

Abstract
We have developed genetic tools for the atypical bacterium Acholeplasma laidlawii. A. laidlawii is a member of the class Mollicutes, which lacks cell walls, has small genomes, and has limited metabolic capabilities, requiring many metabolites from their hosts. Several of these traits have facilitated the development of genome transplantation for some Mollicutes, consequently enabling the generation of synthetic cells. Here, we propose the development of genome transplantation for A. laidlawii. We first investigated a donor–recipient relationship between two strains, PG-8A and PG-8195, through whole-genome sequencing. We then created multihost shuttle plasmids and used them to optimize an electroporation protocol. We also evolved a superior strain for DNA uptake via electroporation. We created a PG-8A donor strain with a Tn5 transposon carrying a tetracycline resistance gene. These tools will enhance Acholeplasma research and accelerate the effort toward creating A. laidlawii strains with synthetic genomes.
Keywords: Acholeplasma laidlawii, electroporation, replicative plasmids, synthetic cell, genome transplantation, strain evolution
Introduction
Mollicutes are a class of cell-wall-lacking bacteria with reduced genomes that evolved toward a parasitic lifestyle with high dependency on a host for metabolites.1Mollicutes can cause human and animal diseases, such as mastitis, arthritis, keratoconjunctivitis, and respiratory infections.2−5 Moreover, Phytoplasmas are plant pathogenic Mollicutes that can cause symptoms such as yellowing, stunting, and phloem necrosis, which can result in decreased crop yields.6 Although Phytoplasmas are economically significant pathogens, laboratory research of these bacteria is technically challenging due to difficulty in propagation of these strains in axenic cultures.1 As such, no genetic tools currently exist for Phytoplasmas, and effective options for the treatment and prevention of plant infections are limited.
One way to overcome this problem is to use whole-genome cloning and transplantation to create hybrid Mollicutes that carry Phytoplasma genomes but also can be propagated under laboratory conditions. The development of two breakthrough methods enabled this technology. The first method is cloning whole bacterial genomes in a eukaryotic host such as Saccharomyces cerevisiae.7 Once cloned or assembled in S. cerevisiae, the yeast genetic toolbox offers species-independent genome editing that often exceeds what is possible in the original bacterial system.8,9 The second method is whole-genome transfer termed “genome transplantation”. This method involves the use of polyethylene glycol (PEG) to deliver a “donor” genome (i.e., one that has been isolated from another bacterium or one that has been cloned or assembled in yeast) into a closely related recipient bacterium,10 although the exact mechanism by which genome transplantation occurs has not yet been determined. The resulting cell will assume genotypic and phenotypic traits conferred by the donor genome. Combining these methods, highly engineered bacterial strains, including synthetic and minimized organisms, have been created.11,12 Although extremely powerful, genome transplantation is currently limited to Mollicutes in the Mycoides clade of Mycoplasma taxonomy.13,14
Since Phytoplasmas are likely too distantly related to Mycoplasmas for genome transplantation to be successful,13 we propose Acholeplasma laidlawii, a closer relative to Phytoplasmas(15) with some existing genetic tools, as a platform for creating hybrid synthetic organisms. Acholeplasmaand Phytoplasma species are among the only Mollicutes to use a standard genetic code, meaning that Phytoplasma genes can be expressed in A. laidlawii without the need for recoding (most Mollicutes use the UGA codon to encode tryptophan). Furthermore, the genome of an A. laidlawii strain has previously been cloned in yeast as a centromeric plasmid, which presents a way to install large Phytoplasma pathways prior to transplantation.16 However, no methods currently exist for the transplantation of a cloned genome back into A. laidlawii. Therefore, we followed the steps that members of the J. Craig Venter Institute initially took to develop genome transplantation using Mycoplasma capricolum, including the development of an OriC-based replicative plasmid and optimizing transformation efficiency of such a plasmid, that formed the basis of developing the successful transplantation protocol. Alternatively, if genome transplantation is not successful for A. laidlawii, optimized transformation protocols could still enable other methods of large-scale genome engineering, such as the bacterial artificial chromosome stepwise insertion synthesis (BASIS) method that has recently been developed for Escherichia coli.17
Multiple A. laidlawii strains have been studied since the late 1900s, and the species as a whole has previously been used as a model system for the study of biological membranes.18−20A. laidlawii PG-8A is perhaps the most extensively studied: this strain has been used as a representative of the Acholeplasma genus, having a sequenced genome, mapped proteome, and characterized promoter structure.21,22 However, despite the extensive research involving this strain, virtually no genetic tools exist for manipulation in vivo, and even reports of plasmid transformation are very limited.23 Other strains of A. laidlawii have been more amenable to genetic manipulation. For instance, strain 8195 is a restriction-deficient derivative of A. laidlawii JA1, which has a history of use for the propagation and study of Mollicute-infecting viruses.24,25 Strain 8195 has more genetic tools available, including transformation, replicative and transposon-bearing plasmids, and heterologous gene expression,26−28 and is therefore a good candidate for a recipient cell that would allow for rebooting synthetic hybrid strains. Despite this past work with strain 8195, many of these tools and the knowledge for using them in A. laidlawii have seemingly been lost from the scientific community.
Here, we show the development of a new OriC plasmid for A. laidlawii 8195, an improved electroporation protocol, and the creation of a PG-8A donor strain with Tn5-transposase insertion. We also show how evolution can be used to create strains that can be more efficiently transformed with plasmids. Our genetic toolbox presented here is the first step toward enabling genome transplantation between A. laidlawii strains PG-8A and 8195, which will ultimately lead to the creation of synthetic hybrid Acholeplasma strains.
Results and Discussion
To enable the creation of A. laidlawii strains driven by synthetic genomes, an efficient way to transfer DNA between various strains needs to be developed. Specifically, whole-genome transfer from bacteria to host organisms and the transplantation of a yeast-cloned donor genome into a recipient bacterium must be established. The genetic tools required for achieving these tasks are listed in Figure 1.
Figure 1.
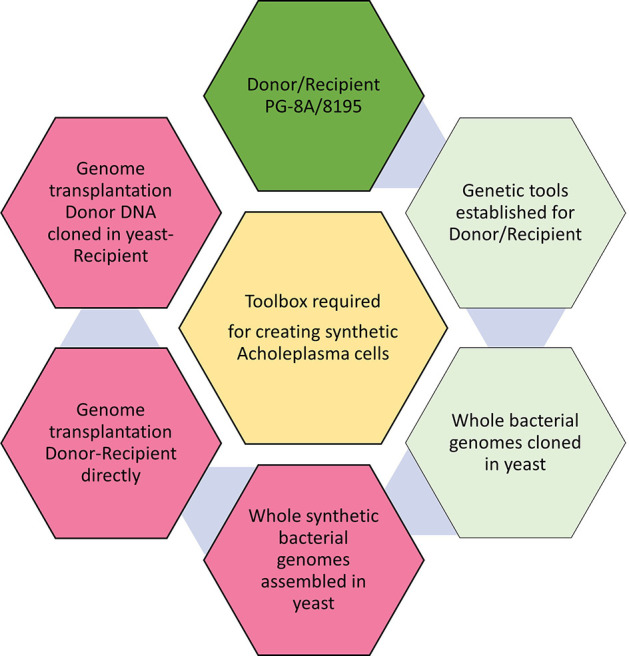
Genetic tools are required for the creation of synthetic Acholeplasma strains. Dark green hexagon: proposed donor/recipient cells are selected. Light green hexagons: some tools exist. Red hexagons: all genetic tools for these tasks need to be developed.
First, we selected two A. laidlawii strains for use as a donor and recipient to initially test genome transplantation from one bacterium to another: PG-8A and 8195, respectively (Figure 2). Both strains produce carotenoids, which result in the yellow color of pelleted cells (Figure 2A, B). Strain PG-8A has a darker pigmentation, which may serve as an initial visual screen following genome transplantation (Figure 2A, B). To gain a better understanding of the genetic differences between A. laidlawii PG-8A and 8195, the genome of each strain was sequenced using the Oxford Nanopore minION platform. The genomes of strains PG-8A and 8195 share 93% sequence identity, and the genomes are similar in size (1.5 Mbp) and have a G/C content of 32%. Furthermore, A. laidlawii strain 8195 has been described to lack restriction and recombination activities,24,29 which makes it a good recipient strain. From our sequencing data, we identified components of a Type I restriction system in strain 8195. However, there is a nonsense mutation in the one restriction subunit identified, which would truncate the predicted protein at amino acid position 794/997. Interestingly, strain 8195 is derived from a parental A. laidlawii strain containing one active restriction system.24 On the other hand, A. laidlawii PG-8A has five documented restriction systems (REBASE)30 that would need to be removed before this strain could be used as a recipient cell; therefore, we proposed to use it first as a donor.
Figure 2.

Proposed donor and recipient Acholeplasma strains for genome transplantation. Cell pellets and genome maps are shown for: (A, C) A. laidlawii PG-8A (donor); (B, D) A. laidlawii 8195 (recipient).
Furthermore, the PG-8A genome was already cloned in yeast,16 which brings us one step closer to creating synthetic genomes in this host strain. In addition to restriction enzymes, Mollicutes contain membrane-associated nucleases that digest exogenous DNA.31−33 The presence of potent extracellular nucleases may be an important consideration for future genome transplantation experiments as the donor genome could be damaged prior to recipient-cell uptake. We developed an assay for easy evaluation of the activity of A. laidlawii nucleases (Figure 3), similar to what has been done with some Mollicutes previously.33 This assay involved incubating live A. laidlawii cells with plasmid DNA, after which the nucleases were inactivated with ethylenediaminetetraacetic acid (EDTA)32 and the cell/DNA mix was visualized on an agarose gel. Indeed, we observed the digestion of extracellular plasmid DNA when incubated with live A. laidlawii cells of either strain (Figure 3). Removal of these nucleases using targeted or random mutagenesis may be necessary to generate a suitable recipient strain.
Figure 3.

An assay demonstrates the potency of A. laidlawii extracellular nucleases. Live A. laidlawii cells were incubated with pUC19 DNA and incubated at 34 °C for 0–90 min prior to inactivation with 1 mM ethylenediaminetetraacetic acid (EDTA). As a control, another set of samples was inactivated with EDTA before incubation. The cell/DNA mixes were visualized on an agarose gel. (A) Results of PG-8A assay. (B) Results of 8195 assay.
With the demonstration that EDTA can inhibit these nucleases, we set out to optimize an electroporation protocol. To this end, we first created a new shuttle plasmid that can replicate in yeast, E. coli, and A. laidlawii. Artificial plasmids have been developed for many Mollicute species by cloning genomic regions containing DnaA boxes into vectors.34 We followed the same strategy by cloning the putative origin of replication (OriC) of A. laidlawii 8195, which contains DnaA boxes upstream of the DnaA gene (Supplementary Figure 1), to create plasmid pAL1 (Figure 4A). For selection in A. laidlawii, we included a tetracycline (TetM) and puromycin resistance gene. In case the plasmid could not be replicated in A. laidlawii, we also included two 500-bp regions of homology to the 8195 genome flanking a homologue of ACL_0117 in strain PG-8A, which is partially toxic to yeast.16,35
Figure 4.

Overview of plasmid transformation to A. laidlawii 8195. (A) Plasmid maps of pAL1 and pNZ18. OriC = Origin of replication, OriT = origin of transfer, AmpR = ampicillin resistance gene, Ori = pMB1 replicon, H1/H2 = regions of homology to A. laidlawii 8195 genome, Puro = puromycin resistance gene, TetM = tetracycline resistance gene, Knt = kanamycin/neomycin resistance gene, and CmR = chloramphenicol resistance gene. Maps were created with BioRender.com. (B) Colony counts of A. laidlawii 8195 following transformation with pAL1 or pNZ18. Bars represent an average colony count from 3 independent experiments. Error bars represent the standard error of the mean. (C) Restriction digests of pAL1 and pNZ18 plasmid DNA recovered from five A. laidlawii transformants. pAL1 original and pNZ18 original were the plasmids used for A. laidlawii transformation.
Next, we transferred pAL1 to A. laidlawii 8195 using previously reported PEG-mediated and electroporation protocols.36 However, in our hands, these protocols were very inefficient and inconsistent. Subsequently, we optimized electroporation by changing volumes, temperatures, and the addition of yeast tRNA and EDTA (Supplementary Table 1), and we also considered what has been done in other protocols and with other Mollicutes.23,37,38 Using the optimized electroporation protocol, we obtained transformation of pAL1 to A. laidlawii with a frequency of 1.56 × 102 colony forming units (CFUs) per 1 μg of DNA. Transformation of pAL1 was only successful when selected for with tetracycline. Although we have observed the sensitivity of A. laidlawii to puromycin, the puromycin marker was not functional (data not shown). The puromycin marker uses a promoter from the Tuf gene of Mycoplasma capricolum, and it is possible that the promoter is weak or is not recognized in A. laidlawii. Alternatively, the TetM promoter and marker were originally isolated from a strain of Streptococcus agalactiae,39 and these elements are known to function in other Mollicutes.37,40 For tetracycline selection, we used 1 μg/mL as this concentration allows for selection and still prevents the appearance of spontaneous mutations during the time when true transformants are selected (after 4–6 days). It is important to note that after 10–12 days, colonies started to appear on our negative control selection plates.
Following our success in transforming pAL1, we attempted our protocol with pNZ18,41 a plasmid that contains a promiscuous Gram-positive replicon (Figure 4A). This plasmid has previously been reported to replicate in A. laidlawii.28 We obtained a transformation frequency of 1.15 × 103 CFUs per 1 μg of DNA (Figure 4C). We selected for transformants with 200 μg/mL neomycin to prevent the appearance of spontaneous mutants, which appeared within a similar time frame to transformed A. laidlawii when selected at concentrations below 60 μg/mL. To enable assembly in yeast of constructs into pNZ18 for delivery to A. laidlawii, we added elements for selection and maintenance in S. cerevisiae. The new plasmid, called pNZ18-CAH, showed similar efficiency of electroporation compared to original pNZ18 (Supplementary Figure 2).
We recovered pAL1 and pNZ18 plasmids from A. laidlawii transformants grown in appropriate selective media by performing DNA isolation and transfer to E. coli to obtain a higher concentration of plasmids. Plasmids recovered from E. coli were digested, and we saw no gross rearrangements in 5/5 clones tested for each plasmid (Figure 4 C, D). It was necessary to recover pNZ18 in E. coli MC1061 rather than in E. coli Epi300; plasmids containing the pSH71 replicon, such as pNZ18, have been noted to have poor transformation efficiencies to RecA-minus E. coli strains.42
When A. laidlawii transformants were propagated in nonselective liquid media, we observed quick loss of both plasmids (Supplementary Table 2). This is a useful feature when propagation of a plasmid is required only for a short period of time, such as the delivery of genome-editing tools (example: Cas9). This experiment also indicated that pAL1 does not experience a high rate of genomic integration either within the OriC or in homology regions. This is consistent with previous unsuccessful attempts to integrate a plasmid into strain 8195, likely because this strain contains a premature stop codon in the RecA gene.29,43
Our transformation frequency of pNZ18 is roughly 30 times higher than what has previously been reported by Sundström and Wieslander for wild type 8195.28 However, following transformation and curing of pNZ18, this group had also reported the isolation of a strain that could be retransformed to a drastically higher frequency relative to wild type 8195, and the reported efficiency with this strain was more than 100 times greater than what we observed.28 Curious if we could recreate such a strain with our own transformation protocol, we subjected wild type A. laidlawii 8195 to four consecutive rounds of electroporation and plasmid curing. During this process, we did not use yeast tRNA or EDTA for electroporation, to evolve the cells for a simplified version of our protocol. We then isolated single colonies from the final pool of transformed cells and tested transformation frequency of several strains (Figure 5 A). Interestingly, we identified strains with noticeably improved electroporation frequencies: for pAL1 and pNZ18, we observed roughly a 200-to-300-fold increase in efficiency with one such strain (Figure 5 B). Plasmids from evolved A. laidlawii could also be recovered in E. coli (data not shown). For the transformation of evolved strains, the concentration of neomycin was reduced to 100 μg/mL, as these strains appeared more sensitive to higher concentrations. Importantly, changing the concentration of neomycin did not appear to increase the transformation efficiency of the wild type strain.
Figure 5.

Creation of an A. laidlawii strain with an improved electroporation frequency. (A) Schematic of the evolution process. Following four rounds of electroporation and plasmid curing, single A. laidlawii colonies were screened for a higher transformation frequency. Created with BioRender.com. (B) Results of electroporation to wild type and a single evolved A. laidlawii 8195 strain with pAL1 and pNZ18. Bars represent the average colony count from 9 biological replicates and 2 technical replicates per strain for pAL1 and 8 biological replicates and 2 technical replicates per strain for pNZ18. Error bars represent standard error of the mean. Statistical significance between wild type and evolved strains (p < 0.05) is indicated by an asterisk.
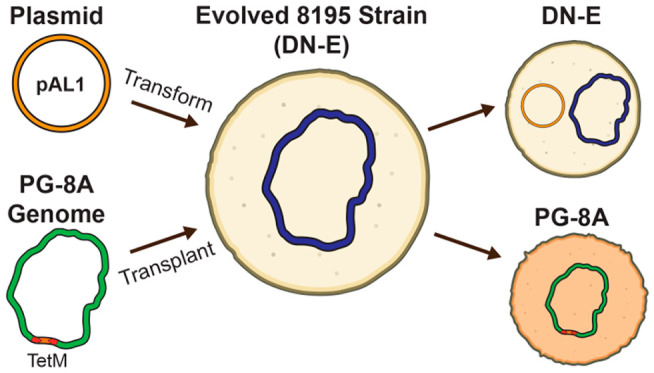
We were interested in seeing what genome-level changes could explain the improvement that was observed in evolved A. laidlawii strains. To this end, we sequenced the genomes of two evolved strains with a high-transformation phenotype and compared differences to the sequence of the wild type genome. We identified 27 common single-nucleotide substitutions and one common insertion between both evolved strains: 24/27 of these substitutions appear within genes, and 22 of them create nonsynonymous codon changes (Supplementary Table 3). Three mutations were confirmed via Sanger Sequencing. We submitted the 24 genes with mutations to the Kyoto Encyclopedia of Genes and Genomes (KEGG)44 mapper tool to identify possible affected pathways or gene networks. Seventeen genes were successfully assigned a KEGG Orthology (KO) and matched to processes including carbohydrate and amino acid metabolism, signaling and cellular processing, and genetic and environmental information processing. While none of these mutations seem immediately apparent in explaining the observed improvement in transformation efficiency, it is possible that a gene of unknown function or a combination of these mutations could be responsible for this observed change. Furthermore, we deposited one of these evolved strains to the American Type Culture Collection (ATCC) under the name A. laidlawii DN-E (Daniel Nucifora–Evolved).
We were curious to see if our OriC-based plasmid could be used to maintain small-to-medium-sized DNA fragments/pathways. To this end, we created a derivative of pAL1 carrying a 10 kb insert from the genome of Mesoplasma florum and transferred the plasmid to an evolved A. laidlawii strain by using electroporation. Following transformation, we tested 3 tetracycline-resistant colonies for the presence of M. florum genomic DNA using PCR, and all 3 colonies screened positive for the inset (Supplementary Figure 3). In summary, we have shown that our electroporation protocol can be used to deliver genetic cargo of approximately 21 kb (pAL1 plasmid with the inset) to an evolved A. laidlawii strain.
Although we have shown electroporation to be a consistent and relatively efficient way to move DNA to A. laidlawii, genome transplantation instead requires the use of PEG to transfer genomes to a recipient. PEG-mediated transformation has previously been established for A. laidlawii,45 but the method developed for this species differs from the traditional transplantation-like protocol, which involves relatively low concentrations of PEG and a longer incubation. We, therefore, aimed to adapt the transplantation-like protocol to A. laidlawii 8195. We first performed a series of pilot experiments to determine a starting protocol for further testing. Shown in Supplementary Table 5 is a summary of our PEG transformation experiments with cultures at various stages of growth and varied protocol parameters. Motivated by our previous success with evolution, many of these experiments were performed with a strain that had been isolated and cured from a previous PEG transformation, although we cannot conclude whether this improved the consistency or efficiency of the transformations. Overall, we performed 88 PEG-mediated transformations to A. laidlawii 8195, and 27 experiments were successful. In future work, parameters from successful transformation will be used for the future optimization of PEG transformation and transplantation protocols. Additionally, several more rounds of PEG transformation/curing could be performed to continue the evolution of an improved recipient cell. Alternatively, a targeted approach, such as using a CRISPR base editor,46 could be used to disrupt genes encoding putative extracellular nucleases or other genetic factors that could be affecting plasmid transformation/genome transplantation.
On the other hand, integration of genetic cassettes into the genome is an important tool on our road to create A. laidlawii driven by synthetic genomes. Therefore, we tested if Tn5 transposase can be used for this purpose. To this end, we first constructed a Tn5 cassette by PCR amplifying the TetM gene with primers that flanked the gene with 19-bp mosaic ends (Figure 6B). The cassette was mixed with EZ-Tn5 Transposase to generate a transposome, which was then electroporated to A. laidlawii. The protocol was the same except that we removed EDTA and yeast tRNA, which may negatively affect the transposase. We obtained transformants for both 8195 and PG-8A (Figure 6A), but in contrast to transformation with plasmid DNA, Tn5 transformations are less consistent (Supplementary Table 4). We did not notice an increase in Tn5 transformation frequency when we used an evolved A. laidlawii strain. We confirmed the presence of the Tn5-TetM transposon in strain PG-8A by passing the transformed strains multiple times to remove any original DNA and then performing PCR analysis (Figure 6C). In future work, we plan to use strain PG-8A transformed with Tn5 as a donor to first establish bacteria-to-bacteria genome transplantation, followed by transplantation of a PG-8A genome that has been cloned in yeast. Furthermore, large-scale mutagenesis could be performed to create a derivative of strain 8195 with reduced or abolished nuclease activity for use as a recipient. Further experiments will be necessary to optimize transposon integration into the genome, and future work using transposon-bearing plasmids may enable a simpler and more cost-effective way to create mutant A. laidlawii libraries.
Figure 6.

Tn5 transposase insertion into A. laidlawii PG-8A to generate a donor strain. (A) Serial dilutions of wild type (WT) PG-8A and one colony transformed with Tn5 transposase (Tn5-TetM) were spot plated on SP-4 plates without selection or SP-4 containing 5 μg/mL tetracycline. (B) Schematic of the Tn5 transposon cassette. The tetracycline resistance gene (TetM) is flanked by 19-bp mosaic ends that are recognized by Tn5 transposase. PCR primers for colony screening that bind to the TetM gene are shown. (C) Confirmation of marker insertion in donor A. laidlawii. PCR amplification with primers that bind to TetM produces a band of the expected size in Tn5-TetM A. laidlawii and the original Tn5 cassette (+ve) but not in the WT strain (−ve).
In conclusion, we have developed an expanded A. laidlawii genetic toolbox, including replicative plasmids, an improved transformation protocol, an improved strain for DNA uptake via electroporation, and a method of genomic integration, which will enable our future work toward the goal of creating synthetic/hybrid strains that should open new possibilities to study Acholeplasmas and possibly Phytoplasmas. In the meantime, our multihost plasmid can be used to clone/assemble genetic pathways in yeast that can be then moved by electroporation to be propagated as episomes in A. laidlawii 8195.
Materials and Methods
Strains and Cultures
A. laidlawii strain 8195 was kindly provided by Dr. Kevin Dyvbig and Dr. James Daubenspeck at the University of Alabama at Birmingham. A. laidlawii strains PG-8A (ATCC 23206) and 8195 were grown in SP-4 media lacking fetal bovine serum (3.5 g/L BBL mycoplasma broth base, 10 g/L tryptone, 5.3 g/L peptone, 8.8 g/L yeast extract, 5 g/L glucose, 0.6 g/L l-glutamine, 1.1 g/L NaHCO3, 2 g/L yestolate, 4 mL 0.5% phenol red solution, adjusted to pH = 7.6 with NaOH and sterilized through filtration). Both strains were initially grown at 34 °C, but slightly better growth was observed at 32 °C, which was used for all remaining experiments. For the PEG transformation experiments, SP-4 was supplemented with 17% horse serum. Solid media was made with 0.95–1% agar and without phenol red solution. In addition to appropriate antibiotics (1 μg/mL tetracycline, 100 μg/mL neomycin, or 200 μg/mL neomycin), SP-4 was always supplemented with 200 u/mL penicillin.
E. coli strain Epi300 (Lucigen, Cat #: LGN-EC300110) and MC1061 (NCBI: txid 1211845) were grown in Luria Broth (LB) (5 g/L yeast extract, 10 g/L tryptone, 10 g/L NaCl) at 37 °C, which was supplemented with 10 μg/mL tetracycline or 10 μg/mL chloramphenicol when appropriate. Solid LB was prepared with 1.5% agar.
S. cerevisiae VL6–48 (ATCC MYA 3666) was grown in 2x YPAD (100 g/L YPD broth, 160 mg/L adenine hemisulfate salt) at 30 °C. Following transformation, S. cerevisiae was instead grown in synthetic drop-out media lacking histidine (2% glucose, 0.5% ammonium sulfate, 60 μg/mL adenine) containing 1 M sorbitol.
Preparation of Tn5 Transposomes
The TetM gene, with its native promoter and terminator, was PCR-amplified from pAL1 using 5′-phosphorylated primers with 19-bp flanking mosaic ends (ME) that are recognized by the Tn5 transposase (Supplementary Table 6). The resulting PCR product was purified and concentrated using EZ-10 Spin Columns (BioBasic). The purified product was diluted to a final concentration of 100 ng/μL in TE buffer, and it was then combined with EZ-Tn5 transposase (Lucigen) as described in the manufacturer’s instructions. Subsequently, 1 μL of the prepared transposome was used for transformation to A. laidlawii.
Transformation
Transformation to A. laidlawii
A. laidlawii culture was grown to OD600 = 0.2–0.25. Prior to harvesting, 1 mL aliquots of culture were pretreated with 50 μL of 100 mM EDTA and incubated at 34 °C for 10 min. Cells were then centrifuged at 15,596g for 10 min at room temperature. Supernatant was discarded, and cell pellets were resuspended in 1 mL of room-temperature wash buffer (272 mM sucrose and 8 mM HEPES, pH = 7.4). Cells were spun as before, and the supernatant was removed. The pellet was resuspended in 100 μL wash buffer and kept on ice for 5 min. Then, 5 μg of pAL1 DNA or 1 μg of pNZ18 DNA (dissolved in ddH2O) and 10 μg of yeast tRNA was added to cells, and the mix was incubated on ice for 2–3 min. The cell/DNA mix was then transferred to a prechilled 2 mm cuvette (VWR) and pulsed in a GenePulser Xcell (Bio-Rad) at 2.5 kV for 5 ms (200 Ω, 25 μF). Cells were recovered in 1 mL of ice-cold SP-4 and kept on ice for 10 min. The cells were then transferred to a 1.5 mL tube and incubated at 34 °C for 2 h before plating. Plates were kept at 34 °C for 4–6 days. For the transformation of transposomes, the EDTA pretreatment and yeast tRNA were omitted. For transformations comparing wild type and evolved A. laidlawii, cells were grown and recovered at 32 °C, only 1 μg of pAL1 DNA was used, and the use of yeast tRNA and EDTA was omitted. When comparing the transformation efficiency of wild type and evolved A. laidlawii, culture growth was instead determined using pH, cultures were transformed at a pH range of 6.6–7.1, cells were grown and recovered at 32 °C, the EDTA pretreatment step was excluded, and only 1 μg of plasmid DNA was used for pAL1 and pNZ18, without any yeast tRNA.
Evolution of A. laidlawii
Wild type A. laidlawii 8195 was subjected to four rounds of electroporation, alternating between pNZ18 and pAL1. Electroporation was performed as described above, but cells were grown and recovered at 32 °C, only 1 μg of DNA was used for each plasmid, and the EDTA pretreatment step and the use of yeast tRNA were excluded. After each round, transformants were pooled in a liquid culture and passed several times without antibiotic selection to promote plasmid curing. Following the fourth round of transformation and passing, serial dilutions of the resulting culture were plated on nonselective SP-4 agar media to obtain single colonies. Colonies were screened for plasmid loss and were tested for improved transformation frequency against the wild type strain.
Nuclease Assay
Cultures of A. laidlawii PG-8A and 8195 were grown to OD600 = 0.2. Cultures were centrifuged at 15,596g for 10 min, and the cell pellet was concentrated in 1/10th the original volume with wash buffer. Next, 20 μL of the concentrated cells were combined with 630 ng of pUC19 DNA in a final volume of approximately 40 μL. For the experimental condition, the cell/DNA mix was incubated at 34 °C for 0, 30, 60, or 90 min. After the appropriate incubation time, EDTA was added to the mix at a final concentration of 1 mM to stop nuclease activity. For the EDTA control, the same amount of EDTA was instead added prior to incubation. For the DNA-only condition, 20 μL of wash buffer was used in place of cells, and EDTA was not added. Following all incubation times, each reaction was run on a 1% TAE agarose gel and imaged with ethidium bromide.
Acknowledgments
This work was supported by the Government of Canada’s New Frontiers in Research Fund (NFRF), [NFRFE-2018-01124]. In addition, research in B.J.K. laboratory is also supported by Natural Sciences and Engineering Research Council of Canada (NSERC), [RGPIN-2018-06172]. Plasmid pNZ18 and sequence/annotation data were kindly provided by the lab of Dr. Leendert Hamoen at the University of Amsterdam. The graphical abstract was created by Emma J. L. Walker.
Data Availability Statement
The genome sequence of A. laidlawii 8195 has been deposited to NCBI (BioProject PRJNA975591). Plasmids pAL1 and pNZ18-CAH are available on Addgene (ID 197285 and ID 203159, respectively). Acholeplasma laidlawii strain DN-E is currently in the process of being deposited to the American Type Culture Collection (ATCC).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.3c00399.
The A. laidlawii 8195 OriC sequence, a summary of electroporation, PEG transformation, and Tn5 transformation experiments, a comparison of mutations between wild type and evolved A. laidlawii strains, a comparison between pNZ18 and pNZ18-CAH transformation efficiencies, and lists of primers used for the study (PDF)
Author Contributions
B.J.K. and D.P.N. conceived the experiments; D.P.N., N.D.M., D.J.G., and B.J.K. conducted the experiments; D.P.N., N.D.M., D.J.G., and B.J.K. analyzed the results; D.P.N. and B.J.K. wrote the paper. All authors edited the paper. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Special Issue
Published as part of ACS Synthetic Biologyvirtual special issue “Synthetic Cells”.
Supplementary Material
References
- Razin S.; Yogev D.; Naot Y. Molecular Biology and Pathogenicity of Mycoplasmas. Microbiol Mol. Biol. Rev. 1998, 62, 1094–1156. 10.1128/MMBR.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites K. B.; Talkington D. F. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2004, 17, 697–728. 10.1128/CMR.17.4.697-728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter S. L.; Franklin R. M.; Baas E. J.; Barile M. F. Epidemic caprine keratoconjunctivitis: experimentally induced disease with a pure culture of Mycoplasma conjunctivae. Infect. Immun. 1977, 18, 816–822. 10.1128/iai.18.3.816-822.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek K.; Nicholas R. A. J.; Szacawa E.; Bednarek D. Mycoplasma bovis Infections—Occurrence, Diagnosis and Control. Pathogens 2020, 9, 640. 10.3390/pathogens9080640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav J. P.; Tomar P.; Singh Y.; Khurana S. K. Insights on Mycoplasma gallisepticum and Mycoplasma synoviae infection in poultry: a systematic review. Anim Biotechnol 2022, 33, 1711–1720. 10.1080/10495398.2021.1908316. [DOI] [PubMed] [Google Scholar]
- Kumari S.; Nagendran K.; Rai A. B.; Singh B.; Rao G. P.; Bertaccini A. Global status of phytoplasma diseases in vegetable crops. Front Microbiol 2019, 10, No. 1349. 10.3389/fmicb.2019.01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benders G. A.; Noskov V. N.; Denisova E. A.; Lartigue C.; Gibson D. G.; et al. Cloning whole bacterial genomes in yeast. Nucleic Acids Res. 2010, 38, 2558–2569. 10.1093/nar/gkq119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran S.; Noskov V. N; Segall-Shapiro T. H; Ma L.; Whiteis C.; Lartigue C.; Jores J.; Vashee S.; Chuang R.-Y. TREC-IN: Gene knock-in genetic tool for genomes cloned in yeast. BMC Genomics 2014, 15, 1180 10.1186/1471-2164-15-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov V. N.; Segall-Shapiro T. H.; Chuang R. Y. Tandem repeat coupled with endonuclease cleavage (TREC): a seamless modification tool for genome engineering in yeast. Nucleic Acids Res. 2010, 38, 2570–2576. 10.1093/nar/gkq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue C.; Glass J. I.; Alperovich N.; Pieper R.; Parmar P. P.; et al. Genome Transplantation in Bacteria: Changing One Species to Another. Science 2007, 317, 632–638. 10.1126/science.1144622. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Glass J. I.; Lartigue C.; Noskov V. N.; Chuang R. Y.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Hutchison C. A. 3rd; Chuang R. Y.; Noskov V. N.; Assad-Garcia N.; Deerinck T. J.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351, aad6253 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- Labroussaa F.; Lebaudy A.; Baby V.; Gourgues G.; Matteau D.; et al. Impact of donor-recipient phylogenetic distance on bacterial genome transplantation. Nucleic Acids Res. 2016, 44, 8501–8511. 10.1093/nar/gkw688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baby V.; Labroussaa F.; Brodeur J.; Matteau D.; Gourgues G.; et al. Cloning and Transplantation of the Mesoplasma florum Genome. ACS Synth. Biol. 2018, 7, 209–217. 10.1021/acssynbio.7b00279. [DOI] [PubMed] [Google Scholar]
- Lim P. O.; Sears B. B. Evolutionary relationships of a plant-pathogenic mycoplasmalike organism and Acholeplasma laidlawii deduced from two ribosomal protein gene sequences. J. Bacteriol. 1992, 174, 2606–2611. 10.1128/jb.174.8.2606-2611.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas B. J.; Tagwerker C.; Yonemoto I. T.; Hutchison C. A. 3rd; Smith H. O. Cloning the Acholeplasma laidlawii PG-8A Genome in Saccharomyces cerevisiae as a Yeast Centromeric Plasmid. ACS Synth. Biol. 2012, 1, 22–28. 10.1021/sb200013j. [DOI] [PubMed] [Google Scholar]
- Zürcher J. F.; Kleefeldt A. A.; Funke L. F. H.; Birnbaum J.; Fredens J.; et al. Continuous synthesis of E. coli genome sections and Mb-scale human DNA assembly. Nature 2023, 619, 555–562. 10.1038/s41586-023-06268-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhaney R. N. The structure and function of the Acholeplasma laidlawii plasma membrane. Biochim. Biophys. Acta 1984, 779, 1–42. 10.1016/0304-4157(84)90002-9. [DOI] [PubMed] [Google Scholar]
- Rilfors L.; Wieslander Å.; Lindblom G.. Regulation and physicochemical properties of the polar lipids in Acholeplasma laidlawii. In Mycoplasma Cell Membranes; Rottem S., Kahane I., Eds.; Springer: New York, Vol. 20, pp 109–166 , 1993. [DOI] [PubMed] [Google Scholar]
- Steinick L. E.; Wieslander Å.; Johansson K. E.; Liss A. Membrane composition and virus susceptibility of Acholeplasma laidlawii. J. Bacteriol. 1980, 143, 1200–1207. 10.1128/jb.143.3.1200-1207.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarev V. N.; Levitskii S. A.; Basovskii Y. I.; Chukin M. M.; Akopian T. A.; et al. Complete genome and proteome of Acholeplasma laidlawii. J. Bacteriol. 2011, 193, 4943–4953. 10.1128/JB.05059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisunov G. Y.; Garanina I. A.; Evsyutina D. V.; Semashko T. A.; Nikitina A. S.; Govorun V. M. Reconstruction of transcription control networks in mollicutes by high-throughput identification of promoters. Front Microbiol 2016, 7, 1977 10.3389/fmicb.2016.01977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrova N. M.; Bevova M. R.; Govorun V. M. Cytotoxic Activity of Mellitin Expressed by Recombinant Vectors in Cells of Acholeplasma laidlawii and Mycoplasma hominis. Genetika 2001, 37, 46–53. [PubMed] [Google Scholar]
- Sladek T. L.; Nowak J. A.; Maniloff J. Mycoplasma Restriction: Identification of a New Type of Restriction Specificity for DNA Containing 5-Methylcytosine. J. Bacteriol. 1986, 165, 219–225. 10.1128/jb.165.1.219-225.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss A.; Maniloff J. Infection of Acholeplasma laidlawii by MVL51 virus. Virology 1973, 55, 118–126. 10.1016/S0042-6822(73)81013-X. [DOI] [PubMed] [Google Scholar]
- Jarhede T. K.; Le Hénaff M.; Wieslander Å. Expression of foreign genes and selection of promoter sequences in Acholeplasma laidlawii. Microbiology (Reading) 1995, 141, 2071–2079. 10.1099/13500872-141-9-2071. [DOI] [PubMed] [Google Scholar]
- Dybvig K.; Cassell G. H. Transposition of gram-positive transposon Tn916 in Acholeplasma laidlawii and Mycoplasma pulmonis. Science 1987, 235, 1392–1394. 10.1126/science.3029869. [DOI] [PubMed] [Google Scholar]
- Sundström T. K.; Wieslander Å. Plasmid transformation and replica filter plating of Acholeplasma laidlawii. FEMS Microbiol Lett. 1990, 72, 147–151. 10.1111/j.1574-6968.1990.tb03879.x. [DOI] [PubMed] [Google Scholar]
- Dybvig K.; Woodard A. Construction of recA Mutants of Acholeplasma laidlawii by Insertional Inactivation with a Homologous DNA Fragment. Plasmid 1992, 28, 262–266. 10.1016/0147-619X(92)90058-I. [DOI] [PubMed] [Google Scholar]
- Roberts R. J.; Vincze T.; Posfai J.; Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010, 38, D234–D236. 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roganti F. S.; Rosenthal A. L. DNases of Acholeplasma spp. J. Bacteriol. 1983, 155, 802–805. 10.1128/jb.155.2.802-805.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack J. D.; Hoffmann P. J. Properties of the Nucleases of Mollicutes. J. Bacteriol. 1982, 152, 538–541. 10.1128/jb.152.1.538-541.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minion F. C.; Jarvill-Taylor K. J.; Billings D. E.; Tigges E. Membrane-associated nuclease activities in mycoplasmas. J. Bacteriol. 1993, 175, 7842–7847. 10.1128/jb.175.24.7842-7847.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbedel S.; Stülke J. Tools for the genetic analysis of Mycoplasma. Int. J. Med. Microbiol 2007, 297, 37–44. 10.1016/j.ijmm.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Cochrane R. R.; Shrestha A.; Severo de Almeida M. M.; Agyare-Tabbi M.; Brumwell S. L.; et al. Superior Conjugative Plasmids Delivered by Bacteria to Diverse Fungi. Biodes Res. 2022, 2022, 9802168 10.34133/2022/9802168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minion F. C.; Kapke P. A.. Transformation of Mycoplasmas. In Mycoplasma Protocols; Miles R. J., Nicholas R. A. J., Eds.; Humana Press, Vol. 104, pp 227–234, 1998. [DOI] [PubMed] [Google Scholar]
- Matteau D.; Pepin M. E.; Baby V.; Gauthier S.; Arango Giraldo M.; Knight T. F.; Rodrigue S. Development of oriC-Based Plasmids for Mesoplasma florum. Appl. Environ. Microbiol. 2017, 83, e03374–16. 10.1128/AEM.03374-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz A.; Just W.; da Silva Cardoso M.; Klotz G. Electroporation-mediated transfection of Acholeplasma laidlawii with mycoplasma virus L1 and L3 DNA. J. Virol 1988, 62, 3050–3052. 10.1128/jvi.62.8.3050-3052.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. E. Plasmid-mediated tetracycline resistance in Campylobacter jejuni: Expression in Escherichia coli and identification of homology with streptococcal class M determinant. J. Bacteriol. 1986, 165, 1037–1039. 10.1128/jb.165.3.1037-1039.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaudin J.; Marais A.; Verdin E.; Duret S.; Foissac X.; et al. Integrative and Free Spiroplasma citri oriC Plasmids: Expression of the Spiroplasma phoeniceum Spiralin in Spiroplasma citri. J. Bacteriol. 1995, 177, 2870–2877. 10.1128/jb.177.10.2870-2877.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos W. M. Gene cloning and expression in lactic streptococci. FEMS Microbiol Lett. 1987, 46, 281–295. 10.1111/j.1574-6968.1987.tb02466.x. [DOI] [Google Scholar]
- De Vos W. M.; Simons G. F. M.. Gene cloning and expression systems in Lactococci. In Genetics and Biotechnology of Lactic Acid Bacteria; Gasson M. J., Vos W. M., Eds.; Springer: Dordrecht, pp 52–105, 1994. [Google Scholar]
- Dybvig K.; Woodard A. Cloning and DNA sequence of a mycoplasmal recA gene. J. Bacteriol. 1992, 174, 778–784. 10.1128/jb.174.3.778-784.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek T. L.; Maniloff J. Polyethylene glycol-dependent transfection of Acholeplasma laidlawii with mycoplasma virus L2 DNA. J. Bacteriol. 1983, 155, 734–741. 10.1128/jb.155.2.734-741.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipoutcha T.; Rideau F.; Gourgues G.; Arfi Y.; Lartigue C.; Blanchard A.; Sirand-Pugnet P. Genome Editing of Veterinary Relevant Mycoplasmas Using a CRISPR-Cas Base Editor System. Appl. Environ. Microbiol. 2022, 88, e00996–22. 10.1128/aem.00996-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome sequence of A. laidlawii 8195 has been deposited to NCBI (BioProject PRJNA975591). Plasmids pAL1 and pNZ18-CAH are available on Addgene (ID 197285 and ID 203159, respectively). Acholeplasma laidlawii strain DN-E is currently in the process of being deposited to the American Type Culture Collection (ATCC).