

Abstract
Incorporation of modified or labeled nucleotides at specific sites in RNAs is critical for gaining insights into the structure and function of RNAs. Preparation of site-specifically labeled large RNAs in amounts suitable for structural or functional studies is extremely difficult using current methodologies. The position-selective labeling of RNA, PLOR, is a recently developed method that makes such syntheses possible. PLOR allows incorporation of various probes, including 2D/13C/15N-isotopic labels, Cy3/Cy5/ Alexa488/Alexa555 fluorescent dyes, biotin and other chemical groups, into specific positions in long RNAs. Here, we describe in detail the use of PLOR to label RNAs at specific segment(s) or discrete sites.
Keywords: PLOR, RNA, Specific labeling
1. Introduction
Knowledge about RNA structures and dynamics is fundamentally important to understand RNA’s functions. First of all, high-resolution structures of macromolecules are invaluable for better understanding their functions in biological systems. NMR and X-ray crystallography are the two main methods used to solve atomic-resolution structures of proteins and RNAs, and account for >97% of the RNA structures in the Protein Data Bank. Specific labeling of RNAs offers several advantages for both of these methods. Selective isotopic labeling of RNAs, especially those larger than 50 nt, can greatly alleviate NMR peak overlap, thereby simplifying spectra interpretation to allow extraction of structures and dynamics information that would otherwise be difficult to obtain [1–8]. With the availability of high-field NMR spectrometers, the use of selectively isotopically labeled RNAs is now one of the primary strategies for RNA structure determination. In X-ray crystallography, the calculation of electron density maps requires determination of the phases of the diffraction data, which is usually done either by molecular replacement (MR) with a known, homologous structure, or by anomalous phasing by incorporating heavy atoms into the crystal [9–16]. Since known, homologous structures are rarely available for RNAs, de novo phasing of RNA crystal data can be achieved through site-specific incorporation of heavy-atom-labeled nucleotides. Secondly, smFRET is a very powerful tool to study conformation space and dynamics. RNA labeled with fluorophores offers the capability to characterize dynamics of individual molecules [17–19] Lastly, RNA molecules are used as biosensors and labeled RNAs with various probes have broad applications in disease diagnosis, substance detection and molecular imaging [20–22].
The most widely used methods to introduce labels into RNAs during synthesis are in vitro transcription and solid-phase chemical synthesis. In vitro transcription has been routinely used to prepare RNAs in large quantity, in which nucleotide types are homogeneously labeled, but is not readily adapted to allow labeling of nucleotides by position [7,23,24]. Chemical synthesis provides greater flexibility in labeling RNA at specific positions. In theory it is possible to use the chemical synthesis method to generate small RNAs selectively labeled at any position, provided that all labeling chemical reagents are commercially available. However, its primary limitations are size and cost [23–26]. Neither of these two methods is practical for preparing multi-milligram amounts of large RNAs with position-specific isotope labels for NMR.
Recently, we reported a method, PLOR, to synthesize selectively-labeled RNA on a milligram scale [27]. We demonstrated PLOR by synthesizing RNAs with various labeling schemes and labeling reagents. Those RNAs include a specifically labeled 104-nt TCV RNA (Fig. 1) from the 3′ untranslated region of turnip crinkle virus [28–30], and a series of 71-nt riboA71 RNAs (Fig. 1) that form the aptamer domain of an adenine riboswitch [29,31–33]. Isotope-labeled (13C, 15N, and 2D), fluorescent (Cy3, Cy5, Alexa 488, and Alexa 555), or biotinylated derivatives of riboA71 were also generated by PLOR, in which the labeled nucleotides were incorporated into a single helix (Fig. 1c and j), a single hairpin (Fig. 1d and f), an internal loop (Fig. 1e), multiple hairpins (Fig. 1g), multiple discrete positions (Fig. 1h, k, and l), or, a single position (Fig. 1i), and subsequently used in NMR and smFRET studies.
Fig. 1.
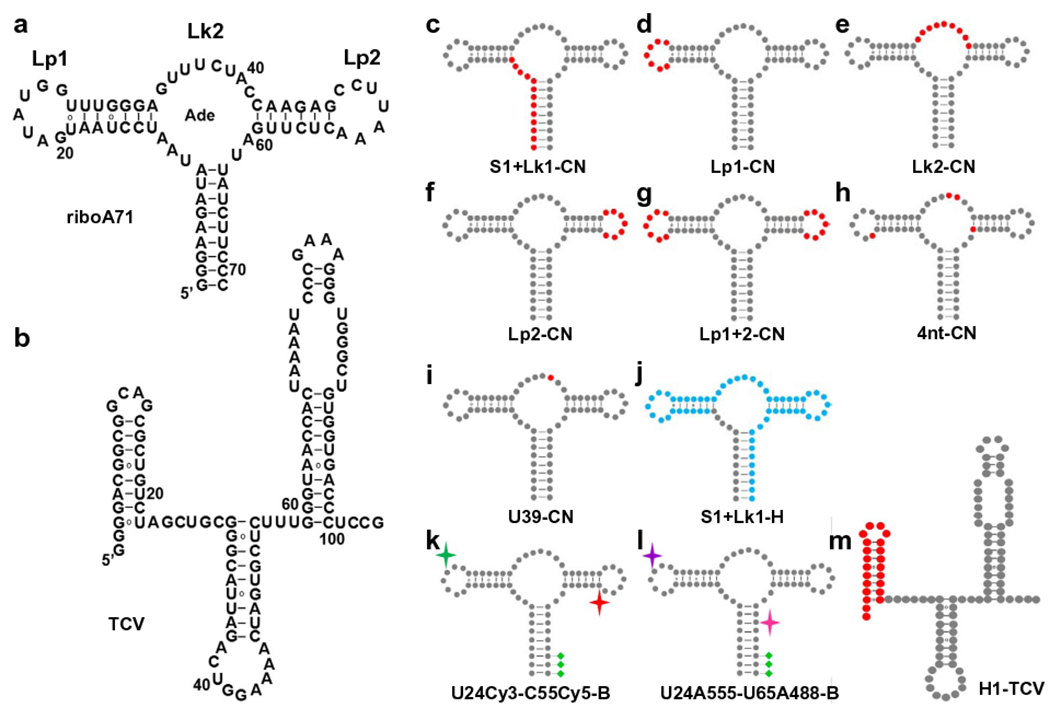
Site-specific labeling of RNA by PLOR. a–b, Secondary structure of a, riboA71,and b, TCV. c–m, riboA71 and TCV with specific labels. Non-labeled residues are shown as grey dots. 13C15N- and 2D-isotopic residues are represented by red and blue dots, respectively, in c (S1 + Lk1-CN-riboA71), d (Lp1-CN-riboA71), e (Lk2-CN-riboA71), f (Lp2-CN-riboA71), g (Lp1 + 2-CN-riboA71), h (4nt-CN-riboA71), i (U39-CN-riboA71), j (S1 + Lk1-H-riboA71), and m (H1-TCV). The fluorescent dyes, Cy3 and Cy5, in k (U24Cy3-C55Cy5-B-riboA71) are represented by green and red stars, respectively. The fluorescent dyes, Alexa 488 and Alexa 555, in l (U24A555-U65A488-B-riboA71) are represented by pink and purple stars, respectively. The green diamonds in k and l represent biotin groups.
2. Theory
In vitro transcription catalyzed by T7 RNA polymerase has a slow, unstable initiation phase, followed by fast chain and highly processive elongation at about 200 nucleotides per second [34,35]. It has been shown that the elongation complex composed of DNA:RNA:T7 RNAP is extremely stable once it is formed, and that transcription by this ternary complex can be paused and resumed [36]. PLOR exploits this ability to pause and resume transcription by the elongation complex by omitting and adding back the NTPs required to proceed pause positions. Polymerase ‘walking’ is not unique to T7 RNA polymerase, such ‘walking’ has been observed for other RNA polymerases to study RNAP mechanism [37]. PLOR combines aspects of both liquid-phase transcription and solid-phase chemical synthesis. The DNA template in PLOR is coupled to a solid support, which allows stepwise buffer and NTP changes. The PLOR reaction is divided into three stages: initiation, elongation, and termination (Fig. 2) [27]. The transcription reaction is initiated by gentle mixing of T7 RNAP, NTPs and bead attached templates in a reaction vessel. One or more types of NTPs are excluded in the reaction mixtures, which causes the RNA polymerase to pause at the positions where it requires the missing NTP(s). After removing the residual NTPs by SPE and rinsing the beads thoroughly, a new NTP mix is added to allow the transcription to be resumed to the next stop. Each of these pause/resume steps constitutes one elongation cycle, and modified NTPs can be introduced during each of these cycles to allow specific labeling of the transcribed RNAs. The number of elongation cycles depends on the labeling scheme and RNA sequence. When labeling at the desired positions is achieved, all four types of NTPs are added simultaneously to complete transcription in the final termination step. The whole process can be repeated a number of times depending on the desired quantity of RNAs. By varying the NTP combinations in the elongation cycles, practically any specific labeling scheme can be performed. This process can be performed automatically using a robotic platform and a customarily made reaction module. As a proof of concept, we have designed and fabricated such a reaction module that performs reactions and executes gentle mixing, washing, filtration cycles at a given temperature driven by a customarily made computer program written in SQL language. The robotic platform was purchased from Zinsser Analytic, Germany [27].
Fig. 2.
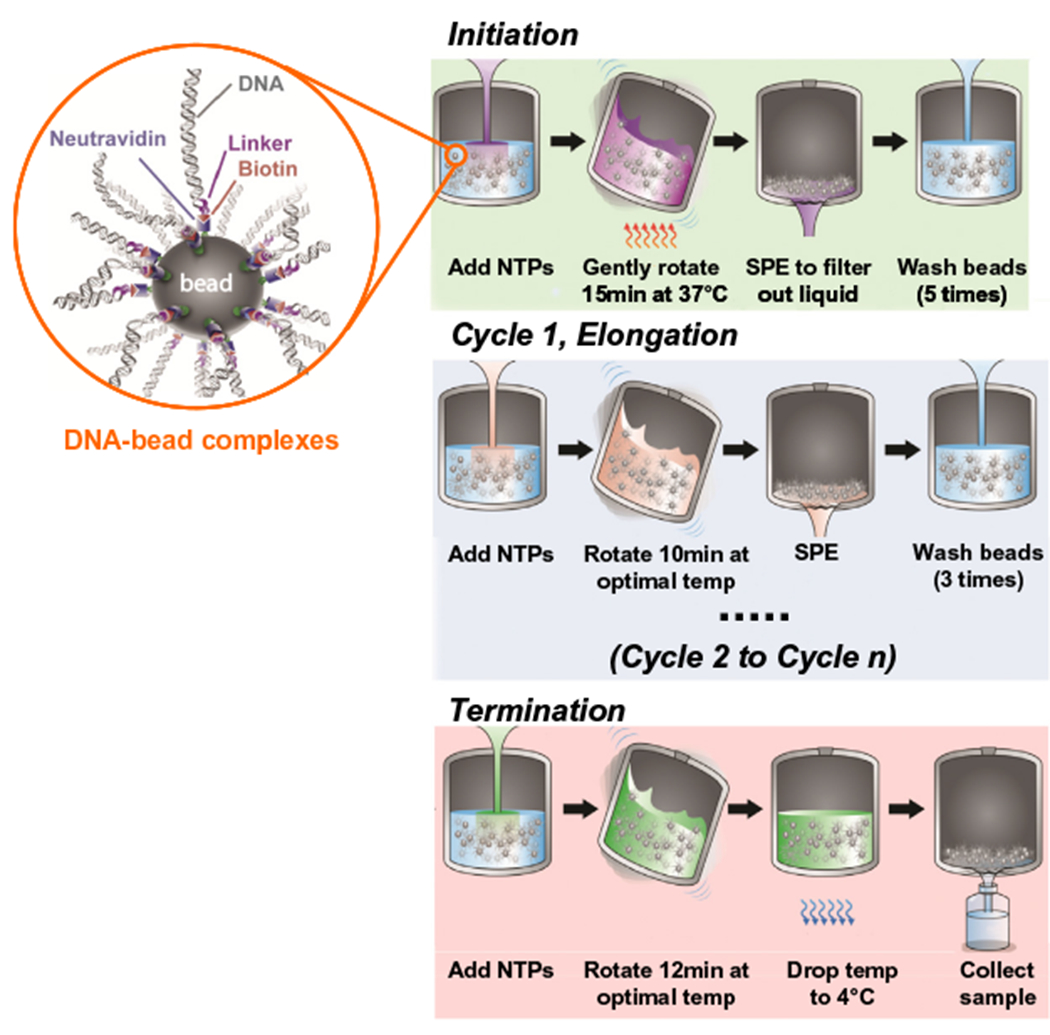
Diagram of PLOR synthesis. The biotin-tagged DNA templates were attached to neutravidin-coated agarose beads (zoomed view shown in orange circle). PLOR is divided into three stages: initiation (green background), elongation (blue background), and termination (red background). In the initiation, an NTP mix (excluding at least one NTP type) and T7 RNAP were added to DNA-beads in the reactor, and gently rotated for 15 min at 37 °C. T7 RNAP was paused at the first position corresponding to the missing NTP. SPE was performed to remove residual NTPs and abortive products, followed by rinsing of the beads 5 times to avoid cross contamination. In cycle 1 of elongation, transcription was resumed for 10 min at room temperature by the addition of a new NTP mix, followed by SPE and 3 bead-rinsing cycles. Transcription was paused and resumed in elongation cycles 2 through n with various NTP combinations according to the desired labeling schemes. In the termination cycle, all four types of NTPs were added, and the reaction was incubated for 12 min to complete synthesis of the desired transcripts, which were collected by elution at 4 °C. This figure is adapted from reference [27].
3. Experiments and procedures
The DNA templates used in the PLOR reactions were generated by PCR, and the forward and reverse primers contained biotin, and 2′-O-methylguanosine (mG), respectively, at the 5′-ends (the template sequences for riboA71 and TCV are listed in Table 1). The biotin at the 5′-end is for immobilization of DNA on the neutravidin-coated agarose beads [38] and mGs at the 5′-end of the coding strand is to reduce the formation of non-templated N + 1 transcripts [39]. The PCR-generated DNAs were then incubated with neutravidin-coated agarose resin at 4 °C. The NTP additions in PLOR vary with labeling schemes [27]. For example, for generating Lp2-CN-riboA71 (single 13C15N-labeled segment, Lp2, Fig. 1f), T7RNAP (20 μM) was mixed with the DNA-beads (20 μM, 25 mL) at 37 °C for 10 min, followed by the addition of ATP (1.92 mM), GTP (1.92 mM) and UTP (192 μM) and incubation for 15 min at 37 °C. The liquid phase in the initiation stage was removed by SPE and the beads were rinsed 5 times to ensure the removal of residual NTPs. ATP (40 μM), CTP (40 μM) and UTP (40 μM) were then added and incubated for 10 min at 25 °C in the reaction vessel, followed by SPE and bead rinsing 3 times. Subsequent elongation cycles and the termination were performed in the same manner as cycle 1 but with different NTP additions for each cycle: ATP(60 μM)/GTP(140 μM)/UTP(160 μM) (cycle 2), ATP(60 μM)/CTP (60 μM)/UTP(20 μM) (cycle 3), ATP(20 μM)/GTP(40 μM) (cycle 4), 13C15N-CTP(40 μM)/13C15N-UTP (40 μM) (cycle 5), 13C15N-ATP (60 μM) (cycle 6), CTP(40 μM)/GTP (20 μM)/UTP (60 μM) (cycle 7), and ATP(40 μM)/CTP(80 μM)/UTP (100 μM). The temperature of the reaction vessel was dropped to 4 °C prior to sample collection. The above protocol was repeated to synthesize various riboA71 and TCV derivatives, with only the elongation cycles and NTP additions varied between different syntheses.
Table 1.
DNA templates for PLOR-generated riboA71 and TCV.
| DNA templates | Sequencea |
|---|---|
| riboA71 non-coding strand | 5′-biotin-TCTGATTCAGCTAGTCCATAATACGACTCACTATAGGGAAGATATAATCCTAATGATATGGTTTGGGAGTTTCTACCAAGAGCCTTAAACTCTTGATTATCTTCCC-3′ |
| riboA71 coding strand | 5′-mGmGGAAGATAATCAAGAGTTTAAGGCTCTTGGTAGAAACTCCCAAACCATATCATTAGGATTATATCTTCCCTATAGTGAGTCGTATTATGGACTAGCTGAATCAGA-3′ |
| TCV non-coding strand | 5′-biotin-TCTGATTCAGCTAGTCCATAATACGACTCACTATAGGGGACGGCGGCAGCGCTGTCTAGCTGCGGGCATTAGACTGGAAAACTAGTGCTCTTTGGGTAACCACTAAAATCCCGAAAGGGTGGGCTGTGGTGACCCTCCG-3′ |
| TCV coding strand | 5′-CGGAGGGTCACCACAGCCCACCCTTTCGGGATTTTAGTGGTTACCCAAAGAGCACTAGTTTTCCAGTCTAATGCCCGCAGCTAGACAGCGCTGCCGCCGTCCCCTATAGTGAGTCGTATTATGGACTAGCTGAATCAGA-3′ |
Residues in the linker regions are italicized. Underlined residues correspond to the T7 promoter. mG refers to 2′-O-methylguanosine.
4. Results and discussion
The 1H13C-TROSY spectra of S1 + Lk1-CN-riboA71 (the first 13 residues in the Helix 1 plus Linker 1 were 1H/13C/15N isotope-labeled in this sample) and Lp2-CN are shown in Fig. 3a and b, respectively [27]. The peaks in the C6-H6 or C8-H8 region of the spectra matched our labeling schemes, and the peaks from the non-labeled residues are invisible, which greatly alleviate peak crowdedness in the spectrum for CN-riboA71 (uniformly 13C15N-labeled riboA71, prepared by conventional in vitro transcription, Fig. 3c). This significantly improved the accuracy of peak identification and analysis. RNA consists of only four building blocks: A, U, G and C. These four building blocks differ only in nucleotide bases but are otherwise identical. Even among the bases, the chemical structures of adenine and guanine are very similar, as are uracil and cytosine. In addition to these similarities, more than 70% of RNA structural blocks are duplexes. All of these chemical and structural similarities result in very similar chemical shift environments, resulting a very narrow chemical shift dispersion and severe NMR signal overlaps. For example, in the case of the aptamer domain of the adenine riboswitch, the aromatic chemical shift signal overlapping among the residues A4, A5, A7, and A9 in Helix 1 was observed in the spectrum of S1 + Lk1-CN-riboA71 (Fig. 3a). To resolve these peaks and confirm their assignments, we produced S1 + Lk1-H by PLOR, in which only Helix 1 and Linker 1 were protonated while the rest of the residues were deuterated. This labeling scheme resulted in a greatly simplified spectrum (Fig. 3d) compared to that of riboA71 (all residues protonated, Fig. 3e), and the strategy of an NOE-walk in a regular helix region was applied for assignment purposes based on sequential connectivity. In addition to being used to isotopically label specific RNA segment(s), PLOR was also applied to place multiple isotope-labeled residues at discrete positions or at a single position for studying RNA dynamics.
Fig. 3.
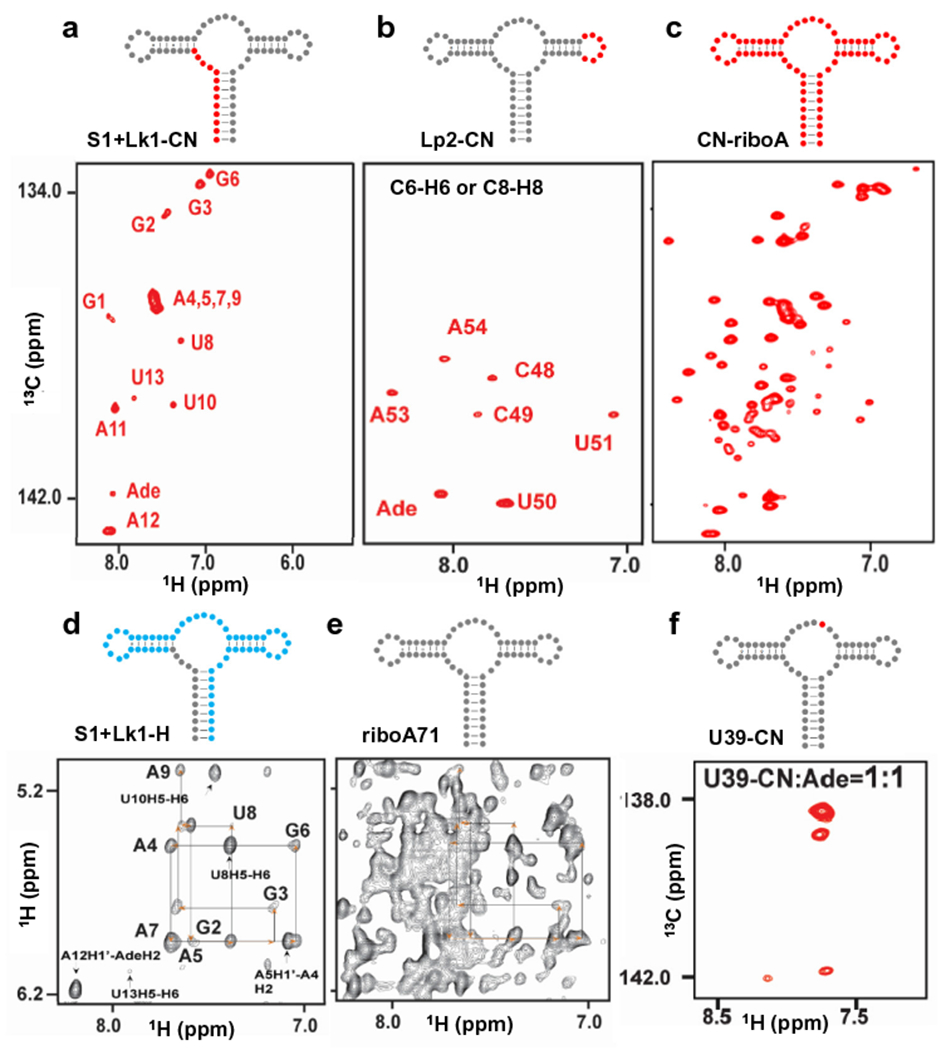
PLOR-generated NMR and FRET samples. a–c, The C6/C8-H6/H8 region of the 1H13C-TROSY spectra of a, S1 + Lk1-CN-riboA71, b, Lp2-CN-riboA71, c, CN-riboA71 (uniformly 13C15N-labeled, synthesized by using conventional in vitro liquid-phase transcription), and f, U39-CN-riboA71. d–e, The H1′/H5-H6/H8 region of the 1H1H-NOESY spectra of d, S1 + Lk1-H-riboA71, and e, riboA71. 13C15N-labeled, 2D-labeled and non-labeled residues are shown in red, blue, and grey dots, respectively. The “Ade” in the assignments refers to the ligand adenine. NMR spectra were collected using an 850 MHz Bruker magnet at 25 °C with 0.3–0.5 mM RNAs in 0.5–5 mM adenine. This figure is adapted from reference [27].
NMR is powerful for detecting multiple conformers of RNA in solution as revealed by the presence of multiple cross peaks associated with the same residues. However, such detection is impractical when signals from other residues are nearby. Therefore, it is critical to be able to detect signals from the residues of interest while signals from other residues are suppressed. With PLOR, it is possible to accomplish this. For example, the imino group of U39 forms a hydrogen bond with the adenine ligand [33,40], so we applied PLOR to introduce a single 1H/13C/15N isotope-labeled residue at this structurally important position to probe the conformations of riboA71 upon binding adenine [27]. Four peaks in the spectrum reveals that four conformations may co-exist in the binding pocket (Fig. 3f). FRET data of the U24Cy3-C55Cy5-B sample generated by PLOR (U24, C55 and 3′-end labeled with Cy3, Cy5, and biotin, respectively.) also supports the conclusion that multiple conformations co-exist and that the high-FRET conformation is formed both in the absence and presence of adenine [41,42].
5. Conclusions
Many new techniques for RNA labeling are constantly being developed, yet the demand is rising in this era full of breakthroughs in RNA [23]. PLOR, a hybrid solid-liquid phase transcription method, combines the robustness of solid-phase synthesis with the efficiency of in vitro liquid-phase transcription for long RNAs. This method is applicable to synthesize long RNAs with discrete labels that are difficult to be obtained with high yields by using enzymatic ligation method. By using PLOR-generated RNAs, we were able to observe chemical signals from only the labeled residues and obscure signals from non-labeled sites. This not only greatly simplifies interpretation of NMR spectra, but also makes it possible to explore details of structure and dynamics in a desired region or position that would otherwise be difficult to study. The capability of PLOR to label discrete residues or a single position in the aptamer domain of the adenine riboswitch allows one to selectively monitor signals from structurally or functionally significant residues and to unambiguously identify the coexistence of conformers. Theoretically, all the labeling schemes described here can be achieved by ligations, however, it is not practical to introduce isotope-labels to multiple discrete segments by using ligations. This can, however, be readily achieved using PLOR. Another distinct advantage of PLOR is that it is able to produce labeled RNA on the milligram scale. Overall yields ([product]/[template]) in the PLOR syntheses for isotopically labeled RNAs decreased with elongation cycle numbers: more cycles led to lower yields, and the yields for generating riboA71 derivatives ranged from 8.5 to 40.3% [27]. The experimental yield for H1-TCV was close to the expected yield that was calculated by using the initiation and elongation efficiencies of the riboA71 system. This indicates that the PLOR yields are not related to RNA length, since TCV contains 104 nt, about 50% longer than riboA71. In principle, PLOR is not limited by RNA size since T7 RNAP has been routinely used to synthesize RNAs ranging from tens to thousands of nucleotides in length [43,44]. Instead, PLOR yields depend primarily on the number of elongation cycles or steps required for achieving a desired labeling scheme, rather than the lengths of the transcripts. However, ‘walking’ the RNA polymerase to a desired site in a long RNA may require more elongation cycles than for a shorter RNA, which can lead to lower yields using the batch methodology in the current version of PLOR.
PLOR differs from a conventional liquid-phase transcription, in which hundreds of rounds of transcriptions are usually carried out on each DNA template. Therefore, PLOR requires significantly more DNA template (for example, 20 μM in the Lp2-CN synthesis) to generate sufficient quantities of labeled RNA for NMR experiments in one round of transcription. Fortunately, DNA templates can be generated readily using efficient PCR protocols. In addition, the templates for PLOR are immobilized on beads, and can be recycled. The ability to reuse the templates allows syntheses to be scaled up (by combining products from multiple PLOR rounds) or used to prepare distinctly labeled RNAs, which makes PLOR both convenient and economical for generating large amounts of specifically-labeled RNAs. The micromolar NTP concentrations used in PLOR are much lower than in conventional in vitro transcription (where they are usually millimolar). Lower NTP concentrations reduces the consumption of expensive reagents, such as isotopically-labeled, fluorescent, or other modified NTPs, and, therefore, reduces the cost of generating selectively labeled RNAs. We further investigated the fidelity of both the sequence and three-dimensional structures of the PLOR-generated RNAs. Superposition of the NMR spectra or crystal structures of the PLOR-generated RNAs with those of the RNAs synthesized by conventional transcription indicates that the RNA samples prepared by using PLOR has the same sequence as designed and the pause/resume synthesis schemes used in PLOR do not affect the folding of these RNAs (Fig. 4).
Fig. 4.
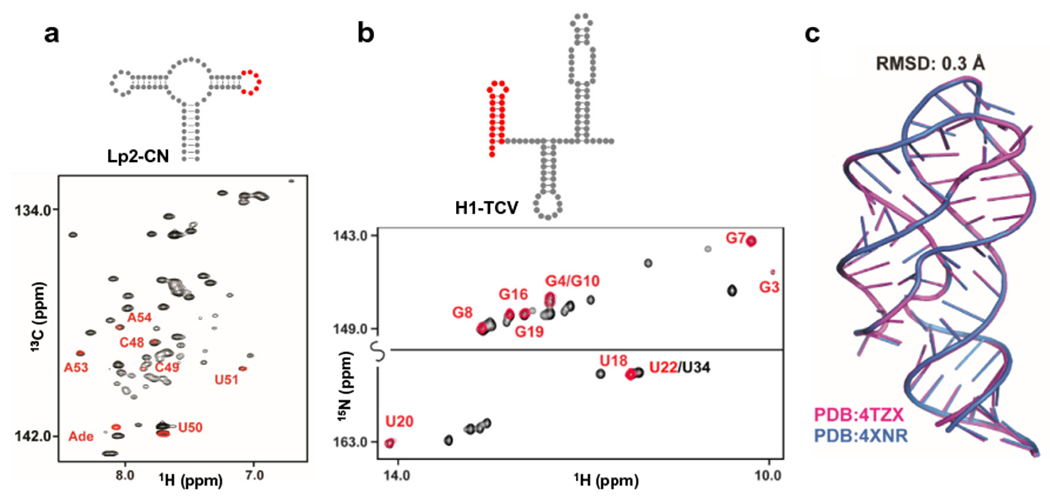
PLOR-generated RNA maintain both sequence and structural fidelity. a, Superposition of the 1H13C-TROSY spectra of CN-riboA71 (black) with Lp2-CN (red). b, Superposition of the 1H15N-TROSY spectra of 104nt-TCV (black) with H1-TCV (red). CN-riboA71 and 104nt-TCV were uniformly labeled by using in vitro liquid-phase transcription, and Lp2-CN and H1-TCV were produced by using PLOR. c, Structural superposition of the riboA71 generated by PLOR (PDB accession number: 4XNR) and by in vitro liquid-phase transcription (PDB accession number: 4TZX). These results support that the RNA synthesized by PLOR fold same as their counterparts synthesized by in vitro liquid-phase transcription. This figure is adapted from reference [27].
The potential applications and utility of an RNA-labeling method depend on its flexibility in allowing a variety of different labels to be placed at desired positions. PLOR has been used to place isotopically labeled, fluorescent, biotinylated, or chemically modified groups at desired positions in RNAs for structural and functional studies. We have also used PLOR to incorporate bromo- or iodo-labeled nucleotides at specific positions in an RNA (data not shown), which is likely to be useful for de novo phasing of diffraction data obtained by X-ray crystallography. The main obstacle of the PLOR method is efficiency, not of the method itself but its implementation: inefficient washing and toleration of RNA polymerase. Inefficient washing can be improved by application of better vacuum filtration or replaced with a microfluidic methodology during bead rinsing. T7 RNAP is used in current PLOR, however, it may not be the most efficient enzyme for PLOR, and efficiency can potentially be improved by using other RNA polymerases that are better-tolerated for bulky modifications. It should be possible to introduce other modified NTPs with bulky groups at specific sites in RNAs by using PLOR as long as the modifications can be tolerated by RNA polymerase.
Acknowledgments
This work was supported by the Intramural Research Programs of the National Cancer Institute. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Abbreviations:
- NMR
nuclear magnetic resonance
- PLOR
position-selective labeling of RNA
- T7 RNAP
T7 RNA polymerase
- nt
nucleotides
- smFRET
single molecule Förster resonance-energy transfer
- SPE
solid-phase extraction
- PCR
polymerase chain reaction
- NOESY
nuclear Overhauser spectroscopy
- TROSY
transverse relaxation optimized spectroscopy
References
- [1].Johnson JE Jr., Julien KR, Hoogstraten CG, Alternate-site isotopic labeling of ribonucleotides for NMR studies of ribose conformational dynamics in RNA, J. Biomol. NMR 35 (2006) 261–274. [DOI] [PubMed] [Google Scholar]
- [2].Nelissen FH, van Gammeren AJ, Tessari M, Girard FC, Heus HA, Wijmenga SS, Multiple segmental and selective isotope labeling of large RNA for NMR structural studies, Nucl. Acids Res 36 (2008) e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alvarado LJ, Longhini AP, LeBlanc RM, Chen B, Kreutz C, Dayie TK, Chemo-enzymatic synthesis of selectively 13C/15N-labeled RNA for NMR structural and dynamics studies, Methods Enzymol. 549 (2014) 133–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Duss O, Lukavsky PJ, Allain FH, Isotope labeling and segmental labeling of larger RNAs for NMR structural studies, Adv. Exp. Med. Biol 992 (2012) 121–144. [DOI] [PubMed] [Google Scholar]
- [5].Wunderlich CH, Juen MA, LeBlanc RM, Longhini AP, Dayie TK, Kreutz C, Stable isotope-labeled RNA phosphoramidites to facilitate dynamics by NMR, Methods Enzymol. 565 (2015) 461–494. [DOI] [PubMed] [Google Scholar]
- [6].Wunderlich CH, Spitzer R, Santner T, Fauster K, Tollinger M, Kreutz C, Synthesis of (6–13C)pyrimidine nucleotides as spin-labels for RNA dynamics,J. Am. Chem. Soc 134 (2012) 7558–7569. [DOI] [PubMed] [Google Scholar]
- [7].Lu K, Miyazaki Y, Summers MF, Isotope labeling strategies for NMR studies of RNA, J. Biomol. NMR 46 (2010) 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Neuner S, Santner T, Kreutz C, Micura R, The “speedy” synthesis of atom-specific 15N imino/amido-labeled RNA, Chem.-Eur. J 21 (2015) 11634–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cate JH, Doudna JA, Solving large RNA structures by X-ray crystallography, Methods Enzymol. 317 (2000) 169–180. [DOI] [PubMed] [Google Scholar]
- [10].Lin L, Sheng J, Huang Z, Nucleic acid X-ray crystallography via direct selenium derivatization, Chem. Soc. Rev 40 (2011) 4591–4602. [DOI] [PubMed] [Google Scholar]
- [11].Keel AY, Rambo RP, Batey RT,Kieft JS,A general strategy to solve the phase problem in RNA crystallography, Structure 15 (2007) 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Du Q, Carrasco N, Teplova M, Wilds CJ, Egli M, Huang Z, Internal derivatization of oligonucleotides with selenium for X-ray crystallography using MAD, J. Am. Chem. Soc 124 (2002) 24–25. [DOI] [PubMed] [Google Scholar]
- [13].Ennifar E, Carpentier P, Ferrer JL, Walter P, Dumas P, X-ray-induced debromination of nucleic acids at the Br K absorption edge and implications for MAD phasing, Acta Crystallogr. Section D Biol. Crystallogr 58 (2002) 1262–1268. [DOI] [PubMed] [Google Scholar]
- [14].Olieric V, Rieder U, Lang K, Serganov A, Schulze-Briese C, Micura R, Dumas P, Ennifar E, A fast selenium derivatization strategy for crystallization and phasing of RNA structures, RNA 15 (2009) 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun HY, Sheng J, Hassan AEA, Jiang SB, Gan JH, Huang Z, Novel RNA base pair with higher specificity using single selenium atom, Nucl. Acids Res 40 (2012) 5171–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moroder H, Kreutz C, Lang K, Serganov A, Micura R, Synthesis, oxidation behavior, crystallization and structure of 2′-methylseleno guanosine containing RNAs, J. Am. Chem. Soc 128 (2006) 9909–9918. [DOI] [PubMed] [Google Scholar]
- [17].Shaw E, St-Pierre P, McCluskey K, Lafontaine DA,Penedo JC, Using sm-FRET and denaturants to reveal folding landscapes, Methods Enzymol. 549 (2014) 313–341. [DOI] [PubMed] [Google Scholar]
- [18].Solomatin S, Herschlag D, Methods of site-specific labeling of RNA with fluorescent dyes, Methods Enzymol. 469 (2009) 47–68. [DOI] [PubMed] [Google Scholar]
- [19].Kruger AC, Birkedal V, Single molecule FRET data analysis procedures for FRET efficiency determination: probing the conformations of nucleic acid structures, Methods 64 (2013) 36–42. [DOI] [PubMed] [Google Scholar]
- [20].Hong H, Goel S, Zhang Y, Cai W, Molecular imaging with nucleic acid aptamers, Curr. Med. Chem 18 (2011) 4195–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Soontornworajit B, Wang Y, Nucleic acid aptamers for clinical diagnosis: cell detection and molecular imaging, Anal. Bioanal. Chem 399 (2011)1591–1599. [DOI] [PubMed] [Google Scholar]
- [22].Pitchiaya S, Heinicke LA, Custer TC, Walter NG, Single molecule fluorescence approaches shed light on intracellular RNAs, Chem. Rev 114 (2014) 3224–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Y, Sousa R, Wang YX, Specific labeling: an effective tool to explore the RNA world, Bioessays 38 (2016) 192–200. [DOI] [PubMed] [Google Scholar]
- [24].Paredes E, Evans M, Das SR, RNA labeling, conjugation and ligation, Methods 54 (2011) 251–259. [DOI] [PubMed] [Google Scholar]
- [25].Caruthers MH, The chemical synthesis of DNA/RNA: our gift to science, J. Biol. Chem 288 (2013) 1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Caruthers MH, A brief review of DNA and RNA chemical synthesis, Biochem. Soc. Trans 39 (2011) 575–580. [DOI] [PubMed] [Google Scholar]
- [27].Liu Y, Holmstrom E, Zhang J, Yu P, Wang J, Dyba MA, Chen D, Ying J, Lockett S, Nesbitt DJ, Ferre-D’Amare AR, Sousa R, Stagno JR, Wang YX, Synthesis and applications of RNAs with position-selective labelling and mosaic composition, Nature 522 (2015) 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zuo X, Wang J, Yu P, Eyler D, Xu H, Starich MR, Tiede DM, Simon AE, Kasprzak W, Schwieters CD, Shapiro BA, Wang YX, Solution structure of the cap-independent translational enhancer and ribosome-binding element in the 3′ UTR of turnip crinkle virus, Proc. Natl. Acad. Sci. USA 107 (2010) 1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang YX, Zuo X, Wang J, Yu P, Butcher SE, Rapid global structure determination of large RNA and RNA complexes using NMR and small-angle X-ray scattering, Methods 52 (2010) 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stupina VA, Meskauskas A, McCormack JC, Yingling YG, Shapiro BA, Dinman JD, Simon AE, The 3′ proximal translational enhancer of Turnip crinkle virus binds to 60S ribosomal subunits, RNA 14 (2008) 2379–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Reining A, Nozinovic S, Schlepckow K, Buhr F, Furtig B, Schwalbe H, Three-state mechanism couples ligand and temperature sensing in riboswitches, Nature 499 (2013) 355–359. [DOI] [PubMed] [Google Scholar]
- [32].Leipply D, Draper DE, Effects of Mg2+ on the free energy landscape for folding a purine riboswitch RNA, Biochemistry 50 (2011) 2790–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ, Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs, Chem. Biol 11 (2004) 1729–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lyakhov DL, He B, Zhang X, Studier FW, Dunn JJ, McAllister WT, Pausing and termination by bacteriophage T7 RNA polymerase,J. Mol. Biol 280 (1998) 201–213. [DOI] [PubMed] [Google Scholar]
- [35].Guo Q, Nayak D, Brieba LG, Sousa R, Major conformational changes during T7RNAP transcription initiation coincide with, and are required for, promoter release, J. Mol. Biol 353 (2005) 256–270. [DOI] [PubMed] [Google Scholar]
- [36].Sohn Y, Shen H, Kang C, Stepwise walking and cross-linking of RNA with elongating T7 RNA polymerase, Methods Enzymol. 371 (2003) 170–179. [DOI] [PubMed] [Google Scholar]
- [37].Levin JR, Krummel B, Chamberlin MJ, Isolation and properties of transcribing ternary complexes of Escherichia coli RNA polymerase positioned at a single template base, J. Mol. Biol 196 (1987) 85–100. [DOI] [PubMed] [Google Scholar]
- [38].Sano T, Vajda S, Cantor CR, Genetic engineering of streptavidin, a versatile affinity tag, J. Chromatogr. B Biomed. Sci. Appl 715 (1998) 85–91. [DOI] [PubMed] [Google Scholar]
- [39].Kao C, Zheng M, Rudisser S, A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase, RNA 5 (1999) 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang J, Ferre-D’Amare AR, Dramatic improvement of crystals of large RNAs by cation replacement and dehydration, Structure 22 (2014) 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lemay JF, Penedo JC, Tremblay R, Lilley DM, Lafontaine DA, Folding of the adenine riboswitch, Chem. Biol 13 (2006) 857–868. [DOI] [PubMed] [Google Scholar]
- [42].Dalgarno PA, Bordello J, Morris R, St-Pierre P, Dube A, Samuel ID, Lafontaine DA, Penedo JC, Single-molecule chemical denaturation of riboswitches, Nucl. Acids Res 41 (2013) 4253–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pokrovskaya ID, Gurevich VV, In vitro transcription: preparative RNA yields in analytical scale reactions, Anal. Biochem 220 (1994) 420–423. [DOI] [PubMed] [Google Scholar]
- [44].Milligan JF, Uhlenbeck OC, Synthesis of small RNAs using T7 RNA polymerase, Methods Enzymol. 180 (1989) 51–62. [DOI] [PubMed] [Google Scholar]