

ABSTRACT
The profiling of gene expression patterns to glean biological insights from single cells has become commonplace over the last few years. However, this approach overlooks the transcript contents that can differ between individual cells and cell populations. In this review, we describe early work in the field of single-cell short-read sequencing as well as full-length isoforms from single cells. We then describe recent work in single-cell long-read sequencing wherein some transcript elements have been observed to work in tandem. Based on earlier work in bulk tissue, we motivate the study of combination patterns of other RNA variables. Given that we are still blind to some aspects of isoform biology, we suggest possible future avenues such as CRISPR screens which can further illuminate the function of RNA variables in distinct cell populations.
Keywords: Alternative splicing, single-cell transcriptomics, isoforms, long-read sequencing technologies
Introduction
The field of single-cell omics has taken the study of gene expression and the resulting inference of cell-type composition, trajectory analysis, and regulatory mechanisms to new levels with hundreds of papers now being published on a yearly basis. The total number of studies is over 1800 at the time of writing [1]. Unlike bulk RNA sequencing, single-cell RNA sequencing employs the labeling of reads with a barcode identifier that denotes their cell of origin. These barcodes can either be attached by isolating single cells in wells, or by employing droplet-based microfluidics. Since 2015, this method has been harnessed to yield genomic and transcriptomic insights about the individual cell, as well as differences between cell-type populations. Somewhat less appreciated, but nevertheless strongly noticed, has been the study of RNA isoforms in single cells.
When discussing RNA, the term “isoform” has been used with some ambiguity. Loosely speaking, the word isoform is used when RNA attributes distinguish different transcripts generated from the same gene. Such RNA attributes – or “RNA variables” [2], as we like to call them, include varied primary sequence elements such as the choice of the transcription start site (TSS), alternative acceptors, donors and selective exon inclusion, as well as the choice of polyadenylation (polyA) site. While not considered in state-of-the-art annotation projects [3–5], polyA-tail length could likewise be considered an isoform-defining element in the primary sequence [6–8], with implications in development and disease. RNA modifications and secondary structure also add to the complexity of RNA molecules. However, in contrast to other RNA variables, RNA modifications and structure are not directly measurable in cDNA. The exception to this rule is RNA editing – where modified adenosine residues are turned into inosine, which are then read as guanosines during reverse transcription – thus representing an RNA modification that can be detected in full-length cDNAs. In our lab, we reserve the term “isoform” to denote long RNA molecules which can harbor multiple alternative RNA variables in tandem. We refer to events involving changes in the state of single RNA variables by specific definitions such as “included alternative exon” or a “modified base”. Thus, the study of RNA isoform expression at single-cell resolution allows for the study of one or multiple of these RNA variables in single cells. Obtaining single-cell isoform readouts gives insights into the complex usage patterns of RNA variables at the level of rare cell types, intermediate cell states, and disease conditions.
In this review, we focus on the merits and pitfalls of analyzing isoform readouts from single cells and cell types and discuss the state of the art on these approaches. We describe studies that have discovered novel RNA variables and preferential usage of isoforms either in specific cell types or under pathological conditions. Throughout the review, we emphasize the need for detecting and devising methods for treating artifacts that may arise either during the library preparation or the sequencing steps. Finally, we discuss the multimodal measurements we can currently detect in tandem with isoform expression and speculate about future innovations that can allow us to study transcription at the single-cell level from more than one angle.
Single-cell resolution – why? Diversity of cell types vs. diversity of single cells
Early work in the field of single-cell sequencing involved deep Illumina sequencing of a small number of cells which were either isolated in wells (Smart-Seq) or captured using microfluidic approaches, such as the Fluidigm C1. This approach allowed researchers to ask how one individual cell differs from another in its molecular makeup. Most of the efforts involving the development of single-cell technologies focused on blood samples due to easy accessibility, the nature of blood being a single-cell suspension, the existence of well-characterized cell types, and their relevance to immunology [9]. The brain was also commonly investigated, conversely motivated by the desire to uncover the complexity of brain cell types and the difficulty in dissociating single cells. Studies querying splicing patterns at the single-cell level suggested that intermediate inclusion levels of exons previously observed in bulk tissue could in large parts be explained by high inclusion levels in some cells and low inclusion levels in others [10,11], often termed exon bimodality. Shalek et al. demonstrated an example of bimodality for an exon in the mRNA of transcriptional regulator Irf7 which is alternatively spliced in bone-marrow derived dendritic cells and can differentially activate two cell populations with distinct transcriptomic signatures [10]. Follow-up studies, while still observing this bimodality for ~20% of exons, showed that unimodality is much more common than multimodality [12]. Other studies since have made the argument that bimodality can be explained by technical artifacts such as information loss in library preparation resulting in low coverage in individual cells [13]. They recommend using stringent normalization and filtering methods to gain biologically meaningful results, or caution against estimating differences in isoform abundances at the single-cell level [14,15]. These studies typically employed short-read data, which does not allow for the identification of full-length isoforms constructed from the combination of multiple RNA variables.
Based on the development of long-read RNA sequencing [16–18], later studies using Pacific Biosciences (PacBio) long-read sequencing for a few individual cells from the oligodendrocytic lineage revealed important biological insight into the vast transcript diversity of single cells and argued against the aforementioned bimodality argument [19]. Karlsson and Linnarson additionally showed that isoform diversity scales up with gene expression; however, they find that coding isoforms are subject to tighter regulation and evolutionary constraints. Additional work using Oxford Nanopore Technologies (ONT) on 7 and 96 single cells revealed differential usage of complex isoforms in B-cell surface receptors, and particularly highlighted a population of cells exhibiting immunotherapy resistance through the loss of a CD19 epitope upon treatment [20]. However, at the time of writing, while useful for understanding the complexity within individual cells, experiments with less than 100 cells would in most cases be deemed inadequate for determining the differences between a-priori unknown cell populations present among these cells.
With the field expanding over the last decade, new protocols and bioinformatic tools have been developed to investigate splicing at the single-cell level; a list of the most recent ones at the time of writing can be found in Tables 1 and 2. We now possess methods to extract splicing information in a variety of distinct settings and therefore query cell-type specific usage of RNA variables. This can be achieved from tens of thousands of fresh individual cells either by long-read sequencing [24,25], using methods to enrich for barcoded, spliced cDNAs from thousands of single nuclei, [31] or by short-read sequencing [13,42]. Using single-cell long-read sequencing, we found splicing patterns distinguishing subtypes of hippocampal inhibitory neurons and granule neuroblasts as early as postnatal day 7 for the calcium/calmodulin dependent protein kinase II beta (Camk2b) gene [34]. Using SMART-seq short-read sequencing data, Booeshaghi et al. showed differential exon usage between glutamatergic and GABAergic neurons in the mouse primary motor cortex for Oxr1, a gene involved in preventing neurodegeneration through oxidation resistance [42]. Similarly, using 10x single-cell RNA-seq data and their computational framework Psix, Buen Abad Najar et al. [13] looked at midbrain dopamine neurons through development. They identified 78 differentially spliced exons that correlated with changes in expression and binding of neuronal splicing factors such as Nova1, Rbfox1 and Mbnl2, which can have downstream regulatory consequences. A more recent computational framework that uses generalized linear models (GLMs) to identify differential splicing across conditions while controlling for length biases in single-cell short read data was developed by Benegas et al. [38]. They used this framework, scQuint, to identify thousands of alternative splicing events in the motor cortex and B-cell development in the bone marrow. This approach is especially useful for leveraging the large amount of droplet-based scRNA-Seq short-read data generated in the last decade.
Table 1.
Wetlab protocols for profiling splicing information from single cells.
| Method | Profiled RNA variables |
Cell number | Sequencing technology to profile splicing | Publication | |||||
|---|---|---|---|---|---|---|---|---|---|
| Exons | Exon junctions | TSS | PolyA | Complete isoforms | Other | ||||
| Smart-Seq2 | Yes | Yes | Yes | Yes | No | - | ~30–160 | Short-read (Illumina) | Picelli et al, 2013 [21] |
| R2C2 | Yes | Yes | Yes | Yes | Majority | - | 96 (extended to 5,000–15,000 in 2022) [22] |
Concatenated long-read (ONT) | Volden et al, 2018 [23] |
| ScISOr-Seq | Yes | Yes | Yes | Yes | Majority | - | ~5,000–15,000 | Long-read (Pacbio, ONT) | Gupta et al, 2018 [24] |
| RAGE-Seq | Yes* | Yes* | Yes* | Yes* | Majority | Targeted T- and B-cell receptor clonotyping * Focused on VDJ |
~5,000–15,000 | Long-read (ONT) | Singh et al, 2019 [25] |
| Smart-seq3 | Yes | Yes | Yes | Yes | No | - | ~3,000–5,000 | Short-read (Illumina) | Hagemann-Jensen et al, 2020 [26] |
| Smart-seq-total | Yes | Yes | Yes | Yes | No | Non poly(A) transcripts | ~200–600 | Short-read (Illumina) | Isakova et al, 2020 [27] |
| ScNaUMI-Seq | Yes | Yes | Yes | Yes | Majority | ~5,000–15,000 | Long-read (ONT) | Lebrigand et al, 2020 [28] | |
| FlsnRNA-Seq | Yes | Yes | Yes | Yes | Minority | - | ~5,000–15,000 | Long-read (ONT) | Long et al, 2021 [29] |
| LR-SplitSeq | Yes | Yes | Yes | Yes | Minority | - | ~1,000 (long-reads) ~37,000 (short reads) |
Long-read (Pacbio) | Rebboah et al, 2021 [30] |
| SnISOr-Seq | Yes | Yes | Yes | Yes | Minority | - | ~5,000–15,000 | Long-read (Pacbio, ONT) | Hardwick et al, 2022 [31] |
Table 2.
Bioinformatic tools to analyze splicing information from single cells.
| Name | Applications | Data type | Publication | Code |
|---|---|---|---|---|
| C3POa | Consensus calling on R2C2 data | Barcoded ONT long reads prepared using R2C2 protocol | https://github.com/christopher-vollmers/C3POa | Volden et al, 2018 [23] |
| snuupy | Barcode/UMI calling, error correction, extract splicing information | Barcoded ONT long reads + 10x reference barcodes and UMIs | https://github.com/ZhaiLab-SUSTech/snuupy | Long et al, 2020 [29] |
| TALON | Processing of long-reads. Single-cell functionality now available | Platform agnostic long read data | https://github.com/mortazavilab/TALON | Wyman et al, 2020 [32] |
| Sierra | Differential transcript usage | 10x short reads | https://github.com/VCCRI/Sierra | Patrick et al, 2020 [33] |
| sicelore | Error-corrected barcode calling | Barcoded ONT long reads + 10x reference barcodes and UMIs | https://github.com/ucagenomix/sicelore-2.1 | Lebrigand et al, 2020 [28] |
| scisorseqr | Perfect-match barcode calling, Isoform identification, differential isoform expression, visualization | Barcoded PacBio/ONT long reads + 10x reference barcodes | https://github.com/noush-joglekar/scisorseqr | Joglekar et al, 2021 [34] |
| ScNapBar | Error corrected barcode calling | Barcoded ONT long reads + 10x reference barcodes and UMIs | https://github.com/dieterich-lab/single-cell-nanopore | Wang et al, 2021 [35] |
| IsoTools | Isoform identification, differential splicing analysis, visualization | Platform agnostic long read data | https://github.com/MatthiasLienhard/isotools | Lienhard et al, 2021 [36] |
| LR-splitpipe | Barcode detection of LR-SplitSeq | Combinatorially barcoded smart-seq2 reads | https://github.com/fairliereese/LR-splitpipe | Rebboah et al, 2021 [30] |
| FLAMES | Barcode and UMI detection, isoform quantification, differential splicing analysis | ONT long read data | https://bioconductor.org/packages/release/bioc/html/FLAMES.html | Tian et al, 2021 [37] |
| scQuint | Differential splicing analysis | 10x 3’ short reads | https://github.com/songlab-cal/scquint | Benegas et al, 2022 [38] |
| spliZ | Differential splicing analysis | 10x 3’ short reads | https://github.com/juliaolivieri/SpliZ_pipeline/ | Olivieri et al, 2022 [39] |
| Psix | Differential alternative splicing events | Platform agnostic short read data | Buen Abad Najar et al, 2022 [13] | |
| IsoQuant | Isoform detection and processing of long-reads. Single-cell functionality now available | Platform agnostic long read data | https://github.com/ablab/IsoQuant | Prjibelski et al, 2023 [40] |
| BLAZE | Barcode detection | ONT long read data | https://github.com/shimlab/BLAZE | You et al, 2023 [41] |
| IsoSeq3 | Barcode calling on PacBio data, | Barcoded PacBio long reads + 10x reference barcodes | https://github.com/PacificBiosciences/IsoSeq | |
| Sockeye | Barcode and UMI calling on Nanopore data | ONT long read data | https://github.com/epi2me-labs/wf-single-cell |
In our own work using a combination of short and long reads, we found the Bin1 gene to be broadly expressed across cell types in the postnatal day 1 (P1) mouse cerebellum, while some of its full-length isoforms were almost perfectly unique to distinct neuronal and glial cell types [24]. A more precise investigation of single-cell isoforms in the P7 mouse prefrontal cortex and hippocampus showed that the majority of brain-region specific isoform expression is caused by one clearly definable cell type [34]. However, for a smaller set of genes, multiple cell types show the same brain-region specific isoform expression patterns [34]. Considering multiple adult brain regions, cell subtypes, and developmental time points, we found that the majority of genes alters isoform expression in at least one of these three dimensions for matched cell types. We furthermore found that isoforms distinguish neuronal subtypes, and clustering on long reads recovered short-read derived cell types and hinted at the possibility of recovering cell states [43]. Long-read clustering was also explored by other groups [28,30,44] who achieved higher throughput of sequencing per single cell, by using library preparation methods such as combinatorial barcoding (LR-SplitSeq), concatenating cDNA molecules (MAS-Iso-Seq), or only using a subsample of cells for long-read sequencing [37]. Bioinformatic tools such as SiCeLoRe [28] leverage error-correction of ONT reads to get higher throughput, and the aforementioned scQuint [38] uses Variational Autoencoders (VAE) for dimensionality reduction of short-read data to visualize cell types as defined by splicing. Since long-read sequencing captures full-length transcripts, including their 5’ ends, we found several examples of other RNA variables such as TSS-mediated isoform regulation, especially in choroid plexus epithelial cells at P7 [34]. Novel TSS were found in key transcription factors such as Smarca4 and Foxp1 during B-cell development, and splicing factors such as Rbfox1 in the motor cortex [38]. Switches in novel alternative TSS usage were also reported during myocyte differentiation, some examples of which were validated with a corresponding increase in chromatin accessibility through Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) at the single-nuclei level [30]. Tian et al. [37] additionally used correlated differential TSS usage in cancer cell lines with open promoter peaks in scATAC-Seq, further demonstrating the power of full-length isoform sequencing.
Sequencing individual nuclei from frozen human-brain tissue also revealed programs of alternative splicing. Prior bulk RNA-seq research in brain tissue had shown that thousands of microexons are highly cell-type specific and regulated by splicing factors such as RBFOX and nSR100/SRRM4, in addition to having a strong link to autism [45–47]. In human cortical single-nuclei data, when comparing exons associated with multiple neurological diseases, we found autism spectrum disorder (ASD) associated exons to exhibit the greatest degree of neuronal and glial specificity [31]. Exons associated with amyotrophic lateral sclerosis (ALS) also showed increased cell-type specificity – although to a lesser extent than those associated with ASD. While neuronal microexons are dysregulated in ASD [46,48,49] in the human frontal cortex, cell-type specificity also extends to longer exons albeit with less strength, and some microexons are also expressed in glial cell types [31]. The potential functions of these exons can be used to inform drug design in targeting specific cell types. As single-cell work is bringing more insight into pathological mechanisms, it is also opening the door for novel disease treatments.
Advancing therapeutics by investigating differential isoform expression
Using single-cell long-read sequencing, differential isoform expression is often observed within cancer cell types. Recent work reported tumor-specific isoform usage as a byproduct of epithelial to mesenchymal transition in the tumor microenvironment [50]. Dondi et al. found over fifty thousand novel isoforms as well as programs of genomic and transcriptional dysregulation. They also described an IGF2BP2:TESPA1 gene fusion found in the cancer cells of a patient which had been mischaracterized as an upregulation of TESPA1 with prior short-read technologies. This highlights how useful single-cell long-read technologies can be when discussing diagnosis and personalized medicine. A manifold of novel full-length isoforms in complex tissues such as brain and blood have also been described, revealing previously unannotated TSS, polyA sites, and alternative exons in excitatory, inhibitory neurons, and glial subtypes [24,34] as well as in B and T cells [22]. Related work showed high diversity of isoforms for the surface receptors that define cell identity and dictate interactions within the immune system. The study of Human Leukocyte Antigen (HLA) isoforms also allowed for the identification of allele-specific isoform expression, including intron retention, alternative exons and polyA sites [51]. Such work is particularly significant not only to understand the immune response to disease, but also for the development of novel therapeutic approaches.
Splicing-based therapeutic approaches have also been discussed in relation to cancer treatments. Data from over 8,000 patients and 32 cancer types demonstrated a 30% increase in alternative splicing events in tumors compared to healthy tissue, with over 900 exon junctions detectable only in tumors [52]. Thus, a novel cancer therapy could employ neoantigens which are tumor splice-site specific. Similarly, other therapies targeting cancer-specific alternative splicing include targeted splicing-factor small molecules, such as pladienolides which block the assembly of the spliceosome [53–55]. Antisense oligonucleotides (ASOs) have been developed to target splicing enhancers or repressors, such as hnRNPA1/A2 to include exon 7 along the SMN2 gene, as a clinically approved treatment for spinal muscular atrophy [56–58]. This work is a prime example of how understanding a fundamental molecular process and its downstream effects is crucial to further the treatment for many diseases, especially in cases where splicing may be cell-type or cell-state specific.
Combination patterns of RNA variables: exhaustive exploration vs. coordination
The above work has revealed interesting observations on individual RNA variables. As isoforms can harbor many RNA variables, the question of how these variables are combined into complete isoforms is of interest to understand the extent of molecular variation. Early studies have shown the coordination of alternative splicing with promoter and polyadenylation site usage, transcription elongation rates, and sequence motifs found in surrounding introns and exons [59–62]. Over the last few years, such works have inspired our lab and others to invest considerable energy into understanding these combination patterns at the single-cell level in complex tissues and in a high throughput manner. Overall, the community has found that many genes harbor at least one pair of coordinated exons [63–65], with examples of mutually exclusive exons found in various pathologies. Such examples include cancer with the coordination of two exons of TMEM16A, a gene encoding a chloride channel overexpressed in many breast cancer tumors [66]. Another well-known spliceopathy is myotonic dystrophy [67] where many pre-mRNA molecules are mispliced, giving rise to fetal isoforms which in turn impair myogenesis. Among others, one example is the two mutually exclusive exons α1 and α2 of the MEF2D gene, a transcription factor essential in muscle cell differentiation and development.
Although coordination effectively limits the variety of isoforms expressed, many genes do contain randomly paired exon pairs, thereby increasing the isoform number per gene considerably. Single-cell long-read approaches have enabled the investigation of how RNA-variable coordination differs among cell types. A surprising observation, made in both mouse [24] and human [31] brain, is that alternative exon pairs with (“distant pairs”) and without (“adjacent pairs”) intervening exons behave differently. Indeed, distant pairs that are coordinated, i.e., distant pairs with a preference for two combinations as opposed to the theoretically possible four, usually achieve coordination in bulk through cell-type specific isoform expression [24,31]. In other words, for mutually associated but distant exons, the both-exon-in isoform characterizes one cell type, while the both-exons-out isoform characterizes another (Figure 1a). For adjacent coordinated exons, however, a different pattern emerges: the preference for two isoforms out of the four possible ones is usually observed the same way in at least one cell type (Figure 1b). Additionally, when coordinated, adjacent exon pairs are predominantly mutually associated, whereas distant ones have a higher tendency to also allow for mutual exclusion [24,31] (Figure 1c).
Figure 1.
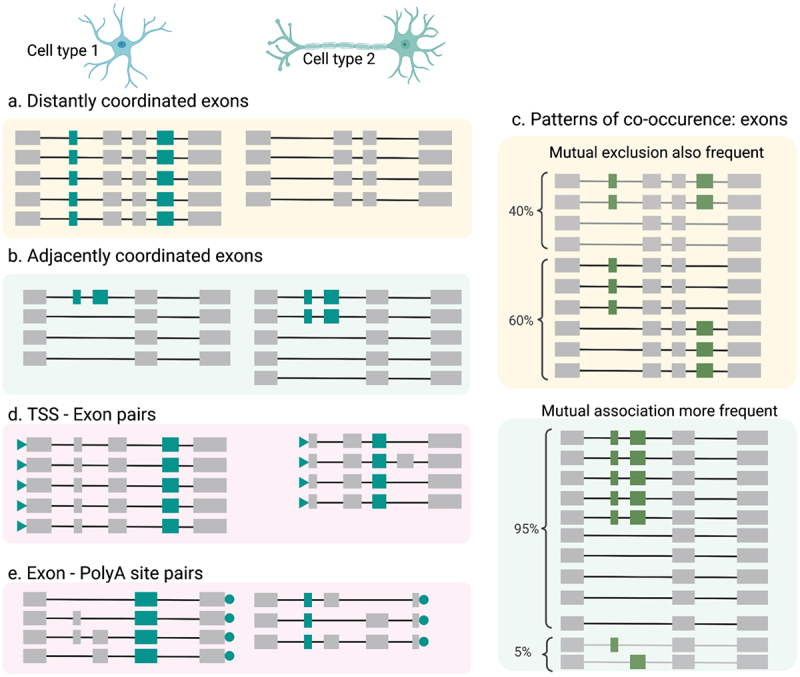
Coordination patterns of RNA variables (a) Distantly coordinated exons exhibit cell-type specificity wherein Cell type 1 (top left) has both exons being included across most/all transcripts whereas Cell type 2 (top right) has both exons being skipped (b) Adjacent coordinated exons tend to be cell-type agnostic, wherein both cell types exhibit two distinct patterns of coordination, albeit to different degrees, (c) Distantly coordinated exons (top) are are frequently mutually exclusive, meaning that transcripts include either the first or the second alternative exon. Adjacent coordinated exons (bottom) on the other hand are primarily mutually associated, meaning that both exons are either included together, or skipped together. (d) Transcription start sites (TSS) usage can also be coordinated with the presence or absence of alternative exons. Cell type 1 displays an upstream canonical start site, whereas Cell type 2 displays an alternative TSS which co-occurs with the existence of the highlighted exon in teal. These patterns are similar to distantly coordinated exons in that they are cell-type specific (e) Same as in (d) but for alternative poly(A) site usage which is also cell type specific.
Of note, previous research has established that promoter structure can influence splicing decisions [60,68–70] and a genome-wide trend was suggested based on the analysis of annotated transcripts [71]. Cramer et al. [60] used multiple genetically engineered constructs to drive transcription of the fibronectin gene from distinct promoters and found differences in the inclusion of an internal exon, depending on the employed promoter. Of course, within complex human tissue, no such genetic engineering can occur. However, human genes can have multiple TSS that are separated by tens of thousands of bases. In these situations, the same gene can be driven from distinct promoters, which makes the logic by Cramer et al. applicable to alternative splicing in complex tissue. Therefore, one would expect to find associations between TSS choice and exon inclusion in genomic data, which we found as coordinated TSS-exon pairs in our human brain single-nuclei isoform data. While these were less prevalent than coordinated exon–exon pairs in our scan, in terms of cell-type specific expression of these pairs, coordinated TSS-exon pairs behaved in majority like distant exon pairs and oppositely to adjacent pairs. In most cases, either the choice of the alternative TSS or the exon is constitutive at the cell-type level (Figure 1d). Therefore, distant coordinated exon pairs, as well as coordinated TSS-exon pairs tend to distinguish cell types, but the resultant isoforms tend to co-occur less within the same cell type [31].
Notably, for most coordination events, we currently ignore whether they mechanistically influence each other, or whether there is an external factor governing the inclusion of both. For two exons in the FN gene, it has been shown that RNA Polymerase II speed influences the inclusion of both exons [61]. It stands to reason that this model may affect other exon pairs as well. However, to what extent this model links to most exon-coordination events is so far unknown.
Given the above considerations about the variability of some splicing events across single cells, for some exon-coordination events, measurements with single-cell and even single-cell-single-nucleotide resolution might considerably advance the field. In this respect, technologies like PERTURB-Seq [72–74], which apply scRNA-seq to CRISPR screens could add important insights to the field. Although the original publications of these techniques investigated immune response in the brain, T-cell receptor induction, as well as unfolded protein stress response, some recent work has focused on RNA variables, notably looking at polyA site usage and polyadenylation regulation [75]. Paired with long-read sequencing, such techniques would gain isoform resolution and could answer questions such as (i) which splicing factors control which exons? (ii) If multiple exons in one gene are affected, are they affected in a coordinated manner or not? Finally, (iii) does the CRISPR-mediated deletion of one exon affect the splicing of introns surrounding the second exon in the same molecule?
Quality, barcode calling, and throughput – important factors in single-cell long-read sequencing
Barcoding approaches were primarily pioneered to facilitate the identification of the cell-of-origin of each RNA molecule to subsequently determine cell-to-cell variability in gene expression [76,77] and then to define cell types in complex tissue. The concept of unique-molecular-identifiers (UMIs) was later added to distinguish PCR duplicates. An unintended, yet interesting consequence is that after PCR, the resulting cDNA mixes contain many copies of the same original RNA molecule. Such cDNA mixes can then be split into pools for multiple experimental procedures. We recently followed this paradigm to get a sequencing readout for the same individual RNA molecule on both the PacBio platform and the ONT system [78]. This highlighted that annotation-diverging NAGNAG acceptors in ONT reads are usually confirmed by the PacBio platform for the same molecule. However, GTNNGT annotation-diverging donors suggested by the ONT reads are almost never confirmed. Such knowledge formed the basis for creating accurate long-read interpretation software [40].
Just like for single-cell short-read sequencing, non-microfluidic approaches have also been explored for high-throughput single-cell long-read sequencing. In combination with long-read sequencing, Rebboah et al. [30] have used a kit from Parse Biosciences, which uses a split-pool approach to allow sequential cDNA barcoding through ligation for higher cell numbers. Furthermore, barcoding techniques can be applied to mounted tissue slices to obtain the location in addition to the spot of origin for an RNA molecule. We leveraged this technique to obtain spatially resolved isoform expression in the brain, and showed clear gradients in isoform choice on a tissue slice during cellular differentiation and postnatal development [34]. Extending this work further, Lebrigand et al. [79] developed a method to obtain higher throughput for spatial isoform information and demonstrated region-specific isoform switching in the olfactory bulb, and provided an A-to-I RNA-editing map in adult mouse brain. They showed that the RNA-editing landscape varies across brain regions, and that the thalamus is an outlier in terms of A-to-I editing, correlating with an increase in the gene expression of editing enzymes adenosine deaminases (ADARs). Accordingly, we recently showed the thalamus being an outlier in terms of regional isoform specificity, as defined by splicing, TSS and poly(A) sites [43]. These studies underscore the importance of considering more than one aspect of RNA biology when studying single-cell gene and isoform expression.
Fundamental to the success of single-cell isoform studies are (i) the number of distinct molecules per cluster or per individual cell that one is able to record, as well as (ii) the accuracy with which barcodes/UMIs can be retrieved. PacBio and ONT, which are currently the two long-read technologies of choice, are both improving. PacBio, which achieves highly accurate reads, is working toward higher throughput using methods of concatenating molecules [44,80]. ONT achieved very strong throughput and averages around 50–100 million cDNA molecules per run of a PromethION flow cell and is improving accuracy with its Q20+ chemistry.
Throughput and quality essentially define the success of recognizing barcodes and UMIs. Given the very high quality on the PacBio system, perfect-match searches for these elements are certainly the most precise approach without substantial losses in recall. On the Nanopore system however, with much higher throughput yet lower quality basecalling, perfect-match searches are prone to strong losses in recall. To circumvent this, methods like rolling-circle amplification were successfully introduced to obtain high accuracy albeit at the cost of throughput [23]. More recently, a number of error-allowing algorithms have been proposed for barcode deconvolution [28,35,41,81]. Some of these approaches use heuristics or network analysis, while others employ deep learning. Of note, to-date, we do not have a perfect understanding of the biases, limitations and advantages of each approach – a topic certainly in need of a closer look.
Single-cell measurements to studying RNA isoforms – what can we measure at this point, what are we yet blind to?
The research described above has allowed us to investigate usage and combination patterns of some RNA variables (Figure 2a) that exist in bulk [63–65] and in single cells [24,31,34]. However, the combination patterns of RNA modifications with themselves as well as the aforementioned TSS, exons, and polyA sites remain relatively under-described in single cells. In bulk, an RNA editing site in the Glur-B gene, which lies one nucleotide away from the exon/intron border of exon 13 has been connected to splicing execution. Splicing of the rat GLUR-B pre-RNA molecule prior to RNA editing has been shown to eliminate all editing of this site while prior editing does not inhibit splicing in vivo. In vitro, additions of adenosine deaminase to GLUR-B pre-RNA molecules shows eradication of splicing around this editing site [82]. This suggests that in vivo interactions between editing and splicing machinery exist such that they sequentially process RNA molecules without impediment. Such an example raises multiple fundamental questions on whether RNA editing, as well as RNA modifications such as m6As or RNA structures can be coordinated genome-wide and at a cell-specific level (Figure 2b). While RNA-editing events can be easily studied using single-cell cDNA sequencing, most other RNA modifications will require more elaborate approaches, as these modifications are lost during the usual cDNA preparation steps. One way of circumventing this problem is the usage of reverse-transcription assays that introduce mismatches in the cDNA upon encountering RNA modifications or unstructured regions, or the induction of in vitro targeted RNA-editing that could then be detected in the cDNA molecule. Analysis of the mismatches with respect to the genome would then essentially allow the localization of modifications and/or RNA structure elements in single-cell cDNAs. An alternative to this approach is to add chemical modifications to non-structured regions and then to deduce the presence of such modifications (and thus the absence of structure) through changes in direct RNA sequencing signal using ONT. This approach has been successfully taken to reveal isoform-specific RNA structures, showing that alternative isoforms may exhibit structural differences in shared exons, which may correlate with differences in translation efficiency [83]. This could in principle be employed in single cells as well – if direct RNA single-cell sequencing was to become possible, a frontier we recently commented on [2].
Figure 2.
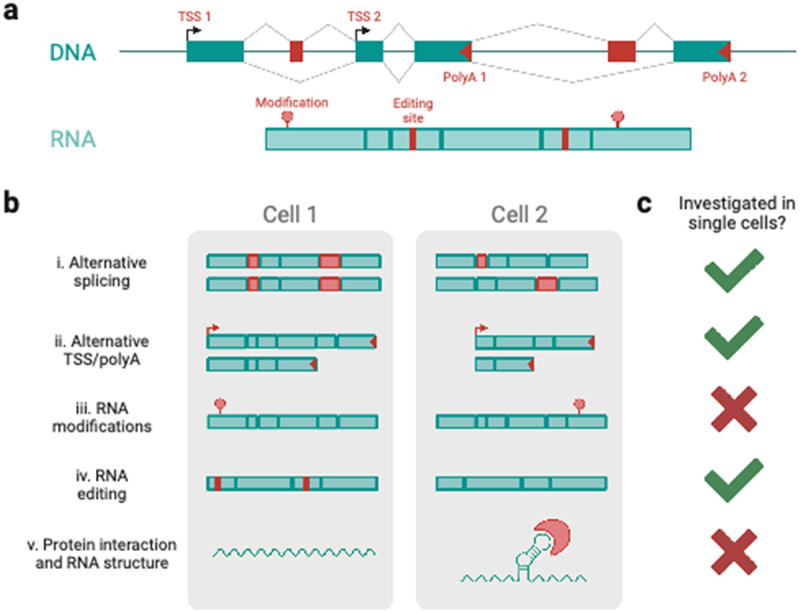
RNA variables in single cells. (a) Schematic of the RNA variables of a gene at the DNA (TSS and polyA site usage, allele-specific sequence) and RNA (nucleotide modifications and editing, alternative splicing, secondary structures) level. (b) Differential RNA variables between hypothetical cells 1 and 2 for i) alternative splicing, ii) alternative TSS and polyA usage, iii) RNA modifications, iv) RNA editing, and v) protein interactions and RNA structure in single cells. (c) Availability of data and techniques to address RNA variables in single cells, as of time of publication (see main text).
Isoform biology is tightly linked to a variety of other factors. Particularly in human tissues, somatic mutations can affect transcriptional outcomes, and SNVs linking to distinct transcriptional profiles have been detected with some success from ONT long-read data [37]. In addition to splicing factors, noncoding RNAs such as snRNAs are intricately linked to the splicing process and microRNAs have been linked to 3’UTR regulation. Moreover, chromatin structure and transcription-factor binding can also influence alternative splicing (see reference [84] for review). Allowing the study of chromatin and RNA in the same single cells, 10 Genomics released their Single-Cell Multiome kit in 2020, which measures both gene expression and chromatin accessibility in single cells. This novel technology expands the field by revealing a fuller picture of what happens inside a single cell, and how variables such as chromatin accessibility, cell-type, age, or disease can influence gene expression. Thus, from an isoform perspective, any multimodal measurement that measures one of the above factors along with the full-length isoforms in a single cell would allow us to decipher the regulatory mechanisms underlying isoform expression and their association with development and disease. Figure 2c highlights the next frontiers to tackle for the field to achieve a fuller picture of isoform biology in single cells.
Conclusion
Alternative splicing is an important step of RNA processing that confers a high degree of specialization to cellular function. In this review, we motivate the need for single-cell measurements of transcript diversity and point out the shortcomings of only focusing on gene counts by highlighting various studies that have identified novel RNA variables and isoform patterns that are specific to cell types and tissues. As a community, we have made significant headway in mapping the transcriptional profiles of common and rare cell types in various tissues, and in the context of disease. However, further technical advances will be needed to map out the splicing patterns and transcriptional diversity on the single-cell front. Given the many sources of noise that can arise in the preparation and sequencing of single-cell splicing libraries, we warn the reader to treat biological results and potential artifacts with caution. Nevertheless, with increasing technological advancements in terms of throughput, accuracy, and read length, we are getting closer to being able to assess isoforms at the single-cell level in a widespread manner. Additionally, with a greater understanding of technical artifacts, we are in a demonstrably better position to come up with analysis methods that circumvent these issues and still yield biologically meaningful results. Finally, we discuss the myriad ways in which we can enhance our understanding of molecular dynamics in single cells by coupling isoform sequencing with readouts of other modalities such as chromatin conformation, RNA modifications, and protein structure, and speculate about other potential avenues of exploration.
Funding Statement
The work was supported by the NIGMS [1R01GM135247-01].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Svensson V, da Veiga Beltrame E, Pachter L.. A curated database reveals trends in single-cell transcriptomics. Database (Oxford). 2020. DOI: 10.1093/database/baaa073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Foord C, Hsu J, Jarroux J, et al. The variables on RNA molecules: concert or cacophony? Answers in long-read sequencing. Nat Methods. 2023;20:20–24. [DOI] [PubMed] [Google Scholar]
- [3].Frankish A, Diekhans M, Ferreira A-M, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–d773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 2012;22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Subtelny AO, Eichhorn SW, Chen GR, et al. Poly(a)-tail profiling reveals an embryonic switch in translational control. Nature. 2014;508:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jalkanen AL, Coleman SJ, Wilusz J.. Determinants and implications of mRNA poly(A) tail size–does this protein make my tail look big? Semin Cell Dev Biol. 2014;34:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Legnini I, Alles J, Karaiskos N, et al. FLAM-seq: full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat Methods. 2019;16:879–886. [DOI] [PubMed] [Google Scholar]
- [9].Acosta J, Ssozi D, van Galen P. Single-cell RNA sequencing to disentangle the blood system. Arterioscler Thromb Vasc Biol. 2021;41:1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shalek AK, Satija R, Adiconis X, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu W, Zhang X. Single-cell alternative splicing analysis reveals dominance of single transcript variant. Genomics. 2020;112:2418–2425. [DOI] [PubMed] [Google Scholar]
- [12].Song Y, Botvinnik OB, Lovci MT, et al. Single-cell alternative splicing analysis with expedition reveals splicing dynamics during neuron differentiation. Mol Cell. 2017;67:148–161 e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Buen Abad Najar CF, Burra P, Yosef N, et al. Identifying cell state-associated alternative splicing events and their coregulation. Genome Res. 2022;32:1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Westoby J, Artemov P, Hemberg M, et al. Obstacles to detecting isoforms using full-length scRNA-seq data. Genome Biol. 2020;21:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Buen Abad Najar CF, Yosef N, Lareau LF. Coverage-dependent bias creates the appearance of binary splicing in single cells. Elife. 2020;9. DOI: 10.7554/eLife.54603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Koren S, Schatz MC, Walenz BP, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sharon D, Tilgner H, Grubert F, et al. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Au KF, Sebastiano V, Afshar PT, et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proc Natl Acad Sci USA. 2013;110:E4821–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Karlsson K, Linnarsson S. Single-cell mRNA isoform diversity in the mouse brain. BMC Genomics. 2017;18:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Byrne A, Beaudin AE, Olsen HE, et al. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun. 2017;8:16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Picelli S, Björklund ÅK, Faridani OR, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. [DOI] [PubMed] [Google Scholar]
- [22].Volden R, Vollmers C. Single-cell isoform analysis in human immune cells. Genome Biol. 2022;23:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Volden R, Palmer T, Byrne A, et al. Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc Natl Acad Sci USA. 2018;115:9726–9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gupta I, Collier PG, Haase B, et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat Biotechnol. 2018;36:1197–1202. [DOI] [PubMed] [Google Scholar]
- [25].Singh M, Al-Eryani G, Carswell S, et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat Commun. 2019;10:3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hagemann-Jensen M, Ziegenhain C, Chen, P, et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat Biotechnol. 2020;38:708–714. [DOI] [PubMed] [Google Scholar]
- [27].Isakova A, Neff N, Quake SR. Single-cell quantification of a broad RNA spectrum reveals unique noncoding patterns associated with cell types and states. Proc Natl Acad Sci USA. 2021;118:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lebrigand K, Magnone V, Barbry P, et al. High throughput error corrected Nanopore single cell transcriptome sequencing. Nat Commun. 2020;11:4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Long Y, Liu Z, Jia J, et al. FlsnRNA-seq: protoplasting-free full-length single-nucleus RNA profiling in plants. Genome Biol. 2021;22:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rebboah E, Reese F, Williams K, et al. Mapping and modeling the genomic basis of differential RNA isoform expression at single-cell resolution with LR-Split-seq. Genome Biol. 2021;22:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hardwick SA, Hu W, Joglekar A, et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat Biotechnol. 2022;40:1082–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wyman D, Balderrama-Gutierrez G, Reese F, et al. A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. bioRxiv. 2020;672931. doi: 10.1101/672931. [DOI] [Google Scholar]
- [33].Patrick R, Humphreys DT, Janbandhu V, et al. Sierra: discovery of differential transcript usage from polyA-captured single-cell RNA-seq data. Genome Biol. 2020;21:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Joglekar A, Prjibelski A, Mahfouz A, et al. A spatially resolved brain region- and cell type-specific isoform atlas of the postnatal mouse brain. Nat Commun. 2021;12:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Q, Bönigk, S., Böhm, V.. Single cell transcriptome sequencing on the Nanopore platform with ScNapBar. RNA. 2021;27:763–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lienhard M, van den Beucken, T. Timmermann, B.. Long-read transcriptome sequencing analysis with IsoTools. bioRxiv; 2021. DOI: 10.1101/2021.07.13.452091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tian L, Jabbari, J. S., Thijssen, R. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol. 2021;22:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Benegas G, Fischer J, Song YS. Robust and annotation-free analysis of alternative splicing across diverse cell types in mice. Elife. 2022;11. DOI: 10.7554/eLife.73520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Olivieri JE, Dehghannasiri R, Salzman J. The SpliZ generalizes ‘percent spliced in’ to reveal regulated splicing at single-cell resolution. Nat Methods. 2022;19:307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Prjibelski AD, Mikheenko, A., Joglekar, A. Accurate isoform discovery with IsoQuant using long reads. Nat Biotechnol; 2023. DOI: 10.1038/s41587-022-01565-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].You Y, Prawer, Y. D., Paoli-Iseppi, D. Identification of cell barcodes from long-read single-cell RNA-seq with BLAZE. Genome Biol. 2023;24:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Booeshaghi AS, Yao, Z., van Velthoven, C.. Isoform cell-type specificity in the mouse primary motor cortex. Nature. 2021;598:195–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Joglekar A, Hu, W., Zhang, B.. Single-cell long-read mRNA isoform regulation is pervasive across mammalian brain regions, cell types, and development. bioRxiv; 2023. DOI: 10.1101/2023.04.02.535281 [DOI] [Google Scholar]
- [44].Al’khafaji AM, Smith, J. T., Garimella, K. V. High-throughput RNA isoform sequencing using programmable cDNA concatenation. bioRxiv. 2021. DOI: 10.1101/2021.10.01.462818 [DOI] [PubMed] [Google Scholar]
- [45].Wang ET, Sandberg, R., Luo, S. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Irimia M, Weatheritt, R. J., Ellis, J. D. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li YI, Sanchez-Pulido L, Haerty W, et al. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015;25:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gonatopoulos-Pournatzis T, Niibori, R., Salter, E. W.. Autism-misregulated eIF4G microexons control synaptic translation and higher order cognitive functions. Mol Cell. 2020;77:1176–1192 e1116. [DOI] [PubMed] [Google Scholar]
- [49].Gonatopoulos-Pournatzis T, Blencowe BJ. Microexons: at the nexus of nervous system development, behaviour and autism spectrum disorder. Curr Opin Genet Dev. 2020;65:22–33. [DOI] [PubMed] [Google Scholar]
- [50].Dondi A, Lischetti, U., Jacob, F., Detection of isoforms and genomic alterations by high-throughput full-length single-cell RNA sequencing for personalized oncology. bioRxiv; 2022. DOI: 10.1101/2022.12.12.520051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cole C, Byrne A, Adams M, et al. Complete characterization of the human immune cell transcriptome using accurate full-length cDNA sequencing. Genome Res. 2020;30:589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kahles A, Lehmann, K. V., Toussaint, N. C.. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211–224 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mizui Y, Sakai, T., Iwata, M. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo). 2004;57:188–196. [DOI] [PubMed] [Google Scholar]
- [54].Folco EG, Coil KE, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011;25:440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sato M, Muguruma, N., Nakagawa, T.. High antitumor activity of pladienolide B and its derivative in gastric cancer. Cancer Sci. 2014;105:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hua Y, Vickers TA, Okunola HL, et al. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Passini MA, Bu, J., Richards, A. M.. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Marasco LE, Dujardin, G., Sousa-Luís, R.. Counteracting chromatin effects of a splicing-correcting antisense oligonucleotide improves its therapeutic efficacy in spinal muscular atrophy. Cell. 2022;185:2057–2070 e2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Helfman DM, Cheley S, Kuismanen E, et al. Nonmuscle and muscle tropomyosin isoforms are expressed from a single gene by alternative RNA splicing and polyadenylation. Mol Cell Biol. 1986;6:3582–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cramer P, Pesce CG, Baralle FE, et al. Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci USA. 1997;94:11456–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Fededa JP, Petrillo, E., Gelfand, M. S. A polar mechanism coordinates different regions of alternative splicing within a single gene. Mol Cell. 2005;19:393–404. [DOI] [PubMed] [Google Scholar]
- [62].Fagnani M, Barash, Y., Ip, J. Y.. Functional coordination of alternative splicing in the mammalian central nervous system. Genome Biol. 2007;8(6):R108. DOI: 10.1186/gb-2007-8-6-r108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tilgner H, Jahanbani, F., Blauwkamp, T. Comprehensive transcriptome analysis using synthetic long-read sequencing reveals molecular co-association of distant splicing events. Nat Biotechnol. 2015;33:736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tilgner H, Jahanbani, F., Gupta, I. Microfluidic isoform sequencing shows widespread splicing coordination in the human transcriptome. Genome Res. 2018;28:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Anvar SY, Allard, G., Tseng, E.. Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol. 2018;19:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ubby I, Bussani, E., Colonna, A.. TMEM16A alternative splicing coordination in breast cancer. Mol Cancer. 2013;12:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Singh RK, Xia, Z., Bland, C. S. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol Cell. 2014;55:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tasic B, Nabholz, C. E., Baldwin, K. K.. Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol Cell. 2002;10:21–33. [DOI] [PubMed] [Google Scholar]
- [69].Pagani F, Stuani C, Zuccato E, et al. Promoter architecture modulates CFTR exon 9 skipping. J Biol Chem. 2003;278:1511–1517. [DOI] [PubMed] [Google Scholar]
- [70].Kolathur KK. Role of promoters in regulating alternative splicing. Gene. 2021;782:145523. [DOI] [PubMed] [Google Scholar]
- [71].Xin D, Hu L, Kong X. Alternative promoters influence alternative splicing at the genomic level. PLoS ONE. 2008;3:e2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Adamson B, Norman, T. M., Jost, M. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1867–1882 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Datlinger P, Rendeiro, A. F., Schmidl, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dixit A, Parnas, O., Li, B.. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853–1866 e1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kowalski MH, Wessels, H. H., Linder, J. S. A. CPA-Perturb-seq: multiplexed single-cell characterization of alternative polyadenylation regulators. bioRxiv; 2023. DOI: 10.1101/2023.02.09.527751 [DOI] [PubMed] [Google Scholar]
- [76].Hashimshony T, Wagner F, Sher N, et al. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–673. [DOI] [PubMed] [Google Scholar]
- [77].Ramskold D, Luo, S., Wang, Y. C. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mikheenko A, Prjibelski AD, Joglekar A, et al. Sequencing of individual barcoded cDnas using Pacific Biosciences and Oxford Nanopore Technologies reveals platform-specific error patterns. Genome Res. 2022;32:726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lebrigand K, Bergenstråhle J, Thrane K, et al. The spatial landscape of gene expression isoforms in tissue sections. Nucleic Acids Res. 2023;51:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zheng Y-F, Chen, Z. C., Shi, Z. X. HIT-scIsoseq: high-throughput and high-accuracy single-cell full-length isoform sequencing for corneal epithelium. bioRxiv; 2020. DOI: 10.1101/2020.07.27.222349 [DOI] [Google Scholar]
- [81].Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bratt E, Ohman M. Coordination of editing and splicing of glutamate receptor pre-Mrna. RNA. 2003;9:309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Aw JGA, Lim, S. W., Wang, J. X. Determination of isoform-specific RNA structure with nanopore long reads. Nat Biotechnol. 2021;39:336–346. [DOI] [PubMed] [Google Scholar]
- [84].Naftelberg S, Schor IE, Ast G, et al. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84:165–198. [DOI] [PubMed] [Google Scholar]