

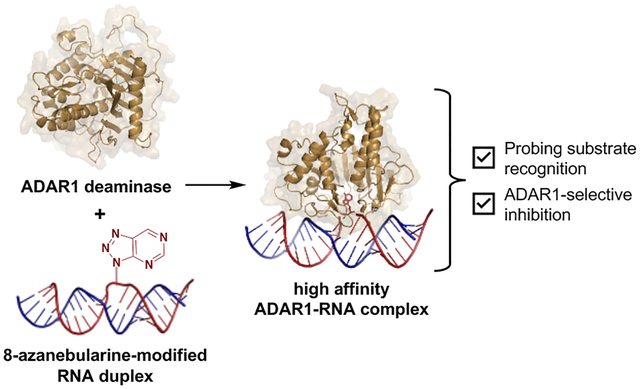
Abstract
Adenosine Deaminases Acting on RNA (ADARs) are RNA editing enzymes that catalyze the hydrolytic deamination of adenosine (A) to inosine (I) in dsRNA. In humans, two biologically active ADARs, ADAR1 and ADAR2, perform this A-to-I editing event. The growing field of nucleotide base editing has highlighted ADARs as promising therapeutic agents while multiple studies have also identified ADAR1’s role in cancer progression. However, the potential for site-directed RNA editing as well as the rational design of inhibitors are being hindered by the lack of detailed molecular understanding of RNA recognition by ADAR1. Here, we designed short RNA duplexes containing the nucleoside analog, 8-azanebularine (8-azaN), to gain insight into molecular recognition by the human ADAR1 catalytic domain. From gel shift and in vitro deamination experiments, we validate ADAR1 catalytic domain’s duplex secondary structure requirement and present a minimum duplex length for binding (14 bp, with 5 bp 5’ and 8 bp 3’ to editing site). These findings concur with predicted RNA-binding contacts from a previous structural model of the ADAR1 catalytic domain. Finally, we establish that neither 8-azaN as a free nucleoside nor a ssRNA bearing 8-azaN inhibit ADAR1 and demonstrate that the 8-azaN-modified RNA duplexes selectively inhibit ADAR1 and not the closely related ADAR2 enzyme.
Graphical Abstract
INTRODUCTION
ADARs (Adenosine Deaminases Acting on RNA) are enzymes involved in A-to-I (adenosine to inosine) editing – a widespread post-transcriptional modification that can exhibit a myriad of effects, from rewriting codon sequences to modifying RNA secondary structures1. ADAR1 and ADAR2 perform this editing activity in humans and are known to bind and edit perfectly matched duplexes (e.g. inverted Alu repeats). However, many endogenous ADAR substrates have helix defects which can either enhance or reduce editing at specific sites2. ADARs are not strictly sequence specific but are dependent on duplex secondary structure and exhibit a local preference for A in an A:C mismatch with a uracil (U) 5’ and a guanosine (G) 3’ to the edit site3–5. The two catalytically active ADARs have distinct yet overlapping substrate specificities4,6,7, hence not all edit sites are efficiently edited by both enzymes. Additionally, the deaminase domains of ADAR1 and ADAR2 are structurally similar, with both having conserved amino acid residues for binding a Zn2+ ion and inositol hexakisphosphate (IP6) near the catalytic site8. These structural elements were found to be crucial for editing activity9. However, an additional zinc binding site that is also important for editing has been identified in the ADAR1 deaminase domain but is absent in ADAR210. An RNA binding loop in the deaminase domain that contacts the substrate 5’ to edit site (5’ binding loop) is also a point of significant sequence divergence between the two ADARs and was shown to contribute to their substrate selectivity11.
Apart from the deaminase domain, human ADARs are also comprised of at least two dsRNA binding domains (dsRBDs) which are known to aid in substrate binding and editing4,9,12. Moreover, a Z-DNA/RNA binding domain (Zß) can be found in ADAR1. The two ADAR1 isoforms, p110 and p150, are identical in sequence except for an additional Z-DNA/RNA binding domain (Zα) at the N terminus of p150. The Zß domain lacks some crucial residues for effective Z nucleic acid (ZNA) binding, but is suggested to play a role in ADAR1 dimerization13. On the other hand, the Zα domain can bind both Z-DNA and Z-RNA and is known to regulate gene expression14 and to facilitate editing15. A recent study has also shown the involvement of Zα in cancer cell survival by sequestering ZNAs that are important ligands of the ZNA binding protein 1 (ZBP1) in order to stimulate a necroptotic response16.
The ability of ADARs to bind and/or edit specific substrates has been associated with growth and development, innate immunity, and overall proper cellular function17. However, aberrant A-to-I editing by ADARs has also been implicated in various cancers and other metabolic and neurological disorders18,19. In particular, studies have shown that ADAR1 deletion results in increased tumor sensitivity to immunotherapy and in eventual cell death for some cancers that are characterized by elevated levels of interferon-stimulated genes20,21. These findings strongly suggest ADAR1 inhibitors as promising candidates for anti-cancer drugs. However, despite all these reports, there are still no reported potent, targeted inhibitors of ADARs to date.
On another note, the recent success of oligonucleotide therapeutics and the emerging field of nucleotide base editing has placed ADAR’s A-to-I editing activity in another therapeutic perspective. Because inosine can be read as guanosine by our translation machinery, ADARs can be harnessed to correct disease-causing G-to-A mutations at the transcript level by recruiting endogenous ADARs to a target edit site using exogenous single-stranded guide oligonucleotides (site-directed RNA editing, SDRE)22–25. Whether as a therapeutic or as a therapeutic target, understanding ADAR’s structure, reaction chemistry, substrate recognition and selectivity is important to control its activity. Several crystal structures of ADAR2 alone or bound to duplex RNA have been made available9,12,26–29; however, our current molecular knowledge of ADAR1 and its interaction with an RNA substrate is lagging. This limits the structure-guided design of inhibitors and guide oligonucleotides for directed editing applications, which is unfortunate given ADAR1’s pathological relevance and ubiquitous expression22,30–32, making it a valuable tool for SDRE.
Much of our lab’s success in characterizing the ADAR2 structure, reaction mechanism, and substrate selectivity can be attributed to the use of the nucleoside analog, 8-azanebularine (8-azaN)12,26,33–36. The 8-azaN nucleobase is hydrated by ADAR to form a product that is a structural mimic of the ADAR reaction intermediate (Fig. 1). Since the 8-azaN hydrate lacks a good leaving group, the 8-azaN reaction cannot proceed forward, hence preventing catalytic turnover. The use of dsRNAs containing 8-azaN at the reactive site allows for the mechanistic trapping of ADAR2-RNA complexes for binding and structural studies12,26–29,33. However, whether this nucleoside analog alone or integrated into an RNA structure can likewise be used for ADAR1 investigations is still unexplored. In this paper, we then sought to employ a panel of short RNA duplexes (≤ 16 bp) bearing 8-azaN to probe the ADAR1 catalytic domain substrate recognition. As in the case for ADAR2, we were able to form high affinity complexes with ADAR1 and the 8-azaN-modified duplexes, which enabled us to study ADAR1-RNA interactions through gel shift and in vitro deamination assays. We confirmed the duplex secondary structure requirement for ADAR1 engagement, as only the duplex containing 8-azaN facilitated binding and both 8-azaN as a free nucleoside and a ssRNA bearing 8-azaN did not. We also showed that an 8-azaN duplex with a high A-U bp content (low TM) does not bind as effectively as an 8-azaN duplex with a high G-C bp content (high TM). Additionally, we found that a 14 bp duplex with 5 bp 5’ and 8 bp 3’ to editing site is the minimum length for ADAR1 recognition. These findings agree with predicted RNA-binding contacts from a previously reported Rosetta model of the ADAR1 catalytic domain10. Lastly, we showed that these short 8-azaN-modified duplexes selectively inhibit ADAR1 and not the closely related ADAR2 enzyme. Together, this work provides new insights into the nature of ADAR1-RNA interactions, new tools for biochemical and biophysical characterization of ADAR1-RNA complexes, and the groundwork for the design of potent and specific modulators of ADAR1.
Fig. 1.
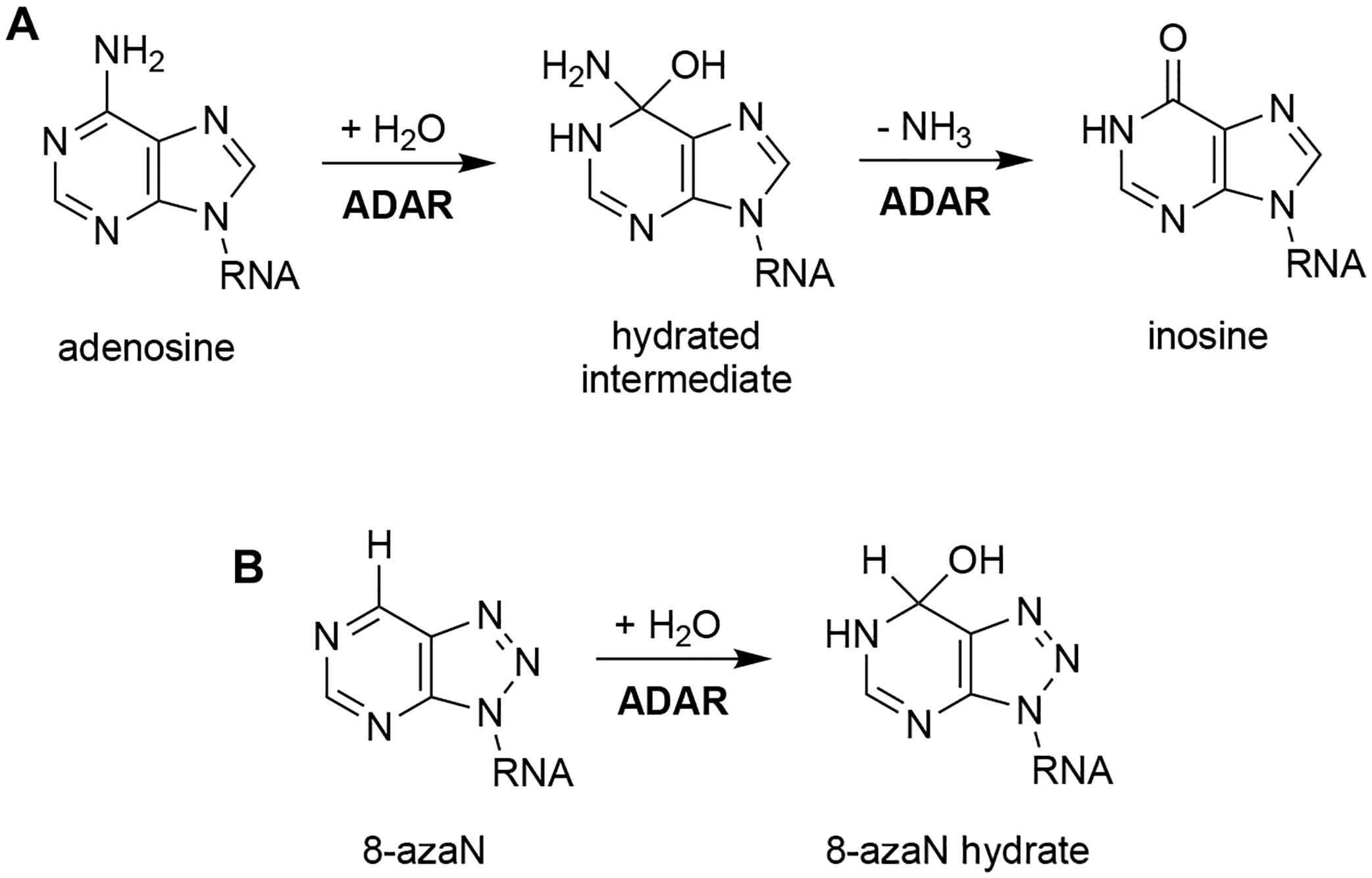
Reaction catalyzed by ADAR. (A) ADAR hydrolytically deaminates adenosine to afford inosine in dsRNA. (B) 8-azaN is hydrated by ADAR to form a product that is a structural mimic of the ADAR reaction intermediate.
MATERIALS AND METHODS
RNA synthesis.
ADAR editing substrates (5-HT2C and NEIL1) were transcribed from a DNA template using HiScribe T7 RNA synthesis kit (NEB). Unmodified and 2’-O-methylated RNA oligonucleotides for generation of short duplexes (≤ 16 bp) were purchased from IDT. RNA oligonucleotides containing 8-azaN were chemically synthesized on a 0.2 μmol scale using an ABI 394 synthesizer. The 8-azaN ribonucleoside phosphoramidite was either purchased from Berry & Associates, Inc. or synthesized in house36. All other phosphoramidites were purchased from Glen Research. Upon synthesis completion, columns were dried overnight under vacuum. The oligonucleotides were then cleaved from the solid support by treatment with 1:3 ethanol/30% NH4OH at 55 °C for 12 h. The supernatant was evaporated to dryness under vacuum, then the pellet was resuspended in anhydrous DMSO. Deprotection was performed by treatment with 55% (v/v) Et3N-3HF at room temperature overnight. The oligonucleotides were then precipitated from the solution by butanol precipitation at −70 °C, desalted using a Sephadex G-25 column, and purified as follows (see Table S1 for sequences).
RNA purification.
All oligonucleotides (purchased or synthesized in-house) were purified using denaturing polyacrylamide gel electrophoresis (PAGE) and visualized by UV shadowing. Bands were excised, crushed, and soaked overnight at 4 °C in a buffer containing 0.5 M NH4OAc and 0.1 mM EDTA. Gel fragments were removed using a 0.2 μm cellulose acetate filter and the oligonucleotides were precipitated from the supernatant by ethanol precipitation at −70 °C. The oligonucleotides were then dried under vacuum, resuspended in nuclease-free water, and quantified by measuring the absorbance at 260 nm. Oligonucleotide masses were confirmed by MALDI-TOF mass spectrometry using a Bruker UltraFlextreme MALDI TOF/TOF mass spectrometer at the UC Davis Mass Spectrometry Facility (see Table S2 for masses).
Synthesis of cross-linked RNA duplexes.
Two ssRNA oligonucleotides each containing 1’-ethynyl-2’-deoxyribose were cross-linked via a bis-azide linker, 1,4-bis(azidomethyl)benzene (1), by copper(I)-catalyzed azide-alkyne cycloaddition or click chemistry37. Bis-azide linker 1 was synthesized according to literature38. The ssRNA containing just the 1’-ethynyl-2’-deoxyribose modification (30 μM) was first clicked with an excess of 1 (5.5 mM) in 10:1 H2O/MeCN to afford an azide-functionalized product. A flat copper bar was added to the mixture, then the reaction was stirred in the dark at room temperature for 1 h. The copper bar was then removed, and the reaction was allowed to continue at room temperature for 2 h. The oligonucleotides were passed through a 3K MW cutoff filter, washed twice with water, then dried under vacuum. The azide-functionalized ssRNA was then purified from the starting material by denaturing PAGE.
The ssRNA containing both 1’-ethynyl-2’-deoxyribose and 8-azaN ribose modifications (20 μM) was then clicked with the complementary azide-functionalized ssRNA (21 μM) in 10:1 H2O/MeCN and 33 mM NaCl to afford the final cross-linked product. A flat copper bar was added to the mixture, then the reaction was stirred in the dark at room temperature for 1 h. The copper bar was then removed, and the reaction was allowed to continue at room temperature for 2 h. The oligonucleotides were passed through a 3K MW cutoff filter, washed twice with water, then dried under vacuum. The cross-linked RNA duplex was then purified from the starting material by denaturing PAGE.
Protein overexpression and purification.
Human ADAR1 p110 (UniProtKB P55265-5), ADAR2 (UniProtKB P78563), and ADAR1d (ADAR1 deaminase domain; UniProtKB P55265, position 886–1221) E1008Q with a C- (ADAR1 p110) or N-terminal (ADAR2 and ADAR1d E1008Q) His10-tag were overexpressed in S. cerevisiae BCY123 as previously described39. Human ADAR1 p150 (UniProtKB P55265-1) with an N-terminal FLAG-tag was purchased from BPS Bioscience. Human ADAR1 p110 was purified by lysing cells in 20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 1000 mM KCl, 30 mM imidazole, 1 mM tris(2-carboxyethyl)phosphine-HCl (TCEP-HCl), 0.05% (v/v) Triton X-100, and 50 μM ZnCl2 using a microfluidizer. The clarified lysate was then passed over a Ni-NTA column using an ÄKTA pure 25 FPLC system. The column was washed with the lysis buffer, then with wash buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 500 mM KCl, 30 mM imidazole, 1 mM TCEP-HCl, and 50 μM ZnCl2). Bound proteins were eluted by gradient elution with imidazole (30 to 400 mM). Fractions containing the target protein were pooled, concentrated, then dialyzed against a storage buffer containing 50 mM Tris-HCl pH 8.0, 10% (v/v) glycerol, 400 mM KCl, 50 mM imidazole, 1 mM TCEP-HCl, and 0.01% (v/v) Nonidet P-40 (NP-40). Protein concentration was determined by running the sample alongside bovine serum albumin (BSA) standards in a sodium dodecyl sulfate-PAGE (SDS-PAGE) gel, followed by SYPRO Orange (Invitrogen) staining. An SDS-PAGE electrophoretogram showing the purity (≥70%) and quality of the protein preparation can be found in Fig. S1.
Human ADAR2 was purified as above except using the following buffers: (1) lysis buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 750 mM NaCl, 35 mM imidazole, 0.01% (v/v) Triton X-100, 1 mM β-mercaptoethanol (BME)); (2) wash buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 300 mM NaCl, 35 mM imidazole, 0.01% (v/v) Triton X-100, 1 mM BME); and (3) storage buffer (20 mM Tris-HCl pH 8.0, 20% (v/v) glycerol, 100 mM NaCl, 1 mM BME).
Human ADAR1d E1008Q was purified as above except using the following buffers: (1) lysis buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 750 mM NaCl, 30 mM imidazole, 1 mM TCEP-HCl, 0.05% (v/v) Triton X-100); (2) wash buffer (20 mM Tris-HCl pH 8.0, 5% (v/v) glycerol, 350 mM NaCl, 30 mM imidazole, 1 mM TCEP-HCl); and (3) storage buffer (50 mM Tris-HCl pH 8.0, 10% (v/v) glycerol, 200 mM KCl, 50 mM imidazole, 1 mM TCEP-HCl, 0.01% (v/v) NP-40).
Preparation of RNA duplex substrates for gel shift assay.
The 16mer 8-azaN or A containing RNA strand was end-labeled with 32P using T4 polynucleotide kinase (NEB). Excess [γ-32P]-ATP was removed by passing the reaction mixture through a Sephadex G-25 column. The labeled oligonucleotides were purified by denaturing PAGE gel as described above, except visualized by storage phosphor autoradiography. The labeled oligonucleotides were then hybridized at a 1:3 ratio to their complement in 10 mM Tris-HCl pH 7.5, 1 mM EDTA, and 100 mM NaCl by heating at 95 °C for 5 min and slow cooling to ≤ 30 °C.
Gel shift assay.
Samples containing 5 nM labeled duplex RNA and 0 to 300 nM enzyme were incubated in 15 mM Tris-HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% glycerol, 0.5 mM dithiothreitol (DTT), 1 μg/mL yeast tRNA, 0.16 U/μL RNAse inhibitor, and 0.2 mg/mL BSA at 30 °C for 30 min. Samples were loaded onto a 6% 80:1 acrylamide/bis-acrylamide gel and electrophoresed under non-denaturing conditions in 1X TBE at 4 °C for 1.5 h. The gels were dried under reduced pressure at 80 °C for 1.5 h, then exposed to storage phosphor imaging plates (Kodak) in the dark. Plates were scanned using a Typhoon imaging system (GE Healthcare) and band intensities were quantified by ImageQuant (GE Healthcare). Data were plotted (fraction RNA bound = protein-RNA / (protein-RNA + free RNA)) and analyzed using Microsoft Excel and GraphPad Prism. The band intensities arising from protein-RNA aggregation in the wells were excluded in final data processing as it does not appreciably change the KD values derived from the binding curves, whether included or excluded from the calculation for fraction of RNA bound to the protein.
Preparation of RNA duplex substrates for deamination assay.
ADAR editing substrates and cross-linked RNA duplexes were allowed to self-anneal in 10 mM Tris-HCl pH 7.5, 1mM EDTA, and 100 mM NaCl by heating at 95 °C for 5 min and slow cooling to ≤ 30 °C. The short intermolecular RNA duplexes were hybridized at a 1:3 ratio of target strand (8-azaN-containing or A/G-containing strand) to complement strand in the same buffer and conditions as above.
In vitro deamination assay.
Deamination assays were performed under single-turnover conditions. Samples containing 100 nM ADAR1 and 0 to 3000 nM short RNA duplex were incubated in 15 mM Tris-HCl pH 7.5, 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% (v/v) NP-40, 4% glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, and 0.16 U/μL RNAse inhibitor at 30 °C for 30 min. The reaction was commenced by addition of 5 nM editing substrate and allowed to incubate at 30 °C for the following times: 15 min for 5-HT2C and 30 min for NEIL1, before quenching with 95 °C water, vortexing, and heating at 95 °C for 5 min. Oligonucleotides were then purified using DNA Clean & Concentrator kit (Zymo) before conversion to cDNA using Access RT-PCR System (Promega). PCR amplicons were purified using 1% agarose gel and submitted for Sanger sequencing (Azenta). Sequencing peak heights at the edit site were quantified using 4Peaks (Nucleobytes). The sequencing traces also served to validate the sequences of the editing substrates. Data were plotted (% editing = [peak height G / (peak height A + peak height G)]*100%) and analyzed using Microsoft Excel and GraphPad Prism.
Deamination assays with ADAR2 were performed as above except in the following buffer conditions: 15 mM Tris-HCl pH 7.5, 60 mM KCl, 3 mM MgCl2, 1.5 mM EDTA, 0.003% (v/v) NP-40, 3% glycerol, 0.5 mM DTT, 1 μg/mL yeast tRNA, and 0.16 U/μL RNAse inhibitor, and for the following reaction times: 10 min for 5-HT2C and 30 min for NEIL1.
Thermal denaturation studies.
Equimolar amounts (1 μM) of intermolecular duplex strand components or 1 μM solution of cross-linked duplex were prepared in 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 100 mM NaCl, and 1.25 μM EvaGreen (Biotium). Samples were placed in a sealed 96-well plate and loaded onto CFX Connect Real-Time PCR Detection System (Bio-Rad). The plate was brought to 95 °C and held at 95 °C for 3 min, then was cooled down to 15 °C at 0.1 °C/s and held at 15 °C for 5 min. The temperature was then slowly brought to 100 °C, with fluorescence (F) measured every 0.2 °C (T). TM was determined as the temperature where -dF/dT is the largest. Data were plotted and analyzed using Microsoft Excel and GraphPad Prism.
RESULTS
A short RNA duplex bearing 8-azaN binds tightly to ADAR1.
From a previous screen for human ADAR substrates in yeast, we identified a number of RNA substrates that are preferentially edited by the human ADAR1 deaminase (hADAR1d) over the human ADAR2 deaminase (hADAR2d)11. One of these substrates, S. cerevisiae HER1 (Hmg2p ER remodeling) mRNA, was relatively small in size and efficiently edited by hADAR1d, deeming it practical and suitable for further investigations of RNA binding by the ADAR1 catalytic domain using chemically modified duplexes. We then designed a 16 bp intermolecular RNA duplex (H16) from a short hairpin stem containing the edit site in the yeast HER1 RNA (Fig. S2) for hADAR1d binding studies (Fig. 2). We have previously shown that the E488Q mutation, which increases hADAR2’s catalytic rate40, also enhances binding of hADAR2d to 8-azaN-containing RNA duplexes and leads to substantially less smearing in gel shift experiments33. This could be due to slower dissociation rates of the complexes formed between the E488Q protein and the 8-azaN duplexes compared to the WT protein. Nevertheless, a similar preference for the 8-azaN-modified duplexes over the unmodified duplexes was observed for both hADAR2d WT and E488Q proteins33. We then proceeded to use the corresponding hyperactive ADAR1 deaminase protein (hADAR1d E1008Q) for gel shift assays with H16 8-azaN or A (Fig. 2). Indeed, we observed a significantly tighter binding with the 8-azaN containing duplex compared to the duplex with adenosine at the editing site, with KD = 21 ± 11 nM for H16 8-azaN and KD > 300 nM for H16 A at the conditions tested. These results clearly demonstrate that 8-azaN can be used to generate high affinity RNA ligands for the ADAR1 deaminase domain and enable studies of ADAR1-RNA interactions.
Fig. 2.
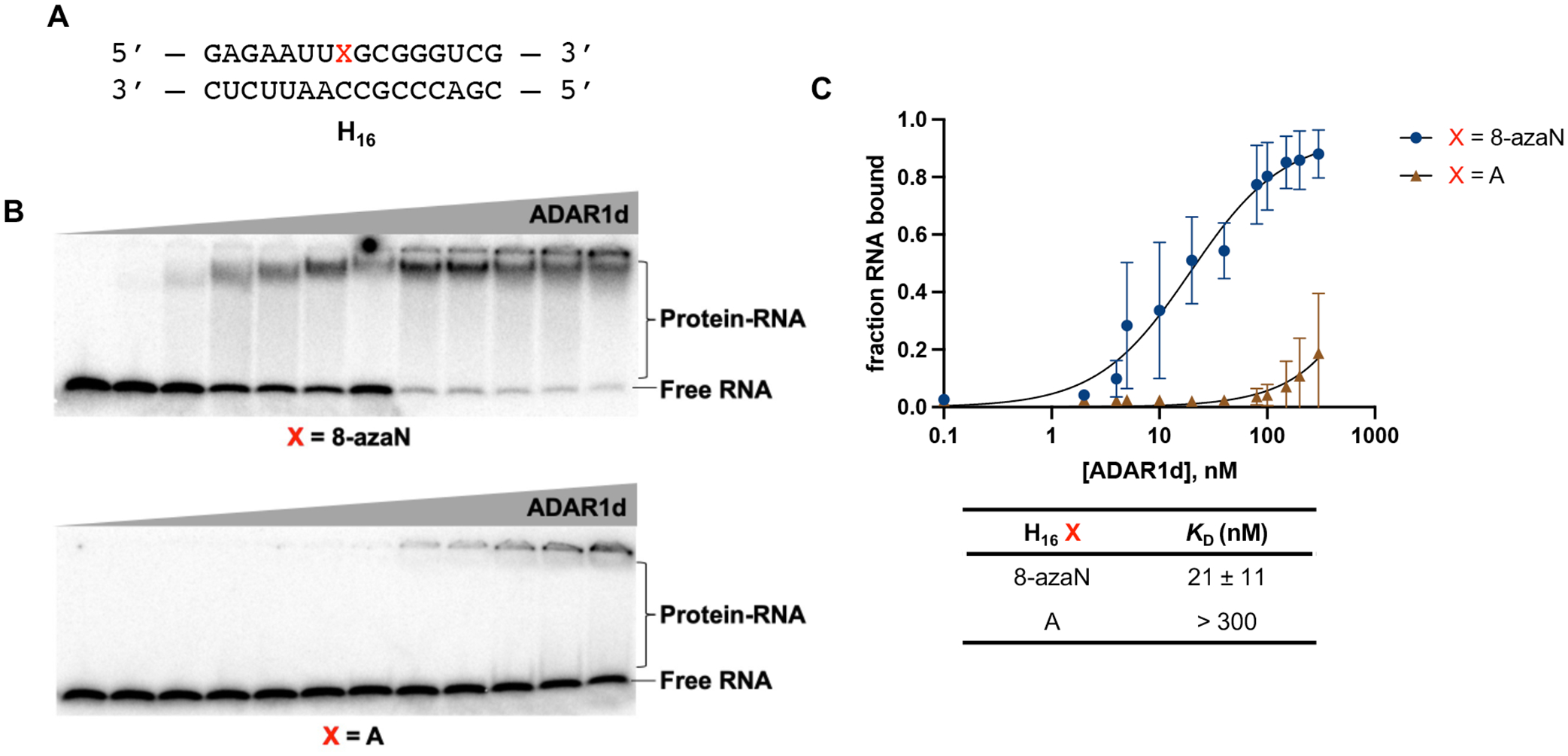
A short RNA duplex bearing 8-azaN binds tightly to ADAR1. (A) 16 bp duplex derived from yeast HER1 RNA. X is A in native target sequence but is varied (8-azaN, A, or G) in the succeeding experiments. (B) Gel shifts of ADAR1d with H16 8-azaN or A at 0, 2, 4, 8, 10, 20, 40, 80, 100, 150, 200, 300 nM ADAR1d E1008Q and 5 nM H16 RNA. (C) Fitted plots of fraction H16 RNA bound vs protein concentration. Data was plotted to the equation: y = A*[x/(KD+x)] where y = fraction H16 RNA bound, x = [ADAR1d]; A = binding endpoint; and KD = dissociation constant. Error bars represent standard deviation from n = 3 technical replicates.
Our findings from the gel shift experiments also led us to consider the use of 8-azaN-modified duplexes as substrate decoys to potentially suppress ADAR1 editing of an RNA target. For this experiment, we turned to use the full length human ADAR1 p110 to gain more biologically relevant insights into ADAR1 inhibition by these modified duplexes. We then set up in vitro deamination reactions with ADAR1 p110 and a fragment of the human 5-hydroxytryptamine receptor 2C (5-HT2C) pre-mRNA, a well-studied ADAR1 substrate, to which we added 0 to 3 μM of H16 8-azaN or A/G control duplexes (Fig. 3). Indeed, our results show that H16 8-azaN inhibited 5-HT2C editing by ADAR1 p110 at the B-site (preferred edit site of ADAR1 in 5-HT2C4, Fig. S3) in a concentration- and 8-azaN-dependent manner, with an estimated IC50 of 13 ± 2 nM (Fig. 3A, Table 1). Titration of the H16 8-azaN duplex also inhibited editing by ADAR1 p110 at a preferred edit site41 (Site 1, Fig. S3) in the human Nei-like DNA glycosylase 1 (NEIL1) pre-mRNA substrate, with an estimated IC50 of 8.9 ± 0.8 nM (Fig. 3A, Table 1). We also tested this assay on another full length human ADAR1 isoform, ADAR1 p150, and observed a similar concentration-and 8-azaN-dependent inhibition of 5-HT2C B-site editing (IC50 = 28 ± 3 nM) (Fig. 3B, Table 1). It is important to note that the A- or G-containing H16 controls also brought about a small concentration-dependent decrease in 5-HT2C and NEIL1 editing by ADAR1 p110 at greater than or equal to 200-fold excess to editing substrate ([H16 A/G] ≥ 1 μM). However, this slight inhibition by the unmodified duplex is not seen in ADAR1 p150 for H16 A. We hypothesize that the additional Z-DNA/RNA binding domain (Zα) in ADAR1 p150 contributes to selective binding to editing substrate RNA over the H16 duplex. This can also explain the two-fold higher IC50 measured for H16 8-azaN in the ADAR1 p150 / 5-HT2C vs the ADAR1 p110 / 5-HT2C reaction.
Fig. 3.
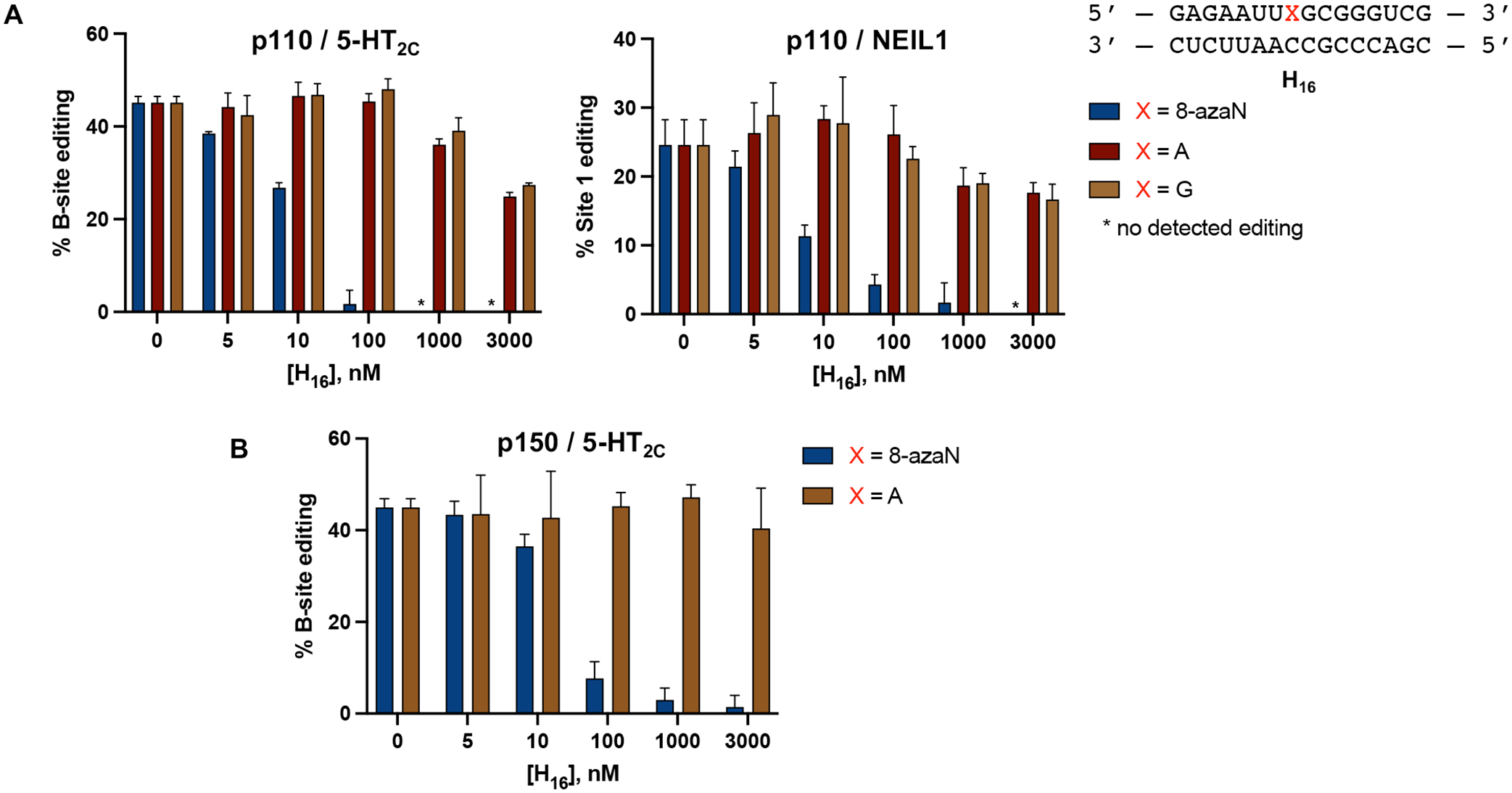
8-azaN-containing duplex inhibits 5-HT2C and NEIL1 editing by ADAR1. (A) Inhibition of ADAR1 p110 by H16 8-azaN tested on two different editing substrates, 5-HT2C (left) and NEIL1 (right). (B) Inhibition of ADAR1 p150 by H16 8-azaN tested on 5-HT2C. In vitro deaminations were performed at the following conditions: 0 – 3 μM H16 8-azaN or A/G control, 100 nM ADAR1 p110 or p150, 5 nM substrate, 15 min (for 5-HT2C) or 30 min (for NEIL1), at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates.
Table 1.
Estimated IC50 values from inhibition experiments with 8-azaN duplexes.
| 8-azaN Duplex | Enzyme | Editing Substrate | IC50 (nM)† |
|---|---|---|---|
| H16 | ADAR1 p110 | 5-HT2C | 13 ± 2 |
| H16 | ADAR1 p110 | NEIL1 | 8.9 ± 0.8 |
| H16 | ADAR1 p150 | 5-HT2C | 28 ± 3 |
| 5’−3’ swap | ADAR1 p110 | 5-HT2C | 15 ± 1 |
| GC-rich | ADAR1 p110 | 5-HT2C | 18 ± 8 |
| AU-rich | ADAR1 p110 | 5-HT2C | 74 ± 3 |
| 2’-OMe | ADAR1 p110 | 5-HT2C | 11.4 ± 0.7 |
| H14 A | ADAR1 p110 | 5-HT2C | 18 ± 4 |
| H14 B | ADAR1 p110 | 5-HT2C | > 3000 |
| H12 A | ADAR1 p110 | 5-HT2C | > 3000 |
| H12 B | ADAR1 p110 | 5-HT2C | > 3000 |
Data from inhibition experiments were plotted to the equation: y = m1+(m2−m1)/[1+(x/m4)m3], where y = % editing; x = log of RNA duplex concentration; m1 = basal response; m2 = maximal response; m3 = slope factor or Hill slope; and m4 = log of IC50 value (see Fig. S4 for IC50 plots). Values reported are the average of three independent measurements ± standard deviation.
Secondary structure requirement for ADAR1 engagement.
To test the importance of the duplex RNA structure for ADAR1 binding, we then repeated the inhibition experiment described above with 8-azaN as a free nucleoside and the 8-azaN-containing single strand component of the H16 8-azaN duplex (Fig. 4). We observed no reduction in 5-HT2C B-site editing by ADAR1 p110 even at the highest concentration tested for 8-azaN nucleoside (1 mM) and 8-azaN ssRNA (3 μM). These results clearly demonstrate the need for 8-azaN to be in a duplex context to effect ADAR1 binding and inhibition.
Fig. 4.
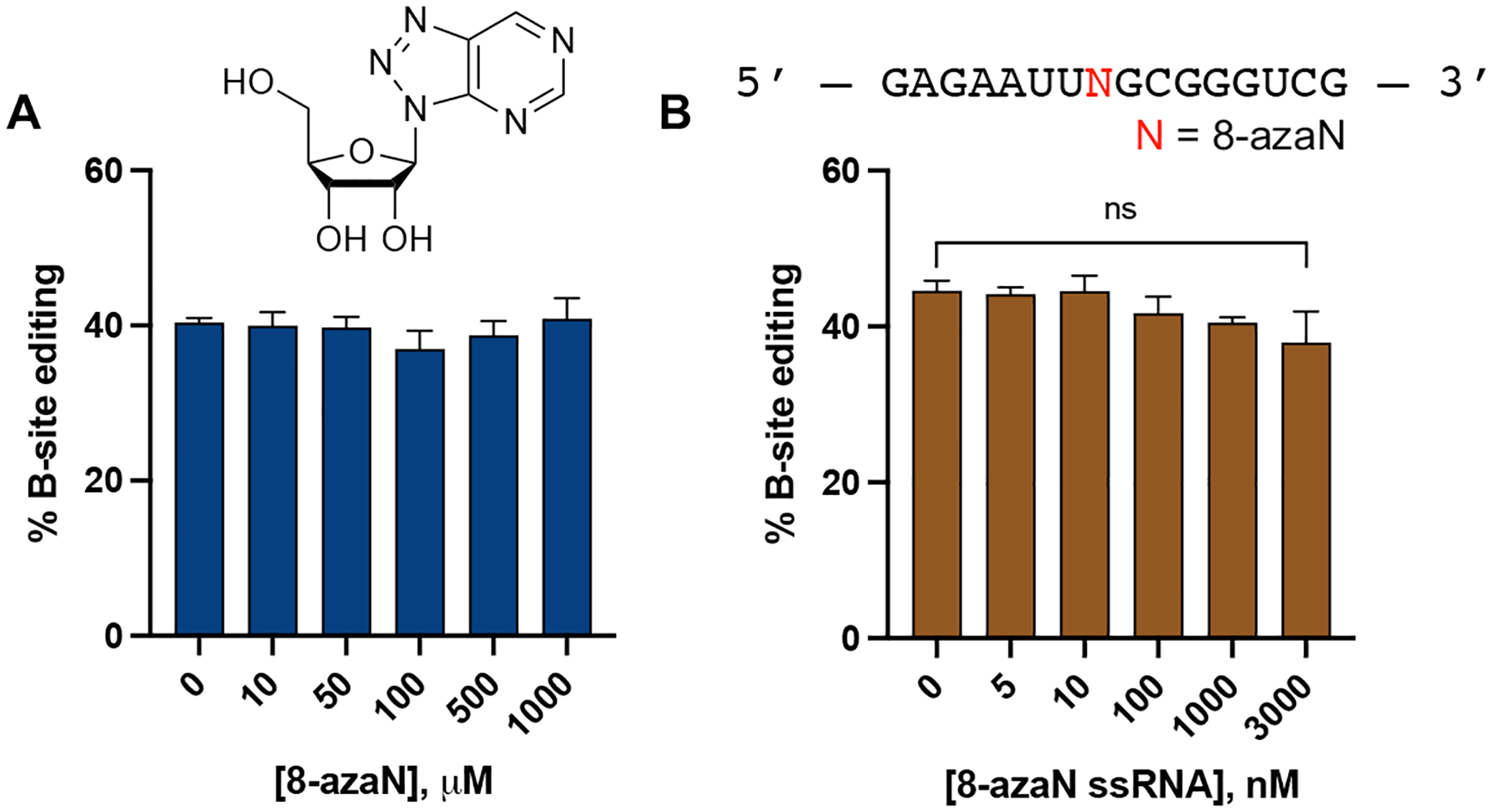
8-azaN as a free nucleoside (A) and an 8-azaN-modified ssRNA (B) do not inhibit ADAR1. In vitro deaminations were performed at the following conditions: 0 – 1 mM 8-azaN or 0 – 3 μM 8-azaN ssRNA, 100 nM ADAR1 p110, 5 nM 5-HT2C, 15 min, at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates. A two-tailed Welch’s t-test was conducted between indicated groups, ns = not significant.
ADARs are known to have a preference for editing adenosines in an A:C mismatch and within a nearest neighbor 5’-U and 3’-G sequence context3–5. However, how the flanking base sequences beyond this 5’-UAG-3’ triplet play a role in ADAR selectivity and recognition is still unclear. Interestingly, the S. cerevisiae GSY1 (glycogen synthase) mRNA, another hADAR1d preferred substrate identified from the yeast screen, shares a common secondary structure surrounding the edit site with HER1 and are both comprised of a short A-U rich (5 bp) hairpin stem on the 5’ side of the edit site11. To test whether this A-U rich region is crucial for binding, we swapped the sequences present on the 5’ and 3’ sides of the H16 8-azaN duplex (5’−3’ swap, Fig. 5A) and titrated the resulting duplex into an ADAR1 p110 / 5-HT2C reaction (Fig. 5B). However, the sequence swapping did not significantly affect binding of the 8-azaN duplex (IC50 = 15 ± 1 nM), suggesting that the short stretch of A-U base pairs 5’ to the edit site is not necessarily a requirement for ADAR1 recognition.
Fig. 5.
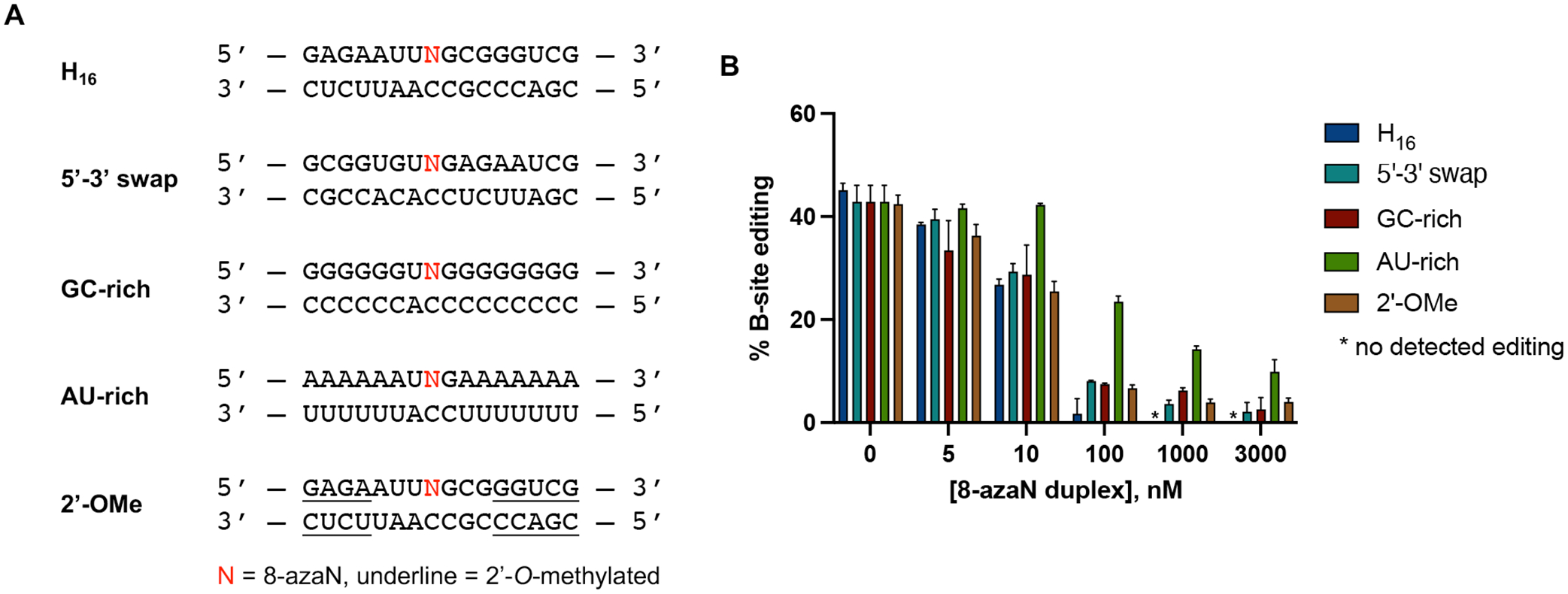
Effect of flanking sequence, G-C content, and 2’-O-methylation on ADAR1 binding. (A) Sequences of 8-azaN duplexes tested. (B) Data from in vitro deaminations performed at the following conditions: 0 – 3 μM 8-azaN duplex, 100 nM ADAR1 p110, 5 nM 5-HT2C, 15 min, at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates.
We next sought to determine the effect of the 8-azaN duplex’s G-C content on ADAR1 binding and subsequent inhibition of target editing. We imagined that increasing duplex stability by increasing G-C bp composition may increase 8-azaN duplex binding and potency. Having an 8-azaN duplex with low G-C content might then have the reverse effect. Therefore, we designed and tested two 8-azaN duplexes with either a higher G-C content (GC-rich) or a higher A-U content (AU-rich) and introduced each of these duplexes to an ADAR1 p110 / 5-HT2C reaction (Fig. 5). Interestingly, the GC-rich duplex inhibited the 5-HT2C substrate deamination with similar potency as the original H16 8-azaN duplex (IC50 = 18 ± 8 nM, Table 1). The AU-rich duplex, on the other hand, suffered a 6-fold decrease in binding affinity (IC50 = 74 ± 3 nM, Table 1). We rationalize that the additional duplex stability brought about by higher G-C content might not exhibit a substantial effect on ADAR1 binding under the conditions used for the deamination assay. The experimental TM values for H16, GC-rich, and AU-rich duplexes (62.5 ± 0.1 °C, 88 ± 1 °C, 25.5 ± 0.1 °C, respectively; Table 2) suggest that the H16 and GC-rich duplexes should predominantly exist in duplex form at 30 °C, while some of the AU-rich duplex species would be single stranded. It is also possible that the expected positive effect of the GC-rich duplex is masked by hindrance of necessary local melting in the duplex for ADAR recognition and reaction (e.g. to facilitate base-flipping around the editing site).
Table 2.
Experimental thermal melting temperatures (TM) for 8-azaN duplexes.
| 8-azaN Duplex | TM (°C)† |
|---|---|
| H16 | 62.5 ± 0.1 |
| GC-rich | 88 ± 1 |
| AU-rich | 25.5 ± 0.1 |
| H14 A | 54.6 ± 0.9 |
| H14 B | 55.1 ± 0.1 |
| XA | 58.0 ± 0.7 |
| XA+ | 81.3 ± 0.1 |
| XB | 20.9 ± 0.5 |
| XB+ | 58.4 ± 0.0 |
TM values were measured at the following conditions: 1 μM duplex, 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 100 mM NaCl, and 1.25 μM EvaGreen. Values reported are the average of three independent measurements ± standard deviation.
Finally, we also studied the effect of 2’-O-methyl modification at nucleotides distal to the editing site (Fig. 5). This modification at the ribose backbone has been shown to increase resistance of oligonucleotides to nucleases42–44. Our results clearly indicate that 2’-O-methylation at the specified positions does not affect ADAR1 binding (Fig. 5B). This is further validated by the comparable IC50 values between the parent H16 duplex and 2’-OMe duplex (IC50 = 11.4 ± 0.7 nM, Table 1).
Minimum duplex length for ADAR1 binding.
The use of short oligonucleotides for biochemical and biophysical studies of nucleic acid binding or modifying enzymes such as ADARs is advantageous for ease of synthesis and incorporation of nucleoside analogs at specific positions in the strand33,45. We then endeavored to determine the minimal 8-azaN duplex length to engage ADAR1 in order to effect editing inhibition. We designed a panel of shortened 8-azaN duplexes derived from the original H16 duplex and tested these duplexes for inhibition of the ADAR1 p110 / 5-HT2C reaction (Fig. 6). Interestingly, we observed that removing two base pairs from the 5’ end of the H16 duplex (5’ and 3’ defined by the 8-azaN-containing strand) did not significantly affect potency of inhibition (IC50, H14 A = 18 ± 4 nM, Table 1). However, removing two base pairs from the 3’ end of H16 resulted in a remarkably weaker inhibitor (IC50, H14 B > 3000 nM, Table 1). We further removed two base pairs from the 5’ end of H14 A to generate H12 A, which resulted in a substantial loss of potency (IC50 > 3000 nM, Table 1). These findings suggest a need for at least 5 bp 5’ and 8 bp 3’ to editing site for an efficient ADAR1-RNA interaction. Finally, removing two base pairs from the 3’ end of H14 A to generate H12 B also led to a notable loss in binding and potency (IC50 > 3000 nM, Table 1). This suggests that the 2 bp at the 3’ end of the parent H16 duplex harbors an important RNA contact site for the protein, consistent with our observation with H14 B. It is also important to note that the difference in potency of the two H14 duplexes is not due to dissimilarity in duplex stability, as shown by the comparable experimental TM values for H14 A and H14 B (54.6 ± 0.9 °C, 55.1 ± 0.1 °C, respectively; Table 2).
Fig. 6.
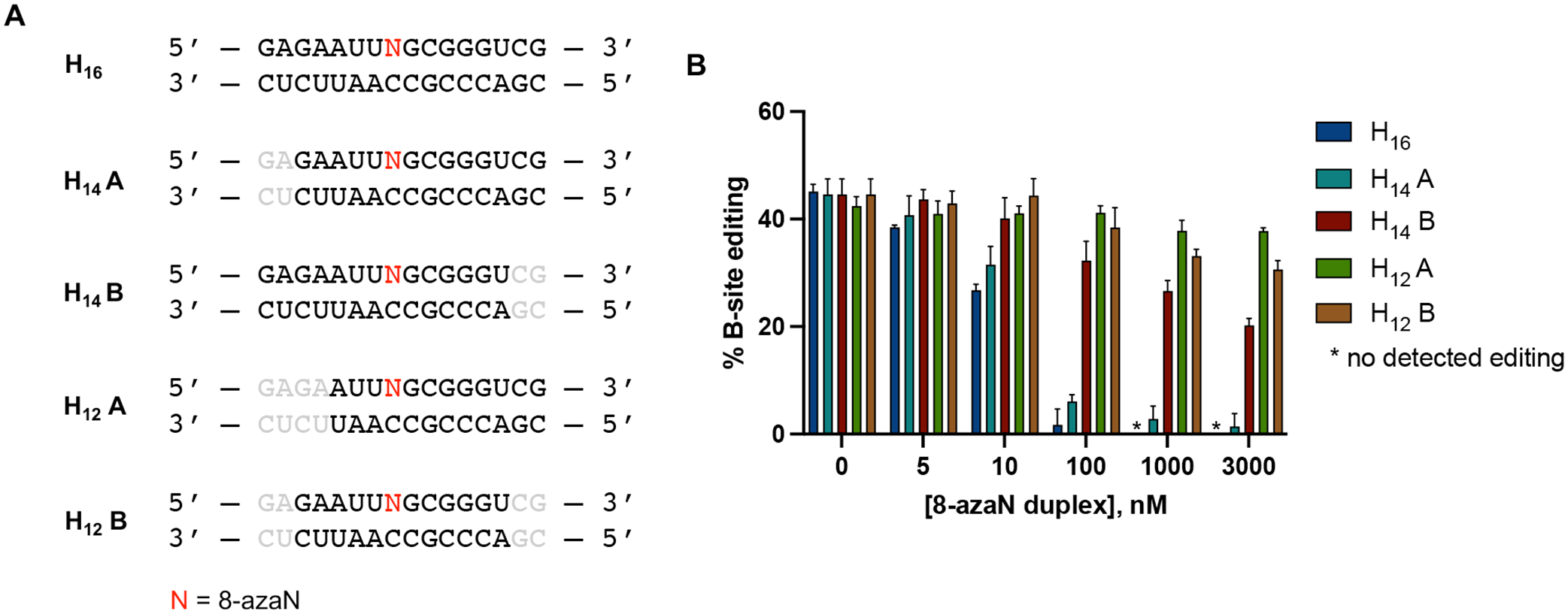
Minimum duplex length for ADAR1 binding. (A) 8-azaN-modified duplexes of different lengths tested for inhibition. Bases omitted from the original H16 8-azaN duplex sequence are in grey. (B) Data from in vitro deaminations performed at the following conditions: 0 – 3 μM 8-azaN duplex, 100 nM ADAR1 p110, 5 nM 5-HT2C, 15 min, at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates.
Previously, we reported a Rosetta homology model of the ADAR1 catalytic domain bound to a dsRNA substrate10. This model suggested that the ADAR1 deaminase domain contacts the RNA duplex mostly at positions proximal to edit site through both target and complement strands (Fig. 7). However, at distal positions, the ADAR1 catalytic domain mostly interacts with the duplex through the complement strand. We then tested the 8-azaN-containing strand components of H12 A and H12 B hybridized to the H14 A complement strand to generate two 12 bp duplexes with 2 nt overhang (H12 A overhang and H12 B overhang) (Fig. 8A). We imagined that the 2 nt overhangs in the complement strand will provide the necessary contact for better ADAR1 engagement than the corresponding blunt-ended 12 bp duplexes. Indeed, titration of H12 A overhang or H12 B overhang resulted in better inhibition of ADAR1 p110 / 5-HT2C reaction compared to H12 A or H12 B (Fig. 8B). However, both duplexes with overhangs are not as potent as H14 A, suggesting that a duplex structure is still needed for more effective contact, especially on the 5’ end of the duplex.
Fig. 7.
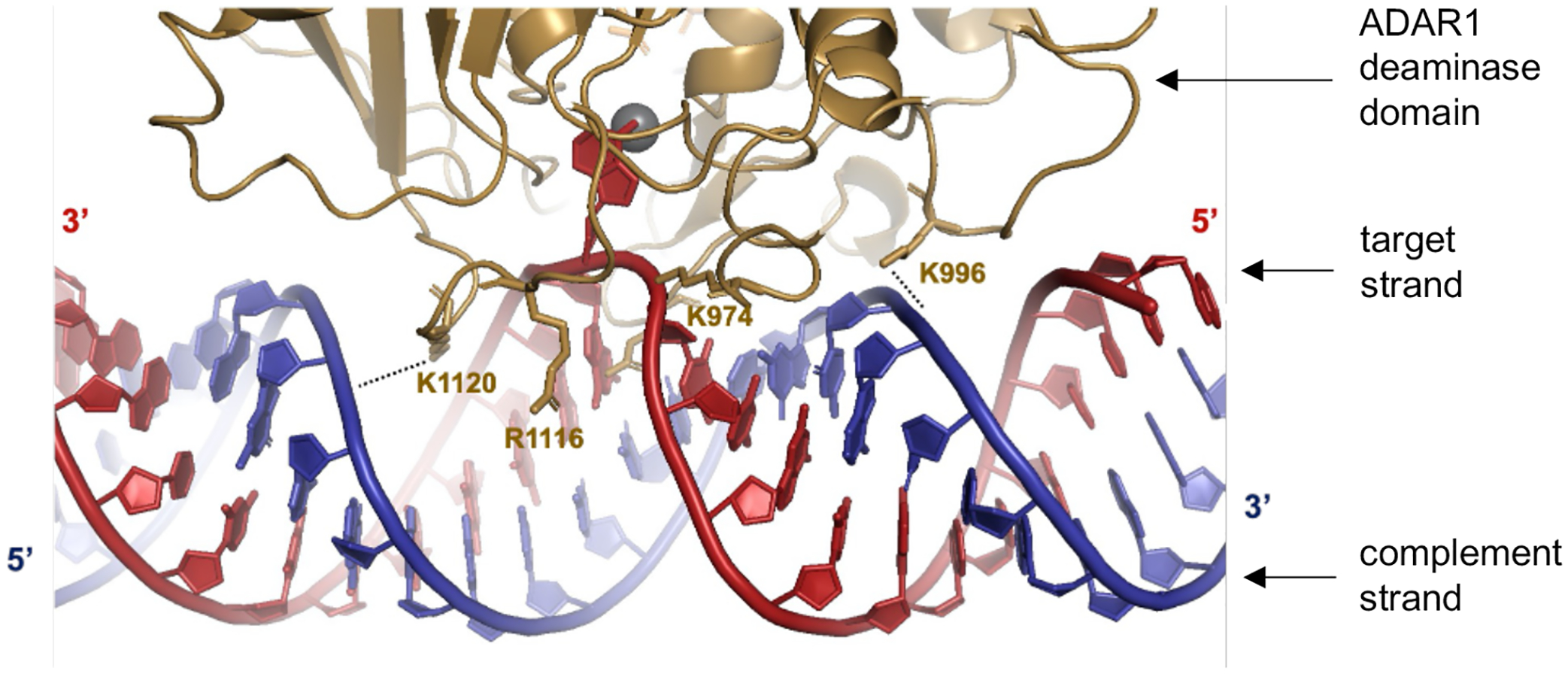
Predicted contacts between ADAR1 deaminase (gold) and an RNA duplex (red = target strand; blue = complement strand) based on a previously reported Rosetta homology model of ADAR1 catalytic domain10. Active site Zn metal (grey sphere).
Fig. 8.
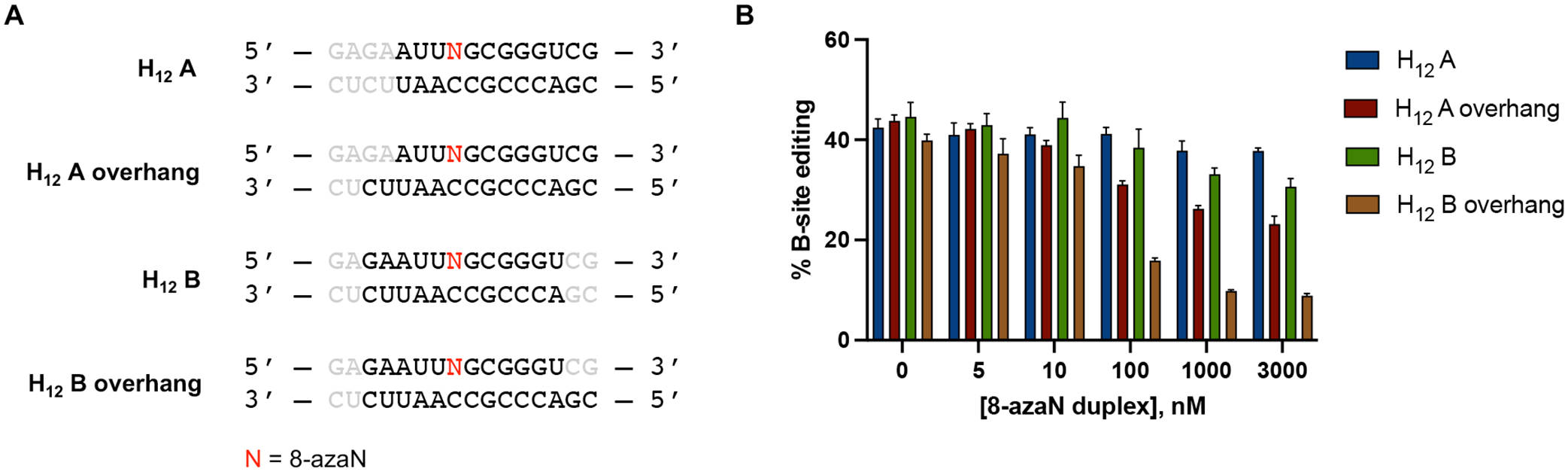
(A) 8-azaN-modified duplexes with overhangs tested for ADAR1 inhibition. Bases omitted from the original H16 8-azaN duplex sequence are in grey. (B) Data from in vitro deaminations performed at the following conditions: 0 – 3 μM 8-azaN duplex, 100 nM ADAR1 p110, 5 nM 5-HT2C, 15 min, at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates.
We then tested the effect of stabilization of these duplexes by generating two covalently cross-linked forms with overhangs (XA+ and XB+) and testing them for inhibition of ADAR1 p110 / 5-HT2C reaction (Fig. 9). We hypothesized that creating an intramolecular duplex via a covalent cross-link would increase duplex stability and subsequent ADAR1 binding, especially for the duplex with shorter 8-azaN-bearing strand (XB+). We expect that the covalent cross-link may not meaningfully cause irregularities in the duplex structure and affect ADAR1 binding as its estimated length from C1’ to C1’ (~13.5 Å, Fig. S5) is very similar to that of a canonical base pair in duplex RNA (~11 Å). While cross-linking indeed drastically increased the experimental TM values for both duplexes (XA to XA+ = 58.0 ± 0.7 °C to 81.3 ± 0.1 °C; XB to XB+ = 20.9 ± 0.5 °C to 58.4 ± 0.0 °C; Table 2), we only found a slight difference in potency of inhibition between XA and XA+ at duplex concentrations ≥ 100 nM and no significant difference in potency of inhibition between XB and XB+ at the conditions tested (Fig. 9B). The minimal increase (XA to XA+) to no increase (XB to XB+) in potency that we observed upon cross-linking could also be explained by a decrease in ADAR1 binding due to the reduction in the duplexes’ conformational flexibility around the site of chemical cross-linking. Nevertheless, these observations agree with our results from the intermolecular duplexes with overhangs in Fig. 8; that while modeling suggests that the ADAR1 deaminase domain contacts the RNA duplex at distal positions to the edit site via the complement strand’s phosphodiester backbone, a duplex structure at the terminal ends of the duplex is still required for efficient interaction.
Fig. 9.
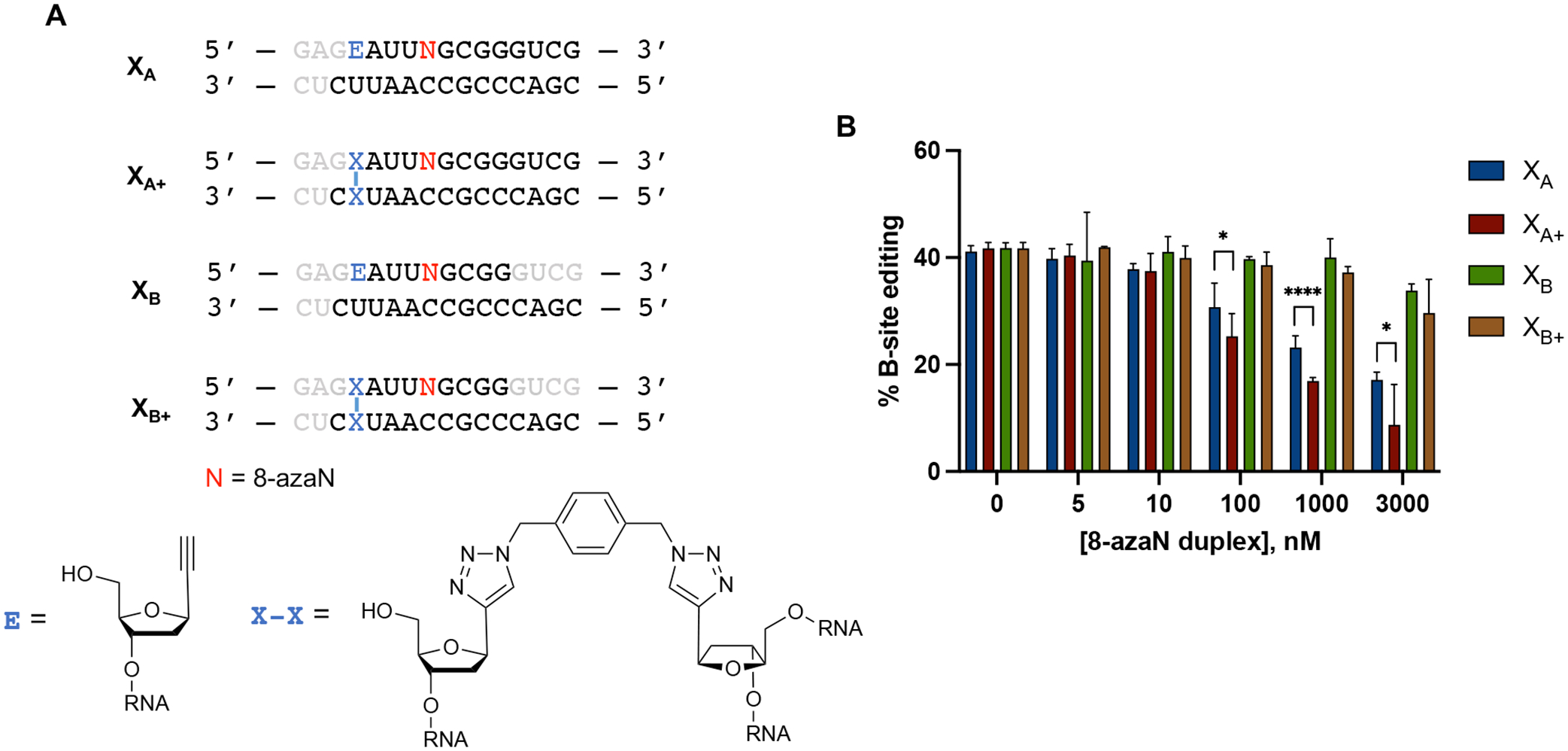
(A) Cross-linked RNA duplexes tested for inhibition. Bases omitted from the original H16 8-azaN duplex sequence are in grey. (B) Data from in vitro deaminations performed at the following conditions: 0 – 3 μM 8-azaN duplex, 100 nM ADAR1 p110, 5 nM 5-HT2C, 15 min, at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates. A two-tailed Welch’s t-test was conducted between indicated groups, * p < 0.05, **** p < 0.0001.
Selective inhibition of ADAR1.
Earlier, we described two hADAR1d preferred substrates, HER1 and GSY1, identified from a screen of hADAR1d substrates in yeast11. These two mRNAs have relatively short (5 bp) stem 5’ to the editing site, which from previous observations, is not long enough for efficient binding to the deaminase domain of human ADAR2 (hADAR2d)11. This suggested the H16 8-azaN duplex may be an ADAR1-specific inhibitor and not inhibit the reaction of ADAR2. This is indeed the case as shown in Fig. 10. We found that concentrations of the H16 8-azaN that clearly inhibit ADAR1 do not affect editing by human ADAR2 of the 5-HT2C D-site (preferred edit site of ADAR2 in 5-HT2C4, Fig. S3) or NEIL1 Site 2 (preferred edit site of ADAR2 in NEIL141, Fig. S3). However, we did observe a reduction of 5-HT2C D-site editing at [H16] = 3 μM and of NEIL1 Site 2 editing at [H16] ≥ 0.1 μM. This decrease in editing, however, does not appear to be 8-azaN-dependent and may be attributed to non-specific binding of the enzyme to the short RNA duplex, hindering access to the editing substrate. Overall, these findings reveal a new strategy for designing effective human ADAR family-specific modulators of ADAR1.
Fig. 10.
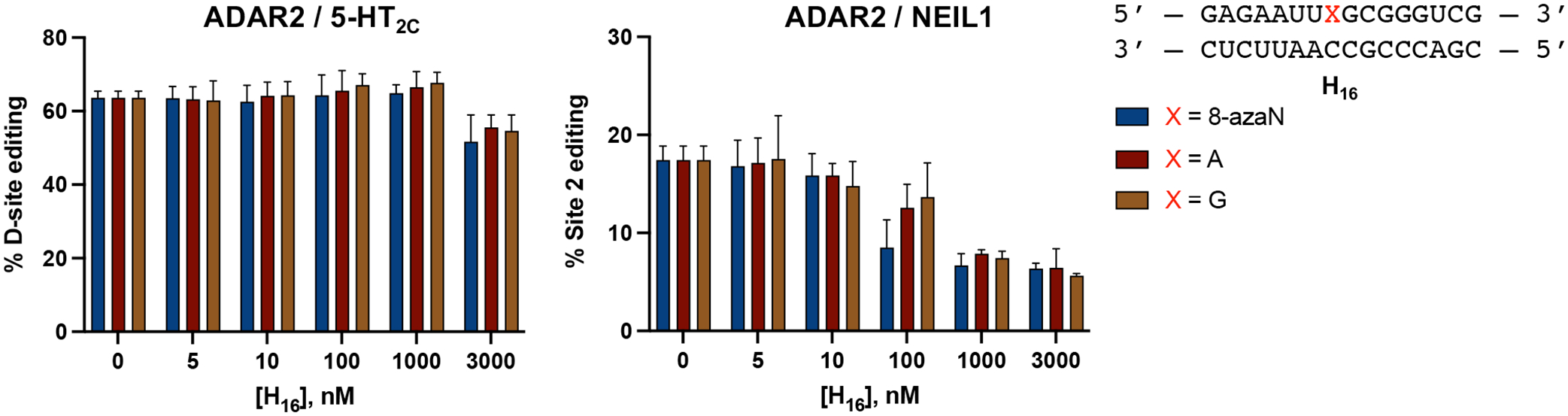
ADAR1-selective inhibition. H16 8-azaN duplex does not inhibit 5-HT2C (left) nor NEIL1 (right) editing by ADAR2. In vitro deaminations were performed at the following conditions: 0 – 3 μM H16 8-azaN or A/G control, 100 nM ADAR2, 5 nM substrate, 10 min (for 5-HT2C) or 30 min (for NEIL1), at 30 °C. Error bars represent standard deviation from n ≥ 3 technical replicates.
DISCUSSION
The nucleoside analog, 8-azaN, is a known inhibitor (IC50 = 1.5 μM) of the nucleoside-modifying enzyme, adenosine deaminase (ADA)46. Although structurally unrelated, ADAs and ADARs can both displace different leaving groups from C6 and the deamination rates of both enzymes are enhanced by 8-aza substitution35. This had driven us to test 8-azaN for ADAR2 inhibition in an earlier study, where we found that 8-azaN as a free ribonucleoside inhibits the ADAR2 reaction poorly with a disappointingly high IC50 value of 15 mM35. Incorporating the nucleoside in a short RNA duplex, on the other hand, dramatically increased binding affinity to a KD = 2 nM36. Since then, this nucleoside analog has been routinely used by our group for acquiring a deeper understanding of ADAR2-RNA recognition12,26,33,34. However, prior to this work, whether 8-azaN could similarly enable studies of the related ADAR1 enzyme was unknown. Here, we show that like ADAR2, ADAR1 forms high affinity complexes with 8-azaN-modified duplexes (Figs. 2–3). We have also established that the 8-azaN as a free ribonucleoside does not inhibit ADAR1 and requires duplex structure for effective binding (Fig. 4).
Previously, we used gel-based assays such as gel shift and endonuclease footprinting experiments to probe ADAR2-RNA interactions and give insight into potential substrates for X-ray crystallography screens12,26,29,33. However, the use of these techniques for ADAR1 studies has been complicated by nonspecific binding from protein aggregates, especially for longer ADAR1 constructs (containing one or more dsRNA or Z-DNA/RNA binding domains). Estimation of relative binding affinities by means of IC50 values in the in vitro deamination assay described here offers an alternative method for biochemical and biophysical investigations of ADAR1-RNA interactions. It is also noteworthy to mention that this assay is compatible with both full length ADAR1 isoforms p110 and p150 (Fig. 3) and does not necessitate the use of the hyperactive mutant to give quantifiable binding measurements with the 8-azaN-modified duplexes. Our ADAR1 inhibition results, especially of the ADAR1 p150 isoform, are also highly relevant given recent findings suggesting p150 as the ADAR1 isoform mainly responsible for cancer progression16,47,48.
Our results from structure-activity relationship studies with the panel of 8-azaN duplexes of different lengths suggest important ADAR1-RNA interactions ~ 4–5 bp 5’ and ~ 7–8 bp 3’ to the editing site (Fig. 6). Importantly, these observations agree with predicted RNA contacts between the ADAR1 deaminase and an RNA duplex from our previously published model of the ADAR1 catalytic domain - RNA complex10 (Fig. 7). The model shows two lysine residues (K996 and K1120) that are in position to contact the phosphodiester backbone of the target strand’s complement. The K996 side chain approaches the duplex between the third and fourth base pair 5’ from the editing site while the side chain of K1120 appears to approach between the seventh and eighth base pair 3’ from the edit site. Additionally, Fig. 11A illustrates the predicted 5’ binding loop contacts of ADAR1 with the RNA duplex 5’ to the edit site. Here, the model suggests that the ADAR1’s 5’ binding loop only makes notable contacts (via K974 and K996) proximal to the editing site (up to ~ 4 bp 5’ from the reactive nucleotide) as the rest of the loop folds away from the RNA to stabilize the second Zn binding site (via H988)10.
Fig. 11.
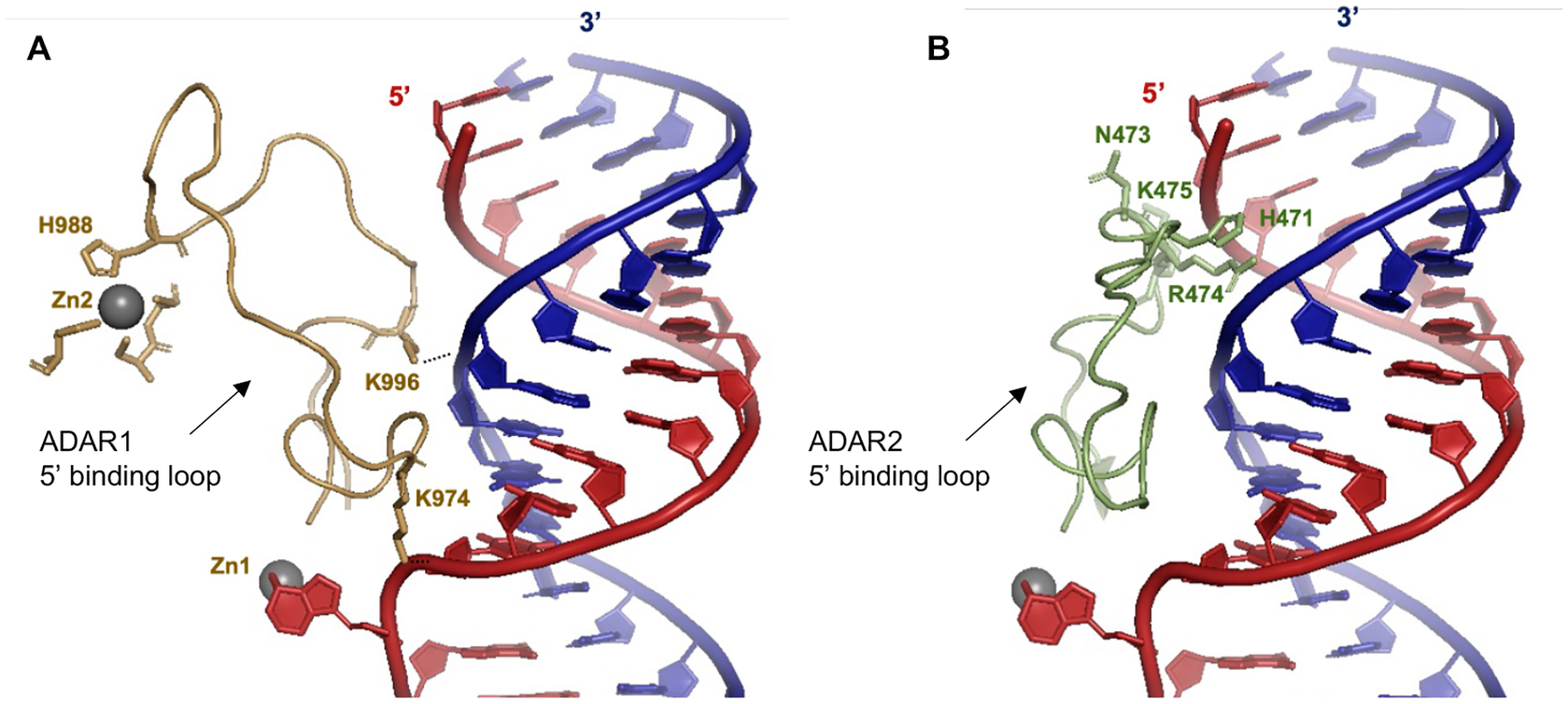
(A) Predicted contacts between the 5’ binding loop of ADAR1 (gold) and an RNA duplex (red = target strand; blue = complement strand) based on a previously reported Rosetta homology model of ADAR1 catalytic domain10. Active site Zn metal (Zn1, grey sphere). ADAR1’s second Zn metal (Zn2, grey sphere) binding site is also shown. (B) Contacts between the 5’ binding loop of ADAR2 (green) and an RNA duplex as shown by X-ray crystallography (PDBID 6VFF12). Active site Zn metal (grey sphere).
It is also important to note that the minimum duplex length that we present here is in agreement with two previously described short ADAR1 substrates from Wang et al.11 and Herbert et al.49. Wang et al. was able to show that a 33 nt hairpin RNA duplex with 5 bp 5’ and 8 bp 3’ to edit site is efficiently edited by hADAR1d11. Herbert et al. also reported that a 15 bp RNA duplex with the edit site at the center and with a single base mismatch at the edit site is sufficient for ADAR1 editing49. These findings are consistent with ADAR1 requiring only a short 5’ duplex to facilitate substrate recognition and reaction. The inability of the H16 8-azaN duplex to effectively bind and inhibit ADAR2 (Fig. 10) is explained by ADAR2 deaminase domain’s requirement for a longer duplex on the 5’ side of the editing site for full contact. This is clearly seen in crystal structures of ADAR2 in complex with an RNA duplex, where a cluster of residues (e.g. H471, N473, R474, K475) in the 5’ binding loop of ADAR2 contact the edited strand of the duplex ~ 10 bp from the edit site (Fig. 11B)12,26,50.
The use of shorter RNA substrates allows for routine incorporation of nucleoside analogs such as 2-aminopurine28,40,51–53 and 8-azanebularine33,34,36,45 for ADAR studies. It also offers an easily modifiable framework for other chemical modifications to enhance targeted delivery, metabolic stability, and binding affinity for cellular studies involving ADARs22–25,28. Our results from inhibition experiments with the 2’-OMe duplex (Fig. 5) and with the cross-linked duplexes (Fig. 9) demonstrate the flexibility for further modification of the 8-azaN duplexes. We anticipate the use of these modified duplexes in experiments where metabolic and chemical stability is advantageous, e.g. for ADAR binding and inhibition studies in cells or to serve as affinity chromatography resins to obtain pure, active fractions of overexpressed or endogenous ADAR1. Overall, our findings in this study provide an essential blueprint for developing effective and targeted modulators of ADAR1.
Supplementary Material
ACKNOWLEDGMENT
We would like to acknowledge Randall Ouye for generating the molecular model of the covalent cross-link using Gaussian 16.
Funding Sources
P.A.B. acknowledges financial support from National Institutes of Health in the form of grant R35GM141907. V.J.M. acknowledges funding from the National Science Foundation Graduate Research Fellowship under grant no. NSF 1650042.
Footnotes
- ADAR1 p110: P55265-5
- ADAR1 p150: P55265-1
- ADAR2: P78563
SUPPORTING INFORMATION
Sequences, masses, and predicted secondary structures of oligonucleotides; gel image of purified proteins; IC50 plots; and molecular model of covalent cross-link
REFERENCES
- (1).Eisenberg E, and Levanon EY (2018) A-to-I RNA editing - Immune protector and transcriptome diversifier. Nat. Rev. Genet 19, 473–490. [DOI] [PubMed] [Google Scholar]
- (2).Lehmann KA, and Bass BL (1999) The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol 291, 1–13. [DOI] [PubMed] [Google Scholar]
- (3).Wong SK, Sato S, and Lazinski DW (2001) Substrate recognition by ADAR1 and ADAR2. Rna 7, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Eggington JM, Greene T, and Bass BL (2011) Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun 2, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li JB, Levanon EY, J. K. Y, Aach J, Xie B, LeProust E, Zhang K, Gao Y, Church GM, Yoon J-K, Aach J, Xie B, LeProust E, Zhang K, Gao Y, and Church GM (2009) Genome-Wide Identification of Human RNA Editing Sites by Parallel DNA Capturing and Sequencing. Science (80-.). 324, 1210–1213. [DOI] [PubMed] [Google Scholar]
- (6).Lehmann KA, and Bass BL (2000) Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39, 12875–12884. [DOI] [PubMed] [Google Scholar]
- (7).Källman AM, Sahlin M, and Öhman M (2003) ADAR2 A→I editing: Site selectivity and editing efficiency are separate events. Nucleic Acids Res. 31, 4874–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang Y, Zheng Y, and Beal PA (2017) Adenosine Deaminases That Act on RNA (ADARs). Enzymes 41, 215–268. [DOI] [PubMed] [Google Scholar]
- (9).Macbeth MR, Schubert HL, VanDemark AF, Lingam AT, Hill CP, and Bass BL (2005) Structural biology: Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science (80-.). 309, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Park SH, Doherty EE, Xie Y, Padyana AK, Fang F, Zhang Y, Karki A, Lebrilla CB, Siegel JB, and Beal PA (2020) High-throughput mutagenesis reveals unique structural features of human ADAR1. Nat. Commun 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang Y, Park SH, and Beal PA (2018) Selective Recognition of RNA Substrates by ADAR Deaminase Domains. Biochemistry 57, 1640–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Thuy-Boun AS, Thomas JM, Grajo HL, Palumbo CM, Park S, Nguyen LT, Fisher AJ, and Beal PA (2020) Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. 48, 7958–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Athanasiadis A, Placido D, Maas S, Brown BA, Lowenhaupt K, and Rich A (2005) The crystal structure of the Zβ domain of the RNA-editing enzyme ADAR1 reveals distinct conserved surfaces among Z-domains. J. Mol. Biol 351, 496–507. [DOI] [PubMed] [Google Scholar]
- (14).Oh DB, Kim YG, and Rich A (2002) Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. Proc. Natl. Acad. Sci. U. S. A 99, 16666–16671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Koeris M, Funke L, Shrestha J, Rich A, and Maas S (2005) Modulation of ADAR1 editing activity by Z-RNA in vitro. Nucleic Acids Res. 33, 5362–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, Wang S, Gautam A, Williams RM, Crawford JC, Peri S, Studitsky V, Beg AA, Thomas PG, Walkley C, Xu Y, Poptsova M, Herbert A, and Balachandran S (2022) ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 606, 594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Erdmann EA, Mahapatra A, Mukherjee P, Yang B, and Hundley HA (2021) To protect and modify double-stranded RNA–the critical roles of ADARs in development, immunity and oncogenesis. Crit. Rev. Biochem. Mol. Biol 56, 54–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kung CP, Maggi LB, and Weber JD (2018) The Role of RNA Editing in Cancer Development and Metabolic Disorders. Front. Endocrinol. (Lausanne) 9, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Slotkin W, and Nishikura K (2013) Adenosine-to-inosine RNA editing and human disease. Genome Med. 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, Buchumenski I, Comstock DE, Brown FD, Ayer A, Kohnle IC, Pope HW, Zimmer MD, Sen DR, Lane-Reticker SK, Robitschek EJ, Griffin GK, Collins NB, Long AH, Doench JG, Kozono D, Levanon EY, and Haining WN (2019) Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, Cherniack AD, Vazquez F, Tsherniak A, Levanon EY, Hahn WC, and Meyerson M (2018) Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Monian P, Shivalila C, Lu G, Shimizu M, Boulay D, Bussow K, Byrne M, Bezigian A, Chatterjee A, Chew D, Desai J, Favaloro F, Godfrey J, Hoss A, Iwamoto N, Kawamoto T, Kumarasamy J, Lamattina A, Lindsey A, Liu F, Looby R, Marappan S, Metterville J, Murphy R, Rossi J, Pu T, Bhattarai B, Standley S, Tripathi S, Yang H, Yin Y, Yu H, Zhou C, Apponi LH, Kandasamy P, and Vargeese C (2022) Endogenous ADAR-mediated RNA editing in non-human primates using stereopure chemically modified oligonucleotides. Nat. Biotechnol 40, 1093–1102. [DOI] [PubMed] [Google Scholar]
- (23).Merkle T, Merz S, Reautschnig P, Blaha A, Li Q, Vogel P, Wettengel J, Li JB, and Stafforst T (2019) Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol 37, 133–138. [DOI] [PubMed] [Google Scholar]
- (24).Qu L, Yi Z, Zhu S, Wang C, Cao Z, Zhou Z, Yuan P, Yu Y, Tian F, Liu Z, Bao Y, Zhao Y, and Wei W (2019) Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol 37, 1059–1069. [DOI] [PubMed] [Google Scholar]
- (25).Nose K, Hidaka K, Yano T, Tomita Y, and Fukuda M (2021) Short-Chain Guide RNA for Site-Directed A-to-I RNA Editing. Nucleic Acid Ther. 31, 58–67. [DOI] [PubMed] [Google Scholar]
- (26).Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, and Beal PA (2016) Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol 23, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Monteleone LR, Matthews MM, Palumbo CM, Thomas JM, Zheng Y, Chiang Y, Fisher AJ, and Beal PA (2019) A Bump-Hole Approach for Directed RNA Editing. Cell Chem. Biol 26, 269–277.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Doherty EE, Wilcox XE, Van Sint Fiet L, Kemmel C, Turunen JJ, Klein B, Tantillo DJ, Fisher AJ, and Beal PA (2021) Rational Design of RNA Editing Guide Strands: Cytidine Analogs at the Orphan Position. J. Am. Chem. Soc 143, 6865–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Doherty EE, Karki A, Wilcox XE, Mendoza HG, Manjunath A, Matos VJ, Fisher AJ, and Beal PA (2022) ADAR activation by inducing a syn conformation at guanosine adjacent to an editing site. Nucleic Acids Res. 50, 10857–10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nishikura K (2016) A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol 17, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kim U, Wang Y, Sanford T, Zeng Y, and Nishikura K (1994) Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. U. S. A 91, 11457–11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Doherty EE, and Beal PA (2022) Oligonucleotide-directed RNA editing in primates. Mol. Ther 30, 2117–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Phelps KJ, Tran K, Eifler T, Erickson AI, Fisher AJ, and Beal PA (2015) Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 43, 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Maydanovych O, and Beal PA (2006) C6-substituted analogues of 8-azanebularine: Probes of an RNA-editing enzyme active site. Org. Lett 8, 3753–3756. [DOI] [PubMed] [Google Scholar]
- (35).Véliz EA, Easterwood LHM, and Beal PA (2003) Substrate analogues for an RNA-editing adenosine deaminase: Mechanistic investigation and inhibitor design. J. Am. Chem. Soc 125, 10867–10876. [DOI] [PubMed] [Google Scholar]
- (36).Haudenschild BL, Maydanovych O, Véliz EA, Macbeth MR, Bass BL, and Beal PA (2004) A transition state analogue for an RNA-editing reaction. J. Am. Chem. Soc 126, 11213–11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pujari SS, Leonard P, and Seela F (2014) Oligonucleotides with “clickable” sugar residues: Synthesis, duplex stability, and terminal versus central interstrand cross-linking of 2′-O-propargylated 2-aminoadenosine with a bifunctional azide. J. Org. Chem 79, 4423–4437. [DOI] [PubMed] [Google Scholar]
- (38).Gubu A, Su W, Zhao X, Zhang X, Fan X, Wang J, Wang Q, and Tang X (2021) Circular Antisense Oligonucleotides for Specific RNase-H-Mediated microRNA Inhibition with Reduced Off-Target Effects and Nonspecific Immunostimulation. J. Med. Chem 64, 16046–16055. [DOI] [PubMed] [Google Scholar]
- (39).Macbeth MR, and Bass BL (2007) Large-Scale Overexpression and Purification of ADARs from Saccharomyces cerevisiae for Biophysical and Biochemical Studies. Methods Enzymol. 424, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kuttan A, and Bass BL (2012) Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U. S. A 109, 3295–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yeo J, Goodman RA, Schirle NT, David SS, and Beal PA (2010) RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl. Acad. Sci. U. S. A 107, 20715–20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Choung S, Kim YJ, Kim S, Park HO, and Choi YC (2006) Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun 342, 919–927. [DOI] [PubMed] [Google Scholar]
- (43).Cummins LL, Owens SR, Risen LM, Lesnik EA, Freier SM, Mc Gee D, Cook CJ, and Cook PD (1995) Characterization of fully 2’-modified oligoribonucleotide hetero-and homoduplex hybridization andnuclease sensitivity. Nucleic Acids Res. 23, 2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Monia BP, Johnston JF, Sasmor H, and Cummins LL (1996) Nuclease resistance and antisense activity of modified oligonucleotides targeted to Ha-ras. J. Biol. Chem 271, 14533–14540. [DOI] [PubMed] [Google Scholar]
- (45).Maydanovych O, Easterwood LHM, Cui T, Véliz EA, Pokharel S, and Beal PA (2007) Probing Adenosine-to-Inosine Editing Reactions Using RNA-Containing Nucleoside Analogs. Methods Enzymol. 424, 369–386. [DOI] [PubMed] [Google Scholar]
- (46).Shewach DS, Krawczyk SH, Acevedo OL, and Townsend LB (1992) Inhibition of adenosine deaminase by azapurine ribonucleosides. Biochem. Pharmacol 44, 1697–1700. [DOI] [PubMed] [Google Scholar]
- (47).Kung CP, Cottrell KA, Ryu S, Bramel ER, Kladney RD, Bao EA, Freeman EC, Sabloak T, Maggi L, and Weber JD (2021) Evaluating the therapeutic potential of ADAR1 inhibition for triple-negative breast cancer. Oncogene 40, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Teoh PJ, Chung TH, Chng PYZ, Toh SHM, and Chng WJ (2020) IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematologica 105, 1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Herbert A, and Rich A (2001) The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc. Natl. Acad. Sci. U. S. A 98, 12132–12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Thomas JM, and Beal PA (2017) How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. BioEssays 39, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Malik TN, Doherty EE, Gaded VM, Hill TM, Beal PA, and Emeson RB (2021) Regulation of RNA editing by intracellular acidification. Nucleic Acids Res. 49, 4020–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Stephens OM, Yi-Brunozzi HY, and Beal PA (2000) Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry 39, 12243–12251. [DOI] [PubMed] [Google Scholar]
- (53).Yi-Brunozzi HY, Stephens OM, and Beal PA (2001) Conformational Changes that Occur during an RNA-editing Adenosine Deamination Reaction. J. Biol. Chem 276, 37827–37833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.