

Abstract
Background
Muscular dystrophies (MDs) are a large, heterogeneous group of degenerative muscle diseases. X‐linked dystrophin‐deficient MD in cats is the first genetically characterized cat model for a human disease and a few novel forms have been identified.
Hypothesis/Objectives
Muscular dystrophy was suspected in a young male domestic shorthair cat. Clinical, molecular, and genetic techniques could provide a definitive diagnosis.
Animals
A 1‐year‐old male domestic shorthair cat presented for progressive difficulty walking, macroglossia and dysphagia beginning at 6 months of age. The tongue was thickened, protruded with constant ptyalism, and thickening and rigidity of the neck and shoulders were observed.
Methods
A complete neurological examination, baseline laboratory evaluation and biopsies of the trapezius muscle were performed with owner consent. Indirect immunofluorescence staining of muscle cryosections was performed using several monoclonal and polyclonal antibodies against dystrophy‐associated proteins. DNA was isolated for genomic analyses by whole genome sequencing and comparison to DNA variants in the 99 Lives Cat Genome Sequencing dataset.
Results and Clinical Importance
Aspartate aminotransferase (687 IU/L) and creatine kinase (24 830 IU/L) activities were increased and mild hypokalemia (3.7 mmol/L) was present. Biopsy samples from the trapezius muscle confirmed a degenerative and regenerative myopathy and protein alterations identified by immunohistochemistry resulted in a diagnosis of a in dystrophin‐deficient form of X‐linked MD. A stop gain variant (c.4849C>T; p.Gln1617Ter) dystrophin was identified by genome sequencing. Precision/genomic medicine efforts for the domestic cat and in veterinary medicine support disease variant and animal model discovery and provide opportunities for targeted treatments for companion animals.
Keywords: animal models, Felis catus, precision medicine, whole genome sequencing
Abbreviations DNA deoxyribonucleic acid
- CK
creatine kinase
- DMD
dystrophin
- FF
superfine
- Gln
glutamic acid
- LoF
loss of function
- MD
muscular dystrophies
- MU
University of Missouri
- NCBI
National Center for Biotechnology Information
- OMIA
online Mendelian inheritance in animals
- TAE
tris(hydroxymethyl)aminomethane—acetic acid—ethylenediaminetetraacetic acid
- Ter
termination amino acid
- WES
whole exome sequencing
- WGS
whole genome sequencing
1. INTRODUCTION
Muscular dystrophies (MD) are a large and heterogeneous group of degenerative muscle diseases. 1 The most common form of MD in people is associated with deficiency of the subsarcolemmal membrane protein dystrophin, referred to as a dystrophinopathy, with many mutations in the dystrophin (DMD) gene having been identified. 2 The most severe form of the disease in people is referred to as Duchenne MD and the milder form as Becker MD. 3 In animals, a spontaneously‐occurring dystrophin‐deficient mouse (mdx) was described in 1984. 4 In dogs, a recent review identified dystrophin‐deficient MD in 16 different dog breeds with 20 different variants in the DMD gene. 5 X‐linked dystrophin‐deficient MD in the Golden retriever, 6 Labrador retriever, 7 , 8 and CXMD dogs in Japan 9 among others 10 has been used as a large animal model of dystrophinopathy in pre‐clinical trials. 11 , 12 In animals, dystrophin‐deficient MD should be referred to as X‐linked dystrophin‐deficient MD and not Duchenne or Becker MD, which refer to the disease in humans.
In contrast, clinical and pathological descriptions of dystrophin‐deficient MD in cats are rare and only three variants have been confirmed. The disease first was described in 1989. 13 In 1992, two 5‐month‐old male domestic shorthair littermates with muscle hypertrophy as a prominent clinical feature of the disease were described, 14 followed in 1994 by the first molecular description of a deletion in the DMD promotor in a male cat with similar clinical and histopathologic findings as the previous reports. 15 Recently, a nonsense variant was identified in a family of Maine coon cats 16 and a missense variant in a family of Maine coon crossbred cats. 17 Herein, we report a highly deleterious variant causing loss of function (LoF) and likely underlying dystrophin‐deficient MD in a random bred cat from the United Kingdom.
Other forms of MD can occur in cats, including congenital MD associated with laminin α2‐deficiency 18 , 19 , 20 and limb‐girdle MD associated with β‐sarcoglycan deficiency, 21 , 22 although the causal gene variants have not yet been identified. Clinical examination cannot distinguish among various myopathies including the MDs, and further diagnostic testing is necessary including evaluation of muscle biopsy samples by histopathology, histochemistry, immunohistochemistry, and measurement of serum creatine kinase (CK) activity. With the improvement of molecular techniques, such as short‐read whole genome sequencing (WGS) and, to some extent, whole exome sequencing (WES), distinguishing among various myopathies is now possible by rapid identification of the underlying variant responsible for causing disease, 23 , 24 including MD in both dogs and cats. 17 , 25
Increased availability of next‐generation sequencing allows identification of new pathogenic genes and the wide spectrum of clinical phenotypes associated with known defective proteins. The deletion in the muscle promoter of DMD was 1 of the earliest genetic variants discovered in the cat. 9 Currently, approximately 185 causal DNA variants are suggested in the cat for a variety of diseases and traits, 26 including specific variants in DMD. As wider use of WGS and WES becomes available at more affordable prices and annotation and assembly of the reference genomes improve, the spectrum of genetic variants resulting in MDs in cats will expand.
2. MATERIALS AND METHODS
2.1. Animals
A 1‐year‐old male indoor domestic shorthair cat (random bred; Fcat‐23286) was referred to a specialty veterinary hospital for progressive difficulty walking, macroglossia, and dysphagia. Because a congenital neuromuscular disease was suspected, a complete neurological examination, baseline laboratory evaluation and muscle biopsy were performed with owner consent. Approximately 5 mL of EDTA whole blood also was collected by venipuncture from an unaffected female littermate for DNA extraction to genotype candidate causal variants with informed consent from the owner.
2.2. Histopathology and immunofluorescent staining
Muscle biopsy samples (unfixed chilled and fixed in neutral buffered formalin) were collected from the trapezius muscle under general inhalational anesthesia. Biopsy samples were shipped by courier service under refrigeration to the University of California, San Diego, Comparative Neuromuscular Laboratory. Upon receipt, the unfixed biopsy sample was flash frozen in isopentane pre‐cooled in liquid nitrogen and then stored at −80°C until further processing using a standard panel of histochemical stains and reactions. 27 Formalin‐fixed biopsy samples were routinely processed into paraffin. Indirect immunofluorescence staining of muscle cryosections was performed using several monoclonal and polyclonal antibodies against dystrophy‐associated proteins as previously described. 16 Immunostaining of the affected cat tissue was compared to archived frozen control feline limb muscle (vastus lateralis).
2.3. Whole genome sequencing and data processing
Deoxyribonucleic acid was isolated from the remaining archived frozen muscle of the affected cat using the DNeasy Blood and Tissue kit (Qiagen, Redwood City, CA) following the manufacturer's protocol. Deoxyribonucleic acid then was submitted to the University of Missouri (MU), evaluated for quality and quantity by agarose gel (1.5% in tris(hydroxymethyl)aminomethane—acetic acid—ethylenediaminetetraacetic acid [TAE]) electrophoresis (80 V for 90 minutes) and visualized with ultra‐violet light after ethidium bromide staining. Approximately 1 μg of high molecular weight DNA was submitted to the MU Genomics Technology Core to construct a 550 base‐pair (bp) sequencing library using a TruSeq DNA PCR‐Free kit (Illumina, San Diego, California). The barcoded case sample library was pooled with approximately 21 additional cat genomic sequencing libraries and loaded onto a single S4 flow cell on a NovaSeq 6000 (Illumina) according to the manufacturer's recommendations to produce approximately 30X sequencing coverage of 150 bp paired‐end reads. Data were processed using a custom Nextflow workflow following best practices for the Genome Analysis toolkit (GATK) version 4.2. 28 , 29 Reads were mapped to Felis_Catus_9.0 (GCF_000181335.3) using Minimap version 2. 30 Duplicate reads were marked using Picard version 2. 31 Specific tools from GATK 4.2 for genotyping, variant database construction, and hard‐filtering were completed as previously described. 32 The produced 22 cat dataset was combined with the previously produced 340 cat variant call file 24 , 32 , 33 and the 61 cat WES dataset for the 99 Lives Cat Genome Sequencing Initiative. 34 , 35 , 36 The National Center for Biotechnology Information (NCBI) RefSeq Felis catus annotation 104 and Ensembl Variant Effect Predictor 37 were used to characterize the variants. Exonic variants and 10 bp flanking each exon were filtered and visualized using VarSeq software (GoldenHelix, Bozeman, Montana). The WES and WGS data are available in the NCBI short read archive under project accession numbers PRJNA308208, PRJNA627536, PRJNA999287 and PRJNA844099 and others (File S1). 38 The case cat is accession SAMN36728792.
2.4. Candidate variant validation
The candidate variant was validated in the case using fluorescence‐based Sanger sequencing. Cat reference assembly F.catus_Fca126_mat1.0 (GCF‐018350175.1; BioProject: PRJNA684600; BioSample: SAMN19729387) and RefSeq Annotation 105 were used to design PCR primers for the DMD stop gain (XM_023249210.1, ENSFCAT00000068370:c.4849C>T; p.Gln1617Ter) using Primer3Plus. 39 The PCR forward primer was 5′‐CACACGGTCATTTCAAAAGC‐3′ and the reverse primer was 5′‐TACGGCACAGACATGGAAAG‐3′. The PCR conditions were optimized for annealing temperature (range 55‐61°C) and MgCl2 concentration (1.5 mM or 2.0 mM; Figure S1A) using 1 U ChoiceTaq DNA polymerase (Denville Scientific, Inc., Metuchen, New Jersey) in 25 μL reactions with 1X PCR buffer supplied by the manufacturer, 0.4 μM each primer, 0.2 mM each nucleotide and 10‐20 ng template to produce a single PCR product with an expected size of 422 bp. The optimized 25 μL PCR conditions were 1.5 mM MgCl2 and had an initial denaturation at 94°C for 3 minutes followed by 35 cycles of 94°C for 30 seconds denaturation, 55°C for 30 seconds annealing, 72°C for 1 minute extension with a final extension at 72°C for 10 minutes. The PCR was conducted on a Veriti thermal cycler (Applied Biosystems, Waltham, Massachusetts). The DMD amplicon was amplified for both the affected male and the unaffected female sibling (Fcat—24202 [326979‐20]). The PCR products were verified by agarose gel (1.5% in TAE) electrophoresis by adding 5 μL PCR product to 1 μL of 6X tracking dye (30% [w/v] glycerol, 0.25% [w/v] bromophenol blue, 0.25% [w/v] xylene cyanol superfine [FF]) and size separation of the products at 100 V for 90 minutes. The fragments were visualized by ethidium bromide staining and recorded using an AlphaImager (Bio‐Rad Laboratories, Inc., Hercules, California). Amplified fragments (Figure S1B) from the remaining PCR product (approximately 20 μL) of the case, the unaffected sibling, and two control cats (laboratory male control Fcat‐4406 and V9.0 cat genome female control Fcat‐12682) were purified using QIAquick PCR Cleanup Kit (Qiagen). Approximately 45 ng was submitted to the MU Genomics Technology Core for Sanger sequencing using a BigDye Terminator v3.1 Cycle sequencing kit (Applied Biosystems) and an ABI 3730XL DNA Analyzer (Applied Biosystems). The generated sequences were aligned to the cat reference genome assembly F.catus_Fca126_mat1.0 and visually inspected using Sequencher 5.1 (GeneCodes, Ann Arbor, Michigan) to confirm amplification of DMD and presence of the variant in the case cat.
2.5. In silico analyses
Genomic tools and resources at the NCBI website, 40 including ClinVar, 41 , 42 the Genome Data Viewer, 43 the Basic Local Alignment Search Tool (BLAST), 44 , 45 and the Genome Aggregation Database (gnomAD) 46 were used to correlate the variant in the cat to DMD variants in humans. The MutPred‐LOF web application was used to evaluate the effect of the LoF variant in both cats and humans. 47 Multi‐species sequence alignment of the exon containing the variant was performed using CLC Sequence Viewer 8 (QIAGEN).
3. RESULTS
3.1. Clinical history
Clinical signs of progressive difficulty walking, a stilted, stiff gait, and macroglossia were apparent beginning at 6 months of age. The tongue was thickened and protruded with constant ptyalism. Thickening and rigidity of the neck and shoulders also were observed (Figure 1). Routine blood test results at the time of presentation showed abnormalities limited to increased aspartate aminotransferase (AST) activity (687 IU/L; reference range, 10‐50 IU/L), markedly increased CK activity (24 830 IU/L; reference range 50‐200 IU/L) and mild hypokalemia (3.7 mmol/L; reference range, 3.8‐5.3 mmol/L). Based on the young age of presentation and markedly increased CK activity, a neuromuscular disease was suspected, and muscle biopsies performed.
FIGURE 1.
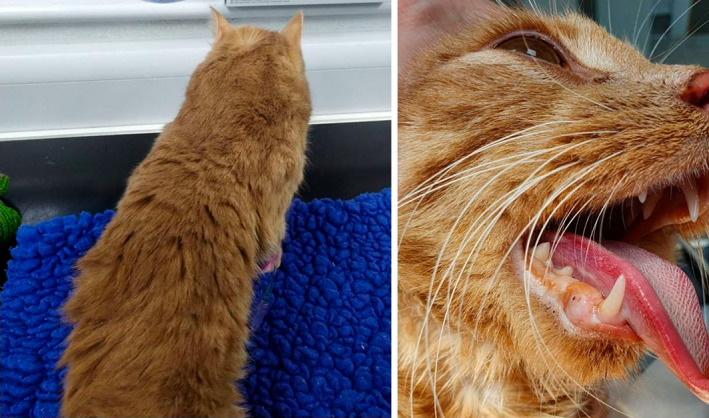
A 1‐year‐old male domestic shorthaired cat with suspected muscular dystrophy. The cat had a 6‐month history of difficulty walking, prominent hypertrophy of the neck muscles (left), progressive dysphagia and a thickened tongue (right). The cat also could not close its mouth.
3.2. Histopathology and immunofluorescent staining
Biopsy samples from the trapezius muscle confirmed a degenerative and regenerative myopathy consistent with a form of MD. To further characterize the form of MD, immunofluorescent staining was performed using various monoclonal and polyclonal antibodies to determine the presence or absence of dystrophy‐associated proteins (Figure 2). Compared to control muscle, sarcolemmal staining in the dystrophic muscle was not detected using antibodies against the rod (DYS1) and C‐terminus (DYS2) of dystrophin. Sarcolemmal staining for utrophin protein (DRP2) was increased in the dystrophic muscle. Regenerating fibers were highlighted by staining for developmental myosin heavy chain (dMHC). Staining intensity for laminin α2 was like the intensity in the control muscle. Staining for collagen 6 highlighted endomysial fibrosis. Based on these findings, a dystrophin‐deficient form of X‐linked MD was diagnosed.
FIGURE 2.
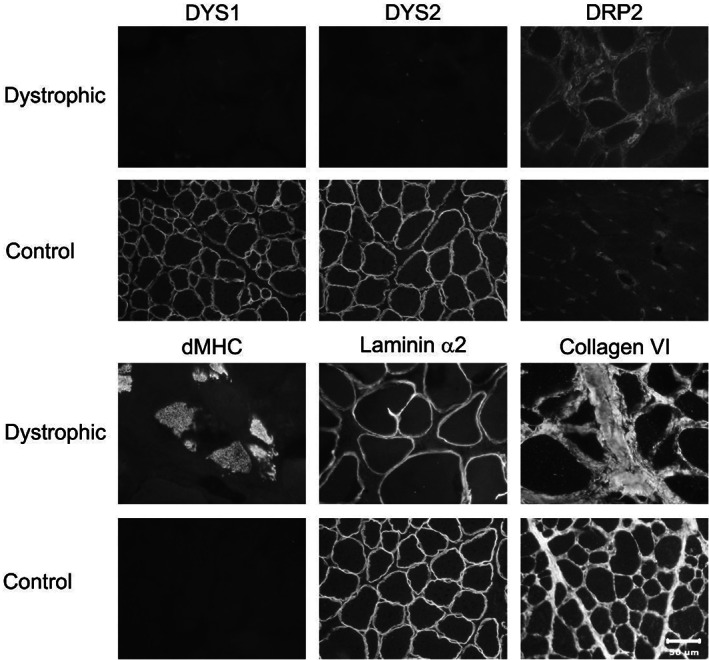
Immunofluorescent staining of cryosections from the trapezius muscle of the dystrophic cat and archived control vastus lateralis muscle was performed for localization of dystrophy‐associated proteins. Antibodies against dystrophy‐associated proteins included those against the rod (DYS1) and carboxy terminus (DYS2) of dystrophin, utrophin (DRP2), developmental myosin heavy chain (dMHC) to demonstrate regenerating fibers, and against laminin α2 and collagen 6. Protein localization for both DYS1 and DYS2 could not be detected and utrophin was increased compared to archived control muscle. Regenerating fibers were highlighted by staining for dMHC. Staining for both laminin α2 and collagen 6 was like control tissue. Bar in lower right image equals 50 μm for all images.
3.3. DNA variant identification
This case presentation was expected to be rare, and both dominant and recessive modes of inheritance were considered during the variant filtering with no a priori consideration of a candidate gene. The 99 Lives dataset has 2 289 566 variants from 413 additional cats, the unique variants for the case cat are presented in Table 1 and File S2A,B. Considering recessive/hemizygous and unique variants with a passing Variant Quality Score Recalibration (VQSR) Tranche score 32 and suspected to alter the gene coding sequences, 71 variants were identified, including a nonsense stop gain in DMD, and 2 missense variants. The DMD stop gain variant (XM_023249210.1, ENSFCAT00000068370:c.4849C>T; p.Gln1617Ter) is at chromosome position X:28201541 in Felis_catus_9.0 and chromosome position X:27949145 in assembly Fca126 (Table 1, File S2A), which is at the beginning of exon 35 of a 13 533 transcript (4511 amino acids) with 78 exons. The variant is suggested to cause LoF, because approximately 65% of the DMD protein is truncated. The variant had 12X sequencing reads for the case, suggesting good sequencing coverage of a region on the X chromosome in a male cat. Considering dominant inheritance and the case as heterozygous, 735 variants were identified including 16 LoF variants, and 132 missense variants (Table 1, File S2B). All variants detected in the 99 Lives dataset for DMD are presented in (File S2C), including the variant recently published in a family of cats 16 and a variant identified in Japan, 48 which are MD cats known to be in the 99 Lives cohort.
TABLE 1.
DNA variants in a cat with a muscular dystrophy.
| Variant | 414 cat variants b | Case—unique variants a | |
|---|---|---|---|
| Hemi‐/homozygous | Heterozygous | ||
| 3′ UTR | 779 313 | 4 | 59 |
| 5′ UTR premature start codon gain | 14 709 | 0 | 1 |
| 5′ UTR | 253 248 | 2 | 19 |
| Disruptive inframe deletion | 3232 | 0 | 0 |
| Disruptive inframe insertion | 7348 | 0 | 0 |
| Downstream gene | 11 812 | 4 | 22 |
| Frameshift | 122 878 | 0 | 14 |
| Inframe deletion | 12 416 | 0 | 6 |
| Inframe insertion | 17 437 | 0 | 2 |
| Initiator codon | 1320 | 0 | 2 |
| Intergenic | 24 | 34 | 180 |
| Intron | 312 834 | 10 | 113 |
| Missense | 404 240 | 2 | 132 |
| Non‐coding exon | 363 539 | 1 | 9 |
| Splice acceptor | 6348 | 0 | 0 |
| Splice donor | 5763 | 0 | 0 |
| Splice region | 99 728 | 2 | 21 |
| Stop gained | 12 325 | 1 (DMD) | 0 |
| Stop lost | 1019 | 0 | 0 |
| Stop retained | 444 | 0 | 0 |
| Synonymous | 692 633 | 4 | 121 |
| Upstream gene variant | 11 315 | 7 | 33 |
| Total variants | 3 133 975 | 71 | 735 |
Exon with flanking 10 bp and passing Tranche scores.
Data from 362 cats with WGS and 62 cats with WES. Includes variants with lower Tranche scores.
For variant validation, a single PCR product of the expected size, 422 bp, was produced (Figure S1A,B). Both control cats, Fcat_4406 (male) and Fcat_12682 (female), were hemizygous/homozygous for the wildtype allele and the affected male cat, Fcat_23286, was hemizygous for the nonsense LoF variant. The normal female sibling did not have the DMD variant (Figure 3). This variant was unique to 414 cats within the 99 Lives dataset. DMD (ENSFCAG00000022242.4) is at position 26 990 371‐29 004 949 on the X chromosome (NC_018741.3) in Felis catus_9.0 reference assembly using Ensembl annotation 104 and at position chromosome X (NC_058386.1):26741260–29120869 in the F._catus_Fca126 assembly using NCBI annotation 105.
FIGURE 3.
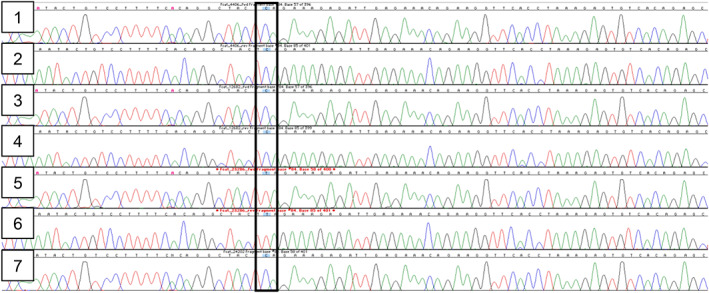
Electropherograms for DMD Sanger sequencing. 1: Control cat—Fcat_4406 coding strand; 2: Control cat—Fcat_4406 complementary strand; 3: Control cat—Fcat_12682 coding strand; 4: Control cat—Fcat_12682 complementary strand; 5: Case cat—Fcat_23286 coding strand; 6: Case cat—Fcat_23286 complementary strand; 7: Sibling cat—Fcat_24202 coding strand. Box indicates region where the case cat is hemizygous for the c.4849C>T variant. Cytosine (C) are blue peaks and thymine (T) are red peaks.
3.4. In silico analyses
The NCBI cat reference assembly F.catus_Fca126 is annotated with 8 identified DMD transcripts in the domestic cat, which code for 78 exons and approximately 4511 amino acids. The cat variant is positioned in exon 35, which has over 93% homology with approximately 13 human DMD transcripts containing exon 35.
The ClinVar (accessed March 27, 2023) database of human deoxyribonucleic acid (DNA) variants (v2.1.1 data set [GRCh37/hg19]) was used to provide comparative genetic support for the function of the cat variant. 41 , 42 The cat DMD c.4849C>T; p.Gln1617Ter variant is analogous to the human p.Gln1618 (NM_004006.3(DMD):c.4852C>T (p.Gln1618Ter)), which is at position X:32365193 in the human genome assembly. In humans, this premature translational stop signal has been observed in familial cases of DMD and is considered pathogenic. 49 Like the cat, the human variant is also a cytosine to thymine transition and changes the codon from CAA to TAA, which is an ochre termination codon. This variant is not present in gnomAD population database, 46 thus additional frequency information in humans was not available.
MutPred‐LOF web application was used to predict the effect of the LoF variant in both cats and humans. The complete cat predicted mRNA (XM_023249210.1) was compared to the predicted mutant mRNA with the stop termination (Q1617X). The predicted score from MutPred_LOF was 0.46141 as compared to the Q1618X variant introduced into human DMD isoform X1 XP_006724531.1, which produced a score of 0.45563. Scores > 0.7 are considered the most deleterious by this prediction application. Multiple species alignments of the region (exon 35) across diverse mammals and zebrafish indicated the region is highly conserved for both nucleotides and amino acids (Figure 4).
FIGURE 4.

Multispecies alignment of Felis catus DMD exon 35. The alignments indicate the position of the DMD stop gain LoF variant (XM_023249210.1, ENSFCAT00000068370:c.4849C>T; p.Gln1617Ter) at chromosome position X:28201541 Felis_catus_9.0 at the beginning of exon 35. (A) Nucleotide alignment for diverse species (DMD affected feline case, normal feline, human, canine, bovine, porcine, murine, and zebrafish) indicating the 100% conservation of the nucleotide and the high conservation of the exon 35, lower histogram. Nucleotides are represented by the different colors. (B) Protein alignments for the same species depicting the termination in exon 35 for the affected cat. The alanine that is 2 amino acids upstream of the amber termination codon is the 5′ start of exon 35. The mammals have nearly 100% amino acid conservation within this region.
4. DISCUSSION
Duchene muscular dystrophy (DMD) is an X‐linked inherited neuromuscular disease in people that is a commonly recognized heritable condition. The gene causing DMD, dystrophin (DMD), is 1 of the larger genes in the mammalian genome and the pooled global DMD birth prevalence has been estimated as 19.8 per 100 000 live male births. 50 The ClinVar database has documented 480 variants as likely pathogenic and 2503 variants as pathogenic for DMD in humans, causing the different forms, severities, and presentations of the disease. 41 , 42 Precise definition of the causal variant is not only vital to the prognosis and counseling for DMD patients and their families, but also defines amenable treatments. Read‐through compounds, such as ataluren, for patients with nonsense (stop codon) mutations, represent exon skipping treatments for DMD. 51 , 52 , 53 The targeted treatments have increased the lifespan of DMD patients by decades. Recently, delandistrogene moxeparvovec, which is an adeno‐associated virus (AAV) vector‐based gene therapy designed to deliver a gene encoding a micro‐dystrophin protein, has been approved in the United States for the treatment of ambulatory DMD pediatric patients and a defined variant in DMD. 54 The ongoing preclinical trials in canine models are supporting the treatment developments and options 55 , 56 , 57 and substantial advances have been made in a variety of treatments. 58 , 59 , 60 , 61 , 62 , 63 , 64 Defining the feline DMD variants supports the use of cats as a biomedical model in preclinical trials, like that of canine models.
Feline X‐linked MD was the first genetically characterized cat model for a human disease (OMIA:001081‐9685). 15 The deletion in the muscle promoter of DMD was characterized by Southern blot analyses, thus the exact extent and positioning of the variant has not been determined. 15 A second, larger deletion involving the promoter region of DMD also has been described in cats, but also not precisely defined because of the limitations of the techniques of discovery. 65 Two additional DMD variants in cats were characterized recently, both in families of Maine coon cats. 16 , 17 A previous report described a nonsense variant in exon 11 of the feline DMD gene, NC_058386.1 (XM_045050794.1): c.1180C>T (p.Arg394*), which was supported by mRNA sequencing and demonstrated the loss of most of the dystrophin protein. 16 These Maine coon cats are part of the 99 Lives cohort, thus the c.1180C>T variant was not present in the current case. Another study described a milder form of MD (Becker‐like; OMIA:001081‐9685), which was a missense variant (c.4186C>T; p.His1396Tyr) also showing familial inheritance in Maine coon cats. 17 This variant was not identified in the 99 Lives cohort of cats, further suggesting its rarity, and supporting causality for disease.
The DMD variant in the present case, c.4849C>T; p.Gln1617Ter, is a LoF variant caused by a nonsense mutation in exon 35 of the 78 exons annotated for cat DMD and likely truncates approximately 65% of the protein. This variant was unique to the case in the 99 Lives cohort, supporting its rarity. The present case is a random bred cat and only 1 sibling could be evaluated. The female sibling did not have the variant, suggesting the LoF variant was transmitted by the queen as a likely heterozygous carrier, or the case cat represented a de novo mutation. ClinVar contains an entry for the human counterpart variant (Q1618X; Variation ID: 1322710)—HGMD CM022951 66 that had the same predicted effect and segregates within a human family with DMD, supporting the cat variant as also pathogenic based on comparative genetic criteria. Loss of function variants are considered 1 of the most pathogenic mutations, but many also can be well tolerated. 47 , 67 Each human genome may contain many stop variants with no observable impact upon phenotype. 68 , 69 However, the noise from poor annotation and poor sequencing techniques is considered part of the false identification of LoF variants that may not actually be present in the genome. In the case of the cat in our report, the variant was suggested as valid by high coverage and high quality of reads at the variant site and further proven by direct Sanger sequencing. Although the genome datasets are less substantial for cats than for humans, the accuracy of the current cat dataset has benefitted from the use of robust technologies and techniques to produce the sequencing data, consistent methods of variant identification based on the tools developed by the Human Genome Project 70 and more robust genome assemblies and annotations. 32 The high conservation of both the nucleotide and amino acid sequences within the region across diverse mammalian species also supports the determination of pathogenicity for the variant. The variant analyses had no a priori consideration of candidate genes. The recessive/hemizygous model identified 71 additional unique variants that would equally qualify for candidates for the disease based on the model selection alone. However, only 2 were within coding regions of genes, both missense variants in the gene Sodium/Potassium‐Transporting ATPase Subunit Beta‐3 (ATPB3) and an uncharacterized gene (LOC101082653). The remaining 16 genes, 10 uncharacterized genes and 34 intergenic variants were in regulatory or uncharacterized regions that would require extensive experimentation and comparative studies to define causality. Similarly, most of the 735 variants identified when considering the dominant model are non‐coding. Most of the LoF frameshift variants are in uncharacterized genes. The gene Titin (TTN) has been associated with MDs, 71 which is a p.Ile31673Val missense variant in the cat case, however the clinical, pathological, and histological data supports a dystrophy deficiency and further investigation of other genes is not warranted.
The young age of this male cat, the markedly increased CK activity, the presence of the dystrophic phenotype in the muscle biopsy sample, and the non‐detectable staining for the rod and carboxy‐terminus of dystrophin with increased utrophin expression in muscle cryosections is consistent with the diagnosis of X‐linked MD. Muscle hypertrophy is a consistent finding in X‐linked dystrophin‐deficient cats, but this finding is not limited to the feline species. 72 Hypertrophy of the gastrocnemius muscle (calf hypertrophy) occurs in Duchenne MD in people and cranial sartorius muscle hypertrophy occurs in the Golden retriever model. 72 Previous studies have determined this finding is true hypertrophy and not pseudohypertropy related to fatty and connective tissue infiltration or to hyperplasia. 72
The expected LoF variant effect, population data, genetic data compared to humans and amino acid conservation within the exon support the identified variant as pathogenic and causing the clinical signs of X‐linked MD in the cat of our report. 73 Precision/genomic medicine efforts for the domestic cat and in veterinary medicine increasingly support disease variant and animal model discovery and provide opportunities for targeted treatments for companion animals.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
The affected cat in this study was privately owned and examined with the consent of the owner. The work involved the use of a nonexperimental animal only. Established internationally recognized high standards (“best practice”) of individual veterinary patient care were followed. The control cat tissues were obtained post‐mortem and did not require IACUC approval. Dr. Lyons has an exemption ACUC protocol 9178 at the University of Missouri for the receiving of samples for genomic applications.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Data S1: Supporting Information.
Data S2: Supporting Information.
Data S3: Supporting Information.
ACKNOWLEDGMENT
Funding provided by EveryCat Health Foundation; Gilbreath McLorn Endowment, College of Veterinary Medicine, University of Missouri; donations to the 99 Lives Project, Winn Feline Foundation, and the George and Phyllis Miller Trust (MT18‐009; MTW18‐009, MT19‐001; Leslie A. Lyons). We appreciate the laboratory assistance of Thomas R. Juba and Yoshi Yu, DVM for the assistance with genetic figures. We thank the contributors of the 99 Lives Cat Genome Sequencing Consortium for access to the variant call file for variant allelic data. Thank you to Dr. Giuseppe Nacci for performing the muscle biopsy on the affected cat. Our appreciation to the owners of the cats that were the subjects of this study for permission to collect samples from their pets. All the sequence variants unique to the affected cat are included in Table S1. The whole genome sequencing data are available in the NCBI short read archive under project accession number PRJNA308208, PRJNA627536, PRJNA999287 and PRJNA844099 and others with this case cat as accession SAMN36728792. Cats of the 99 Lives Project—signalment, contacts, SRA accessions and membership list are presented in File S1.
Shelton GD, Tucciarone F, Guo LT, Coghill LM, Lyons LA. Precision medicine using whole genome sequencing identifies a novel dystrophin (DMD) variant for X‐linked muscular dystrophy in a cat. J Vet Intern Med. 2024;38(1):135‐144. doi: 10.1111/jvim.16971
REFERENCES
- 1. Dubowitz V, Sewry CA, Oldfors A. Classification of neuromuscular disorders. Muscle biopsy: A Practical Approach. 5th ed. Philadelphia, PA: Saunders Elsevier; 2021. [Google Scholar]
- 2. Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509‐517. [DOI] [PubMed] [Google Scholar]
- 3. Hoffman EP, Kunkel LM. Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron. 1989;2:1019‐1029. [DOI] [PubMed] [Google Scholar]
- 4. Bulfield G, Siller WG, Wight PA, et al. X chromosome‐linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shelton GD, Minor KM, Friedenberg SG, et al. Current classification of canine Muscular dystrophies and identification of new variants. Genes. 2023;14:1557‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Birch SM, Lawlor MW, Conlon TJ, et al. Assessment of systemic AAV‐microdystrophin gene therapy in the GRMD model of Duchenne muscular dystrophy. Sci Transl Med. 2023;15:eabo1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Le Guiner C, Servais L, Montus M, et al. Long‐term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun. 2017;8:16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amoasii L, Hildyard JCW, Li H, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koo T, Okada T, Athanasopoulos T, et al. Long‐term functional adeno‐associated virus‐microdystrophin expression in the dystrophic CXMDj dog. Gene Med. 2011;13:497‐506. [DOI] [PubMed] [Google Scholar]
- 10. Shin JH, Pan X, Hakim CH, et al. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol Ther. 2013;21:750‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cooper BJ, Winand NJ, Stedman H, et al. The homologue of the Duchenne locus is defective in X‐linked muscular dystrophy of dogs. Nature. 1988;334:154‐156. [DOI] [PubMed] [Google Scholar]
- 12. Kornegay JN. The golden retriever model of Duchenne muscular dystrophy. Skelet Muscle. 2017;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carpenter JL, Hoffman EP, Romanul FC, et al. Feline muscular dystrophy with dystrophin deficiency. Am J Pathol. 1989;135:909‐919. [PMC free article] [PubMed] [Google Scholar]
- 14. Gaschen FP, Hoffman EP, Gorospe JR, et al. Dystrophin deficiency causes lethal muscle hypertrophy in cats. J Neurol Sci. 1992;110:149‐159. [DOI] [PubMed] [Google Scholar]
- 15. Winand NJ, Edwards M, Pradhan D, et al. Deletion of the dystrophin muscle promoter in feline muscular dystrophy. Neuromuscul Disord. 1994;4:433‐445. [DOI] [PubMed] [Google Scholar]
- 16. Beckers E, Cornelis I, Bhatti SFM, et al. A nonsense variant in the DMD gene causes X‐linked muscular dystrophy in the Maine coon cat. Animals (Basel). 2022;12:2928‐2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hilton S, Christen M, Bilzer T, et al. Dystrophin (DMD) missense variant in cats with Becker‐type muscular dystrophy. Int J Mol Sci. 2023;24:3192‐3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Awamura Y, Uchida K, Arikawa‐Hirasawa E. Long‐term follow‐up of laminin alpha2 (merosin)‐deficient muscular dystrophy in a cat. J Feline Med Surg. 2008;10:274‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Brien DP, Johnson GC, Liu LA, et al. Laminin alpha 2 (merosin)‐deficient muscular dystrophy and demyelinating neuropathy in two cats. J Neurol Sci. 2001;189:37‐43. [DOI] [PubMed] [Google Scholar]
- 20. Poncelet L, Resibois A, Engvall E, et al. Laminin alpha2 deficiency‐associated muscular dystrophy in a Maine coon cat. J Small Anim Pract. 2003;44:550‐552. [DOI] [PubMed] [Google Scholar]
- 21. Bouillon J, Taylor SM, Vargo C, et al. Beta‐sarcoglycan‐deficient muscular dystrophy presenting as chronic bronchopneumonia in a young cat. JFMS Open Rep. 2019;5:2055116919856457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salvadori C, Vattemi G, Lombardo R, et al. Muscular dystrophy with reduced beta‐sarcoglycan in a cat. J Comp Pathol. 2009;140:278‐282. [DOI] [PubMed] [Google Scholar]
- 23. Gandolfi B, Grahn RA, Creighton EK, et al. COLQ variant associated with Devon Rex and Sphynx feline hereditary myopathy. Anim Genet. 2015;46:711‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kopke MA, Shelton GD, Lyons LA, et al. X‐linked myotubular myopathy associated with an MTM1 variant in a Maine coon cat. J Vet Intern Med. 2022;36:1800‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nghiem PP, Bello L, Balog‐Alvarez C, et al. Whole genome sequencing reveals a 7 base‐pair deletion in DMD exon 42 in a dog with muscular dystrophy. Mamm Genome. 2017;28:106‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Online Mendelian Inheritance in Animals, OMIA. Accessed October 18, 2023. https://www.omia.org
- 27. Dubowitz V, Sewry CA, Oldfors A. Histological and histochemical stains and reactions. Muscle biopsy: A Practical Approach. 5th ed. Philadelphia, PA: Saunders Elsevier; 2021. [Google Scholar]
- 28. Van der Auwera G, O'Connor B. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. Sebastopol, CA: O'Reilly Media; 2020:439. [Google Scholar]
- 29. Di Tommaso P, Chatzou M, Floden EW, et al. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35:316‐319. [DOI] [PubMed] [Google Scholar]
- 30. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094‐3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Broad Institute Picard Tools . https://broadinstitute.github.io/picard/
- 32. Buckley RM, Davis BW, Brashear WA, et al. A new domestic cat genome assembly based on long sequence reads empowers feline genomic medicine and identifies a novel gene for dwarfism. PLoS Genet. 2020;16:e1008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lyons LA, Buckley RM, Harvey RJ, et al. Mining the 99 Lives Cat Genome Sequencing Consortium database implicates genes and variants for the Ticked locus in domestic cats (Felis catus). Anim Genet. 2021;52:321‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katz ML, Buckley RM, Biegen V, et al. Neuronal ceroid lipofuscinosis in a domestic cat associated with a DNA sequence variant that creates a premature stop codon in CLN6 G3 (Bethesda). 2020;10:2741‐2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodney AR, Buckley RM, Fulton RS, et al. A domestic cat whole exome sequencing resource for trait discovery. Sci Rep. 2021;11:7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lyraki M, Hibbert A, Langley‐Hobbs S, et al. CTSK variant implicated in suspected pyknodysostosis in a domestic cat. JFMS Open Rep. 2022;8:20551169221137536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodney AR, Skidmore ZL, Grenier JK, et al. Genomic landscape and gene expression profiles of feline oral squamous cell carcinoma. Front Vet Sci. 2023;10:1079019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Untergasser A, Nijveen H, Rao X, et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71‐W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. National Library of Medicine, National Center for Biotechnology Information (NCBI) . 2023. https://www.ncbi.nlm.nih.gov/
- 41. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980‐D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Landrum MJ, Chitipiralla S, Brown GR, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835‐D844. Accessed July 26, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rangwala SH, Kuznetsov A, Ananiev V, et al. Accessing NCBI data using the NCBI Sequence Viewer and Genome Data Viewer (GDV). Genome Res. 2021;31:159‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Z, Schwartz S, Wagner L, et al. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203‐214. [DOI] [PubMed] [Google Scholar]
- 45. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:403‐410. [DOI] [PubMed] [Google Scholar]
- 46. Chen S, Francioli LC, Goodrich JK, et al. A genome‐wide mutational constraint map quantified from variation in 76,156 human genomes. Nature. 2023. doi: 10.1038/s41586-023-06045-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kymberleigh A, Pagel VP, Lin GN, et al. When loss‐of‐function is loss of function: assessing mutational signatures and impact of loss‐of‐function genetic variants. Bioinformatics. 2017;33:i389‐i398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muto H, Yu Y, Lyons LA, et al. Identification of a paradoxical novel DMD genetic nonsense variant in the mild clinical course of feline X‐linked muscular dystrophy. J Vet Intern Med. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dolinsky LC, de Moura‐Neto RS, Falcao‐Conceicao DN. DGGE analysis as a tool to identify point mutations, de novo mutations and carriers of the dystrophin gene. Neuromuscul Disord. 2002;12:845‐848. [DOI] [PubMed] [Google Scholar]
- 50. Crisafulli S, Sultana J, Fontana A, et al. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta‐analysis. Orphanet J Rare Dis. 2020;15:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bello L, Pegoraro E. Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy. Acta Myol. 2016;35:122‐127. [PMC free article] [PubMed] [Google Scholar]
- 52. Landfeldt E, Sejersen T, Tulinius M. A mini‐review and implementation model for using ataluren to treat nonsense mutation Duchenne muscular dystrophy. Acta Paediatr. 2019;108:224‐230. [DOI] [PubMed] [Google Scholar]
- 53. Salmaninejad A, Jafari Abarghan Y, Bozorg Qomi S, et al. Common therapeutic advances for Duchenne muscular dystrophy (DMD). Int J Neurosci. 2021;131:370‐389. [DOI] [PubMed] [Google Scholar]
- 54. Hoy SM. Delandistrogene Moxeparvovec: First approval. Drugs. 2023;83:1323‐1329. [DOI] [PubMed] [Google Scholar]
- 55. Barraza‐Flores P, Fontelonga TM, Wuebbles RD, et al. Laminin‐111 protein therapy enhances muscle regeneration and repair in the GRMD dog model of Duchenne muscular dystrophy. Hum Mol Genet. 2019;28:2686‐2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song Y, Morales L, Malik AS, et al. Non‐immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med. 2019;25:1505‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martin PT, Zygmunt DA, Ashbrook A, et al. Short‐term treatment of golden retriever muscular dystrophy (GRMD) dogs with rAAVrh74.MHCK7.GALGT2 induces muscle glycosylation and utrophin expression but has no significant effect on muscle strength. PLoS One. 2021;16:e0248721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilton‐Clark H, Yokota T. Recent trends in antisense therapies for Duchenne Muscular Dystrophy. Pharmaceutics. 2023;15:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patterson G, Conner H, Groneman M, et al. Duchenne muscular dystrophy: Current treatment and emerging exon skipping and gene therapy approach. Eur J Pharmacol. 2023;947:175675. [DOI] [PubMed] [Google Scholar]
- 60. Chemello F, Olson EN, Bassel‐Duby R. CRISPR‐editing therapy for Duchenne Muscular Dystrophy. Hum Gene Ther. 2023;34:379‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fatehi S, Marks RM, Rok MJ, et al. Advances in CRISPR/Cas9 genome editing for the treatment of muscular dystrophies. Hum Gene Ther. 2023;34:388‐403. [DOI] [PubMed] [Google Scholar]
- 62. Chang M, Cai Y, Gao Z, et al. Duchenne muscular dystrophy: pathogenesis and promising therapies. J Neurol. 2023;270:3733‐3749. [DOI] [PubMed] [Google Scholar]
- 63. Gupta S, Sharma SN, Kundu J, et al. Morpholino oligonucleotide‐mediated exon skipping for DMD treatment: Past insights, present challenges and future perspectives. J Biosci. 2023;48:38. [PubMed] [Google Scholar]
- 64. Happi Mbakam C, Tremblay JP. Gene therapy for Duchenne muscular dystrophy: an update on the latest clinical developments. Expert Rev Neurother. 2023;23:905‐920. [DOI] [PubMed] [Google Scholar]
- 65. Gambino AN, Mouser PJ, Shelton GD, et al. Emergent presentation of a cat with dystrophin‐deficient muscular dystrophy. J Am Anim Hosp Assoc. 2014;50:130‐135. [DOI] [PubMed] [Google Scholar]
- 66. Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database (HGMD((R))): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. MacArthur DG, Tyler‐Smith C. Loss‐of‐function variants in the genomes of healthy humans. Hum Mol Genet. 2010;19:R125‐R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sulem P, Helgason H, Oddson A, et al. Identification of a large set of rare complete human knockouts. Nat Genet. 2015;47:448‐452. [DOI] [PubMed] [Google Scholar]
- 70. Genomes Project C , Abecasis GR, Altshuler D, et al. A map of human genome variation from population‐scale sequencing. Nature. 2010;467:1061‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Misaka T, Yoshihisa A, Takeishi Y. Titin in muscular dystrophy and cardiomyopathy: Urinary titin as a novel marker. Clin Chim Acta. 2019;495:123‐128. [DOI] [PubMed] [Google Scholar]
- 72. Kornegay JN, Childers MK, Bogan DJ, et al. The paradox of muscle hypertrophy in muscular dystrophy. Phys Med Rehabil Clin N Am. 2012;23:149‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting Information.
Data S2: Supporting Information.
Data S3: Supporting Information.