

Abstract
Since their discovery almost two decades ago, IL-17-producing CD4+ T cells (Th17 cells) have been implicated in the pathogenesis of multiple autoimmune and inflammatory disorders. In addition, Th17 cells have been found to play an important role in tissue homeostasis, especially in the intestinal mucosa. Recently, the use of single-cell technologies along with fate-mapping and various mutant mouse models has led to substantial progress in the understanding of Th17 cell heterogeneity in tissues and of Th17 cell plasticity leading to alternative T cell states and differing functions. In this review, we discuss the heterogeneity of Th17 cells and the role of this heterogeneity in diverse functions of Th17 cells from homeostasis to tissue inflammation. In addition, we discuss Th17 cell plasticity and its incorporation into the current understanding of T cell subsets and alternative views as to the role of Th17 cells in autoimmune and inflammatory diseases.
Introduction
CD4+ T helper cells are essential regulators of adaptive immune responses. In the 1980s, CD4+ T helper cells were classified into Th1 and Th2 subsets based on their distinct expression of cytokines and effector functions [1]. Th1 cells are differentiated with IL-12, produce IFNγ and control intracellular pathogens, whereas Th2 cells are differentiated with IL-4 and IL-2, express IL-4, IL-5, and IL-13, and protect from extracellular pathogens, including parasites, but also promote development of allergic diseases. Th1 cells were originally thought to be the main T-cell population driving organ-specific autoimmunity. However, findings that IFNγ-, IFNγ -receptor-, IL-12p35-, and IL-12 receptor β2 -deficient mice developed exacerbated experimental autoimmune encephalomyelitis (EAE) challenged the concept of Th1 cells as the pathogenic population in autoimmunity [2–5]. In the early 2000s, after the discovery of the novel cytokine IL-23 [6], Cua and colleagues demonstrated that IL-23, and not IL-12, is the cytokine required for the induction of EAE [7]. In a follow-up study in 2005, Cua and colleagues suggested that IL-23 promotes expansion of a novel IL-17-producing T-cell population capable of inducing autoimmunity upon adoptive transfer [8]. In 2005, we proposed that indeed IL-17 producing T cells represented an independent T cell subset, but that IL-23 was not the differentiating factor that induces Th17 cells and instead acts on previously differentiated Th17 cells, stabilizing their effector functions [9]. While IL-23 induced IL-17 expression in activated T cells, this required prior induction of IL-23R through STAT3-mediated signaling initiated by IL-6 or IL-21 and amplified by IL-23 itself. Th17 cells were established as an independent T helper cell subset with the discovery of signature cytokines IL-17A, IL-17F, and IL-22, lineage-specific differentiation factors, and dedicated or “master” transcription factors [10–16]. In 2006, the cytokines TGF-β and IL-6 were described as differentiation factors required to polarize naïve T cells into Th17 cells [13–15]. Discovery that the orphan nuclear receptor RORγt was expressed in a subset of intestinal CD4+ T cells and that it was required for expression of IL-17 by both these cells and in vitro differentiated Th17 cells cemented its role as the “master transcription factor” for the Th17 lineage [16]. Although RORγt was both necessary and sufficient for at least part of the Th17 gene expression program, multiple additional transcription factors were shown to be required for the expression of IL-17, including STAT3, IRF4, and BATF [17–20].
It is well-established that T helper cells have diverse capabilities beyond their conventionally assigned functions. This has only been reinforced by the recent application of single cell genomics to CD4+ T cells in homeostatic and proinflammatory microenvironments, which has revealed a previously unappreciated complexity of the T helper cell subsets [21]. It is therefore difficult to reconcile such functional diversity with a recently proposed reclassification of T helper cell subsets based on function: providing help to phagocytes (cellular immunity- Th1 cells), to class switching and antibody production by B cells (humoral immunity- Th2 cells), and regulating non-immune cells at barrier sites (mucosal immunity- Th17 cells) [22]. Indeed, Th1 cells are well-known to also regulate B cells and humoral immunity [23, 24], Th2 cells regulate multiple non-immune tissue cells at mucosal sites [25], and Th17 cells may contribute to production of high-affinity IgA antibodies and to IgG2a and IgG3 class switching [26, 27]. As our appreciation of the multiple capabilities of T helper cells continues to grow, it may be preferable to adopt a more nuanced classification of the cells, based on expression of gene modules that distinguish between various T cell subsets, in place of the Th1, Th2 or Th17 nomenclature. Indeed, the distinction of “homeostatic” versus “pathogenic” Th17 cells, discussed in detail below, may be only one example of the important functional diversity within a single CD4+ T cell subset.
The discovery of Th17 cells in mouse models signaled an important shift in studies of T-cell-mediated inflammatory diseases. It was rapidly appreciated that Th17 differentiation and function are evolutionarily conserved, which led to several successful efforts to target the pathway and to multiple new therapies. In particular, anti-IL-17 pathway antibodies are effective in the treatment of inflammatory conditions, including plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis [28]. Initial double-blind clinical trials showed that anti-IL-17 antibody secukinumab was also effective in reducing new magnetic resonance imaging (MRI) lesions, cumulative lesion load, and expanded disability status scale (EDSS) scores in multiple sclerosis (MS) patients [29, 30]. However, some animal model studies proposed that IL-17 and Th17 cells are not required to drive autoimmune tissue inflammation [31, 32], and that other T cells, particularly GM-CSF-producing T cells, are the primary inducers of autoimmune tissue inflammation [33–36]. Moreover, analysis of Th17 cells in different tissue sites has revealed that Th17 cells may exhibit distinct transcriptional profiles associated with either homeostatic or pathogenic functions [37–43]. Thus, Th17 cells, besides being highly pathogenic and able to induce autoimmunity, have also been implicated in mediating tissue homeostasis, regulating tissue integrity by protecting mucosal barriers from potentially invasive microbes, particularly extracellular bacteria and fungi, influencing the composition of the commensal microbiota at body surfaces, and modulating epithelial permeability to diverse nutrients and metabolites [44–48]. This homeostatic function of Th17 cells likely underlies the finding that blockade of IL-17 in Crohn’s disease is not only ineffective but can exacerbate colitis [28, 49]. Additionally, animal model studies have suggested that distinct Th17-inducing commensal microbes contribute to exacerbated autoimmune disease at extra-intestinal sites, potentially mediated by the mucosa-derived Th17 cells [50–52]. Overall, the mechanisms by which tissue Th17 cells mediate divergent functions, i.e., promoting tissue homeostasis while contributing to the development of autoimmune tissue inflammation, and the roles of the IL-17 cytokines and Th17 cell plasticity in autoimmunity remain unclear.
In this review, we discuss the in vitro and in vivo heterogeneity of Th17 cells and the role of this heterogeneity in the function of Th17 cells in homeostasis and autoimmunity. In addition, we review and discuss divergent views as to the role of Th17 cells in autoimmune disease and specifically plasticity and diversity of Th17 cells and their relationship to other T cell states.
Heterogeneity of in vitro and in vivo differentiated Th17 cells
The concept of Th17 cell heterogeneity came originally from analyses in which different cytokine combinations that induce bona fide IL-17-producing Th17 cells in in vitro cultures had diverse abilities to induce autoimmune disease following adoptive transfer [39, 40, 53]. In vitro polarization of naïve CD4+ T helper cells with IL-6 and TGF-ß generates Th17 cells that produce IL-17 but induce little or no tissue inflammation or autoimmunity (non-pathogenic Th17 cells), whereas IL-6, IL-1ß and IL-23 or IL-6, TGF-ß, and IL-23 differentiate Th17 cells that are highly potent in transferring disease (pathogenic Th17 cells) [39, 53]. In subsequent studies, the transcriptional signatures distinguishing in vitro-differentiated non-pathogenic Th17 cells and pathogenic Th17 cells were defined [53, 54]. Although both subtypes expressed IL-17, non-pathogenic Th17 cells additionally expressed the immunoregulatory genes Il10, Il9, Maf, and Ahr, whereas pathogenic Th17 cells were characterized by the expression of proinflammatory genes including Csf2 (gene encoding GM-CSF), Ifng, Tbx21, Il23r and Gzmb. These analyses led to the discovery and successful validation of multiple novel Th17 cell regulators [55–58]. By undertaking high density, temporal expression analysis of differentiating Th17 cells, regulatory networks for Th17 cells were constructed, which confirmed the central role of RORγt in differentiating Th17 cells [20, 59], but also identified a number of novel regulators that formed mutually antagonistic modules, which, while promoting Th17 cell fate, suppress differentiation to other T cell states [20]. Furthermore, by integrating single-cell ATAC-seq and single-cell RNA-seq, we generated a regulatory network across npTh17, pTh17, and Th1 cells to reveal the underlying regulators and mechanisms that drive the non-pathogenic and pathogenic Th17 subsets [60]. The network predicted the pathogenic Th17- and Th1- program to be regulated by the transcription factors ATF3, BHLHE40, FOS, NR4A1, EOMES, and T-BET. Predicted regulators of the non-pathogenic- and general- Th17 program included RORγt, ETS1, BACH2, FOSL2, and RBPJ. We validated BACH2 as a novel regulator of Th17 pathogenicity that promoted immunomodulatory programs and restrained pro-inflammatory Th1-like programs in Th17 cells. In a study analyzing the transcriptional programs of human Th17-IL-10+ and Th17-IL-10− cells, BACH2 was predicted to regulate the transcriptional program of the Th17-IL-10+ subset [61]. Moreover, multiple single-nucleotide polymorphisms (SNPs) in the BACH2 locus are associated with human autoimmune diseases and a protective allele for MS disease correlates with an increase of BACH2 expression in CD4+ T cells [60, 62]. Hence, in humans BACH2 might be an important regulator of homeostatic Th17 cells that could be exploited for the treatment of human autoimmune diseases.
The divergent transcriptomic profiles of in vitro differentiated pathogenic and non-pathogenic Th17 cells have also been observed in differentiated T cells induced by diverse intestinal commensal microbes. Thus, intestinal non-pathogenic/homeostatic Th17 cells specific for segmented filamentous bacteria (SFB) have a transcriptome that differs markedly from the pro-inflammatory Th17 cells elicited by Helicobacter hepaticus (H. hepaticus) in mice defective for induced intestinal Treg cells [43]. The gene expression profiles of these cells resemble those described for the in vitro differentiated pathogenic Th17 cells, suggesting that genes identified in the in vitro studies could serve as good therapeutic targets [53, 54, 63].
Interestingly, human T cell subsets have been identified that are transcriptionally similar to murine in vitro-differentiated non-pathogenic and pathogenic Th17 cells. While in mice evidence for distinct types of Th17 cells came from studies of microbiota-specific T cells and inflammatory disease models, human Th17 cell heterogeneity was identified based on the roles of different Th17 populations in immunity against fungi and bacteria [64, 65]. Th17 memory cells (initially defined as CCR4+CCR6+) specific for Candida albicans (C. albicans) produced IL-17 and IFNγ and no IL-10, and thus resemble pathogenic Th17 cells. In contrast, Staphylococcus aureus (S. aureus)-specific Th17 cells produced IL-17 and IL-10, but no IFNγ, and thus resemble non-pathogenic/homeostatic Th17 cells [66]. Analysis of CXCR3+ cells identified two T-BET-expressing populations, one that is CCR6−, and represents Th1 cells, and another that is also CCR6+, and is dependent on expression of RORγt [64, 65]. It is now appreciated that the CXCR3+CCR6+ cells, named Th1* cells, resemble pathogenic Th17 cells and are required to control infection with both C. albicans and mycobacteria, including the attenuated Bacillus Calmette-Guerin (BCG) [64, 67]. Indeed, genetic defects in humans in the IL-17/IL-17-receptor axis, STAT3, and RORγt, resulting in dysfunctional Th17 cells, are associated with chronic mucocutaneous candidiasis and severe disease following BCG immunization [67–69]. Patients with mucocutaneous candidiasis also show increased susceptibility to S. aureus infections. Hence, the non-pathogenic/homeostatic and pathogenic Th17 cell phenotypes studied in the mouse models may mirror the physiological roles of human Th17 cell subsets that are important for immunity against distinct bacteria and fungi.
Tissue Th17 cells during homeostasis
Environmental influence on tissue Th17 cells
Although Th17 cells were originally described as drivers of tissue inflammation, at steady-state Th17 cells are most abundant at mucosal barriers such as the intestinal lamina propria, where they play important roles in mediating tissue homeostasis and in preventing microbial invasion and controlling infections at barrier sites. The Th17 cell-signature cytokines IL-17A, IL-17F, and IL-22 induce the expression of antimicrobial proteins and the formation of tight junctions by intestinal epithelial cells and have been implicated in regulation of IgA responses [70, 71]. The constant exposure of the intestine to environmental agents including microbiota, diet, and metabolites has a significant influence on the intestinal Th17 population (Figure 1). A number of seminal studies have established the ability of specific microbes to induce intestinal Th17 cells. Colonization of germ-free mice with SFB, but not other Clostridium or Bacteroides species, induced SFB-specific Th17 cells, largely in the terminal ileum, where SFB binds to the intestinal epithelium [72]. Adhesion to the epithelium is thought to be essential for the induction of Th17 cells, as rat-derived SFB, that does not adhere to the small intestinal epithelium in mouse hosts, failed to induce intestinal Th17 cells [73]. In addition, with the use of adhesion-defective mutants, the induction of intestinal Th17 cells by Citrobacter rodentium (C. rodentium) and Escherichia coli O157 was shown to be dependent on their physical interaction with the intestinal epithelium. SFB antigen delivery across the intestinal epithelium involves a novel vesicular compartment and the GTPase CDC42 [74], and results in induction of the homeostatic Th17 cell program in naïve antigen-specific CD4+ T cells in the draining mesenteric lymph nodes. These Th17 cells distributed throughout the length of the intestine, but IL-17 cytokine production was largely confined to ileal Th17 cells, in part in response to epithelium-derived serum amyloid A proteins 1 and 2 (SAA1/2) [75]. SAA induction is one component of an intestinal epithelial cell program induced locally by IL-22 derived from SFB-activated ILC3s, thus explaining the basis of the localized effector response. Interestingly, the induction of Th17 cells by SFB in the small intestine does not induce a proinflammatory reaction in the intestine, but instead induces a homeostatic Th17 population that supports intestinal barrier function [37, 43]. The barrier-protective function of Th17 cells is mainly mediated by the expression of their signature cytokines IL-17 and IL-22, that are also expressed by intestinal γδ T cells and ILC3s, making it difficult to distinguish the contribution of the different immune cell populations to tissue homeostasis. Transcriptomic and fate-mapping studies indicate that at homeostasis the main source of IL-17A is Th17 cells, while most IL-22 is derived from ILC3s [76, 77]. However, during C. rodentium infection, while early phase defense is mediated by ILC3-derived IL-22, protection from systemic dissemination and survival require IL-22 production by T cells [78].
Figure 1: Th17 cell heterogeneity in intestinal homeostasis.
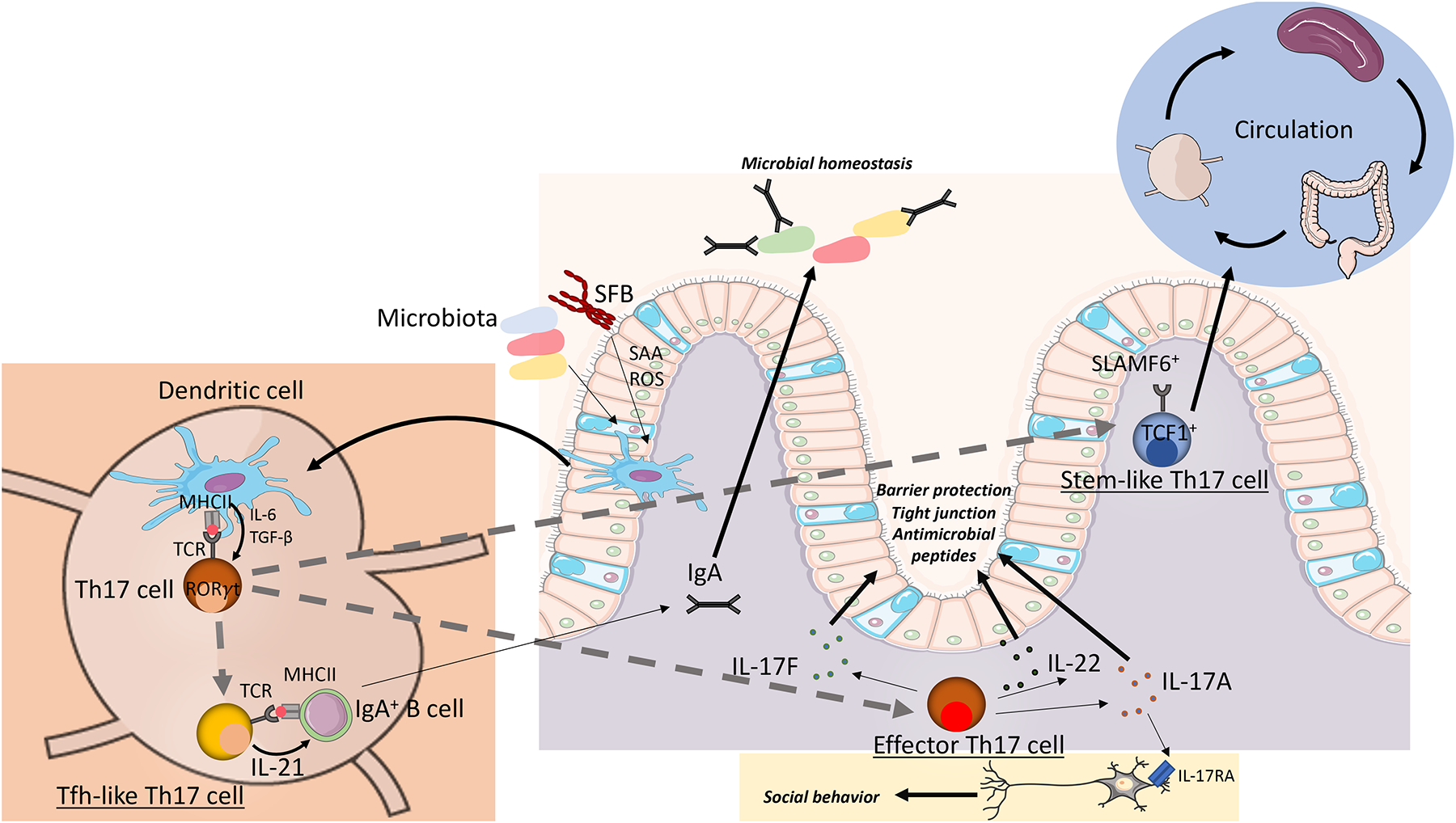
Schematic depicting the role of intestinal Th17 cells in intestinal homeostasis. Specific microbiota, including epithelial-adhering bacteria, induce the differentiation of antigen-specific, naïve CD4+ T cells to Th17 cells [71]. By differences in the APC or intra-tissue plasticity, the Th17-polarized T cell clones can give rise to different Th17 subpopulations. One subpopulation includes SLAMF6+ stem-like Th17 cells that circulate through lymphoid organs and intestinal tissues and are maintained by the gut microbiota [38]. Effector Th17 cells express high levels of IL-17A, IL-17F, and IL-22 which directly support the barrier function of the intestinal epithelium by enhancing the expression of tight junction proteins and antimicrobial peptides [70, 71]. In addition, intestinal IL-17A has been shown to support social behavior by directly signaling through neurons [82]. Tfh-like Th17 cells induce IgA-production by B cells in mesenteric lymph nodes and Peyer’s patches, thereby supporting microbial homeostasis [26, 38].
To analyze the immunomodulatory effects of human gut microbes, mice were mono-colonized with human bacterial species and analyzed for effects on the host immune system. These analyses identified multiple human bacterial species, including Bifidobacterium adolescentis and Staphylococcus saprophyticus, that induced intestinal Th17 cells [79, 80]. In addition to the microbiota, intestinal mycobiota and protists, including Tritrichomonas musculis, were shown to induce intestinal Th17 cells that likely promote homeostatic functions [81–83].
Diet and metabolism have also been shown to influence the phenotype of intestinal Th17 cells. A high-salt diet increases the number of Th17 cells in the intestine and exacerbates Th17-dependent autoimmunity [84, 85]. The increase in salt concentration induces the expression of serum/glucocorticoid-regulated kinase 1 (SGK1) that deactivates FOXO1 and thereby promotes the expression of IL-23R and the generation of pathogenic Th17 cells [84]. In addition, high levels of salt in the diet have been shown to have profound effects on microbiota which in turn may indirectly affect the development of Th17 cells [86]. In a recent study by Kawano et al., sugar in mouse diets was found to induce metabolic disease by disrupting Th17-inducing microbiota leading to a loss of intestinal Th17 cells [87]. Intestinal Th17 cells protected against metabolic disease by regulating epithelial lipid absorption. Lipids have also been shown to regulate the differentiation and phenotype of Th17 cells. Long-chain fatty acids (LCFAs) promote the differentiation of Th17 cells and enhance the severity of EAE, whereas dietary short-chain fatty acids (SCFAs) induce intestinal Treg cells and ameliorate EAE [88]. Th17 cells utilize acetyl-CoA carboxylase 1 (ACC1)-mediated de novo fatty acid synthesis to synthesize phospholipids for cellular membranes [89]. ACC1-deletion in mice results in inhibition of Th17 cell-driven autoimmune disease. CD5 antigen-like (CD5L), a regulator of lipid metabolism, was selectively expressed by Th17 cells differentiated in vitro under non-pathogenic conditions, and was shown to inhibit Th17 cells to attain a pathogenic Th17 cell phenotype [57]. In addition, bile acid metabolites were shown to regulate the differentiation of Th17 cells and Treg cells, and may control their balance in the intestine. While the secondary bile acids 3-oxo-lithocholic acid (LCA) and iso-LCA inhibit the differentiation of intestinal Th17 cells by direct binding to RORγt, isoalloLCA enhances the differentiation of intestinal Treg cells [90]. Defined human gut microbes were found to produce 3-oxoLCA and iso-LCA that inhibit Th17 differentiation, thereby potentially directly regulating inflammatory disorders in the intestine including IBD [91]. Interestingly, centenarians, who have decreased susceptibility to chronic inflammation and infectious diseases, were found to have distinct gut microbiomes enriched for microorganisms that produce these secondary bile acids, further supporting a potential beneficial role of the bile-acid producing microbes [92]. A recent publication by Chen et al. showed that the nuclear xenobiotic receptor constitutive androstane receptor (CAR) induces the expression of bile acid detoxifying enzymes/transporters and the anti-inflammatory cytokine IL-10 in T cells specifically in the small intestine lamina propria [93]. These data suggest a direct role of CAR in detoxifying bile acids and preventing inflammation in the small intestine, resulting in the induction of nonpathogenic/homeostatic Th17 cells and offering a potential novel approach in the treatment of Crohn’s disease.
The metabolic states of immune cells were recently shown to regulate cell phenotype and function. Multiple studies on Th17 cells have highlighted the distinct metabolic mechanisms underlying different Th17 cell phenotypes. Intestinal SFB-induced homeostatic Th17 cells are characterized by a resting aerobic profile primarily dependent on OXPHOS metabolism, which is typical for quiescent T cells [42]. Differentiation of these cells proceeds even if they lack expression of GPI1 and, hence, have reduced glycolytic flux [94]. In contrast, C. rodentium infection-induced Th17 cells with a pro-inflammatory phenotype show enrichment in aerobic glycolysis and oxidative phosphorylation. Consistent with this, the differentiation of proinflammatory Th17 cells in both colitis and EAE models was dependent on intact glycolytic flux due to the relatively hypoxic environment in the setting of inflammation.
The existence of metabolically distinct Th17 cell populations was further substantiated by recent studies with the EAE model. A Th17 cell subset with stem-like features, resembling in vitro differentiated, non-pathogenic Th17 cells, was found to have low anabolic metabolism, while another subset with Th1-like features had high metabolic activity [95]. Disruption of mTORC1 signaling or anabolic metabolism inhibited differentiation of the Th1-like phenotype cells and ameliorated autoimmunity. This may be due to an inability of the stem-like cells to trans-differentiate towards the more pro-inflammatory phenotype. To study cellular metabolism based on single-cell transcriptomes and flux balance analysis, we and others developed a novel algorithm called Compass [96]. Using Compass analysis, polyamine metabolism was identified as a major regulator of T helper cell fate direction, epigenetic programming, and pathogenic potential. Increased glycolysis and polyamine biosynthesis was detected in in vitro differentiated pathogenic Th17 cells, whereas non-pathogenic Th17 cells exhibited enhanced fatty acid oxidation (FAO). Blockade of the polyamine pathway by perturbation of ornithine decarboxylase 1 (ODC1) or spermidine/spermine N1 acetyltransferase 1 (SAT1), rate-limiting enzymes of biosynthesis and catabolism in the polyamine pathway, respectively, alleviated EAE disease. This weakened pathogenic potential of Th17 cells was accompanied by a remodeled transcriptome and epigenome toward a Treg-like program, highlighting the importance of the polyamine pathway in Th17 pathogenicity and in Th17 cell lineage fidelity. In a parallel study performed by the laboratory of Erika Pearce, the polyamine pathway was further identified as generally important for T helper cell polarization [97]. ODC1 deficiency resulted in the failure of CD4+ T cells to acquire subset-specific cytokine and transcription factor expression. Furthermore, perturbation of the polyamine-hypusine axis led to changes in histone acetylation, inhibiting accurate CD4+ T cell polarization. Collectively, the two studies demonstrate an essential function of polyamine metabolism in regulating CD4+ T cell fate decisions. Interestingly, in pathogenic settings in vivo, Th17 cells can acquire features of Th1 cells [76] and Treg cells have been suggested to convert to Th17 cells [98]. Hence, since the polyamine pathway is critical for T cell subset fidelity, limited polyamine availability and inhibited polyamine biosynthesis in pathogenic settings may promote the trans-differentiation of Th17 cells to other T cell subsets that have reduced potential for pathogenesis.
Changes in physiological conditions including hypoxia and temperature also regulate Th17 phenotype. Immune cells in lymphoid organs and in tissues are exposed to different levels of oxygen, which can directly influence their phenotype and function. The transcription factor hypoxia-inducible factor 1α (HIF1α) plays an essential role in the response to hypoxia and is required for the differentiation of Th17 cells [99, 100]. At the same time, HIF1α mediates glycolytic activity and FOXP3 degradation, thereby preventing Treg differentiation. Hence, low oxygen availability is a direct regulator of the Th17/Treg balance. In a recent study, temperature was shown to have an influence on the differentiation and phenotype of Th17 cells [101]. Febrile temperature enhanced the production of cytokines by Th17 cells in vitro and promoted pathogenic gene expression and pro-inflammatory function in vivo. In contrast, cold exposure was found to lead to ameliorated neuroinflammation in EAE with decreased frequencies of Th17 cells in the CNS, likely due to reduced priming by monocytes [102]. These results imply that temperature plays an important role in regulating the proinflammatory program of Th17 cells, with implications for infections and autoimmune diseases.
Heterogeneity of tissue Th17 cells during homeostasis
Several studies have reported evidence of Th17 cell plasticity under physiological conditions in vivo. Fate-mapping studies using IL-17-fate reporter mice showed IL-23R-dependent differentiation of IL-17-producing cells into encephalitogenic IFNγ-producing Th1/Th17 cells [37, 76]. However, it is unclear whether this represents plasticity of homeostatic Th17 cells differentiating into pathogenic Th1-like cells upon exposure to IL-23 or if there is a distinct Th17 cell population with potential to become pathogenic in the presence of IL-23 (see further discussion in later sections). Peyer’s patch Th17 cells were also proposed to trans-differentiate into T follicular helper (Tfh) cells that promote IgA production by germinal center B cells [26]. However, a more recent study reported normal Peyer’s patch IgA production to oral immunization even when Th17 cells were abrogated, and suggested that the earlier study reflected sustained IgA plasma cell function following SFB-induced Th17 cell expansion [103]. Th17 cells were additionally shown to upregulate expression of the anti-inflammatory cytokine IL-10 and acquire a T regulatory type 1 (Tr1) cell phenotype during the resolution of intestinal inflammation and re-establishment of homeostasis [104, 105]. This phenotypic transition may thus reflect trans-differentiation of pathogenic Th17 cells to cells with a regulatory or anti-inflammatory function.
To generate an atlas reflecting the diversity of Th17 cells within and across tissues at homeostasis, we profiled Th17 cells from multiple tissues of naïve IL-17-fate reporter mice by single-cell RNA-seq [38]. Similar to tissue Treg cells [106] and tissue macrophages [107], tissue Th17 cells were found to exhibit tissue-specific transcriptomic signatures. These distinct tissue signatures could indicate distinct environmental cues in the tissue microenvironment that drive the adaptation of the transcriptome and thereby the cellular function of immune cells in various tissue microenvironments (Figure 2). In recent years, immunometabolism has emerged as a field describing how metabolic states regulate phenotype and function of immune cells [108]. Hence, in tissues, we propose that available nutrients and metabolites are likely to have a strong impact on immune cell transcriptome and functional phenotype. In addition, tissue Th17 cell signatures are likely influenced by growth factors, oxygen levels, and cellular interactions that potentially affect their transcriptome and function. Tissue Th17 signatures in the spleen and lymphoid tissues included genes associated with a stem-like T-cell state (Tcf7, Il7r, Ccr7 etc.), whereas genes associated with activation of Th17 cells (Cd44, Cd69, Il17a etc.) were upregulated in intestinal tissues. Analysis of the variation among Th17 cells within each tissue revealed distinct functional clusters of Th17 cells that, interestingly, were transcriptionally similar to clusters in other tissues, indicating the presence of similar Th17 heterogeneity even in different tissue sites. In particular, Tfh-like clusters, effector-like clusters, stem-like clusters, proliferating clusters, interferon-stimulated genes (ISGs)-expressing clusters and Treg clusters were identified (Figure 1). While previous studies described the presence of RORγt+ Treg cells at homeostasis [109, 110], the relationship of these Th17-like Treg cells to other Treg and Th17 cells remained unclear. Using TCR clonality analysis, the Th17-like Treg clusters were found to be clonally distinct from those of non-Treg Th17 clusters, indicating that, at homeostasis, Th17-like Treg cells likely differentiate from other Treg cells or naïve CD4+ T cells, and not from Th17 cells. This is likely explained by the specific induction of distinct intestinal Th17 populations and Treg cells by gut microbiota [71, 72]. In contrast to the Treg cells, tissue Th17 cells were highly clonally related within a tissue, suggesting intra-tissue plasticity and transitions between subpopulations [38].
Figure 2: Drivers of tissue-specific signatures of immune cells.
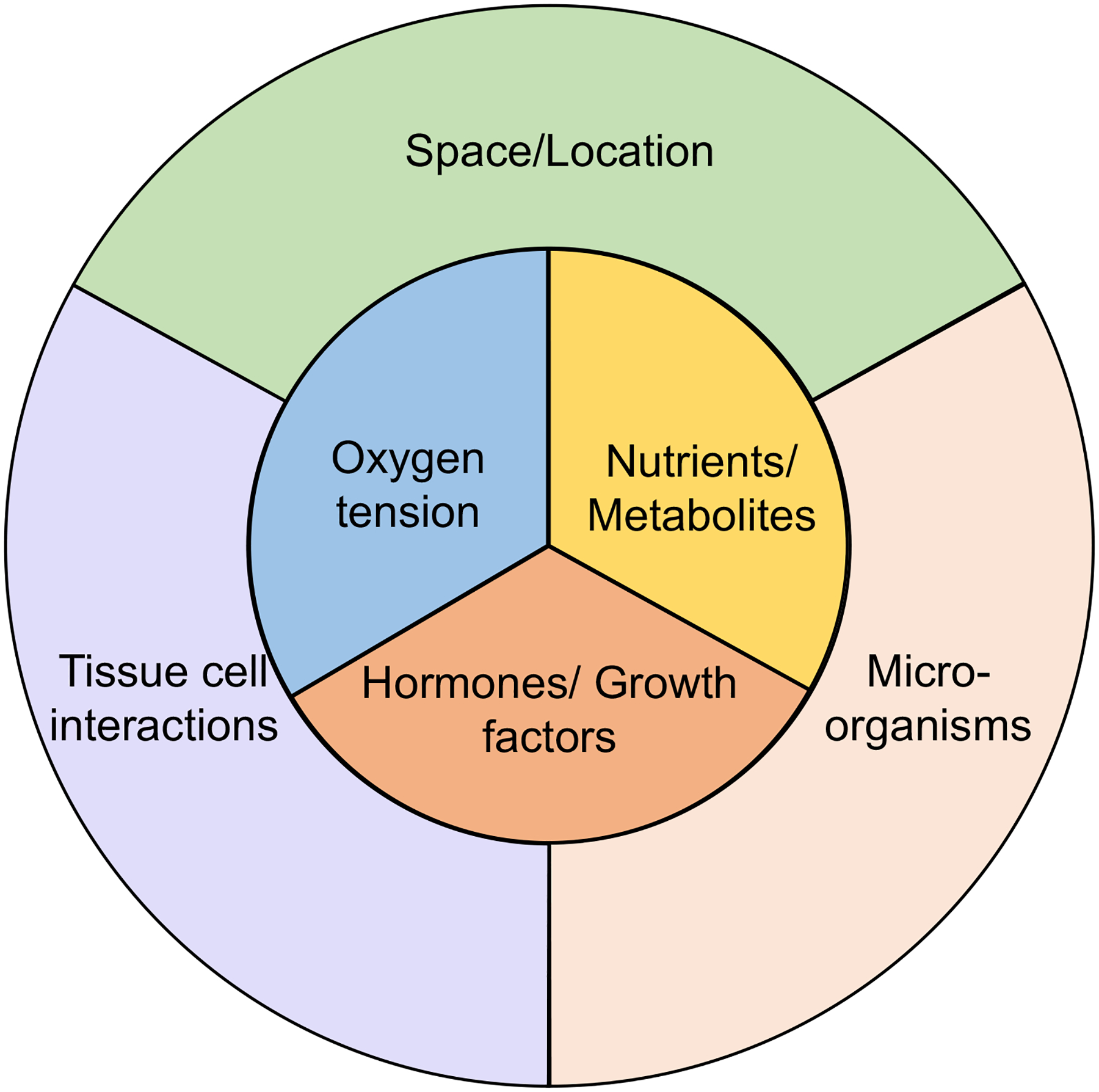
Tissue immune cells, including macrophages [107], Treg cells [106] and Th17 cells [38], acquire specific transcriptomic signatures depending on the tissue they are resident in. Indirect (outer circle) and direct (inner circle) factors likely shape these tissue-specific signatures of immune cells. Indirect factors include the space and location of the cell in the tissue, the interactions with tissue cells, and the presence of microorganisms. Direct factors influencing the tissue signatures are oxygen availability in the tissue, present nutrients and metabolites shaping the cell metabolism, and hormones and growth factors that directly bind and signal through their receptors on the immune cells.
The role of Th17 cells in autoimmunity and tissue inflammation
Th17 cells in animal models of autoimmune disease
The discovery of IL-23 as an essential cytokine for induction of EAE and programming of IL-17-producing T cells that induce autoimmunity upon adoptive transfer [8] paved the way for studying the role of Th17 cells in autoimmunity. Multiple follow-up studies in mouse models supported the role of Th17 cells as important drivers of autoimmunity. In EAE, a mouse model for MS, Th17 cells were shown to be potent inducers of autoimmunity. IL-17A-deficient mice exhibit significantly attenuated EAE disease, whereas IL-17F-deficient mice show marginally reduced EAE [111, 112]. Therapeutic neutralization of IL-17A ameliorates disease in EAE [113]. In addition, the transfer of in vitro differentiated pathogenic Th17 cells expressing a transgenic TCR specific for the MOG35–55 peptide (2D2 cells [114]) is sufficient to induce severe disease in recipient mice [115]. Some reports have suggested that the expression of IL-17A and F by T cells was not required for the development of EAE [31], which was based on studies in which IL-17A was neutralized with an antibody in IL-17F-deficient mice. However, when using genetically deficient IL-17A and IL-17F mice, our own unpublished studies and those of others have shown that IL-17A/F double-deficient mice were highly resistant to the development of EAE [116]. To explain this discrepancy, the Waisman group suggested that the disease resistance was due to changes in the microbiota mediated by loss of IL-17A/F in the mice [116]. In the study they showed that overexpression of IL-17A by intestinal epithelial cells was sufficient to reestablish EAE susceptibility in IL-17A/F-knockout mice and therefore IL-17 production by T cells was not pre-requisite for the development of EAE. However, in this study it was unclear whether the IL-17A overexpression in the intestine remained local or led to systemic elevation of IL-17A levels, thereby impacting blood-brain-barrier (BBB) function and affecting trafficking and the phenotype of immune cells that promote encephalitogenicity. Moreover, in a recent study from one of our labs (unpublished), we found that IL-17A/F-deficient MOG-TCR transgenic 2D2 cells lacked the ability to induce disease in Rag1-deficient recipient mice, whereas 2D2 T cells from wildtype mice induced severe disease, which supports the idea that IL-17 produced by T cells is critical for the induction of EAE, independently of changes in the microbiota. These results highlight the important role of IL-17 expression in Th17 cells for their pathogenic effector functions. Hence, additional analysis will be required to understand the role of IL-17 expression in T cells and in the intestine in the development of autoimmune diseases.
In experimental models of psoriasis, Th17 cells are thought to be the main pathogenic T cell population driving disease. The Th17-associated cytokines IL-23, IL-22 and IL-17A have been shown to play essential roles in these models and in the pathogenesis of psoriasis [117–119]. IL-23, produced by macrophages and dendritic cells, drives the differentiation and expansion of IL-22+ and IL-17A+ Th17 cells that activate the surrounding tissue cells including keratinocytes and fibroblasts, thereby mediating tissue inflammation. In addition to Th17 cells, dermal γδ T cells respond to IL-23 and are a major source of IL-17A, IL-17F, and IL-22 cytokines in psoriatic lesions, making them additional contributors to disease pathogenesis [120, 121].
IL-17A has been shown to have a key pathogenic role in murine arthritis models. IL-17-deficient mice are protected against collagen-induced arthritis (CIA) [122] and from spontaneous arthritis in IL-1Rα−/− mice [123]. Blockade of IL-17 or the IL-17 receptor inhibits the development of arthritis in mice [124].
The role of Th17 cells in mouse models of inflammatory bowel disease (IBD) is controversial, which is likely due to the dichotomous nature of intestinal Th17 cells, as they are mediators of tissue homeostasis yet can become drivers of tissue inflammation [37]. In the T cell transfer model of colitis, disease induction is not dependent on IL-17A-expression by T cells [125, 126], although loss of both IL-17A and IL-17F abrogated T cell pathogenicity [127]. In fact, some studies show a protective function for IL-17A in colitis [128, 129], possibly due to the role of IL-17A in promoting intestinal epithelial barrier integrity [130]. However, multiple studies have shown that IL-23 and RORγt, which are essential mediators of Th17 differentiation, are required for colitis [127, 131, 132]. In the Il10−/− model of colitis, in which IL-10-deficient mice develop spontaneous microbiota-dependent colitis, Th17 cells were identified as the main immune population mediating the inflammatory immune response [43, 133]. Collectively, these data suggest that Th17 cells are major disease drivers in the transfer model and Il10−/− model of colitis.
Th17 cells in human autoimmune diseases
The importance of Th17 cells in autoimmunity, initially demonstrated in multiple mouse models, has been validated by targeting the pathway in human inflammatory and autoimmune diseases. The most striking efficacy of such therapies is in psoriasis, for which multiple agents targeting this pathway are now in the clinic [28, 29]. In psoriatic skin lesions, the majority of CD4+ and CD8+ T cells, and γδ T cells, express IL-17 [134], and elevated IL-17A, IL-22 and IL-23 levels were found in patients with active psoriasis [135]. After the identification of the IL-23/IL-17 axis as an important mediator of pathogenesis in preclinical models, ustekinumab, an inhibitor of p40 targeting both IL-23 and IL-12, was first shown to be efficacious in treating psoriasis [136]. More recently, selective IL-23 inhibitors (tildrakizumab, risankizumab, and guselkumab) that specifically target the IL-23 p19 subunit were found to be more effective than TNF inhibitors [137] and IL-23/IL-12 inhibitors [28, 138]. IL-17A and IL-17RA inhibitors (secukinumab, ixekizumab, and brodalumab) are approved for the treatment of moderate to severe plaque psoriasis and show superior efficacy to previously approved therapies for psoriasis, including TNF inhibitors and IL-23/IL-12 inhibitors, resulting in disease-free skin in up to 42% of patients [28, 29, 139, 140].
Psoriatic arthritis (PsA), which shares clinical features with psoriasis and ankylosing spondylitis, is characterized by skin and joint inflammation. Similar to psoriasis, IL-17 pathway inhibitors (secukinumab and ixekizumab) have been shown to be effective in the treatment of PsA [141, 142]. Moreover, secukinumab has also been approved for treatment of ankylosing spondylitis [143], demonstrating a role of IL-17 in the pathology of multiple human autoimmune and inflammatory diseases.
IL-17 was also found to be highly expressed in active MS lesions [144] and Th17 cells mediate blood-brain barrier disruption by the expression of IL-17 and IL-22 [145]. In therapeutic interventions the inhibitor of IL-12+23, ustekinumab, showed no efficacy in relapsing remitting MS (RRMS) [146]. However, this antibody inhibits the generation of both Th1 and Th17 cells and therefore cannot be equated with pure Th17 blockade. On the other hand, the IL-17A inhibitor secukinumab was found to be effective in reducing active lesions in patients with RRMS in a double-blind clinical trial (NCT01051817), but was not pursued further because of other competing orally available drugs [29, 30].
Multiple studies found an association of IL-17 with rheumatoid arthritis (RA). Elevated levels of IL-17 and IL-17R were detected in tissues and synovial fluids from RA patients and synovial IL-17 levels were found to correlate with disease progression [147–149]. However, despite the rationale from preclinical mouse and human studies, targeting IL-17 in RA was found to be only modestly effective. IL-17A inhibition was superior to placebo and improved symptoms in patients non-responsive to TNF targeting, suggesting a pathogenic role for IL-17A in RA [150–152]. However, in this study, TNF-targeting abatacept was found to be superior to secukinumab in efficacy.
In IBD, increased levels of IL-17 and IL-23 were found in colon biopsies from patients and multiple single nucleotide polymorphisms in IL23R were associated with susceptibility to IBD [153, 154]. However, blockade of IL-17A in Crohn’s patients was found to be ineffective and instead resulted in exacerbated colitis leading to study termination [29, 49]. Recent clinical trials testing selective inhibition of IL-23 in Crohn’s disease have shown promising results [155, 156]. These clinical results suggest that IL-23-specific inhibition may prevent the development of pathogenic T cells, while allowing the IL-23 independent production of IL-17A, potentially from homeostatic Th17 cells, to maintain intestinal epithelial barrier integrity [130, 157].
Tissue Th17 cell heterogeneity during autoimmunity
Th17 cell plasticity during autoimmunity
Under inflammatory conditions, tissue Th17 cells have been reported to exhibit plastic changes in their effector programs. Use of cell fate reporter mice showed that T cells that initially express IL-17 (ex-Th17 cells) can subsequently express proinflammatory cytokines, including IFNγ and GM-CSF [38, 76]. Especially the trans-differentiation of Th17 cells (IL-17A+ IFNγ−) to ex-Th17 Th1 cells that lose ability to produce IL-17 but continue to express IFNγ (IL-17A− IFNγ+) have been implicated in pathogenicity of autoimmune diseases. In the T cell transfer model of colitis, the trans-differentiation of Th17 cells to Th1-like cells has been shown to be required for their ability to induce disease [158, 159]. In human autoimmune diseases, IL-17A- and IFNγ-expressing T cells are preferentially found at sites of inflammation and correlate with pathogenicity [160–165].
A recent study from one of our laboratories, using fate reporter mice, provides strong in vivo support for the notion that homeostatic Th17 cells differentiate into inflammatory Th17 cells that mediate autoimmune disease. Using single cell genomics, we identified a homeostatic IL-17A/F+ population, marked by SLAMF6, that gives rise to a pathogenic IFNγ+ GM-CSF+ population marked by CXCR6 during EAE [38], suggesting a direct in vivo relationship between these cells. In the CNS, the pathogenic CXCR6 population gives rise to multiple different, clonally related Th17 subpopulations, including quiescent-, migratory-, proliferating-, proinflammatory- and CD8-like cells, revealing intra-tissue plasticity and/or transitions between subpopulations of Th17 cells. Interestingly, the CD8-like cells expressed both Cd4 and CD8+ T cell markers and were also recently reported in human IBD, and may thus represent a common phenomenon in T-cell-driven autoimmune tissue inflammation [166, 167].
A key driver of the Th17 cell transition towards a pathogenic phenotype is the cytokine IL-23. In the absence of IL-23, Th17 cells lose the ability to up-regulate IFNγ and GM-CSF [76]. IL-23 was shown to induce PRDM1, that, together with RORγt and STAT3, enhances the expression of pro-inflammatory molecules including IL-23R, IL17A, and GM-CSF [168]. We identified IL-23-signaling as a main driver of the conversion of homeostatic IL-17A/F+ SLAMF6+ cells to pathogenic IFNγ+ GM-CSF+ CXCR6+ cells in EAE [38]. Thus, multiple studies support IL-23 as a key mediator of the generation of pathogenic IFNγ+ GM-CSF+ Th17 cells. In addition to its effect on Th17 cells, IL-23 induces expression of cytokines that can have protective or proinflammatory functions in multiple cell types, including γδ T cells, ILC3s, and MAIT cells [169–172]. In a recent study, we found a small fraction of Th1 cells to express IL-23R. When differentiated with IL-12 and IL-21, Th1 cells express IL-23R [173]. IL-23 signaling in Th1 cells is critical for their ability to become colitogenic, with upregulation of several effector molecules that are not induced by IL-23 in Th17 cells. This suggests that IL-23R signaling is more broadly relevant than previously thought and that it may also provide an important target for inhibition of the pathogenicity of multiple effector cells that mediate autoimmunity.
Despite many reports showing a direct relationship between Th17 cells and GM-CSF+ T cells, some publications have proposed the existence of an independent GM-CSF-expressing T helper cell subset, termed ThGM cells [22, 33, 174, 175]. Multiple studies have described an important pathogenic function of GM-CSF in neuroinflammation. Similar to IL-17-deficient mice, GM-CSF-deficient mice are resistant to EAE [176] and GM-CSF+ T cells are enriched in patients with MS [36]. Specific ablation of GM-CSF expression in T cells during EAE leads to unchanged invasion of T cells into the CNS, but the infiltrating cells induce a significantly milder disease with reduced numbers of CNS-invading monocyte-derived cells [33]. However, in the 2D2 T cell transfer EAE model, Csf2−/− 2D2 cells transfer similar disease to wildtype 2D2 cells, whereas deletion of Bhlhe40 (required for expression of Csf2) leads to complete loss of disease (unpublished). Moreover, minimal evidence exists that ThGM cells exhibit a novel T helper program that is independent of other T helper cell subsets. To date, no master transcription factor for GM-CSF+ T cells has been identified and no distinct mechanisms or differentiation conditions have been identified that specifically induce GM-CSF in naïve CD4+ T cells. Most importantly, fate-mapping studies need to be performed to show that encephalitogenic GM-CSF-producing T cells do not reflect induction of the cytokine in other T helper cell subsets. In fact, during EAE, ~80% of CNS-infiltrating CD4+ T cells were found to originate from the Th17 lineage and ~40% of these cells expressed IFNγ and ~40% expressed GM-CSF, indicating that a great proportion of GM-CSF+ T cells during EAE are derived from IL-17A-expressing cells [76]. Moreover, our recent in vivo fate-mapping studies show that in EAE there is significant TCR clonal sharing between homeostatic IL-17A/F+ Th17 cells (SLAMF6+) and pathogenic GM-CSF+ cells (CXCR6+). There was also in vivo conversion of SLAMF6+ cells to CXCR6+ cells upon adoptive transfer, suggesting that, during EAE, pathogenic Th17 cells arise by the conversion of homeostatic Th17 cells to pathogenic Th17 cells [38]. Together, these data suggest that under pro-inflammatory conditions (IL-23, IL-1β, etc.) GM-CSF is generally up-regulated in T cells. This conclusion is further supported by the findings that GM-CSF+ T cells co-express a great variety of other cytokines including IL-2, IL-3, IL-4, IL-6, IL-13, IL-17A, IL-22, IFNγ, and TNF [33, 36, 76, 175]. Therefore, GM-CSF does not seem to mark T cells with a unique program that is distinct from programs directed by the key master transcription factors for Th1 (T-BET), Th2 (GATA3), and Th17 (RORγt) cells. Hence, we propose GM-CSF expression as part of a general pathogenic program together with other pro-inflammatory molecules such as BHLHE40, IFNγ, and CXCR6 that can be induced across multiple T helper cell subsets [33, 38, 177]. (Figure 3). Conversely, expression of IL-10 and co-inhibitory molecules marks a general anti-inflammatory module that can be induced by IL-27 in any T cell subset [105, 178–180].
Figure 3: Th1 and Th17 cell subsets in tolerance and inflammation.
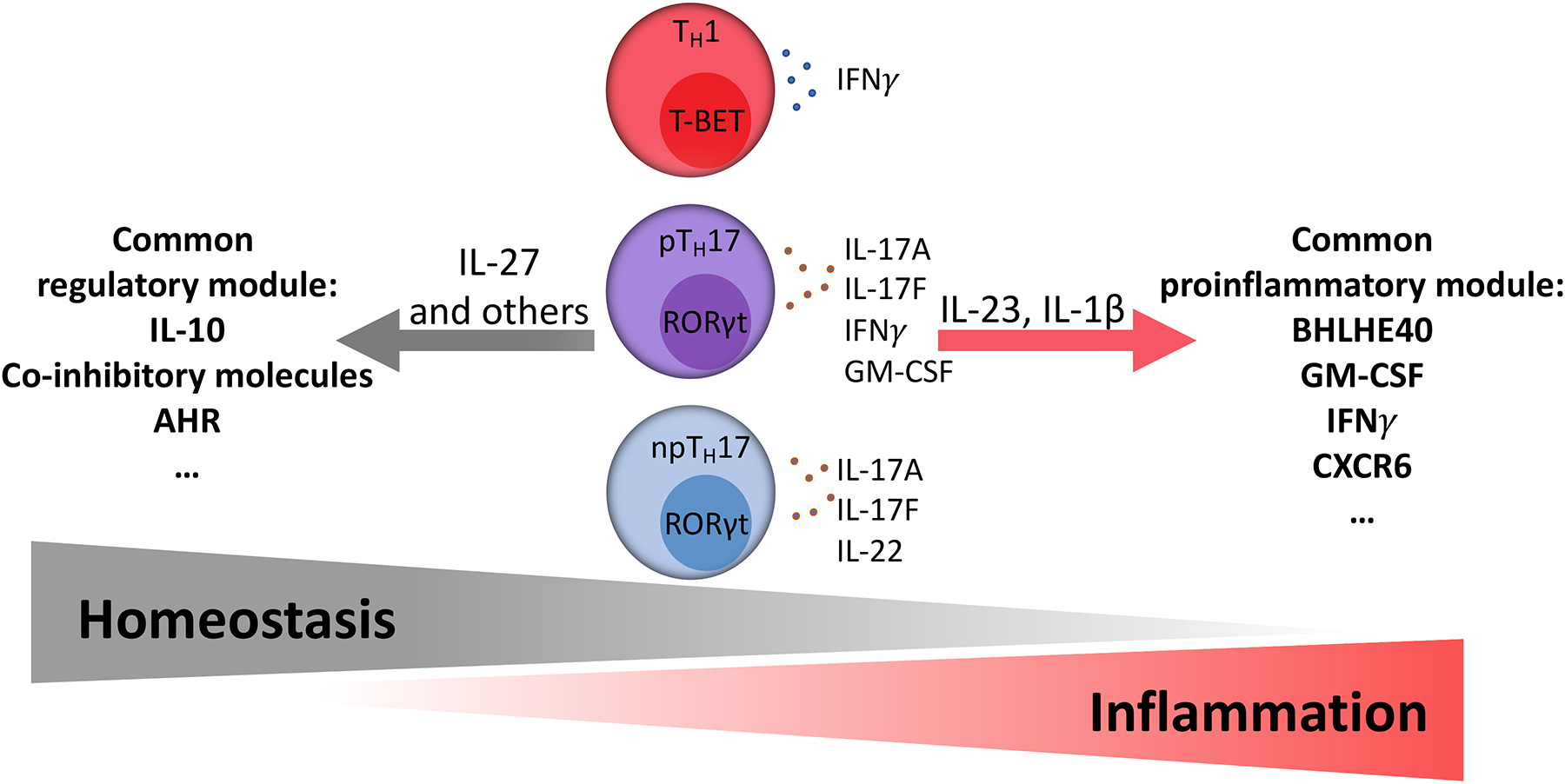
CD4+ T helper cell subsets, including Th1 cells, Th2 cells, and Th17 cells, are characterized by the expression of signature cytokines and master transcription factors. In addition, during inflammation or resolution of inflammation common pro-inflammatory or regulatory modules can be induced in the cells, as shown for Th1 [173] and Th17 cells [53]. IL-23 and IL-1ß induce a common pro-inflammatory module including Bhlhe40, GM-CSF, IFNγ, and CXCR6 [33, 38, 177]. In contrast, IL-27 induces the expression of a common regulatory module that includes anti-inflammatory molecules such as IL-10 and co-inhibitory molecules [105, 180]. The color coding and positioning of the cell types represents pTh17 cells as an intermediate cell type between npTh17 cells and Th1 cells [60]. pTh17= pathogenic Th17 cell, npTh17= non-pathogenic Th17 cell.
Intestinal Th17 cells and autoimmunity
Intestinal Th17 cells are important mediators of intestinal epithelial barrier integrity [130, 157], however, they have also been implicated in driving intestinal inflammation and, interestingly, in regulating autoimmune diseases outside of the intestine (Figure 4). In the intestine, Th17 cytokines IL-17A, IL-17F, and IL-22 were shown to mediate protective functions, but the requirement for IL-23 and RORγt in mouse models of colitis indicates the potential involvement of Th17 cells in inducing intestinal inflammation as well [127, 131, 132]. In Helicobacter hepaticus- dependent colitis, pathobiont-specific inflammatory Th17 cells are major mediators of intestinal inflammation [43]. Expansion of these pro-inflammatory Th17 cells is restrained by H. hepaticus-specific RORγt+FOXP3+ regulatory T cells (iTreg) that are dependent on the expression of the transcription factor c-MAF. Recent data suggest that the induction of these iTreg cells is mediated by a novel population of RORγt+ antigen presenting cells (APCs) [181–183]. Treg-inducing activity of these RORγt+ APCs requires their expression of the MHC class II antigen presentation machinery, CCR7, and αvβ8 integrin that releases active TGF-β from its latent form. The RORγt+ cells have been proposed to be a subset of type 3 innate lymphoid cells (ILC3) or a recently described novel bone marrow-derived AIRE+ cell with features of both dendritic cell and medullary thymic epithelial cells [181–183]. In contrast, induction of the H. hepaticus-specific pathogenic Th17 cells is mediated by APCs that do not require CCR7 and do not express CD11c. While the precise identity of these APCs is not yet known, they are distinct from APCs required for the induction of homeostatic Th17 cells by SFB, as those cells are dependent on CCR7 and express CD11c [182] (unpublished).
Figure 4: Th17 cells in intestinal and extra-intestinal autoimmunity.
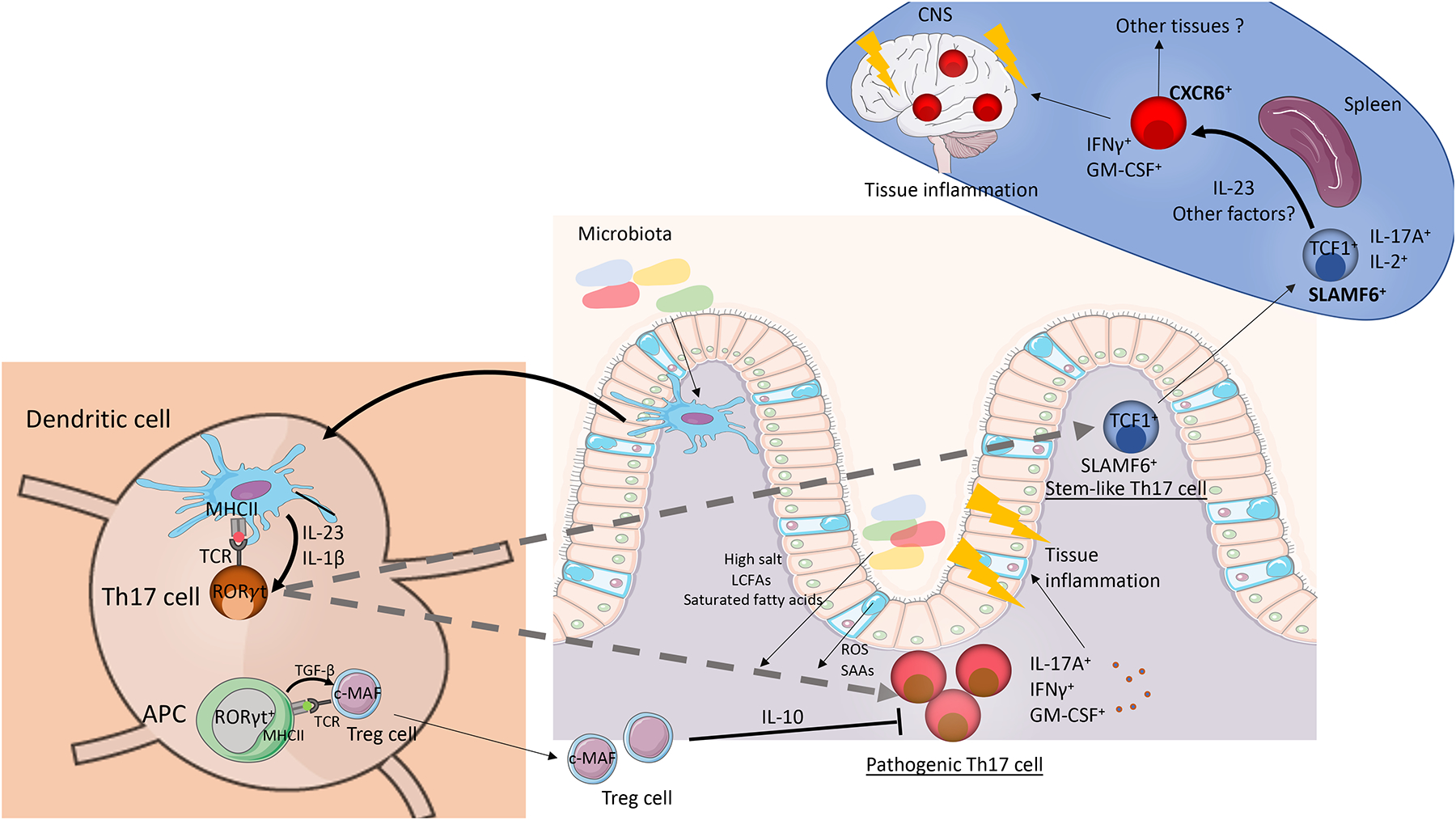
Schematic of the role of intestinal Th17 cells in autoimmune tissue inflammation in the intestine and in extra-intestinal organs. Specific microbiota induce the generation of intestinal Th17 cells [71]. Some of the Th17 cells can give rise to the SLAMF6+ stem-like Th17 subpopulation that circulates through lymphoid organs and intestinal tissues [38]. Under the presence of IL-23 signaling and presentation of auto-antigens by APCs in the spleen, the SLAMF6+ population can give rise to CXCR6+ pathogenic Th17 cells that express high levels of GM-CSF and IFNγ. This CXCR6+ pathogenic Th17 population migrates directly to the inflamed tissue where it mediates autoimmune tissue inflammation. In the intestine diverse factors promote the generation of pathogenic Th17 cells that express high levels of IL-17A, GM-CSF, and IFNγ and drive tissue inflammation. High salt, LCFAs, and saturated fatty acids from the diet and the microbiota and the expression and secretion of SAAs and reactive oxygen species (ROS) by intestinal epithelial cells favor a pathogenic phenotype in Th17 cells [37, 71]. In addition, the secretion of pro-inflammatory cytokines IL-1β and IL-23 by myeloid cells differentiates pathogenic Th17 cells. The pathogenic Th17 population is regulated by a pool of intestinal Treg cells that are dependent on the transcription factor c-MAF [43]. In Helicobacter hepaticus- dependent intestinal colitis, the induction of the H. hepaticus-specific Treg cells is mediated by a recently identified population of RORγt+ APCs, that are equipped to release TGF-β [181–183].
In addition to pro-inflammatory functions in the intestine, the intestinal Th17 population has been implicated in driving extra-intestinal autoimmune diseases. Multiple studies have highlighted a critical role of the intestinal microbiota in the development of extra-intestinal autoimmune diseases [88, 184–186]. The intestinal microbiota is required for the development of EAE, as germ-free mice are protected against EAE [50, 184]. The transfer of intestinal microbiota from patients with MS into mice exacerbates EAE [186]. In a recent study two intestinal microorganisms were found to act together to promote EAE disease [185]. Erysipelotrichaceae acted as an adjuvant that induced Th17 cells in the small intestine and was synergistic in promoting EAE with Lactobacillus reuteri, which possesses peptides that show molecular mimicry with MOG. Mice co-colonized with both strains developed more severe EAE disease than germ-free or mono-colonized mice. Interestingly, the induction of intestinal Th17 cells by gut microbes has been suggested as the mechanism of microbiota-dependent exacerbation of EAE disease in multiple studies [50, 184, 185, 187]. Indeed, in multiple autoimmune disease mouse models, including EAE, autoimmune arthritis, and autoimmune renal disease, the microbiota-specific induction of intestinal Th17 cells was found to be associated with worsened autoimmune disease at extra-intestinal sites [50–52]. However, until recently, the mechanisms by which intestinal Th17 cells influence extra-intestinal autoimmunity remained unclear. One study by Hiltensperger et al. [188] performed site-specific in vivo labeling of T cells in EAE and identified inguinal lymph node-derived T cells (i-T cells) as exhibiting a distinct transcriptomic signature from mesenteric lymph node-derived T cells (m-T cells). While i-T cells are marked by CXCR6 and infiltrate white matter, m-Tcells are marked by P2RX7 and migrate to the gray matter, indicating that the priming-site of T cells regulates their function. In our recent study we combined fate-mapping with single-cell profiling of the transcriptomes and TCR clonotypes of Th17 cells during EAE and identified a homeostatic, stem-like TCF1+ IL-17+ SLAMF6+ Th17 cell population that traffics to the intestine where it is maintained by the microbiota [38]. Interestingly, this intestinal SLAMF6+ population shares TCRs with encephalitogenic GM-CSF+ IFNγ+ CXCR6+ T cells, suggesting that it may provide a reservoir for the IL-23-driven generation of the pathogenic cells. These findings provide a plausible mechanism for how homeostatic intestinal Th17 cells participate in extra-intestinal autoimmune disease. In the future it will be important to identify antigen specificities of the TCRs shared between the SLAMF6 and CXCR6 populations to identify the pathogenic targets and to explore the possibility of the involvement of cross-reactivity to microbiota in tissue-specific autoimmunity. In addition, it will be interesting to reveal the identity of the APCs inducing the different types of Th17 cells and whether homeostatic Th17 cells with potential to further differentiate into pathogenic Th17 cells are primed by the same or different APC subsets. Our data indicate that APCs that direct the differentiation of SFB-specific homeostatic Th17 cells are distinct from those that direct H. hepaticus-specific colitogenic Th17 cells. It is possible that Th17 cells primed by one type of APC can acquire pathogenic functions in the presence of IL-23 upon subsequent stimulation by the same or a different type of APC. It will also be important to understand whether similar mechanisms induce pathogenic Th17 cells in autoimmune inflammation in other tissues besides the CNS. Moreover, if the stem-like, non-pathogenic Th17 cells in the intestine can be further validated as a reservoir for the generation of effector T cells across autoimmune diseases, the population might contribute to the chronicity and relapses observed across many human autoimmune conditions [189]. Hence, the targeting of the conversion of stem-like homeostatic to pathogenic T cells in autoimmunity could yield a novel treatment approach for prevention of relapses. On an additional note, the homeostatic SLAMF6+ cell population shows multiple parallels in transcriptional phenotype and function to previously described stem-like CD8+ T cells in tumors and in response to LCMV infection. Interestingly, anti-tumor immunity [190] and responses to check-point blockade therapy [191–195] have recently been shown to be influenced and regulated by the gut microbiota. Hence, the conversion of stem-like intestinal CD8+ T cells to effector T cells driving anti-tumor immunity could be a mechanism by which gut microbiota influences anti-tumor immunity and response to checkpoint blockade therapy. Future studies will be required to explore these novel directions.
Conclusion and perspective
Th17 cells are an important cell type that exerts both beneficial or pathogenic functions under different conditions and at multiple tissue sites. In the last few years, the use of single-cell technologies has revealed a significant amount of heterogeneity within the Th17 population in vivo, shedding light on mechanisms that underlie the diversity between pathogenic Th17 and homeostatic Th17 states. Furthermore, plasticity has been identified as major characteristic of the Th17 lineage, including the upregulation of discrete effector modules in response to environmental stimuli in different tissues. At homeostasis Th17 cells were found to exhibit strong tissue-specific signatures. In future studies it will be interesting to understand if these tissue-specific signatures represent specialized functional roles acquired by Th17 cells in different tissue microenvironments, similar to tissue Treg cells [106] and tissue macrophages [107].
The role of Th17 cells in autoimmune diseases has been controversial, partly due to the proposal that GM-CSF+ cells make up a pathogenic T cell subset distinct from Th17 cells. However, fate-mapping studies revealed an in vivo relationship of IL-17-expressing Th17 cells and GM-CSF+ T cells, indicating that GM-CSF+ cells are either current Th17 cells or ex-Th17 cells. These analyses highlight the importance of fate-mapping studies and/or TCR clonotype tracing in understanding the trajectory of T cell phenotype development. In future studies, it will be interesting to understand the role of the Th17 program in the context of pathogenic GM-CSF+ IFNγ+ T cells. Why do these cells go through a Th17 trajectory and is that required for their pathogenic potential? Moreover, in addition to IL-23, what are the main drivers converting homeostatic Th17 cells to pathogenic Th17 cells in vivo? Finally, is there a requirement for a stem-like or homeostatic pool of Th17 cells to supply further differentiated pathogenic cells at inception and during the chronic phase of inflammation?
In addition to autoimmunity, Th17 cells have also been implicated in playing a role in cancer. However, current evidence for the role of Th17 cells in cancer is contradictory. While some studies suggest that Th17 cells contribute to carcinogenesis by promoting chronic inflammation, fostering angiogenesis, tumor cell proliferation and migration, other studies suggest an anti-tumor function of Th17 cells by enhancing cytotoxic T cell and NK cell responses and neutrophil infiltration [196–202]. The differences in Th17 cell functions in tumors seem to be tumor context-dependent. The comprehension of the processes controlling anti- and pro-tumor functions of Th17 cells will likely provide important insight for developing effective cancer immunotherapy.
Our understanding of the dichotomy within the Th17 cellular compartment creates opportunities for improved strategies for the treatment of autoimmune and inflammatory diseases. The exacerbation of IBD following IL-17 blockade [29, 49] highlights the need for specific targeting of the pathogenic Th17 population, while leaving Th17 cells that mediate barrier functions and homeostasis intact. Indeed, strategies focused on the transition of homeostatic Th17 cells to pathogenic Th17 cells, beyond IL-23 blockade, could represent a novel approach that would leave homeostatic functions intact while limiting tissue damage mediated by pathogenic Th17 cells.
Acknowledgments
We would like to thank Mary Collins for critical feedback on the manuscript. The authors thank laboratory members of the Littman and Kuchroo laboratories for contributions to some of the discussed studies. This work was supported by National Institutes of Health grants R01NS045937, R01NS30843, R01AI144166, P01AI073748, P01AI039671, and P01AI056299 (to V.K.K.) R01AI158687 and RO1CA255635 (to D.R.L.) and A.S. was supported by a German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes) PhD fellowship.
Footnotes
Competing interests
V.K.K. is cofounder of Celsius Therapeutics, Tizona Therapeutics, Larkspur Biosciences and Bicara Therapeutics. His interests are reviewed and managed by the Brigham and Women’s Hospital and Partners Healthcare in accordance with their conflict of interest policies. D.R.L. is co-founder of Vedanta Biosciences and Immunai, on advisory boards of Chemocentryx and Imidomics, and on the board of directors of Pfizer, Inc. All other authors declare no competing interests.
References
- 1.Mosmann TR and Coffman RL, TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol, 1989. 7: p. 145–73. [DOI] [PubMed] [Google Scholar]
- 2.Krakowski M and Owens T, Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol, 1996. 26(7): p. 1641–6. [DOI] [PubMed] [Google Scholar]
- 3.Tran EH, Prince EN, and Owens T, IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol, 2000. 164(5): p. 2759–68. [DOI] [PubMed] [Google Scholar]
- 4.Gran B, et al. , IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol, 2002. 169(12): p. 7104–10. [DOI] [PubMed] [Google Scholar]
- 5.Zhang GX, et al. , Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol, 2003. 170(4): p. 2153–60. [DOI] [PubMed] [Google Scholar]
- 6.Oppmann B, et al. , Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity, 2000. 13(5): p. 715–25. [DOI] [PubMed] [Google Scholar]
- 7.Cua DJ, et al. , Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature, 2003. 421(6924): p. 744–8. [DOI] [PubMed] [Google Scholar]
- 8.Langrish CL, et al. , IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med, 2005. 201(2): p. 233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals the role of IL-23 in expanding pathogenic Th17 cells and the capability of Th17 cells to induce autoimmunity, paving the road for the analysis of the role of Th17 cells in autoimmune diseases.
- 9.Bettelli E and Kuchroo VK, IL-12- and IL-23-induced T helper cell subsets: birds of the same feather flock together. J Exp Med, 2005. 201(2): p. 169–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aggarwal S, et al. , Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem, 2003. 278(3): p. 1910–4. [DOI] [PubMed] [Google Scholar]
- 11.Harrington LE, et al. , Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol, 2005. 6(11): p. 1123–32. [DOI] [PubMed] [Google Scholar]
- 12.Park H, et al. , A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol, 2005. 6(11): p. 1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veldhoen M, et al. , TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity, 2006. 24(2): p. 179–89. [DOI] [PubMed] [Google Scholar]
- 14.Bettelli E, et al. , Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature, 2006. 441(7090): p. 235–8. [DOI] [PubMed] [Google Scholar]
- 15.Mangan PR, et al. , Transforming growth factor-beta induces development of the T(H)17 lineage. Nature, 2006. 441(7090): p. 231–4. [DOI] [PubMed] [Google Scholar]
- 16.Ivanov II, et al. , The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell, 2006. 126(6): p. 1121–33. [DOI] [PubMed] [Google Scholar]; This study identifies RORγt as the master transcription factor of the Th17 lineage.
- 17.Brustle A, et al. , The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol, 2007. 8(9): p. 958–66. [DOI] [PubMed] [Google Scholar]
- 18.Schraml BU, et al. , The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature, 2009. 460(7253): p. 405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou L and Littman DR, Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol, 2009. 21(2): p. 146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yosef N, et al. , Dynamic regulatory network controlling TH17 cell differentiation. Nature, 2013. 496(7446): p. 461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zemmour D, Kiner E, and Benoist C, CD4(+) teff cell heterogeneity: the perspective from single-cell transcriptomics. Curr Opin Immunol, 2020. 63: p. 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuzlak S, et al. , Repositioning TH cell polarization from single cytokines to complex help. Nat Immunol, 2021. 22(10): p. 1210–1217. [DOI] [PubMed] [Google Scholar]
- 23.Smith KM, et al. , Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol, 2000. 165(6): p. 3136–44. [DOI] [PubMed] [Google Scholar]
- 24.Vazquez MI, Catalan-Dibene J, and Zlotnik A, B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine, 2015. 74(2): p. 318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gieseck RL 3rd, Wilson MS, and Wynn TA, Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol, 2018. 18(1): p. 62–76. [DOI] [PubMed] [Google Scholar]
- 26.Hirota K, et al. , Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol, 2013. 14(4): p. 372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitsdoerffer M, et al. , Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A, 2010. 107(32): p. 14292–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwicky P, Unger S, and Becher B, Targeting interleukin-17 in chronic inflammatory disease: A clinical perspective. J Exp Med, 2020. 217(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel DD and Kuchroo VK, Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity, 2015. 43(6): p. 1040–51. [DOI] [PubMed] [Google Scholar]
- 30.Havrdova E, et al. , Activity of secukinumab, an anti-IL-17A antibody, on brain lesions in RRMS: results from a randomized, proof-of-concept study. J Neurol, 2016. 263(7): p. 1287–95. [DOI] [PubMed] [Google Scholar]
- 31.Haak S, et al. , IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest, 2009. 119(1): p. 61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGinley AM, et al. , Interleukin-17A Serves a Priming Role in Autoimmunity by Recruiting IL-1beta-Producing Myeloid Cells that Promote Pathogenic T Cells. Immunity, 2020. 52(2): p. 342–356 e6. [DOI] [PubMed] [Google Scholar]
- 33.Komuczki J, et al. , Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1beta. Immunity, 2019. 50(5): p. 1289–1304 e6. [DOI] [PubMed] [Google Scholar]
- 34.Rasouli J, et al. , A distinct GM-CSF(+) T helper cell subset requires T-bet to adopt a TH1 phenotype and promote neuroinflammation. Sci Immunol, 2020. 5(52). [DOI] [PubMed] [Google Scholar]
- 35.Codarri L, et al. , RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol, 2011. 12(6): p. 560–7. [DOI] [PubMed] [Google Scholar]
- 36.Galli E, et al. , GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med, 2019. 25(8): p. 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stockinger B and Omenetti S, The dichotomous nature of T helper 17 cells. Nat Rev Immunol, 2017. 17(9): p. 535–544. [DOI] [PubMed] [Google Scholar]
- 38.Schnell A, et al. , Stem-like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell, 2021. 184(26): p. 6281–6298 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first analysis of combined single-cell RNA-sequencing and TCR-sequencing of tissue Th17 cells during homeostasis and EAE, identifying a stem-like, intestinal Th17 population that gives rise to pathogenic, effector Th17 cells.
- 39.Ghoreschi K, et al. , Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature, 2010. 467(7318): p. 967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGeachy MJ, et al. , TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol, 2007. 8(12): p. 1390–7. [DOI] [PubMed] [Google Scholar]
- 41.Esplugues E, et al. , Control of TH17 cells occurs in the small intestine. Nature, 2011. 475(7357): p. 514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Omenetti S, et al. , The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity, 2019. 51(1): p. 77–89 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu M, et al. , c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature, 2018. 554(7692): p. 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blaschitz C and Raffatellu M, Th17 cytokines and the gut mucosal barrier. J Clin Immunol, 2010. 30(2): p. 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kinugasa T, et al. , Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology, 2000. 118(6): p. 1001–11. [DOI] [PubMed] [Google Scholar]
- 46.Cao AT, et al. , Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol, 2012. 189(9): p. 4666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernandez-Santos N and Gaffen SL, Th17 cells in immunity to Candida albicans. Cell Host Microbe, 2012. 11(5): p. 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bedoya SK, et al. , Th17 cells in immunity and autoimmunity. Clin Dev Immunol, 2013. 2013: p. 986789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hueber W, et al. , Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut, 2012. 61(12): p. 1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]; An important clinical study showing that IL-17 blockade in Crohn’s disease is not only ineffective but exacerbates disease.
- 50.Lee YK, et al. , Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A, 2011. 108 Suppl 1: p. 4615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes that the induction of intestinal Th17 cells with SFB exacerbates EAE disease, providing evidence for a role of the intestinal Th17 population in extra-intestinal autoimmune disease.
- 51.Wu HJ, et al. , Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity, 2010. 32(6): p. 815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krebs CF, et al. , Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity, 2016. 45(5): p. 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee Y, et al. , Induction and molecular signature of pathogenic TH17 cells. Nat Immunol, 2012. 13(10): p. 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the differentiation conditions and transcriptomic signature of pathogenic Th17 cells.
- 54.Gaublomme JT, et al. , Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell, 2015. 163(6): p. 1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first single-cell RNA-sequencing study of CD4+ T cells analyzing heterogeneity of in vitro- and in vivo-derived Th17 cells to identify novel regulators of Th17 cells.
- 55.Meyer Zu Horste G, et al. , RBPJ Controls Development of Pathogenic Th17 Cells by Regulating IL-23 Receptor Expression. Cell Rep, 2016. 16(2): p. 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kishi Y, et al. , Protein C receptor (PROCR) is a negative regulator of Th17 pathogenicity. J Exp Med, 2016. 213(11): p. 2489–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang C, et al. , CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell, 2015. 163(6): p. 1413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meyer Zu Horste G, et al. , Fas Promotes T Helper 17 Cell Differentiation and Inhibits T Helper 1 Cell Development by Binding and Sequestering Transcription Factor STAT1. Immunity, 2018. 48(3): p. 556–569 e7. [DOI] [PubMed] [Google Scholar]
- 59.Ciofani M, et al. , A validated regulatory network for Th17 cell specification. Cell, 2012. 151(2): p. 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thakore PI, et al. , The chromatin landscape of Th17 cells reveals mechanisms of diversification of regulatory and pro-inflammatory states. bioRxiv, 2022: p. 2022.02.26.482041. [Google Scholar]
- 61.Aschenbrenner D, et al. , An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat Immunol, 2018. 19(10): p. 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.International Multiple Sclerosis Genetics, C., Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science, 2019. 365(6460). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JY, et al. , Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell, 2020. 180(1): p. 79–91 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sallusto F, Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol, 2016. 34: p. 317–34. [DOI] [PubMed] [Google Scholar]
- 65.Acosta-Rodriguez EV, et al. , Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol, 2007. 8(6): p. 639–46. [DOI] [PubMed] [Google Scholar]
- 66.Zielinski CE, et al. , Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature, 2012. 484(7395): p. 514–8. [DOI] [PubMed] [Google Scholar]
- 67.Okada S, et al. , IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science, 2015. 349(6248): p. 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Puel A, et al. , Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science, 2011. 332(6025): p. 65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma CS, et al. , Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med, 2008. 205(7): p. 1551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weaver CT, et al. , The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol, 2013. 8: p. 477–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Honda K and Littman DR, The microbiota in adaptive immune homeostasis and disease. Nature, 2016. 535(7610): p. 75–84. [DOI] [PubMed] [Google Scholar]
- 72.Ivanov II, et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell, 2009. 139(3): p. 485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows the specific induction of intestinal Th17 cells by SFB.
- 73.Atarashi K, et al. , Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell, 2015. 163(2): p. 367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ladinsky MS, et al. , Endocytosis of commensal antigens by intestinal epithelial cells regulates mucosal T cell homeostasis. Science, 2019. 363(6431). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sano T, et al. , An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell, 2015. 163(2): p. 381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hirota K, et al. , Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol, 2011. 12(3): p. 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes the first IL-17-fate-mapping mouse and shows extensive plasticity of Th17 cells during EAE, demonstrating that different pro-inflammatory cytokines are expressed by ex-Th17 cells.
- 77.Ahlfors H, et al. , IL-22 fate reporter reveals origin and control of IL-22 production in homeostasis and infection. J Immunol, 2014. 193(9): p. 4602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Basu R, et al. , Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity, 2012. 37(6): p. 1061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan TG, et al. , Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A, 2016. 113(50): p. E8141–E8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Geva-Zatorsky N, et al. , Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell, 2017. 168(5): p. 928–943 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li XV, Leonardi I, and Iliev ID, Gut Mycobiota in Immunity and Inflammatory Disease. Immunity, 2019. 50(6): p. 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leonardi I, et al. , Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chudnovskiy A, et al. , Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell, 2016. 167(2): p. 444–456 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu C, et al. , Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature, 2013. 496(7446): p. 513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleinewietfeld M, et al. , Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature, 2013. 496(7446): p. 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilck N, et al. , Salt-responsive gut commensal modulates TH17 axis and disease. Nature, 2017. 551(7682): p. 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kawano Y, et al. , Microbiota imbalance induced by dietary sugar disrupts immune-mediated protection from metabolic syndrome. Cell, 2022. 185(19): p. 3501–3519 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haghikia A, et al. , Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity, 2015. 43(4): p. 817–29. [DOI] [PubMed] [Google Scholar]
- 89.Berod L, et al. , De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med, 2014. 20(11): p. 1327–33. [DOI] [PubMed] [Google Scholar]
- 90.Hang S, et al. , Bile acid metabolites control TH17 and Treg cell differentiation. Nature, 2019. 576(7785): p. 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Paik D, et al. , Human gut bacteria produce TauEta17-modulating bile acid metabolites. Nature, 2022. 603(7903): p. 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sato Y, et al. , Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature, 2021. 599(7885): p. 458–464. [DOI] [PubMed] [Google Scholar]
- 93.Chen ML, et al. , CAR directs T cell adaptation to bile acids in the small intestine. Nature, 2021. 593(7857): p. 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu L, et al. , Niche-Selective Inhibition of Pathogenic Th17 Cells by Targeting Metabolic Redundancy. Cell, 2020. 182(3): p. 641–654 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karmaus PWF, et al. , Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature, 2019. 565(7737): p. 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wagner A, et al. , Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell, 2021. 184(16): p. 4168–4185 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Puleston DJ, et al. , Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell, 2021. 184(16): p. 4186–4202 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Komatsu N, et al. , Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med, 2014. 20(1): p. 62–8. [DOI] [PubMed] [Google Scholar]
- 99.Shi LZ, et al. , HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med, 2011. 208(7): p. 1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dang EV, et al. , Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell, 2011. 146(5): p. 772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X, et al. , Febrile Temperature Critically Controls the Differentiation and Pathogenicity of T Helper 17 Cells. Immunity, 2020. 52(2): p. 328–341 e5. [DOI] [PubMed] [Google Scholar]
- 102.Spiljar M, et al. , Cold exposure protects from neuroinflammation through immunologic reprogramming. Cell Metab, 2021. 33(11): p. 2231–2246 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gribonika I, et al. , Peyer’s patch TH17 cells are dispensable for gut IgA responses to oral immunization. Sci Immunol, 2022. 7(73): p. eabc5500. [DOI] [PubMed] [Google Scholar]
- 104.Gagliani N, et al. , Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature, 2015. 523(7559): p. 221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang H, et al. , An IL-27-Driven Transcriptional Network Identifies Regulators of IL-10 Expression across T Helper Cell Subsets. Cell Rep, 2020. 33(8): p. 108433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Munoz-Rojas AR and Mathis D, Tissue regulatory T cells: regulatory chameleons. Nat Rev Immunol, 2021. 21(9): p. 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lavin Y, et al. , Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell, 2014. 159(6): p. 1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Spiljar M and Kuchroo VK, Metabolic regulation and function of T helper cells in neuroinflammation. Semin Immunopathol, 2022. [DOI] [PubMed] [Google Scholar]
- 109.Sefik E, et al. , MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science, 2015. 349(6251): p. 993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pratama A, et al. , Developmental and cellular age direct conversion of CD4+ T cells into RORgamma+ or Helios+ colon Treg cells. J Exp Med, 2020. 217(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Komiyama Y, et al. , IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol, 2006. 177(1): p. 566–73. [DOI] [PubMed] [Google Scholar]
- 112.Yang XO, et al. , Regulation of inflammatory responses by IL-17F. J Exp Med, 2008. 205(5): p. 1063–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hofstetter HH, et al. , Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol, 2005. 237(2): p. 123–30. [DOI] [PubMed] [Google Scholar]
- 114.Bettelli E, et al. , Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med, 2003. 197(9): p. 1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jager A, et al. , Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol, 2009. 183(11): p. 7169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Regen T, et al. , IL-17 controls central nervous system autoimmunity through the intestinal microbiome. Sci Immunol, 2021. 6(56). [DOI] [PubMed] [Google Scholar]
- 117.Zheng Y, et al. , Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature, 2007. 445(7128): p. 648–51. [DOI] [PubMed] [Google Scholar]
- 118.Ma HL, et al. , IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest, 2008. 118(2): p. 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li B, et al. , The role of Th17 cells in psoriasis. Immunol Res, 2020. 68(5): p. 296–309. [DOI] [PubMed] [Google Scholar]
- 120.Cai Y, et al. , Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity, 2011. 35(4): p. 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pantelyushin S, et al. , Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest, 2012. 122(6): p. 2252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nakae S, et al. , Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol, 2003. 171(11): p. 6173–7. [DOI] [PubMed] [Google Scholar]
- 123.Nakae S, et al. , IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci U S A, 2003. 100(10): p. 5986–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lubberts E, IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine, 2008. 41(2): p. 84–91. [DOI] [PubMed] [Google Scholar]
- 125.Noguchi D, et al. , Blocking of IL-6 signaling pathway prevents CD4+ T cell-mediated colitis in a T(h)17-independent manner. Int Immunol, 2007. 19(12): p. 1431–40. [DOI] [PubMed] [Google Scholar]
- 126.Izcue A, et al. , Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity, 2008. 28(4): p. 559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leppkes M, et al. , RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology, 2009. 136(1): p. 257–67. [DOI] [PubMed] [Google Scholar]
- 128.Ogawa A, et al. , Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol, 2004. 110(1): p. 55–62. [DOI] [PubMed] [Google Scholar]
- 129.O’Connor W Jr., et al. , A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol, 2009. 10(6): p. 603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li J, Casanova JL, and Puel A, Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol, 2018. 11(3): p. 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Uhlig HH, et al. , Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity, 2006. 25(2): p. 309–18. [DOI] [PubMed] [Google Scholar]
- 132.Kullberg MC, et al. , IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med, 2006. 203(11): p. 2485–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yen D, et al. , IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest, 2006. 116(5): p. 1310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Teunissen MB, et al. , Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol, 1998. 111(4): p. 645–9. [DOI] [PubMed] [Google Scholar]
- 135.Fotiadou C, et al. , IL-17A, IL-22, and IL-23 as Markers of Psoriasis Activity: A Cross-sectional, Hospital-based Study. J Cutan Med Surg, 2015. 19(6): p. 555–60. [DOI] [PubMed] [Google Scholar]
- 136.Papp KA, et al. , Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet, 2008. 371(9625): p. 1675–84. [DOI] [PubMed] [Google Scholar]
- 137.Reich K, et al. , Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet, 2017. 390(10091): p. 276–288. [DOI] [PubMed] [Google Scholar]
- 138.Gordon KB, et al. , Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet, 2018. 392(10148): p. 650–661. [DOI] [PubMed] [Google Scholar]
- 139.Langley RG, et al. , Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med, 2014. 371(4): p. 326–38. [DOI] [PubMed] [Google Scholar]; Two clinical trials demonstrating high efficacy of secukinumab in psoriasis with superior efficacy to previously approved psoriasis therapies.
- 140.Griffiths CE, et al. , Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet, 2015. 386(9993): p. 541–51. [DOI] [PubMed] [Google Scholar]
- 141.McInnes IB, et al. , Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 2015. 386(9999): p. 1137–46. [DOI] [PubMed] [Google Scholar]
- 142.Mease PJ, et al. , Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis, 2017. 76(1): p. 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Baeten D, et al. , Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N Engl J Med, 2015. 373(26): p. 2534–48. [DOI] [PubMed] [Google Scholar]
- 144.Lock C, et al. , Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med, 2002. 8(5): p. 500–8. [DOI] [PubMed] [Google Scholar]
- 145.Kebir H, et al. , Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med, 2007. 13(10): p. 1173–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Segal BM, et al. , Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol, 2008. 7(9): p. 796–804. [DOI] [PubMed] [Google Scholar]
- 147.Ziolkowska M, et al. , High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol, 2000. 164(5): p. 2832–8. [DOI] [PubMed] [Google Scholar]
- 148.Honorati MC, et al. , High in vivo expression of interleukin-17 receptor in synovial endothelial cells and chondrocytes from arthritis patients. Rheumatology (Oxford), 2001. 40(5): p. 522–7. [DOI] [PubMed] [Google Scholar]
- 149.Kirkham BW, et al. , Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum, 2006. 54(4): p. 1122–31. [DOI] [PubMed] [Google Scholar]
- 150.Hueber W, et al. , Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med, 2010. 2(52): p. 52ra72. [DOI] [PubMed] [Google Scholar]
- 151.Genovese MC, et al. , A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol, 2014. 66(7): p. 1693–704. [DOI] [PubMed] [Google Scholar]
- 152.Blanco FJ, et al. , Secukinumab in Active Rheumatoid Arthritis: A Phase III Randomized, Double-Blind, Active Comparator- and Placebo-Controlled Study. Arthritis Rheumatol, 2017. 69(6): p. 1144–1153. [DOI] [PubMed] [Google Scholar]
- 153.Fujino S, et al. , Increased expression of interleukin 17 in inflammatory bowel disease. Gut, 2003. 52(1): p. 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Duerr RH, et al. , A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science, 2006. 314(5804): p. 1461–3. [DOI] [PMC free article] [PubMed] [Google Scholar]; Genome-wide association study in inflammatory bowel disease identifying the IL23R gene highly associated with disease.
- 155.Feagan BG, et al. , Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet, 2017. 389(10080): p. 1699–1709. [DOI] [PubMed] [Google Scholar]
- 156.Sands BE, et al. , Efficacy and Safety of MEDI2070, an Antibody Against Interleukin 23, in Patients With Moderate to Severe Crohn’s Disease: A Phase 2a Study. Gastroenterology, 2017. 153(1): p. 77–86 e6. [DOI] [PubMed] [Google Scholar]
- 157.Lee JS, et al. , Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity, 2015. 43(4): p. 727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Harbour SN, et al. , Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A, 2015. 112(22): p. 7061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee YK, et al. , Late developmental plasticity in the T helper 17 lineage. Immunity, 2009. 30(1): p. 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Annunziato F, et al. , Phenotypic and functional features of human Th17 cells. J Exp Med, 2007. 204(8): p. 1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kebir H, et al. , Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol, 2009. 66(3): p. 390–402. [DOI] [PubMed] [Google Scholar]
- 162.Nistala K, et al. , Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A, 2010. 107(33): p. 14751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mazzoni A, et al. , Biological and clinical significance of T helper 17 cell plasticity. Immunology, 2019. 158(4): p. 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Reinert-Hartwall L, et al. , Th1/Th17 plasticity is a marker of advanced beta cell autoimmunity and impaired glucose tolerance in humans. J Immunol, 2015. 194(1): p. 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Paroni M, et al. , Recognition of viral and self-antigens by TH1 and TH1/TH17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J Allergy Clin Immunol, 2017. 140(3): p. 797–808. [DOI] [PubMed] [Google Scholar]
- 166.Tom MR, et al. , Novel CD8+ T-Cell Subsets Demonstrating Plasticity in Patients with Inflammatory Bowel Disease. Inflamm Bowel Dis, 2016. 22(7): p. 1596–608. [DOI] [PubMed] [Google Scholar]
- 167.Smillie CS, et al. , Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell, 2019. 178(3): p. 714–730 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jain R, et al. , Interleukin-23-Induced Transcription Factor Blimp-1 Promotes Pathogenicity of T Helper 17 Cells. Immunity, 2016. 44(1): p. 131–142. [DOI] [PubMed] [Google Scholar]
- 169.Malik S, Want MY, and Awasthi A, The Emerging Roles of Gamma-Delta T Cells in Tissue Inflammation in Experimental Autoimmune Encephalomyelitis. Front Immunol, 2016. 7: p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Takatori H, et al. , Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med, 2009. 206(1): p. 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Buonocore S, et al. , Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature, 2010. 464(7293): p. 1371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang H, et al. , IL-23 costimulates antigen-specific MAIT cell activation and enables vaccination against bacterial infection. Sci Immunol, 2019. 4(41). [DOI] [PubMed] [Google Scholar]
- 173.Pawlak M, et al. , Induction of a colitogenic phenotype in Th1-like cells depends on interleukin-23 receptor signaling. Immunity, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sheng W, et al. , STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res, 2014. 24(12): p. 1387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ingelfinger F, De Feo D, and Becher B, GM-CSF: Master regulator of the T cell-phagocyte interface during inflammation. Semin Immunol, 2021. 54: p. 101518. [DOI] [PubMed] [Google Scholar]
- 176.McQualter JL, et al. , Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med, 2001. 194(7): p. 873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lin CC, et al. , Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat Commun, 2014. 5: p. 3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fitzgerald DC, et al. , Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol, 2007. 8(12): p. 1372–9. [DOI] [PubMed] [Google Scholar]
- 179.Awasthi A, et al. , A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol, 2007. 8(12): p. 1380–9. [DOI] [PubMed] [Google Scholar]
- 180.Chihara N, et al. , Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature, 2018. 558(7710): p. 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang J, et al. , Single-cell multiomics defines tolerogenic extrathymic Aire-expressing populations with unique homology to thymic epithelium. Sci Immunol, 2021. 6(65): p. eabl5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kedmi R, et al. , A RORgammat(+) cell instructs gut microbiota-specific Treg cell differentiation. Nature, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Akagbosu B, et al. , Novel antigen presenting cell imparts Treg-dependent tolerance to gut microbiota. Nature, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Berer K, et al. , Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature, 2011. 479(7374): p. 538–41. [DOI] [PubMed] [Google Scholar]; This article shows the involvement of the intestinal microbiota in the development of EAE.
- 185.Miyauchi E, et al. , Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature, 2020. 585(7823): p. 102–106. [DOI] [PubMed] [Google Scholar]
- 186.Berer K, et al. , Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A, 2017. 114(40): p. 10719–10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cosorich I, et al. , High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv, 2017. 3(7): p. e1700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hiltensperger M, et al. , Skin and gut imprinted helper T cell subsets exhibit distinct functional phenotypes in central nervous system autoimmunity. Nat Immunol, 2021. 22(7): p. 880–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rosenblum MD, Remedios KA, and Abbas AK, Mechanisms of human autoimmunity. J Clin Invest, 2015. 125(6): p. 2228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zitvogel L, et al. , Cancer and the gut microbiota: an unexpected link. Sci Transl Med, 2015. 7(271): p. 271ps1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vetizou M, et al. , Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science, 2015. 350(6264): p. 1079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sivan A, et al. , Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science, 2015. 350(6264): p. 1084–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Routy B, et al. , Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science, 2018. 359(6371): p. 91–97. [DOI] [PubMed] [Google Scholar]
- 194.Matson V, et al. , The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science, 2018. 359(6371): p. 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gopalakrishnan V, et al. , Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science, 2018. 359(6371): p. 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang JP, et al. , Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol, 2009. 50(5): p. 980–9. [DOI] [PubMed] [Google Scholar]
- 197.Sun Y, et al. , IL-17/miR-192/IL-17Rs regulatory feedback loop facilitates multiple myeloma progression. PLoS One, 2014. 9(12): p. e114647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huang Q, et al. , IL-17 Promotes Angiogenic Factors IL-6, IL-8, and Vegf Production via Stat1 in Lung Adenocarcinoma. Sci Rep, 2016. 6: p. 36551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lv L, et al. , The accumulation and prognosis value of tumor infiltrating IL-17 producing cells in esophageal squamous cell carcinoma. PLoS One, 2011. 6(3): p. e18219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lin Y, et al. , Interleukin-17 is a favorable prognostic marker for colorectal cancer. Clin Transl Oncol, 2015. 17(1): p. 50–6. [DOI] [PubMed] [Google Scholar]
- 201.Asadzadeh Z, et al. , The paradox of Th17 cell functions in tumor immunity. Cell Immunol, 2017. 322: p. 15–25. [DOI] [PubMed] [Google Scholar]
- 202.Knochelmann HM, et al. , When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol, 2018. 15(5): p. 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]