

Abstract
Polygenic risk scores (PRS) are increasingly used to estimate the personal risk of a trait based on genetics. However, most genomic cohorts are of European populations, with a strong under-representation of non-European groups. Given that PRS poorly transport across racial groups, this has the potential to exacerbate health disparities if used in clinical care. Hence there is a need to generate PRS that perform comparably across ethnic groups. Borrowing from recent advancements in the domain adaption field of machine learning, we propose FairPRS - an Invariant Risk Minimization (IRM) approach for estimating fair PRS or debiasing a pre-computed PRS. We test our method on both a diverse set of synthetic data and real data from the UK Biobank. We show our method can create ancestry-invariant PRS distributions that are both racially unbiased and largely improve phenotype prediction. We hope that FairPRS will contribute to a fairer characterization of patients by genetics rather than by race.
Keywords: Polygenic Risk Scores, Fairness, Racial Disparity, Invariant Risk Minimization, Machine Learning, Precision medicine
1. Introduction
Genome wide association studies (GWAS) were developed for finding statistical associations between single nucleic polymorphisms (SNPs) and phenotype traits. Later, these associations were then aggregated into a score – a polygenic (risk, for diseases) score (PRS) – for predicting traits.1 PRS became extremely popular due to its promise of harnessing one’s genome to act as a biomarker for personalizing medical risk estimation. This capacity for personalization can also translate to heterogeneity on the population level with PRS helping to identify subpopulations that are at higher risk of disease.2
Unfortunately, PRSs are plagued by many issues. Primarily GWAS cohorts strongly suffer from a lack of sample diversity. For example, 79% of all participants in the NHGRI-EBI GWAS catalog3 are of European descent despite being only 16% of the global population.4 The under-representation of minority groups in cohorts leads to inferior PRS because PRS derived from European ancestry tend to perform poorly in genetically diverse populations and even within other admixed European populations.5 As a simple example, polygenic scores for height predict all Africans to be shorter than Europeans, contrary to empirical evidence.6 Thus, using PRS for precision medicine in its current form may exacerbate health disparities until the lack of representation is solved.4
Reducing racial bias in genomic prediction may contribute to more equitable healthcare for all. But to establish health equity in precision medicine we require better genetic cohorts whose multi-ethnic representation matches real life. This solution, however, is resource heavy and is long-term. Meanwhile, we can apply advances in machine intelligence to mitigate bias in trait prediction from PRS.
There is prior work on using computational frameworks for making PRS generalize better across subgroups. These include deconvoluting ancestry and partial PRS computation,7 computing ancestry-specific PRS to showcase their utility as predictors across different populations,8 or enabling more accurate effect size estimation by leveraging linkage disequilibrium diversity with GWAS summary statistics.9 Advances in machine learning such as using transfer learning-based methods10 and deep learning based methods have been applied to make PRS more portable across ancestries.11 However, either some of these methods assume part of the background genome is still of European origin7,10 or consider pre-computed associated markers as input to reduce search space which can contain significant bias or spurious associations.
In this work, we apply a domain-adaptation-based paradigm called Invariant Risk Minimization (IRM)12 in the context of PRS. We consider the problem of generalizability of PRS as an out of distribution generalization problem, a common machine learning problem where models are developed in one domain but are deployed in another.13 IRM’s goal is to generate invariant predictors given multiple training domains. In our context, these different domains are adapted to be the different ancestry groups, therefore allowing for race-invariant phenotype prediction from PRS. Our goal is then to learn a generalizable PRS that contains as little ancestry information as possible, while still accurately predicting the phenotype of interest.
We present FairPRS, a framework for finding and mitigating bias in PRS which improves generalizability across populations and make it portable while increasing the prediction accuracy of the phenotype of interest. FairPRS is robust across both rigorous simulation studies involving arbitrary population structure and pre-computed PRS obtained from UK Biobank (UKB).14
2. Methods
FairPRS offers an entire pipeline from genetic data to trait prediction. It has three possible access points for input: genotypes, genotypes with summary statistics, or a pre-computed PRS. We will explain the FairPRS framework herein, followed by the autoencoder architecture, training, and evaluation phases of the pipeline. Thereafter, we will discuss the simulation and real data used in the study for evaluating FairPRS including computational details.
2.1. FairPRS framework
The FairPRS pipeline is designed for ease and customization with multiple access points based on the user needs. Moreover, the pipeline can be run for a user-determined number of iterations for all or specific portions. The first stage focuses on processing the genotype data towards PRS computation. It allows to calculate the summary statistics, from GWAS, and principal components (PCs) of the genotype data. The PCs can be used as covariates for the GWAS and as input to the FairPRS model. The summary statistics are computed using PLINK v2.015 and the PCs are efficiently calculated for large scale data using TeraPCA.16 Next, the pipeline allows starting at the PRS computation step if the user has previously calculated the summary statistics. The betas are extracted from the summary statistics and used for PRS computation through PRSice217 in the validation cohort.
Lastly, the third stage is the FairPRS model which uses the pipeline-computed or user-provided PRS and PCs as input, while the phenotype and the PRS will be used for the training supervision. The model is implemented as a dual task autoencoder and MLP as shown in Figure 1. Briefly, first, the data is encoded into a shared latent representation. The latent representation is then fed into two tasks in parallel: decoding the PRS input and predicting the phenotype. The losses are then combined with the ancestry information to obtain the IRM loss. The fair PRS estimates are obtained from the PRS decoder output. A key point in this step is the automatic multi-thread hyperparameter tuning per iteration with allows the pipeline to train high-performing models in an efficient manner. After the model training and evaluation, the average performance over the iterations is reported and a dictionary with all the results per iteration is saved for further analysis and reproducibility purposes.
Fig. 1.
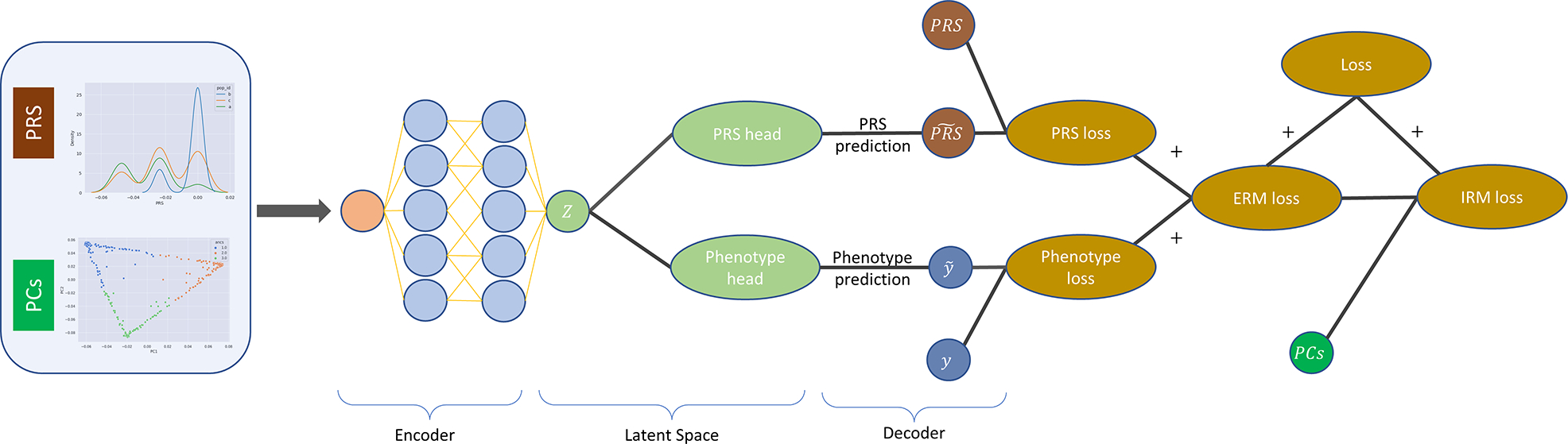
Pipeline of FairPRS outlining the input variables: pre-computed PRS and genetic structure as represented by PCs from test data, the autoencoder used with IRM loss for learning the fair PRS output estimates with negligible ancestry influence.
2.1.1. Implementation and Evaluation
Detailed architecture
The encoder is a single layer with ReLU activation, and latent space size determined as a hyperparameter. Both the PRS decoder and the phenotype prediction head perform a 10% dropout and then apply a single linear layer. The ERM loss is obtained by adding the two MSE losses with equal weight. The final loss is a weighted sum of the ERM and IRM losses, with the weight being a hyperparameter. Adam method was used for optimization.18 The framework is implemented using PyTorch 1.11.19
Training
The proposed model allows using regression losses for the double task network and employs multiple environments corresponding to the number of populations present in the PRS data. An automatic hyper-parameter search with parallel trials is used while training to fine-tune the model in a more efficient manner. The random search of hyperparameters was done for the learning rate (log-uniform [10−5,0.1]), the dimension of the latent space (uniform from 2i : i ∈ [2,9]), and the relative weight of the IRM loss (uniform [0.5,1.5]). The search space was defined based on preliminary experiments allowing for a wide search without a prohibitively computationally expensive search space. Tuning was done using Ray Tune.20 UK Biobank data was also randomly split to train (70%), validation (20%), and test (10%) sets. The best hyperparameter configuration was selected based on a validation set and was subsequently used for evaluation.
Evaluation
To test the model against a baseline in a fair way, both the original PRS and those resulting from the model were regressed separately against the outcome using ordinary least squares. The covariate-adjusted coefficients of determination (adjustedR2 scores) for both models are reported. Regression was done in Python using statsmodels.21 Results per iteration are computed to finally report the mean performance across all iterations.
2.2. Data
FairPRS was evaluated on multiple simulated and real datasets. The simulated datasets included a wide array of configurations and were generated using the data simulator in a previous work.22 Additionally, UK Biobank enhanced PRS (ePRS-UKB) for multiple phenotypes were used to further evaluate the model in real-world scenarios across different disease outcomes.
Simulated data
Three models for simulating genetic datasets with arbitrary population structure: Balding-Nichols (BN), Prichard-Stephens-Donelly (PSD), and 1000 Genomes Project (TGP) with 3 variance proportion configurations for genetic, environment and noise, , totaling in 9 different simulation scenarios were used to evaluate FairPRS. We used three populations for BN and PSD and ten populations for TGP. For each, model we generated 10 iterations resulting in 90 different datasets. The 3 proportions configurations used were . The number of causal SNPs was set at 5% for all simulated datasets. Moreover, for all configurations the simulated datasets included 100,000 SNPs, 10,000 samples for GWAS, 1000 for PRS training and 400 for PRS testing.
Real data
PRS and ancestry data were obtained from the UKB for further model validation.23 ePRS-UKB for 6 different conditions across 104,231 multi-ethnic individuals were used in our analysis, these are height, body mass index (BMI), glycated hemoglobin (HBA1C), high-density lipoprotein cholesterol (HDL), and low-density lipoprotein cholesterol (LDL).
3. Results
3.1. Simulated data
FairPRS consistently achieved higher or comparable phenotype prediction accuracy with respect to the original PRS computed by PRSice2,17 measured in terms of adjusted R2 after correcting for top eight principal components (PCs) computed by TeraPCA16 (Supplementary Figure 1). FairPRS achieved better results on all models across all simulation scenarios (Figure 2), each run with 10 iteration for reproducibility. Kolmogorov-Smirnov (KS) two-sample tests, a goodness of fit test of equality of the original vs. observed PRS distributions were done to test the null hypothesis of whether the two distributions were sampled from the same unknown distribution. This resulted in very low p-values (p < 10−160) across all simulation scenarios which rejected the null hypothesis that the FairPRS distributions and the original PRS distribution were sampled from the same distribution (see Supplementary Table 1). The KS tests were done using SciPy package in python. The Net Reclassification Index (NRI), a percentage score reflecting the directional change and the difference in adjusted R2 by using FairPRS on top of pre-computed PRS using PRSice2 shows that as the genetic variance increases in contribution to the phenotype, the NRI also increases (Figure 3). Hence, when a pre-computed PRS is augmented with FairPRS not only do we observe a higher R2 across all the simulation scenarios, but we also obtain a relatively unbiased PRS estimate with negligible ancestry influence.
Fig. 2.
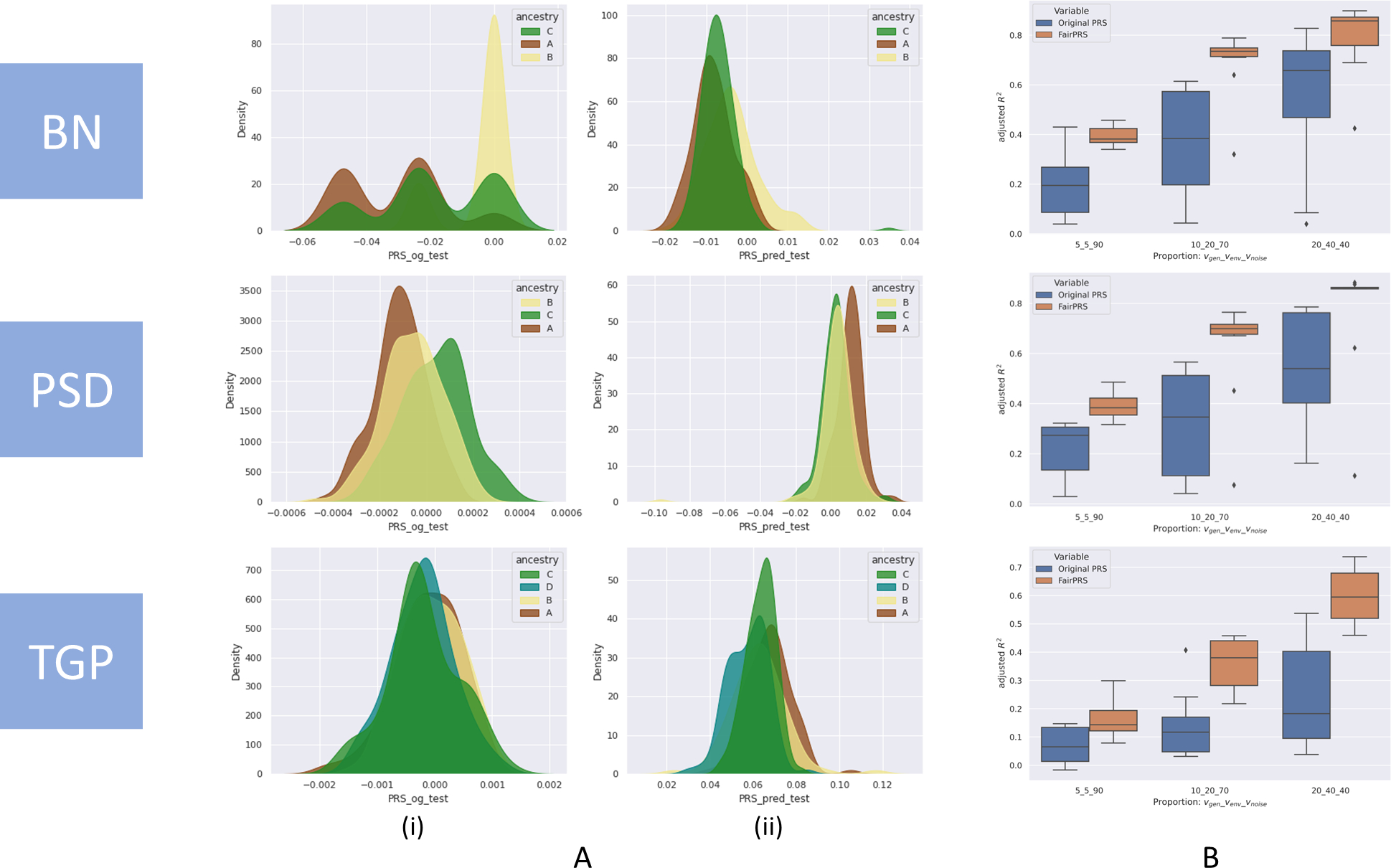
Simulation study results for three simulation models, BN, PSD and TGP. A. Distributions of ancestry-specific PRS computed by (i) PRSice2 and (ii) FairPRS. B. Box-and-whisker plot of adjusted R2 between the phenotype and PRS computed by PRSice2 and FairPRS across the variance proportions for .
Fig. 3.
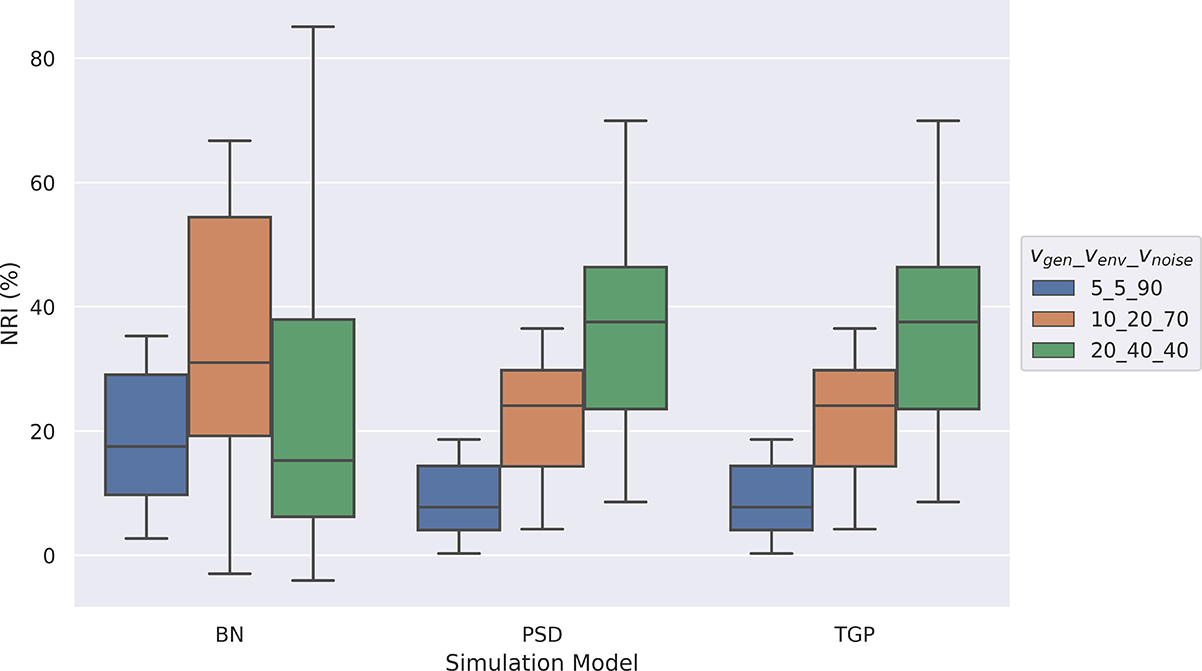
Box-and-whisker plot of NRI (%) between the phenotype and PRS after using FairPRS from pre-computed PRS, across the variance proportions for .
3.2. Real data
To demonstrate how FairPRS estimates real-world traits, we applied it on UKB-ePRS across six traits as mentioned above. FairPRS achieves considerably higher R2 compared to the pre-computed ePRS-UKB for all traits analyzed (Figure 4).
Fig. 4.
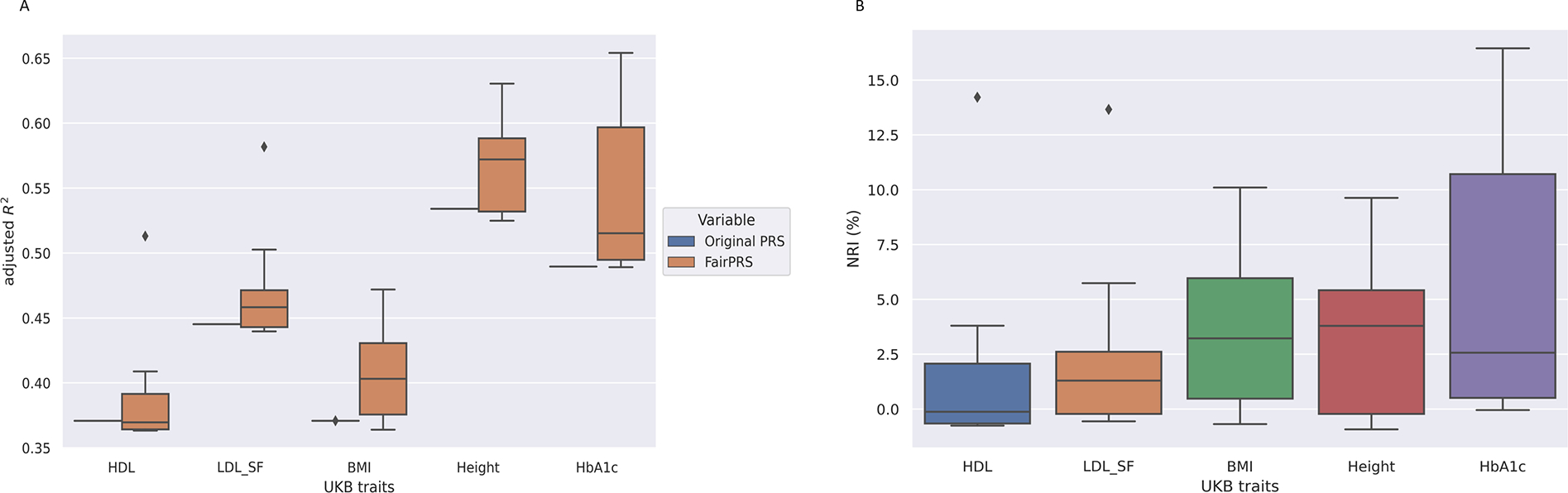
Applying FairPRS on UKB-ePRS estimates. A. Box-and-whisker plot of adjusted R2 between the UKB traits and PRS computed by PRSice2 and FairPRS. B. Box-and-whisker plot of NRI (%) of adjusted R2 between the phenotype and PRS after using FairPRS from pre-computed PRS.
We compared FairPRS with another recent transfer learning approach, TL-PRS10 and found that on the demo data set made available by TL-PRS, FairPRS performed similarly in predicting the phenotype after correcting for covariates.
We further examined the variance explained within each ancestry group. Figure 5 shows that, for all traits, FairPRS achieves increased performances among white, mixed, black ancestry groups while performing marginally better in the Asian ancestry group. HDL cholesterol is decreased in black with marginal increase in other populations as it is known to have a protective effect on Black British.24
Fig. 5.
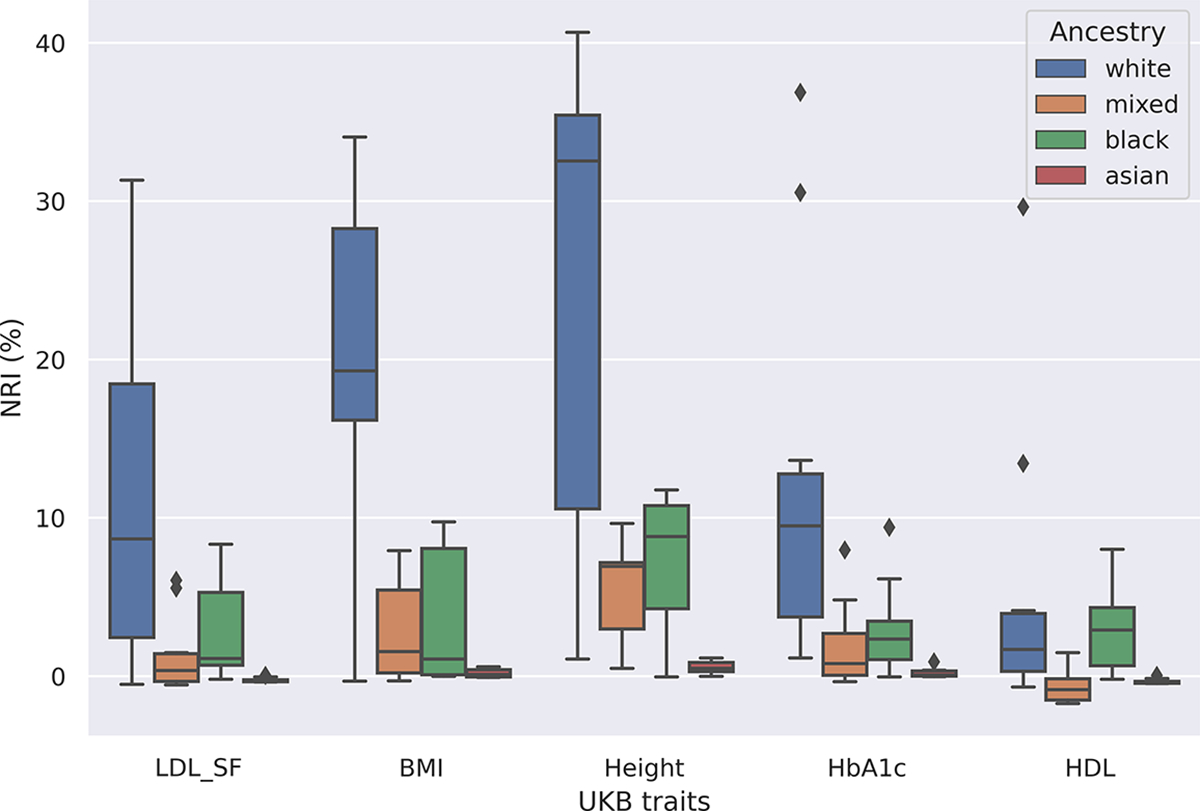
Applying FairPRS on UKB-ePRS estimates. Box-and-whisker plot of NRI (%) of adjusted R2 between the phenotype and PRS after using FairPRS from pre-computed PRS per ancestry group.
FairPRS was run 10 times for each ePRS-UKB trait analyzed for reproducibility and hyperparameter tuning. The NRI was computed by the percentage difference in R2 when using FairPRS vs. pre-computed PRS. Maximum NRI was observed in glycated hemoglobin (HbA1c) which is a biomarker for Type 2 Diabetes and has been shown to have high predictive accuracy for PRS. This was followed by BMI and Height, respectively which are very well-studied in terms of phenotypic variance explained by PRS.25 KS test for the two PRS distributions, FairPRS and pre-computed ePRS-UKB also resulted in the rejection of the null hypothesis (see Supplementary Table 2) and demonstrated that FairPRS learns a domain invariant distribution different from its input. This shows how FairPRS can result in better predictive accuracy in large biobanks such as UKB and can be integrated into precision medicine efforts.
4. Discussion
In this work, we combined notions from classical genetics: the polygenic risk scores (PRS), with notions from machine learning and domain adaptation. We developed a model that applies an Invariant Risk Minimization (IRM) approach to estimating PRS. Using both synthetic data and pre-computed PRS from the UK Biobank, we obtained PRS that are indistinguishable across races, while improving overall prediction accuracy in terms of adjusted R2 and NRI.
Our results show that performance also improved within ancestry groups in the UK Biobank data. Predictive performance improved for all ancestry groups, except Asians (east and south), for whom the performance was equivalent to the ePRS.23 The fact that improvement in accuracy did not come at the expense of either group is reassuring, suggesting FairPRS is safe in the sense it might not cause more harm than using regular PRS.
Despite their potential, GWAS are often plagued by the over-representation of European ancestry populations in their cohorts. If left uncorrected, this disproportional representation of population structure can lead to spurious associations and might only be able to explain a small fraction of heritability, among others issues.26 As PRS are computed from GWAS summary statistics, PRS inherits many of these drawbacks which contribute to its poor generalizability and transferability across populations due to the underlying influence of LD structure and environmental factors.2 Our method for finding fair estimates of PRS based on domain adaptation learns ancestry-invariant estimates which provide both qualitative and quantitative advantages.
Domain adaptation is a sub-field of machine learning focusing on model performance across multiple domains. The simplest driving example is when the distribution of data used for development, shifts during the deployment of the model. For example, using images of Swiss cows in the grassy Alps for training, while deploying the model to identify cows on the sandy beaches of Corsica.12,27 By having training data from multiple such sources and by training in an environment-aware approach - as with IRM, we can reduce the number of spurious correlations our model learns, like the grassy Alpine background.
In this work, we extend the notion of “domains” to different population ancestries. We apply the IRM framework, a form of supervised domain adaptation, to adjust the pre-computed PRS scores to be ancestry ignorant. Intuitively, we try to learn the most phenotype-predictive PRS, while forcing ourselves to ignore (or “forget”) any residual race information. Using IRM means we encourage the model to learn only information that is shared across ancestries. By constraining the PRS distribution of ancestries to coalesce, we ensure that when using the PRS for phenotype prediction, we get equal performance across ancestries. Thus, leading to a fairer PRS.
Different ancestries do exhibit disparities in health-related measures, and, therefore, different phenotypic distributions. However, these differences are rarely inherently biological. More often they are the result of how different ethnic subgroups interact with the healthcare system differently.28,29 (More formally, race disparities are more of an acquisition shift, rather than population or prevalence shift30). Consequently, forcing to disentangle race information from genetic information will (at least partially) remove race bias and will lead to a fairer usage of genetic data when assessing genetic risk.
Nonetheless, IRM is not limitations-free. First, more generally, IRM includes a challenging bi-level optimization that can fail if test data are too dissimilar to the training data.31 To counter that, more advanced flavors of IRM have been subsequently developed.32 In this work, we used the original formulation since we observe all environments (ancestries) during training, guaranteeing that test-time environments are indeed similar to training-time ones. Secondly, We also encountered difficulties when modeling binary traits, probably due to combining a cross-entropy loss for the classification task with a mean squared error for the continuous PRS reconstruction, which operate on different scales, requiring an additional hyper-parameter to weigh between them and further complicating the training process. Substituting the cross-entropy loss with an MSE, which is equivalent to a Brier score33 objective, lead to smoother training, but not necessarily better performance. We aim to fix this part of the model in future to obtain similar performance in binary traits as we observed in continuous traits. Thirdly, we saw a performance deterioration after increasing the number of expected environments and having not all of these present in the dataset, e.g., when having six expected ancestries in UKB experiments. However, in the real world we usually only have two to four ancestries that present relevant population structure, so while this was observed, it might be less of concern. An exciting future research direction is delving deeper into the interplay of FairPRS with local-ancestry based methods which highlights population sub-structure.
Limitations notwithstanding, FairPRS can be used as a tool to find unbiased estimates of pre-computed PRS or from GWAS summary statistics which would better predict the phenotype of interest. Unlike other methods which adjusts for admixed populations or LD interactions in computing PRS9,10 and needs summary statistics, LD information, etc. FairPRS can work with pre-computed PRS as well as summary statistics, making it easier to work with.
FairPRS estimates can be used as a step forward to achieve equity in precision medicine and evaluating disease risk in large clinical cohorts. It can be extensively used for out-of-sample prediction with pre-computed PRS to obtain ancestry-robust PRS which transport better across ancestries and datasets. In future work, we want to compare the performance of PRS computed by state-of-the-art methods and ancestry-robust FairPRS and evaluate their portability to other ancestries.
As the use of PRS is being advocated in clinical care, FairPRS can be an important tool to achieve equity in healthcare as well as further our understanding of true genetic causes of disease risk. We hope that FairPRS will contribute to a fairer characterization of patients by genetics rather than by race.
Supplementary Material
Acknowledgements
This work was funded by IBM. We would like to thank Kenney Ng for helping with data access.
Footnotes
Code Availability A Pytorch based implementation of FairPRS, along with scripts, descriptions and sample data to run experiments are available at https://github.com/ComputationalGenomics/FairPRS
Supplementary Material Supplementary material is hosted in the Supplementary directory in https://github.com/ComputationalGenomics/FairPRS
Data Availability
Simulated data is made available upon request. UKB-ePRS are available from UK Biobank.
References
- 1.Dudbridge F, Power and predictive accuracy of polygenic risk scores, PLoS genetics 9, p. e1003348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De La Vega FM and Bustamante CD, Polygenic risk scores: a biased prediction?, Genome medicine 10, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J et al. , The new nhgri-ebi catalog of published genome-wide association studies (gwas catalog), Nucleic acids research 45, D896 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM and Daly MJ, Clinical use of current polygenic risk scores may exacerbate health disparities, Nature genetics 51, 584 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamiza AB, Toure SM, Vujkovic M, Machipisa T, Soremekun OS, Kintu C, Corpas M, Pirie F, Young E, Gill D et al. , Transferability of genetic risk scores in african populations, Nature Medicine, 1 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD and Kenny EE, Human demographic history impacts genetic risk prediction across diverse populations, The American Journal of Human Genetics 100, 635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marnetto D, Pärna K, Läll K, Molinaro L, Montinaro F, Haller T, Metspalu M, Mägi R,Fischer K and Pagani L, Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals, Nature communications 11, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fritsche LG, Ma Y, Zhang D, Salvatore M, Lee S, Zhou X and Mukherjee B, On cross-ancestry cancer polygenic risk scores, PLoS genetics 17, p. e1009670 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruan Y, Lin Y-F, Feng Y-CA, Chen C-Y, Lam M, Guo Z, He L, Sawa A, Martin AR, Qin S et al. , Improving polygenic prediction in ancestrally diverse populations, Nature Genetics 54, 573 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Z, Fritsche LG, Smith JA, Mukherjee B and Lee S, The construction of multi-ethnic polygenic risk score using transfer learning, medRxiv (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gyawali PK, Guen YL, Liu X, Tang H, Zou J and He Z, Improving genetic risk prediction across diverse population by disentangling ancestry representations, arXiv preprint arXiv:2205.04673 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arjovsky M, Bottou L, Gulrajani I and Lopez-Paz D, Invariant risk minimization, arXiv preprint arXiv:1907.02893 (2019). [Google Scholar]
- 13.Shen Z, Liu J, He Y, Zhang X, Xu R, Yu H and Cui P, Towards out-of-distribution generalization: A survey, arXiv preprint arXiv:2108.13624 (2021). [Google Scholar]
- 14.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M et al. , Uk biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age, PLoS medicine 12, p. e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM and Lee JJ, Second-generation PLINK: rising to the challenge of larger and richer datasets, GigaScience 4, p. 7 (February 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bose A, Kalantzis V, Kontopoulou E-M, Elkady M, Paschou P and Drineas P, Terapca: a fast and scalable software package to study genetic variation in tera-scale genotypes, Bioinformatics 35, 3679 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Choi SW and O’Reilly PF, Prsice-2: Polygenic risk score software for biobank-scale data, Gigascience 8, p. giz082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kingma DP and Ba J, Adam: A method for stochastic optimization, arXiv preprint arXiv:1412.6980 (2014). [Google Scholar]
- 19.Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, Killeen T, Lin Z, Gimelshein N, Antiga L, Desmaison A, Kopf A, Yang E, DeVito Z, Raison M, Tejani A, Chilamkurthy S, Steiner B, Fang L, Bai J and Chintala S, Pytorch: An imperative style, high-performance deep learning library, in Advances in Neural Information Processing Systems 32, eds. Wallach H, Larochelle H, Beygelzimer A, d’Alché-Buc F, Fox E and Garnett R (Curran Associates, Inc., 2019) pp. 8024–8035. [Google Scholar]
- 20.Liaw R, Liang E, Nishihara R, Moritz P, Gonzalez JE and Stoica I, Tune: A research platform for distributed model selection and training, arXiv preprint arXiv:1807.05118 (2018). [Google Scholar]
- 21.Seabold S and Perktold J, Statsmodels: Econometric and statistical modeling with python, in Proceedings of the 9th Python in Science Conference, eds. van der Walt Stéfan and Millman Jarrod, (61) (n.p, 2010). [Google Scholar]
- 22.Bose A, Burch MC, Chowdhury A, Paschou P and Drineas P, Clustrat: a structure informed clustering strategy for population stratification, in International Conference on Research in Computational Molecular Biology, (Springer, 2020). [Google Scholar]
- 23.Thompson DJ, Wells D, Selzam S, Peneva I, Moore R, Sharp K, Tarran WA, Beard EJ, Riveros-Mckay F, Palmer D et al. , Uk biobank release and systematic evaluation of optimised polygenic risk scores for 53 diseases and quantitative traits, medRxiv (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batty GD, Gale CR, Kivimäki M, Deary IJ and Bell S, Comparison of risk factor associations in uk biobank against representative, general population based studies with conventional response rates: prospective cohort study and individual participant meta-analysis, bmj 368 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, Distefano M, Senol-Cosar O, Haas ME, Bick A et al. , Polygenic prediction of weight and obesity trajectories from birth to adulthood, Cell 177, 587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loos RJ, 15 years of genome-wide association studies and no signs of slowing down, Nature Communications 11, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beery S, Van Horn G and Perona P, Recognition in terra incognita, in Computer Vision – ECCV 2018, eds. Ferrari V, Hebert M, Sminchisescu C and Weiss Y(Springer International Publishing, Cham, 2018). [Google Scholar]
- 28.Kaufman JS, Dolman L, Rushani D and Cooper RS, The contribution of genomic research to explaining racial disparities in cardiovascular disease: a systematic review, American journal of epidemiology 181, 464 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Obermeyer Z, Powers B, Vogeli C and Mullainathan S, Dissecting racial bias in an algorithm used to manage the health of populations, Science 366, 447 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Castro DC, Walker I and Glocker B, Causality matters in medical imaging, Nature Communications 11, 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenfeld E, Ravikumar P and Risteski A, The risks of invariant risk minimization, arXiv preprint arXiv:2010.05761 (2020). [Google Scholar]
- 32.Ahuja K, Shanmugam K, Varshney KR and Dhurandhar A, Invariant risk minimization games, in Proceedings of the 37th International Conference on Machine Learning, ICML’20 (JMLR.org, 2020). [Google Scholar]
- 33.Brier GW et al. , Verification of forecasts expressed in terms of probability, Monthly weather review 78, 1 (1950). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Simulated data is made available upon request. UKB-ePRS are available from UK Biobank.