

Introduction
Incidental radiological abnormalities of the adrenal glands can be identified in between 1 in 10 and 1 in 20 CT images in adults1. The current challenge is to institute cost-efficient clinical pathways, to avoid excessive investigations in patients with benign non-functional adrenal tumours, to ensure that functional tumours are identified and treated, and to avoid missing the very rare but extremely aggressive adrenocortical carcinomas (ACCs).
The relative rarity of malignant adrenal tumours makes it very difficult to organize randomised controlled trials (RCTs) to address areas of uncertainty, so most of the guidelines published in recent years have been based on level III–V evidence. In this context, it is important to review the information from recent cohort studies to ensure that the management of contemporaneous patients is in line with up-to-date evidence.
The European Society of Endocrine Surgeons (ESES) has dedicated the 2023 symposium to discussing the management of advanced endocrine malignant tumours. This paper summarizes current knowledge and discusses areas of uncertainty related to the management of ACC, malignant phaeochromocytoma, and adrenal metastases. The emphasis is on data published in the past decade, and on changes to surgical practice since the previous similar publication from ESES in 20122 and after the publication of most recent clinical guidelines from the European Network for the Study of Adrenal Tumors (ENSAT), the European Society for Endocrinology, the North American Neuroendocrine Tumor Society (NANETS), and the European Society of Medical Oncology3–9.
Methods
A working group established by ESES reviewed the current guidelines and undertook a literature search of the PubMed database focused on several clinical questions that were used as the heading for each section of this document. Papers deemed relevant were scrutinized and additional references cross-checked if not already retrieved through the initial PubMed search.
Throughout the text, quotations from current guidelines are formatted in italic. Recent publications are quoted to strengthen the validity of each recommendation. All the new recommendations made by the working group appear in box, and are classified using the GRADE (Grading of Recommendations, Assessment, Development, and Evaluations) evidence profile10 as strong or weak, in favour or against an intervention (Table 1). Strong recommendations suggest that almost all individuals would choose that intervention. Weak recommendations imply that there is likely to be a variation in the decision that informed individuals are likely to make. Therefore, patient engagement in a shared decision-making process is essential in such instances.
Table 1.
GRADE (Grading of Recommendations, Assessment, Development, and Evaluations) levels of evidence
| Certainty | Symbol | What it means |
|---|---|---|
| Very low | ⊕○○○ | The true effect is probably markedly different from the estimated effect |
| Low | ⊕⊕○○ | The true effect might be markedly different from the estimated effect |
| Moderate | ⊕⊕⊕○ | The true effect is probably close to the estimated effect |
| High | ⊕⊕⊕⊕ | There is high confidence in that the true effect is similar to the estimated effect |
GRADE, Grading of Recommendations, Assessment, Development, and Evaluations.
The findings of this paper were discussed in the plenary session of the 2023 ESES meeting, and a vote was taken on several areas of uncertainty or without enough published data. The result of each vote is reported.
Adrenocortical cancer
ACC is an exceedingly rare tumour, with an estimated incidence of 1–2 per million population per year, and an overall 5-year survival rate of only 30 per cent. The survival rate ranges from 60 to 80 per cent for tumours confined to the adrenal gland, from 35 to 50 per cent for locally advanced disease, and much lower for patients with metastatic disease (below 30 per cent)3.
Preoperative diagnosis of adrenocortical cancer
Clinical and biological assessment
Current recommendations are that patients with suspected ACC undergo a detailed hormonal work-up to identify potential autonomous excess of glucocorticoids, sex hormones, mineralocorticoids, and adrenocortical steroid hormone precursors3.
Clinical history and examination should explore the presence of symptoms and signs of adrenal hormone excess or pressure symptoms created by large non-functional tumours. If there is compression/obstruction of the inferior vena cava (IVC) or renal vein, collateral circulation on the abdomen, leg oedema or a hydrocele can be observed (Fig. 1). Irrespective of symptoms and resting blood pressure, a phaeochromocytoma must be excluded in all patients by demonstrating normal values of urinary or blood metanephrines (MNs).
Fig. 1.
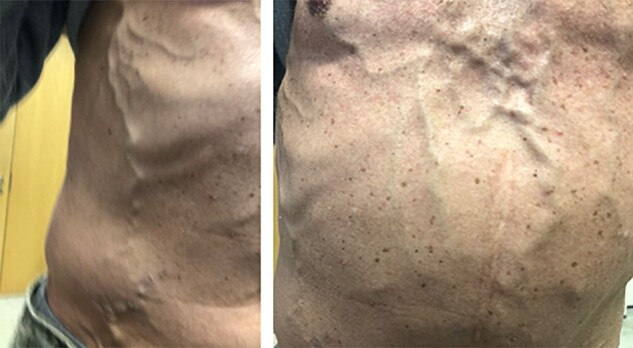
Clinical signs of inferior vena cava obstruction from a large adrenal tumour
According to local facilities, biochemical assessment follows the above guidelines. In addition, urine steroid profiling is increasingly being used in many centres. Mass spectrometry-based urinary steroid metabolite profiling allows identification of numerous steroid compounds that are produced specifically by malignant adrenal tumours. The largest study11 on this topic used data from over 2000 patients presenting with an adrenal mass, and demonstrated that a triple-test strategy was very accurate in diagnosing ACC in the presence of tumour size larger than 4 cm, suspicious radiological appearance, and abnormal urinary steroid profile.
The ESES recommendation is to include urine steroid profile in the diagnostic work-up of all patients suspected of having an ACC (⊕⊕⊕○).
Cross-sectional imaging
The criteria for suspicion of malignancy on unenhanced CT should be changed from 10 to 20 Hounsfield units (HU). In an analysis11 of over 2000 patients, there was no ACC among 1300 patients with tumours smaller than 4 cm and less than 20 HU, but there were 95 patients with ACC among 247 with tumours larger than 4 cm and over 20 HU (risk 39 per cent).
An imaging score was proposed recently based on analysis of 56 patients with histologically proven ACC and 156 with lipid-poor cortical adenomas. The score is built on differences between the groups in nine parameters: size, attenuation, thin and thick rim enhancement patterns, heterogeneity, calcification, necrosis, fat infiltration, and lymph node prominence. The score had 100 per cent sensitivity for the exclusion of ACC, 80 per cent specificity, 66 per cent positive predictive value (PPV), and 100 per cent negative predictive value (NPV,) with an area under the curve of 0.97412. The utility of this score should be confirmed in future studies.
Stage IV metastatic disease is expected in almost one-fifth of patients with ACC, most frequently involving the lung (approximately 45 per cent), liver (about 42 per cent), and, less commonly in bone13. Thoracic–abdominal–pelvic CT allows planning of the surgical approach and assessment of distant metastases. Hepatic MRI or vena cava MRI can be added if hepatic metastases or vena cava infiltration are suspected3.
The ESES recommendation is to perform thoracic–abdominal–pelvic CT in all patients with suspected ACC (⊕⊕⊕⊕). The criteria for suspicion of malignancy on unenhanced CT should be changed from 10 to 20 HU (⊕⊕⊕○).
Role of preoperative PET and new tracers in detecting metastatic disease
Routine [18F]fluorodeoxyglucose (FDG) PET–CT was not recommended by the ESE guidelines in 20183, but more recent data challenge this decision. There is a correlation between standardized uptake value (SUV) ratio, malignancy, and Weiss score that can contribute to the differentiation between benign and malignant adrenal lesions in patients presenting with a large adrenal mass14. In a prospective multicentre study15, the optimal threshold value of tumour maximum SUV (SUVmax) to liver SUVmax was over 1.5, and provided 87 per cent sensitivity, 86 per cent specificity, 57 per cent PPV, and 97 per cent NPV. Therefore [18F]FDG PET–CT complements adrenal washout CT in the evaluation of adrenal masses. This recommendation is reinforced by data from a recent meta analysis16.
There is limited experience with the use of [11C]metomidate, a new PET tracer with specificity for adrenocortical cells. This compound has a very short half-life and so cannot be made available commercially. As an alternative, imaging with metomidate-labelled with 123I (iodometomidate) can demonstrate primary and metastatic adrenocortical lesions with high specificity. Moreover, it can identify patients suitable for specific, targeted radioactive treatment17. As [11C]metomidate imaging is increasingly being used to lateralize aldosterone secretion in patients with hyperaldosteronism, its accessibility could improve and it might become a standard test for patients with suspected ACC.
The ESES recommendation is that [18F]FDG PET–CT should be considered in all patients with suspected ACC (⊕⊕⊕⊕).
Adrenal biopsy
Current recommendations do not support the use of an adrenal biopsy in the diagnostic work-up of patients with suspected ACC unless there is evidence of metastatic disease that precludes surgery and histopathologic proof is required to inform oncological management3.
For the diagnosis of ACC, biopsy is unhelpful and can have a negative impact on the outcome as it compromises the chances of subsequent R0 resection. In a study18 of 1410 patients with non-metastatic ACC registered in the National Cancer Database (NCDB), adrenal biopsy was associated with decreased overall survival in patients with T1/T2 tumours. Median overall survival was significantly shorter among 88 patients who had a biopsy than among the 742 who had a clinical diagnosis only (55 and 104 months respectively). Patients who had biopsy of T1/T2 tumours had a survival similar to that of patients with T3 tumours diagnosed clinically.
The ESES recommendation is to restrict adrenal biopsy only to patients with tumours deemed inoperable as a diagnostic test before starting palliative mitotane chemotherapy. Before any adrenal biopsy, a full biochemical work-up should exclude phaeochromocytoma (⊕⊕⊕⊕).
Age-related outcomes
The new prognostic score S-GRAS uses age 50 years as part of the risk stratification19, whereas many nomograms equate age with a parallel increase in the scores attributed. Based on an analysis20 of 3262 patients registered in the NCDB, those aged less than 55 years with ENSAT I–II disease had significantly better 5-year survival than older patients (0.6 versus 0.2). Using 60 years as a threshold between younger and older patients in a cohort of 876 patients, others21 found that a lower proportion of older patients underwent surgery, regional lymph node surgery, and chemotherapy than younger patients. Moreover, among those who underwent surgery, older patients had inferior overall survival (30 versus 68 per cent) and cancer specific survival (40 versus 73 per cent).
The ESES recommendation is that age should be taken into consideration when discussing ACC outcome (⊕⊕○○).
Controlling severe hypercortisolism pharmacologically before adrenalectomy
Severe hypercortisolism can lead to life-threatening co-morbidities such as uncontrolled hypokalaemia, hypertension, diabetes, and opportunistic infections. Medical treatment consists of steroidogenesis inhibitors or a single treatment with osilodrostat, and this can decrease the 24-h urinary free cortisol level rapidly. Medical treatment should be provided at high doses, with the possibility of a block-and-replace regimen being monitored during inpatient admission in an expert endocrinology department. This should lead to rapid improvement of co-morbidities and could mitigate perioperative risks.
Two studies reported the efficacy of a combination of steroidogenesis inhibitors. Ketoconazole and metyrapone were administered to control hypercortisolism rapidly, whereas the maximal efficacy of mitotane was achieved later. In the first study22 of 14 patients, ketoconazole and metyrapone led to a decrease in urinary free cortisol levels from 40- to 3.2-fold the upper limit of the value at the end of the first week. In the second study23 of 10 patients, urinary free cortisol levels decreased dramatically after 24–48 h in all patients, and seven patients achieved normal levels. In both studies, the overall tolerance was good. Etomidate infusion can be used for rapid control of severe hypercortisolism in patients with significant preoperative instability, biochemical disturbances or psychosis who cannot proceed immediately with surgery24. To date, no randomized study has assessed the benefits of anticortisol treatment before surgery for ACC.
The ESES recommendation is to discuss the feasibility of medical control of severe hypercortisolism within the local multidisciplinary team (MDT). Attempts to normalize cortisol levels should not create undue delay to surgical treatment (⊕⊕○○; 80 per cent of ESES members voted agree or strongly agree).
Is open adrenalectomy necessary for all patients with adrenocortical carcinoma?
Current recommendation is to perform a complete en bloc resection of all adrenal tumours suspected to be ACC including the peritumoral/periadrenal retroperitoneal fat. Eunucleation and partial adrenal resection is recommended against enucleation and partial adrenal resection for suspected ACC. If adjacent organs are suspected to be invaded, en bloc resection is recommended3.
The above recommendations remain a valid guide for those involved in ACC surgery, and they create the background for the ongoing debate about the role of laparoscopic adrenalectomy as a reasonable alternative approach to malignant adrenal tumours. Units with large-volume practice have published excellent results after laparoscopic surgery25, but such outcomes might not be transferable to surgeons or hospitals with limited experience in treating patients with ACC. Owing to the rarity of the disease and specific challenges of individual cases, an RCT is not feasible, and decisions are informed by the results of several literature reviews and meta-analyses that have provided similar conclusions (Table 2).
Table 2.
Recent studies, systematic reviews, and meta-analyses comparing open and laparoscopic/ minimally invasive adrenalectomy
| Reference | Study type |
No. of patients | Findings | Conclusion |
|---|---|---|---|---|
| 26 | Meta-analysis, 11 studies | Total 1617 MIA 472 OA 1145 |
OA had lower rate of positive resection margin than laparoscopic surgery (OR 1.52, 95% c.i. 1.10 to 2.10) OA had more favourable overall survival (OR 0.56, 0.44 to 0.72) and recurrence-free (OR 0.60, 0.42 to 0.85) rates than laparoscopic surgery at 3 years |
OA should still be considered the standard operative approach; however, LA could be regarded as an effective and safe operation for selected patients with ACC when appropriate laparoscopic expertise exists |
| 27 | Meta-analysis, 15 studies | Total 2207 | MIA had earlier recurrence and more positive surgical margins (RR 1.56) and peritoneal recurrence (RR 2.63) Overall recurrence (RR 1.07) and local recurrence (RR 1.33) were comparable Surgical approaches did not differ in overall survival. |
OA is the standard treatment, but MIA approaches could be offered for selected patients with ACC |
| 28 | Meta-analysis, 9 studies | Total 797 MIA 240 OA 557 |
There was no difference in overall recurrence rate between LA and OA (RR 1.09), whereas development of peritoneal carcinomatosis was more common after LA (RR 2.39, 95% c.i. 1.41 to 4.04) No difference could be found for time to recurrence and cancer-specific mortality |
OA should still be considered the standard surgical management of ACC LA can offer a shorter hospital stay and possibly a faster recovery MIA should be offered only to carefully selected patients to avoid jeopardizing the oncological outcome |
| 29 | Meta-analysis, 11 studies | No differences in rate of positive surgical margins, disease-free survival, and overall survival between OA and LA in localized disease More aggressive and open surgery performed in high-volume centres Higher local recurrence and distant metastasis rates, and a shorter time to recurrence seen in low-volume centres |
LA for localized ACC is safe and effective when performed by expert surgeons in high-volume centres Patients with more extensive tumours should have open surgery |
|
| 30 | Review, 14 studies | Total 2574 MIA 795 OA 1779 |
Six studies considered OA to be superior to MIA, whereas eight studies reported that MIA is as effective as OA in highly selected cases | OA remains the standard approach for management of ACC. MIA may play a role in selected patients (high-volume institutions, experienced surgeons) |
| 31 | Cohort study | MIA 364 OA 182 | MIA had similar positive surgical margins and overall survival to OA, irrespective of tumour size |
MIA, minimally invasive adrenalectomy; OA, open adrenalectomy; LA, laparoscopic adrenalectomy; ACC, adrenocortical carcinoma; RR, relative risk; OR, odds ratio.
Irrespective of the surgical approach, the oncological principles should not be breached. The aim of the operation is to ensure local control of disease by achieving negative resection margins (R0). In a study32 of 1973 patients registered in the NCDB, 1 in 10 patients had positive margins, more likely in the presence of extra-adrenal extension (HR 4.92), lymph node metastases (HR 2.64), and distant metastases (HR 1.53), and there was no significant difference in margin status between patients who had an open versus minimally invasive procedure. Several other studies33–39 reported the completeness of resection after laparoscopic and open surgical approaches, and found no clear evidence against laparoscopic surgery.
Conversion from laparoscopic to open adrenalectomy appears to have a negative impact on overall survival. Based on records of 196 patients undergoing attempted minimally invasive adrenalectomy (MIA) for ACC, one-fifth required conversion to open adrenalectomy and these patients had significantly reduced overall survival compared with those who had a successful MIA or planned open resection40. These findings reinforce the idea that there is little to be gained by attempting a laparoscopic resection unless there is high likelihood of achieving an uncompromised R0 resection and extraction of the specimen without fragmentation/rupture of the tumour.
The debate regarding whether open adrenalectomy should be offered to all patients with suspected ACC is restricted to the choice between open and laparoscopic transperitoneal resection. There are no published data on the use of retroperitoneoscopic adrenalectomy for patients with ACC because the number of surgeons confident with this procedure is relatively small, and the operation becomes increasingly challenging for tumour larger than 4–6 cm. In addition, it is likely that the role of robotic adrenalectomy for ACC surgery will become more relevant in the coming years. The most recent analysis of the NCDB39 showed that one-fifth of patients had robotic operations (128 patients versus 416 who underwent laparoscopic adrenalectomy). The intraoperative conversion rate was lower among robotic compared with laparoscopic adrenalectomies (8 versus 18 per cent), and operations that required conversion had a greater margin positivity rate, longer hospital stay, and were associated with poor overall survival. The authors concluded that, if a surgeon is not planning an open adrenalectomy for adrenal malignancy, robotic adrenalectomy might become the preferred minimally invasive approach for resectable adrenal tumours. This initial experience reported from the USA is yet to be validated by other national registries.
It is generally agreed, but difficult to prove, that choosing the operating surgeon is more important that the choice of surgical approach. This is highlighted in the conclusion of a recent review: ‘…surgeon's expertise is more important than surgical technique to determine outcome. Even a state-of-the-art surgery cannot however prevent disease recurrence that is determined mainly by specific tumour characteristics’41.
Based on the acknowledged volume–outcome correlation for adrenal surgery42,43, a national framework for improving delivery of endocrine services in England under the acronym GIRFT (Getting it Right for Time) recently recommended that patients with ACC should be treated in centres performing a minimum of 12 adrenal operations per year. Implementing such changes would benefit service delivery in most countries.
The ESES recommendation is to refer patients with large adrenal tumours with signs of local invasion for open adrenalectomy under the care of surgeons with high-volume practice (⊕⊕⊕⊕).
Patients with small tumours (less than 6 cm) can be considered for minimally invasive surgery if the surgeon is confident that a complete resection can be achieved (⊕⊕○○).
With increasing experience with robotic adrenalectomy, the choice of surgical approach for tumours larger than 6 cm would be decided based on individual expertise (⊕⊕⊕○).
Multiorgan resection for adrenocortical carcinoma and extent of surgery
Current recommendation suggests against the routine resection of the ipsilateral kidney in the absence of direct renal invasion3.
The need for ipsilateral nephrectomy in the treatment of ACC is arguable as the adrenal tumour is rarely seen to infiltrate the kidney. In many patients, the position of the tumour in relation to the renal vessels, however, makes it more likely to ensure a radical excision (Fig. 2). An analysis of 52 patients showed that the 22 who underwent a nephrectomy had larger tumours (mean(s.d.) 120(42) versus 85(38) mm), and were more likely to have a formally assessed N status, suggesting an oncological benefit of more extensive resection (R. Mihai, unpublished data). In a retrospective study44 of 41 patients with stage II ACC, the addition of nephrectomy did not modify the oncological results in terms of recurrence-free and overall survival (P = 0.3). Thus, when there are no signs of ACC local invasion, surgeons should make every effort to preserve the kidney.
Fig. 2.

Renal involvement requiring nephrectomy for en bloc resection
a Cross sectional imaging demonstrating relation of renal vessels with the tumour. b FDG-PET image demonstrating presence of tumour thrombus into the left renal vein up to its confluence into the inferior vena cava.
For left-sided tumours, splenectomy can be considered if the splenic vessels run in contact with the upper pole of a large adrenal tumour. Once a large surgical specimen has been created during en bloc resection, utmost care needs to be taken to recognize accurately the distorted anatomy as rare but life-threatening complications can occur if the superior mesenteric artery is injured.
For right-sided tumours, partial resection of the IVC can be considered if there are signs of local invasion or thrombus. Because of the rarity of ACC, there are no controlled trials addressing the benefit of such an aggressive approach. As R0 surgical resection is the only curative option, IVC resection should be discussed in a referral centre. A recent publication45 detailed the technical aspects of such operations and the support provided by cardiac surgeons. The multicentre ESES survey46 published in 2012 reported favourable outcomes in patients with IVC invasion. In a more recent retrospective study47 of 11 such patients, three had extensive invasion that required cardiac bypass for retrieval of atrial thrombus. Four of six patients with a large amount of tumour thrombus died within 48 months, suggesting very poor prognosis.
The ESES recommendation in the event of IVC invasion is that patients should be referred to a high-volume centre with a MDT before considering the tumour to be unresectable (⊕⊕⊕○).
Limitations and benefits of lymphadenectomy for adrenocortical carcinoma
Current recommendations suggest that routine locoregional lymphadenectomy should be performed with adrenalectomy for highly suspected or proven ACC. It should include (as a minimum) the periadrenal and renal hilum nodes. All suspicious or enlarged lymph nodes identified on preoperative imaging should be removed4.
Moreover, that routine locoregional lymphadenectomy should be performed with adrenalectomy for highly suspected or proven ACC. It should include (as a minimum) the periadrenal and renal hilum nodes. All suspicious or enlarged lymph nodes identified on preoperative imaging or intraoperatively should be removed3.
This topic is being discussed increasingly, but there has been minimal progress in defining the technique for lymphadenectomy during radical adrenalectomy. Therefore, the intraoperative management of regional lymph nodes remains highly variable and surgeon-dependent. The anatomical borders of lymphadenectomy for right-sided tumours would be the IVC, the upper border of the right renal vein and renal artery, and the upper pole of the kidney. The superior limit of dissection would depend on the degree of liver mobilization created during tumour dissection, but on many occasions could extend along the IVC towards the diaphragm. On the left side, the border would be the aorta, the upper border of renal vessels and the diaphragm. Despite attempts to resect all soft tissue in the area described (that is to attempt radical en bloc resection and lymphadenectomy), it remains possible that no lymph nodes will be found on histopathological examination.
Resecting lymph nodes below the renal vessels is not necessary in the absence of radiological evidence of extensive disease. Records of 897 patients who underwent margin-negative resection for ACC showed that 147 (16 per cent) had lymph nodes examined. Lymph node harvest and lymph node metastasis were associated with more advanced tumours, open operation, and resection at an academic facility48. Nodal staging provides important prognostic information because median overall survival is incrementally worse with increasing number of positive lymph nodes (88 months for patients with N0 disease, 35 months for those with 1–3 positive nodes, and 16 months for patients with more than four positive nodes)48,49.
Lymphadenectomy appeared to be associated with improved survival in an analysis of over 380 patients registered in the NCDB (HR 0.82, 95 per cent c.i. 0.67 to 0.99)49. These findings were subsequently confirmed in a meta-analysis29 that identified a shorter time to recurrence when no proper lymphadenectomy was performed, and a trend towards better recurrence-free survival and disease-specific survival after lymph node dissection.
The ESES recommendation is that, owing to the lack of definitive guidance on the extent and technique for lymphadenectomy, a prospective multicentre trial should define the strategy for enhanced lymph node yield during open adrenalectomy and should assess the additional morbidity from extensive para-aortic dissection (⊕⊕⊕○; 80 per cent of 131 ESES members voted agree or strongly agree).
In patients with suspected ACC, total adrenalectomy including periadrenal fat should be performed (⊕⊕⊕⊕).
Role of neoadjuvant chemotherapy
Borderline resectable adrenal tumour is a concept introduced by clinicians at the MD Anderson Cancer Center, USA, to describe ACCs with characteristics at presentation that argue against immediate surgery because of an unacceptable risk of morbidity or death, incomplete resection or recurrence. Of 15 patients who had borderline resectable ACCs and received neoadjuvant therapy (mitotane and etoposide or cisplatin-based chemotherapy (EDP-M)), 13 went on to have surgical resection, of whom five had a partial response, seven had stable disease, and one had progressive disease. Median disease-free survival for patients with borderline resectable ACC was 28 months, compared with 13 months for 38 patients who had upfront surgery. Five-year overall survival rates were similar at 65 and 50 per cent, respectively50.
A similar approach was followed in other centres, which confirmed that EDP-M therapy can lead to a decrease in lesion size such that the tumour subsequently becomes resectable51,52. The most recent publication53 on a large cohort of 58 patients treated using the EDP-M protocol showed that 26 of 55 underwent surgery, of whom 13 became disease-free.
The ESES recommendation is that all patients with ACC that is deemed inoperable should be referred to a regional centre that can provide expertise in multiple surgical specialties working in an established multidisciplinary environment with oncologists. Restaging of the tumour is recommended after neoadjuvant chemotherapy and the feasibility of surgery reconsidered after 3–6 months of neoadjuvant treatment (⊕⊕⊕○; 94 per cent of 112 ESES members voted agree or strongly agree).
Role of adrenal surgery in patients with adrenocortical carcinoma presenting with metastatic disease
Current recommendation for patients presenting at time of initial diagnosis with limited intra-abdominal metastases, surgical therapy is suggested if complete resection of all lesions seems feasible. In case of limited extra-abdominal lesions, adrenal tumour resection in conjunction with therapy aiming at long-term tumour control of the other lesions is suggested. In all patients, the recommendation is to start mitotane therapy as soon as clinically possible3. The routine use of adrenal surgery is recommended against in case of widespread metastatic disease at the time of first diagnosis3.
Based on the above recommendations, it is generally assumed that surgery is not beneficial for patients presenting with metastatic disease. A different decision might be reached for those with severe Cushing’s syndrome and limited metastatic burden as they are likely to benefit from the removal of the adrenal primary tumour in order to reduce the severity of hypercortisolism before instituting chemotherapy.
The decision to offer surgery to patients with metastatic non-functional tumours is generally controversial, but there is growing evidence that it is preferable to operate than to offer palliative treatment alone (Table 3). The speed of disease progression and the site/extent of metastatic deposits need to be considered. In a small study57 of 12 patients, the growth rates of metastatic ACC lesions varied with the host organ, with the volume doubling time per organ system being shorter in the liver (27 days) than the lungs (90 days) and lymph nodes (95 days).
Table 3.
Cohort studies reporting benefits of surgery in patients with metastatic adenocortical carcinoma
| Reference | No. of patients | Findings | Conclusion | |
|---|---|---|---|---|
| 54 | 1993–2014 13 institutions of US Adrenocortical Carcinoma Group |
Synchronous metastasectomy 26 (31%) Metachronous metastasectomy 58 (69%) |
Patients with synchronous disease who had R0 resections had improved survival versus those who had R1/2 resections The metachronous group had prolonged median survival after the index resection (86 versus 17 months) and metastasectomy (37 versus 17 months) |
Selected patients with metastatic ACC may benefit from metastasectomy Patients with metachronous metastasectomy have a more durable survival benefit than those undergoing synchronous metastasectomy |
| 55 | 1973–2014 SEER database |
Total 290 Primary-site surgery 118 No primary-site surgery 172 |
Primary-site surgery significantly improved both overall (HR 0.41, 95% c.i. 0.30 to 0.57; P < 0.01) and cancer-specific (HR 0.41, 0.29 to 0.57; P < 0.01) survival | Primary-site surgery in patients with metastatic ACC significantly improved overall and cancer-specific survival |
| 56 | 9 referral centres (American–Australian–Asian Adrenal Alliance) | Total 239 CRS group 128 No-CRS group 111 |
After a mean follow-up of 67 months, patients in no-CRS group had a greater risk of death than those in CRS group (HR 3.18, 2.34 to 4.32) | CRS of the primary tumour in patients with metastatic ACC is associated with prolonged survival |
ACC, adrenocortical carcinoma; SEER, Surveillance, Epidemiology, and End Results; CRS, cytoreductive surgery.
The ESES recommendation is that cytoreductive surgery is to be considered for all patients with ACC presenting with metastatic disease, in particular those with functional tumours. Such operations have to be restricted to high-volume centres where surgery can be undertaken with minimal morbidity (⊕⊕⊕○; 92 per cent of 99 ESES members voted agree or strongly agree).
Surgery for local recurrence
Current recommendation is that in patients with recurrent disease and a disease-free interval of at least 12 months, in whom a complete resection/ablation seems feasible, surgery or alternatively other local therapies are recommended3.
Local recurrence develops in up to 75 per cent of patients. Recent studies58 have suggested a benefit in terms of disease-free survival and overall survival for patients with recurrent ACC undergoing reoperation. In a retrospective multicentre study59 of 55 patients with local ACC recurrence, those who underwent reoperation had significantly better median overall survival after recurrence than non-operated patients (91 versus 15 months), especially if the initial disease-free interval was greater than 12 months. Furthermore, in a meta-analysis60 of 11 studies, 573 patients who underwent reoperation after recurrence had significatively better overall survival and survival after recurrence than the 391 who received only non-surgical treatments. Patients with multiple recurrences, shorter disease-free interval, and R1/R2 resections tended to benefit less from reoperations.
The most recent publication61 on this topic reported a vast experience of 106 patients with ACC recurrence treated at a single centre in Italy, and showed that two-thirds of the patients became disease-free and attained a second recurrence-free survival of 15 (i.q.r. 6–64) months. Margin status RX and R1, an increase in Ki-67 percentage, and recurrence in multiple organs were associated with an increased mortality risk, whereas adjuvant mitotane treatment and longer time to first recurrence were associated with reduced risk. Recurrence in multiple organs and systemic treatment of recurrence had a negative impact on survival after treatment of recurrence.
The ESES recommendation is that, in the absence of metastatic disease, surgery or an alternative other local therapy for local recurrence of ACC can be considered, especially in patients with a disease-free survival interval greater than 12 months (⊕⊕⊕○).
Surgery and local treatment for metastases from adrenocortical carcinoma
ACC metastases are present at the time of diagnosis in up to one-third of patients. In addition, about 64 per cent of those without metastases at the time of diagnosis develop recurrence (local, regional or distant) and, of these, 72 per cent have only one site of recurrence62. As systemic therapy (mitotane, chemotherapy) has limited impact, complete resection remains the only potentially curative treatment for metastatic ACC. Thus, in patients with oligometastases or recurrence after 12 months, surgical options should be discussed by expert teams. Although prospective data on this topic are limited, expert consensus recommends surgery or alternative local therapies in such patients.
Most ACC metastases are located in the lung, liver, and lymph nodes, and rarely in bone. Lung metastases occur in over 40 per cent of patients62 and can be treated with good results, either by surgery, thermoablation or radiofrequency therapy if isolated63. Liver metastases occur in over 40 per cent of patients. Surgery remains a good option, especially if it can be performed during the initial operation, but a retrospective study64 reported high recurrence (80–100 per cent) and morbidity (50 per cent) rates. In a multicentre study65, patients with a non-functioning primary tumour, small number of liver metastases, longer disease-free interval, extrahepatic disease, and R0 resection had extended survival. In multivariable analysis, only prolonged disease-free interval and non-functioning primary tumour were associated with a clear survival benefit after hepatic resection for metastatic ACC65. Alternative treatments include radiofrequency ablation (RFA), transarterial embolization (TACE) or selective internal radiation therapy (SIRT). In a series of 23 patients66, six who received TACE or SIRT experienced significantly longer overall survival than those who were not treated (32 versus 10 months). Recently, decreasing size of metastases and liver disease-free survival of up to 2 years were reported after the use of 90Y SIRT for liver metastases of ACC combined with systemic or other local treatment (RFA, surgery, chemoembolization)67–69. Bones metastases are rare, and found in only 10 per cent of patients; data are poor regarding this location and limited mostly to case reports. In a retrospective international multicentre study70 that included 156 patients with bone metastases among 1129 with ACC, most patients had mitotane associated with chemotherapy and only 23 per cent had bone surgery.
A recent single-centre experience61 of 106 patients with ACC recurrence found a single lesion in 36 per cent, multiple lesions in a single organ in 21 per cent, and metastases affecting multiple organs in 43 per cent of patients. Recurrence-free survival was significantly longer in those with a single metastasis. These findings support the use of locoregional treatments to treat disease recurrence.
The ESES recommendation is that surgical options for resection of ACC metastases in addition to systemic therapy should always be considered in a MDT discussion, and active treatment should be favoured (⊕⊕⊕○).
Risk stratification after resection of adrenocortical carcinoma
Current recommendation is to use of the Weiss system for the distinction of benign and malignant adrenocortical tumours. The pathology report of a suspected ACC should at least contain the following information: Weiss score (including the exact mitotic count), exact Ki-67 index, resection status, and pathological tumour stage (indicating invasion or not of the capsule and/or surrounding tissue and organs) and nodal status.
Standard clinical and pathological parameters
Patients at low risk of recurrence are those who have undergone R0 resection for localized disease (stage I–II ACC), and low-grade tumour (Ki-67 index below 10 per cent). Patients at very high risk of recurrence are those with at least one of the following: Ki-67 index above 30 per cent, large venous tumour thrombus, R1–R2 resection or stage IV ACC41.
Nomograms
Several nomograms have been designed based on large data sets, but the prediction made by each of the these for individual patients showed poor correlation71, and they have not been incorporated into clinical decisions-making protocols.
S-GRAS score
A combined score (S-GRAS) based on age, hormone- or tumour-related symptoms at presentation, ENSAT stage, R status, and Ki-67 index was constructed based on information on 940 patients with ACC19, and validated against a large Surveillance, Epidemiology, and End Results data set72. This score can stratify patients with different outcomes, and could guide the selection of those who might benefit from postoperative adjuvant therapy.
Inflammation-based scores
In a retrospective analysis73 of 90 patients with advanced ACC treated with mitotane (40 patients) or EDP ± mitotane (50), a pretreatment neutrophil-to-lymphocyte ratio (NLR) of at least 5 and platelet-to-lymphocyte ratio (PLR) of at least 190 predicted shorter overall survival in multivariable analysis. NLR was also associated with shorter time to progression. The findings mirror a previous report74 of a group of 57 patients who underwent ACC resection with curative intent, in whom the median NLR was 4.6 and median PLR 186, and in whom indices above the median values were strongly associated with shorter overall survival (HR 2.24 for high NLR and 4.02 for high PLR).
The ESES recommendation is that the impact of S-GRAS and inflammation-based scores on therapeutic decisions should be assessed prospectively in future cohort studies (⊕⊕○○).
Genetic testing and disease subtype-based risk stratification
Current recommendation for adults with ACC, is to perform at least a basic clinical genetic evaluation, exploring personal and family history for evidence of a hereditary predisposition syndrome. The panel does not recommend for or against genetic tumour testing for somatic alterations3.
In adults, ACC has been reported in patients with multiple endocrine neoplasia (MEN), familial adenomatous polyposis coli, and neurofibromatosis type 1 (NF1). The evidence associating ACC with these syndromes is, however, less well substantiated. This is in stark contrast with paediatric ACCs, of which up to 80 per cent are associated with TP53 mutations as part of Li–Fraumeni syndrome. As such, genetic screening in adults with ACC is not performed nor recommended.
A decade ago, a review75 published in Nature reported several transcriptomic (mRNA and microRNA expression profiles), epigenomic (DNA methylation profiles), and genomic (DNA mutations and chromosomal alterations) differences between benign and malignant tumours with potential to stratify the prognosis of ACC, but such complex analysis has not (yet) influenced clinical care.
High-throughput characterization of ACC using multiomics approaches, including exome sequencing, methylation arrays, RNA sequencing, and microRNA sequencing, has demonstrated that ACC is composed of three distinct molecular subtypes (so-called COC1, COC2, and COC3) with significant clinical heterogeneity. This could explain why some patients with apparently successful radical resection of ACC might still have poor outcomes76. In the future, such information could change the approach to clinical trials. For patients with slow-growing disease (COC1 subtype), practice-changing clinical data through traditional phase I–III clinical trials may emerge only after a decade or more of enrolment. For patients with rapidly growing disease (COC2–COC3 subtypes), this approach is viable only in the relapse/recurrence setting, with high risk of trial failure. Advances in preclinical modelling of ACC could therefore improve clinical trial outcomes by prioritizing therapeutic agents that have greater potential to provide therapeutic benefit.
In addition to the conventional type, the three different histological subtypes of oncocytic, myxoid, and sarcomatoid ACC have been classified by the 2022 WHO classification of endocrine tumours77. These represent a very small minority as the currently published number of cases is under 200. They are important because histopathological diagnosis of their malignancy remains a challenge, despite use of the Lin–Weiss–Bisceglia criteria78,79, and their outcome might be different. In these subgroups of tumours, oncocytic ACC is slightly more common and is defined as a tumour with more than 90 per cent oncocytic cells77. Survival analysis in oncocytic ACC showed conflicting results, and an ongoing ENSAT study aims to address some of the current uncertainties.
The ESES recommendation is that, at present, no specific treatment can be defined for specific histological subtypes and patients should be treated independently of this parameter (⊕⊕○○).
Need for multidisciplinary team assessment in care of patients with adrenocortical carcinoma/adrenal tumour
A study80 of the impact of introducing a MDT discussion for all patients in a single centre compared 14 patients treated before the establishment of a formal adrenal MDT with 33 patients discussed by the MDT. Among patients with stage III–IV disease, there was longer median overall survival (31 versus 4 months) and longer progression-free survival (27 versus 3 months) for those discussed by the MDT, demonstrating significant gains in terms of survival. This topic has not been addressed in formal studies, but the overall expert opinion of oncologists and surgeons is that MDT discussion is a mandatory component of the care package for patients with advanced malignancy.
The ESES recommendation is that all patients with a suspicion of or confirmed diagnosis of ACC should be referred to a (regional) centre recognized for its multidisciplinary care based on previous experience with dealing with such patients, and ability to provide the entire spectrum of complex diagnostic and therapeutic options (⊕⊕⊕⊕).
All adrenal tumours deemed inoperable should be referred to a regional centre that can provide expertise in multiple surgical specialties working in an established multidisciplinary environment with oncologists (⊕⊕⊕⊕; 92 per cent of 148 ESES members voted agree or strongly agree).
Malignant phaeochromocytoma and paraganglioma
Based on the 2022 WHO classification of endocrine and neuroendocrine tumours81, phaeochromocytoma is described as a neuroendocrine neoplasm that originates from the chromaffin cells of the adrenal medulla (adrenal paraganglioma (PGGL)). Previously, PGGLs were defined as extra-adrenal phaeochromocytomas. More importantly, in the 2022 WHO classification81, all PGGLs and phaeochromocytomas are considered as malignant neoplasms with variable metastatic potential, a different emphasis from the historical assumption that the overall risk of malignancy in PGGLs is about 10 per cent.
Predicting the likelihood of malignant paragangliomas
A recent retrospective multicentre study82 reported that metastatic PGGLs (33 per cent of 582 patients) were significantly more common in males and younger patients, and presented more often with large, extra-adrenal, multifocal tumours, and with higher prevalence of SDHB (succinate dehydrogenase complex iron sulphur subunit B) mutations.
Age
Younger patients are more at risk. In one study83, patients with metastatic disease were significantly younger than those without metastases (mean age 41 versus 48 years), and the proportion of patients aged 35 years or less was significantly higher in the metastatic group (39 versus 17 per cent). In contrast, in a retrospective review84 of 272 PGGLs, older age at primary diagnosis was a strong predictor of rapid progression (death within 5 years of initial presentation).
Tumour size
Large tumour size is a potential independent predictor of aggressiveness84,85. Average size of tumours (6.5 versus 4.5 cm) and the mean proportion of tumours at least 6 cm in size (57 versus 30 per cent) were significantly greater among those with metastasis compared with the non-metastatic group83. Patients with metastatic disease have primary tumours over 5 cm in size in up to 76 per cent of cases86. The 5-year risk of new events is approximately 26 per cent if the size of the primary tumour exceeds 150 mm5.
Extra-adrenal location
Extra-adrenal location is a risk factor for metastatic disease86,87 and is associated with twice the risk of death from disease compared with that for adrenal phaeochromocytomas, making this a strong predictor of aggressiveness and decreased survival7. The presence of a PGGL was also significantly more common in the metastatic group than in the non-metastatic group (21.7 versus 7.4 per cent), albeit this was statistically significant only in univariable analysis83. Similarly, patients with recurrent disease tend to have a higher rate of PGGLs (16.7 versus 7.5 per cent)85.
Biochemical profile suggestive of malignancy
High values of 3-methoxytyramine (3-MT, the O-methylated metabolite of dopamine) raise the suspicion of metastatic disease or a possible SDHx gene mutation82,88. Measuring plasma 3-MT concentration after strict pretest conditions (supine rest and an overnight fast before blood sampling) is superior to measurement of urinary 3-MT89. As the assay for 3-MT is not available widely, it was not included in current guidelines7. In a prospective multicentre study90 of 213 PGGLs, measurement of plasma 3-MT only modestly improved the detection rate, yet it was useful for the detection of head–neck PGGLs. On the other hand, the inclusion of 3-MT measurement helps towards discrimination of true-positive results.
Genetic testing
Current guidelines state that all patients with PGGL to be considered for genetic testing5. All patients with primary or metastatic PGGLs to be genetically tested for germinative mutations7.
There are currently more than 20 driver genes known to be implicated in phaeochromocytoma/PGGL. Based on their mechanism of action, these genes are classified into three clusters91,92 (Table 4). The majority of metastatic tumours are associated with SDHB gene mutations, and less frequently with NF1, SDHA, HIF2A, MAX, and FH gene mutations91,92.
Table 4.
Genotype–phenotype correlation in paragangliomas
| Cluster 1 Pseudohypoxic cluster |
Cluster 2 TK-linked signalling pathways |
Cluster 3 Wnt signalling |
|
|---|---|---|---|
| Genetic pathways | Krebs’ cycle-related genes (SDHx) and hypoxia-signalling pathway genes (VHL/EPAS1-related genes) | RET, NF1, HRAS, MAX | Wnt signalling |
| Tumour location | Mostly extra-adrenal | Mostly within adrenal | |
| Biochemical phenotype | Noradrenergic/dopaminergic | Adrenergic | Unknown catecholamine phenotype |
| Clinical phenotype | Better clinical outcome than cluster 1 (low metastatic risk: ∼4%) | Aggressive phenotype |
TK, tyrosine kinase; SDHx, succinate dehydrogenase gene – enzyme complex composed of four subunits encoded by four separate genes called SDH-A, SDH-B, SDH-C and SDH-D recognised under the generic label of SDHx; VHL, von Hippel-Lindau gene; EPAS1, endothelias PAS domain protein 1, often known as HIF2A (the gene encoding protein called hypoxia-inducible factor 2-alpha, HIF-2α); RET gene, REarranged during Transfection gene, proto-oncogene encodes a receptor tyrosine kinase for members of the glial cell line-derived neurotrophic factor (GDNF) family of extracellular signalling molecules; NF1, neurofibromatosis type 1 gene; MAX, myc-associated factor X gene.
Recent guidelines93 have provided detailed advice for genetic counselling in patients with phaeochromocytoma/PGGL. Early knowledge of genetic status has an immediate impact on patient outcomes, because this triggers a specialized surveillance/screening programme of mutation carriers. One study94 compared the management and outcome of 221 patients diagnosed with PGGL carrying mutations in SDHx or VHL (Von Hippel–Lindau), who were informed of their positive genetic status either within the first year or more than 7 years after the initial PGGL diagnosis. Fewer patients were lost to follow-up in the group with early genetic testing than in the group tested later (9 versus 72 per cent respectively). Moreover, during follow-up, the former group developed smaller new PGGLs with less metastatic spread. In addition, patients in this group who developed metastases had a better 5-year survival rate than patients who did not undergo genetic testing at diagnosis. In closely monitored individuals, the SDHB mutation loses its significance as a prognostic factor for worse overall survival86.
The risk of new events is approximately twice as high in patients with genetic or syndromic PGGLs than among those with sporadic disease5. Genetic mutations are the strongest predictor of disease recurrence94,95. The recurrence pattern is also influenced by the type of mutation; there is a higher local recurrence risk for those with SDHA, SDHB, and MEN2B mutations, and a higher metastatic risk associated with SDHB, VHL, and MEN2A mutations95. Germline mutations are not associated with worse overall survival than that of patients with sporadic disease96. In the coming years, it is expected that results from genomic high-throughput platforms will elucidate optimal therapeutic options based on molecular biomarkers in PGGL97.
Carriers of the SDHB genetic mutation are at highest risk of developing metastatic disease (25–50 per cent by the age of 60 years)98, yet less than half of patients with metastatic PGGL have SDHB mutations and a large retrospective study did not confirm the prognostic role of the SDHB mutation86. In a study of 140 patients with PGGLs, of whom 94 had genetic testing and 36 (38 per cent) had a mutation detected, the presence of a mutation was associated with younger age, smaller tumour size, and bilateral adrenal tumours95. Disease recurrence developed at a median of 5.4 (i.q.r. 2.8–11.0) years after treatment in 21 patients (15 per cent), of whom 14 had a mutation in a susceptibility gene.
With increased accessibility and affordability, there has been a shift from phenotype-driven mutation analysis to broad genetic screening for up to 10 genes in all patients with phaeochromocytoma/PGGL. The use of multigene panel testing in 1727 individuals with suspicion of hereditary PGGL showed a very high yield in those with and without established risk factors. Overall, 27 per cent of individuals had a pathogenic or likely pathogenic variant, 9 per cent had a variant of uncertain significance, and 63 per cent had a negative result. Pathogenic variants were identified in SDHB (40 per cent), SDHD (21 per cent), SDHA (10 per cent), VHL (8 per cent), SDHC (7 per cent), RET (4 per cent), and MAX (4 per cent) genes. These findings support universal testing of all individuals with PGGL using a multigene panel99, but accessibility is highly variable between centres and countries. For this reason, voting within the ESES forum (123 participants) led to a split opinion regarding the need for genetic testing in all patients with phaeochromocytoma/PGGL; 50 per cent voted agree/strongly agree, 18 per cent were neutral, and 32 per cent voted disagree/strongly disagree. No recommendation was therefore made.
In addition to these data, complex genomic profiling showed that the risk of metastasis and time to progression correlate with tumour mutational burden, microsatellite instability, mutations in Krebs’ cycle genes, the TERT gene (telomerase reverse transcriptase) and ATRX gene (α-thalassemia/mental retardation syndrome X-linked), and somatic copy number alteration profiles, and a signature of 26 genes was expressed differentially between primary tumours of patients with and without metastatic disease100. None of these tests are available for use in clinical decision-making pathways.
Criteria for malignancy in paraganglioma
Current recommendation recommendations define malignancy of phaeochromocytoma/PGGL as the presence of metastasis in lymph node or other distant sites5.
In contrast with the above statement from the 2016 guidelines, according to the 2022 WHO classification81, PGGLs are no longer classified as benign and malignant, because any lesion can have metastatic potential and there are no clear-cut features that can predict metastatic behaviour. The WHO classification neither endorses the various scoring systems for malignant potential (Table 5) nor discourages their use.
Table 5.
Risk scores proposed for estimating the risk of malignancy in phaeochromocytomas and paragangliomas
| Score | Reference | Full name | Criteria, interpretation | Summary of findings |
|---|---|---|---|---|
| PASS | 101 | Phaeochromocytoma of the Adrenal gland Scaled Score | Histological features (with considerable interobserver variation), biologically aggressive behaviour (PASS ≥ 4) versus tumours that behave in a benign fashion (PASS < 4) | Both PASS ≥ 4 (binary variable) and high values of PASS (considered as continuous variable) correlate with disease recurrence85 A systematic review of the literature and meta-analysis included phaeochromocytomas (105 malignant, 705 benign) and PGGLs (13 malignant, 29 benign), and concluded that PASS can identify patients with an exceptionally low risk of future metastases, rather than primarily identify those at risk of disseminated disease102 |
| GAPP | 103,104 | Grading for Adrenal Phaeochromocytoma and Paraganglioma | Excludes some non-specific histological parameters and adds immunohistochemical (Ki-67) and biochemical (catecholamine type) parameters Well differentiated tumours (score ≤ 2), moderately differentiated tumours (score 3–6), and poorly differentiated tumours (score 7–10) |
Validated in a cohort of 143 patients. Higher GAPP score is associated with metastatic PCC/PGGL. Good concordance and significantly less interobserver variability than the PASS score105 |
| M-GAPP | 106,107 | Molecular Grading for Adrenal Phaeochromocytoma and Paraganglioma | Combines SDHB immunocytochemistry with several factors with high correlation within GAPP score | M-GAPP is worse than the GAPP grading system in specificity, sensitivity, and accuracy rate106 Neither GAPP nor M-GAPP grading systems have a credible prediction accuracy rate for metastatic tumours107 |
| COPPS | 108 | COmposite Phaeochromocytoma/paraganglioma Prognostic Score | Based on pathological features (tumour size, necrosis, and vascular invasion) and loss of PS100 (sustentacular cells) and SDHB immunostaining Score ≥ 3 correlates with high metastatic risk, with 100% sensitivity and 95% specificity |
High sensitivity and positive prediction rate for non-metastatic PGGL (100%). Lower in prediction accuracy rate for metastatic PGGL (47.4%)108 |
| SGAP | 85 | Size, Genetic, Age, and PASS | Age (≤ 35 years), tumour size (> 50 mm), PASS ≥ 3, and genetic germline mutations in known susceptibility genes 3 classes: low risk (score 0–2), intermediate risk (3–4) and high-risk (5–8) |
Patients with SGAP score 5–8 have a markedly increased risk of recurrence > 60% after 10 years Insufficient available data to create different predictive models to distinguish between the risk of local relapse, a new primary tumour, and distant metastases |
| ASES | 83 | Age, Size, Extra-adrenal location, Secretory type | Discriminatory ability for malignant potential was moderate (AUC 0.735), but negative predictive value was 97% for a cut-off point of 2 |
Although not validated in multicentre studies, the simplicity of this scoring system could make it a useful tool for preliminary assignation of patients for follow-up |
| AJCC staging system (8th edition) | 109 | TNM staging T1 < 5 cm T2 > 5 cm T3 locally invasive N1 regional lymph node metastases M1 M2 M3 |
All sympathetic extra-adrenal PGGLs (≤5 cm) are at least T2, stage II tumours N1 implies stage III, making it equivalent to primary tumour invasion into surrounding tissues (T3) M category stratifies prognosis taking in account that up to 20% of patients with metastatic PGGL present with bone metastasis only and have longer overall survival than those with metastasis to other sites |
In 458 patients (MD Anderson Cancer Center, Texas, USA) 10-year overall survival rates correlated with TNM stage; probabilities were 0.844 (95% c.i. 0.768 to 0.928) for stage I tumours, 0.792 (0.726 to 0.865) for stage II, 0.595 (0.435 to 0.813) for stage III, and 0.221 (0.127 to 0.384) for stage IV Compared with stage I, HRs for death were 1.50 (95% c.i. 0.87 to 2.57) for stage II, 2.85 (1.45 to 5.63) for stage III, and 8.88 (5.16 to 15.29) for stage IV96 |
PGGL, paraganglioma; PCC, phaeochromocytoma; SDHB, succinate dehydrogenase complex iron sulphur subunit B; AUC, area under the curve.
Several complementary histological criteria for malignancy have been proposed, but none has been accepted widely.
Cell proliferation (Ki-67 index)
According to a NANETS group analysis7, it remains unclear whether Ki-67 index is predictive of aggressive disease or not. Moreover, a recommendation for cut-off value for the Ki-67 index cannot be made, because there are no definitive studies in PGGLs. A correlation between high Ki-67 and the presence of metastatic disease has been reported in some studies110.
Depletion of sustentacular cells
These cells are usually present in PGGLs and create a pattern of small nests (zellballen) that are S-100 protein-positive. Depletion in the density of sustentacular cells has been reported for metastatic PGGLs111.
Molecular markers
Several molecular markers have been related to metastatic PGGLs, but data are limited and require validation in large cohorts of patients107.
Immunohistochemistry
SDHB mutation status can be detected by immunohistochemistry (IHC) as negative or weak diffuse staining112. Thus, SDHB IHC could be used as a surrogate for the identification of SDHx-mutated tumours when genetic testing is not available. Because IHC can be incongruent in some instances, the NANETS group7 did not reach consensus on whether to recommend SDHB IHC staining in all PGGL pathology specimens.
The ESES recommendation is that risk stratification for recurrence is based on the genetic profile and one of the clinical scores outlined in Table 5 when planning the intensity of follow-up after phaeochromocytoma/PGGL surgery (⊕⊕○○).
Assessment of patients with malignant/metastatic paraganglioma by functional imaging
In addition to their ability to detect and localize the disease, the tracers used in functional imaging allow better molecular characterization of tumours. This is useful for planning targeted therapy with [131I]metaiodobenzylguanidine (MIBG), or peptide receptor radionuclide therapy (PRRT) with 177Lu-labelled DOTATATE92. Because [123I]MIBG scintigraphy is widely available it has been used extensively in the initial assessment of phaeochromocytoma/PGGL. Its clinical benefits remain debatable113, and a recent systematic review114 failed to demonstrate a substantial benefit of functional imaging over CT/MRI only. Owing to its high specificity, it can be used as a complementary diagnostic test in patients with uncertain biochemical results. The ability to detect additional PGGLs is limited by the fact that many extra-adrenal tumours are MIBG-negative.
Current recommendations suggest to screen for metastatic tumours by [18F]FDG PET–CT, if possible, preoperatively in patients with PGGAs; in patients with phaeochromocytomas and elevated (that is 3-fold above the normal range) levels of 3-MT; and in patients carrying germline mutations of the SDHB gene5.
In many centres, [18F]FDG is the only tracer available for use in PET, and for this reason the above guidelines focus on its use (Fig. 3). Several other tracers are used for phaeochromocytoma/PGGL but accessibility remains challenging. 18F-labelled DOPA PET–CT has been used in the detection of metastatic lesions in patients with PGGL, but has lower sensitivity in patients with SDHx mutations115. 68Ga-labelled DOTATATE PET has detection rate superior to that of [18F]FDG PET and could improve the detection rate up to 100 per cent in metastatic PGGLs116. A meta-analysis117 has suggested that 68Ga-labelled DOTA-somatostatin analogue (SST) PET–CT may provide a higher detection rate for metastatic PGGLs, both in their SDHx forms and in sporadic forms, than [18F]FDOPA or [18F]FDG PET. Where available, 68Ga-labelled DOTA-SSA PET–CT should be prioritized over [18F]FDG PET–CT115.
Fig. 3.
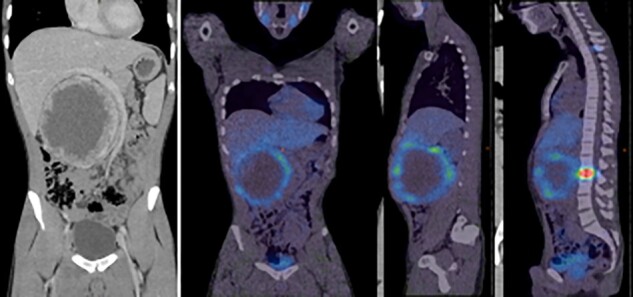
PET images of patient with malignant phaeochromocytoma with bone metastases
Current recommendation is that somatostatin receptor (SSTR) PET–CT should be first-line functional imaging modality when suspicions of metastatic PGGL arise. In the absence of this imaging method, [18F]FDG PET–CT may be a useful alternative, most especially in patients with SDHB mutations7,8.
It should also be mentioned that sometimes a single imaging modality cannot detect all tumour locations. There are patients with MIBG-positive and -negative tumours and, correspondingly, DOTATOC PET-positive and -negative tumours. Therefore, in selected patients, the imaging modalities can be combined to demonstrate all lesions.
Access to the new tracers for PET is highly variable and so no specific recommendations can be made. Each institution will have to decide on the best imaging protocols based on locoregional availability of expertise in nuclear medicine.
Recurrence after paraganglioma surgery
Historically, characterization of phaeochromocytoma as the ‘10 per cent tumour’ included the assumption that 10 per cent of phaeochromocytomas could recur. A meta-analysis118 of 13 studies including 430 patients described a pooled recurrence rate after curative surgery of 3 (95 per cent c.i. 2 to 6) per cent (I2 = 0 per cent), with a weighted mean(s.d.) time to recurrence of 49(31) months and a weighted mean follow-up of 77 months. This very low recurrence rate influences follow-up strategies for patients with truly sporadic phaeochromocytomas. Data from the most recent publications on this topic data are summarized in Table 6.
Table 6.
Studies reporting risk of recurrence in patients with paraganglioma
| Reference | No. of patients | Findings |
|---|---|---|
| 106 | 72 | Metastases occurred in 15 patients (21%) followed up for a median duration of 43 (i.q.r. 6–81) months after the initial operation. This included 5 (6.9%) with synchronous and 10 (13.9%) with metachronous metastases |
| 84 | 272 | 65% of patients developed metachronous metastases at a median of 5.5 (range 0.3–53) years after the primary tumour diagnosis; 29 patients had rapid disease progression (survival < 5 years since the primary tumour diagnosis) and 188 patients had indolent disease (alive for at least 5 years since diagnosis). Median time from diagnosis to metachronous metastasis was 0.5 (i.q.r. 0.4–2.4) years in the first group and 6.2 (0.4–53.4) years in the latter group |
| 83 | 333 | Metastasis rates were 19.7 per 10 000 person-months (236.7 per 10 000 person-years) in PGGL, including 16.9 per 10 000 person-months in phaeochromocytoma and 47.4 per 10 000 person-months in PGGL (P = 0.039, log rank test) |
| 86 | 169 | Median time between initial diagnosis and identification of metastases was 43 (range 0–614) months. Metastases were diagnosed within the first year in 79 patients (47%). A delayed diagnosis above 5 and 10 years was observed in 47 (28%) and 26 (15%) patients respectively |
| 85 | 242 | Cumulative incidence of recurrence at end of follow-up (12 years) was 21.5%. Median recurrence time was 3 years. Recurrence risk was 2.5 (95% c.i. 1.1 to 5.5)% at 1 year and 12.3 (95% c.i. 8.7 to 17.8)% at 5 years after surgery |
| 82 | 639 | Patients with metastatic PGGLs had a median metastasis-free interval of 4 (range 1–25) years and those with HNPGGLS of 7 (2–29) years, with a statistically significant difference between groups (P = 0.015) |
| 41 | 177 | Patients with a SGAP score of 5–8 had a markedly increased risk of recurrence of > 60% after 10 years |
| 119 | 170 | The majority (95%) of phaeochromocytomas/sPGGLs were considered apparently benign at time of diagnosis. Overall risk of recurrence was 13% at 10 years and 33% at 30 years Risk of new tumour recurrence was higher in patients with hereditary tumours, but still significant in patients with apparently sporadic variants (20-year risk: 38 versus 6.5% respectively; P < 0.001) Risk of metastatic recurrence was higher in patients with locally aggressive tumours at diagnosis, but also present in those with apparently benign variants (5-year risk: 100 versus 1% respectively; P < 0.001) Lifelong follow-up is required for hereditary phaeochromocytoma/sPGGL and for apparently benign and sporadic tumours at diagnosis |
PGGL, paraganglioma; HNPGGLS, head & neck paragangliomas; SGAP, Size, Genetic, Age, and Phaeochromocytoma of the Adrenal gland Scaled Score; sPGGL, sporadic paragangliomas.
These studies have confirmed that metastases can occur after a 5-year event-free interval and sometimes even after 10 years, justifying the follow-up of patients who have had surgery for PGGL for at least 10 years, if not more, maybe lifelong5,7. Because the different recurrence predictors are not superimposable, there is no clear subgroup of patients with PGGLs in whom follow-up can be interrupted safely.
Adopting lifelong clinical, biochemical, and radiological surveillance after surgery for phaeochromocytomas/PGGLs would have a massive impact on resources and service provision, and the solution to this dilemma would vary between centres and countries. The discussion in the ESES forum led to a vote with 78 per cent agreeing/strongly agreeing to the following recommendation.
The ESES recommendation is that the structure, duration, and provision of follow-up after successful resection of phaeochromocytoma/PGGL remain influenced by established local agreements, and should be shared between surgeon and endocrinologist.
Follow-up protocols after resection of paraganglioma
Current recommendation is that in patients with PGGL who underwent an apparently full resection of the primary tumour, the risk of new events persists long term and is even higher in patients with genetic or syndrome diseases5.
New events can be classified as: disease persistence owing to incomplete tumour resection or tumour spillage during surgery; local disease recurrence from a microscopically incomplete resection; development of new tumours in the contralateral adrenal gland, in the remnant after subtotal adrenalectomy or a new PGGL; or metastatic disease and tumours in other organs such as renal cancer (in patients with VHL syndrome) or medullary thyroid carcinoma (in patients with MEN2). Persistence versus recurrence is defined according to perioperative biochemical and imaging tests. Each of these negative outcomes can be monitored by different approaches.
Biochemical follow-up
Current recommendation is to measure plasma or urinary levels of MN and 3-MT 2–6 weeks after recovery from surgery in patients who had elevated MN levels preoperatively5,8; assaying plasma or urinary MN and 3-MT every year5,8. In individuals who had primary PGGLs that were secreting, at least annual testing of plasma-free or 24-h urine fractionated MNs7.
After complete resection of MN- or 3-MT-producing tumours, the identification of raised hormone levels strongly suggests persistent disease, which should prompt imaging tests to confirm the presence and location of residual tissue. Recurrent PGGLs are associated with higher levels of noradrenaline (norepinephrine) and lower levels of adrenaline (epinephrine)88,89. Increased MN or 3-MT levels correlate with a new event, presenting as metastasis, recurrence or a new tumour. Furthermore, patients operated for inactive PGGLs might develop new biochemically active tumours, especially those with hereditary disease5. There is evidence that plasma 3-MT is the most accurate biomarker for the discrimination of metastatic disease88, but 3-MT testing is very limited in North America and so NANETS7 does not recommend its routine use either for diagnosis or screening purposes.
Current recommendation is to measure plasma chromogranin A (CgA) every year for patients operated on for MN- and 3-MT-negative, and CgA-positive PGGLs8.
This recommendation from 2016 is based on a study showing that SDHB mutation carriers with PGGL may exhibit normal MN and raised CgA levels in plasma. Recent studies120,121 have investigated the value of MN and CgA levels in follow-up, and the above recommendation remains valid.
Current recommendation is to measure CgA levels 2–6 weeks after recovery from surgery in patients with normal preoperative MN and 3-MT levels, and elevated preoperative CgA levels5.
Plasma CgA level is an alternative marker for diagnosis and monitoring of the functional activity of patients with PGGL associated with normal preoperative levels of MN and 3-MT. High levels of CgA are present both in metastatic and in non-metastatic forms of phaeochromocytoma/PGGL, but the absence of a raised CgA level should not be used to rule out PGGLs122. CgA may be used as a biochemical marker for postoperative follow-up of patients with primary or metastatic PGGLs with SDHB mutations123. The NANETS study group7 did not recommend routine use of CgA monitoring in all patients with primary or metastatic PGGL because of the high false-positive rate caused by concomitant diseases (renal or hepatic) and some treatments (proton pump inhibitors).
Radiological follow-up
Current recommendation is to perform an imaging test 3 months after an allegedly complete tumoral resection in only three categories of patients: patients with increased levels of MN or 3-MT immediately after operation (to confirm the suspicion of residual disease), patients who preoperatively had normal levels of MN or 3-MT (as an alternative evaluation method for the possible residual disease), and patients who had not had their MN and 3-MT levels measured before surgery (these patients can be part of any of the previous categories)7,8.
Follow-up of PGGLs can be done with either whole-body CT–MRI or whole-body functional imaging. Nuclear imaging is more sensitive than anatomical imaging124. Furthermore, PGGL-specific functional imaging is minimally influenced by post-treatment sequelae, enabling accurate diagnosis that could be missed by anatomical imaging. Still, functional imagining is not available widely and is not cost-effective.
Current recommendation suggests to screen for local or metastatic recurrences or new tumours with imaging tests every 1–2 years5,8.
Imaging tests are the only option where there are no reliable biochemical markers. The recommended time interval for screening remains arbitrary, because there are no observational or randomized studies that support any particular interval. For the purpose of minimizing radiation exposure, thoraco–abdomino–pelvic MRI is preferred.
68Ga-labelled DOTA-SSA PET–CT can be used as first choice in metastatic PGGL, whereas [18F]FDOPA PET–CT can be the second choice in patients with no or an unknown SDHB mutation, or [18F]FDG PET the second choice in those with a SDHB mutation; [123I]MIBG scintigraphy is used only if [131I]MIBG therapy is being considered115.
The NANETS working group7 recommended against the routine use of functional imaging in all patients with primary PGGL; however, if metastatic disease is strongly suspected, the significant majority of them recommended SSTR PET–CT (if available) as a first-line functional imaging modality given its high sensitivity.
In future years, it is likely that functional imaging will be decided based on the known mutational background of each tumour92. The suggested protocol includes SSTR for cluster 1A, and FDOPA for cluster 1B and cluster 2.
The ESES recommendation is that, considering the complex decisions regarding functional imaging, each centre should have an internal protocol based on the availability of different tracers, costs, and feasibility of regional referrals.
Duration of follow-up
Current recommendation is that the follow-up of all patients operated for a PGGL is at least 10 years, in order to monitor local or metastatic and new tumour recurrences5. A lifelong annual follow-up should be offered to patients considered to be at high risk of developing new events5.
There are no randomized studies addressing the appropriate duration of follow-up, and the tests that should be used to detect and monitor new tumours or recurrences. The current recommendations are based on the results of the analysis of the PGGL cases in the preliminary meta-analysis8 published between 1980 and 2011, and those from the ENSAT database5. The incidence of new events is low, about 1 in 100 person-years, but over 40 per cent of new events are malignant recurrences, and new events may occur after a 5-year event-free interval.
Non-surgical interventions for paraganglioma
Radiopharmaceutical therapy with [I131]MIBG
There are no agreed standards to define the indication for [I131]MIBG therapy. In general, it is indicated for patients with advanced metastatic PGGL requiring systemic treatment7. Because many patients with biochemical evidence of recurrence have indolent disease, the presence of metastases alone is not sufficient in the absence of objective evidence of disease progression or symptoms that cannot be controlled conservatively. In addition, there is insufficient evidence to recommend the routine use of [131I]MIBG in the adjuvant or neoadjuvant setting. There are only anecdotal reports suggesting its use as a bridge to resectability, but dramatic anatomical responses are unlikely and so its benefits remain questionable in this context7.
When MIBG therapy is being considered, positive diagnostic [123I]MIBG imaging is required, as only patients with MIBG-avid disease are candidates for therapy. The amount of uptake required to predict responsiveness to this therapy is poorly defined125. Because of these limitations, it was proposed that the metabolic tumour volume (MTV) and total lesion glycolysis (TLG) derived from [18F]FDG PET can better predict the prognosis of patients with unresectable PGGLs. Overall survival was significantly shorter in the high-MTV group and the high-TLG group, with no significant difference between the high- and low-SUVmax groups126.
It is outside the scope of these recommendations to detail the reasoning for establishing two treatment protocols for MIBG therapy, labelled as low dose (low specific activity, LSA) or high dose (HSA), but a few comments are necessary. LSA [131I]MIBG is used in most centres but the regimens vary among hospitals127. A retrospective systematic review and meta-analysis128 of 17 studies that included 243 patients with malignant paragangliomas (mPGGLs) treated with LSA [131I]MIBG showed oncological effects (such as tumour stabilization), symptomatic improvement, and a substantial reduction in catecholamine secretion. This review was subsequently criticized because only four studies used Response Evaluation Criteria in Solid Tumours (RECIST), no studies undertook individual tumour dosimetry to optimize dose delivery, and some of the proposed doses were known to have a high risk of severe systemic toxic effects127. HSA [131I]MIBG is recommended in the USA because it is the only regimen approved by the Food and Drug Administration (FDA)125 and therefore recommended by the NANETS working group in 2021. In a prospective phase II trial129 of HSA [131I]MIBG in 68 patients with advanced, symptomatic (hypertensive) PGGLs, 25 per cent had a durable reduction in baseline antihypertensive medication use, 92 per cent achieved RECIST partial response or stable disease, and 68 per cent of patients with a raised CgA level at baseline had at least a 50 per cent reduction. No direct therapeutic comparisons of LSA versus HSA [131I]MIBG have been done, but an analysis of published data reported that the therapeutic responses overlap125. The choice, therefore, is likely to be based on cost, availability, and ability to provide the treatment as inpatient (as is compulsory for HSA regimens) or outpatient (as is possible for LSA regimens).
Peptide receptor radionuclide therapy
Current recommendation based on the recommendation of the European Society of Hypertension133 on the management of patients with PGGL with SDHD mutation to use targeted radionuclide therapy as the first-line systemic therapy for SSTR-positive or [123I]MIBG-positive metastatic tumours with moderate-to-high tumour burden and without evidence of rapidly progressive disease9.
177Lu-labelled DOTATATE is the only PRRT therapeutic agent approved by the FDA and the European Medicines Agency for gastroenteropancreatic neuroendocrine tumours, and guidelines130 for its use have been published. Although 177Lu-labelled DOTATATE has not been approved for PGGL treatment, there is a rationale for its off-label use for patients with relevant symptoms, high tumour burden or progressive disease.
A prerequisite for PRRT therapy is tumour avidity on SSTR imaging. Other PPRT options, such as use of 90Y-labelled DOTATOC and 90Y-labelled DOTATATE, are not available commercially, and their use is limited to certain academic centres.
Experience in the use of PRRT for treatment of PGGLs is limited, but early results in small studies have shown benefit and limited toxicity. A meta-analysis131 of 12 observational studies found similar response rates between 90Y-labelled DOTATOC and 177Lu-labelled DOTATATE, with 25 (95 per cent c.i. 19 to 32) per cent of patients demonstrating an objective response, 61 per cent a clinical response, and 84 per cent biochemical improvement. Estimated overall survival was 55 months and mean progression-free survival was 37 months. In an analysis of reports describing five or more patients receiving 90Y-labelled or 177Lu-labelled DOTA SST for metastatic or inoperable PGGLs, treatment led to partial regression and stable disease, as well as significant biochemical and symptomatic responses. Currently there is an ongoing prospective phase II clinical trial (NCT03206060) of patients with sporadic or SDHx-related metastatic or inoperable PGGLs, with clear evidence of progression, receiving 177Lu-labelled DOTATATE in four cycles. Preliminary data showed that, after two cycles of treatment, almost half of the lesions were stable in 11 patients.
A recent expert opinion132 on this topic concluded, based on a Delphi consensus, that [131I]MIBG and 177Lu-labelled DOTATATE are the most appropriate treatments for both non-functional and functional PGGLs with uncontrolled hormonal symptoms.
Targeted molecular therapy
Tyrosine kinase inhibitors are a therapeutic option for patients with malignant PGGL, especially those with tumours that do not express the noradrenaline transporter (MIBG non-avid), mixed tumours, and patients with contraindications to MIBG therapy (bone marrow suppression owing to bone metastases) or for patients with a high tumour burden or rapid disease progression7.
In a meta-analysis134 of seven studies with 160 patients, the pooled proportion of partial response, stable disease, and disease control rate was 0.32, 0.52, and 0.86 respectively. The combined median progression-free survival in six studies was 9 months. Larger clinical trials are expected in the future. Axitinib, pazopanib, cabozantinib, lenvatinib, and sunitinib are currently being, or have been, evaluated in phase II clinical trials (Table 7).
Table 7.
Clinical trials of tyrosine kinase inhibitors for the management of metastatic paragangliomas
| Drug tested | Trial number | Findings |
|---|---|---|
| Axitinib | NCT01967576 | Recruited 14 patients; preliminary results derived from 11 patients showed 4 with a PR (36%) and 6 with SD (55%); only 1 patient did not respond to therapy |
| Pazopanib | NCT1340794 | Terminated early owing to poor patient recruitment and moderate efficacy |
| Lenvatinib | NCT03008369 | Terminated owing to slow accrual rate |
| Cabozantinib | NCT02302833 | Recruited 17 patients; preliminary results in 11 patients identified 4 with a PR (37%) and 6 with SD (55%). PFS was 16 months |
| Sunitinib | NCT01371201 (FIRSTMAPPP trial) | Recruited 74 patients (including 32% with SDHB mutations)—metastases in the distant lymph nodes (73%), bone (65%), lung (51%), and liver (49%) After a median follow-up of 27 months, PFS at 12 months was 36% (versus 19% in placebo group). Serious adverse events were reported in 54% of patients receiving sunitinib versus 49% of those receiving placebo, and included asthaenia/fatigue (18 versus 3%) and hypertension (10 versus 6%) |
| Sunitinib | NCT00843037 (SNIPP trial) | Recruited 25 patients (23 with mPGGLs and 2 with unresectable primary tumours without metastases). Results showed PR in 3 patients (13%) and SD in 16 (70%). Six patients with SD had tumour regression (12–27%). DCR was 83 (95% c.i. 56 to 93)% and PFS was 13.4 (5.3 to 24.6) months. All patients with SDHx PGGLs had a PR or SD |
PR, partial response; SD, stable disease; PFS, progression-free survival; mPGGL, malignant paragangliomas; DCR, disease control rate; PGGL, paragangliomas.
Outside the NCT03008369 clinical trial, lenvatinib used in 11 patients from the Mayo Clinic, for a median duration of 15 months, led to a progression-free survival of 15 months and an overall survival rate of 80 per cent at 12 months. Carbozantinib is currently in clinical use (off-label) for patients with PGGL. Based on the results of the FIRSTMAPP trial, it is expected that sunitinib will be the first-line option in patients with progressive malignant phaeochromocytomas/PGGLs. Current recommendation is to use tyrosine kinase inhibitors (for example, sunitinib) or temozolomide in patients with progressing tumours who are not eligible for PRRT or [131I]MIBG or following progression to radionuclide therapy or chemotherapy with cyclophosphamide, vincristine, and dacarbazine (CVD)9.
Chemotherapy
Current recommendation that in patients for whom chemotherapy with CVD is not tolerated, not wished for by the patient, or if there are contraindications to CVD, tyrosine kinase inhibitors (for example sunitinib) or temozolomide can be used as alternative agents while carefully evaluating their adverse effects9.
Currently, chemotherapy with CVD is the most established and longest studied therapy for aggressive and rapidly progressive PGGLs91. Although based on small, single-arm and/or retrospective studies without any RCTs, chemotherapy with CVD is the recommended first-line therapy for rapidly progressing disease or a high visceral tumour burden7,9, and the second-line therapy if there is progression or a high tumour burden after targeted radionuclide therapy9.
Temozolomide is used to treat specific types of brain cancer (for example malignant glioma, anaplastic astrocytoma) and has been used for the treatment of metastatic phaeochromocytoma/PGGL in adults and children135. It has been proposed as first-line alternative to CVD chemotherapy or as maintenance therapy, especially in patients with mutated SDHB92. Two retrospective studies136,137 and several case reports138 have demonstrate the efficacy of temozolomide in patients with mPGGLs, including those with SDHx mutations. Although the first of the two retrospective studies showed partial responses only in patients with SDHB mutations, the second analysis suggested a response to temozolomide also when there were SDHx mutations. An ongoing prospective, multi-institutional phase II trial (NCT04394858) is currently evaluating temozolomide versus temozolomide and olaparib for advanced PGGLs. An important objective of the trial is to assess whether biomolecular markers are associated with clinical outcome.
Immune therapy
Current recommendation state that immunotherapy may bring benefits to subgroups of patients with progressive mPGGLs and that immunotherapy to be limited to clinical trials at this time7.
In a phase II clinical trial139 of only 11 patients with progressive mPGGL, pembrolizumab had an objective response rate of 9 per cent; progression-free survival was 5.7 months and the non-progression rate was 40 per cent. The clinical benefit rate (patients with an objective response or stable disease for at least 4 months) was 73 per cent. An ongoing phase II clinical trial (NCT04924075) is evaluating belzutifan (an FDA-approved HIF-2α inhibitor) monotherapy in patients with advanced PGGLs and VHL disease-associated tumours; the trial has completed recruitment and outcome data are expected soon.
Radiotherapy
Current recommendation states that radiotherapy is a non-invasive therapy that can be effective for unresectable mPGGL disease, relieving pain, preventing pathological fracture, and spinal cord compression, with good local control rates. If bone metastases are in weight-bearing bones, we recommend radiation to those sites for stability7.
There are no prospective data evaluating the use of radiotherapy for metastatic PGGLs, but numerous case studies have reported local control and fair toxicity of fractionated stereotactic body radiation therapy (SBRT) for adrenal metastases in selected patients (Table 8).
Table 8.
Recent papers reporting efficacy of radiotherapy in metastatic paraganglioma
| Reference | No. of patients | Findings |
|---|---|---|
| 140 | 24 patients with 47 lesions—bone (85%), abdominal (6%), and central nervous system (9%) | Symptomatic control rates of up to 81% after 3D conformal EBRT |
| 141 | 41 patients with 107 sites treated | Symptomatic improvement in 94% of patients treated with EBRT Median EBRT dose 40 (range 6.5–70) Gy. Median biologically effective dose using α/β = 10 (biological effective dose) was 53 (9–132). Median follow-up 3.8 (0.04–41.5) years; mean follow-up 9.7 years. Overall survival at 5 years 65%: 79% for those treated with curative intent and 50% for patients treated with palliative intent (P = 0.028) |
| 142 | 7 patients with 17 spinal metastases | Local control rate 93.7% |
| 143 | 14 patients with 17 lesions at various locations | Hypofractionated intensity-modulated RT (15 lesions in 12 patients) and conventional fractionated RT (in 2 patients). For 8 patients with locally advanced primary tumours or recurrent in situ tumours, local control was achieved in 100%, and none had developed recurrence or distant metastasis after RT at last follow-up (median 29 months) In 12 patients with catecholamine-related syndromes, 91% of symptomatic lesions improved after RT and an over 50% decline in catecholamines was reported |
3D, three-dimensional; RT, radiotherapy; EBRT, external beam radiotherapy.
Ablation procedures
Current recommendation states that percutaneous image-guided thermal (radiofrequency or cryo) ablation is effective for symptom control and prevention of skeletal-related events from oligometastatic mPGGL and should be performed under α-blockade and monitored anaesthesia care, because release of vasoactive hormones is expected7.
Chemical ablation with ethanol under CT guidance has been reported in isolated phaeochromocytoma case series, but the technique remains controversial, the ethanol volume has not been standardized, and the distribution of ethanol within the tumour is unpredictable owing to septa. Furthermore, it is associated with major haemodynamic adverse events, and was not recommended by the 2021 NANETS expert group7.
Studies of RFA and microwave ablation of phaeochromocytomas are also restricted to isolated case reports. A retrospective review from the Mayo Clinic144 reported 31 patients with a total of 123 metastases (63 osseous, 54 liver, and 6 in other locations), treated with a combination of RFA (42 procedures), cryoablation (23 procedures), or ethanol injection (4 procedures) under adrenergic blockade. There was a 94 per cent success rate for the ablation techniques and labile postablation haemodynamics were observed in only six sessions. The same group later reported that, despite preoperative adrenergic blockade and metyrosine, there were large BP oscillations during ablation for both functional and non-functional metastases145.
Embolization
Current recommendation is that there is no evidence supporting routine preoperative embolization for abdominopelvic PGGL and chemoembolization can be performed safely for local control/symptom management from PGGL liver metastases7.
The above is agreed upon because the favourable experience in head–neck PGGL is not unanimously accepted146, and has not been replicated in phaeochromocytomas and abdominal PGGL.
Adrenal metastases
Metastases to the adrenal glands occur in a variety of neoplastic diseases, commonly lung, renal, colorectal cancers and melanoma, and occasionally in breast carcinoma, hepatocellular carcinoma, thyroid cancer, carcinoma of the bladder, lymphoma, seminoma of the testis, and osteogenic sarcoma.
The incidence of adrenal metastases has increased in recent decades owing to prolonged overall survival in patients undergoing cancer treatment that allows extensive radiological evaluation for tumour recurrence. As more patients are identified, more are being referred for surgery. In this context, the most recent audit report147 from the UK Registry of Endocrine and Thyroid Surgery showed that adrenalectomy for metastatic disease increased from 48 procedures in 2006–2010 to 239 in 2016–2020.
Diagnosis of adrenal metastases
Adrenal metastases are usually asymptomatic, unilateral, and most commonly discovered on radiological imaging.
In patients with an adrenal incidentaloma, the diagnosis of adrenal metastasis is rare among those who do not have a history of extra-adrenal malignancy, and the risk ranges from 20 to 70 per cent in those with a history of extra-adrenal malignancy.
Because the adrenal glands are common sites of metastatic disease, an adrenal mass discovered in a patient with personal history of cancer represents a metastasis in over 75 per cent of cases. In a study148 involving 208 patients with adrenal incidentalomas, 19 (9 per cent) had metastatic adrenal lesions and, of these, 10 (5 per cent) had bilateral adrenal lesions. In another study149 that included 211 adrenal masses detected during standard contrast-enhanced CT, 19 per cent of the patients had a metastasis on final histology.
The diagnostic work-up for adrenal masses follows the same principles as those for patients with known or unknown extra-adrenal malignancy, and should include hormone assessment and morphological/functional imaging. Non-contrast or contrast-enhanced CT is the most commonly used imaging technique to detect adrenal metastases. Diagnosis is based on irregular shape, high Hounsfield counts (over 20 HU) on non-contrast CT, and high enhancement after administration of intravenous contrast. The cut-off of over 10 HU at non-contrast CT has high sensitivity but low specificity for detecting malignancy, with PPVs of 70–80 per cent150. MRI has high sensitivity (89–99 per cent) and specificity (60–93 per cent)150. Functional imaging with [18F]FDG PET–CT has good sensitivity of 82 per cent and specificity of 96 per cent, but several benign adrenal tumours (functional adenomas, benign phaeochromocytoma) may be FDG-positive. Moreover, false-negative results may be found in a few subtypes of extra-adrenal malignancies with low uptake, such as renal cancers, neuroendocrine tumours, and necrotic or haemorrhagic metastases151.
Biopsy is not recommended routinely152. When performed, its expected sensitivity and specificity are 87 and 96 per cent respectively153. The benefits of biopsy are restricted to the rare patients for whom its results would change the oncological management, such as choosing systemic therapy based on the biopsy material. Before biopsy of an adrenal mass is performed, biochemical hormone screening with 24-h urinary MNs is necessary to exclude a phaeochromocytoma.
Current recommendation states that indications for adrenal biopsy are limited to those masses not conclusively characterized on conventional imaging techniques, for which a biopsy result would affect the therapeutic decisions152.
The ESES recommendation is to avoid adrenal biopsy in the majority of patients, in particular those who are surgical candidates for resection of the adrenal metastasis, and those for whom a primary adrenal tumour remains in the differential diagnosis (⊕⊕⊕⊕).
Indication for surgery for adrenal metastases
Evidence-based guidelines for adrenal metastasectomy are yet to exist because of the rapid changes in effective systemic therapies for various cancers as well as differences across healthcare systems regarding abilities to assess the affordability of such treatments. Most decisions regarding referral for adrenalectomy are discussed in organ-specific MDT meetings before referral of the patient to an adrenal surgeon. These decisions are influenced by the performance status of the patient, response to previous systemic therapies, the disease-free interval after treatment of the primary tumour, and whether the metastasis is documented as being synchronous or metachronous with the primary tumour.
Based on the data available, adrenalectomy is the procedure of choice when the adrenal metastasis is isolated, the operation is feasible without major morbidity, and the primary tumour is (or was) radically resectable154,155. Large series of adrenal metastasectomies are summarized in Table 9.
Table 9.
Recent large series of patients who underwent adrenal metastasectomy
| Reference | No. of patients | Primary cancer | Surgical approach | Median overall survival (months)* |
Factors associated with improved outcome | Factors associated with worse outcome |
|---|---|---|---|---|---|---|
| 154 | 317 | Lung, colorectal, renal, breast melanoma, other | MIA 146 Open 171 |
29 | Metachronous isolated metastasis from RCC Operative treatment of primary tumour Complete operative resection Laparoscopic adrenalectomy |
Synchronous adrenal metastases, presence of extra-adrenal disease, extended adrenalectomy, incomplete operative resection (R1/R2), duration of hospital stay, and tumour size |
| 156 | 45 | Liver (HCC, CCC, sarcoma), lung, kidney, stomach, oesophageal, NET, colonic, ovarian, melanoma, others | MIA 6 Open 39 |
14 | R0 resection Renal cell carcinoma |
R1/R2 resection Primary tumours of upper gastrointestinal tract |
| 157 | 36 | Lung, breast, colorectal, kidney, thyroid, melanoma, ovarian | MIA 36 | 26 | Postadrenalectomy DFI | |
| 158 | 39 | NSCLC, RCC, colonic | Open 39 | 18 | Colorectal cancer | NSCLC Larger size of metastasis |
| 159 | 42 | Renal, lung, melanoma, breast, Other | MIA 38 Open 4 |
56 | RCC | Primary lung cancer and melanoma |
| 160 | 263 | Lung, RCC, melanoma, sarcoma, colorectal, oesophageal, breast, thyroid, ovarian, oropharyngeal, NET, endometrium, others | MIA 149 Open 114 |
53 | Lung primary tumour (longer DFS, but shortest OS) | Larger adrenal tumour size Presence of extra-adrenal oligometastatic disease at initial presentation Chemotherapy as definitive treatment for primary tumour Adjuvant chemotherapy Oligometastatic extra-adrenal disease at time of adrenalectomy R1/R2 resection |
| 161 | 95 | Colorectal, lung, melanoma, RCC, breast, other | MIA 53 Open 42 |
20 | Colorectal cancer | Lung cancer Melanoma |
| 155 | 395 | Renal, lung, colorectal, other | MIA 237 Open 158 | 28 | Renal and colorectal cancer | Lung cancer Increasing numbers of Charlson Co-morbidity Index factors Tumour size > 50 mm Presence of extra-adrenal metastases Open surgical approach R2 resection |
| 162 | 119 | Lung | MIA 84 Open 35 |
47 | Ipsilateral metastases Adjuvant or primary chemotherapy |
Radiotherapy of primary tumour Extra-adrenal metastases at adrenalectomy Small cell histology |
| 163 | 43 | Lung | MIA 19 Open 24 |
1-year OS 71% 2-year OS 53% |
None reported |
*Unless indicated otherwise. MIA, minimally invasive adrenalectomy; RCC, renal cell carcinoma; HCC, hepatocellular carcinoma; CCC, cholangiocarcinoma; NET, neuroendocrine tumour; DFI, disease-free interval; NSCLC, non-small cell lung cancer; DFS, disease-free survival; OS, overall survival.
In a large multicentre European study154 including 317 patients undergoing adrenal metastasectomy, it was found that the removal of adrenal metastases, particularly metachronous isolated metastasis from renal cell carcinoma (RCC), was associated with favourable long-term outcomes (median survival 84 months). Hence, as a general criterion, patient selection for adrenalectomy should consider the site and pathology of the primary tumour, the progression-free interval after surgical or oncological treatment, and the disease burden in terms of oligometastatic and systemic disease164.
Survival depending on primary tumour type
The 5-year overall survival rate was 31 per cent in a study155 of 435 patients who underwent adrenal metastasectomy between 2000 and 2018. Factors associated with poor survival included tumour size (over 50 mm; HR 1.79), radicality of the operation (non-radical pR2 resection; HR 3.57), open surgical approach (HR 1.33), and lung cancer origin (HR 1.77). More detailed analysis of the impact of primary tumour sites has been reported in large cohort studies.
Renal cancer adrenal metastases
Approximately 30 per cent of patients undergoing radical surgery for RCC develop metachronous metastases during their lifetime, with a rate of ipsilateral adrenal metastasis of up to 10 per cent165. By contrast, contralateral adrenal metastases occur rarely (1 per cent) and bilateral adrenal metastases are limited to about 20 cases reported in the literature. Adrenalectomy for RCC metastases is associated with better overall survival than that for other tumour types. In a study of 1635 patients with RCC, those with a solitary adrenal metastasis achieved a significant tumour-specific survival benefit from adrenal metastasectomy166. In a multicentre study160, median overall survival in patients undergoing adrenal metastasectomy for RCC was 59 months, with a 5-year survival probability of 40 per cent.
Lung cancer adrenal metastases
Adrenal gland metastasis is common in non-small cell lung cancer (NSCLC), with rates ranging from 18 to 42 per cent in autopsy series. The prevalence of isolated adrenal metastasis is, however, relatively low in patients with resectable lung cancer (up to 7 per cent). Low metastatic burden indicates a better prognosis, and local treatment of the primary tumour and all metastases, with or without addition of standard systemic treatment, can lead to long-term disease control and 5-year overall survival rates of between 13 and 56 per cent167. In a recent multicentre series160 of 269 patients, those with lung metastasis had improved disease-free survival compared with patients with other forms of cancer.
A review of 11 papers reported a median survival rate of 24 months and approximately one-third of these patients being 5-year survivors168. Results from more recent series are encouraging, as a reflection of improvements in systemic therapy. Median overall survival of 77 months was reported in 59 French patients from eight centres who had extensive preoperative staging to rule out extra-adrenal disease169. Shorter median overall survival of 47 months (80 per cent at 1 year; 35 per cent at 5 years) was observed in 122 patients operated in six centres in the USA, without strict selection criteria162. Longer overall survival was associated with ipsilateral metastasis (HR 0.55) and adjuvant chemotherapy (HR 0.35), whereas shorter overall survival was associated with extra-adrenal metastases at adrenalectomy (HR 3.52) and lung radiotherapy (HR 3.37).
A retrospective multicentre study170 showed no difference between recurrence or survival rates among 32 patients with synchronous and 11 with metachronous disease, but others have found that metachronous metastases occurring at over 6 months after initial treatment for lung cancer are likely to have a better prognosis. Significantly longer median OS was found in patients with metachronous metastatic disease compared with synchronous disease (31 versus 12 months) across 10 publications with a total of 114 patients undergoing adrenal metastasectomy for NSCLC171.
Only a few studies have compared adrenal metastasectomy against non-surgical treatments. A small study172 of nine patients with isolated adrenal metastasis showed that five patients who underwent adrenalectomy had a longer mean survival and mean disease-free interval than those who underwent palliative treatment (22 versus 3.5 months, and 7.5 versus 3.5 months, respectively). Similarly, a higher 5-year survival rate was reported among 20 patients undergoing adrenal metastasectomy compared with 17 non-surgically treated patients (34 versus 0 per cent respectively)173.
Melanoma
Adrenalectomy for metastatic melanoma secures better overall survival than non-operative management (Fig. 4). In a single-institution series174 of 91 patients with stage IV melanoma, median overall survival was 29 months after adrenalectomy and 9 months after non-operative management.
Fig. 4.
Metastatic melanoma to adrenal gland
In recent years, the increased efficacy of immune therapy for metastatic melanoma has created a new paradigm. Some metastatic sites respond well, such as lung metastases, but adrenal metastases fail to respond because these are highly resistant to immune checkpoint inhibitors175. In this context, the concept of sanctuary site for metastatic growth despite systemic therapy is evolving. In a recent series176 of 15 such patients, after a median follow-up of 24 months, 7 had no evidence of disease, 6 had progression with eventual death, whereas another patient had stable disease with maintenance therapy.
Surgical approach for adrenal metastases
Adrenal metastasectomy should be an R0 resection achieved by an en bloc removal of the entire adrenal gland together with part of the perinephric fat. The preferred surgical approach for adrenal metastases is minimally invasive surgery, but the data summarized in Table 5 show that open surgery is performed in a large proportion of these patients. Contraindications to minimally invasive surgery are the presence of locally advanced adrenal lesions and/or invasion or thrombus in major vessels. Even though the surgical treatment may be more challenging because of previous surgical procedures and adverse effects of systemic treatments, unilateral minimally invasive adrenalectomy is a safe procedure with a very low morbidity rate and long-term outcomes similar to those of open adrenalectomy. Whether a laparoscopic or retroperitoneoscopic approach is used is based on local expertise available in each centre. It is likely that robotic surgery will be considered for such patients in the future, but currently there are no published data to compare its efficacy.
Bilateral adrenalectomy for metastases
Surgical treatment is rarely indicated in patients with bilateral adrenal metastases owing to a high risk of multiple synchronous metastases. Concerns about additional morbidity related to adrenal insufficiency following bilateral adrenalectomy have to be balanced by the fact that bilateral adrenal involvement is likely to procreate adrenal insufficiency. Only a few bilateral adrenalectomies for adrenal metastases in NSCLC have been reported in the literature158,177,178.
The ESES recommendation is that decisions regarding bilateral adrenalectomy should be restricted to a small number of patients after full discussion within the MDT meeting and consideration of patients’ views (⊕⊕⊕⊕).
Subtotal adrenalectomy is rarely an option in such patients; because of the extent of adrenal involvement, it is unlikely to allow preservation of an adrenal remnant large enough to avoid adrenal insufficiency while mitigating the risk of local recurrence (⊕⊕○○).
Non-surgical treatments for adrenal metastases
SBRT is an effective non-invasive approach, although the number of studies using SBRT to treat adrenal metastases remains limited. Of 24 patients followed for more than 3 months, 15 had a partial response, four had a stable response, four had disease progression, and only one patient had a complete response179. In another study180 of 20 patients, the 1-year overall survival rate was 78 per cent and there was 100 per cent local control. Although the SBRT experience remains limited, some units have reported a doubling in the number of patients treated since 2016163. A pooled analysis181 of 1006 patients reported in 39 studies noted very good local control (82 per cent at 1 year, 63 per cent at 2 years), with reasonable overall survival (66 and 42 per cent respectively) after receiving a median biological equivalent dose of 67 Gy. Dose escalation between 60 and 100 Gy appeared to be associated with improved local control.
Transarterial chemoembolization is technically difficult owing to the complex vascular anatomy of the adrenal gland, and differences in vascularity patterns of adrenal metastases from different primary malignancies play a crucial role in the efficacy of this technique.
Stereotactic minimally invasive techniques including cryoablation, and thermal ablation of adrenal metastases with RFA or microwave ablation, have the potential for locoregional control, but experience remains very limited and efficacy unproven.
When is an adrenal tumour unresectable?
Radical surgery provides local control of disease for patients with ACC and malignant phaeochromocytoma, and might be the only option for patients with large adrenal metastases. The size of the tumour, its anatomical relationships, and the radiological suspicion of local invasion can lead to a decision to refuse surgery in some patients with advanced disease. In the absence of metastatic disease, it is imperative that all such patients are discussed with clinicians in a regional referral centre that can provide multidisciplinary surgical input, including endocrine, vascular, hepatic, and thoracic surgeons (as deemed necessary) before considering a tumour unresectable. Furthermore, neoadjuvant chemotherapy can facilitate surgery in patients who on presentation were considered to have a borderline (un)resectable tumour.
Extensive liver invasion, tumour encasing the origin of the superior mesenteric artery, and diffuse intraperitoneal metastases would be accepted as clear contraindications to surgery. Local invasion into the ipsilateral kidney or colon, vascular invasion into renal veins or IVC, and large tumour thrombus in the IVC can all be addressed during surgery if preoperative planning ensures that the structure of the surgical team is appropriate.
The ESES recommendation is that all adrenal tumours deemed inoperable should be referred to recognized expert surgeons or to a regional centre that can provide expertise in multiple surgical specialties working in an established multidisciplinary environment with oncologists (⊕⊕⊕○).
Conclusion
Most of the recommendations made in the present guidelines have been reinforced by recent publications, and progress has been made in several areas with an impact on clinical practice.
For ACC, there has been a proposed change to 20 HU as a threshold for suspicion of malignancy (replacing the 10-HU threshold used previously), and the role of urinary steroid profiling is increasingly being recognized. The use of neoadjuvant chemotherapy is encouraged for borderline resectable tumours, and the evidence in favour of adrenalectomy in patients presenting with metastatic disease has grown. Therefore, patients diagnosed with stage IV disease could be candidates for surgery.
There has been a significant change in the definition of phaeochromocytoma and PGGL that highlights the risk of recurrence/malignancy. Genetic testing has been proven to be beneficial for all patients as it can/should influence decisions regarding follow-up strategies, but concerns are acknowledged about the limited access to such tests. A large number of non-surgical treatments are being explored for metastatic disease.
Adrenal metastases are being resected in increasing numbers and more data have been published in favour of metastasectomy. Non-surgical methods are expanding, but their efficacy should be assessed against the long-term results of surgical metastasectomy.
The working group and the ESES membership emphasized the need for a multidisciplinary approach to all (complex) adrenal cases, and the benefits of referring patients with tumours that appear unresectable for discussion to an expert surgical centre recognized for expertise in this field, and where multispecialty surgical and oncological input can be provided for patients with advanced adrenal tumours.
Contributor Information
Radu Mihai, Churchill Cancer Centre, Oxford University Hospital NHS Foundation Trust, Oxford, UK.
Carmela De Crea, Centro di Ricerca in Chirurgia delle Ghiandole Endocrine e dell’Obesità, Università Cattolica del Sacro Cuore, Rome, Italy; Endocrine Surgery Unit, Hospital Fatebenefratelli Isola Tiberina—Gemelli Isola, Rome, Italy.
Carole Guerin, Department of Endocrine and Metabolic Surgery, Aix-Marseille University, Hôpital de La Conception, Marseille, France.
Francesca Torresan, Endocrine Surgery Unit, Department of Surgery, Oncology and Gastroenterology, University of Padua, Padua, Italy.
Orhan Agcaoglu, Department of General Surgery, Koc University School of Medicine, Istanbul, Turkey.
Razvan Simescu, Department of General and Endocrine Surgery, Medlife-Humanitas Hospital, Cluj-Napoca, Romania.
Martin K Walz, Department of Surgery and Minimally Invasive Surgery, Kliniken Essen-Mitte, Essen, Germany.
Funding
The authors have no funding to declare.
Author contributions
Radu Mihai (Conceptualization, Data curation, Formal analysis, Project administration, Supervision, Writing—original draft, Writing—review & editing), Carmela De Crea (Writing—original draft, Writing—review & editing), Carole Guerin (Data curation, Writing—original draft, Writing—review & editing), Francesca Torresan (Data curation, Writing—original draft, Writing—review & editing), Orhan Agcaoglu (Data curation, Writing—original draft, Writing—review & editing), Razvan Simescu (Data curation, Writing—original draft, Writing—review & editing), and Martin K. Walz (Conceptualization, Data curation, Writing—review & editing)
Disclosure
The authors declare no conflict of interest.
References
- 1. Hanna FWF, Hancock S, George C, Clark A, Sim J, Issa BGet al. Adrenal incidentaloma: prevalence and referral patterns from routine practice in a large UK university teaching hospital. J Endocr Soc 2022;6:bvab180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Henry JF, Peix JL, Kraimps JL. Positional statement of the European Society of Endocrine Surgeons (ESES) on malignant adrenal tumors. Langenbecks Arch Surg 2012;397:145–146 [DOI] [PubMed] [Google Scholar]
- 3. Fassnacht M, Dekkers OM, Else T, Baudin E, Berruti A, de Krijger Ret al. European Society of Endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018;179:G1–G46 [DOI] [PubMed] [Google Scholar]
- 4. Gaujoux S, Mihai R; Joint Working Group of ESES and ENSAT . European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma. Br J Surg 2017;104:358–376 [DOI] [PubMed] [Google Scholar]
- 5. Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JWet al. European Society of Endocrinology clinical practice guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016;174:G1–G10 [DOI] [PubMed] [Google Scholar]
- 6. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MHet al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915–1942 [DOI] [PubMed] [Google Scholar]
- 7. Fishbein L, Del Rivero J, Else T, Howe JR, Asa SL, Cohen DLet al. The North American Neuroendocrine Tumor Society Consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas 2021;50:469–493 [DOI] [PubMed] [Google Scholar]
- 8. Fassnacht M, Assie G, Baudin E, Eisenhofer G, de la Fouchardiere C, Haak HRet al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020;31:1476–1490 [DOI] [PubMed] [Google Scholar]
- 9. Taieb D, Wanna GB, Ahmad M, Lussey-Lepoutre C, Perrier ND, Nolting Set al. Clinical consensus guideline on the management of phaeochromocytoma and paraganglioma in patients harbouring germline SDHD pathogenic variants. Lancet Diabetes Endocrinol 2023;11:345–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guyatt GH, Oxman AD, Sultan S, Glasziou P, Akl EA, Alonso-Coello Pet al. GRADE guidelines: 9. Rating up the quality of evidence. J Clin Epidemiol 2011;64:1311–1316 [DOI] [PubMed] [Google Scholar]
- 11. Bancos I, Taylor AE, Chortis V, Sitch AJ, Jenkinson C, Davidge-Pitts CJet al. Urine steroid metabolomics for the differential diagnosis of adrenal incidentalomas in the EURINE-ACT study: a prospective test validation study. Lancet Diabetes Endocrinol 2020;8:773–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yalon T, Yalon M, Assaf D, Lenartowicz K, Foster T, Lyden Met al. Differentiating between adrenocortical carcinoma and lipid-poor cortical adenoma: a novel cross-sectional imaging-based score. Surgery 2023;173:35–42 [DOI] [PubMed] [Google Scholar]
- 13. Sinclair TJ, Gillis A, Alobuia WM, Wild H, Kebebew E. Surgery for adrenocortical carcinoma: when and how? Best Pract Res Clin Endocrinol Metab 2020;34:101408. [DOI] [PubMed] [Google Scholar]
- 14. Amodru V, Taieb D, Guerin C, Paladino NC, Brue T, Sebag Fet al. Large adrenal incidentalomas require a dedicated diagnostic procedure. Endocr Pract 2019;25:669–677 [DOI] [PubMed] [Google Scholar]
- 15. Guerin C, Pattou F, Brunaud L, Lifante JC, Mirallie E, Haissaguerre Met al. Performance of 18F-FDG PET/CT in the characterization of adrenal masses in noncancer patients: a prospective study. J Clin Endocrinol Metab 2017;102:2465–2472 [DOI] [PubMed] [Google Scholar]
- 16. Kim SJ, Lee SW, Pak K, Kim IJ, Kim K. Diagnostic accuracy of 18F-FDG PET or PET/CT for the characterization of adrenal masses: a systematic review and meta-analysis. Br J Radiol 2018;91:20170520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khan TS, Sundin A, Juhlin C, Langstrom B, Bergstrom M, Eriksson B. 11C-metomidate PET imaging of adrenocortical cancer. Eur J Nucl Med Mol Imaging 2003;30:403–410 [DOI] [PubMed] [Google Scholar]
- 18. Naffouje SA, Sabesan A, Hallanger-Johnson J, Kirtane K, Gonzalez RJ, Mullinax J. Adrenal biopsy, as a diagnostic method, is associated with decreased overall survival in patients with T1/T2 adrenocortical carcinoma: a propensity score-matched analysis. J Surg Oncol 2021;124:1261–1271 [DOI] [PubMed] [Google Scholar]
- 19. Elhassan YS, Altieri B, Berhane S, Cosentini D, Calabrese A, Haissaguerre Met al. S-GRAS score for prognostic classification of adrenocortical carcinoma: an international, multicenter ENSAT study. Eur J Endocrinol 2021;186:25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Asare EA, Wang TS, Winchester DP, Mallin K, Kebebew E, Sturgeon C. A novel staging system for adrenocortical carcinoma better predicts survival in patients with stage I/II disease. Surgery 2014;156:1378–1385; discussion 1385–1396 [DOI] [PubMed] [Google Scholar]
- 21. He S, Huang X, Zhao P, Zhang P. The prognosis difference between elderly and younger patients with adrenocortical carcinoma. Front Genet 2023;13:1029155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corcuff JB, Young J, Masquefa-Giraud P, Chanson P, Baudin E, Tabarin A. Rapid control of severe neoplastic hypercortisolism with metyrapone and ketoconazole. Eur J Endocrinol 2015;172:473–481 [DOI] [PubMed] [Google Scholar]
- 23. Kamenicky P, Droumaguet C, Salenave S, Blanchard A, Jublanc C, Gautier JFet al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab 2011;96:2796–2804 [DOI] [PubMed] [Google Scholar]
- 24. Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol 2012;167:137–143 [DOI] [PubMed] [Google Scholar]
- 25. Mihai R. Diagnosis, treatment and outcome of adrenocortical cancer. Br J Surg 2015;102:291–306 [DOI] [PubMed] [Google Scholar]
- 26. Nakanishi H, Miangul S, Wang R, El Haddad J, El Ghazal N, Abdulsalam FAet al. Open versus laparoscopic surgery in the management of adrenocortical carcinoma: a systematic review and meta-analysis. Ann Surg Oncol 2023;30:994–1005 [DOI] [PubMed] [Google Scholar]
- 27. Hu X, Yang WX, Shao YX, Dou WC, Xiong SC, Li X. Minimally invasive versus open adrenalectomy in patients with adrenocortical carcinoma: a meta-analysis. Ann Surg Oncol 2020;27:3858–3869 [DOI] [PubMed] [Google Scholar]
- 28. Autorino R, Bove P, De Sio M, Miano R, Micali S, Cindolo Let al. Open versus laparoscopic adrenalectomy for adrenocortical carcinoma: a meta-analysis of surgical and oncological outcomes. Ann Surg Oncol 2016;23:1195–1202 [DOI] [PubMed] [Google Scholar]
- 29. Langenhuijsen J, Birtle A, Klatte T, Porpiglia F, Timsit MO. Surgical management of adrenocortical carcinoma: impact of laparoscopic approach, lymphadenectomy, and surgical volume on outcomes—a systematic review and meta-analysis of the current literature. Eur Urol Focus 2016;1:241–250 [DOI] [PubMed] [Google Scholar]
- 30. Rossi L, Becucci C, Ambrosini CE, Puccini M, Vasquez MC, Gjeloshi Bet al. Surgical management of adrenocortical carcinoma: a literature review. J Clin Med 2022;11:5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ginsburg KB, Castro Bigalli AA, Schober JP, Perlman D, Handorf EA, Chen DYTet al. Association of tumor size and surgical approach with oncological outcomes and overall survival in patients with adrenocortical carcinoma. Urol Oncol 2022;40:455.e19–455.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Skertich NJ, Tierney JF, Chivukula SV, Babazadeh NT, Hertl M, Poirier Jet al. Risk factors associated with positive resection margins in patients with adrenocortical carcinoma. Am J Surg 2020;220:932–937 [DOI] [PubMed] [Google Scholar]
- 33. Brix D, Allolio B, Fenske W, Agha A, Dralle H, Jurowich Cet al. Laparoscopic versus open adrenalectomy for adrenocortical carcinoma: surgical and oncologic outcome in 152 patients. Eur Urol 2010;58:609–615 [DOI] [PubMed] [Google Scholar]
- 34. Cooper AB, Habra MA, Grubbs EG, Bednarski BK, Ying AK, Perrier NDet al. Does laparoscopic adrenalectomy jeopardize oncologic outcomes for patients with adrenocortical carcinoma? Surg Endosc 2013;27:4026–4032 [DOI] [PubMed] [Google Scholar]
- 35. Fossa A, Rosok BI, Kazaryan AM, Holte HJ, Brennhovd B, Westerheim Oet al. Laparoscopic versus open surgery in stage I–III adrenocortical carcinoma—a retrospective comparison of 32 patients. Acta Oncol 2013;52:1771–1777 [DOI] [PubMed] [Google Scholar]
- 36. Miller BS, Ammori JB, Gauger PG, Broome JT, Hammer GD, Doherty GM. Laparoscopic resection is inappropriate in patients with known or suspected adrenocortical carcinoma. World J Surg 2010;34:1380–1385 [DOI] [PubMed] [Google Scholar]
- 37. Mir MC, Klink JC, Guillotreau J, Long JA, Miocinovic R, Kaouk JHet al. Comparative outcomes of laparoscopic and open adrenalectomy for adrenocortical carcinoma: single, high-volume center experience. Ann Surg Oncol 2013;20:1456–1461 [DOI] [PubMed] [Google Scholar]
- 38. Calcatera NA, Hsiung-Wang C, Suss NR, Winchester DJ, Moo-Young TA, Prinz RA. Minimally invasive adrenalectomy for adrenocortical carcinoma: five-year trends and predictors of conversion. World J Surg 2018;42:473–481 [DOI] [PubMed] [Google Scholar]
- 39. Hue JJ, Ahorukomeye P, Bingmer K, Drapalik L, Ammori JB, Wilhelm SMet al. A comparison of robotic and laparoscopic minimally invasive adrenalectomy for adrenal malignancies. Surg Endosc 2022;36:5374–5381 [DOI] [PubMed] [Google Scholar]
- 40. Delozier OM, Stiles ZE, Deschner BW, Drake JA, Deneve JL, Glazer ESet al. Implications of conversion during attempted minimally invasive adrenalectomy for adrenocortical carcinoma. Ann Surg Oncol 2021;28:492–501 [DOI] [PubMed] [Google Scholar]
- 41. Parasiliti-Caprino M, Bioletto F, Lopez C, Bollati M, Maletta F, Caputo Met al. From SGAP-model to SGAP-score: a simplified predictive tool for post-surgical recurrence of pheochromocytoma. Biomedicines 2022;10:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mihai R, Donatini G, Vidal O, Brunaud L. Volume–outcome correlation in adrenal surgery—an ESES consensus statement. Langenbecks Arch Surg 2019;404:795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gray WK, Day J, Briggs TWR, Wass JAH, Lansdown M. Volume–outcome relationship for adrenalectomy: analysis of an administrative dataset for the Getting It Right First Time Programme. Br J Surg 2021;108:1112–1119 [DOI] [PubMed] [Google Scholar]
- 44. Porpiglia F, Fiori C, Daffara FC, Zaggia B, Ardito A, Scarpa RMet al. Does nephrectomy during radical adrenalectomy for stage II adrenocortical cancer affect patient outcome? J Endocrinol Invest 2016;39:465–471 [DOI] [PubMed] [Google Scholar]
- 45. Greco R, Tsappa I, Mihai R, Petrou M. Surgical management of adrenal tumours extending into the right atrium. Gland Surg 2019;8:S53–S59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mihai R, Iacobone M, Makay O, Moreno P, Frilling A, Kraimps JLet al. Outcome of operation in patients with adrenocortical cancer invading the inferior vena cava—a European Society of Endocrine Surgeons (ESES) survey. Langenbecks Arch Surg 2012;397:225–231 [DOI] [PubMed] [Google Scholar]
- 47. Ciancio G, Farag A, Gonzalez J, Gaynor JJ. Adrenal tumors of different types with or without tumor thrombus invading the inferior vena cava: an evaluation of 33 cases. Surg Oncol 2021;37:101544. [DOI] [PubMed] [Google Scholar]
- 48. Deschner BW, Stiles ZE, DeLozier OM, Drake JA, Tsao M, Glazer ESet al. Critical analysis of lymph node examination in patients undergoing curative-intent resection for adrenocortical carcinoma. J Surg Oncol 2020;122:1152–1162 [DOI] [PubMed] [Google Scholar]
- 49. Tseng J, DiPeri T, Chen Y, Shouhed D, Ben-Shlomo A, Burch Met al. Adrenocortical carcinoma: the value of lymphadenectomy. Ann Surg Oncol 2022;29:1965–1970 [DOI] [PubMed] [Google Scholar]
- 50. Bednarski BK, Habra MA, Phan A, Milton DR, Wood C, Vauthey Net al. Borderline resectable adrenal cortical carcinoma: a potential role for preoperative chemotherapy. World J Surg 2014;38:1318–1327 [DOI] [PubMed] [Google Scholar]
- 51. Kobayakawa Y, Hamamoto S, Kamisawa H, Okada S, Taguchi K, Naiki Tet al. Long-term survival of a patient with refractory advanced adrenocortical carcinoma after combination chemotherapy with paclitaxel and carboplatin plus mitotane. IJU Case Rep 2022;5:288–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kiesewetter B, Riss P, Scheuba C, Mazal P, Kretschmer-Chott E, Haug Aet al. Management of adrenocortical carcinoma: are we making progress? Ther Adv Med Oncol 2021;13:17588359211038409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lagana M, Grisanti S, Cosentini D, Ferrari VD, Lazzari B, Ambrosini Ret al. Efficacy of the EDP-M scheme plus adjunctive surgery in the management of patients with advanced adrenocortical carcinoma: the Brescia experience. Cancers (Basel) 2020;12:941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prendergast KM, Smith PM, Tran TB, Postlewait LM, Maithel SK, Prescott JDet al. Features of synchronous versus metachronous metastasectomy in adrenal cortical carcinoma: analysis from the US adrenocortical carcinoma database. Surgery 2020;167:352–357 [DOI] [PubMed] [Google Scholar]
- 55. Wang S, Gao WC, Chen SS, Bai L, Luo L, Zheng XGet al. Primary site surgery for metastatic adrenocortical carcinoma improves survival outcomes: an analysis of a population-based database. Onco Targets Ther 2017;10:5311–5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Srougi V, Bancos I, Daher M, Lee JE, Graham PH, Karam JAet al. Cytoreductive surgery of the primary tumor in metastatic adrenocortical carcinoma: impact on patients’ survival. J Clin Endocrinol Metab 2022;107:964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fuller SN, Shafiei A, Venzon DJ, Liewehr DJ, Mauda Havanuk M, Ilanchezhian MGet al. Tumor doubling time using CT volumetric segmentation in metastatic adrenocortical carcinoma. Curr Oncol 2021;28:4357–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang F, Liu Z, Liang J, Tang Y, Liu S, Zhou Cet al. Operative intervention for recurrence of adrenocortical carcinoma: a single-center experience. Surgery 2021;169:1131–1138 [DOI] [PubMed] [Google Scholar]
- 59. Simon G, Pattou F, Mirallie E, Lifante JC, Nomine C, Arnault Vet al. Surgery for recurrent adrenocortical carcinoma: a multicenter retrospective study. Surgery 2017;161:249–256 [DOI] [PubMed] [Google Scholar]
- 60. Zhang F, Liu Z, Feng D, Tang Y, Liu S, Wu Ket al. Reoperation for recurrent adrenocortical carcinoma: a systematic review and pooled analysis of population-based studies. Front Surg 2022;9:781406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Calabrese A, Puglisi S, Borin C, Basile V, Perotti P, Pia Aet al. The management of postoperative disease recurrence in patients with adrenocortical carcinoma: a retrospective study in 106 patients. Eur J Endocrinol 2023;188:118–124 [DOI] [PubMed] [Google Scholar]
- 62. Tran TB, Maithel SK, Pawlik TM, Wang TS, Hatzaras I, Phay JEet al. Clinical score predicting long-term survival after repeat resection for recurrent adrenocortical carcinoma. J Am Coll Surg 2016;223:794–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Veltri A, Basile D, Calandri M, Bertaggia C, Volante M, Porpiglia Fet al. Oligometastatic adrenocortical carcinoma: the role of image-guided thermal ablation. Eur Radiol 2020;30:6958–6964 [DOI] [PubMed] [Google Scholar]
- 64. Baur J, Buntemeyer TO, Megerle F, Deutschbein T, Spitzweg C, Quinkler Met al. Outcome after resection of adrenocortical carcinoma liver metastases: a retrospective study. BMC Cancer 2017;17:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ayabe RI, Narayan RR, Ruff SM, Wach MM, Lo W, Nierop PMHet al. Disease-free interval and tumor functional status can be used to select patients for resection/ablation of liver metastases from adrenocortical carcinoma: insights from a multi-institutional study. HPB (Oxford) 2020;22:169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Owen DH, Patel S, Wei L, Phay JE, Shirley LA, Kirschner LSet al. Metastatic adrenocortical carcinoma: a single institutional experience. Horm Cancer 2019;10:161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Makary MS, Krishner LS, Wuthrick EJ, Bloomston MP, Dowell JD. Yttrium-90 microsphere selective internal radiation therapy for liver metastases following systemic chemotherapy and surgical resection for metastatic adrenocortical carcinoma. World J Clin Oncol 2018;9:20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lin TY, Lin KY, Kuo HY, Huang KH, Wang CY, Lin YLet al. Yttrium-90 selective internal radiation therapy plus cryoablation for recurrent adrenocortical carcinoma with liver metastases. J Endocr Soc 2022;6:bvac091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lu S, Dhillon J, Johnson JH, El-Haddad G. Yttrium-90 radioembolization of isolated hepatic adrenocortical carcinoma metastases with negative surgical pathology. EJNMMI Res 2021;11:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Berruti A, Libe R, Lagana M, Ettaieb H, Sukkari MA, Bertherat Jet al. Morbidity and mortality of bone metastases in advanced adrenocortical carcinoma: a multicenter retrospective study. Eur J Endocrinol 2019;180:311–320 [DOI] [PubMed] [Google Scholar]
- 71. de Jong MC, Khan S, Christakis I, Weaver A, Mihai R. Comparative performances of nomograms and conditional survival after resection of adrenocortical cancer. BJS Open 2021;5:zraa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lin W, Dai J, Xie J, Liu J, Sun F, Huang Xet al. S-GRAS score performs better than a model from SEER for patients with adrenocortical carcinoma. Endocr Connect 2022;11:e220114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mangone A, Altieri B, Detomas M, Prete A, Abbas H, Asia Met al. Inflammation-based scores as predictors of treatment response in advanced adrenocortical carcinoma. Endocr Relat Cancer 2023;30:e220372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. de Jong MC, Mihai R, Khan S. Neutrophil-to-lymphocyte ratio (NLR) and platelet-to-lymphocyte ratio (PLR) as possible prognostic markers for patients undergoing resection of adrenocortical carcinoma. World J Surg 2021;45:754–764 [DOI] [PubMed] [Google Scholar]
- 75. Assie G, Jouinot A, Bertherat J. The ‘omics’ of adrenocortical tumours for personalized medicine. Nat Rev Endocrinol 2014;10:215–228 [DOI] [PubMed] [Google Scholar]
- 76. Lerario AM, Mohan DR, Hammer GD. Update on biology and genomics of adrenocortical carcinomas: rationale for emerging therapies. Endocr Rev 2022;43:1051–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mete O, Erickson LA, Juhlin CC, de Krijger RR, Sasano H, Volante Met al. Overview of the 2022 WHO classification of adrenal cortical tumors. Endocr Pathol 2022;33:155–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kanitra JJ, Hardaway JC, Soleimani T, Koehler TJ, McLeod MK, Kavuturu S. Adrenocortical oncocytic neoplasm: a systematic review. Surgery 2018;164:1351–1359 [DOI] [PubMed] [Google Scholar]
- 79. Renaudin K, Smati S, Wargny M, Al Ghuzlan A, Aubert S, Leteurtre Eet al. Clinicopathological description of 43 oncocytic adrenocortical tumors: importance of Ki-67 in histoprognostic evaluation. Mod Pathol 2018;31:1708–1716 [DOI] [PubMed] [Google Scholar]
- 80. Tizianel I, Caccese M, Torresan F, Lombardi G, Evangelista L, Crimi Fet al. The overall survival and progression-free survival in patients with advanced adrenocortical cancer is increased after the multidisciplinary team evaluation. Cancers (Basel) 2022;14:3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol 2022;33:90–114 [DOI] [PubMed] [Google Scholar]
- 82. Pamporaki C, Prodanov T, Meuter L, Berends AMA, Bechmann N, Constantinescu Get al. Determinants of disease-specific survival in patients with and without metastatic pheochromocytoma and paraganglioma. Eur J Cancer 2022;169:32–41 [DOI] [PubMed] [Google Scholar]
- 83. Cho YY, Kwak MK, Lee SE, Ahn SH, Kim H, Suh Set al. A clinical prediction model to estimate the metastatic potential of pheochromocytoma/paraganglioma: ASES score. Surgery 2018;164:511–517 [DOI] [PubMed] [Google Scholar]
- 84. Hamidi O, Young WF Jr, Iniguez-Ariza NM, Kittah NE, Gruber L, Bancos Cet al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab 2017;102:3296–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Parasiliti-Caprino M, Lucatello B, Lopez C, Burrello J, Maletta F, Mistrangelo Met al. Predictors of recurrence of pheochromocytoma and paraganglioma: a multicenter study in Piedmont, Italy. Hypertens Res 2020;43:500–510 [DOI] [PubMed] [Google Scholar]
- 86. Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar Let al. Prognosis of malignant pheochromocytoma and paraganglioma (MAPP-prono study): a European Network for the study of adrenal tumors retrospective study. J Clin Endocrinol Metab 2019;104:2367–2374 [DOI] [PubMed] [Google Scholar]
- 87. Cho JW, Lee YM, Sung TY, Yoon JH, Chung KW, Hong SJ. Factors related to improved clinical outcomes associated with adrenalectomy for metachronous adrenal metastases from solid primary carcinomas. Surg Oncol 2018;27:18–22 [DOI] [PubMed] [Google Scholar]
- 88. Eisenhofer G, Deutschbein T, Constantinescu G, Langton K, Pamporaki C, Calsina Bet al. Plasma metanephrines and prospective prediction of tumor location, size and mutation type in patients with pheochromocytoma and paraganglioma. Clin Chem Lab Med 2020;59:353–363 [DOI] [PubMed] [Google Scholar]
- 89. Eisenhofer G, Prejbisz A, Peitzsch M, Pamporaki C, Masjkur J, Rogowski-Lehmann Net al. Biochemical diagnosis of chromaffin cell tumors in patients at high and low risk of disease: plasma versus urinary free or deconjugated O-methylated catecholamine metabolites. Clin Chem 2018;64:1646–1656 [DOI] [PubMed] [Google Scholar]
- 90. Rao D, Peitzsch M, Prejbisz A, Hanus K, Fassnacht M, Beuschlein Fet al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol 2017;177:103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Crona J, Lamarca A, Ghosal S, Welin S, Skogseid B, Pacak K. Genotype–phenotype correlations in pheochromocytoma and paraganglioma: a systematic review and individual patient meta-analysis. Endocr Relat Cancer 2019;26:539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nolting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss Met al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev 2022;43:199–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Garcia-Carbonero R, Matute Teresa F, Mercader-Cidoncha E, Mitjavila-Casanovas M, Robledo M, Tena Iet al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin Transl Oncol 2021;23:1995–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Buffet A, Ben Aim L, Leboulleux S, Drui D, Vezzosi D, Libe Ret al. Positive impact of genetic test on the management and outcome of patients with paraganglioma and/or pheochromocytoma. J Clin Endocrinol Metab 2019;104:1109–1118 [DOI] [PubMed] [Google Scholar]
- 95. Wang H, Papachristos AJ, Gill AJ, Clifton-Bligh R, Aniss AM, Glover Aet al. Genotype–phenotype correlations and clinical outcomes in 155 cases of pheochromocytoma and paraganglioma. World J Surg 2023;47:690–698 [DOI] [PubMed] [Google Scholar]
- 96. Jimenez C, Ma J, Roman Gonzalez A, Varghese J, Zhang M, Perrier Net al. TNM staging and overall survival in patients with pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab 2022;107:1239–1246 [DOI] [PubMed] [Google Scholar]
- 97. Cascon A, Calsina B, Monteagudo M, Mellid S, Diaz-Talavera A, Curras-Freixes Met al. Genetic bases of pheochromocytoma and paraganglioma. J Mol Endocrinol 2023;70:e220167. [DOI] [PubMed] [Google Scholar]
- 98. Andrews KA, Ascher DB, Pires DEV, Barnes DR, Vialard L, Casey RTet al. Tumour risks and genotype–phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet 2018;55:384–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Horton C, LaDuca H, Deckman A, Durda K, Jackson M, Richardson MEet al. Universal germline panel testing for individuals with pheochromocytoma and paraganglioma produces high diagnostic yield. J Clin Endocrinol Metab 2022;107:e1917–e1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Calsina B, Pineiro-Yanez E, Martinez-Montes AM, Caleiras E, Fernandez-Sanroman A, Monteagudo Met al. Genomic and immune landscape of metastatic pheochromocytoma and paraganglioma. Nat Commun 2023;14:1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Thompson LD. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002;26:551–566 [DOI] [PubMed] [Google Scholar]
- 102. Stenman A, Zedenius J, Juhlin CC. The value of histological algorithms to predict the malignancy potential of pheochromocytomas and abdominal paragangliomas—a meta-analysis and systematic review of the literature. Cancers (Basel) 2019;11:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi Net al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer 2014;21:405–414 [DOI] [PubMed] [Google Scholar]
- 104. Kimura N, Takekoshi K, Naruse M. Risk stratification on pheochromocytoma and paraganglioma from laboratory and clinical medicine. J Clin Med 2018;7:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wachtel H, Hutchens T, Baraban E, Schwartz LE, Montone K, Baloch Zet al. Predicting metastatic potential in pheochromocytoma and paraganglioma: a comparison of PASS and GAPP scoring systems. J Clin Endocrinol Metab 2020;105:e4661–e4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Koh JM, Ahn SH, Kim H, Kim BJ, Sung TY, Kim YHet al. Validation of pathological grading systems for predicting metastatic potential in pheochromocytoma and paraganglioma. PLoS One 2017;12:e0187398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang Y, Li M, Deng H, Pang Y, Liu L, Guan X. The systems of metastatic potential prediction in pheochromocytoma and paraganglioma. Am J Cancer Res 2020;10:769–780 [PMC free article] [PubMed] [Google Scholar]
- 108. Pierre C, Agopiantz M, Brunaud L, Battaglia-Hsu SF, Max A, Pouget Cet al. COPPS, a composite score integrating pathological features, PS100 and SDHB losses, predicts the risk of metastasis and progression-free survival in pheochromocytomas/paragangliomas. Virchows Arch 2019;474:721–734 [DOI] [PubMed] [Google Scholar]
- 109. Abdel-Rahman O. Assessment of the AJCC staging system of pheochromocytomas/paragangliomas. Endocrine 2022;75:239–243 [DOI] [PubMed] [Google Scholar]
- 110. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson ARet al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017;31:181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Powers JF, Tischler AS. Immunohistochemical staining for SOX10 and SDHB in SDH-deficient paragangliomas indicates that sustentacular cells are not neoplastic. Endocr Pathol 2020;31:307–309 [DOI] [PubMed] [Google Scholar]
- 112. Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018;72:106–116 [DOI] [PubMed] [Google Scholar]
- 113. Rao D, van Berkel A, Piscaer I, Young WF, Gruber L, Deutschbein Tet al. Impact of 123I-MIBG scintigraphy on clinical decision making in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2019;104:3812–3820 [DOI] [PubMed] [Google Scholar]
- 114. Brito JP, Asi N, Gionfriddo MR, Norman C, Leppin AL, Zeballos-Palacios Cet al. The incremental benefit of functional imaging in pheochromocytoma/paraganglioma: a systematic review. Endocrine 2015;50:176–186 [DOI] [PubMed] [Google Scholar]
- 115. Taieb D, Hicks RJ, Hindie E, Guillet BA, Avram A, Ghedini Pet al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2019;46:2112–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Xu S, Pan Y, Zhou J, Ju H, Zhang Y. Integrated PET/MRI with 68Ga-DOTATATE and 18F-FDG in pheochromocytomas and paragangliomas: an initial study. Clin Nucl Med 2022;47:299–304 [DOI] [PubMed] [Google Scholar]
- 117. Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of 68Ga-DOTA-conjugated somatostatin receptor-targeting peptide PET in detection of pheochromocytoma and paraganglioma: a systematic review and metaanalysis. J Nucl Med 2019;60:369–376 [DOI] [PubMed] [Google Scholar]
- 118. Holscher I, van den Berg TJ, Dreijerink KMA, Engelsman AF, Nieveen van Dijkum EJM. Recurrence rate of sporadic pheochromocytomas after curative adrenalectomy: a systematic review and meta-analysis. J Clin Endocrinol Metab 2021;106:588–597 [DOI] [PubMed] [Google Scholar]
- 119. Torresan F, Beber A, Schiavone D, Zovato S, Galuppini F, Crimi Fet al. Long-term outcomes after surgery for pheochromocytoma and sympathetic paraganglioma. Cancers (Basel) 2023;15:2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bílek R, Zelinka T, Vlček P, Duškova J, Michalský D, Novák Ket al. Radioimmunoassay of chromogranin A and free metanephrines in diagnosis of pheochromocytoma. Physiol Res 2017;66:S397–S408 [DOI] [PubMed] [Google Scholar]
- 121. Parisien-La Salle S, Provencal M, Bourdeau I. Chromogranin A in a cohort of pheochromocytomas and paragangliomas: usefulness at diagnosis and as an early biomarker of recurrence. Endocr Pract 2021;27:318–325 [DOI] [PubMed] [Google Scholar]
- 122. Bilek R, Vlcek P, Safarik L, Michalsky D, Novak K, Duskova Jet al. Chromogranin A in the laboratory diagnosis of pheochromocytoma and paraganglioma. Cancers (Basel) 2019;11:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zuber S, Wesley R, Prodanov T, Eisenhofer G, Pacak K, Kantorovich V. Clinical utility of chromogranin A in SDHx-related paragangliomas. Eur J Clin Invest 2014;44:365–371 [DOI] [PubMed] [Google Scholar]
- 124. Kong G, Schenberg T, Yates CJ, Trainer A, Sachithanandan N, Iravani Aet al. The role of 68Ga-DOTA-octreotate PET/CT in follow-up of SDH-associated pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2019;104:5091–5099 [DOI] [PubMed] [Google Scholar]
- 125. Carrasquillo JA, Chen CC, Jha A, Pacak K, Pryma DA, Lin FI. Systemic radiopharmaceutical therapy of pheochromocytoma and paraganglioma. J Nucl Med 2021;62:1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Takenaka J, Watanabe S, Abe T, Hirata K, Uchiyama Y, Kimura Ret al. Prognostic value of [18F]FDG-PET prior to [131I]MIBG treatment for pheochromocytoma and paraganglioma (PPGL). Ann Nucl Med 2023;37:10–17 [DOI] [PubMed] [Google Scholar]
- 127. Jimenez C, Erwin W, Chasen B. Targeted radionuclide therapy for patients with metastatic pheochromocytoma and paraganglioma: from low-specific-activity to high-specific-activity Iodine-131 Metaiodobenzylguanidine. Cancers (Basel) 2019;11:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. van Hulsteijn LT, Niemeijer ND, Dekkers OM, Corssmit EP. 131I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf) 2014;80:487–501 [DOI] [PubMed] [Google Scholar]
- 129. Pryma DA, Chin BB, Noto RB, Dillon JS, Perkins S, Solnes Let al. Efficacy and safety of high-specific-activity 131I-MIBG therapy in patients with advanced pheochromocytoma or paraganglioma. J Nucl Med 2019;60:623–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Hope TA, Abbott A, Colucci K, Bushnell DL, Gardner L, Graham WSet al. NANETS/SNMMI procedure standard for somatostatin receptor-based peptide receptor radionuclide therapy with 177Lu-DOTATATE. J Nucl Med 2019;60:937–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Satapathy S, Mittal BR, Bhansali A. Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: a systematic review and meta-analysis. Clin Endocrinol (Oxf) 2019;91:718–727 [DOI] [PubMed] [Google Scholar]
- 132. Capdevila J, Grande E, Garcia-Carbonero R, Simo M, Del Olmo-Garcia MI, Jimenez-Fonseca Pet al. Position statement on the diagnosis, treatment, and response evaluation to systemic therapies of advanced neuroendocrine tumors, with a special focus on radioligand therapy. Oncologist 2022;27:e328–e339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb Det al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens 2020;38:1443–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhou Y, Cui Y, Zhang D, Tong A. Efficacy and safety of tyrosine kinase inhibitors in patients with metastatic pheochromocytomas/paragangliomas: a systematic review and meta-analysis. J Clin Endocrinol Metab 2022;107:1667–167835106590 [Google Scholar]
- 135. Urquhart C, Fleming B, Harper I, Aloj L, Armstrong R, Hook Let al. The use of temozolomide in paediatric metastatic phaeochromocytoma/paraganglioma: a case report and literature review. Front Endocrinol (Lausanne) 2022;13:1066208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hadoux J, Favier J, Scoazec JY, Leboulleux S, Al Ghuzlan A, Caramella Cet al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer 2014;135:2711–2720 [DOI] [PubMed] [Google Scholar]
- 137. Perez K, Jacene H, Hornick JL, Ma C, Vaz N, Brais LKet al. SDHx mutations and temozolomide in malignant pheochromocytoma and paraganglioma. Endocr Relat Cancer 2022;29:533–544 [DOI] [PubMed] [Google Scholar]
- 138. Tong A, Li M, Cui Y, Ma X, Wang H, Li Y. Temozolomide is a potential therapeutic tool for patients with metastatic pheochromocytoma/paraganglioma—case report and review of the literature. Front Endocrinol (Lausanne) 2020;11:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Jimenez C, Subbiah V, Stephen B, Ma J, Milton D, Xu Met al. Phase II clinical trial of pembrolizumab in patients with progressive metastatic pheochromocytomas and paragangliomas. Cancers (Basel) 2020;12:2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Vogel J, Atanacio AS, Prodanov T, Turkbey BI, Adams K, Martucci Vet al. External beam radiation therapy in treatment of malignant pheochromocytoma and paraganglioma. Front Oncol 2014;4:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Breen W, Bancos I, Young WF Jr, Bible KC, Laack NN, Foote RLet al. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv Radiat Oncol 2018;3:25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Mesko S, Deegan BJ, D’Souza NM, Ghia AJ, Chapman BV, Amini Bet al. Spine stereotactic radiosurgery for metastatic pheochromocytoma. Cureus 2019;11:e4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gu Z, Hu K, Liang Y, Zhang F, Tong A, Hou X. Favorable outcome in advanced pheochromocytoma and paraganglioma after hypofractionated intensity modulated radiotherapy. J Endocrinol Invest 2023;46:477–485 [DOI] [PubMed] [Google Scholar]
- 144. Kohlenberg J, Welch B, Hamidi O, Callstrom M, Morris J, Sprung Jet al. Efficacy and safety of ablative therapy in the treatment of patients with metastatic pheochromocytoma and paraganglioma. Cancers (Basel) 2019;11:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Deljou A, Kohlenberg JD, Weingarten TN, Bancos I, Young WF Jr, Schroeder DRet al. Hemodynamic instability during percutaneous ablation of extra-adrenal metastases of pheochromocytoma and paragangliomas: a case series. BMC Anesthesiol 2018;18:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Robertson V, Poli F, Hobson B, Saratzis A, Ross Naylor A. A systematic review and meta-analysis of the presentation and surgical management of patients with carotid body tumours. Eur J Vasc Endovasc Surg 2019;57:477–486 [DOI] [PubMed] [Google Scholar]
- 147. Aspinal S, Mihai R, Kinsman R, Walton P. Sixth National Audit Report of the United Kingdom Registry of Endocrine and Thyroid Surgery. https://www.baets.org.uk/wp-content/uploads/BAETS-Sixth-Audit-Report-FINAL.pdf
- 148. Kasperlik-Zeluska AA, Roslonowska E, Slowinska-Srzednicka J, Migdalska B, Jeske W, Makowska Aet al. Incidentally discovered adrenal mass (incidentaloma): investigation and management of 208 patients. Clin Endocrinol (Oxf) 1997;46:29–37 [DOI] [PubMed] [Google Scholar]
- 149. Song JH, Grand DJ, Beland MD, Chang KJ, Machan JT, Mayo-Smith WW. Morphologic features of 211 adrenal masses at initial contrast-enhanced CT: can we differentiate benign from malignant lesions using imaging features alone? AJR Am J Roentgenol 2013;201:1248–1253 [DOI] [PubMed] [Google Scholar]
- 150. Dinnes J, Bancos I, Ferrante di Ruffano L, Chortis V, Davenport C, Bayliss Set al. Management of endocrine disease: imaging for the diagnosis of malignancy in incidentally discovered adrenal masses: a systematic review and meta-analysis. Eur J Endocrinol 2016;175:R51–R64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ansquer C, Scigliano S, Mirallie E, Taieb D, Brunaud L, Sebag Fet al. 18F-FDG PET/CT in the characterization and surgical decision concerning adrenal masses: a prospective multicentre evaluation. Eur J Nucl Med Mol Imaging 2010;37:1669–1678 [DOI] [PubMed] [Google Scholar]
- 152. Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev Aet al. Management of adrenal incidentalomas: European Society of Endocrinology clinical practice guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016;175:G1–G34 [DOI] [PubMed] [Google Scholar]
- 153. Bancos I, Tamhane S, Shah M, Delivanis DA, Alahdab F, Arlt Wet al. Diagnosis of endocrine disease: the diagnostic performance of adrenal biopsy: a systematic review and meta-analysis. Eur J Endocrinol 2016;175:R65–R80 [DOI] [PubMed] [Google Scholar]
- 154. Moreno P, de la Quintana Basarrate A, Musholt TJ, Paunovic I, Puccini M, Vidal Oet al. Adrenalectomy for solid tumor metastases: results of a multicenter European study. Surgery 2013;154:1215–1222; discussion 1222–1223 [DOI] [PubMed] [Google Scholar]
- 155. Vlk E, Ebbehoj A, Donskov F, Poulsen PL, Rashu BS, Bro Let al. Outcome and prognosis after adrenal metastasectomy: nationwide study. BJS Open 2022;6:zrac047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Hornstein I, Schwarz C, Ebbing S, Hoppe-Lotichius M, Otto G, Lang Het al. Surgical resection of metastases to the adrenal gland: a single center experience. Langenbecks Arch Surg 2015;400:333–339 [DOI] [PubMed] [Google Scholar]
- 157. Puccini M, Panicucci E, Candalise V, Ceccarelli C, Neri CM, Buccianti Pet al. The role of laparoscopic resection of metastases to adrenal glands. Gland Surg 2017;6:350–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Samsel R, Cichocki A, Roszkowska-Purska K, Papierska L, Koalasińska-Ćwikła A, Karpeta Eet al. Adrenal metastases—long-term results of surgical treatment, single-centre experience. Contemp Oncol (Pozn) 2020;24:29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ramsingh J, O’Dwyer P, Watson C. Survival outcomes following adrenalectomy for isolated metastases to the adrenal gland. Eur J Surg Oncol 2019;45:631–634 [DOI] [PubMed] [Google Scholar]
- 160. Wachtel H, Roses RE, Kuo LE, Lindeman BM, Nehs MA, Tavakkoli Aet al. Adrenalectomy for secondary malignancy: patients, outcomes, and indications. Ann Surg 2021;274:1073–1080 [DOI] [PubMed] [Google Scholar]
- 161. Metman MJH, Vietor CL, Seinen AJ, Berends AMA, Hemmer PHJ, Kerstens MNet al. Outcomes after surgical treatment of metastatic disease in the adrenal gland; valuable for the patient? Cancers (Basel) 2021;14:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Krumeich LN, Roses RE, Kuo LE, Lindeman BM, Nehs MA, Tavakkoli Aet al. Survival after adrenalectomy for metastatic lung cancer. Ann Surg Oncol 2022;29:2571–2579 [DOI] [PubMed] [Google Scholar]
- 163. van Vliet C, Dickhoff C, Bahce I, Engelsman AF, Hashemi SMS, Haasbeek CJAet al. Treatment patterns for adrenal metastases using surgery and SABR during a 10-year period. Radiother Oncol 2022;170:165–168 [DOI] [PubMed] [Google Scholar]
- 164. Zaborowski AM, Prichard RS. Adrenalectomy for metastases. Br J Surg 2022;109:1030–1031 [DOI] [PubMed] [Google Scholar]
- 165. Parkin C AG, Louie-Johnsun M. Solitary metastasis of renal cell carcinoma to the adrenal gland: treatment outcomes following laparoscopic retroperitoneal adrenalectomy. J Urol Surg 2021;8:185–190 [Google Scholar]
- 166. Siemer S, Lehmann J, Kamradt J, Loch T, Remberger K, Humke Uet al. Adrenal metastases in 1635 patients with renal cell carcinoma: outcome and indication for adrenalectomy. J Urol 2004;171:2155–2159; discussion 2159 [DOI] [PubMed] [Google Scholar]
- 167. Juan O, Popat S. Ablative therapy for oligometastatic non-small cell lung cancer. Clin Lung Cancer 2017;18:595–606 [DOI] [PubMed] [Google Scholar]
- 168. Beitler AL, Urschel JD, Velagapudi SR, Takita H. Surgical management of adrenal metastases from lung cancer. J Surg Oncol 1998;69:54–57 [DOI] [PubMed] [Google Scholar]
- 169. De Wolf J, Bellier J, Lepimpec-Barthes F, Tronc F, Peillon C, Bernard Aet al. Exhaustive preoperative staging increases survival in resected adrenal oligometastatic non-small-cell lung cancer: a multicentre study. Eur J Cardiothorac Surg 2017;52:698–703 [DOI] [PubMed] [Google Scholar]
- 170. Porte H, Siat J, Guibert B, Lepimpec-Barthes F, Jancovici R, Bernard Aet al. Resection of adrenal metastases from non-small cell lung cancer: a multicenter study. Ann Thorac Surg 2001;71:981–985 [DOI] [PubMed] [Google Scholar]
- 171. Tanvetyanon T, Robinson LA, Schell MJ, Strong VE, Kapoor R, Coit DGet al. Outcomes of adrenalectomy for isolated synchronous versus metachronous adrenal metastases in non-small-cell lung cancer: a systematic review and pooled analysis. J Clin Oncol 2008;26:1142–1147 [DOI] [PubMed] [Google Scholar]
- 172. Higashiyama M, Doi O, Kodama K, Yokouchi H, Imaoka S, Koyama H. Surgical treatment of adrenal metastasis following pulmonary resection for lung cancer: comparison of adrenalectomy with palliative therapy. Int Surg 1994;79:124–129 [PubMed] [Google Scholar]
- 173. Raz DJ, Lanuti M, Gaissert HC, Wright CD, Mathisen DJ, Wain JC. Outcomes of patients with isolated adrenal metastasis from non-small cell lung carcinoma. Ann Thorac Surg 2011;92:1788–1792; discussion 1793 [DOI] [PubMed] [Google Scholar]
- 174. Flaherty DC, Deutsch GB, Kirchoff DD, Lee J, Huynh KT, Lee DYet al. Adrenalectomy for metastatic melanoma: current role in the age of nonsurgical treatments. Am Surg 2015;81:1005–1009 [PMC free article] [PubMed] [Google Scholar]
- 175. Borgers JSW, Tobin RP, Torphy RJ, Vorwald VM, Van Gulick RJ, Amato CMet al. Melanoma metastases to the adrenal gland are highly resistant to immune checkpoint inhibitors. J Natl Compr Canc Netw 2021;19:53–63 [DOI] [PubMed] [Google Scholar]
- 176. Zippel D, Yalon T, Nevo Y, Markel G, Asher N, Schachter Jet al. The non-responding adrenal metastasis in melanoma: the case for minimally invasive adrenalectomy in the age of modern therapies. Am J Surg 2020;220:349–353 [DOI] [PubMed] [Google Scholar]
- 177. Li Y, Ji Z, Wang D, Xie Y. Bilateral adrenal metastasis of renal cell carcinoma 4 years after radical nephrectomy: a case report and review of literature. Medicine (Baltimore) 2021;100:e26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Mao JJ, Dages KN, Suresh M, Bancos I. Presentation, disease progression and outcomes of adrenal gland metastases. Clin Endocrinol (Oxf) 2020;93:546–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Chawla S, Chen Y, Katz AW, Muhs AG, Philip A, Okunieff Pet al. Stereotactic body radiotherapy for treatment of adrenal metastases. Int J Radiat Oncol Biol Phys 2009;75:71–75 [DOI] [PubMed] [Google Scholar]
- 180. Katoh N, Onishi H, Uchinami Y, Inoue T, Kuriyama K, Nishioka Ket al. Real-time tumor-tracking radiotherapy and general stereotactic body radiotherapy for adrenal metastasis in patients with oligometastasis. Technol Cancer Res Treat 2018;17:1533033818809983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Chen WC, Baal JD, Baal U, Pai J, Gottschalk A, Boreta Let al. Stereotactic body radiation therapy of adrenal metastases: a pooled meta-analysis and systematic review of 39 studies with 1006 patients. Int J Radiat Oncol Biol Phys 2020;107:48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]