator and a potential therapeutic target. Life Sci. 2020;242:117206. [DOI] [PubMed] [Google Scholar]
- 79. Saleh M, Rüschenbaum S, Welsch C, Zeuzem S, Moradpour D, Gouttenoire J, et al. Glycogen synthase kinase 3β enhances hepatitis C virus replication by supporting miR-122. Front Microbiol. 2018;9:2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schwartsburd PM. Catabolic and anabolic faces of insulin resistance and their disorders: a new insight into circadian control of metabolic disorders leading to diabetes. Future Sci OA. 2017;3:FSO201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.