

Summary
The dominant risk factors for late-onset Alzheimer’s disease (AD) are advanced age and the APOE4 genetic variant. To examine how these factors alter neuroimmune function, we generated an integrative, longitudinal single-cell atlas of brain immune cells in AD model mice bearing the three common human APOE alleles. Transcriptomic and chromatin accessibility analyses identified a reactive microglial population defined by concomitant expression of inflammatory signals and cell-intrinsic stress markers whose frequency increased with age and APOE4 burden. An analogous population was detectable in brains of human AD patients, including in cortical tissue using multiplexed spatial transcriptomics. This population, which we designate terminally inflammatory microglia (TIMs), exhibited defects in amyloid-β clearance and altered cell-cell communication during aducanumab treatment. TIMs may represent an exhausted-like state for inflammatory microglia in the AD milieu that contributes to AD risk and pathology in APOE4 carriers and the elderly, thus presenting a potential therapeutic target for AD.
Introduction
Alzheimer’s disease (AD) is an incurable neurodegenerative disease and the predominant form of dementia, characterized by progressive synaptic dysfunction, neuronal loss, and cognitive decline1,2. Excessive accumulation of the amyloid-β (Aβ) peptide and aggregates of hyperphosphorylated tau proteins are the major pathological features of the Alzheimer’s brain, followed by neuroinflammation, which is thought to be driven primarily by microglial cells1–3. Microglia are yolk sac-derived myeloid cells and the dominant immune population of the brain4–6. While microglia are central mediators of classical neuroinflammation, they are also a highly heterogeneous population, present in the AD brain in distinct states that can be differentially beneficial or detrimental to disease progression7–14. Nonetheless, the factors determining whether a given microglial population either constrains or contributes to AD pathology remain poorly defined.
Apolipoprotein E (APOE) is a secreted protein named for its central role in lipid trafficking with three common human isoforms: APOE2, APOE3, and APOE4. Although these alleles only vary at two amino acid sites, they are nonetheless strongly associated with differential risk for several diseases, including hyperlipidemia and atherosclerosis15. APOE is of particular interest in human health due to its role as the single largest monoallelic risk factor for late-onset AD (LOAD), with APOE4 increasing risk and APOE2 reducing risk relative to APOE3, the most common allele in the population16. APOE is also a regulator of immunity more broadly, with roles in anti-tumor immunity17,18, the response to SARS-CoV-2 infection19, and multiple other inflammatory contexts20. Still, the mechanisms by which brain immune cells are jointly modulated by aging and distinct APOE alleles are not well understood.
We aimed to characterize the immune cellular changes in the AD brain driven by aging and distinct APOE alleles by generating a single-cell atlas of immune cells from brains of AD mice bearing either APOE2, APOE3, or APOE4 alleles at distinct ages. Our atlas encompasses mice from 10 weeks of age to ~2 years of age, an understudied super-elderly state. By combining two global genome-wide modalities across multiple timepoints in AD progression, we systematically profiled the complex dynamics of the neuroimmune system and leveraged this data to interrogate its emergent biological properties. We identified a population of microglia expressing a signature of inflammatory and stress signaling markers whose frequency was enriched by age and APOE4 genotype. These microglia are present in human AD, exhibit impaired capacity for Aβ clearance, and appear to be key participants in the microglial response to aducanumab, an approved AD treatment. These findings identify a putative exhausted-like state for microglia in AD and a potential target cell for future therapeutic intervention.
Results
Single-Cell RNA Sequencing Identifies a Terminally Inflammatory Microglial State in AD Mice
We crossed 5×FAD mice, a common murine model of AD progression, with mice bearing human APOE2, APOE3, or APOE4 alleles in the murine Apoe locus (hereafter denoted AD*APOE2, AD*APOE3, and AD*APOE4). We flow-sorted Cd45+ cells from the hippocampal and cortical regions of these APOE-homozygote 5×FADhet mice at 10 weeks and 20 weeks of age, as well as of APOE3- and APOE4-homozygote 5×FADhet mice aged to 96 weeks of age (~2 years, equivalent to a human octogenarian), pooling cells from 3–6 mice per group. We then performed 10X single-cell RNA sequencing using either 3’v3 or 5’v2 capture technology, yielding an atlas of 30,868 single-cells after filtering and quality control (Fig. 1A). We sorted microglia specifically from hippocampal and cortical tissue due to their central roles in AD pathology and progression3,21–23. Reciprocal principal component analysis mediated integration24 followed by clustering identified 17 clusters, including one major cluster representing ~80% of single-cell profiles that expressed microglial markers8 such as P2ry12 and Tmem119 (Fig. 1B, Fig. S1A). Subclustering this microglial group generated 20 discrete microglial subpopulations, including clusters corresponding to the disease-associated microglia (DAM) state25 that were identified by high expression of genes such as APOE and Cst7 (Fig. 1C). We augmented these 20 clusters by leveraging a k-nearest neighbors (KNN) approach26 to survey the single-cell manifold for small, highly connected groups of cells with unique expression signatures. After hierarchical statistical evaluation, a single high-confidence microcluster of 11 cells expressing a transcriptional program including Il34 and Ano1 passed thresholding (Fig. S1B–C). We manually annotated this cluster as Il34+ microglia, arriving at a final set of 21 clusters. To establish that the clusters identified in this pipeline are meaningful and not spurious products of overclustering, we trained a 100-ply random forest classifier on a 4-fold cross-validation scheme over 25 iterations on the raw counts data from each cluster, using the classifier to produce a pairwise confusion matrix between each combination of microglial subclusters. Cells were rarely misassigned by this classifier (Fig. S1D), supporting the final clustering generated by this approach.
Figure 1: The age- and APOE isoform-dependent AD neuroimmune atlas.
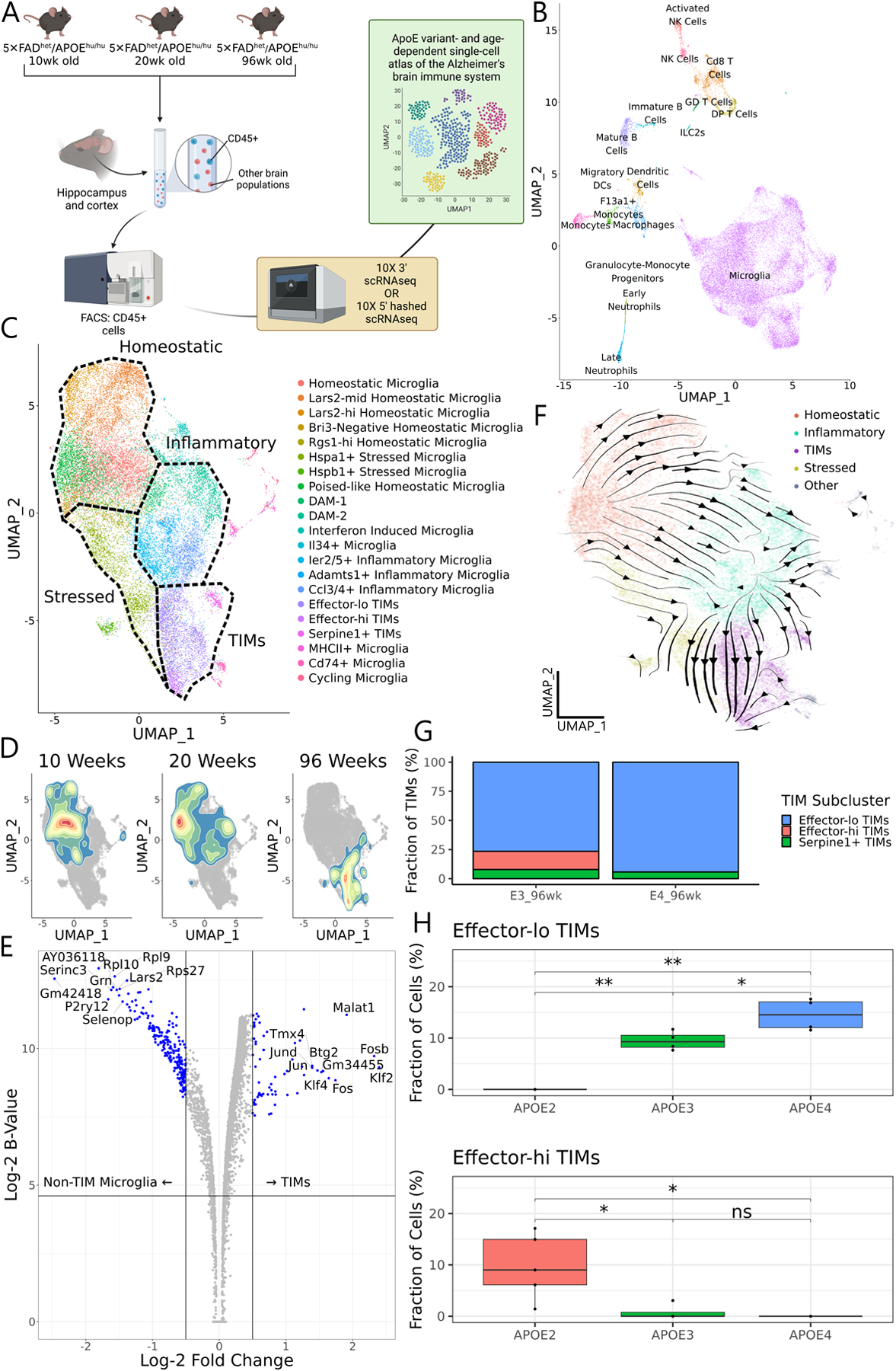
(A) Schematic of the workflow used to generate the atlas, generated from n = 3–6 animals per age and genotype. (B) UMAP of all cells in the atlas. (C) Subclustering and UMAP of microglia only. (D) 2D density plots overlaid on the microglial UMAP showing cell distributions at 10, 20, and 96 weeks of age. (E) Volcano plot of differentially expressed genes between TIMs and non-TIM microglia. The B-statistic is the log-odds that a gene is differentially expressed. Statistics were calculated using voom normalization and empirical Bayesian estimation through the limma package. (F) CellRank-calculated velocity streams on data from 20wk- and 96wk-year-old mice. Streams were estimated by a custom kernel based on splicing dynamics, connectivity, and CytoTRACE. Cells are embedded on the same UMAP manifold as in (C). (G) Stacked barplot of TIM subpopulations in the atlas from AD*APOE3 and AD*APOE4 animals at 96 weeks of age. (H) Boxplots of the proportion of effector-lo and effector-hi TIMs in bulk sequencing samples of 60-week-old AD*APOE animals, estimated by in silico decomposition with the atlas as a reference. Significance evaluated by Welch’s t-test.
We next asked whether some clusters were over- or under-represented in any samples. While microglia appeared uniformly distributed by genotype in UMAP space after integration, microglia from 96-week-old mice showed a clear shift toward clusters marked by concomitant expression of inflammatory genes such as S100a8 and S100a9 and immediate early response genes such as Fos, Jun, and Egr127 (Fig. 1D–E, Fig. S1E); we denote these clusters terminally inflammatory microglia, or TIMs (justification below). TIMs were almost exclusive to samples from 96-week-old mice and particularly enriched in AD mice bearing the APOE4 genotype (45% of all microglia from AD*APOE3 at 96 weeks, 69% of all microglia from AD*APOE4 at 96 weeks, and <1% from all other samples, Fig. S1F). Importantly, TIMs were not a low-quality population or a group of doublets, as a spline-based miQC28 approach scored cells from TIM clusters as less likely to be low-quality (p = 6.1×10−33 by Mann-Whitney-Wilcoxon nonparametric test, Fig. S1G) than all other microglial clusters, even canonical populations such as DAMs and homeostatic microglia.
Inflammatory microglia, the activated counterpart of homeostatic microglia, accumulate in the AD milieu due to persistent AD-associated neuroinflammation14,29. Given the high frequency of TIMs in aged samples and their near absence in younger mice, we reasoned that they may represent a more advanced stage for inflammatory microglia in the AD brain. To this end, we generated splice-aware alignments30 and performed directed fate mapping, using CellRank31 to construct a composite transition kernel. Splicing dynamics are powerful features for the study of cell transitions, as the ratio between spliced and unspliced products can inform whether genes are being stably expressed, are being induced, or are being repressed at the time of library preparation. However, we elected to design the kernel based heavily on connectivity and CytoTRACE32 with a comparatively minimal contribution from splicing dynamics, as the cell transitions being captured in our data occur on a much longer timescale than that of splicing regulation. Consistent with TIMs representing a terminal state, CellRank identified a robust flow from homeostatic microglia through acutely and chronically inflammatory clusters and into the age-specific population (Fig. 1F). Based on the above observations, we termed these cells terminally inflammatory microglia, or TIMs.
We noted that TIMs partitioned into two major subpopulations, one that maintained high expression of effector markers and one that instead exhibited higher expression of stress markers; we termed these effector-hi TIMs and effector-lo TIMs, respectively (Fig. 1C, Fig. S1A). The atlas suggested that effector-lo TIMs might be enriched in AD*APOE4 and that effector-hi TIMs might be enriched in AD*APOE3 (Fig. 1G), but the statistical power to confirm this was lacking. We similarly questioned whether AD*APOE2, which was absent from the atlas at the 96-week timepoint, would display shifts in TIM subpopulations. To explore these questions, we aged a cohort of AD*APOE2, AD*APOE3, and AD*APOE4 mice (n = 5 per genotype) to 60 weeks of age and separately sorted Cd45+ cells from the hippocampal and cortical regions of each brain. We then generated bulk RNAseq libraries from each of the 15 samples and used our single-cell atlas as a reference to perform in silico decomposition33 of the bulk counts (Fig. 1H, Fig. S1H). Consistent with our atlas, we found that AD*APOE4 mice were enriched for effector-lo TIMs compared to their AD*APOE3 counterparts. Interestingly, while effector-hi TIMs were not increased in AD*APOE3 compared to AD*APOE4, again consistent with our atlas, we found that essentially all TIMs from AD*APOE2 mice were effector-hi in transcriptional signature. These findings demonstrate that TIMs exhibit heterogeneity and that this heterogeneity is modulated in the context of AD by APOE genotype.
TIMs are Defined by Distinct Transcription Factors, Cell-Cell Contacts, and Metabolic Pathways
To better characterize the drivers of TIMs and other microglial populations in our atlas, we used Single-Cell Regulatory Network Inference and Clustering (SCENIC)34 to perform gene regulatory network reconstruction (Fig. 2A). Hierarchical clustering of the most highly variable regulons recapitulated known features of microglial biology, such as the enrichment of an Irf7-driven regulon in a subcluster of microglia expressing interferon-associated genes35 and Hif1a scoring highly in the late-stage DAM-2 cluster36. We found that a regulon defined by Smad4, which is required for microglial differentiation and development37, was significantly depleted in DAMs relative to all other microglia in our atlas (Fig. S2A), suggesting that DAMs may acquire their phenotype through quasi-reversion to a border-associated macrophage-like state. Meanwhile, TIMs were strongly associated both with a suite of inflammatory regulons driven by NF-κB38 and CEB/P39 family transcription factors and with multiple AP-1 family transcription factors (Fig. 2B, Fig. S2B). Interestingly, TIMs were strongly enriched for some inflammatory regulons (Nfkb2, Irf1, Irf3, Irf4) but depleted for others (Irf5, Irf7, Irf9, Stat1/2) relative to homeostatic microglia (Fig. S2C), indicating potential selective regulation of specific inflammatory pathways. We additionally found that effector-lo TIMs were distinguished from effector-hi TIMs by increased utilization of these AP-1 and Klf-family factors and comparatively weaker regulation through regulons such as Sox4 and Tcf4 (Fig. 2C).
Figure 2: TIMs are defined by a unique transcriptional program, interactome, and metabolic state.
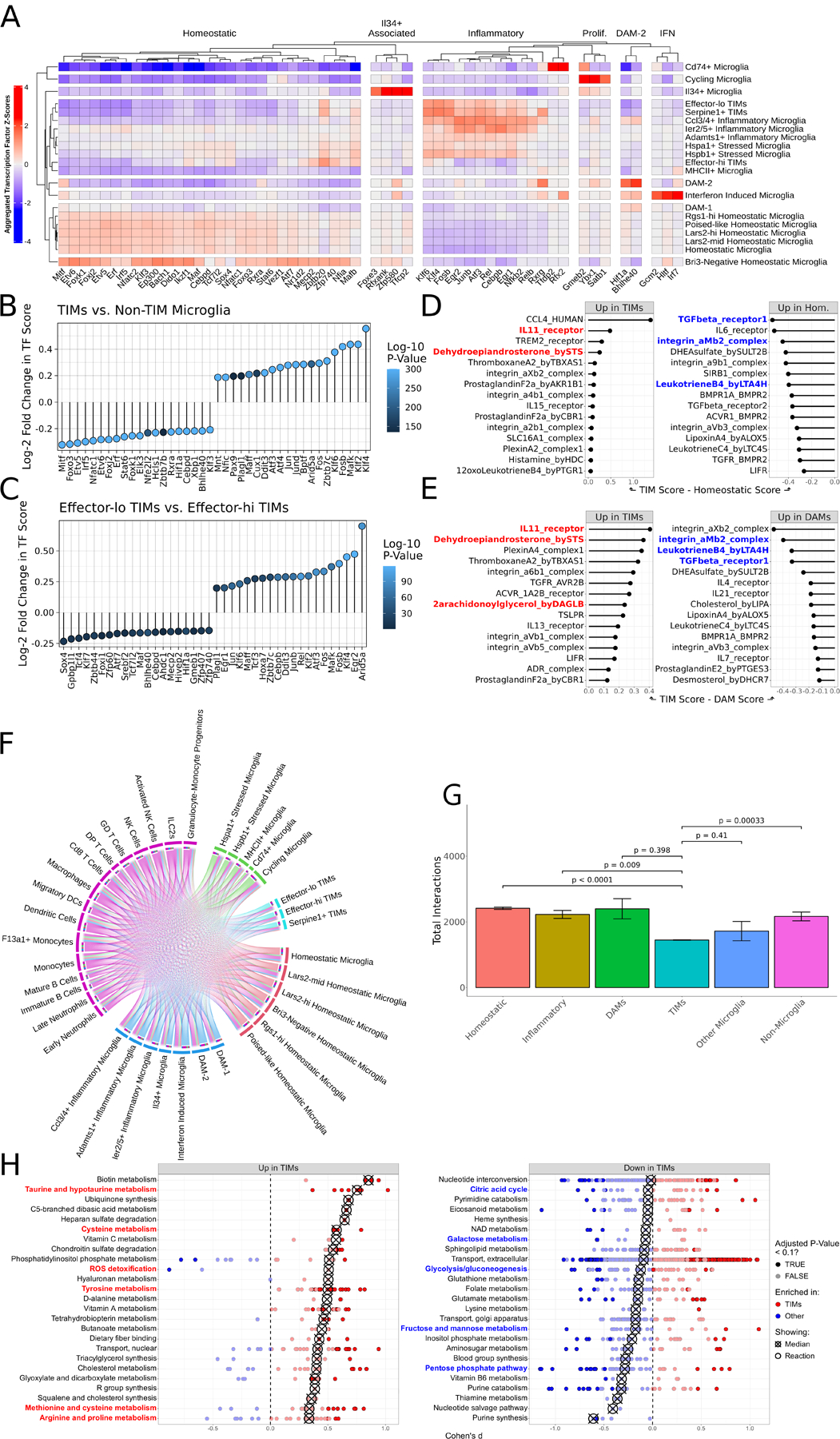
(A) Heatmap of SCENIC-derived regulons per cluster, filtered to most highly variable transcription factors. (B) Lollipop plot of differentially enriched SCENIC-derived regulons between TIMs and all non-TIM microglia. Positive values indicate increased strength in TIMs. (C) Same as (B) but comparing effector-lo TIMs to effector-hi TIMs. Positive values indicate increased strength in effector-lo TIMs. (D) CellPhoneDB scores for ligand:receptor complexes, comparing TIMs and homeostatic microglia. (E) Same as (D) but comparing TIMs and DAMs. (F) Circos plot of the atlas interactome. Size of outermost bars represents number of interactions, divided into cluster-by-other and other-by-cluster. (G) Barplot showing the total number of interactions predicted to be made by each cluster. Superclusters follow the same division as in (F). Bars are means ± standard error, significance evaluated by Welch’s t-test. (H) Dot plot of the Cohen’s d of Compass scores for metabolic pathways, comparing TIMs to non-TIM microglia. Each point represents a reaction within the larger subsystem; subsystems are sorted by median enrichment value. Medians are indicated by crossed points.
While our brain immune atlas is dominated by microglia, it also includes several other immune populations that commingle with microglia in the AD milieu. We consequently aimed to model these contacts using cell-cell interaction imputation via CellPhoneDB40. We compared ligand:receptor complexes with high predicted importance between TIMs and both homeostatic (Fig. 2D) and DAM (Fig. 2E) clusters, nominating IL11, dehydroepiandrosterone (DHEA), and 2-arachidonoylglycerol (2-AG) as strongly enriched while integrin αMβ2 (MAC-1) and leukotriene B4 were strongly depleted in TIMs relative to both. IL11, DHEA, and 2-AG have all been implicated as attenuators of microglial activation and mediators of remyelination and repair programs41–43. Likewise, MAC-1 has been nominated as a key contributor to microglial activation and neurodegeneration in both AD44 and other neurodegenerative conditions such as Parkinson’s disease45, while leukotriene B4 is an established contributor to neutrophil infiltration and tissue damage46. Together, this shift in the interactome of TIMs supports a bias towards decreased inflammation and reduced activation in comparison to other inflammatory populations like DAMs. Interestingly, TGFβ signaling was also predicted to be depleted in TIMs compared to both DAMs and homeostatic microglia despite TGFβ being a potent anti-inflammatory microglial cytokine47, suggesting an alternative mechanism by which TIMs acquire a less inflammatory phenotype. Also consistent with the view that TIMs skew less inflammatory in their cell state, we found that TIMs were predicted to be significantly less promiscuous in their cell interaction networks than all other microglial populations (Fig. 2F–G), indicating a diminished ability to contribute to inflammation in their local environment.
We next aimed to leverage scUTRquant48, a pipeline that performs 3’UTR-sensitive alignment, to study differential 3’UTR length and intronic polyadenylation events in our data. Given our particular interest in TIMs and their relative enrichment in AD*APOE4 mice over AD*APOE3 mice, we aligned and compared reads from AD*APOE3 and AD*APOE4 96-week-old mice. Several genes passed differential enrichment thresholding after bootstrapping, including immune-related genes (Il13ra1, Apobec3a), chromatin remodelers (Rbbp4), and a striking number of endocytosis-related and lysosomal genes (Glmp, Rab7, Abcg1, Tubb5, Atp2b4, Ppt1) with significantly differentially regulated 3’UTR lengths and internal polyadenylation events between the two APOE genotypes (Fig. S2D–F). Moreover, given the function of metabolism in supporting microglial states, we surmised that TIMs would be defined by differential metabolic phenotypes when compared to other microglia. We therefore inferred the metabolic state of all microglia using Compass49, a flux balance analysis algorithm that estimates a model of bidirectional flow of all metabolic reactions in Recon2 weighted by the expression level of key metabolic enzymes (Fig. 2H). Interestingly, pathways in cellular energetics such as the citric acid cycle, glycolysis/gluconeogenesis, the pentose phosphate pathway, and the metabolism of sugars such as galactose, fructose, and mannose were depleted in TIMs, suggesting an energetic deficiency unique to this population. Meanwhile, TIMs were enriched for pathways in ROS detoxification and in the metabolism of several amino acids, including taurine, hypotaurine, cysteine, tyrosine, methionine, arginine, and proline, implicating altered glutathione and amino acid metabolism as key features of the TIM state.
Single-Cell Multiome Sequencing Uncovers Mediators of Transcriptional Regulation in TIMs
Collectively, these data suggested that TIMs are a distinct population of microglia that arises exclusively in aged mice and with higher frequency in AD*APOE4 than in AD*APOE3. However, our atlas lacked resolution in the period between 20 weeks and 96 weeks, when TIMs transition from essentially absent to nearly dominant, limiting our ability to predict the determinants of TIM emergence. Moreover, the atlas was restricted only to transcriptomic space, reducing the confidence with which claims about gene regulation could be made, and was generated by integrating both 3’ and 5’ libraries, an approach that makes assumptions about capture efficiency and library preparation. We therefore complemented the atlas by using 10X snRNAseq/scATACseq multiome sequencing to jointly characterize the transcriptome and the chromatin accessibility landscape of Cd45+ cells sorted from the hippocampal and cortical regions of AD*APOE4 mice aged to 60 weeks. We elected to perform multiome sequencing on samples from AD*APOE4 mice to maximize the likelihood that we would detect TIMs at this intermediate timepoint. After filtering and quality control50, the resulting library consisted of 5,081 single-cell profiles across 32,285 RNA species and 134,523 differentially accessible regions. Joint representation of RNA and ATAC features followed by clustering identified 23 clusters, 8 of which were microglial subclusters (Fig. 3A). Confirming our results from the age- and APOE isoform-dependent atlas, one cluster in these 60-week-old mice was defined by a gene expression program corresponding to that of TIMs and was present at a frequency of 7.3% of all microglia, more frequently than in 20-week-old mice (0.3%) and less frequently than was detected in 96-week-old mice (69%) (Fig. S3A–B). These findings indicate that TIMs are already present at robustly detectable levels by 60 weeks of age and continue to accumulate thereafter in an age-dependent manner. Reassuringly, both RNA and ATAC modalities had high concordance in markers for TIMs such as Egr1 and Btg2 (Fig. S3C) and for well-established markers in other clusters such as APOE for DAMs, Skap1 for T cells, Ngp for neutrophils, and Ebf1 for B cells (Fig. S3D). Further validating results from the atlas, transcription factor footprinting of CEBPD, a predicted regulon in DAMs, and NFKB2, a predicted regulon in TIMs, showed higher Tn5 insertion enrichment in their respective clusters (Fig. 2A, Fig. S3E).
Figure 3: Multiome sequencing of AD*APOE4 mice at 60 weeks of age nominates regulatory features of TIMs.
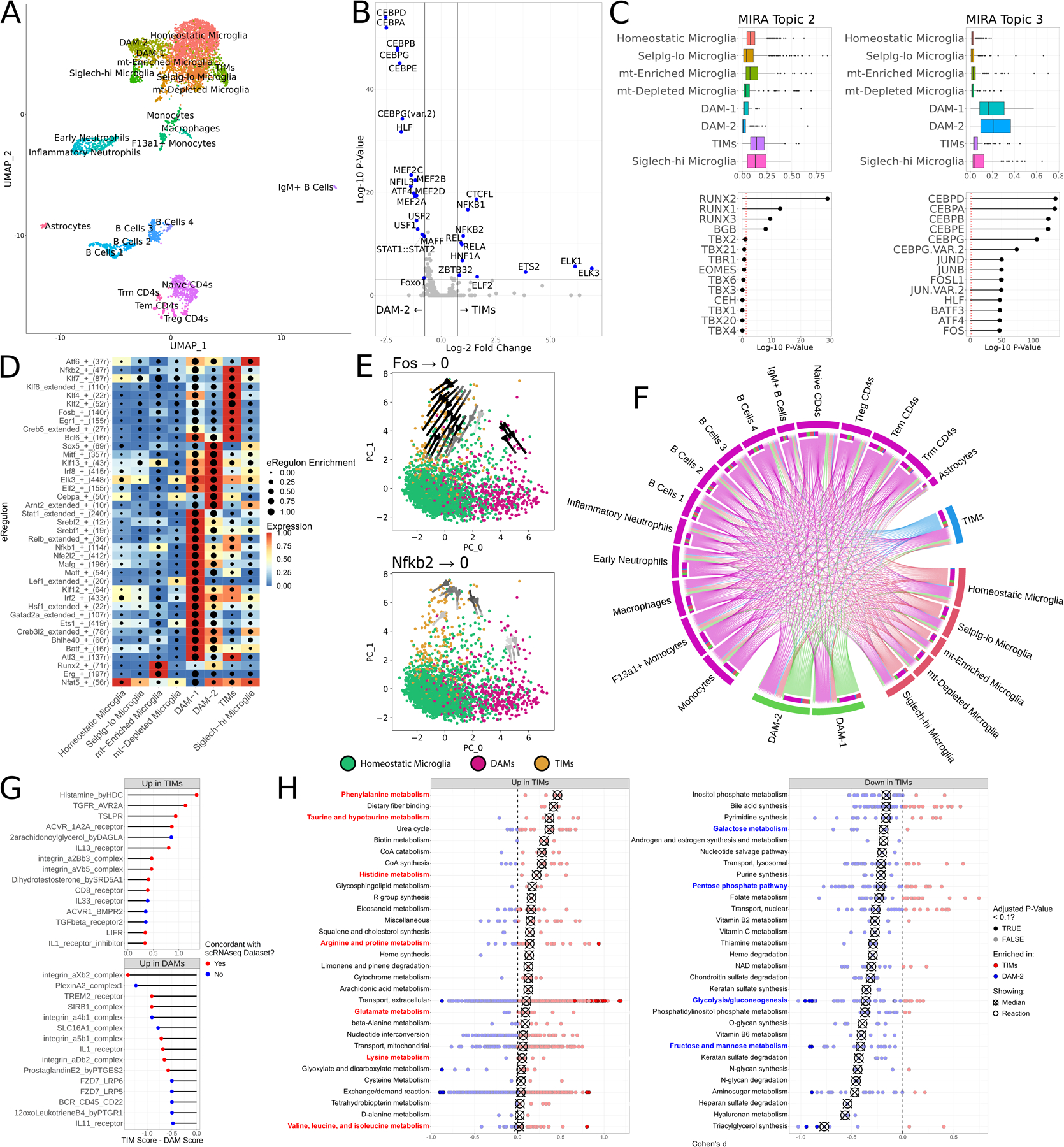
(A) Joint UMAP of RNA and ATAC features from the multiome library. (B) Volcano plot of chromVAR motif accessibility between TIMs and DAM-2 cells. (C) Scores for two topics derived from latent Dirichlet allocation of ATAC features and their associated transcription factors. (D) Heatmap of eRegulon enrichment and expression across microglial clusters. (E) Perturbation simulation plots ablating Fos and Nkfb2. Expression of the respective transcription factor was set to 0 and the gene regulatory networks were re-initialized to generate new expression profiles for each cell. Cells are projected in a PCA space defined by the gene regulatory net. Arrow shade indicates the magnitude of the transition flow. (F) Circos plot of the multiome dataset interactome. Size of outermost bars represents number of interactions, divided into cluster-by-other and other-by-cluster. (G) CellPhoneDB scores for ligand:receptor complexes, comparing TIMs and DAMs. Points are colored by whether the complex was also found to be differentially enriched in the atlas. (H) Dot plot of the Cohen’s d of Compass scores for metabolic pathways, comparing TIMs to DAM-2s. Each point represents a reaction within the larger subsystem; subsystems are sorted by median enrichment value. Medians are indicated by crossed points.
To leverage chromatin accessibility information from the multiome dataset, we used chromVAR51 to unbiasedly profile the chromatin landscape and infer motif accessibilities in each cell. In addition to the motifs we previously identified by footprinting, ETS family motifs were nominated as uniquely enriched in TIMs compared to other clusters (Fig. 3B). As a more sophisticated alternative to naïve enrichment, we also performed latent Dirichlet allocation-based topic modeling52 of RNA and ATAC features, identifying 11 RNA topics and 20 ATAC topics that captured the dynamics of multiomic regulation across the entire dataset (Fig. 3C). Of these, ATAC topic 2 (defined by RUNX factors) was enriched in TIMs while ATAC topic 3 (defined by both CEB/P factors and AP-1 factors) was enriched in DAMs.
We then aimed to capture the gene regulatory networks undergirding the accumulation of TIMs by using the SCENIC+ suite53 to model transcription factor “enhancer regulons”, or eRegulons, contributing to cell state in each cluster (Fig. 3D). Consistent with the above-mentioned results, TIMs scored highly for Nfkb2 and Fos eRegulons, while DAMs scored highly for Cebpa and Sox5 eRegulons. We reasoned that these regulatory networks were strong drivers of their respective cell clusters and that their ablation would be sufficient to drive these two cell populations back to a homeostatic state. To test this, we performed computational perturbation simulations of the SCENIC+ gene regulatory network by artificially setting expression of the given transcription factor to 0, recalculating expression of all other genes based on the SCENIC+ gene regulatory network, and embedding the resulting cells on a PCA manifold constructed from eRegulon information. Ablation of Fos and Nfkb2 signaling produced a robust flow from TIMs back onto the homeostatic cluster (Fig. 3E), while ablation of Sox5 likewise generated a flow from DAMs back onto the homeostatic cluster (Fig. S3F). Interestingly, ablation of other regulons associated with the TIM state such as Klf4 did not produce concordant shifts in gene regulatory network status (Fig. S3G), suggesting that Fos and Nfkb2 may be required for TIM maintenance while Klf4 and other factors might be dispensable once the TIM state is attained.
Seeking to replicate results from the integrated atlas, we modeled cell-cell interactions in the multiome dataset with CellPhoneDB. In agreement with findings from the atlas, we identified fewer interactions made by TIMs than by either homeostatic microglia or DAMs (Fig. 3F). Moreover, many of the differentially enriched complexes from the atlas, such as DHEA, TGFβ, and 2-AG, were among the strongest contributors to the microglial interactome in the multiome dataset (Fig. S3H). A direct comparison of TIMs to DAMs in the multiome dataset identified a number of ligand:receptor pairs that were differentially enriched in each, the bulk of which were concordant in directionality with results from the larger dataset (Fig. 3G). The most strongly enriched ligand in TIMs in the multiome dataset, histamine, was concordant with the atlas and is implicated in microglial inflammation54,55, consistent with the view of TIMs as an inflammatory state. Meanwhile, DAMs were strongly enriched for TREM2, a known driver of DAM progression9 and again concordant with the atlas. We similarly aimed to model the metabolic state of microglia in the multiome dataset using Compass to explore pathways with differential flux between TIMs and DAMs. Corroborating results from the atlas, TIMs were markedly enriched for metabolic pathways of multiple amino acids, including taurine, hypotaurine, phenylalanine, histidine, arginine, proline, glutamate, lysine, cystine, alanine, valine, leucine, and isoleucine, while energetic pathways such as glycolysis, pentose phosphate pathway, and the metabolism of fructose, mannose, and galactose were strongly depleted in TIMs compared to in DAMs (Fig. 3H).
TIMs are Pervasive in Late-Stage Human AD and are Enriched by Age and APOE4
Previous efforts utilizing murine models to describe microglial subpopulations in AD have frequently been constrained by the absence of equivalent populations in human samples; for instance, DAMs are observed across murine AD models but have no single analogue in human AD56. We consequently asked whether the TIM phenotype was unique to our aged mouse model or if it might also be detectable in single-cell data gathered from human patients. To address this question, we used Seurat anchor integration for label transfer to project microglia from ten previously published snRNAseq human brain datasets57–66 onto our murine multiome data (Fig. 4A). TIMs were identified in all ten datasets at frequencies comparable to that of the multiome reference; moreover, TIMs were identified whether tissue was processed by enzymatic digestion or by cold Dounce homogenization, indicating that the TIM state is not exclusively a product of ex vivo stress during sample preparation (Fig. 4B). We next leveraged these integrations to query the accompanying metadata and explore the impact of various factors on the frequency of TIMs. Consistent with our expectations, TIMs were more abundant in samples from patients with AD than in those without disease (Fig. 4C), in patients with more advanced disease as measured by Braak stage, a histopathological proxy for AD progression (Fig. 4D), and in patients carrying an APOE4 allele compared to their APOE3/APOE3 counterparts (Fig. 4E). Importantly, the frequency of TIMs did not correlate with post-mortem interval (Fig. 4F), suggesting that TIMs are unlikely to be an artifact of sample preparation. To better model the interplay of all these factors, we built a multiple linear regression model of TIM frequency across all individuals in the second ROSMAP dorsolateral prefrontal cortex project, a repository of snRNAseq data from the dorsolateral prefrontal cortex of 465 human AD donors; again consistent with our observations, we found that APOE4 allele count and Braak score were strong predictors of TIM frequency while other factors such as postmortem interval had no predictive power (Fig. 4G). Strikingly, we also observed that male sex was nearly as strong a modifier of TIM frequency as APOE4 allele count in this dataset, suggesting the existence of a sex-specific phenotype controlling the emergence of this unique cell state. We note that while age at death was not a robust predictor of TIM frequency, >91% of samples in the dataset are from individuals over the age of 80, making it a poorly informative parameter in this analysis. Finally, we performed label transfer between our larger single-cell atlas and microglia from the Tabula Muris Senis, a single-cell atlas of mouse tissues across age67. We found that the frequency of TIMs increased monotonically from 3-month-old to 24-month-old mice, in agreement with the view that TIMs accumulate with age (Fig. S4). These findings suggest that TIMs are also present at robustly detectable levels in human brains and that their frequency follows the same trends as in mice with respect to age, disease, and presence of the APOE4 allele.
Figure 4: TIMs are detected in publicly available human snRNAseq datasets.
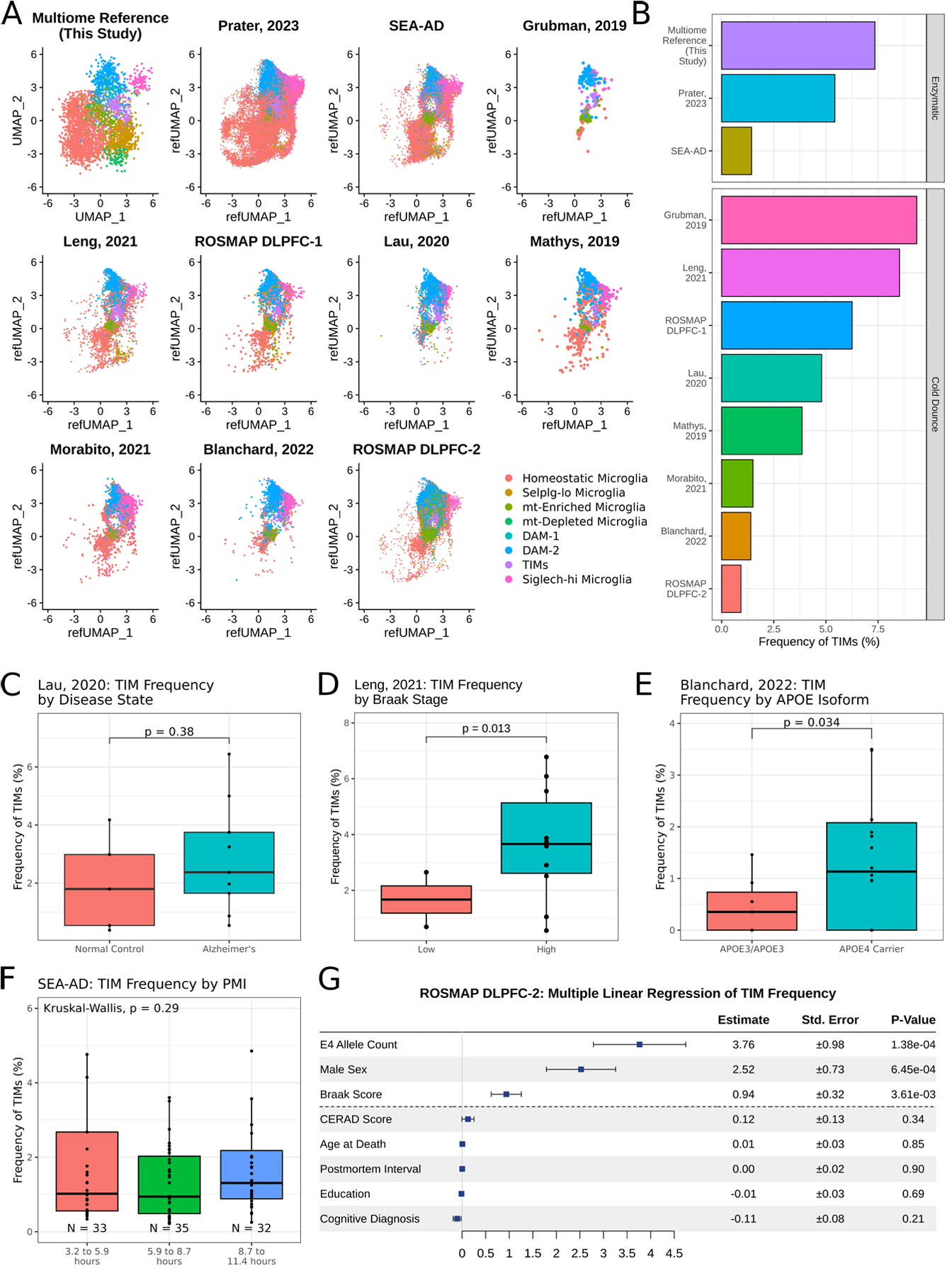
(A) UMAP projection of microglia from ten publicly available human snRNAseq datasets after anchor integration onto the data acquired in this study. (B) Barplot of TIM frequency in each dataset. (C) Boxplot of TIM frequency in data projected from Lau, 2020, grouped by disease state. Significance evaluated by Welch’s t-test. (D) Boxplot of TIM frequency in data projected from Leng, 2021, grouped by Braak stage, a measure of disease severity. Significance evaluated by Welch’s t-test. (E) Boxplot of TIM frequency in data projected from Blanchard, 2022, grouped by either amyloid-β burden or by presence of an APOE4 allele. Significance evaluated by Welch’s t-test. (F) Boxplot of TIM frequency in data projected from the Seattle AD Brain Atlas, grouped by post-mortem interval. Significance evaluated by Kruskal-Wallis test. (G) Multiple linear regression of TIM frequency by metadata provided in the second ROSMAP dorsolateral prefrontal cortex (n = 465).
The TIM Spatial Niche is Enriched for Aβ Plaques and Inflammatory Cells in the Cortex
To further examine TIMs in the context of human AD, we used the Xenium platform to perform multiplexed in situ transcriptomics on cortical tissue samples from six human AD donors, three homozygous for APOE4 and three homozygous for APOE3. After quality control and filtering68, the resulting dataset covered 494,376 individual cells over a panel of 266 genes. We leveraged our computational label transfer of previously published human snRNAseq datasets to identify putative markers of human TIMs during the annotation process (Fig. S5A), yielding 22 clusters covering both neuronal and glial populations (Fig. S5B–C). Critically, we observed a clear population of TIMs with robust representation across both genotypes (Fig. 5A). Consistent with our murine data, we found that donors homozygous for APOE4 were enriched for TIMs relative to those homozygous for APOE3 (22.7% vs. 5.2%; Fig. 5B). We generated a cell-cell spatial neighborhood matrix across all six sections, confirming that our data had captured the expected architecture of the cortex (Fig. S5D). We then investigated the spatial niche of TIMs within the tissue by calculating the increased probability of observing a given cell type near a TIM compared to the null distribution. We found that TIMs are about 50% more likely to be found next to other TIMs than would be expected based on their frequency, while L5 neurons and two astrocyte populations with high copy number of APP or APOE transcripts were 10–20% more likely than expected to be found next to TIMs (Fig. 5C, Fig. S5E). We repeated the same analysis in a genotype-aware fashion, revealing that TIMs in APOE4 donors were more likely to cluster with neurons and these two astrocyte populations, whereas TIMs in APOE3 donors were more often adjacent to oligodendrocytes, the majority of which reside in the white matter (Fig. 5D). These data suggest that the APOE4 genotype is associated both with an increased frequency of TIMs and an increased proximity of those TIMs to grey matter. We then stained the same tissue sections used for Xenium with methoxy-X04, a fluorescent stain specific for Aβ plaques. Comparing results from methoxy-X04 staining to our Xenium annotations revealed that TIMs could be detected in regions containing high Aβ burden (Fig. 5E), suggesting a potential role for TIMs in Aβ-related pathogenesis.
Figure 5: TIMs are enriched in the cortical layers of human AD patients bearing ApoE4.
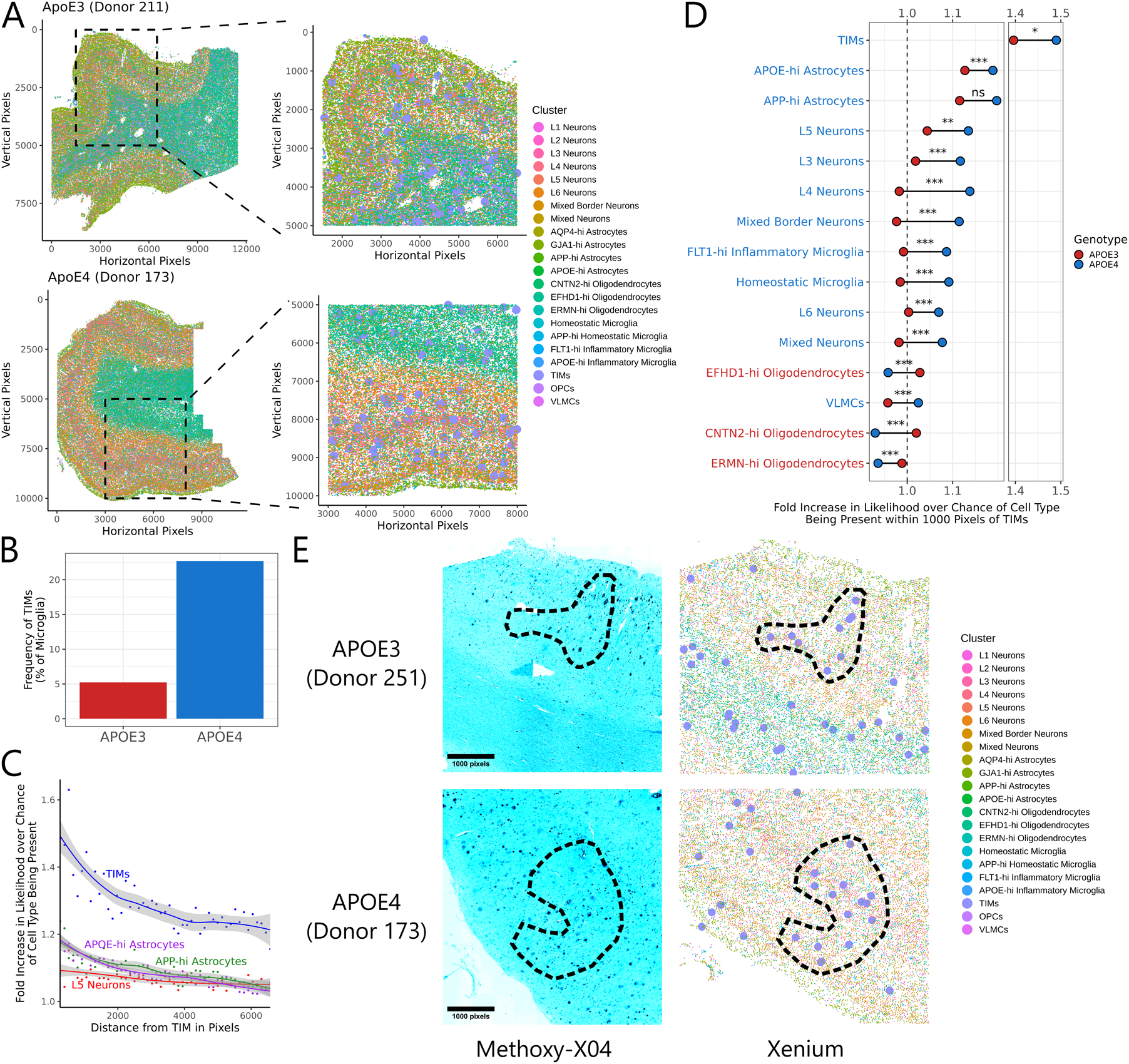
(A) Spatial scatter plot of cell annotations in two representative sections out of the six subjected to Xenium analysis. At left is the full section, at right is a zoomed inset of the indicated region. TIMs are marked by larger point sizes in the zoomed inset for clarity. (B) Barplot of the fraction of microglia from each genotype annotated as a TIM. (C) Smoothed trendlines of the increased likelihood of finding a given cell type within a circle of the indicated radius centered on a TIM compared to over base expectation. Only the top four most enriched clusters are shown. (D) Barbell plot showing the increased likelihood of finding a given cell type within a circle of the indicated radius centered on a TIM compared to base expectation, separated by genotype. Significance evaluated by Welch’s t-test. Clusters are colored by which genotype shows higher enrichment around TIMs. (E) Representative fluorescence micrographs of cortical tissue sections from APOE3 and APOE4 donors after post-Xenium staining with methoxy-X04 (a stain for Aβ) and accompanying annotations from Xenium data. TIMs are marked by larger point sizes in the Xenium annotations for clarity. Circled regions indicate areas of high overlap between TIMs and Aβ plaques.
TIMs Exhibit Defective Capacity for Aβ Phagocytosis and Clearance
Collectively, our findings suggested that the TIM state might have consequences on microglial function and phenotype. To test this prediction, we harvested cells from the hippocampal and cortical regions of AD*APOE2, AD*APOE3, and AD*APOE4 mice at 60 weeks of age and incubated them ex vivo with fluorescently labeled and oligomerized amyloid-β 1–42 (Aβ). We employed a pH-insensitive fluorophore, HiLexa 488, to ensure that fluorescent signal would be stable after cellular uptake. We then sorted Cd45+ cells, splitting each sample into two tubes based on Aβ uptake as measured by fluorescent signal, before performing post-sort cell hashing followed by 10X 5’v2 scRNAseq (Fig. 6A). Filtering, quality control, and computational integration resulted in a dataset of 12,613 cells, 76% of which fell into one of 12 microglial subclusters and 24% of which fell into one of 17 smaller non-microglial populations (Fig. 6B). Comparing the relative distribution of microglia from Aβ-hi and Aβ-lo pools revealed that the Aβ-lo population was qualitatively shifted away from homeostatic clusters and towards TIMs (Fig. 6C). We then examined the relative proportion of cells in each cluster that originated in the Aβ-hi pool per genotype to identify which populations were most or least capable of Aβ uptake (Fig. 6D). Two populations, one of homeostatic microglia enriched for ribosomal transcripts (χ20.05 = 1.88×10−58) and one of microglia marked by interferon responsive genes (χ20.05 = 2.70×10−17) were consistently overrepresented in the Aβ-hi pool, though interferon induced microglia from AD*APOE4 were less so than their AD*APOE2 and AD*APOE3 counterparts. Meanwhile, both effector-lo TIMs (χ20.05 = 6.87×10−27) and effector-hi TIMs (χ20.05 = 1.58×10−14) were underrepresented in the Aβ-hi pool. Strikingly, AD*APOE4 was defined by both a more severe underrepresentation of effector-lo TIMs in the Aβ-hi pool – suggestive of a more exacerbated defect in Aβ clearance by this population in this genotype – alongside a higher frequency of effector-lo TIMs overall, revealing a compounding effect in which AD*APOE4 is overburdened with a population of particularly impaired cells.
Figure 6: TIMs are functionally impaired in capacity for amyloid-β clearance.
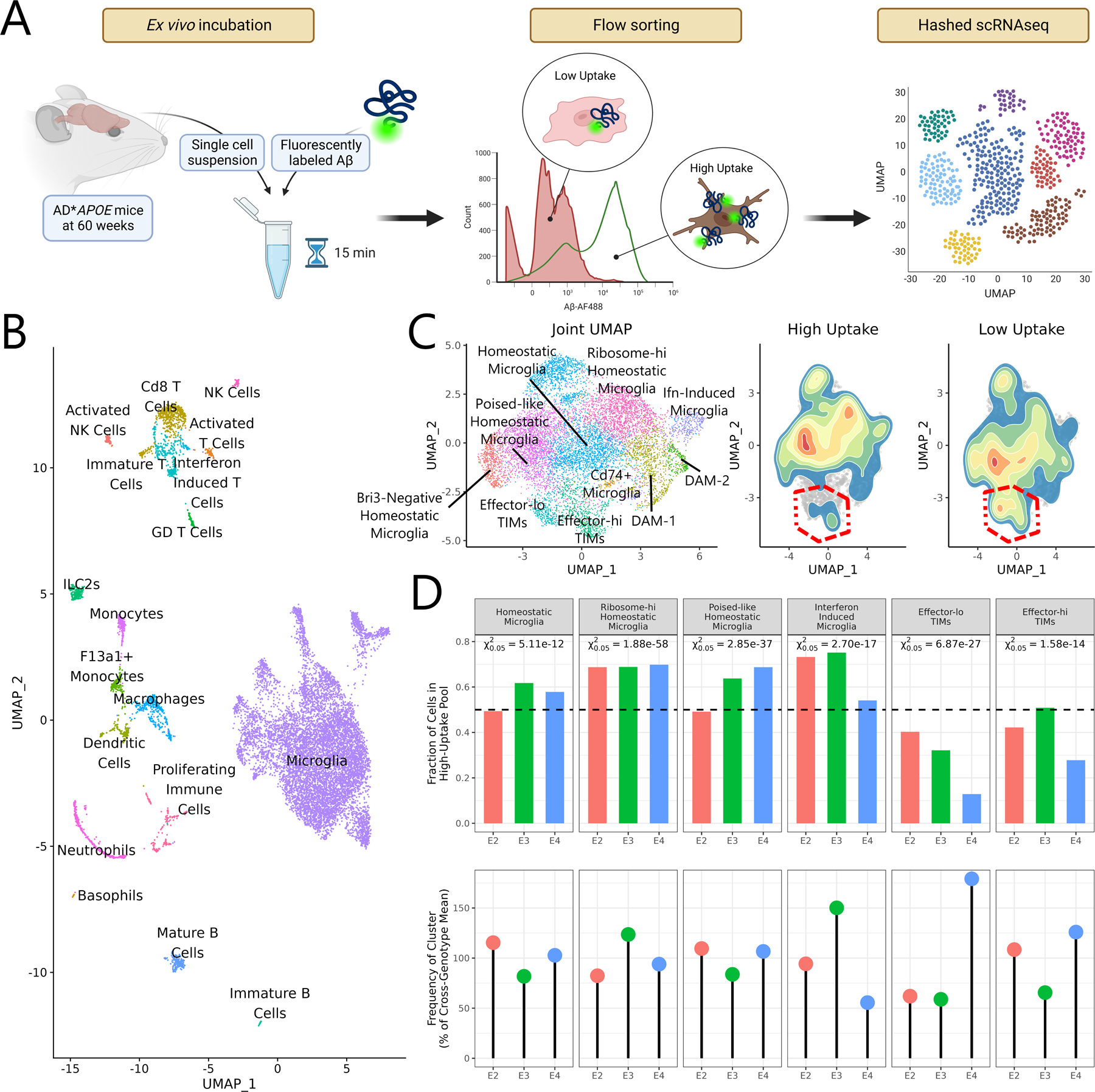
(A) Schematic of the experimental strategy to characterize microglial capacity for ex vivo Aβ uptake. (B) UMAP generated from all cells sequenced after the Aβ uptake experiment. (C) Joint subclustering UMAP of all microglial cells in the dataset and 2D density plots overlaid on the microglial UMAP showing cell distributions from the high uptake and low uptake populations. TIM clusters are outlined in red. Note that TIMs, and particularly effector-lo TIMs, are depleted in the high uptake fraction. (D) Barplot of the fraction of cells from each genotype and cluster in the high uptake pool and dotplot showing the fraction of cells from each genotype in the given cluster. Degree of over- or underrepresentation in the high uptake pool was evaluated using a chi-square test on the null expectation of an even split. P-values are reported at the α = 0.05 threshold.
The Frequency and Interactome of TIMs are Modulated by Aducanumab Treatment
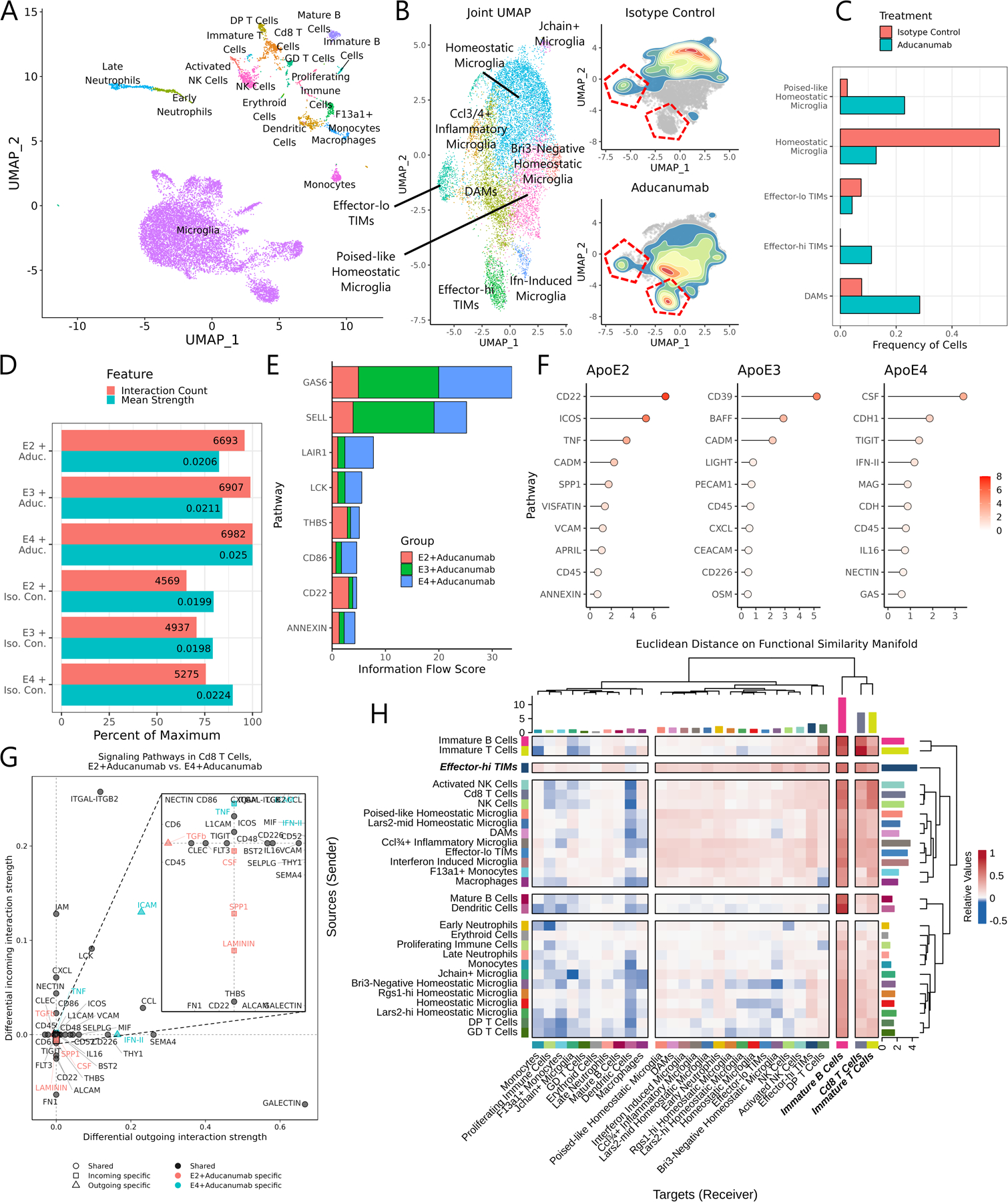
While AD remains incurable, aducanumab is a recently developed and approved monoclonal antibody therapeutic that aims to slow disease progression by selectively binding to Aβ aggregates69. It is believed that these aggregates are then cleared by Fc-receptor mediated binding with myeloid cells. While clinical trials showed modest improvements in cognition driven by treatment, patients nonetheless experienced significant side-effects such as neuroinflammation and edema70. Given aducanumab’s strong impact on myeloid cells in the AD brain, we wondered how it would influence the emergence and behavior of TIMs in late-stage disease. We treated 60-week-old AD*APOE2, AD*APOE3, and AD*APOE4 mice with daily intraperitoneal injections of aducanumab or of an isotype control for five days before performing single-cell sequencing of Cd45+ cells from their hippocampal and cortical regions (Fig. 7A). Processing and annotation produced a dataset of 13,483 cells, 80% of which were microglia (Fig. 7B). Subclustering to microglia revealed that cells from animals treated with isotype control were predominantly homeostatic, acutely inflammatory, or effector-lo TIMs while those from animals treated with aducanumab were instead largely DAMs, effector-hi TIMs, and poised-like homeostatic microglia (Fig. 7C–D). Poised-like homeostatic microglia are differentiated from homeostatic microglia by higher expression of MHC-I genes and Cd52 (Fig. S6A), a gene signature associated with the microglial response to demyelination or injury4.
Figure 7: Aducanumab treatment profoundly alters the landscape of immune cells in the AD milieu.
(A) UMAP of all cells from the unified aducanumab dataset. (B) Joint subclustering UMAP of all microglial cells in the dataset and 2D density plots overlaid on the microglial UMAP showing cell distributions from the aducanumab-treated and isotype control-treated populations. TIM clusters are outlined in red. (C) Barplot of the frequency of microglial clusters in aducanumab-treated and isotype control-treated populations. (D) Barplot of the number of predicted interactions and the mean predicted interaction strength from each of the six samples in the aducanumab dataset, as estimated by CellChat. (E) Stacked barplot showing the total information flow predicted by CellChat through each signaling pathway. (F) Lollipop plot showing pathways with highest differential regulation between aducanumab-treated and isotype control-treated samples in each genotype, quantified by Euclidean distance on the joint functional similarity manifold produced by CellChat embedding. (G) Dotplot of differentially enriched signaling pathways in Cd8 T cells between AD*APOE2 and AD*APOE4 aducanumab-treated animals by both incoming and outgoing signal strength. Pathways are color- and shape-coded by directionality and sample specificity. Positive numbers indicate a greater strength in AD*APOE4. (H) Heatmap showing the mean difference in incoming and outgoing signaling between aducanumab-treated and isotype control-treated animals. Positive numbers indicate a greater strength in aducanumab-treated animals. Note that the strongest increases in outgoing signaling are in inflammatory microglia and especially in effector-hi TIMs.
We used CellChat71 to better understand the shifts in the interactome driven by both aducanumab and APOE isoform across these samples, finding that aducanumab treatment and APOE4 were both associated with a higher number of predicted interactions and a stronger average predicted interaction strength (Fig. 7D). Querying across specific pathways identified key pathways with both high activity and differential utilization across APOE isoform (Fig. 7E); for instance, Gas6, a major driver of inflammation implicated in Aβ plaque clearance72, was more active after aducanumab treatment in APOE3 and APOE4, while Cd22, a potent inhibitor of microglial phagocytosis73, was more active in APOE2 after treatment. These findings suggest that aducanumab treatment was less able to drive microglial responses to Aβ in APOE2 animals than in APOE3 or APOE4 counterparts. We then projected all ligand-receptor complexes onto a unified functional similarity manifold and determined the pairwise Euclidean distance between pathways from aducanumab-treated and isotype control-treated animals of the same genotype (Fig. 7F), revealing that APOE2 mice exhibited the strongest shifts in signaling in Cd22, while APOE3 and APOE4 instead had shifts in Cd39, a regulator of microglial migration74, and Csf1r, a key microglial survival factor75.
Next, we examined interactome shifts within specific cell types driven by aducanumab treatment. Strikingly, aducanumab massively expanded the total interactome of adaptive immune cells across all three genotypes, particularly Cd8 T cells and immature B cells (Fig. S6B), both of which inhabit the dura of the meninges76–78. Moreover, the interactome of these cell types was expanded in APOE4 animals to a greater extent than in APOE2 or APOE3 animals. A closer examination of the Cd8 T cell interactome revealed that this was driven primarily by increases in inflammatory signaling through type-II interferons along with elevated cell-cell adhesion via ICAMs (Fig. 7G). Similar pathways were also modulated in immature B cells, effector-lo TIMs, and effector-hi TIMs (Fig. S6C). Finally, we explored which cell types might be driving differential T and B cell signaling after aducanumab treatment by separating signaling into senders and receivers and finding the mean difference in signaling strength between aducanumab-treated and isotype control-treated animals across all APOE isoforms (Fig. 7H). As expected, inflammatory microglial clusters showed the strongest increase in signal sending after aducanumab treatment. Remarkably, though, the largest growth in signaling after aducanumab treatment came from effector-hi TIMs, suggesting that this population may be particularly critical while coordinating cell-cell communication networks during inflammation. Combined with the higher frequency of effector-hi TIMs in aducanumab-treated animals, these findings highlight effector-hi TIMs as central orchestrators of the adaptive immune response to aducanumab therapy.
Discussion
Microglia display remarkable diversity, particularly within the inflammatory milieu of disease states. In this study, we present a single-cell atlas of microglial gene expression in AD in an age- and APOE isoform-dependent manner. Just as single-cell techniques revealed unappreciated heterogeneity in macrophage state beyond the M1-M2 paradigm79, these data define a microglial state beyond the homeostatic-inflammatory binary characterized by futile inflammation and impaired response to chronic stimuli. We refer to this population as TIMs and show that its frequency is increased by APOE4 and age. TIMs are distinguished from DAMs and other previously described microglial states in several respects, including their concomitant expression of inflammatory markers and cell-intrinsic stress signatures, their distinct chromatin accessibility landscape, and their unique metabolic state and interactome.
A transcriptomic signature with some similarity to that of TIMs has been presumed to represent an ex vivo stress signature induced by enzymatic processing80. However, TIMs are robustly detectable in snRNAseq datasets of human AD, irrespective of whether single-cell suspensions were prepared by enzymatic processing or by cold Dounce homogenization. While we cannot exclude enzymatic digestion as a potential confounder of transcriptional state, our analysis shows that TIMs are not purely an artifact of sample preparation. Moreover, even under the interpretation that the TIM phenotype is exacerbated by ex vivo conditions, our data indicates that microglia from aged brains are more strongly predisposed towards stochastically adopting this state and that this transition is genetically modulated by APOE genotype. We consequently propose that TIMs represent a bona fide microglial program and suggest that this state may have previously been underappreciated due to its age-dependent accumulation.
Many of the characteristics of TIMs, most notably their decreased utilization of glycolytic pathways and their simultaneous expression of both pro- and anti-inflammatory signals, are also observed in T cell exhaustion, a phenotypic state that is similarly induced by chronic stimulation and inflammation and is marked by impaired capacity to respond to those inflammatory signals. Likewise, TIMs are characterized by elevated expression of Serpine1, a marker of senescence whose expression in microglia is associated with Aβ pathology and inflammation81. This is further supported by the existence of a relatively minor population of TIMs with particularly high expression of Serpine1 whose frequency is not affected by APOE genotype but is modified by age in the same way as other TIM states. These observations frame TIMs as a potential microglial equivalent of exhausted T cells, posing the possibility that TIMs represent a terminal state for activated microglia marked by a less inflammatory, functionally impaired, exhausted-like state82. We note that this microglial exhaustion is distinct from microglial depletion, a chronic, systemic decrease in microglial frequency that is a suggested therapeutic strategy against AD83.
In the context of the inflammatory AD milieu, these dysfunctional microglia may contribute to AD pathology, potentially explaining elements of AD evolution across stages of severity. Moreover, the greater frequency of TIMs in APOE4 carriers may contribute to their increased susceptibility to Alzheimer’s hallmarks, earlier disease onset, and differential rate of disease progression84. We speculate that this may occur through TIM accumulation resulting in a concomitant loss of homeostatic microglia, thereby interfering with normal constraint of AD progression and accelerating disease pathology. Alternatively, TIMs may have an active role in disruptive cell signaling or secretion of factors that lead to detrimental disease outcomes; ligands such as IL11, DHEA, 2-AG, and histamine, all of which were strongly enriched in TIMs in our data by cell-cell interaction imputation, are worthy of further exploration in this regard.
Epidemiological studies have long noted that AD incidence is considerably higher for females than for males85. While this is partially explained by the longer average lifespan of females, other factors, including post-menopausal changes in hormones and sex-specific differences in immune responses, are also implicated in this phenomenon86–88. However, comparatively little is understood with respect to putative drivers of AD that might be unique to males. One possibility is raised by a meta-analysis of the impact of herpes zoster vaccination on dementia, which revealed that while vaccination is protective against development of dementia later in life, this effect is stronger in females than in males89. Our finding that TIMs are enriched in males suggests a potential basis for this phenotype: the aged male neuroimmune system might be less responsive to the effects of historical immune interventions such as vaccination due to its higher burden of dysfunctional microglia. Combined with the observation that sex-specific AD risk in APOE4 carriers is particularly exacerbated between the ages of 65 and 7590, the approximate equivalent human age range to the period in which TIM emergence is most accelerated in our murine model, we posit that TIM frequency may be a cell-type-based risk factor that partially describes the incidence and pathology of AD in males.
Our results also underscore the presence of two distinct classes of TIMs in the AD milieu: effector-lo TIMs are defined predominantly by stress markers, while effector-hi TIMs maintain expression of genes relevant for immune crosstalk and function. While APOE4-bearing animals are strongly enriched for effector-lo TIMs, APOE2-bearing animals instead enrich for effector-hi TIMs. Our functional exploration of TIMs reveals that both subtypes show defects in Aβ clearance and that this is exacerbated in effector-lo TIMs. Meanwhile, effector-hi TIMs appear to be dominant contributors to the immune interactome during aducanumab treatment. Additional work is needed to clarify the differential roles of these TIM subtypes during aging, in AD, and at physiological steady state, as well as the mechanism by which APOE variants bias TIMs towards different states.
Pioneering work in immunology revealed that T cells can undergo exhaustion, characterized by increased expression of inhibitory receptors and inappropriate activation in response to stimulus; this discovery laid the groundwork for immunotherapies that leverage antibody-based therapeutics to promote effective T cell responses91,92. In much the same way, we postulate that a better understanding of the exhausted-like microglial phenotype we describe herein could enable a new treatment paradigm for AD predicated not on directly reducing neuroinflammation but instead on the reprogramming of microglia towards a more homeostatic state. Further functional characterization of TIMs and their ramifications in situ over the course of AD pathology may therefore uncover novel regulatory nodes and therapeutic avenues for the treatment and prevention of AD.
Limitations of Study
While the single-cell atlas generated in this work enables the exploration of deep questions in microglial heterogeneity, its statistical power is limited by the fact that one library was generated for each genotype and age. Moreover, while our analysis of human datasets identified a strong correlation between TIM frequency and sex, our murine work was underpowered with respect to this question. Future work will be necessary to validate these findings. Additionally, our computational modeling of the TIM gene regulatory network nominated a suite of AP-1 factors as being critical for either the establishment or the maintenance of the TIM state. In vivo studies modulating these transcription factors can conclusively determine necessity and sufficiency in their regulation of TIM emergence. Finally, our work reveals that TIMs are ubiquitous in murine and human AD, but whether TIMs also play a role in non-pathological aging or in other chronic neuroinflammatory conditions such as multiple sclerosis, Parkinson’s disease, and brain tumors remains to be described. Computational and experimental techniques can be leveraged to explore these questions, extending our findings to microglial biology beyond the scope of AD.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sohail Tavazoie (Sohail.Tavazoie@rockefeller.edu).
Materials availability
The AD*APOE mouse lines generated in this study will be made available for academic use upon reasonable request.
Data and code availability
Collectively, all new sequencing data generated in this study are available on GEO under SuperSeries GSE239999. All raw data, Cell Ranger outputs, and processed Seurat and Signac structures for the atlas and multiome dataset are available on GEO under accession number GSE225503. All raw data, Cell Ranger outputs, and processed Seurat structures for the aducanumab treatment experiments are available on GEO under accession number GSE239975. All raw data, Cell Ranger outputs, and processed Seurat structures for the Aβ uptake experiments are available on GEO under accession number GSE239974. All raw data and counts matrices from bulk sequencing of aged AD*APOE mice are available on GEO under accession number GSE239977. All Xenium raw data, the processed Squidpy structure, and full-slide scans after post-Xenium methoxy-X04 staining are available on Zenodo at 10.5281/zenodo.8206638. The ROSMAP DLPFC-1 and DLPFC-2 data used in this analysis are available at Synapse under accession codes syn16780177 and syn31512863.
All R and Python code necessary to reproduce the analysis and figures in this manuscript has been annotated and uploaded to Github at https://github.com/alonmillet/apoe-ad-age-atlas.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model Details
C57BL/6 (strain 000664) and 5×FAD (strain 34840) mice were purchased from the Jackson Laboratories and maintained in our facilities. Mice bearing knock-in of human APOE2, APOE3, or APOE4 in the murine Apoe locus [B6.129P2-Apoetm1(APOE*2)Mae N9 (APOE2), B6.129P2-Apoetm2(APOE*3)Mae N8 (APOE3) and B6.129P2-Apoetm3(APOE*4)Mae N8 (APOE4)] were purchased from Taconic Biosciences and maintained in our facilities. Mice expressing each APOE allele were crossed with 5×FAD hemizygous mice to generate the final AD*APOE line. 5×FAD hemizygous*APOE homozygous mice were used in this study. Mice were weaned at the third postnatal week, genotyped in-house and kept on a 12 h/12 h light/dark cycle (lights on at 7:00) with access to food and water ad libitum.
Both female and male mice were used for experiments. Animal care and experimentation were according to NIH guidelines and were approved by the Institutional Animal Care and Use Committee at The Rockefeller University (protocols #20010 and #23007).
Method Details
Brain immune cell isolation
Brain immune cells were isolated as previously described93–96 with minor modifications. Briefly, mice were anesthetized with a ketamine/xylazine cocktail and perfused with 25 ml of Ca2+/Mg2+-free DPBS (Sigma). Cortex and hippocampus were removed and placed in FACS buffer (PBS containing 5% FBS and 10 mM HEPES), minced with scissors, and incubated with 80 U/mL of collagenase D (Roche) at 37°C for 30 min. Collagenase was inactivated by adding 10 mM EDTA for an additional 5min incubation at 37°C. Digested material was passed through a 70μm cell strainer and centrifuged at 1500rpm for 10min. The pellet was then resuspended in 7mL of 38% Percoll (Sigma) followed by a centrifugation at 2000rpm for 30 min. Nonspecific binding to Fc receptors was blocked by incubation with a CD16- and CD32-specific antibody (BioLegend) for 15min. Lastly, cells were washed and stained with an anti-CD45 antibody (BioLegend) and 0.05 μg/mL DAPI (Sigma), then washed and resuspended in FACS buffer. Cells were sorted on a FACSAria-II flow cytometer (Becton Dickinson) for singlet DAPI– Cd45+ cells.
Single-cell library preparation
For samples from 96-week-old mice, sorted CD45+ cells from each genotype were washed, strained through a 40μm strainer (Flowmi), and resuspended in PBS containing 0.04% BSA at ~1000 cells/μL. Libraries were prepared using 10X Genomics 3’v3 chemistry per manufacturer’s instructions and sequenced on an Illumina NovaSeq SP. Six mice were pooled for the APOE3 library while four mice were pooled for the APOE4 library.
For samples from 10-week-old and 20-week-old mice, cells were also stained with 0.5μg of TotalSeq-C mouse hashing antibody (BioLegend) per genotype before cell sorting. Sorted CD45+ cells from each genotype were then pooled at equal cell numbers, washed, strained through a 40μm strainer (Flowmi), and resuspended in PBS containing 0.04% BSA at ~1500 cells/μL for superloading as previously described97. Libraries were prepared using 10X Genomics 5’v2 chemistry with Feature Barcoding per manufacturer’s instructions and sequenced on an Illumina NovaSeq SP. Three mice were pooled together per genotype for each library.
Aducanumab treatment and library preparation
For the aducanumab treatment experiment, 60-week-old mice were treated intraperitoneally with 0.5mg of either aducanumab (Cardinal Health) or human IgG isotype control (Invitrogen #02–7102) daily for 5 days prior to immune cell isolation and library prep as for the 10-week-old and 20-week-old mice, ultimately pooling cells at ~2000 cells/μL for superloading. Samples were sequenced on an Illumina NovaSeq S2. Three mice were pooled together per genotype for each library.
Multiome library preparation
Brain immune cells were isolated as described above from two 60-week-old AD*APOE4 mice. Sorted CD45+ cells were centrifuged at 500rcf for 5min at 4°C and resuspended in 100μL lysis buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 0.01% Tween-20, 0.01% Nonidet P40 substitute, 0.001% digitonin, 1% BSA, 1mM DTT, and 1U/μL Sigma Protector RNase inhibitor in nuclease-free water). Cells were lysed for 3min on ice. Lysis was stopped by addition of 1mL wash buffer (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mM MgCl2, 1% BSA, 0.1% Tween-20, 1mM DTT, and 1U/μL Sigma Protector RNase inhibitor in nuclease-free water) followed by centrifugation at 500rcf for 5min at 4°C. Cells were washed twice more in wash buffer, then resuspended at 3000 nuclei/μL in diluted nuclei buffer (10X Genomics nuclei buffer diluted with 1mM DTT and 1U/μL Sigma Protector RNase inhibitor in nuclease-free water). Libraries were prepared using 10X Genomics Single Cell Multiome ATAC + Gene Expression chemistry per manufacturer instructions. The GEX library was sequenced on an Illumina Nextseq P2, while the ATAC library was sequenced on an Illumina NovaSeq S1.
Bulk sequencing of neuroimmune cells
Brain immune cells (Cd45+) were isolated from five 60-week-old mice per genotype as for single-cell library preparation but sorted directly into 500μL of Buffer RLT (Qiagen). Each brain was sorted into a separate tube rather than bulking genotypes together. RNA was isolated using the RNeasy Micro Kit (Qiagen) and used as input for library preparation using the SMART-Seq v4 Low Input workflow (Takara Bio). Samples were sequenced on an Illumina NovaSeq SP. The resulting FASTQs were processed with Salmon98 to generate per-sample counts matrices, followed by in silico decomposition against the atlas using Bisque33.
Paraffin embedded sections of human brains
Human brain sections of postmortem age-matched Alzheimer’s disease APOE3/APOE3 carriers and APOE4/APOE4 carriers were obtained from the Carroll A. Campbell, Jr. Neuropathology Laboratory at the Medical University of South Carolina, which is part of the South Carolina Alzheimer’s Disease Research Center. The experiments involving these sections were reviewed and approved by the IRB committee of the Medical University of South Carolina (eIRB Pro0012869). This study met the “Not Human Research” criteria defined by the Code of Federal Regulations (45CFR46).
APOE genotyping
APOE alleles were genotyped as previously described99. In short, genomic DNA was extracted from 10–20g pieces of brain tissue using the E.Z. 96 Tissue DNA Kit (Omega Bio-Tek). The APOE locus was amplified using the forward primer 5’-ACAGAATTCGCCCCGGCCTGGTACAC-3’ and the reverse primer 5’-TAAGCTTGGCACGGCTGTCCAAGGA-3’ with the following thermocycling settings: 94°C 3min, (94°C 30s → 60°C 30s → 72°C 1min) × 35 cycles, 72°C 2min, 12°C hold. Amplicons were digested by addition of 0.165μL each of AflII (New England Biolabs) and HaeII (New England Biolabs) and incubation for 120min at 37°C. The resulting digests were run on a 4% agarose gel to differentiate between the characteristic banding patterns.
Xenium library preparation
Sections were prepared from FFPE blocks of cortical tissue from human AD donors, acquired from the Carroll A. Campbell, Jr. Neuropathology Laboratory, which is part of the South Carolina Alzheimer’s Disease Research Center. Sample preparation and imaging was performed as per manufacturer recommendations using the Human Brain Gene Expression Panel (10X Genomics).
Methoxy-X04 staining
After the Xenium workflow, slides were subjected to the manufacturer’s quencher removal protocol used for post-Xenium H&E staining. After quencher removal, slides were stained for 10min in 30μM methoxy-X04 (Tocris Bioscience) solution in 40% EtOH, adjusted to pH 10 with NaOH. Slides were dipped briefly in water 5× before being differentiated for 2min in 0.2% NaOH in 80% EtOH. Sections were then stained with 1:10,000 STYOX Green (ThermoFischer) in HBSS for 10min to mark nuclei. Slides were washed 3× in water before being imaged in 405nm and 488nm channels.
Aβ uptake experiment
Single-cell suspensions were generated from the hippocampal and cortical regions of the brains of 60-week-old mice as previously described. Aβ 1–42, HiLexa™ Fluor 488-labeled (Anaspec #AS-65627) was used for Aβ oligomer preparation (denoted AβO-488). Aβ oligomers were prepared according to Ledo et al., 2020100. Cells were treated with 1μM of AβO-488 per 15 min at RT before being washed and sorted on a FACSAria-II and gated for singlet DAPI− Cd45+ cells. Within this population, cells were sorted into either AβO-488-hi or AβO-488-lo pools depending on whether they were in the top or bottom 50% of AF488 signal. Sorted cells were spun down at 300rcf for 7min at 4°C, resuspended in 100μL FACS buffer containing 0.5μg of TotalSeq-C mouse hashing antibody, and incubated on ice for 15 min. Cells were then washed twice more in FACS buffer before counting, pooling at equal numbers, and resuspending at ~2000 cells/μL for single-cell library preparation with superloading using 10X Genomics 5’v2 chemistry per manufacturer instructions. Three mice were pooled together per genotype for each library.
Library analysis
Counts matrices were produced using Cell Ranger version 6.1.1 with alignment against mm10 as a reference genome. For multiplexed samples, hashed demultiplexing was performed using Cell Ranger multi, with read R2 and pattern 5PNNNNNNNNNN(BC). Hashing sequences used for each sample are provided in Supplemental Table 1.
All code used for subsequent bioinformatic analysis is provided; briefly, data from single-cell libraries was loaded into Seurat101, filtered by QC metrics such as fraction of reads with mitochondrial origin, number of reads, and number of unique features using MiQC28, and integrated by Seurat rPCA integration to generate a single dataset encompassing cells from 10-week, 20-week, and 96-week samples (E2_10wk: 3,494 cells; E3_10wk: 3,523 cells; E4_10wk: 3,344 cells; E2_20wk: 2,997 cells; E3_20wk: 3,685 cells; E4_20wk: 4,634 cells; E3_96wk: 6,745 cells; E4_96wk: 2,446 cells). This structure was clustered and manually annotated to arrive at the final, analyzed data structure. Data from the aducanumab and Aβ libraries were analyzed and processed using the same method as was used for the atlas.
Data from the multiome library was loaded into Signac50 and filtered manually by QC metrics such as fraction of reads with mitochondrial origin, number of GEX reads, number of ATAC reads, number of unique features, TSS enrichment, and nucleosome signal. Peaks were called using MACS2102. The resulting GEX and peak information were used to generate a joint UMAP representation via Seurat’s weighted nearest neighbor approach. After clustering and manual annotation, peaks were re-called within each cluster to identify rare, cluster-specific peaks to arrive at the final, analyzed data structure.
For all downstream analysis, please refer to the accompanying code documentation.
Spatial transcriptome analysis
Preprocessing and cell segmentation of Xenium data was performed by the onboard pipelines on the Xenium machine. The resulting counts matrices were loaded into Squidpy68, log-normalized, and filtered to cells with at least 5 transcripts and to genes with expression in at least 5 cells. As all libraries were generated from the same source and on the same machine run, data were merged without computational integration. This data was clustered and manually annotated to arrive at a final data structure. Markers for each cluster were identified by filtering the microglial atlas to only the genes probed by the utilized Xenium panel and using conventional differential gene analysis methods such as the FindMarkers function to identify putative markers of each population.
Single-cell density plots
Density plots were generated by retrieving the raw UMAP cell embeddings from the Seurat structure. The ggplot::stat_density_2d function was then used to calculate the two-dimensional kernel density estimated with an axis-aligned bivariate normal kernel evaluated on a square grid.
RNA velocity analysis
Raw reads from the single-cell library were realigned against the mouse transcriptome in splice-aware fashion using the kallisto bustools kb-python workflow30. Spliced and unspliced matrices were then imported into Python alongside barcodes, UMAP coordinates, and cluster information previously calculated as described above. Matrices were filtered to remove empty droplets and merged with UMAP and cluster data before being processed with scVelo103 in dynamical modeling mode. The top 100 genes were corrected for differential kinetics before the calculation of velocity for each individual cell.
The velocity calculated by scVelo was then used as an input for CellRank31 to construct a custom kernel. Alongside the velocity kernel, a connectivity kernel based on k-nearest neighbors, a pseudotime kernel, and a kernel built from CytoTRACE32 were included at a ratio of 2%, 90%, 4%, and 4%, respectively. GPCCA was used to estimate flow through the kernel via Schur decomposition with the Krylov method on 10 components, with five terminal macrostates identified. This kernel and estimation were used for downstream analysis.
Ligand-receptor analysis
For the microglial atlas and the multiome dataset, CellPhoneDB40 was used to estimate ligand-receptor interactions. Raw counts were normalized to counts per 10,000 per cell and gene names were converted to human orthologs using the MGI human-mouse ortholog dataset. CellPhoneDB was run in statistical_analysis mode against the CellPhoneDBv3 database using default parameters. Statistical significance was evaluated by CellPhoneDB’s internal bootstrapping method and only significant interactions were used for downstream analysis.
For the aducanumab treatment dataset, CellChat71 was used to estimate ligand-receptor interactions. For each sample in the integrated dataset, a CellChat object was generated, overexpressed genes and interactions were identified, and communication probabilities and pathways were computed. These CellChat objects were then lifted onto the same cluster labels using the liftCellChat method before being merged for later analysis. All interactions were estimated from raw counts against the CellChatDB.mouse object with default parameters.
Transcription factor program analysis
SCENIC34 and SCENIC+53 were used to perform transcription factor regulatory program estimation on single-cell and multiome data, respectively. For SCENIC, GRNBoost2 was used to identify coexpressed modules from the raw counts data. RCisTarget was then used to identify regulons before AUCell was used to estimate the activity of each regulon at single-cell resolution. The output matrix of regulons by cells was used for downstream plotting and analysis. Differential module analysis was performed by importing the regulon-by-cell matrix as a Seurat assay and performing conventional differential expression analysis via Wilcoxon rank sum test.
For SCENIC+, raw counts were imported along with cluster annotations. RNA counts were log-normalized, scaled, and filtered to highly variable genes. Raw ATAC reads were imported and peaks were called normalized to the size of each chromosome using MACS2 before being filtered using the Aerts Lab mm10 blacklist. Consensus peaks were converted to a bed file for downstream analysis. Processed RNA and ATAC data were subjected to QC controlling for fragment count, FRIP, TSS enrichment, and duplication rate to filter to high-quality cells. CisTopic was used to perform latent Dirichlet allocation topic modeling before PyCisTarget was used to identify enriched motifs and differentially accessible regions. These results were used as inputs to the SCENIC+ algorithm against a list of all known mouse transcription factors from the HUST database. Estimated gene regulatory networks were used for downstream analysis. For in silico perturbation modeling, the plot_perturbation_effect_in_embedding function was used to re-estimate transcriptional profiles after setting the expression of a given transcription factor to 0.
Multiome topic modeling
The MIRA suite52 was used to perform topic modeling and latent Dirichlet allocation on multiome RNA and ATAC data. Raw RNA and ATAC counts were filtered to genes with expression at least 15 cells and normalized to 1000 total counts per cell. Genes were separated into highly variable genes based on dispersion for training of each topic model. These models were then used to generate a joint representation of RNA and ATAC features with a box-cox transformation strength of 0.33 selected by examining the quality of the resulting UMAPs, per MIRA documentation recommendations. Clusters were renamed to match clusters from the Seurat-generated joint representation before per-cell topic scores for all RNA and ATAC topics were exported and used for downstream analysis.
Bulk sequencing decomposition
Bisque33 was used to decompose bulk RNA sequencing counts into estimates of population frequencies. After reads were aligned with Salmon, counts were merged into a single table for later analysis. Raw counts from the integrated single-cell atlas were used as a reference for Bisque decomposition via the SeuratToExpressionSet function. The bulk table was then decomposed with no prespecified markers. The resulting proportion estimates were used for later plotting and analysis.
Metabolic state estimation
Compass49 was used to estimate the flux through each metabolic reaction in Recon2 in the atlas and multiome datasets. In order to decrease computational demands and to reduce the impact of signal sparsity, fifty cells for the atlas or ten cells for the multiome dataset from each cluster were randomly pseudobulked together. Raw counts were then exported and used as input to Compass on all Mus musculus reactions with default parameters. The IBM ILOG CPLEX Optimization Studio was used with an academic license to solve flux balance analysis.
Quantification and Statistical Analysis
R version ≥4.1 and Python version ≥3.7 were used for data analysis. FlowJo software (Tree Star) was used for analysis of flow cytometry data. No statistical methods were used to predetermine sample sizes; sample sizes were determined according to data reported in previous publications. Animals in the same litter were randomly assigned to different experimental groups and blinded to experimenters.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TruStain FcX | BioLegend | Cat#101319 |
| Alexa Fluor® 700 anti-mouse CD45 Antibody | BioLegend | Cat#103127 |
| TotalSeq™-C0302 anti-mouse Hashtag 2 Antibody | BioLegend | Cat#155863 |
| TotalSeq™-C0303 anti-mouse Hashtag 3 Antibody | BioLegend | Cat#155865 |
| TotalSeq™-C0304 anti-mouse Hashtag 4 Antibody | BioLegend | Cat#155867 |
| TotalSeq™-C0305 anti-mouse Hashtag 5 Antibody | BioLegend | Cat#155869 |
| TotalSeq™-C0306 anti-mouse Hashtag 6 Antibody | BioLegend | Cat#155871 |
| TotalSeq™-C0308 anti-mouse Hashtag 8 Antibody | BioLegend | Cat#155875 |
| Biological Samples | ||
| Brain sections from postmortem human AD donors | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase D | Roche | Cat#11088858001 |
| Percoll | Sigma | Cat#P1644-100ML |
| DAPI | Sigma | Cat#D8417-1MG |
| 40um Strainer | Flowmi | Cat#BAH136800040-50EA |
| Aducanumab | Cardinal Health | Cat#64406-0101-01 |
| Human IgG Isotype Control | Invitrogen | Cat#02-7102 |
| Sigma Protector RNase Inhibitor | Sigma | Cat#3335402001 |
| Buffer RLT | QIAGEN | Cat#79216 |
| AflII | New England Biolabs | Cat#R0520S |
| HaeII | New England Biolabs | Cat#R0107S |
| Human Brain Gene Expression Panel | 10X Genomics | Cat#1000599 |
| Methoxy-X04 | Tocris Bioscience | Cat#4920 |
| SYTOX Green | ThermoFischer | Cat#S7020 |
| Aβ 1-42, HiLexa™ Fluor 488-labeled | Anaspec | Cat#AS-65627 |
| Critical Commercial Assays | ||
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| SMART-Seq v4 Low Input Kit | Takara Bio | Cat#634894 |
| E.Z. 96 Tissue DNA Kit | Omega Bio-Tek | Cat#D1196-00 |
| Deposited Data | ||
| Raw data, Cell Ranger outputs, and processed Seurat and Signac structures for atlas and multiome dataset | This paper | GSE225503 |
| Raw data, Cell Ranger outputs, and processed Seurat and structures for aducanumab experiments | This paper | GSE239975 |
| Raw data, Cell Ranger outputs, and processed Seurat and structures for Aβ uptake experiments | This paper | GSE239974 |
| Raw data and counts matrices from bulk sequencing of aged AD*APOE mice | This paper | GSE239977 |
| Xenium raw data, processed Squidpy structure, and full-slide scans from methoxy-X04 imaging | This paper | 10.5281/zenodo.8206638 |
| Data from Blanchard et al.57 | Synapse | syn38120890 |
| Data from Mathys et al.58 | Synapse | syn18485175 |
| Data from Morabito et al.59 | Synapse | syn22079621 |
| Data from Lau et al.60 | GEO | GSE157827 |
| Data from Leng et al.61 | Synapse | syn21788402 |
| Data from Grubman et al.62 | GEO | GSE138852 |
| Data from Gabitto et al.63 | brain-map.org | UMSVXTDIAZTAFKGE43T |
| Data from Prater et al.66 | Synapse | syn51272688 |
| ROSMAP DLPFC-1 (Data from Green et al.)64 | Synapse | syn16780177 |
| ROSMAP DLPFC-2 (Data from Cain et al.)65 | Synapse | syn31512863 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Jackson Laboratory | Cat#000664 |
| 5×FAD | Jackson Laboratory | Cat#34840 |
| B6.129P2-Apoetm1(APOE*2)Mae N9 | Taconic Biosciences | Cat#1547 |
| B6.129P2-Apoetm2(APOE*3)Mae N8 | Taconic Biosciences | Cat#1548 |
| B6.129P2-Apoetm3(APOE*4)Mae N8 | Taconic Biosciences | Cat#1549 |
| Oligonucleotides | ||
| APOE F: ACAGAATTCGCCCCGGCCTGGTACAC | IDT | N/A |
| APOE R: TAAGCTTGGCACGGCTGTCCAAGGA | IDT | N/A |
| Software and Algorithms | ||
| Analysis scripts | This paper | https://github.com/alonmillet/apoe-ad-age-atlas |
| R 4.1 | R Foundation for Statistical Computing | RRID:SCR_001905 |
| Python 3.7 | Python Software Foundation | RRID:SCR_008394 |
| FlowJo | Tree Star | RRID:SCR_008520 |
| ILOG CPLEX Optimization Studio | IBM | N/A |
| CellRanger | 10X Genomics | RRID:SCR_023221 |
| Seurat | Hao et al.101 | RRID:SCR_007322 |
| miQC | Hippen et al.28 | RRID:SCR_022697 |
| MACS2 | Zhang et al.102 | RRID:SCR_013291 |
| Squidpy | Palla et al.68 | N/A |
| kb-python | Melsted et al.30 | RRID:SCR_018213 |
| scVelo | Bergen et al.103 | RRID:SCR_018168 |
| CellRank | Lange et al.31 | RRID:SCR_022827 |
| CytoTRACE | Gulati et al.32 | RRID:SCR_022828 |
| CellPhoneDB | Efremova et al.40 | RRID:SCR_017054 |
| CellChat | Jin et al.71 | RRID:SCR_021946 |
| SCENIC | Aibar et al.34 | RRID:SCR_017247 |
| SCENIC+ | González-Blas et al.53 | N/A |
| MIRA | Lynch et al.52 | N/A |
| Bisque | Jew et al.33 | N/A |
| Salmon | Patro et al.98 | RRID:SCR_017036 |
| Compass | Wagner et al.49 | N/A |
Acknowledgments
This work was supported in part by R35CA274446 and the Reem Kayden Award. A.M. was supported by NIH T32 GM132083. We thank the Rockefeller Transgenic and Reproductive Core for assistance with IVF, the Rockefeller Genomics Core for assistance with sequencing, the Rockefeller Flow Cytometry Core for assistance with FACS, the Weill Cornell Epigenomics Core for their assistance with multiome sequencing, the Memorial Sloan Kettering Single Cell Analytics Innovation Lab for assistance with Xenium, the Memorial Sloan Kettering Molecular Cytology Core for assistance with methoxy-X04 imaging, the Rockefeller University veterinary technician team for assistance with mouse work, and Dr. Eric Hamlett for his assistance with acquisition and preparation of human brain sections. We thank the Carroll A. Campbell, Jr. Neuropathology Laboratory at the Medical University of South Carolina for providing human brain sections. We thank Alexandra Pinzaru and King Faisal Yambire for their invaluable comments during manuscript preparation. The graphical abstract and Figures 1A, 5A, and S1B were prepared using Biorender.com.
Footnotes
Declaration of Interests
S.F.T. is a cofounder, shareholder and member of the scientific advisory board of Inspirna.
References
- 1.Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurology 14, 388–405. 10.1016/s1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long JM, and Holtzman DM (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J Cell Biol 217, 459–472. 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2018). Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6. 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (New York, N.Y.) 330, 841–845. 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SEW, Pollard JW, et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (New York, N.Y.) 336, 86–90. 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 7.St-Pierre M-K, VanderZwaag J, Loewen S, and Tremblay M-È (2022). All roads lead to heterogeneity: The complex involvement of astrocytes and microglia in the pathogenesis of Alzheimer’s disease. Front Cell Neurosci 16, 932572. 10.3389/fncel.2022.932572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paolicelli RC, Sierra A, Stevens B, Tremblay M-E, Aguzzi A, Ajami B, Amit I, Audinat E, Bechmann I, Bennett M, et al. (2022). Microglia states and nomenclature: A field at its crossroads. Neuron 110, 3458–3483. 10.1016/j.neuron.2022.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, and Amit I (2018). Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081. 10.1016/j.cell.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Heneka MT, Kummer MP, Stutz A, and Delekate A (2013). NLRP3 is activated in Alzheimer/’s disease and contributes to pathology in APP/PS1 mice. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ennerfelt H, Frost EL, Shapiro DA, Holliday C, Zengeler KE, Voithofer G, Bolte AC, Lammert CR, Kulas JA, Ulland TK, et al. (2022). SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 185, 4135–4152.e22. 10.1016/j.cell.2022.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S, Sudan R, Peng V, Zhou Y, Du S, Yuede CM, Lei T, Hou J, Cai Z, Cella M, et al. (2022). TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 185, 4153–4169.e19. 10.1016/j.cell.2022.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sierra A, Abiega O, Shahraz A, and Neumann H (2013). Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 7, 6. 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khalil YA, Rabès J-P, Boileau C, and Varret M (2021). APOE gene variants in primary dyslipidemia. Atherosclerosis 328, 11–22. 10.1016/j.atherosclerosis.2021.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Liu C-C, Liu C-C, Kanekiyo T, Xu H, and Bu G (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9, 106–118. 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, Kurth I, Andreu-Agullo C, Derbyshire ML, Posada J, et al. (2018). LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 172, 825–840.e18. 10.1016/j.cell.2017.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostendorf BN, Bilanovic J, Adaku N, Tafreshian KN, Tavora B, Vaughan RD, and Tavazoie SF (2020). Common germline variants of the human APOE gene modulate melanoma progression and survival. Nat Med 26, 1048–1053. 10.1038/s41591-020-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostendorf BN, Patel MA, Bilanovic J, Hoffmann H-H, Carrasco SE, Rice CM, and Tavazoie SF (2022). Common human genetic variants of APOE impact murine COVID-19 mortality. Nature 611, 346–351. 10.1038/s41586-022-05344-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Initiative ADN, Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roussarie J-P, Yao V, Rodriguez-Rodriguez P, Oughtred R, Rust J, Plautz Z, Kasturia S, Albornoz C, Wang W, Schmidt EF, et al. (2020). Selective Neuronal Vulnerability in Alzheimer’s Disease: A Network-Based Analysis. Neuron. 10.1016/j.neuron.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzioras M, McGeachan RI, Durrant CS, and Spires-Jones TL (2023). Synaptic degeneration in Alzheimer disease. Nat Rev Neurol 19, 19–38. 10.1038/s41582-022-00749-z. [DOI] [PubMed] [Google Scholar]
- 23.Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, and Holtzman DM (2011). Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 14, 750–756. 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Lubatti G, Stock M, Iturbide A, Segura MLRT, Tyser R, Theis FJ, Srinivas S, Torres-Padilla M-E, and Scialdone A (2022). CIARA: a cluster-independent algorithm for the identification of markers of rare cell types from single-cell RNA seq data. Biorxiv, 2022.08.01.501965. 10.1101/2022.08.01.501965. [DOI] [Google Scholar]
- 27.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hippen AA, Falco MM, Weber LM, Erkan EP, Zhang K, Doherty JA, Vähärautio A, Greene CS, and Hicks SC (2021). miQC: An adaptive probabilistic framework for quality control of single-cell RNA-sequencing data. Plos Comput Biol 17, e1009290. 10.1371/journal.pcbi.1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onuska KM (2020). The Dual Role of Microglia in the Progression of Alzheimer’s Disease. J Neurosci 40, 1608–1610. 10.1523/jneurosci.2594-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, Min K.H. (Joseph), Beltrame E. da V., Hjörleifsson KE, Gehring J, and Pachter L (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat Biotechnol 39, 813–818. 10.1038/s41587-021-00870-2. [DOI] [PubMed] [Google Scholar]
- 31.Lange M, Bergen V, Klein M, Setty M, Reuter B, Bakhti M, Lickert H, Ansari M, Schniering J, Schiller HB, et al. (2022). CellRank for directed single-cell fate mapping. Nat Methods 19, 159–170. 10.1038/s41592-021-01346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411. 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jew B, Alvarez M, Rahmani E, Miao Z, Ko A, Garske KM, Sul JH, Pietiläinen KH, Pajukanta P, and Halperin E (2020). Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun 11, 1971. 10.1038/s41467-020-15816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086. 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 36.Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, Rossello FJ, Simmons R, Buckberry S, Landin DV, et al. (2021). Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun 12, 3015. 10.1038/s41467-021-23111-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brioschi S, Belk JA, Peng V, Molgora M, Rodrigues PF, Nguyen KM, Wang S, Du S, Wang W-L, Grajales-Reyes GE, et al. (2023). A Cre-deleter specific for embryo-derived brain macrophages reveals distinct features of microglia and border macrophages. Immunity. 10.1016/j.immuni.2023.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, Fan L, Khawaja RR, Liu B, Zhan L, Kodama L, Chin M, Li Y, Le D, Zhou Y, et al. (2022). Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat Commun 13, 1969. 10.1038/s41467-022-29552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Zhu B, Liu Y, Zhang X, Yin J, Li X, Jiang L, Hodges AP, Rosenthal SB, Zhou L, et al. (2020). Multi-omic comparison of Alzheimer’s variants in human ESC–derived microglia reveals convergence at APOE. J Exp Med 217, e20200474. 10.1084/jem.20200474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc 15, 1484–1506. 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 41.Maheshwari A, Janssens K, Bogie J, Haute CVD, Struys T, Lambrichts I, Baekelandt V, Stinissen P, Hendriks JJA, Slaets H, et al. (2013). Local Overexpression of Interleukin-11 in the Central Nervous System Limits Demyelination and Enhances Remyelination. Mediat Inflamm 2013, 685317. 10.1155/2013/685317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexaki VI, Fodelianaki G, Neuwirth A, Mund C, Kourgiantaki A, Ieronimaki E, Lyroni K, Troullinaki M, Fujii C, Kanczkowski W, et al. (2018). DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol Psychiatr 23, 1410–1420. 10.1038/mp.2017.167. [DOI] [PubMed] [Google Scholar]
- 43.Mecha M, Yanguas-Casás N, Feliú A, Mestre L, Carrillo-Salinas F, Azcoitia I, Yong VW, and Guaza C (2019). The endocannabinoid 2-AG enhances spontaneous remyelination by targeting microglia. Brain Behav Immun 77, 110–126. 10.1016/j.bbi.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 44.Zhang D, Hu X, Qian L, Chen S-H, Zhou H, Wilson B, Miller DS, and Hong J-S (2011). Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflamm 8, 3. 10.1186/1742-2094-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao H-M, Zhou H, Zhang F, Wilson BC, Kam W, and Hong J-S (2011). HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci Official J Soc Neurosci 31, 1081–1092. 10.1523/jneurosci.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hijioka M, Futokoro R, Ohto-Nakanishi T, Nakanishi H, Katsuki H, and Kitamura Y (2020). Microglia-released leukotriene B4 promotes neutrophil infiltration and microglial activation following intracerebral hemorrhage. Int Immunopharmacol 85, 106678. 10.1016/j.intimp.2020.106678. [DOI] [PubMed] [Google Scholar]
- 47.Spittau B, Dokalis N, and Prinz M (2020). The Role of TGFβ Signaling in Microglia Maturation and Activation. Trends Immunol 41, 836–848. 10.1016/j.it.2020.07.003. [DOI] [PubMed] [Google Scholar]
- 48.Fansler MM, Zhen G, and Mayr C (2021). Quantification of alternative 3′UTR isoforms from single cell RNA-seq data with scUTRquant. Biorxiv, 2021.11.22.469635. 10.1101/2021.11.22.469635. [DOI] [Google Scholar]
- 49.Wagner A, Wang C, Fessler J, DeTomaso D, Avila-Pacheco J, Kaminski J, Zaghouani S, Christian E, Thakore P, Schellhaass B, et al. (2021). Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 184, 4168–4185.e21. 10.1016/j.cell.2021.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R (2021). Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341. 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lynch AW, Theodoris CV, Long HW, Brown M, Liu XS, and Meyer CA (2022). MIRA: joint regulatory modeling of multimodal expression and chromatin accessibility in single cells. Nat Methods 19, 1097–1108. 10.1038/s41592-022-01595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.González-Blas CB, Winter SD, Hulselmans G, Hecker N, Matetovici I, Christiaens V, Poovathingal S, Wouters J, Aibar S, and Aerts S (2023). SCENIC+: single-cell multiomic inference of enhancers and gene regulatory networks. Nat. Methods, 1–13. 10.1038/s41592-023-01938-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, Zhang X, Zhang Y, Qu C, Zhou X, and Zhang S (2020). Histamine Induces Microglia Activation and the Release of Proinflammatory Mediators in Rat Brain Via H1R or H4R. J Neuroimmune Pharm 15, 280–291. 10.1007/s11481-019-09887-6. [DOI] [PubMed] [Google Scholar]
- 55.Ferreira R, Santos T, Gonçalves J, Baltazar G, Ferreira L, Agasse F, and Bernardino L (2012). Histamine modulates microglia function. J Neuroinflamm 9, 90. 10.1186/1742-2094-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun 11, 6129. 10.1038/s41467-020-19737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blanchard JW, Akay LA, Davila-Velderrain J, Maydell D. von, Mathys H, Davidson SM, Effenberger A, Chen C-Y, Maner-Smith K, Hajjar I, et al. (2022). APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779. 10.1038/s41586-022-05439-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morabito S, Miyoshi E, Michael N, Shahin S, Martini AC, Head E, Silva J, Leavy K, Perez-Rosendahl M, and Swarup V (2021). Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat Genet 53, 1143–1155. 10.1038/s41588-021-00894-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lau S-F, Cao H, Fu AKY, and Ip NY (2020). Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. P Natl Acad Sci Usa 117, 25800–25809. 10.1073/pnas.2008762117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci 24, 276–287. 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22, 2087–2097. 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 63.Gabitto MI, Travaglini KJ, Rachleff VM, Kaplan ES, Long B, Ariza J, Ding Y, Mahoney JT, Dee N, Goldy J, et al. (2023). Integrated multimodal cell atlas of Alzheimer’s disease. bioRxiv, 2023.05.08.539485. 10.1101/2023.05.08.539485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Green GS, Fujita M, Yang H-S, Taga M, McCabe C, Cain A, White CC, Schmidtner AK, Zeng L, Wang Y, et al. (2023). Cellular dynamics across aged human brains uncover a multicellular cascade leading to Alzheimer’s disease. bioRxiv, 2023.03.07.531493. 10.1101/2023.03.07.531493. [DOI] [Google Scholar]
- 65.Cain A, Taga M, McCabe C, Green GS, Hekselman I, White CC, Lee DI, Gaur P, Rozenblatt-Rosen O, Zhang F, et al. (2023). Multicellular communities are perturbed in the aging human brain and Alzheimer’s disease. Nat. Neurosci 26, 1267–1280. 10.1038/s41593-023-01356-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prater KE, Green KJ, Mamde S, Sun W, Cochoit A, Smith CL, Chiou KL, Heath L, Rose SE, Wiley J, et al. (2023). Human microglia show unique transcriptional changes in Alzheimer’s disease. Nat. Aging 3, 894–907. 10.1038/s43587-023-00424-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Consortium T.T.M., Almanzar N, Antony J, Baghel AS, Bakerman I, Bansal I, Barres BA, Beachy PA, Berdnik D, Bilen B, et al. (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. 10.1038/s41586-020-2496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palla G, Spitzer H, Klein M, Fischer D, Schaar AC, Kuemmerle LB, Rybakov S, Ibarra IL, Holmberg O, Virshup I, et al. (2022). Squidpy: a scalable framework for spatial omics analysis. Nat. Methods 19, 171–178. 10.1038/s41592-021-01358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56. 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 70.Haeberlein SB, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, Dent G, Hansson O, Harrison K, Hehn C. von, et al. (2022). Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis 9, 197–210. 10.14283/jpad.2022.30. [DOI] [PubMed] [Google Scholar]
- 71.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 12, 1088. 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Owlett LD, Karaahmet B, Le L, Belcher EK, Dionisio-Santos D, Olschowka JA, Elliott MR, and O’Banion MK (2022). Gas6 induces inflammation and reduces plaque burden but worsens behavior in a sex-dependent manner in the APP/PS1 model of Alzheimer’s disease. J. Neuroinflammation 19, 38. 10.1186/s12974-022-02397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, et al. (2019). CD22 blockade restores homeostatic microglial phagocytosis in aging brains. Nature 568, 187–192. 10.1038/s41586-019-1088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matyash M, Zabiegalov O, Wendt S, Matyash V, and Kettenmann H (2017). The adenosine generating enzymes CD39/CD73 control microglial processes ramification in the mouse brain. PLoS ONE 12, e0175012. 10.1371/journal.pone.0175012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, et al. (2014). Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 82, 380–397. 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, et al. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mesquita SD, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, Contarino C, Onengut-Gumuscu S, Farber E, Raper D, et al. (2018). Functional aspects of meningeal lymphatics in aging and Alzheimer’s disease. Nature 560, 185–191. 10.1038/s41586-018-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schafflick D, Wolbert J, Heming M, Thomas C, Hartlehnert M, Börsch A-L, Ricci A, Martín-Salamanca S, Li X, Lu I-N, et al. (2021). Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat. Neurosci 24, 1225–1234. 10.1038/s41593-021-00880-y. [DOI] [PubMed] [Google Scholar]
- 79.Ma R-Y, Black A, and Qian B-Z (2022). Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol 43, 546–563. 10.1016/j.it.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 80.Marsh SE, Walker AJ, Kamath T, Dissing-Olesen L, Hammond TR, Soysa T.Y. de, Young AMH, Murphy S, Abdulraouf A, Nadaf N, et al. (2022). Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat Neurosci 25, 306–316. 10.1038/s41593-022-01022-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hu Y, Fryatt GL, Ghorbani M, Obst J, Menassa DA, Martin-Estebane M, Muntslag TAO, Olmos-Alonso A, Guerrero-Carrasco M, Thomas D, et al. (2021). Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Reports 35, 109228. 10.1016/j.celrep.2021.109228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kummer MP, Ising C, Kummer C, Sarlus H, Griep A, Vieira‐Saecker A, Schwartz S, Halle A, Brückner M, Händler K, et al. (2021). Microglial PD‐1 stimulation by astrocytic PD‐L1 suppresses neuroinflammation and Alzheimer’s disease pathology. Embo J 40, e108662. 10.15252/embj.2021108662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 10, 3758. 10.1038/s41467-019-11674-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sando SB, Melquist S, Cannon A, Hutton ML, Sletvold O, Saltvedt I, White LR, Lydersen S, and Aasly JO (2008). APOE ε4 lowers age at onset and is a high risk factor for Alzheimer’s disease; A case control study from central Norway. Bmc Neurol 8, 9. 10.1186/1471-2377-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seshadri S, Wolf PA, Beiser A, Au R, McNulty K, White R, and D’Agostino RB (1997). Lifetime risk of dementia and Alzheimer’s disease: The impact of mortality on risk estimates in the Framingham Study. Neurology 49, 1498–1504. 10.1212/wnl.49.6.1498. [DOI] [PubMed] [Google Scholar]
- 86.Coales I, Tsartsalis S, Fancy N, Weinert M, Clode D, Owen D, and Matthews PM (2022). Alzheimer’s disease-related transcriptional sex differences in myeloid cells. J. Neuroinflammation 19, 247. 10.1186/s12974-022-02604-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klein SL, and Flanagan KL (2016). Sex differences in immune responses. Nat. Rev. Immunol 16, 626–638. 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 88.Xiong J, Kang SS, Wang Z, Liu X, Kuo T-C, Korkmaz F, Padilla A, Miyashita S, Chan P, Zhang Z, et al. (2022). FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 603, 470–476. 10.1038/s41586-022-04463-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eyting M, Xie M, Heß S, Heß S, and Geldsetzer P (2023). Causal evidence that herpes zoster vaccination prevents a proportion of dementia cases. medRxiv, 2023.05.23.23290253. 10.1101/2023.05.23.23290253. [DOI] [Google Scholar]
- 90.Neu SC, Pa J, Kukull W, Beekly D, Kuzma A, Gangadharan P, Wang L-S, Romero K, Arneric SP, Redolfi A, et al. (2017). Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-analysis. JAMA Neurol. 74, 1178–1189. 10.1001/jamaneurol.2017.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 92.Leach DR, Krummel MF, and Allison JP (1996). Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 271, 1734–1736. 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 93.Ledo JH, Zhang R, Mesin L, Mourão-Sá D, Azevedo EP, Troyanskaya OG, Bustos V, and Greengard P (2020). Lack of a site-specific phosphorylation of Presenilin 1 disrupts microglial gene networks and progenitors during development. Plos One 15, e0237773. 10.1371/journal.pone.0237773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mass E, Jacome-Galarza CE, Blank T, Lazarov T, Durham BH, Ozkaya N, Pastore A, Schwabenland M, Chung YR, Rosenblum MK, et al. (2017). A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 549, 389–393. 10.1038/nature23672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, and Gan W-B (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Garré JM, Silva HM, Lafaille JJ, and Yang G (2017). CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-α. Nat Med 23, 714–722. 10.1038/nm.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224. 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sadeghi HM, Sabzghabaee AM, Mousavian Z, Saadatnia M, Shirani S, and Moazen F (2011). Polymorphism of Apo lipoprotein E gene and the risk of multiple sclerosis. J. Res. Méd. Sci. : Off. J. Isfahan Univ. Méd. Sci 16, 1519–1524. [PMC free article] [PubMed] [Google Scholar]
- 100.Ledo JH, Liebmann T, Zhang R, Chang JC, Azevedo EP, Wong E, Silva HM, Troyanskaya OG, Bustos V, and Greengard P (2020). Presenilin 1 phosphorylation regulates amyloid-β degradation by microglia. Mol Psychiatr, 1–16. 10.1038/s41380-020-0856-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Collectively, all new sequencing data generated in this study are available on GEO under SuperSeries GSE239999. All raw data, Cell Ranger outputs, and processed Seurat and Signac structures for the atlas and multiome dataset are available on GEO under accession number GSE225503. All raw data, Cell Ranger outputs, and processed Seurat structures for the aducanumab treatment experiments are available on GEO under accession number GSE239975. All raw data, Cell Ranger outputs, and processed Seurat structures for the Aβ uptake experiments are available on GEO under accession number GSE239974. All raw data and counts matrices from bulk sequencing of aged AD*APOE mice are available on GEO under accession number GSE239977. All Xenium raw data, the processed Squidpy structure, and full-slide scans after post-Xenium methoxy-X04 staining are available on Zenodo at 10.5281/zenodo.8206638. The ROSMAP DLPFC-1 and DLPFC-2 data used in this analysis are available at Synapse under accession codes syn16780177 and syn31512863.
All R and Python code necessary to reproduce the analysis and figures in this manuscript has been annotated and uploaded to Github at https://github.com/alonmillet/apoe-ad-age-atlas.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.