

Abstract
Background:
Calcific aortic stenosis (CAS) is the most common valvular heart disease in older adults and has no effective preventive therapies. Genome-wide association studies (GWAS) can identify genes influencing disease and may help prioritize therapeutic targets for CAS.
Methods:
We performed a GWAS and gene association study of 14,451 CAS cases and 398,544 controls in the Million Veteran Program (MVP). Replication was performed in MVP, Penn Medicine Biobank, Mass General Brigham Biobank, BioVU, and BioMe, totaling 12,889 cases and 348,094 controls. Causal genes were prioritized from genome-wide significant (GWS) variants using Polygenic Priority Score gene localization, expression quantitative trait locus colocalization, and nearest gene methods. CAS genetic architecture was compared to that of atherosclerotic cardiovascular disease (ASCVD). Causal inference for cardiometabolic biomarkers in CAS was performed using Mendelian randomization and GWS loci were further characterized through phenome-wide association study (PheWAS).
Results:
We identified 23 GWS lead variants in our GWAS representing 17 unique genomic regions. Of the 23 lead variants, 14 were significant in replication, representing 11 unique genomic regions. Five replicated genomic regions were previously known risk loci for CAS (PALMD, TEX41, IL6, LPA, FADS) and six were novel (CEP85L, FTO, SLMAP, CELSR2, MECOM, CDAN1). Two novel lead variants were associated in non-Whites (p < 0.05): rs12740374 (CELSR2) in Black and Hispanic individuals, and rs1522387 (SLMAP) in Black individuals. Of the 14 replicated lead variants, only two (rs10455872 [LPA], rs12740374 [CELSR2]) were also significant in ASCVD GWAS. In Mendelian randomization, lipoprotein (a) [Lp(a)] and LDL-cholesterol were both associated with CAS, but the association between LDL-cholesterol and CAS was attenuated when adjusting for Lp(a). PheWAS highlighted varying degrees of pleiotropy, including between CAS and obesity at the FTO locus. However, the FTO locus remained associated to CAS after adjusting for body-mass index and maintained a significant independent effect on CAS in mediation analysis.
Conclusions:
We performed a multi-ancestry GWAS in CAS and identified 6 novel genomic regions in the disease. Secondary analyses highlighted the roles of lipid metabolism, inflammation, cellular senescence, and adiposity in the pathobiology of CAS and clarified the shared and differential genetic architectures of CAS with ASCVDs.
Keywords: calcific aortic stenosis, genome-wide association study, genomics, lipoprotein (a)
INTRODUCTION
Aortic stenosis is a structural heart disorder resulting from hemodynamic obstruction at the aortic valve. Calcific aortic stenosis (CAS) is the most common etiology of aortic stenosis and is estimated to afflict 12.6 million individuals worldwide, including 1,841 cases per 100,000 adults over the age of 70 years1. CAS is a progressive inflammatory process with overlapping risk factors for atherosclerotic cardiovascular disease (ASCVD)2–4. CAS is managed expectantly until reaching a severe stage when either surgical or transcatheter aortic valve replacement is considered, with a global burden of over 370,000 aortic valve replacements performed annually5,6. In the absence of valve replacement surgery, mortality for severe, symptomatic aortic stenosis is over 50% at two years5,7. In contrast to coronary artery disease (CAD) or heart failure, other common cardiovascular conditions with high morbidity and mortality, there are no effective medical therapies to slow the progression or reverse the disease course of CAS.
Human genetics may be used to nominate putative causal mechanisms underlying CAS. Large-scale genetic studies have previously elucidated candidate genes in the pathogenesis of CAS. The most robustly replicated of these is LPA, encoding lipoprotein (a) [Lp(a)], which is also linked to subclinical valvular calcification4,8. Mendelian randomization studies support the hypothesis that Lp(a) is a causal risk factor for CAS9. Similar studies support a role for Lp(a) in ASCVD, and clinical trials are underway to test the hypothesis that Lp(a)-lowering will reduce ASCVD risk10. Other candidate genes implicated in prior genome-wide association studies (GWAS) with CAS include PALMD, ALPL, NAV1, IL6, TEX41, and FADS1/211–14. Further genetic discovery in CAS is challenged mainly by relatively modest numbers of cases in prior studies and limited ancestral diversity. Furthermore, while ASCVD and CAS are comorbid with Lp(a) as a putative shared genetic factor, the extent to which arterial vascular and valvular clinical diseases are driven by common and distinct genetics is not well understood.
We performed a multi-ancestry discovery GWAS analysis in the Million Veteran Program (MVP), a large and diverse biobank using data from national Veteran Affairs hospitals. Using MVP data, we replicate five previously discovered genomic regions in CAS and identify six additional novel genomic regions that replicate in independent samples through a multi-ancestry meta-analysis. We further characterize the phenotypic consequences of these genome-wide significant (GWS) variants using phenome-wide association study (PheWAS). Finally, we detail the biomarkers, genes, and biologic pathways relevant to the genetic architecture of CAS and describe the similarities and differences between CAS and other ASCVDs using Mendelian randomization, gene association analysis, expression quantitative trait loci (eQTL) colocalization, and by exploring shared overlap with other large population atherosclerotic disease GWAS research.
METHODS
Data Availability
The full summary statistics for the discovery MVP CAS GWAS are publicly available in dbGaP (accession phs001672.v5.p1). Additional data supporting the manuscript, including replication of GWS single-nucleotide polymorphisms (SNPs), are available upon request from the corresponding author (COD). Data contributed by CARDIoGRAMplusC4D are available online (http://www.cardiogramplusc4d.org/). Data contributed by MEGASTROKE are available online (https://www.megastroke.org/). Data contributed by Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) are available on dbGaP (accession phs000930.v1.p1). Data contributed for single tissue cis-QTL are available online (https://www.gtexportal.org/home/). Data contributed by the Genetic Investigation of Anthropomorphic Traits (GIANT) consortium are available online (https://portals.broadinstitute.org/collaboration/giant/index.php/Main_Page). Data contributed by the International Consortium of Blood Pressure GWAS (ICBP) are on dbGaP (accession phs000585.v2.p1).
Ethics Approval
This study received ethics approval by the Veterans Affairs central institutional review board (IRB) under IRB protocol 16–06.
Population and Phenotyping
The MVP is a large biobank with enrollment beginning in 2011 consisting of individuals aged 19 years and older in the Veterans Affairs Healthcare system15. A CAS phenotype was developed in the MVP using a combination of International Classification of Diseases (ICD) 9/10 and Current Procedural Terminology (CPT) codes (Table S1) which was validated by manual adjudication of a random sample of 191 charts (99 cases and 92 controls) evaluating for the presence of hemodynamically significant tricuspid aortic stenosis.
Statistical Analysis
Statistical methods are explained throughout the following sections. Additional detail is available in the Expanded Methods/Results. A schematic overview of study design is presented in Figure S1.
Genome-Wide Association Study
Consented individuals in the MVP were genotyped using a customized Affymetrix Axiom Biobank Array. GWAS was performed using logistic regression in PLINK separately for each Harmonized Ancestry and Race/Ethnicity16 group (White, Black, and Hispanic) with adjustment for age, sex, and five ancestry-specific principal components, and filtered for variants with ancestry specific minor allele frequency (MAF) of greater than 1%. Multi-ancestry results were determined using fixed effects inverse variance weighted meta-analysis using GWAMA 2.2.217. GWS was met for variants with a p-value < 5×10−8. GWS lead SNPs and genomic regions were annotated on the web-based platform Functional Mapping and Annotation of Genome-Wide Association Studies [FUMA]18.
Replication and Meta-Analysis
SNPs identified in either multi-ancestry or ancestry-specific analyses as the lead SNP for a given genomic region were carried forward for replication. Replication cohorts included MVP, Penn Medicine Biobank (PMBB), Massachusetts General Brigham Biobank (MGBB), BioVU (Vanderbilt University), and BioME (Mt. Sinai). Phenotypes and details of genotyping and imputation for replication cohorts, including MVP, PMBB, MGBB, BioVU, and BioME are described in the Expanded Methods/Results. Replication results were meta-analyzed using fixed-effects inverse-variance weighted meta-analysis and considered significant if meeting a Bonferroni-corrected p-value for the number of unique loci assessed, n = 17 (p < 0.05/17 = 0.0029).
Causal Gene Prioritization
All GWS variants were annotated by location (intergenic, intron, exon) and nearest gene using the single nucleotide polymorphism database (dbSNP) of nucleotide sequence information. Causal genes were also inferred using Polygenic Priority Score (PoPS) Gene Localization, eQTL colocalization, and gene analysis in Multi-Marker Analysis of GenoMic Annotation (MAGMA).
Phenome-Wide Association Study and Mendelian Randomization
We performed PheWAS for ICD-9/10 clinical traits in the United Kingdom Biobank (UKB) using Phecodes19, as well as for select quantitative traits available in the UKB ascertained at baseline. Genotype-phenotype associations were considered significant if they had a p-value < 2.4×10−5 (=0.05/2057 traits present in both MVP and UKBB PheWAS).
Mendelian randomization was performed for select biomarkers previously identified as risk factors or pathobiological factors for CAS. We included serum lipid traits (total cholesterol, LDL cholesterol [LDL-C], triglycerides, Lp(a), apolipoprotein B [ApoB], HDL cholesterol [HDL-C], and apolipoprotein A-1 [ApoA-1]), serum phosphate and calcium, hemoglobin A1c (HA1c), c-reactive protein (CRP), body mass index (BMI), and blood pressure indices (systolic blood pressure, diastolic blood pressure, and pulse pressure)20–23. Summary statistics from GWAS for BMI were obtained from the GIANT consortium GWAS24. Summary statistics from GWAS for blood pressure were obtained from the ICBP25. Summary statistics from GWAS for remaining biomarkers were performed in the UKB and obtained from the Neale laboratory data repository [http://www.nealelab.is/uk-biobank/ accessed on February 15th, 2022]. There was no overlap in samples between exposure GWAS and outcome GWAS.
IL6 Pathway Polygenic Transcriptome Analysis and Colocalization
To disentangle the genetics of interleukin 6 (IL6) pathway mediated inflammation in CAS and CAD we developed a series of Polygenic Transcriptome Scores (PTS) for select inflammatory genes and evaluated their effect on CAS and CAD. We chose inflammatory genes that are biologically closely related to the NLRP3/IL-1β/IL6R inflammasome (specifically IFNG, AIM2, NLRP3, IL1B, IL18, and IL6R) based on review of established biological pathways26 and high protein-protein interaction networks provided by STRING (https://string-db.org/). We performed colocalization experiments using GWAS summary statistics from our MVP CAS discovery GWAS effort (for CAS) and CARDIoGRAMplusC4D (for CAD) and select single tissue cis-QTL data from GTEx to compare differential expression of IL6 and IL6R between CAS and CAD.
RESULTS
Population and Phenotyping
There were a total of 14,304,921 variants passing quality control as described, with a MAF > 1% in the MVP discovery cohort. After applying all inclusion and exclusion criteria, there were 12,395 White cases (mean ± standard deviation age 73.2±10 years, 97% male) and 287,787 White controls (age 63.4±14 years, 93% male), 1,445 Black cases (age 67.6±10 years, 95% male) and 79,229 Black controls (age 57.2±12 years, 87% male), and 611 Hispanic cases (age 69.4±11 years, 95% male) and 31,458 Hispanic controls (age 54.5±16 years, 91% male) (Table S2).
Results for genotyping and imputation in replication datasets, including for MVP, PMBB, BioME, BioVU, and MGBB are available in the Expanded Methods/Results. Across all the replication datasets, there were 11,433 White cases and 254,839 White controls, 1,110 Black cases and 64,397 Black controls, and 346 Hispanic cases and 28,858 Hispanic controls (Table S2). In comparison to the discovery cohort, replication cohorts had a similar distribution of age between cases and controls but included a greater proportion of female cases.
Sensitivity for our ICD-9/10 aortic stenosis phenotype was 0.99, specificity was 0.81, positive predictive value (PPV) was 0.78, and negative predictive value (NPV) was 0.99 (Table S3). After review of the 22 individuals identified as having CAS by ICD/procedure code but without aortic stenosis on adjudication, 15 of these (68%) were identified as having aortic stenosis based on at least two ICD-9 codes (424.1) but no ICD-10 code (I35.0) for aortic valve stenosis. Chart review clarified that these participants had aortic insufficiency without aortic stenosis.
GWAS Discovery and Replication
A multi-ancestry GWAS meta-analysis of CAS from White, Black, and Hispanic MVP participants resulted in 72 genome-wide significant SNPs, representing 10 unique genomic regions (Table S4). Of these, six genomic regions were previously established risk loci for CAS, with lead SNPs including rs7543130 (PALMD), rs2246363 (TEX41), rs3753782 (ALPL), rs10455872 (LPA), rs1474347 (IL6), and rs174533 (MYRF/FADS). The remaining four genomic regions were putatively novel with lead SNPs including rs12740374 (CELSR2), rs1522388 (SLMAP), rs56853411 (NCK1), and rs12206973 (CEP85L) (Table S5, Figure 1). The genomic control lambda value for the multi-ancestry GWAS was 1.013.
Figure 1: Hudson Plot of Multi-Ancestry GWAS findings (top) and MAGMA Gene Association Analysis (bottom).
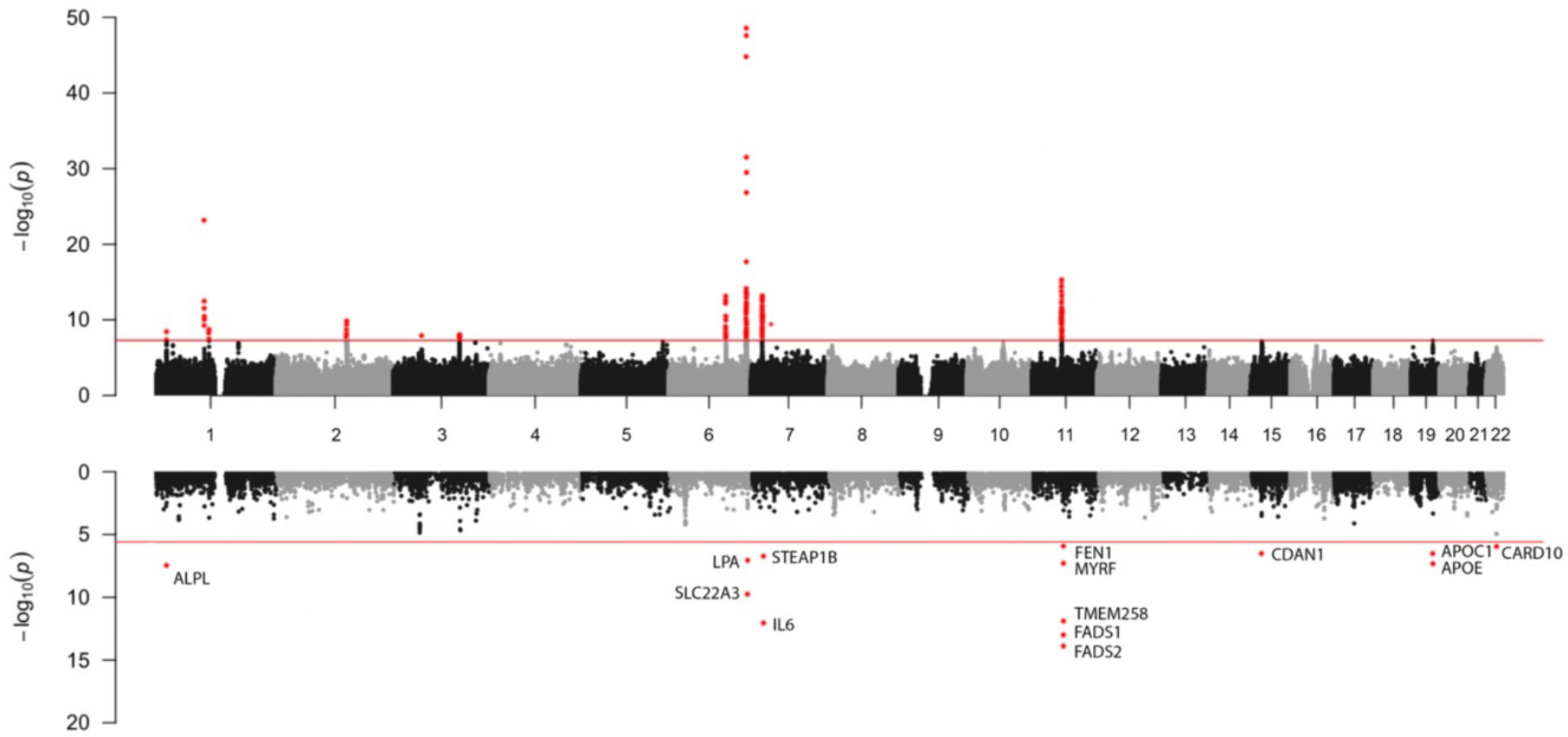
Hudson plot with multi-ancestry GWAS Manhattan plot (top) with genome-wide significant variants (p-value < 5×10−08) labeled in red and a gene analysis of multi-ancestry GWAS data (bottom) using Multi-Marker Analysis of Genomic Annotation (MAGMA) with gene wide significant results (19,021 protein coding genes, p-value < 2.6 × 10−6) labeled in red.
In White individuals, there were a total of 58 genome-wide significant SNPs, representing 14 unique genomic regions (Table S4, Table S5, Figure S2A). Of these, six were previously established genomic regions for CAS with lead SNPs including rs11166276 [PALMD], rs3753782 [ALPL], rs2246363 [TEX41], rs10455872 [LPA], rs13311155 [IL6], rs174533 [MYRF/FADS]). The remaining eight genomic regions were putatively novel and included the lead SNPs rs59030006 (MECOM), rs6493062 (CDAN1), rs742152 (CARD10), rs11647020 (FTO), rs1277930 (PSRC1), rs1522388 (SLMAP), rs34682748 (NCK1), and rs117202424 (CEP85L). The genomic control lambda value for the GWAS of White individuals was 1.082 (Q-Q plots are presented in Figure S3A–D).
There were two genome-wide significant SNPs representing two genomic regions identified in GWAS of Hispanic individuals (Table S4, Table S5, Figure S2B). Both were putatively novel: rs116418597 (SLIT2) and rs61849569 (MPP7). There were two genome-wide significant SNPs representing one genomic region in GWAS among Black individuals, rs112270619 (LOC105377714), which was putatively novel (Table S4, Table S5, Figure S2C). The genomic control lambda value for the GWAS of Hispanic individuals was 1.011. The genomic control lambda value for the GWAS of Black individuals was 1.006. In Firth logistic regression analysis of lead SNPs for Hispanic and Black individuals, effect estimates were similar, however the strength of association was attenuated (Table S6).
In conditional analysis of White individual GWAS results, there were 24 independently associated variants with joint association P-values < 5×10−8. Conditional analysis revealed seven independent associations at the CESLR2 genomic region, five independent associations at the TEX41 genomic region, two independent associations at the LPA genomic region, and three independent associations at the CDAN1 genomic region. Care should be taken in interpreting the presence of multiple independent associations at the LPA genomic region due to challenges of linkage disequilibrium calculation in the presence of common structural variants27. There were no additional independent associations identified for Hispanic or Black individuals (Table S7).
In total, 23 unique lead SNPs were carried forward in replication, representing 17 unique genomic regions. Of these, there were a total of 14 lead SNPs (representing 11 unique genomic regions) that met a Bonferroni corrected p-value for the number of unique genomic regions analyzed in replication (0.05/17 = 0.0029) (Table 1). Six were novel in CAS: rs12740374 (CELSR2), rs1522387 (SLMAP), rs117202424 (CEP85L), rs59030006 (MECOM), rs6493062 (CDAN1), and rs11647020 (FTO). Two established risk variants and two novel variants had at least nominal significance in Black individual meta-analysis: rs10455872 (LPA), rs174533 (MYRF), rs12740374 (CELSR2), and rs1522387 (SLMAP) (Figure S4A–D). Similarly, two established risk variants and one novel variant had at least nominal significance in Hispanic individual meta-analysis: rs2246363 (TEX41), rs1474347 (IL6), rs12740374 (CELSR2) (Figure S4E–H).
Table 1:
Discovery and Replication GWAS Results
| Chr:Pos | rsID | EA/ NEA |
Pop | EAF EUR | EAF AFR | EAF HIS | Discovery OR |
Discovery P-Value |
Replication OR |
Replication P-Value |
Annotation | Nearest Gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1:100049785 | rs7543130 | A/C | Multi | 0.50 | 0.30 | 0.24 | 1.14 [1.11–1.17] | 6.2 × 10−24 | 1.11 [1.08–1.15] | 1.06 × 10−10 | Intergenic | PALMD |
| 1:109817590 | rs12740374 | G/T | Multi | 0.78 | 0.80 | 0.75 | 1.10 [1.06–1.14 | 1.6 × 10−09 | 1.06 [1.03–1.10] | 2.51 × 10−04 | 3’ UTR | CELSR2 |
| 2:145783282 | rs2246363 | A/G | Multi | 0.75 | 0.81 | 0.93 | 1.11 [1.08–1.14] | 1.4 × 10−10 | 1.10 [1.05–1.14] | 2.20 × 10−06 | Intron | TEX41 |
| 3:57946096 | rs1522387 | A/G | Multi | 0.57 | 0.44 | 0.55 | 1.08 [1.05–1.10] | 1.0 × 10−08 | 1.09 [1.05–1.12] | 5.14 × 10−07 | Intergenic | SLMAP |
| 6:161010118 | rs10455872 | G/A | Multi | 0.07 | 0.04 | 0.01 | 1.42 [1.35–1.48] | 2.7 × 10−49 | 1.37 [1.30–1.44] | 7.76 × 10−34 | Intron | LPA |
| 7:22768124 | rs1474347 | C/A | Multi | 0.42 | 0.21 | 0.16 | 1.10 [1.07–1.13] | 6.7 × 10−14 | 1.09 [1.06–1.13] | 2.27 × 10−07 | Intron | IL6 |
| 11:61549025 | rs174533 | G/A | Multi | 0.67 | * | 0.92 | 1.12 [1.09–1.15] | 4.6 × 10−12 | 1.08 [1.04–1.12] | 1.95 × 10−05 | Intron | MYRF |
| 3:169207475 | rs59030006 | T/C | White | 0.51 | 0.52 | 0.51 | 1.08 [1.05–1.11] | 1.9 × 10−08 | 1.04 [1.02–1.08] | 0.0021 | Intron | MECOM |
| 6:118821706 | rs117202424 | A/G | White | 0.05 | 0.03 | 0.01 | 1.25 [1.18–1.32] | 4.4 × 10−15 | 1.11 [1.04–1.18] | 8.59 × 10−04 | Intron | CEP85L |
| 15:43017919 | rs6493062 | G/A | White | 0.81 | 0.82 | 0.34 | 1.10 [1.06–1.14] | 8.1 × 10−09 | 1.06 [1.02–1.11] | 0.0026 | Intron | CDAN1 |
| 16:538123990 | rs11647020 | T/C | White | 0.52 | 0.35 | 0.18 | 1.09 [1.05–1.11] | 6.0 × 10−09 | 1.06 [1.03–1.10] | 2.24 × 10−04 | Intron | FTO |
Note that discovery OR [95% CI] and P-values were calculated in the population described in the population column
Variant did not pass genotype quality control measures for this genetic ancestry
EA: effect allele
NEA: non-effect allele
Pop: Population
EAF: effect allele frequency
Multi: multi-ancestry
UTR: un-translated region
Evaluation for Pleiotropy Among Cardiovascular Relevant Traits
To explore whether any GWS variant in either multi-ancestry or ancestry-specific GWAS modifies risk for other atherosclerotic traits, we first determined the associations between CAS GWS variants and relevant atherosclerotic trait analyses including CARDIoGRAMplusC4D for CAD28, MEGASTROKE for stroke29, and coronary artery, descending thoracic aortic, and abdominal aortic calcification phenotypes in CHARGE30,31 (Figure S5). Of 11 queried variants only two variants, rs10455872 (LPA) and rs12740374 (CELSR2), had associations with atherosclerotic traits (CAD and coronary artery calcification) meeting a Bonferroni corrected P-value of < 0.0009 (0.05/[5 traits x 11 variants]).
We subsequently performed a sensitivity analysis whereby all associations for GWS loci in the discovery MVP cohort were repeated with additional adjustments for CAD and PAD. We observed 45% of individuals with CAS having CAD and 23% of individuals with CAS having PAD (Table S8). Despite correction for other atherosclerotic traits, the effect size, direction, and strength of association were similar for all GWS variants except for two variants at LPA which had attenuated effect estimates after adjusting for both CAD and PAD (Figure S6). We further compared associations between lead SNPs and CAS in subsets of individuals with and without CAD (we excluded variants with significant ASCVD pleiotropy such as those near LPA, CELSR2/SORT1, or FTO) (Table S9). Effect estimates were not significantly different between the two subgroups (9 of 22 variants had a higher effect estimate in the group without CAD, exact binomial test p-value of 0.52).
We performed several sensitivity analyses with alternate case definitions for CAS. Given that a number of false positive cases adjudicated during chart review had aortic insufficiency without CAS, we performed a sensitivity analysis repeating association analyses for all replicated GWS loci after excluding individuals with aortic insufficiency (AI) (14% of CAS individuals had AI, removing these individuals resulted in 12,387 CAS cases and 395,060 CAS controls, Table S8). Of 11 GWS loci with significant replication, two lost GWS after excluding AI (rs1522387 [SLMAP]: OR 1.08 [1.05–1.10], p-value 7.0 × 10−8; rs59030006 [MECOM]: OR 1.07 [1.04–1.10], p-value 1.5 × 10−6) although the effect estimates remained materially unchanged (Table S10). We additionally evaluated associations for all lead SNPs against a case definition including only individuals with aortic valve replacement (2,555 cases/280,067 controls in White individuals, 224 cases/77,891 controls in Black individuals, 717 cases/30,951 controls in Hispanic individuals). Effect estimates were similar though with an attenuated strength of association, however all associations met at least nominal significance with the exception of rs6493062 (CDAN1) (Table S11). Finally, we performed lookups for all lead SNPs in a meta-analysis of Framingham Heart Study (FHS)/Multiethnic Study of Atherosclerosis (MESA)/Age, Gene/Environment Susceptibility (AGES) aortic valve calcification GWAS4. Only rs10455872 (LPA) and rs174533 (MYRF) achieved at least nominal significance in association to aortic valve calcification (Table S12). Concordance of the direction of effect for lead SNPs in our MVP CAS GWAS and GWAS for aortic valve calcium was significant (18 of 24 variants had concordant direction of effect estimates, exact binomial test p-value of 0.004).
To clarify the relative contribution of BMI to the association of rs11647020 (FTO) and CAS, we adjusted the association for rs11647020 (FTO) and CAS with log-transformed BMI. The association remained statistically significant, but we observed a slightly reduced effect estimate and strength of association after including BMI as a covariate (discovery OR 1.09 [1.05–1.11], p-value 6.0 × 10−9 versus BMI adjusted OR 1.06 [1.04–1.09], p-value 9.6 × 10−8). In mediation analysis of BMI and rs11647020, the mediator BMI explained a substantial but modest proportion, 19% [15–28, 95% CI], of the total effect of rs11647020 on CAS. In contrast, mediation analysis of rs12740374/rs1277930 (both near SORT1) and LDL-C demonstrated that the mediator LDL-C explained a higher proportion of the total effect of each variant on CAS (27% [20–44, 95% CI] and 26% [19–41, 95% CI], respectively) (Table S13).
An evaluation for pleiotropy was performed by PheWAS for both binary clinical traits and quantitative biomarkers (Figure 2, Figure S6, Table S14). Notable pleiotropy was demonstrated for the rs10455872 risk alle (LPA) with atherosclerotic traits including increased risk of myocardial infarction, PAD, and cerebrovascular disease, as well as elevation in Lp(a). The rs174533 risk allele (MYRF/FADS) was associated with lower plasma levels of docosahexanoic acid and omega-3 fatty acids but higher plasma levels of triglycerides and linoleic acid. Of the novel GWS variants, notable pleiotropy was demonstrated for the rs12740374 risk allele (CELSR2) with hyperlipidemia and increased risk of atherosclerotic traits including coronary atherosclerosis and myocardial infarction. The rs11647020 risk allele (FTO) was associated with an increased risk of adiposity-related traits including obesity, diabetes, and hypertension. The rs2246363 risk allele (TEX41) was associated with increased systolic and diastolic blood pressure as well as higher plasma hemoglobin concentration. The rs117202424 risk allele (CEP85L) was associated with an increased risk of paroxysmal tachycardia and had a suggestive association to higher baseline pulse rate (p-value 9.3 × 10−3). The rs1522387 risk allele (SLMAP) was associated with higher plasma platelet counts.
Figure 2: Heatmap of Select Phenome-Wide Association Results in White Individuals from the UK Biobank and Replication Million Veteran Program Cohorts.

Heatmap of phenome-wide association results for both binary clinical traits and quantitative biomarkers. Binary clinical trait Phenome-Wide Association Study (PheWAS) was performed using the Denny PheWAS method and represents inverse-variance weighted meta-analysis of results from the Million Veteran Program and UK Biobank. Quantitative traits were rank-normal transformed prior to association analysis in the UK Biobank. The heatmap is scaled using Z-scores for all analyses and organized using hierarchical clustering via the Ward method.
Mendelian Randomization of Cardiovascular Biomarkers against CAS
Mendelian randomization was performed for 15 cardiometabolic biomarkers against CAS, as described previously (Figure 3). Of 15 biomarkers tested, six met a Bonferroni p-value threshold of 0.0033 (0.05/15). These were total cholesterol, LDL-C, Lp(a), apoB, triglycerides, and phosphate. Associations for total cholesterol, LDL-C, and Lp(a) were robust to Egger regression, a sensitivity analysis accounting for pleiotropy (Table S15). Multivariable Mendelian randomization analysis performed using instruments for LDL-C, Lp(a), triglycerides, and HDL demonstrated independent associations between LDL-C and Lp(a) with CAS, but not for triglycerides or HDL-C (Table S16). While the effect estimates for LDL-C and Lp(a) with CAS were similar in single exposure Mendelian randomization (LDL-C OR 1.29 [1.14–1.46] versus Lp(a) 1.28 [1.19–1.25]), the LDL-C effect estimate was attenuated in multivariable analysis including both LDL-C and Lp(a) as exposures (LDL-C OR 1.14 [1.05–1.46] versus Lp(a) 1.27 [1.23–1.32]).
Figure 3: Mendelian Randomization of Cardiometabolic Biomarkers in the UK Biobank and Risk of Aortic Stenosis.
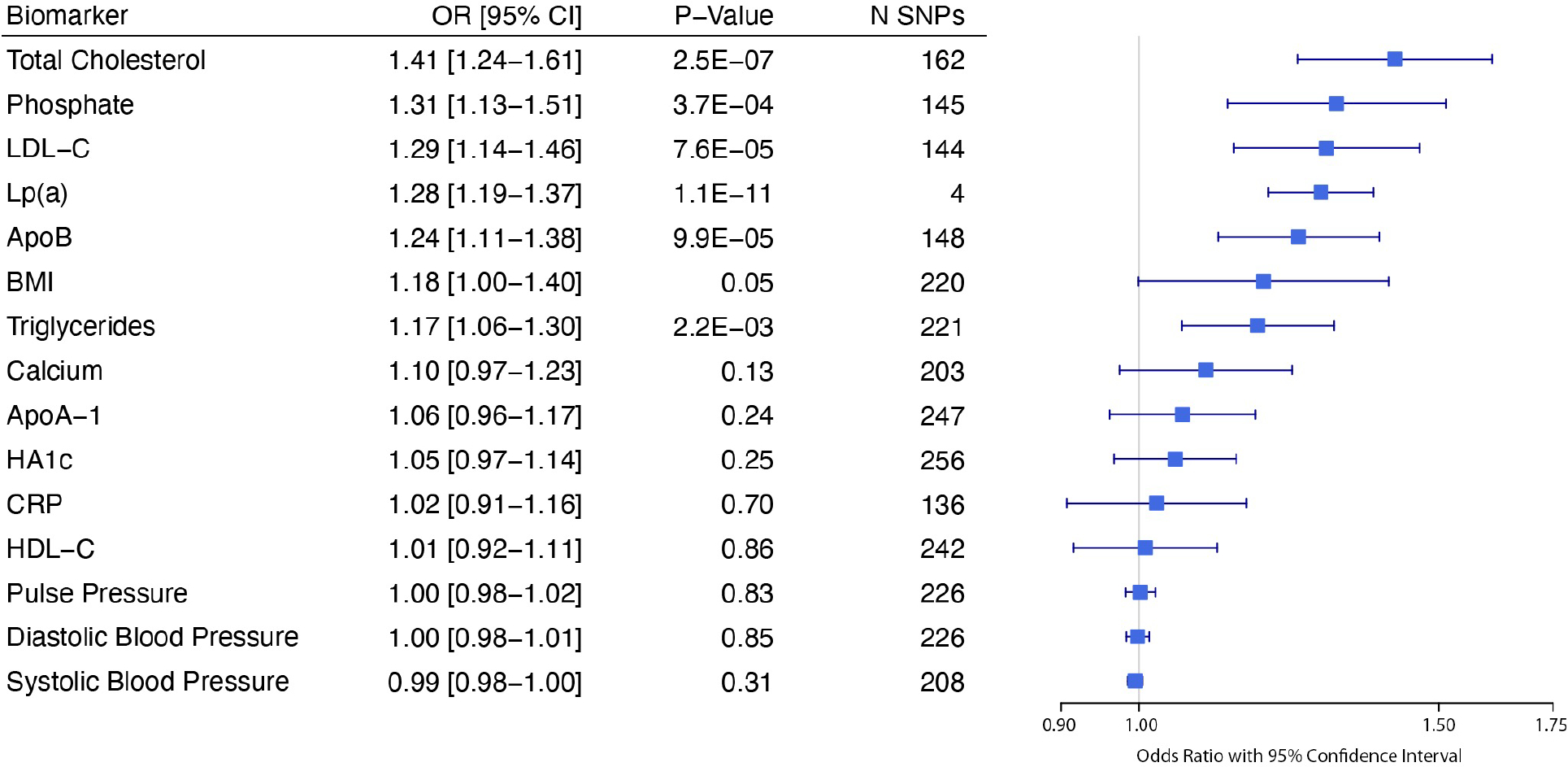
Forest plot of Mendelian randomization experiments for select exposures using genetic instruments built in the UK Biobank against an outcome of aortic stenosis applied in the Million Veteran Program.
Causal Gene Prioritization
Nearest gene annotations (Table 1) were generally concordant with results of gene association analysis (Figure 1, bottom; Figure S2A–C, bottom) with the exception that gene association analysis uniquely identified significant gene-phenotype associations between CAS and APOC1 (encoding apolipoprotein C1) as well as between CAS and APOE (encoding apolipoprotein E). PoPS gene localization identified several additional putative causal genes for novel variants including SORT1 (rs12740374, nearest gene CELSR2), FLNB (rs1522388, nearest gene SLMAP), SLC35F1 (rs117202424, nearest gene CEP85L), STARD9 (rs6493062, nearest gene CDAN1), and RBL2 (rs11647020, nearest gene FTO) (Table S17). There were several colocalization results with posterior probability of a single shared causal variant (pp.H4) > 0.50. Results of interest included liver expression of SORT1 for rs12740374 and rs1277930 (pp.H4 of 0.99), subcutaneous adipose tissue expression of AC073072.5 for rs13311155 (pp.H4 of 0.99) and lung expression of ACO73072.5 for rs1474347 (pp.H4 of 0.76), whole blood expression of FLNB for rs1522387 (pp.H4 of 0.64), and aortic arterial expression of CDAN1 for rs6493062 (pp.H4 of 0.89) (Table S18).
Predicted Gene Expression Analyses
A PTS predicting increased expression of IL6 was significantly associated with decreased risk of CAS (beta −0.47, p-value 3.5×10−6) (Table S19). The A allele of rs2228145 (IL6R Asp358Ala, which increases plasma levels of soluble IL6R32) was nominally associated with increased risk of CAS (beta 0.03, p-value 0.02). In contrast, the A allele of rs2228145 (IL6R Asp358Ala) was strongly associated with increased risk of CAD (beta 0.03, p-value 1.6×10−15), whereas the PTS for IL6 was only nominally associated with risk of CAD (beta 0.11, p-value 0.03). PTS predicting increased expression of STAT3 (beta −0.06, p-value 4.6 × 10−7) and TNF (beta −0.04, p-value 8.0×10−3) were associated with decreased risk of CAD but not CAS.
In colocalization analysis of IL6 and CAS, two tissues had a posterior probability of a single shared causal variant (pp.H4) of > 0.50 (Table S20). These were tibial arterial tissue (0.52) and cultured fibroblasts (0.57). Colocalization of IL6R and CAS demonstrated only one tissue with pp.H4 > 0.50 (aortic arterial tissue, 0.66). In contrast, there were no colocalization results between IL6 and CAD with pp.H4 > 0.50. Colocalization for IL6R and CAD was notable for pp.H4 in whole blood of 0.86 and in left atrial appendage of 0.71.
Notable associations between tissue-level eQTL data and CAS include whole blood expression of FADS1 with CAS (p-value 2.7 × 10−14), left atrial appendage tissue expression of FEN1 with CAS (p-value 7.1 × 10−13), and tibial arterial tissue expression of FADS2 with CAS (p-value 7.1 × 10−13) all near the MYRF gene locus. Also notable was subcutaneous adipose tissue expression of STEAPB1 with CAS (2.6 × 10−11) which is near rs1474347 (closest to IL6), aortic arterial tissue expression of CDAN1 with CAS (p-value 7.4 × 10−8) and left ventricular expression of TRIOBP (near CARD10) with CAS (p-value 8.1 × 10−9) (Table S21).
DISCUSSION
We performed a large, multi-ancestry GWAS of CAS in the MVP with replication in several biobanks amounting to analyses of over 27,000 individuals with CAS, representing the largest CAS GWAS effort to date. In multi-ancestry analysis, we replicated five previously identified genomic regions in CAS (PALMD, TEX41, LPA, IL6, FADS/MYRF) and identified six novel genomic regions (SLMAP, CELSR2, CEP85L, MECOM, CDAN1, FTO). Two novel lead SNPs demonstrated at least nominal significance in non-white replication: rs12740374 (CELSR2) in both Black and Hispanic individuals and rs1522387 (SLMAP) in Black individuals. Through secondary analyses, including Mendelian randomization, gene association analysis, PoPS, eQTL colocalization, and PheWAS, we prioritized causal genes and characterized their potential functional relevance in CAS pathobiology. These data highlight the shared and differential genetic architecture between CAS and other atherosclerotic traits, particularly CAD, and suggest potential therapeutic targets to modify the natural history of disease.
CAS shares many risk factors with atherosclerotic vascular disease and has a proposed similar pathobiology involving intimal accumulation of apoB-containing particles, inflammation, and calcification. Much of the shared genetic architecture between CAS and CAD is manifest by shared risk loci in lipid metabolism. Accordingly, we demonstrated a novel association between the CELSR2-PSRC1-SORT1 gene locus and CAS, which is a well described risk locus for CAD28. PoPS gene localization and eQTL colocalization prioritized the gene SORT1, encoding sortilin, which binds apolipoprotein B-100 and modulates very low density LDL-C secretion via lysosomal degradation33. Prior genetic and translational research in CAS implicates LDL-C, in addition to Lp(a), in disease pathogenesis34,35. Genetic variation accounting for lifetime elevations in both LDL-C and Lp(a) is similarly well described for CAD in multiple prior genetic and epidemiologic studies36–38. We noted that while LDL-C and Lp(a) were both independently associated with CAS in multivariable Mendelian randomization, the effect estimate and strength of association for LDL-C was attenuated when compared to Lp(a). Lp(a) is thought to be pathogenic in CAS in part due to the presence of oxidized phospholipids, which are covalently bound to the apolipoprotein (a) moiety of Lp(a) and which mediate pro-inflammatory pathways when present in the valve intima39,40. The vast majority of circulating oxidized phospholipids are bound to Lp(a) rather than LDL-C or other apoB containing particles34. Among other mechanisms, oxidized phospholipids may help explain the difference in risk observed between Lp(a) and LDL-C for CAS. It will be important to develop therapies that address both the contribution of LDL-C and Lp(a)/oxidized phospholipids to maximally impact CAS risk.
Inflammation is a well-described treatment target in atherosclerosis, with multiple clinical trials such as CANTOS, COLCOT, and LoDoCo2 demonstrating a reduction in cardiovascular risk for individuals treated with agents modifying the NLRP3/IL-1β/IL6 pathway of innate immunity41–43. IL6 (encoding the IL6 ligand), IL6R (encoding the IL6 receptor, IL6R), or gene products modulating IL6 metabolism (such as histone deacetylase 9, HDAC-9) are present as GWS in GWAS for CAS14, CAD28, PAD44, and ischemic stroke29. In contrast to CAD, where the dominant relationship between inflammation and disease appears to be modified by IL6R rather than the IL6 ligand, our PRS analysis suggests that this relationship is reversed in CAS with the IL6 ligand, rather than IL6R, maintaining the strongest association to disease. This is supported by our colocalization experiments with a stronger posterior probability for a single shared causal variant observed between tissues expressing IL6 and CAS than between tissues expressing IL6R and CAS, and further supported by recent clinical data in which elevated plasma levels of IL6 are demonstrated to be strongly associated with death in individuals with aortic stenosis45. Colocalization experiments for rs13311155 and rs1474347 (both near IL6) highlighted expression of AC073072.5, encoding the long non-coding RNA IL6 antisense 1 (IL6-AS1) in multiple tissues, a finding also observed in prior colocalization efforts in CAS14. IL6-AS1 is hypothesized to function as a transcriptional regulator of IL6 and may explain how lead variants near IL6 ultimately translate to differential plasma expression of the IL6 ligand46. Our PTS results suggested that increased tissue-specific IL6 expression was strongly associated with decreased risk of CAS, which, while opposite expectation, underscores complexity of IL6 signaling and need for further study.
Adiposity is increasingly recognized as a causal risk factor for aortic stenosis47, with multiple proposed mechanisms including structural heart changes in obesity, hypertension leading to valvular endothelial injury, and increased plasma concentrations of atherogenic particles such as triglyceride rich lipoproteins known to impact progression of CAS22. We identified a novel association between rs11647020, a variant in intron 1 of the FTO gene, with CAS. Since its discovery in 2007, the fat mass and obesity associated (FTO) gene has maintained a position as arguably the most important obesity locus, with the largest effect size and greatest explained variance for obesity among European genetic ancestry individuals48. FTO gene variants are demonstrated to be associated with a number of cardiometabolic diseases including CAD, however the effect seems to be primarily mediated through BMI49.
We observed that rs11647020 maintained a significant association to CAS even after adjustment for BMI and that a substantial proportion of the total effect of rs11647020 on CAS was independent of BMI in mediation analysis. The functional mechanism of FTO remains unknown but is thought to impact pathways controlling food intake48. Both PoPS and colocalization experiments prioritized rs11647020 as impacting expression of RBL2, encoding the retinoblastoma-like 2 protein (Rbl2) which is approximately 270,000 base pairs from FTO. A similar finding is reported in a 2010 study evaluating human tissue expression data of the variant rs8050136 (also in intron 1 of FTO and in moderate LD with rs11647020 in European individuals, R2 0.57), with the observation that rs8050136 was strongly associated with RBL2, but not FTO, gene expression50. Rbl2 is hypothesized to influence telomere regulation with Rbl2 deficient mouse embryos observed to have markedly elongated telomeres51. Telomere-driven cellular senescence has previously been observed in CAS52, providing a possible functional mechanism whereby intronic FTO variants may impact CAS pathobiology independently of BMI through RBL2 mediated telomere dysregulation. As BMI-associated alleles influence weight over time53, we cannot rule out the possibility of residual confounding for the persistent association of FTO with CAS independent of cross-sectional BMI.
A large fraction of individuals with CAS do not have clinical atherosclerotic cardiovascular disease. Up to 55% of individuals in MVP with CAS have no CAD and 77% with CAS have no PAD, highlighting the existence of complementary pathways. The novel loci rs59030006 (MECOM), rs1522387 (SLMAP), rs117202424 (CEP85L), and rs6493062 (CDAN1) were not observed to have associations with other atherosclerotic traits in either PheWAS or atherosclerotic trait GWAS interrogations. Further elucidating the mechanisms by which these loci impact CAS pathobiology offers distinct opportunities for CAS-specific risk modulation.
MECOM, identified by both nearest gene and PoPS as the most likely causal gene for rs59030006, encodes MDS1 and EVI1 Complex Loc, and is an established risk locus in hematologic malignancies, hypertension, and pre-eclampsia54. MECOM is recognized to play a role in arterial endothelial cell identity during embryogenesis55. We observed that MECOM had a suggestive association between aortic arterial tissue expression and CAS in S-MultiXcan analysis and was associated with both congenital anomalies of the great vessels and increased systolic blood pressure in PheWAS. Valve endothelial cells (VECs) are central to the proposed pathogenesis of CAS, with disruption resulting in endothelial-to-mesenchymal transition and adoption of an osteogenic phenotype56.
SLMAP, nearest to the novel lead SNP rs1522387, encodes Sarcolemma Membrane Associated Protein, which is involved in cardiomyocyte contraction and associated clinically with the Brugada syndrome and heart failure. Colocalization experiments for rs1522387 prioritized expression of FLNB, encoding filamin B, in whole blood and FLNB-AS1, encoding filamin B antisense 1, in tibial arterial tissue. Filamin B is a protein deficient in various bony abnormalities including bone mineral density, perhaps implicating a role for rs1522387 in valvular calcification57.
CEP85L, nearest to the novel locus rs117202424, encodes Centrosomal Protein 85 Like, which is the top eQTL gene for the rs4946333 variant identified in prior GWAS for atrial fibrillation58. rs117202424 had a significant association with paroxysmal tachycardia (and suggestive association to baseline pulse rate) in our PheWAS. PoPS gene localization for rs117202424 prioritized SLC35F1, encoding solute carrier family 35 member F1, which is observed in GWAS for heart rate, QT interval, and left ventricular diastolic dimension59,60. While SLC35F1 is primarily expressed in the brain, there are human data suggesting that this protein is expressed in cardiac tissue61, however, its functional mechanism in cardiovascular disease remains unknown. It is noteworthy that SLC35F1 is less than 100 kb from PLN, encoding phospholamban, which is implicated in the pathogenesis of non-ischemic dilated cardiomyopathy whereby PLN aggregates collect in cardiomyocytes62.
We identified several putative causal genes for the variant rs6493062, including CDAN1 (encoding codanin-1, nearest gene, strongest association by MAGMA and with pp.H4 of 0.89 for expression in aortic arterial tissue), STARD9 (encoding StAR Related Lipid Transfer Domain Containing 9, identified as most likely causal gene by PoPS), and URB1 (encoding URB1 Ribosome Biogenesis Homolog, identified by MAGMA). Further study is necessary to better clarify the gene(s) through which rs6493062 modifies the risk of CAS.
Among the study limitations, our primary phenotype is ICD-9/10/CPT generated, and as made apparent by chart review, PPV was modest (0.78) in comparison to NPV (0.99). This result can be partly attributed to the inclusion of veterans with AI instead of aortic stenosis due to a more generalized ICD-9 definition for aortic valve disease (424.1) that included both aortic stenosis and AI. Aortic stenosis and AI were subsequently coded differently and more precisely in ICD-10. When we performed additional adjustment for aortic insufficiency, two fell below GWS (SLMAP and MECOM), however the effect estimates were materially unchanged and we suspect that the loss of GWS is at least partly explained by loss of power. Further study is required to disentangle whether mixed aortic valve disease represents a unique pathobiology distinct from severe or late-stage CAS. Other limitations of the current study include limited power in non-White populations despite significantly larger sample sizes of non-White participants relative to prior efforts, and the MVP’s predominantly male population. Finally, while our study is more ancestrally diverse compared to prior CAS GWAS efforts, we anticipate that future GWAS meta-analysis with the inclusion of additional non-White individuals will lead to improved understanding of CAS genetic architecture.
In summary, we performed a large, multi-ancestry GWAS of CAS and identified 14 unique lead SNPs encompassing 5 previously identified genomic regions and 6 novel genomic regions in CAS. Two of the novel lead SNPs demonstrated at least nominal significance in non-White individual meta-analysis. We characterized the phenotypic consequences of these lead SNPs through PheWAS and demonstrate that while there is some overlap with CAD and coronary calcification, most identified CAS risk loci do not share pleiotropy with ASCVDs. Pathways highlighted in the pathobiology of CAS include lipid metabolism, inflammation, cellular senescence, calcification, and adiposity.
Supplementary Material
Clinical Perspective.
What is New?
In a large multi-ancestry GWAS of CAS in the Million Veteran Program, we identified six novel genomic regions in the disease including CEP85L, FTO, SLMAP, CELSR2, MECOM, and CDAN1.
In spite of epidemiologic overlap between CAS and atherosclerotic cardiovascular diseases, the genetic architecture of CAS appears distinct, with lead SNPs remaining significant even after adjustment for coronary artery disease and peripheral artery disease.
What are the Clinical Implications?
Results from this study may help prioritize gene and/or pathway targets for medical therapies in CAS.
Funding Sources
Vanderbilt University Medical Center’s BioVU projects are supported by numerous sources: institutional funding, private agencies, and federal grants. These include NIH funded Shared Instrumentation Grant S10OD017985, S10RR025141, and S10OD025092; Clinical and Translational Science Awards Program grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711. SMD is supported by IK2-CX001780. PN is supported by grants from NIH (R01HL142711, 148050, R01HL151283, R01HL127564, U01HG011719, R01HL151152). GMP is supported by grants from NIH (R01HL142711 and R01HL127564). AMS is supported by T32HG010464. LCW was supported by NIH grant 1R01HL139731 and American Heart Association (AHA) grant 18SFRN34110082. PWilson/K Cho are supported by the Veterans Affairs Merit Award BX004821. This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by award BX004821. This Publication does not represent the views of the Department of Veteran Affairs or the United States Government.
Non-standard Abbreviations and Acronyms:
- CAS
calcific aortic stenosis
- ASCVD
atherosclerotic cardiovascular disease
- CAD
coronary artery disease
- Lp(a)
lipoprotein (a)
- GWAS
genome-wide association study
- MVP
Million Veteran Program
- GWS
genome-wide significant
- PheWAS
phenome-wide association study
- eQTL
expression quantitative trait locus
- SNP
single nucleotide polymorphism
- ICD
international classification of diseases
- CPT
current procedural terminology
- MAF
minor allele frequency
- GIANT
Genetic Investigation of Anthropomorphic Traits
- ICBP
International Consortium of Blood Pressure
- PMBB
Penn Medicine Biobank
- MGBB
Mass General Brigham Biobank
- BioVU
Vanderbilt University Biorepository
- BioME
Mount Sinai Hospital Biorepository
- PoPs
Polygenic Priority Score
- MAGMA
Multi-Marker Analysis of GenoMic Annotation
- CHARGE
Cohorts for Heart and Aging Research in Genomic Epidemiology
- LDL-C
low density lipoprotein cholesterol
- HDL-C
high density lipoprotein cholesterol
- ApoB
apolipoprotein B
- ApoA-1
apolipoprotein A1
- HA1c
hemoglobin A1c
- CRP
c-reactive protein
- BMI
body mass index
- UKB
United Kingdom Biobank
- IL6
interleukin 6
- PTS
Polygenic Transcriptome Scores
- PPV
positive predictive value
- NPV
negative predictive value
- AI
aortic insufficiency
Footnotes
Conflict of Interest Disclosures
SMD receives research support from RenalytixAI and personal consulting fees from Calico Labs, outside the scope of the current research. PN reports personal consulting fees from Amgen, Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Genentech, Novartis, and TenSixteen Bio, investigator-initiated grants from Apple, AstraZeneca, and Boston Scientific, is a co-founder of TenSixteen Bio, equity in TenSixteen Bio, geneXwell, and Vertex, and spousal employment at Vertex, all unrelated to the present work. CJO is an employee of Novartis Institutes for Biomedical Research. LCW receives research support from IBM to the Broad Institute.
Supplemental Materials
References
- 1.Yadgir S, Johnson CO, Aboyans V, Adebayo OM, Adedoyin RA, Afarideh M, Alahdab F, Alashi A, Alipour V, Arabloo J, et al. Global, Regional, and National Burden of Calcific Aortic Valve and Degenerative Mitral Valve Diseases, 1990–2017. Circulation. 2020;141:1670–1680. doi: 10.1161/CIRCULATIONAHA.119.043391 [DOI] [PubMed] [Google Scholar]
- 2.Dweck MR, Jones C, Joshi NV, Fletcher AM, Richardson H, White A, Marsden M, Pessotto R, Clark JC, Wallace WA, et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation. 2012;125:76–86. doi: 10.1161/CIRCULATIONAHA.111.051052 [DOI] [PubMed] [Google Scholar]
- 3.Smith JG, Luk K, Schulz CA, Engert JC, Do R, Hindy G, Rukh G, Dufresne L, Almgren P, Owens DS, et al. Association of low-density lipoprotein cholesterol-related genetic variants with aortic valve calcium and incident aortic stenosis. JAMA. 2014;312:1764–1771. doi: 10.1001/jama.2014.13959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otto CM, Prendergast B. Aortic-valve stenosis--from patients at risk to severe valve obstruction. N Engl J Med. 2014;371:744–756. doi: 10.1056/NEJMra1313875 [DOI] [PubMed] [Google Scholar]
- 6.Blaser MC, Kraler S, Luscher TF, Aikawa E. Multi-Omics Approaches to Define Calcific Aortic Valve Disease Pathogenesis. Circ Res. 2021;128:1371–1397. doi: 10.1161/CIRCRESAHA.120.317979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carabello BA, Paulus WJ. Aortic stenosis. The Lancet. 2009;373:956–966. doi: 10.1016/s0140-6736(09)60211-7 [DOI] [PubMed] [Google Scholar]
- 8.Chen HY, Dufresne L, Burr H, Ambikkumar A, Yasui N, Luk K, Ranatunga DK, Whitmer RA, Lathrop M, Engert JC, et al. Association of LPA Variants With Aortic Stenosis: A Large-Scale Study Using Diagnostic and Procedural Codes From Electronic Health Records. JAMA Cardiol. 2018;3:18–23. doi: 10.1001/jamacardio.2017.4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emdin CA, Khera AV, Natarajan P, Klarin D, Won HH, Peloso GM, Stitziel NO, Nomura A, Zekavat SM, Bick AG, et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J Am Coll Cardiol. 2016;68:2761–2772. doi: 10.1016/j.jacc.2016.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, Tardif JC, Baum SJ, Steinhagen-Thiessen E, Shapiro MD, Stroes ES, Moriarty PM, Nordestgaard BG, et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N Engl J Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239 [DOI] [PubMed] [Google Scholar]
- 11.Chen HY, Cairns BJ, Small AM, Burr HA, Ambikkumar A, Martinsson A, Theriault S, Munter HM, Steffen B, Zhang R, et al. Association of FADS1/2 Locus Variants and Polyunsaturated Fatty Acids With Aortic Stenosis. JAMA Cardiol. 2020;5:694–702. doi: 10.1001/jamacardio.2020.0246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helgadottir A, Thorleifsson G, Gretarsdottir S, Stefansson OA, Tragante V, Thorolfsdottir RB, Jonsdottir I, Bjornsson T, Steinthorsdottir V, Verweij N, et al. Genome-wide analysis yields new loci associating with aortic valve stenosis. Nat Commun. 2018;9:987. doi: 10.1038/s41467-018-03252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theriault S, Gaudreault N, Lamontagne M, Rosa M, Boulanger MC, Messika-Zeitoun D, Clavel MA, Capoulade R, Dagenais F, Pibarot P, et al. A transcriptome-wide association study identifies PALMD as a susceptibility gene for calcific aortic valve stenosis. Nat Commun. 2018;9:988. doi: 10.1038/s41467-018-03260-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Theriault S, Dina C, Messika-Zeitoun D, Le Scouarnec S, Capoulade R, Gaudreault N, Rigade S, Li Z, Simonet F, Lamontagne M, et al. Genetic Association Analyses Highlight IL6, ALPL, and NAV1 As 3 New Susceptibility Genes Underlying Calcific Aortic Valve Stenosis. Circ Genom Precis Med. 2019;12:e002617. doi: 10.1161/CIRCGEN.119.002617 [DOI] [PubMed] [Google Scholar]
- 15.Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol. 2016;70:214–223. doi: 10.1016/j.jclinepi.2015.09.016 [DOI] [PubMed] [Google Scholar]
- 16.Liao KP, Sun J, Cai TA, Link N, Hong C, Huang J, Huffman JE, Gronsbell J, Zhang Y, Ho YL, et al. High-throughput multimodal automated phenotyping (MAP) with application to PheWAS. J Am Med Inform Assoc. 2019;26:1255–1262. doi: 10.1093/jamia/ocz066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magi R Software GWAMA- software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, Crawford DC. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xia C, Lei W, Hu Y, Yang H, Zeng X, Chen M. Association of serum levels of calcium phosphate and vitamin D with risk of developing aortic stenosis: the UK Biobank cohort. Eur J Prev Cardiol. 2022. doi: 10.1093/eurjpc/zwac016 [DOI] [PubMed] [Google Scholar]
- 21.Mundal LJ, Hovland A, Igland J, Veierod MB, Holven KB, Bogsrud MP, Tell GS, Leren TP, Retterstol K. Association of Low-Density Lipoprotein Cholesterol With Risk of Aortic Valve Stenosis in Familial Hypercholesterolemia. JAMA Cardiol. 2019;4:1156–1159. doi: 10.1001/jamacardio.2019.3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaltoft M, Langsted A, Nordestgaard BG. Triglycerides and remnant cholesterol associated with risk of aortic valve stenosis: Mendelian randomization in the Copenhagen General Population Study. Eur Heart J. 2020;41:2288–2299. doi: 10.1093/eurheartj/ehaa172 [DOI] [PubMed] [Google Scholar]
- 23.Kronenberg F Lipoprotein(a) and aortic valve stenosis: work in progress. Eur Heart J. 2022. doi: 10.1093/eurheartj/ehac436 [DOI] [PubMed] [Google Scholar]
- 24.Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet. 2018;27:3641–3649. doi: 10.1093/hmg/ddy271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ Res. 2020;126:1260–1280. doi: 10.1161/CIRCRESAHA.120.315937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zekavat SM, Ruotsalainen S, Handsaker RE, Alver M, Bloom J, Poterba T, Seed C, Ernst J, Chaffin M, Engreitz J, et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat Commun. 2018;9:2606. doi: 10.1038/s41467-018-04668-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, Wunderer F, Smith AV, Wong Q, Pechlivanis S, et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. 2019;51:1580–1587. doi: 10.1038/s41588-019-0514-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Donnell CJ, Kavousi M, Smith AV, Kardia SL, Feitosa MF, Hwang SJ, Sun YV, Province MA, Aspelund T, Dehghan A, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124:2855–2864. doi: 10.1161/CIRCULATIONAHA.110.974899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafiq S, Frayling TM, Murray A, Hurst A, Stevens K, Weedon MN, Henley W, Ferrucci L, Bandinelli S, Corsi AM, et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007;8:552–559. doi: 10.1038/sj.gene.6364414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conlon DM. Role of sortilin in lipid metabolism. Curr Opin Lipidol. 2019;30:198–204. doi: 10.1097/MOL.0000000000000598 [DOI] [PubMed] [Google Scholar]
- 34.Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31:440–450. doi: 10.1097/HCO.0000000000000300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nazarzadeh M, Pinho-Gomes AC, Bidel Z, Dehghan A, Canoy D, Hassaine A, Ayala Solares JR, Salimi-Khorshidi G, Smith GD, Otto CM, et al. Plasma lipids and risk of aortic valve stenosis: a Mendelian randomization study. Eur Heart J. 2020;41:3913–3920. doi: 10.1093/eurheartj/ehaa070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erqou S Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA. 2009;302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perrot N, Theriault S, Dina C, Chen HY, Boekholdt SM, Rigade S, Despres AA, Poulin A, Capoulade R, Le Tourneau T, et al. Genetic Variation in LPA, Calcific Aortic Valve Stenosis in Patients Undergoing Cardiac Surgery, and Familial Risk of Aortic Valve Microcalcification. JAMA Cardiol. 2019;4:620–627. doi: 10.1001/jamacardio.2019.1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clarke R Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. N Engl J Med. 2009;361. [DOI] [PubMed] [Google Scholar]
- 39.Leibundgut G, Scipione C, Yin H, Schneider M, Boffa MB, Green S, Yang X, Dennis E, Witztum JL, Koschinsky ML, et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res. 2013;54:2815–2830. doi: 10.1194/jlr.M040733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsson M Presence of Oxidized Low Density Lipoprotein in Nonrheumatic Stenotic Aortic Valves. Arterioscler Thromb Vasc Biol. 1999. [DOI] [PubMed] [Google Scholar]
- 41.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 42.Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381:2497–2505. doi: 10.1056/NEJMoa1912388 [DOI] [PubMed] [Google Scholar]
- 43.Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, The SHK, Xu XF, Ireland MA, Lenderink T, et al. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383:1838–1847. doi: 10.1056/NEJMoa2021372 [DOI] [PubMed] [Google Scholar]
- 44.Klarin D, Lynch J, Aragam K, Chaffin M, Assimes TL, Huang J, Lee KM, Shao Q, Huffman JE, Natarajan P, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nature medicine. 2019;25:1274–1279. doi: 10.1038/s41591-019-0492-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vidula MK, Orlenko A, Zhao L, Salvador L, Small AM, Horton E, Cohen JB, Adusumalli S, Denduluri S, Kobayashi T, et al. Plasma biomarkers associated with adverse outcomes in patients with calcific aortic stenosis. Eur J Heart Fail. 2021;23:2021–2032. doi: 10.1002/ejhf.2361 [DOI] [PubMed] [Google Scholar]
- 46.Yi E, Zhang J, Zheng M, Zhang Y, Liang C, Hao B, Hong W, Lin B, Pu J, Lin Z, et al. Long noncoding RNA IL6-AS1 is highly expressed in chronic obstructive pulmonary disease and is associated with interleukin 6 by targeting miR-149–5p and early B-cell factor 1. Clin Transl Med. 2021;11:e479. doi: 10.1002/ctm2.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaltoft M, Langsted A, Nordestgaard BG. Obesity as a Causal Risk Factor for Aortic Valve Stenosis. J Am Coll Cardiol. 2020;75:163–176. doi: 10.1016/j.jacc.2019.10.050 [DOI] [PubMed] [Google Scholar]
- 48.Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2014;10:51–61. doi: 10.1038/nrendo.2013.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fall T The Role of Adiposity in Cardiometabolic Traits- A Mendelian Randomization Analysis. PLOS Medicine. 2013;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jowett JB, Curran JE, Johnson MP, Carless MA, Goring HH, Dyer TD, Cole SA, Comuzzie AG, MacCluer JW, Moses EK, et al. Genetic variation at the FTO locus influences RBL2 gene expression. Diabetes. 2010;59:726–732. doi: 10.2337/db09-1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y, Hambly BD, McLachlan CS. FTO associations with obesity and telomere length. J Biomed Sci. 2017;24:65. doi: 10.1186/s12929-017-0372-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurz DJ, Kloeckener-Gruissem B, Akhmedov A, Eberli FR, Buhler I, Berger W, Bertel O, Luscher TF. Degenerative aortic valve stenosis, but not coronary disease, is associated with shorter telomere length in the elderly. Arterioscler Thromb Vasc Biol. 2006;26:e114–117. doi: 10.1161/01.ATV.0000222961.24912.69 [DOI] [PubMed] [Google Scholar]
- 53.Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, Distefano M, Senol-Cosar O, Haas ME, Bick A, et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell. 2019;177:587–596 e589. doi: 10.1016/j.cell.2019.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinthorsdottir V, McGinnis R, Williams NO, Stefansdottir L, Thorleifsson G, Shooter S, Fadista J, Sigurdsson JK, Auro KM, Berezina G, et al. Genetic predisposition to hypertension is associated with preeclampsia in European and Central Asian women. Nat Commun. 2020;11:5976. doi: 10.1038/s41467-020-19733-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCracken IR, Dobie R, Bennett M, Passi R, Beqqali A, Henderson NC, Mountford JC, Riley PR, Ponting CP, Smart N, et al. Mapping the developing human cardiac endothelium at single cell resolution identifies MECOM as a regulator of arteriovenous gene expression. Cardiovasc Res. 2022. doi: 10.1093/cvr/cvac023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aikawa E The Developmental Origin of Calcific Aortic Stenosis. N Engl J Med. 2022;386. [DOI] [PubMed] [Google Scholar]
- 57.Xu Q Comparative analysis of the two extremes of FLNB-mutated autosomal dominant disease spectrum- from clinical phenotypes to cellular and molecular findings. Am J Transl Res. 2018;10. [PMC free article] [PubMed] [Google Scholar]
- 58.Hsu J, Gore-Panter S, Tchou G, Castel L, Lovano B, Moravec CS, Pettersson GB, Roselli EE, Gillinov AM, McCurry KR, et al. Genetic Control of Left Atrial Gene Expression Yields Insights into the Genetic Susceptibility for Atrial Fibrillation. Circ Genom Precis Med. 2018;11:e002107. doi: 10.1161/CIRCGEN.118.002107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eijgelsheim M, Newton-Cheh C, Sotoodehnia N, de Bakker PI, Muller M, Morrison AC, Smith AV, Isaacs A, Sanna S, Dorr M, et al. Genome-wide association analysis identifies multiple loci related to resting heart rate. Hum Mol Genet. 2010;19:3885–3894. doi: 10.1093/hmg/ddq303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Avery CL, Wassel CL, Richard MA, Highland HM, Bien S, Zubair N, Soliman EZ, Fornage M, Bielinski SJ, Tao R, et al. Fine mapping of QT interval regions in global populations refines previously identified QT interval loci and identifies signals unique to African and Hispanic descent populations. Heart Rhythm. 2017;14:572–580. doi: 10.1016/j.hrthm.2016.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vasan RS. Genetic Variants Associated With Cardiac Structure and Function. JAMA. 2009;302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. 2022;19:151–167. doi: 10.1038/s41569-021-00608-2 [DOI] [PubMed] [Google Scholar]
- 63.Loh PR, Danecek P, Palamara PF, Fuchsberger C, Y AR, H KF, Schoenherr S, Forer L, McCarthy S, Abecasis GR, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klarin D, Busenkell E, Judy R, Lynch J, Levin M, Haessler J, Aragam K, Chaffin M, Haas M, Lindstrom S, et al. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat Genet. 2019;51:1574–1579. doi: 10.1038/s41588-019-0519-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fang H, Hui Q, Lynch J, Honerlaw J, Assimes TL, Huang J, Vujkovic M, Damrauer SM, Pyarajan S, Gaziano JM, et al. Harmonizing Genetic Ancestry and Self-identified Race/Ethnicity in Genome-wide Association Studies. Am J Hum Genet. 2019;105:763–772. doi: 10.1016/j.ajhg.2019.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J, Ferreira T, Morris AP, Medland SE, Genetic Investigation of ATC, Replication DIG, Meta-analysis C, Madden PA, Heath AC, Martin NG, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–375, S361–363. doi: 10.1038/ng.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balduzzi S, Rucker G, Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid Based Ment Health. 2019;22:153–160. doi: 10.1136/ebmental-2019-300117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–784. doi: 10.1093/bioinformatics/btu704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karlson EW, Boutin NT, Hoffnagle AG, Allen NL. Building the Partners HealthCare Biobank at Partners Personalized Medicine: Informed Consent, Return of Research Results, Recruitment Lessons and Operational Considerations. J Pers Med. 2016;6. doi: 10.3390/jpm6010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.BioME BioBank Program. https://icahn.mssm.edu/research/ipm/programs/biome-biobank. 2022. Accessed 5/6/22.
- 71.Weeks EM, Ulirsch JC, Cheng NY, Trippe BL, Fine RS, Miao J, Patwardhan TA, Kanai M, Nasser J, Fulco CP, et al. Leveraging Polygenic Enrichments of Gene Features to Predict Genes Underlying Complex Traits and Diseases. medRxiv. 2020. doi: 10.1101/2020.09.08.20190561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuber V, Grinberg NF, Gill D, Manipur I, Slob EAW, Patel A, Wallace C, Burgess S. Combining evidence from Mendelian randomization and colocalization: Review and comparison of approaches. Am J Hum Genet. 2022. doi: 10.1016/j.ajhg.2022.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Y, Ding J, Reynolds LM, Lohman K, Register TC, De La Fuente A, Howard TD, Hawkins GA, Cui W, Morris J, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet. 2013;22:5065–5074. doi: 10.1093/hmg/ddt356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, Torstenson ES, Shah KP, Garcia T, Edwards TL, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full summary statistics for the discovery MVP CAS GWAS are publicly available in dbGaP (accession phs001672.v5.p1). Additional data supporting the manuscript, including replication of GWS single-nucleotide polymorphisms (SNPs), are available upon request from the corresponding author (COD). Data contributed by CARDIoGRAMplusC4D are available online (http://www.cardiogramplusc4d.org/). Data contributed by MEGASTROKE are available online (https://www.megastroke.org/). Data contributed by Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) are available on dbGaP (accession phs000930.v1.p1). Data contributed for single tissue cis-QTL are available online (https://www.gtexportal.org/home/). Data contributed by the Genetic Investigation of Anthropomorphic Traits (GIANT) consortium are available online (https://portals.broadinstitute.org/collaboration/giant/index.php/Main_Page). Data contributed by the International Consortium of Blood Pressure GWAS (ICBP) are on dbGaP (accession phs000585.v2.p1).