

Abstract
Skeletal stem/progenitor cell (SSPC) senescence is a major cause of decreased bone regenerative potential with aging, but the causes of SSPC senescence remain unclear. In this study, we revealed that macrophages in calluses secrete prosenescent factors, including grancalcin (GCA), during aging, which triggers SSPC senescence and impairs fracture healing. Local injection of human rGCA in young mice induced SSPC senescence and delayed fracture repair. Genetic deletion of Gca in monocytes/macrophages was sufficient to rejuvenate fracture repair in aged mice and alleviate SSPC senescence. Mechanistically, GCA binds to the plexin-B2 receptor and activates Arg2-mediated mitochondrial dysfunction, resulting in cellular senescence. Depletion of Plxnb2 in SSPCs impaired fracture healing. Administration of GCA-neutralizing antibody enhanced fracture healing in aged mice. Thus, our study revealed that senescent macrophages within calluses secrete GCA to trigger SSPC secondary senescence, and GCA neutralization represents a promising therapy for nonunion or delayed union in elderly individuals.
Subject terms: Metabolic bone disease, Bone quality and biomechanics
Introduction
Osteoporosis is characterized by low bone mass and destroyed microarchitecture, resulting in an increased risk of fracture.1 Epidemiological studies show that in the year 2000, there were estimated to be nearly 9 million osteoporotic fractures each year on a global scale, suggesting that almost one in two women and one in five men will experience a fracture in their lifetime from the age of 50 years.2 Additionally, decreased bone regenerative potential and a high risk of malunion, delayed union and nonunion in elderly individuals contribute to long-term disability or even death.3,4 However, the etiology of age-related impaired skeletal regenerative capacity remains incompletely understood.
Aged-related skeletal deterioration and impaired fracture healing are related to the accumulation of senescent cells, which are characterized by permanent cell cycle arrest, apoptosis resistance and a senescence-associated secretory phenotype (SASP).5–8 Targeting cellular senescence using pharmacological and genetic approaches prevents age-related bone loss in mice.9 Farr et al. revealed that myeloid cells within bone marrow and osteocytes in bone matrix become senescent and secrete SASP factors during aging.10 It has also been demonstrated that bone marrow adipocyte lineage cells undergo rapid cellular senescence after glucocorticoid treatment and secrete SASP factors to distribute cellular senescence of vascular endothelial cells and osteoprogenitors in the metaphysis of growing bone, leading to decreased peak bone mass, architectural deterioration, and increased fracture risk.11,12 Antagonizing the senescence of these cells improves glucocorticoid-induced bone deterioration.11 Our previous study found that senescent immune cells accumulated in the bone marrow and secreted grancalcin (GCA), which suppressed bone turnover and promoted marrow fat accumulation.13 Thus, accumulated senescent cells in the bone microenvironment play a significant role in skeletal aging, and eliminating senescent cells and/or blunting their SASP factors can prevent or delay age-related bone loss. Recent studies also suggested that the accumulation of senescent cells impaired fracture healing, and removing senescent cells by genetic and pharmacological approaches in aged mice improved fracture repair.14–16 However, the effects and underlying mechanisms of senescent cells in fracture healing during aging remain elusive.
Skeletal stem/progenitor cells (SSPCs) serve as osteochondro-precursors in the callus during fracture healing.17 Josephson et al. reported that the number of SSPCs decreases with age, leading to poor bone regeneration in elderly individuals in a population cohort.18 Studies have shown that SSPCs exert senescent phenotypes in aged mice, accompanied by decreased proliferation ability and diminished osteochondrogenic activity.18–20 The function of SSPCs within the callus is at least partially affected by local microenvironmental cues. Liu et al. reported that senescent cells in fracture calluses secrete SASP factors to inhibit the growth and proliferation of callus-derived mesenchymal progenitor cells and ultimately impair bone regeneration.14 However, the identification of a prosenescent niche, including primary senescent cells and prosenescent factors, within the callus that is responsible for SSPC senescence and dysfunction is far from clear.
Chronic inflammation is thought to be a major cause of the impaired regenerative capacity of the skeleton during aging, in contrast to well-balanced inflammation, which is needed for successful bone repair.21 The interaction between macrophages and SSPCs is critical for bone regeneration.22 Macrophages phagocytose necrotic cells and tissue debris at the fracture site and initiate the recruitment of periosteal mesenchymal stem cells, bone marrow mesenchymal stem cells, and vascular progenitor cells.23,24 Activated macrophages secrete chemokines to control the migration of SSPCs to calluses.25,26 Aging negatively affects the inflammatory response during fracture healing.21 Parabiosis and bone marrow transplantation studies have shown that macrophages from young mice secrete factors that rejuvenate fracture repair in aged mice.27,28 In contrast, macrophages from aged mice delay fracture healing in young mice.29 However, the factors secreted by aged macrophages remain elusive. Our previous studies have shown that senescent immune cells, including macrophages, release GCA to induce age-related bone loss.13 However, the effects of GCA secreted by infiltrating macrophages in calluses on fracture healing during aging need to be further explored.
Here, we show that macrophages release abundant quantities of GCA during aging to induce SSPC senescence, leading to the dysfunction of SSPCs and delayed fracture healing. Genetic deletion of Gca in monocytes/macrophages is sufficient to rejuvenate fracture repair in aged mice. In addition, Arg2-mediated mitochondrial dysfunction contributes to SSPC senescence induced by the GCA-plexin-B2 axis. Depletion of Plxnb2 in SSPCs leads to SSPC dysfunction and senescence, thus impairing fracture healing in young mice. Moreover, GCA neutralization markedly enhances fracture healing ability in aged mice. Thus, our study reveals that senescent macrophages within the callus secrete GCA to induce SSPC senescence and impair fracture healing during aging, and local injection of a GCA-neutralizing antibody represents a promising therapy for impaired fracture repair in elderly individuals.
Results
Senescent SSPCs accumulate in calluses and impair fracture healing in aged mice
To detect changes in fracture healing ability during aging, aged mice (18 months old) and young mice (3 months old) were subjected to transverse mid-diaphyseal femoral fractures, and healing was measured. Consistent with previous reports,5 aged mice showed obviously impaired fracture regeneration, as evidenced by smaller calluses and discontinuous cortical bone detected by microcomputed tomography (micro-CT) analysis, compared with young mice (Fig. S1a, b). Histological analysis indicated that aged mice displayed smaller areas of cartilage in the callus (Fig. S1c, d). Skeletal stem/progenitor cells (SSPCs), especially callus SSPCs, are indispensable for successful bone regeneration.30–33 The decreased number and function of SSPCs have been identified as potential causes of impaired tissue regeneration during aging.18 To identify the SSPC populations in calluses, we performed single-cell RNA sequencing (scRNA-seq) on callus tissues at different timepoints post-fracture.34 Periosteal stem cells were rarely detected before fracture (Day 0), increased at 3 days post-fracture (Day 3), and peaked at 7 days post-fracture (Day 7) (Fig. 1a and Fig. S1e, f). To label callus SSPCs in vivo, we compared the markers previously identified in labeled SSPCs,31–33,35–39 including Ctsk, Prrx1, Pdgfrα, Pdgfrβ, Nes, LepR, Mx1 and Gli1, and found that the SSPCs with Ctsk expression accounted for the highest proportion of SSPCs in the fracture callus (Fig. 1b and Fig. S2a). Previous studies reported that Ctsk lineage cells in the periosteum were the major contributors to chondrocytes and osteoblasts during fracture healing.31 Thus, we selected Ctsk+ SSPCs for the subsequent study of fracture healing. To determine the senescence of SSPCs in calluses with age, we performed fluorescence staining in situ using antibodies against Ctsk and senescence markers (p21 and γH2AX) and found that the number of senescent Ctsk+ SSPCs was increased significantly in the calluses of aged mice compared with young mice (Fig. 1c, d and Fig. S2b, c). Previous studies found that the number of senescent cells increased in the fracture calluses of aged mice and caused inflammatory and senescent phenotypes.18–20 To explore the profile of senescent SSPCs in calluses during aging, we performed scRNA-seq on purified callus SSPCs from young (4-month-old) and aged (21-month-old) mice.20 Dominik et al. generated a senescence gene set (“SenMayo”) of 125 genes to identify senescent cells.40 SenMayo was validated to identify senescent cells at the single-cell level in murine bone/bone marrow and was confirmed to outperform existing senescence gene sets. Thus, we chose SenMayo to evaluate these callus SSPCs from young (4-month-old) and aged (21-month-old) mice. We found that in aged mice, senescence-associated genes were more highly enriched, i.e., had higher enrichment scores in the osteogenic and proliferating progenitors compared to those of young mice (Fig. 1e). Additionally, these callus progenitors showed decreased gene-set enrichment associated with stemness and osteogenesis (Fig. 1f–h). These data suggest that impaired skeletal regenerative capacity is accompanied by the dysfunction and senescence of SSPCs during aging.
Fig. 1.

Senescent SSPCs accumulate in calluses and impair fracture healing in aged mice. a, b Uniform manifold approximation and projection (UMAP) plot showing 12 distinct clusters of cells identified (control group and on Days 3, 7, and 14). b Feature plots showing the expression distribution of marker genes of SSPC subsets on Day 7. Expression levels for each cell are color-coded and overlaid onto the UMAP plot. c, d Representative IF images in the periosteum of fracture callus at 10 days post-fracture, immunostained with Ctsk (green) and p21 (red) or γH2AX (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. e–h Boxplots demonstrating that aged SSPCs positively correlate with gene sets for senescence and negatively correlate with gene sets for stemness by the Wilcoxon test. Data are presented as the means ± SDs
Grancalcin secreted by senescent macrophages in calluses induces SSPC senescence and delays bone regeneration
Next, we wondered which type of cells are the primary senescent cells that propagate senescence in SSPCs. The scRNA-seq data revealed that immune cells, particularly macrophages, responded quickly to fracture, and the population of macrophages peaked earlier than that of SSPCs in calluses (Fig. 2a). To explore whether macrophages accumulated in the callus become senescent with aging, we performed fluorescence staining in situ using antibodies against biomarkers for senescence-associated changes and F4/80+ macrophages in young and aged mice. We found that the percentage of p21+F4/80+ cells, as well as γH2AX+F4/80+ cells, increased in calluses with age (Fig. 2b and Fig. S3a–c). This observation prompted us to speculate that senescent macrophages at the fracture site secrete proinflammatory or prosenescence factors to create an inflammatory and senescent niche that leads to SSPC senescence. Previously, we reported that grancalcin (GCA) is mainly secreted by senescent macrophages and suppresses bone formation and resorption, leading to skeletal aging.13 In this study, we observed a higher level of GCA at the bone injury site in aged mice on 5 days post-fracture (5 dpf) than in young mice (Fig. 2c) (GEO: GSE99388). In addition, we compared GCA secreted by macrophages in the calluses of young and aged mice by performing fluorescence staining in situ using antibodies against GCA and F4/80. The number of GCA-expressing macrophages was higher in aged mice than in young controls (Fig. 2d, e). To determine whether GCA could induce SSPC senescence and lead to delayed fracture repair, we placed hydrogels containing human rGCA or PBS locally to transverse mid-diaphyseal femoral fractures of young mice. We detected a higher level of GCA at the bone injury site in mice administered rGCA than in PBS-treated controls (Fig. S3d, e). By micro-CT analysis, we observed a significantly reduced callus size in mice with administration of rGCA in comparison with PBS-treated controls at 10 dpf and 21 dpf (Fig. 2f–h). Smaller islands of cartilage and reduced woven bone area at the fracture site were detected in the calluses of rGCA-treated mice versus controls by Safranin O/Fast green staining analysis (Fig. 2i–k). Next, we measured the effects of rGCA on SSPC senescence. Immunofluorescence staining showed an increased number of senescent SSPCs in the calluses of rGCA-treated mice compared with PBS-treated mice, as characterized by increased p21- and γ-H2AX-positive SSPCs (Fig. 2l, m and Fig. S3f, g). To further test the direct effects of GCA on SSPC senescence, we harvested primary mouse SSPCs and treated these cells with rGCA. qPCR analysis revealed that SSPCs treated with rGCA showed higher expression of genes associated with senescence and SASP factors (Fig. 3a, b). Western blotting analysis and SA-β-gal staining also demonstrated that rGCA treatment led to SSPC senescence (Fig. 3c, d). We then performed RNA sequencing on SSPCs treated with PBS/rGCA. Consistent with previous results, the key senescence genes Cdkn2a, Cdkn2c, and Cdkn2d and the “SenMayo” gene set displayed highly significant enrichment in rGCA-treated SSPCs (Fig. 3e, f). In addition, we observed a diminished osteochondrogenic capacity of SSPCs with rGCA administration in contrast to the PBS-treated controls (Fig. 3g–j). Taken together, these data indicate that GCA secreted by senescent macrophages induces SSPC senescence, impairs the function of SSPCs and delays fracture healing.
Fig. 2.

Aged macrophages in fracture calluses release GCA and impair bone regeneration. a Stacked bar chart showing the percentages of each cell type within callus tissue quantified on Day 0, Day 3, Day 7, and Day 14 post-fracture based on UMAP distribution. b Representative IF images of fracture calluses immunostained with F4/80 (green) and p21 (red) antibodies and counterstained with DAPI (blue) (n = 4). Scale bars indicate 100 µm. c Bioinformatics analysis of Gca gene expression levels in calluses from 5-month-old and 25-month-old mice at 5 days post-fracture (n = 5). d Representative IF images of fracture calluses immunostained with F4/80 (green) and GCA (red) antibodies and counterstained with DAPI (blue) (n = 4). Scale bars indicate 100 µm. e Quantification of GCA+F4/80+ cells (n = 4). f Representative micro-CT images of fractured femurs from PBS- and rGCA-treated mice at 10 dpf and 21 dpf. g, h The callus index of fractured femurs from PBS- and rGCA-treated mice at 10 dpf and 21 dpf (n = 5). i–k Safranin O staining showed cartilage callus formation and woven bone area from PBS- and rGCA-treated mouse fractured femurs at 10 dpf and 21 dpf (n = 4–6). Scale bar indicates 100 μm. l Representative IF images in the periosteum of the fracture callus at 10 days post-fracture, immunostained with Ctsk (green) and p21 (red) and counterstained with DAPI (blue). Scale bars indicate 100 µm. m Quantification of the number of p21- and Ctsk-positive cells per mm2 tissue area (N. p21+Ctsk+ cells) by Welch’s t test (n = 5). Data are presented as the means ± SDs. Unpaired t test, *P < 0.05 and **P < 0.01
Fig. 3.

rGCA accelerates SSPC senescence and dysfunction. a, b RT‒PCR analysis of the gene expression of senescence markers and SASP factors in SSPCs (n = 3). p16 by Welch’s t test. c Western blotting analysis of p16, p21 and GCA protein expression in PBS- or rGCA-treated SSPCs. d SA-β-Gal staining of SSPCs treated with PBS and rGCA. e GSEA plots demonstrating that SSPCs treated with rGCA positively correlate with gene sets for senescence (SenMayo). f Heatmap showing increased expression of INK family genes in rGCA-treated cells relative to PBS-treated cells. g RT‒PCR analysis of the gene expression of chondrogenesis markers in SSPCs (n = 3). h RT‒PCR analysis of the gene expression of osteogenesis markers in SSPCs (n = 3). i, j Representative images of ALP staining (i) and Alizarin Red staining (j) of SSPCs treated with PBS or rGCA. Scale bar indicates 100 μm. Data are presented as the means ± SDs. Unpaired t test. *P < 0.05, **P < 0.01 and ***P < 0.001
Genetic deletion of Gca in macrophages mitigates SSPC senescence and rejuvenates fracture repair in aged mice
The above data prompted us to test whether the deletion of Gca in macrophages rejuvenates fracture repair in aged mice. We crossed Gcaflox/flox mice with Lyz2-Cre mice to generate mice with deletion of the Gca gene in myeloid lineage cells (Gca-Lyz2-CKO) (Fig. 4a).41,42 The number of GCA-expressing macrophages in the calluses of Gca-Lyz2-CKO mice was decreased by 61.9% compared with that in Gcaflox/flox mice (Fig. S4a, b). We observed significantly increased cartilage formation in aged (18-month-old) Gca-Lyz2-CKO mice compared to age-matched Gcaflox/flox control mice (WT) (Fig. 4b, c). Subsequently, micro-CT reconstruction of fractured bones demonstrated a robust bony hard callus in aged Gca-Lyz2-CKO mice at 21 dpf (Fig. 4d, e). Safranin O/Fast green staining analysis revealed a larger callus size and more bone trabeculae in calluses from aged Gca-Lyz2-CKO mice than in those from control mice (Fig. 4f, g). To determine whether Gca knockout in fracture calluses would affect SSPC senescence, we performed immunofluorescence in situ in fracture calluses at 10 dpf. As expected, the number of senescent SSPCs decreased significantly in the calluses of Gca-Lyz2-CKO mice compared with Gca flox/flox mice (Fig. 4h–j). In addition, an increased number of osteocalcin (Ocn)-positive osteoblastic cells was observed in the calluses of Gca-Lyz2-CKO mice compared with those of Gca flox/flox mice (Fig. 4k, l). These results show that deletion of Gca in macrophages is able to mitigate SSPC senescence in calluses and promote fracture repair in aged mice.
Fig. 4.

Genetic deletion of Gca in macrophages rejuvenates fracture repair in aged mice. a Schematic of experiments on Gca-Lyz2-CKO mice. b, c Safranin O staining showing cartilage callus formation in aged Gca-Lyz2-CKO mice and Gcaflox/flox control mice (WT) at 10 dpf (n = 3). Scale bar indicates 100 μm. d Representative micro-CT images of fractured femurs in Gca-Lyz2-CKO mice and WT mice at 21 dpf. e The callus index of fractured femurs from Gca-Lyz2-CKO mice and WT mice at 21 dpf (n = 4–6). f, g Safranin O staining showing the woven bone area of aged Gca-Lyz2-CKO mice and Gcaflox/flox control mice (WT) at 21 dpf (n = 3). Scale bar indicates 100 μm. h Representative IF images in the periosteum of the fracture callus at 10 days post-fracture, immunostained with Ctsk (green) and p21 (red) or γH2AX (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. i, j Quantification of the number of p21- and Ctsk-positive cells per mm2 tissue area (N. p21+Ctsk+ cells) by Welch’s t test and the number of γH2AX- and Ctsk-positive cells per mm2 tissue area (N. γH2AX+Ctsk+ cells) (n = 4). k Representative IF images of fracture calluses at 21 days post-fracture, immunostained with Ocn (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. l Quantification of the number of Ocn-positive cells per mm2 tissue area (N. Ocn+ cells) (n = 4). Data are presented as the means ± SDs. Unpaired t test, *P < 0.05, **P < 0.01 and ***P < 0.001
GCA binds to the plexin-B2 receptor to induce SSPC senescence
Next, we further explored the downstream signaling that mediates the effects of GCA on SSPCs. Previously, we found that plexin-B2 (PLXNB2) is a functional receptor of GCA in BMSCs regulating osteogenesis.13,43 Additionally, it has been reported that PLXNB2 is expressed in vascular endothelial cells and protects bone blood vessels from senescence.11 Thus, we speculate that PLXNB2 may also mediate the effects of GCA on SSPC senescence. PLXNB2 belongs to the plexin family.44 We analyzed the distribution of plexin family members in callus SSPCs at different timepoints following fracture by conducting scRNA-seq.34 Notably, we found that the expression of Plxnb2 was highest in SSPCs compared with other members of the plexin family (Fig. 5a and Fig. S5a). Then, we divided SSPCs into Plxnb2-positive SSPCs and Plxnb2-negative SSPCs (Fig. S5b). We found that Plxnb2-positive SSPCs showed higher expression of genes associated with stemness, stem cell differentiation and skeletal development than Plxnb2-negative SSPCs (Fig. 5b–d and Fig. S5c). In contrast, senescence-related genes, such as Cdkn2a, Cdkn2b, Cdkn2c, and Cdkn2d, were enriched in Plxnb2-negative callus SSPCs (Fig. 5e–h). To validate the direct effects of PLXNB2 on SSPCs, SSPCs were subjected to Plxnb2 siRNA transfection, and the senescence phenotype of SSPCs was then measured. Higher expression of senescence-related genes and an increased number of SA-β-gal+ cells were detected in the SSPCs transfected with Plxnb2 siRNA relative to control siRNA-treated SSPCs (Fig. 5i–l and Fig. S5d). We then examined whether knockdown of Plxnb2 would lead to the diminished osteochondrogenic capacity of SSPCs. We found that SSPCs transfected with Plxnb2 siRNA showed decreased expression of chondrogenic genes (Sox9, Col2a1 and Acan) and osteogenic genes (Sp7, Runx2, Alp and Bglap) (Fig. 5m, n).
Fig. 5.

Knockout of Plxnb2 accelerates SSPC senescence and dysfunction. a Feature plots showing the expression distribution of the plexin family on Day 7. Expression levels for each cell are color-coded and overlaid onto the UMAP plot. b–d Violin plots demonstrating that Plxnb2+ SSPCs positively correlate with gene sets for stemness. e–h Violin plots demonstrating that Plxnb2- SSPCs positively correlate with INK family genes. i, j QPCR analysis of senescence marker and SASP factor gene expression in SSPCs (n = 3–4). p16 and Il1 by Welch’s t test. k Western blotting analysis of p16, p21 and PLXNB2 protein expression in siRNA-NC- or siRNA-Plxnb2-transfected SSPCs. l SA-β-Gal staining of siRNA-NC- or siRNA-Plxnb2-transfected SSPCs. Scale bar indicates 100 μm. m RT‒PCR analysis of chondrogenesis marker gene expression in SSPCs (n = 4). n RT‒PCR analysis of osteogenic marker gene expression in SSPCs (n = 4). Bglap by Welch’s t test. Data are presented as the means ± SDs. Unpaired t test. *P < 0.05, **P < 0.01, ***P < 0.001 and ns indicates no significant difference
To further investigate the effects of PLXNB2 in SSPCs on fracture repair during aging in vivo, we obtained mice with deletion of the Plxnb2 gene in Ctsk+ SSPCs by crossing Plxnb2flox/flox mice with Ctsk-Cre mice. Plxnb2-Ctsk-CKO mice and control mice were subjected to transverse mid-diaphyseal femoral fractures. Fracture repair was impaired in Plxnb2-Ctsk-CKO mice compared to control mice. Micro-CT analysis indicated a smaller and less mineralized callus in Plxnb2-Ctsk-CKO mice compared with control littermates (Fig. 6a, b and Fig. S6a). Additionally, we performed histological analysis in Plxnb2-Ctsk-CKO mice and control mice. We found that the area of cartilage and woven bone in calluses was significantly decreased in Plxnb2-Ctsk-CKO mice at 10 dpf and 21 dpf (Fig. 6c–e). To further exclude the effects of PLXNB2 in Ctsk+ non-SSPCs on fracture healing, we generated a mouse model with knockout of the Plxnb2 gene in Prrx1+ SSPCs (Plxnb2-Prrx1-CKO mice) according to the high expression of Prrx1 genes in SSPCs in calluses, as shown in Fig. 1b. Smaller calluses and smaller areas of cartilage and woven bone were also observed in Plxnb2-Prrx1-CKO mice than in control mice (Fig. 6f–j and Fig. S6b).
Fig. 6.

Deletion of Plxnb2 in Ctsk+ and Prrx1+ cells impairs fracture repair in adult mice. a Representative micro-CT images of fractured femurs from Plxnb2-Ctsk-CKO mice and Plxnb2flox/flox control mice at 21 dpf. b The callus index of fractured femurs from Plxnb2-Ctsk-CKO mice and Plxnb2flox/flox mice at 21 dpf (n = 3–6). c–e Safranin O staining showing cartilage callus formation and woven bone area of fractured femurs from Plxnb2-Ctsk-CKO mice and Plxnb2flox/flox mice at 10 dpf and 21 dpf (n = 3–4). Scale bar indicates 100 μm. f Representative micro-CT images of fractured femurs from Plxnb2-Prrx1-CKO mice and Plxnb2flox/flox control mice at 21 dpf. g The callus index of fractured femurs from Plxnb2-Prrx1-CKO mice and Plxnb2flox/flox mice at 21 dpf (n = 3–6). h–j Safranin O staining showing cartilage callus formation and woven bone area of fractured femurs from Plxnb2-Prrx1-CKO mice and Plxnb2flox/flox mice at 10 dpf and 21 dpf (n = 3–5). Scale bar indicates 100 μm. k Representative IF images of fracture calluses at 10 days post-fracture, immunostained with GFP (green) and p21 (red) or H2AXγ (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. l, m Quantification of the number of p21- and GFP-positive cells per mm2 tissue area (N. p21+GFP+ cells) and the number of γH2AX- and GFP-positive cells per mm2 tissue area (N. γH2AX+GFP+ cells) (n = 3). n Representative IF images of fracture calluses at 21 days post-fracture, immunostained with Ocn (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. o Quantification of the number of Ocn-positive cells per mm2 tissue area by Welch’s t-test (N. Ocn+ cells) (n = 3–4). Data are presented as the means ± SDs. Unpaired t test, *P < 0.05 and **P < 0.01
To trace the changes in Ctsk lineage cells at the fracture site in response to Plxnb2 deficiency in vivo, we crossed Plxnb2-Ctsk-CKO mice with YFP reporter mice to generate Ctsk-Cre; Plxnb2 flox/flox; YFPflox/+ mice (Plxnb2Ctsk; YFPfl/+), and Ctsk-Cre; YFPfl/+ mice served as controls. Costaining of GFP with senescence markers (p21 and γ-H2AX) showed that the number of senescent GFP+ SSPCs increased significantly in the calluses of mice with Plxnb2 knockdown in Ctsk+ lineage cells (Fig. 6k–m and Fig. S6c). In addition, a decreased number of Ocn-positive osteoblasts was observed in the calluses of Plxnb2-Prrx1-CKO mice compared with Plxnb2flox/flox mice (Fig. 4n, o). Thus, these data suggested that depletion of Plxnb2 leads to SSPC senescence and impaired fracture repair.
The GCA-PLXNB2 axis may promote SSPC senescence through Arg2-mediated mitochondrial dysfunction
To identify the downstream pathway of PLXNB2 signaling regulated by GCA in SSPCs, we performed RNA sequencing on SSPCs treated with rGCA and SSPCs transfected with siRNA-Plxnb2. The analysis of data from rGCA-treated versus PBS-treated SSPCs identified 59 upregulated genes and 39 downregulated genes (Fig. 7a). Additionally, we found 142 genes with upregulated expression and 129 genes with downregulated expression in SSPCs transfected with siRNA-Plxnb2 compared with controls (Fig. 7a). To identify shared downstream factors of rGCA-treated SSPCs and siRNA-Plxnb2-transfected SSPCs, we performed collective analysis of the combined data from rGCA- and PBS-treated SSPCs and siRNA-Plxnb2- and siRNA-control-transfected SSPCs. A total of 7 differentially expressed factors with at least a 2-fold change were identified, including Oasl1, Colgalt2, Slco4a1, Arg2, Adora3, Nyx and Isg15 (Fig. S7a). Among them, arginase-II (Arg2) has been reported to promote endothelial cell senescence and vascular smooth muscle cell senescence.45–47 Arg2 deficiency plays an important role in lifespan extension in mice.48 Thus, we chose Arg2 for further study. We verified significantly increased mRNA and protein levels of Arg2 in rGCA-treated and siRNA-Plxnb2-transfected SSPCs (Fig. 7b–d). To test whether Arg2 could influence SSPC senescence, we treated SSPCs with H2O2 to induce senescence and transfected SSPCs with siRNA-Arg2. We observed decreased senescence-related gene expression in SSPCs transfected with Arg2-siRNA compared to the control siRNA group (Fig. 7e, f and Fig. S7b-f). Arg2 is a mitochondrial enzyme and has been reported to be involved in mitochondrial dysfunction.49–51 To better dissect the role of mitochondrial dysfunction in GCA-induced SSPC senescence, we compared differentially expressed genes (DEGs) associated with mitochondrial function in SSPCs treated with rGCA or PBS. Gene Ontology (GO) analysis revealed that these DEGs were highly related to the “mitochondrial electron transport, cytochrome c to oxygen”, “mitochondrial ATP synthesis coupled electron transport”, and “mitochondrial respiratory chain complex IV” pathways, which were notable for their close links to mitochondrial function (Fig. 7g). We then examined key genes related to mitochondrial biogenesis and glycolysis in vitro and found sharply decreased expression in SSPCs treated with rGCA (Fig. 7h and Fig. S7g). In addition, rGCA treatment decreased the mitochondrial transmembrane potential (Fig. 7i, j). These data suggest that Arg2-mediated mitochondrial dysfunction may be an important mechanism of SSPC senescence regulated by the GCA-PLXNB2 axis.
Fig. 7.

The GCA-PLXNB2 axis may promote SSPC senescence through the mitochondrial enzyme Arg2. a Statistics of differentially expressed genes in the rGCA and/or siRNA-Plxnb2 groups ( ≥ 2-fold change). b Volcano plots showing increased Arg2 expression in the rGCA and/or siRNA-Plxnb2 groups ( > 2-fold change). c, d Western blotting analysis of Arg2 protein expression in the rGCA and/or siRNA-Plxnb2 groups. e RT‒PCR analysis of senescence marker gene expression in siRNA-NC and siRNA-Arg2 SSPCs (n = 3). f Western blotting analysis of Arg2 and p21 protein expression in the siRNA-NC and siRNA-Arg2 groups. g GO analysis of differentially expressed genes related to mitochondrial function. h RT‒PCR analysis of Tfam and mtNd1 gene expression in PBS- and rGCA-treated SSPCs (n = 3). Tfam by Welch’s t test. i Fluorescence microscopy image showing the JC-1 probe in PBS- and rGCA-treated SSPCs. Scale bars indicate 20 µm. j Mitochondrial membrane potential was measured using JC1 staining by Welch’s t test. Data are presented as the means ± SDs. Unpaired t test, *P < 0.05 and **P < 0.01
GCA-neutralizing antibody restores SSPC function and bone regeneration in aged mice
The above data indicate that GCA secreted by senescent macrophages accelerates SSPC senescence and impairs fracture repair. We then tested whether a neutralizing antibody against GCA (GCA-NAb) could promote fracture healing in aged mice. Our previous studies screened a GCA-NAb and confirmed its therapeutic potential for treating bone loss in old mice.13 To further investigate the therapeutic effects of GCA-NAb in fracture repair during aging, we treated aged mice with GCA-NAb locally at the fracture sites twice a week for 21 days. Consistent with our findings that genetic ablation of Gca is beneficial to fracture repair, we found that treatment with GCA-NAb restored fracture repair in aged mice. Micro-CT analysis demonstrated that GCA-NAb improved the formation of mineralized calluses relative to vehicle-treated controls (Fig. 8a, b). We then performed SOFG staining to detect the area and morphology of the fracture site. Relative to that of the vehicle-treated mice, the size and density of newly formed woven bone were significantly increased in GCA-NAb-treated mice (Fig. 8c, d). Furthermore, compared to the control mice, GCA-NAb-treated mice showed a decreased number of Arg2-positive cells in the callus region (Fig. 8e, f). We found that the numbers of p21+Ctsk+ periosteal stem cells and γH2AX+Ctsk+ periosteal stem cells were decreased in GCA-NAb-treated mice compared with vehicle-treated mice (Fig. 8g–i). Furthermore, GCA-NAb-treated mice showed an increased number of Ocn-positive osteoblasts in the callus (Fig. 8j, k). Collectively, these data show that the local application of GCA-NAb promotes bone-forming capacity and restores bone regeneration in aged mice.
Fig. 8.

GCA-neutralizing antibody restores bone regeneration in aged mice. a Representative micro-CT images of fractured femurs from GCA-NAb- and PBS-treated mice at 21 dpf. b The callus index of fractured femurs from GCA-NAb- and PBS-treated mice at 21 dpf (n = 5). c, d Safranin O staining showing the woven bone area in calluses of fractured femurs from GCA-NAb- and PBS-treated mice at 21 dpf (n = 4). Scale bar indicates 100 μm. e Representative IF images of fracture calluses at 21 days post-fracture, immunostained with Arg2 (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. f Quantification of the number of Arg2-positive cells per mm2 tissue area (N. Arg2+ cells) (n = 5). g Representative IF images of the periosteum of fracture calluses immunostained with Ctsk (green) and p21 (red) or γH2AX (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. h, i Quantification of the number of p21- and Ctsk-positive cells per mm2 tissue area (N. p21+Ctsk+ cells) by Welch’s t test and the number of γH2AX- and Ctsk-positive cells per mm2 tissue area (N. γH2AX+Ctsk+ cells) (n = 4). j Representative IF images of fracture calluses at 21 days post-fracture, immunostained with Ocn (red) antibodies and counterstained with DAPI (blue). Scale bars indicate 100 µm. k Quantification of the number of Ocn-positive cells per mm2 tissue area (N. Ocn+ cells) (n = 5). Data are presented as the means ± SDs. Unpaired t test, *P < 0.05, **P < 0.01
Discussion
In this study, we revealed that the dysfunction and senescence of SSPCs induced by macrophage-derived GCA lead to impaired fracture healing in aged mice. Local treatment with rGCA in young mice delays fracture repair, and depletion of Gca in macrophages partially rejuvenates fracture repair in aged mice. We showed that GCA binds to the PLXNB2 receptor and activates Arg2-mediated mitochondrial dysfunction, resulting in SSPC senescence and defective osteochondrogenic capacity (Fig. 9). In addition, we show that the application of GCA-NAb improves bone regeneration in aged mice.
Fig. 9.
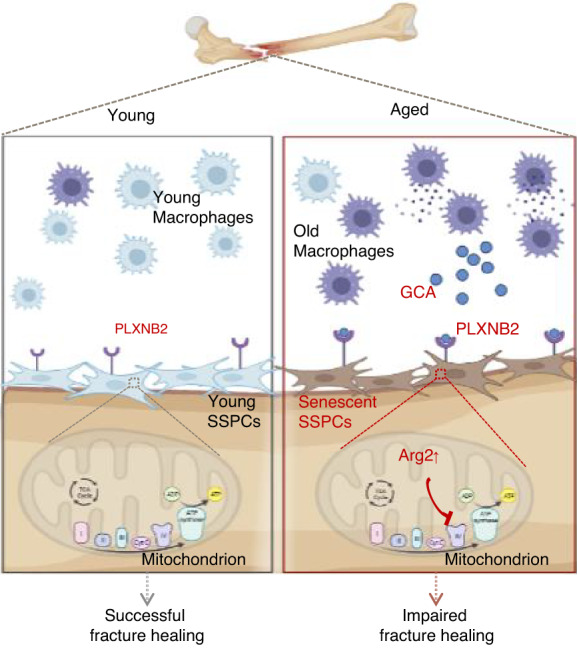
Graphical abstract of impaired fracture healing in aged mice. Schematic model for GCA-PLXNB2 transcription signaling in impaired fracture healing in aged mice
Our previous studies have found that GCA suppresses bone turnover and is mainly produced by proinflammatory and senescent subtypes of neutrophils and macrophages.13 In the current study, we investigated the role of GCA in fracture repair during aging. We demonstrate that deletion of the Gca gene in monocyte/macrophage lineage cells (Gca-Lyz2-CKO) mitigates SSPC senescence in the calluses of aged mice. The number of senescent cells at the fracture sites is significantly increased compared with that at nonfractured sites, indicating that acute injury may also induce cell senescence.14 Considering that GCA+ immune cells infiltrate the callus in response to fracture and that more GCA+ immune cells in the callus are observed during aging, we speculate that macrophages are activated in acute injury and secrete abundant GCA during aging. In addition, GCA+ immune cells might also migrate to the fracture site from bone marrow in aged mice. Although GCA secreted by senescent macrophages shows a negative impact on fracture healing, we recognize that GCA+ macrophages may have multiple effects on bone regeneration since ablation of macrophages also impairs fracture healing.52,53 In this study, we focused on the exact role of GCA in SSPC senescence and fracture repair, but gaining further insight into the role of GCA+ immune cells in age-related impaired bone regeneration will deepen the understanding of senescent immune cells in tissue repair.
PLXNB2 belongs to the plexin family, which has been reported to modulate neurogenesis, angiogenesis, immune response, tumorigenesis, and tissue regeneration.54 Our previous study illustrated that PLXNB2 is a functional receptor of GCA in BMSCs and plays a critical role in the osteogenic differentiation of BMSCs. PLXNB2 is widely expressed in a variety of cells and is implicated in regulating cell proliferation and migration.55 Additionally, plexin-B2 has been reported to protect vascular endothelial cells in the metaphysis from senescence.11 In this study, we found that the expression of Plxnb2 was highest among the members of the plexin family in callus SSPCs. However, the effects of Plxnb2 in bone regeneration have not yet been reported. Here, we show for the first time that depletion of Plxnb2 leads to SSPC senescence, dysfunction of osteogenic and chondrogenic differentiation and impaired fracture healing. It is possible that other members of the plexin family, such as PLXNA1, with high expression in callus SSPCs also exert important effects on bone regeneration, which is worthy of future study.
In this study, we selected Ctsk as a marker of SSPCs to investigate the role of callus SSPCs in vivo, which is consistent with a previous study showing that periosteal SSPCs express Ctsk.31,56 Ctsk was initially identified in mature osteoclasts. To exclude the possible effects of Plxnb2 deletion in osteoclasts on fracture repair, we used another mouse model with depletion of Plxnb2 in Prrx1+ SSPCs to confirm the critical effects of Plxnb2 in SSPCs. Both Plxnb2-Ctsk-CKO mice and Plxnb2-Prrx1-CKO mice showed impaired bone regeneration ability, which indicated the vital roles of PLXNB2 in maintaining the number and function of SSPCs during fracture healing. In addition to callus SSPCs inducing periosteal bone formation, endochondral bone formation, intramembranous bone formation and bone remodeling also contribute to fracture repair.57 Whether plexin-B2 is also expressed in other cell types in bone, such as endothelial cells, chondrocytes, osteoblasts, and osteocytes, thus playing a significant role in fracture repair, is an interesting topic for further investigation.
In summary, our findings highlight the critical role of the GCA-PLXNB2 axis in the crosstalk of macrophages and SSPCs in bone regeneration during aging. Whether age-related GCA accumulation affects additional biological processes involved in tissue repair requires further study. Nevertheless, our study supports the concept that senescent macrophages release GCA to induce SSPC secondary senescence and delay bone regeneration during aging, and targeting GCA may be a good strategy for promoting fracture repair in elderly individuals.
Materials and methods
Mice
The Gca-floxed mice and Plxnb2-floxed mice were constructed utilizing CRISPR/Cas9 technology at BIORAY LABORATORIES (China). Lyz2-Cre mice, Ctsk-Cre mice, Prrx1-Cre mice and YFP reporter mice were purchased from the Jackson Laboratory. All mice were kept on a C57BL/6 J background and maintained in a standard, specific pathogen-free facility of the Laboratory Animal Research Center of Central South University at a controlled temperature (22 °C–24 °C) with a 12 h dark/light cycle (07:00 to 19:00 light on), with standard food (Hunan SJA Laboratory Animal Company. China) and water provided ad libitum and environmental enrichments. Age- and sex-matched littermates were used as control mice. We crossed Gca-floxed mice with Lyz2-Cre mice to generate Gca-Lyz2-CKO mice and Gcaflox/flox mice. Ctsk-Cre mice or Prrx1-Cre mice were crossed with Plxnb2-floxed mice. The offspring were intercrossed to generate Plxnb2-Ctsk-CKO mice and Plxnb2-floxed mice or Plxnb2-Prrx1-CKO mice and Plxnb2-floxed mice. The genotypes of the mice were determined by PCR analyses of genomic DNA extracted from mouse tail snips.
Bicortical femoral fracture
Mice were anesthetized using pentobarbital. The patella was dislocated laterally to expose the femoral condyles, and a 25-gauge regular bevel needle (BD BioSciences) was inserted to stabilize the femur. A transverse fracture was created in the mid-diaphysis using scissors. Muscles were reapproximated, and the skin was closed using a 6/0 nylon suture.
Hydrogel placement
We purchased GelMA hydrogel from Engineering For Life Company. rGCA or PBS was added to the hydrogel solution at a concentration of 750 nmol/L. Solutions were exposed to ultraviolet light for 30 s in volumes of 10 μL each to obtain factor-loaded hydrogels.
Microcomputed tomography
The fractured femora from mice were dissected and fixed for 24 h with 4% paraformaldehyde and then scanned and analyzed with high-resolution μCT (Skyscan 1172, Bruker MicroCT, Kontich, Belgium).58 The callus index (CI) of fractured bone was calculated as the maximum diameter of the callus divided by the diameter of the adjacent diaphysis.
Histochemistry and immunofluorescence staining
Fractured bone was harvested from mice after euthanasia, fixed in 10% formalin for 12 h, and decalcified in 10% EDTA for 21 days. For histochemistry staining, the fractured bones were embedded in paraffin. Five-millimeter-thick longitudinally oriented bone sections were stained with Safranin O-Fast green (SOFG) to quantify the cartilage area and woven bone area. For immunofluorescence staining, we prepared cryo-embedding medium composed of 2% polyvinylpyrrolidone (PVP), 20% sucrose and 8% gelatin in 1× PBS. The fractured bones were embedded with cryo-embedding medium, and 7 µm cryosections were obtained. Bone sections were processed for antigen retrieval by digestion with 0.05% trypsin at 37 °C for 15 min and then incubated with primary antibodies against Ctsk (ab19027, 1:200, Abcam), p21 (sc-6246, 1:100, Santa Cruz), γ-H2AX (sc-517348, 1:100, Santa Cruz), GFP (ab6673, 1:200, Abcam), Arg2 (sc-393496, 1:100, Santa Cruz), GCA (PA5–77127, 1:200, Invitrogen), F4/80 (ab6640, 1:400, Abcam) and Ocn (M137, 1:500, Takara) overnight at 4 °C, followed by incubation with FITC- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch, 1:200). Nuclei were counterstained with DAPI (Sigma).
SSPC isolation and culture
Femurs were dissected, cleaned of soft tissue and cut into pieces using microscissors. Then, the tissue was digested in 2.2 mg/mL collagenase II buffer at 37 °C for 60 min. The cell suspension was passed through a 70 μm nylon filter, washed in growth medium (α-MEM containing 10% FBS and 1% penicillin/streptomycin) and pelleted at 1 000 r/min. Hematopoietic lineage cells were depleted via ACK lysis for 5 min. The cells were washed again in growth medium (α-MEM containing 10% FBS and 1% penicillin/streptomycin) and pelleted at 1 000 r/min. The cell pellet was resuspended in growth medium (α-MEM containing 10% FBS and 1% penicillin/streptomycin) and plated in a 10 cm dish. All cells were grown at 37 °C in a 5% CO2 humid atmosphere.
Osteogenic differentiation assay
For osteoblast differentiation, SSPCs treated with 100 nmol/L recombinant human grancalcin protein (rGCA, NBP1-50967, Novus Biologicals Inc) or transfected with siRNA-Plxnb2 (RiboBio, China) or control SSPCs were cultured in 6/12-well plates with α-MEM containing 10% fetal bovine serum, 0.1 mmol/L dexamethasone, 10 mmol/L β-glycerol phosphate, and 50 mmol/L ascorbate-2-phosphate for 21 days. Then, the cells were stained with 2% Alizarin Red S (Sigma‒Aldrich) at pH 4.2 to evaluate cell matrix mineralization.
Chondrogenic differentiation assay
For chondrogenic differentiation, SSPCs treated with PBS or 100 nmol/L recombinant human grancalcin protein (rGCA, NBP1-50967, Novus Biologicals Inc) or transfected with siRNA-Plxnb2 (RiboBio, China) or control SSPCs were cultured in 6/12-well plates with DMEM containing 10% fetal bovine serum, 0.1 μmol/L dexamethasone, 50 μg/mL L-ascorbic acid and 50 mg/mL ITS.
Western blotting analysis
Total cell lysates were separated by SDS‒PAGE and blotted on polyvinylidene difluoride membranes (Millipore). The membranes were incubated with specific antibodies against PLXNB2 (PA5-47880, 1:1 000, Invitrogen), p16 (ab211542, 1:1 000, Sigma), p21 (ZRB1141, 1:1 000, Sigma), Arg2 (A6355, 1:1 000, ABclonal), GCA (PA5-77127, 1:800, Invitrogen), and GAPDH (5174, 1:2 000, Cell Signaling Technology) and then reprobed with appropriate horseradish peroxidase-conjugated secondary antibodies. Blots were visualized by enhanced chemiluminescence (ECL Kit; Amersham Biosciences).
qRT‒PCR analysis
qRT‒PCR analysis was performed using a Roche Molecular Light Cycler (Roche) as previously described.59 Total RNA from cells was isolated using TRIzol reagent (Invitrogen). Reverse transcription was performed using 1 mg total RNA and Super-Script II (Invitrogen). Amplification reactions were set up in 25 mL reaction volumes containing SYBR Green PCR Master Mix (PE Applied Biosystems), a 1 mL volume of cDNA, and amplification primers.
Mitochondrial membrane potential assay
SSPCs were incubated with PBS or 100 nmol/L recombinant human grancalcin protein (rGCA, NBP1-50967, Novus Biologicals Inc.) for 48 h. Cells were stained using the Mitochondrial Membrane Potential Assay Kit with JC-1 (Beyotime).
RNA sequencing
The RNA-sequencing assay was performed by Personalbio. RNA was extracted from PBS- or rGCA-treated SSPCs and siRNA-Plxnb2- or siRNA-NC-transfected SSPCs after 36 h of treatment. Differential gene expression analyses were conducted using DESeq with the criteria of fold change >2 and P-value < 0.05. The RNA sequencing data used in this article are available on the Sequence Read Archive website with the BioProject accession number PRJNA987851.
Statistical analysis
Data are presented as the mean ± SD. A Shapiro‒Wilk test for normality was performed on all datasets. Homogeneity was confirmed by a comparison of variances test. Parametric data were analyzed using an appropriate two-sided Student’s t test or Welch’s t test when two groups were being compared. Nonparametric data were analyzed with a Mann‒Whitney U test when two groups were being compared. Differences were considered significant at P < 0.05. The sample size for in vivo and in vitro experiments was based on previous experience. All the samples were randomly assigned, and none of the experiments in the study were performed in a blinded fashion. For both in vitro and in vivo experiments, no initial exclusion criteria were used, and no animals or replicates were excluded from the study.
Supplementary information
Acknowledgements
This work was supported by the National Key R&D Program of China (Project No. 2019YFA0111900 to C.J.L. and Y.J., 2022YFC3601900 to G.H.L., 2022YFC3601903 to X.H.L., and 2022YFC3601905), the National Natural Science Foundation of China (Grant Nos. 82261160397, 82272560, 81922017 to C.J.L. and 81930022, 91749105 to X.H.L.), the NSFC/RGC Joint Research Scheme, the Research Grants Council (UGC) of the Hong Kong Special Administrative Region and the National Natural Science Foundation of China (NSFC/RGC Project No. N_CUHK483/22 to Y.J.), the Hunan Provincial Science and Technology Department (2023JJ30896 to C.J.L.), the Key Research and Development Program of Hunan Province (2022SK2023 to C.J.L.), and the Science and Technology Innovation Program of Hunan Province (2023RC1027 to C.J.L., 2022RC1009 to J.W, and 2022RC3075 to C.Z.).
Author contributions
C.J.L. and N.Y.Z. designed the experiments; N.Y.Z. carried out most of the experiments and drafted the manuscript; R.L., L.L., Y.C.S., and Y.R.J. helped to conduct animal experiments; W.Z.H., M.H., and Y.L.X. helped in sample collection; M.Y., Q.G., Y.H., T.S., and Y.X. helped in data analysis; Y.J., W.S.W., J.W., C.Z., G.H.L., and X.H.L. helped in manuscript revision and proofreading; C.J.L. and XH.L. supervised the study; C.J.L. designed the experiments, analyzed the results, interpreted the data, and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Xiang-Hang Luo, Email: xianghangluo@hotmail.com.
Chang-Jun Li, Email: lichangjun@csu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41413-023-00309-1.
References
- 1.Lane, J. M., Russell, L. & Khan, S. N. Osteoporosis. Clin. Orthop Relat. Res. 139–150 (2000). [DOI] [PubMed]
- 2.Curtis EM, et al. Epidemiology of fractures in the United Kingdom 1988-2012: variation with age, sex, geography, ethnicity and socioeconomic status. Bone. 2016;87:19–26. doi: 10.1016/j.bone.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tarrant SM, Balogh ZJ. The global burden of surgical management of osteoporotic fractures. World J. Surg. 2020;44:1009–1019. doi: 10.1007/s00268-019-05237-y. [DOI] [PubMed] [Google Scholar]
- 4.Ekegren CL, Edwards ER, de Steiger R, Gabbe BJ. Incidence, costs and predictors of non-union, delayed union and mal-union following long bone fracture. Int. J. Environ. Res. Public Health. 2018;15:2845. doi: 10.3390/ijerph15122845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saul D, Khosla S. Fracture healing in the setting of endocrine diseases, aging, and cellular senescence. Endocr. Rev. 2022;43:984–1002. doi: 10.1210/endrev/bnac008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang CL, Liu B, Wan M. “Bone-SASP” in skeletal aging. Calcif. Tissue Int. 2023;113:68–82. doi: 10.1007/s00223-023-01100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khosla S, Farr JN, Monroe DG. Cellular senescence and the skeleton:pathophysiology and therapeutic implications. J. Clin. Invest. 2022;132:e154888. doi: 10.1172/JCI154888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wan M, Gray-Gaillard EF, Elisseeff JH. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021;9:41. doi: 10.1038/s41413-021-00164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farr JN, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017;23:1072–1079. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farr JN, et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner Res. 2016;31:1920–1929. doi: 10.1002/jbmr.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, et al. Osteoclasts protect bone blood vessels against senescence through the angiogenin/plexin-B2 axis. Nat. Commun. 2021;12:1832. doi: 10.1038/s41467-021-22131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, et al. Oxylipin-PPARgamma-initiated adipocyte senescence propagates secondary senescence in the bone marrow. Cell Metab. 2023;35:667–684. doi: 10.1016/j.cmet.2023.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li CJ, et al. Senescent immune cells release grancalcin to promote skeletal aging. Cell Metab. 2021;33:1957–1973. doi: 10.1016/j.cmet.2021.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, et al. Age-associated callus senescent cells produce TGF-beta1 that inhibits fracture healing in aged mice. J. Clin. Invest. 2022;132:e148073. doi: 10.1172/JCI148073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saul D, et al. Modulation of fracture healing by the transient accumulation of senescent cells. Elife. 2021;10:e69958. doi: 10.7554/eLife.69958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Q, et al. Deletion of p16 accelerates fracture healing in geriatric mice. Am. J. Transl. Res. 2021;13:11107–11125. [PMC free article] [PubMed] [Google Scholar]
- 17.Chan CK, et al. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Josephson AM, et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA. 2019;116:6995–7004. doi: 10.1073/pnas.1810692116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambrosi TH, et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature. 2021;597:256–262. doi: 10.1038/s41586-021-03795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin X, et al. Aged callus skeletal stem/progenitor cells contain an inflammatory osteogenic population with increased IRF and NF-kappaB pathways and reduced osteogenic potential. Front. Mol. Biosci. 2022;9:806528. doi: 10.3389/fmolb.2022.806528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark D, Nakamura M, Miclau T, Marcucio R. Effects of aging on fracture healing. Curr. Osteoporos. Rep. 2017;15:601–608. doi: 10.1007/s11914-017-0413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pajarinen J, et al. Mesenchymal stem cell-macrophage crosstalk and bone healing. Biomaterials. 2019;196:80–89. doi: 10.1016/j.biomaterials.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bastian O, et al. Systemic inflammation and fracture healing. J. Leukoc. Biol. 2011;89:669–673. doi: 10.1189/jlb.0810446. [DOI] [PubMed] [Google Scholar]
- 24.Wu AC, Raggatt LJ, Alexander KA, Pettit AR. Unraveling macrophage contributions to bone repair. Bonekey Rep. 2013;2:373. doi: 10.1038/bonekey.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitaori T, et al. Stromal cell-derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum. 2009;60:813–823. doi: 10.1002/art.24330. [DOI] [PubMed] [Google Scholar]
- 26.Ringe J, et al. Towards in situ tissue repair: human mesenchymal stem cells express chemokine receptors CXCR1, CXCR2 and CCR2, and migrate upon stimulation with CXCL8 but not CCL2. J. Cell Biochem. 2007;101:135–146. doi: 10.1002/jcb.21172. [DOI] [PubMed] [Google Scholar]
- 27.Baht GS, et al. Exposure to a youthful circulaton rejuvenates bone repair through modulation of beta-catenin. Nat. Commun. 2015;6:7131. doi: 10.1038/ncomms8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vi L, et al. Macrophage cells secrete factors including LRP1 that orchestrate the rejuvenation of bone repair in mice. Nat. Commun. 2018;9:5191. doi: 10.1038/s41467-018-07666-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark D, et al. Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell. 2020;19:e13112. doi: 10.1111/acel.13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, et al. PDGFRalpha reporter activity identifies periosteal progenitor cells critical for bone formation and fracture repair. Bone Res. 2022;10:7. doi: 10.1038/s41413-021-00176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Debnath S, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. 2018;562:133–139. doi: 10.1038/s41586-018-0554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeffery EC, Mann TLA, Pool JA, Zhao Z, Morrison SJ. Bone marrow and periosteal skeletal stem/progenitor cells make distinct contributions to bone maintenance and repair. Cell Stem Cell. 2022;29:1547–1561. doi: 10.1016/j.stem.2022.10.002. [DOI] [PubMed] [Google Scholar]
- 33.Duchamp de Lageneste O, et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat. Commun. 2018;9:773. doi: 10.1038/s41467-018-03124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, et al. Single-cell RNA sequencing reveals B cells are important regulators in fracture healing. Front. Endocrinol. (Lausanne) 2021;12:666140. doi: 10.3389/fendo.2021.666140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H, et al. Prrx1 marks stem cells for bone, white adipose tissue and dermis in adult mice. Nat. Genet. 2022;54:1946–1958. doi: 10.1038/s41588-022-01227-4. [DOI] [PubMed] [Google Scholar]
- 36.Sun J, et al. Histone demethylase LSD1 is critical for endochondral ossification during bone fracture healing. Sci. Adv. 2020;6:eaaz1410. doi: 10.1126/sciadv.aaz1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao B, et al. Macrophage-lineage TRAP+ cells recruit periosteum-derived cells for periosteal osteogenesis and regeneration. J. Clin. Invest. 2019;129:2578–2594. doi: 10.1172/JCI98857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ortinau LC, et al. Identification of functionally distinct Mx1+ αSMA+ periosteal skeletal stem cells. Cell Stem Cell. 2019;25:784–796. doi: 10.1016/j.stem.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, et al. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat. Commun. 2017;8:2043. doi: 10.1038/s41467-017-02171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saul D, et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 2022;13:4827. doi: 10.1038/s41467-022-32552-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi J, Hua L, Harmer D, Li P, Ren G. Cre driver mice targeting macrophages. Methods Mol. Biol. 2018;1784:263–275. doi: 10.1007/978-1-4939-7837-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, et al. PLEXIN-B2 promotes the osteogenic differentiation of human bone marrow mesenchymal stem cells via activation of the RhoA signaling pathway. Cell Signal. 2019;62:109343. doi: 10.1016/j.cellsig.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 44.Pascoe HG, Wang Y, Zhang X. Structural mechanisms of plexin signaling. Prog. Biophys. Mol. Biol. 2015;118:161–168. doi: 10.1016/j.pbiomolbio.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yepuri G, et al. Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell. 2012;11:1005–1016. doi: 10.1111/acel.12001. [DOI] [PubMed] [Google Scholar]
- 46.Xiong Y, Yu Y, Montani JP, Yang Z, Ming XF. Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: implications for atherosclerotic plaque vulnerability. J. Am. Heart Assoc. 2013;2:e000096. doi: 10.1161/JAHA.113.000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le AN, et al. DRG2 depletion promotes endothelial cell senescence and vascular endothelial dysfunction. Int. J. Mol. Sci. 2022;23:2877. doi: 10.3390/ijms23052877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong Y, Yepuri G, Montani JP, Ming XF, Yang Z. Arginase-II deficiency extends lifespan in mice. Front. Physiol. 2017;8:682. doi: 10.3389/fphys.2017.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shosha E, et al. Endothelial arginase 2 mediates retinal ischemia/reperfusion injury by inducing mitochondrial dysfunction. Mol. Metab. 2021;53:101273. doi: 10.1016/j.molmet.2021.101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Touyz RM. Linking LOX-1 and arginase II through mitochondria: a novel paradigm in endothelial dysfunction. Circ. Res. 2014;115:412–414. doi: 10.1161/CIRCRESAHA.114.304550. [DOI] [PubMed] [Google Scholar]
- 51.Pandey D, et al. OxLDL triggers retrograde translocation of arginase2 in aortic endothelial cells via ROCK and mitochondrial processing peptidase. Circ. Res. 2014;115:450–459. doi: 10.1161/CIRCRESAHA.115.304262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hozain S, Cottrell J. CDllb+ targeted depletion of macrophages negatively affects bone fracture healing. Bone. 2020;138:115479. doi: 10.1016/j.bone.2020.115479. [DOI] [PubMed] [Google Scholar]
- 53.Schlundt C, et al. Macrophages in bone fracture healing: their essential role in endochondral ossification. Bone. 2018;106:78–89. doi: 10.1016/j.bone.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 54.Yu W, et al. Plexin-B2 mediates physiologic and pathologic functions of angiogenin. Cell. 2017;171:849–864. doi: 10.1016/j.cell.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saha B, Ypsilanti AR, Boutin C, Cremer H, Chedotal A. Plexin-B2 regulates the proliferation and migration of neuroblasts in the postnatal and adult subventricular zone. J. Neurosci. 2012;32:16892–16905. doi: 10.1523/JNEUROSCI.0344-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zou N, Liu R, Li C. Cathepsin K+ non-osteoclast cells in the skeletal system: function, models, identity, and therapeutic implications. Front. Cell Dev. Biol. 2022;10:818462. doi: 10.3389/fcell.2022.818462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Einhorn TA, Gerstenfeld LC. Fracture healing: mechanisms and interventions. Nat. Rev. Rheumatol. 2015;11:45–54. doi: 10.1038/nrrheum.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie H, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat. Med. 2014;20:1270–1278. doi: 10.1038/nm.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xiao YZ, et al. Reducing hypothalamic stem cell senescence protects against aging-associated physiological decline. Cell Metab. 2020;31:534–548 . doi: 10.1016/j.cmet.2020.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.