

Abstract
This study investigated the role of Alpha-tocopherylquinone (TQ) in regulating the intestinal immune system and the underlying mechanisms. In the experimental dextran sodium sulfate and T cell-mediated colitis models, TQ significantly reduced the mRNA levels of interleukin (IL)-6, IL-1β, IL-17A, IL-23, and tumor necrosis factor (TNF)-α and the abundance of proinflammatory macrophages, T helper (Th)17 cells, and ILC3s in the colons of wild-type mice. TQ also prevented lipopolysaccharide (LPS)-induced activation of NFκB and signal transducer and activator of transcription (Stat)-3 pathways in the human macrophage U937 cells. Pharmacological inhibition or CRISPR-Cas-9-mediated knockout of Aryl hydrocarbon Receptor (AhR) prevented the anti-inflammatory effects of TQ in the LPS-treated U937 cells. Furthermore, TQ reduced the mRNA levels of the LPS-induced pro-inflammatory cytokines in the WT but not Ahr−/− mice splenocytes. TQ also reduced IL-6R protein levels and IL-6-induced Stat-3 activation in Jurkat cells and in vitro differentiation of Th17 cells from wild-type but not Ahr−/− mice naive T cells. Additionally, TQ prevented the pro-inflammatory effects of LPS on macrophages and stimulation of T cells in human PBMCs and significantly reduced the abundance of tumor necrosis factor-α, IL-1β, and IL-6hi inflammatory macrophages and Th17 cells in surgically resected Crohn’s disease (CD) tissue. Our study shows that TQ is a naturally occurring, non-toxic, and effective immune modulator that activates AhR and suppresses the Stat-3-NFκB signaling.
INTRODUCTION
Homeostasis of the immune system forms a key foundation of host health. Carefully orchestrated and stringently regulated signaling pathways that activate and subsequently inactivate the innate and adaptive branches of the immune system mediate robust protection from foreign antigens while protecting against bystander self-damage. In diseases with immune dysregulation like rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease (IBD), including Crohn’s disease (CD), and ulcerative colitis (UC), however, these homeostatic regulatory circuits are often compromised by the loss of the inhibitory signaling of the immune system, resulting in persistent, unresolved inflammation1,2.
Aberrant activation and loss of regulation of the IL-6 signaling axis have been implicated in inflammatory conditions including IBD, Systemic Lupus Erythematosus, etc3. Interleukin (IL)-6 is a pleiotropic cytokine that is produced by multiple immune cell types and serves as a key regulator of the signal relay between the innate and humoral immune responses. IL-6 cis- or transsignaling activates the Janus kinase/signal transducer (Jak) and activator of transcription (Stat) kinases and alters the transcriptional state of the target cells to initiate inflammation4. Moreover, other than being a standalone regulator of proinflammatory responses, IL-6 is one of the key precursors for the differentiation of T helper (Th)17 cells, a special subset of the CD4+ T cells that secrete IL-17A/F and IL-22 as their signature cytokines. IL-6, combined with tumor growth factor-β, and IL-23 promotes the differentiation of naive T cells into the Th17 lineage while inhibiting the development of the anti-inflammatory Treg cells5. Th17 cells serve a key role in multiple immunological responses against pathogens and tissue repair; however, like IL-6, unregulated proliferation and the hyperactivation of Th17 cells have been attributed to several diseases with immune dysregulation, such as IBD6,7.
As an environmental sensor, the AhR binds to a variety of synthetic and natural compounds and acts as a class I, basic helix-loop-helix transcriptional regulator8. Upon activation, the AhR dimerizes with the AhR nuclear translocator protein, which then binds xenobiotic-responsive element (XRE; also known as dioxin response element) sites in gene regulatory regions, regulating the expression of a diverse set of genes. Genetic deletion studies have demonstrated important functions of AhR in the immune responses of innate immune cells, like macrophages, and in adaptive immunity, like its regulatory role in T cell differentiation. Furthermore, AhR activation has been shown to improve colitis outcomes in animal IBD models9–20. According to a current hypothesis, decreased intestinal AhR expression or defective AhR activation contributes to intestinal barrier dysfunction, immune dysregulation, and dysbiosis, thus favoring chronic inflammation and IBD21.
Vitamin E derivatives are believed to have several clinical benefits, including regulation of oxidative stress and cell death22, although the molecular basis of the many physiological functions of vitamin E remains unknown. Alpha-tocopherylquinone (TQ) is a quinone-structured, non-toxic, less reactive, cell membrane integrity-promoting oxidation product of vitamin E23,24. In this study, we examined the immunomodulatory role of TQ. Our studies show that TQ has remarkable immunomodulatory abilities, including inhibiting pro-inflammatory IL-6 production by macrophages and IL-17A/F production by effector CD4+ T cells and preventing the differentiation of naive CD4+ T cells to Th17 cells. Consequently, TQ significantly reduced intestinal inflammation and Th17 abundance in experimental colitis. Additionally, TQ reduced IL-17A/F production by effector T cells from CD patient-derived tissues. We also show that TQ activates AhR, and the immunomodulatory effect of TQ is AhR-dependent. Thus, our study shows that TQ offers a naturally occurring, non-toxic, potential tool for use as an anti-inflammatory agent in immune-mediated disease conditions.
RESULTS
TQ treatment decreases the production of inflammatory cytokines in mouse colon in experimental colitis models
In our previous studies, we have observed that TQ enhances the intestinal epithelial tight junction barrier in an AhR-dependent mechanism and ameliorates disease activity and inflammation in various models of experimental colitis25. The role of TQ in the regulation of intestinal immune responses is not known. Thus, we investigated the effect of TQ on the production of inflammatory cytokines in experimental colitis. TQ administration significantly reduced the DSS-induced increase in the levels of IL-1β, IL-6, IL-17A, IL-23, and tumor necrosis factor (TNF)-α transcripts during the acute and chronic DSS colitis (Figs. 1A and 1B) and T cell-mediated colitis (Fig. 1C). Having observed this anti-inflammatory effect of TQ in the different experimental colitis models, we next directly assessed the abundance of pro-inflammatory inflammatory macrophages, Th17 cells, and ILC3s26, cells that have been implicated in the development and pathology of IBD, in the mouse colons. We observed that TQ administration in both the acute DSS (Figs. 2A–C) and chronic DSS colitis models (Figs. 2D–F) significantly reduced the abundance of all three cell types within the colons of WT mice. Taken together, our data show that administration of TQ dampens excessive colonic immune response in experimental colitis.
Fig 1.
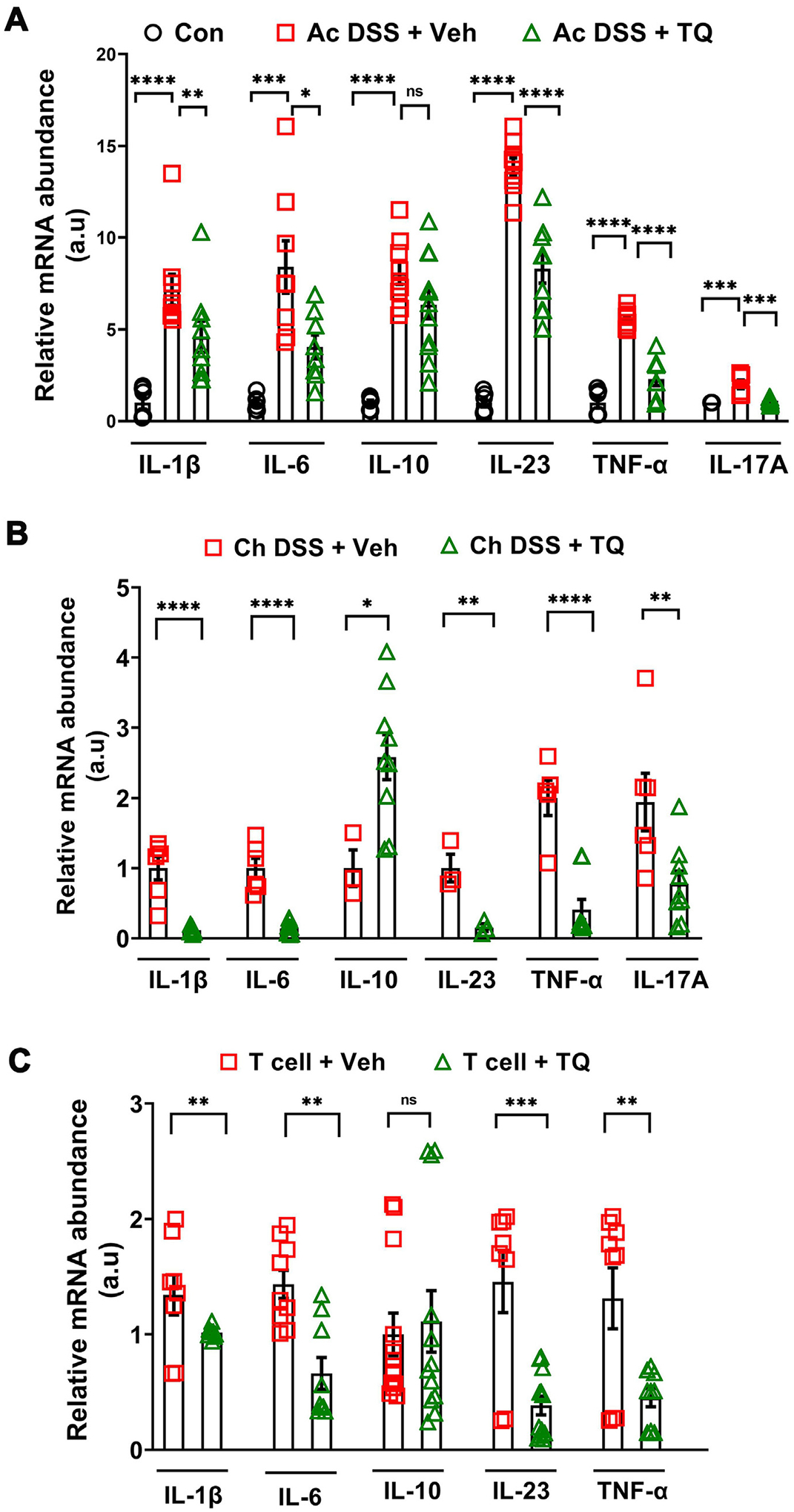
TQ ameliorates colonic inflammation in WT mice. (A) In the acute DSS (Ac DSS) model of colitis, TQ reduced the mRNA levels of key inflammatory cytokines IL-1β, IL-6, IL-17A, IL-23, and TNF-α in the colons of WT B6 mice. TQ, however, had no effect on IL-10 mRNA levels in this acute DSS colitis model. (B) In the chronic DSS (Ch DSS) model of colitis, TQ reduced the mRNA levels of key inflammatory cytokines IL-1β, IL-6, IL-23, TNF-α, and IL-17A in the colons of WT B6 mice compared to vehicle (Veh) administered mice. In this model of colitis, TQ also increased the levels of IL-10 mRNA. (C) In the T cell colitis model, TQ gavaging reduced the mRNA levels of the inflammatory cytokines IL-1β, IL-6, IL-23, and TNF-α in the colons from Rag−/− B6 mice. Like the Ac DSS model, TQ had no significant effect on IL-10 mRNA levels in this model of colitis. Data represented as mean ± SEM, n ≥ 3/group from two independent experiments. Student’s t-test or one-way analysis of variance with Tukey’s post test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns = not significant; a.u = arbitrary units.
Fig. 2.
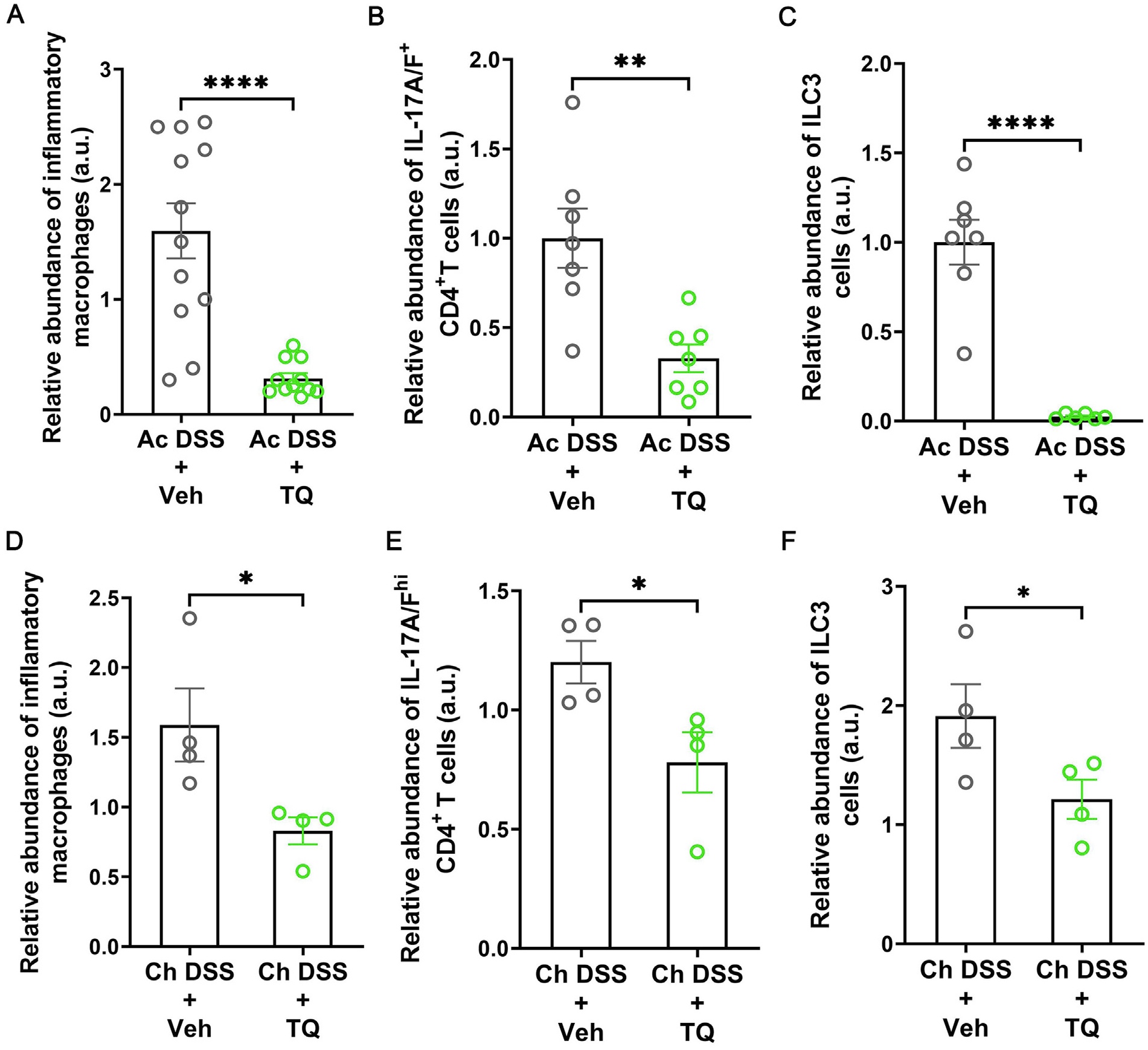
TQ ameliorates intestinal inflammation by reducing the abundance of inflammatory cell types. In acute DSS colitis (Ac DSS), administration of TQ along with DSS reduced the abundance of IL-6, TNF-α, and IL-1β -enriched inflammatory macrophages (A), IL-17A/F enriched RORγt+ CD4+ T cells (B), and IL-17A/F+ IL-22+ CD4− innate lymphoid cells type 3 (ILC3) (C) in the colons of WT mice. In chronic DSS colitis (Ch DSS), administration of TQ along with DSS reduced the abundance of IL-6, TNF-α, and IL-1β -enriched inflammatory macrophages (D), IL-17A/F enriched RORγt+ CD4+ T cells (E), and IL-17A/F+ IL-22+ CD4− innate lymphoid cells type 3 (ILC3) (F) in the colons of WT mice. The flow cytometry data are represented as the ratio of the absolute number of IL-6, and TNF-α - enriched inflammatory macrophages to the total number of macrophages (A and C) or the ratio of the absolute number of IL-17A-enriched CD4+ T cells to the total number of CD4+ T cells (B and D) or IL-17A and IL-17F-enriched cells to the total RORγt+ CCR6+ cells (C and F), normalized to the average values of similar ratio of the vehicle-treated control (Veh) mice. Data represented as mean ± SEM. n ≥ 3/group from two independent experiments for Ac DSS and n = 4/group for Ch DSS. Student’s t-test. *p < 0.05, **p < 0.01, ****p < 0.0001.
We next sought to directly investigate the anti-inflammatory role of TQ by using murine splenocytes. We observed that activation of the splenocytes by lipopolysaccharide (LPS) or a cocktail of PMA and ionomycin significantly increased the mRNA levels of the inflammatory cytokines IL-1β, IL-6, IL-17A, and IL-23, which was prevented upon TQ treatment (Fig. 3A). We next assessed if this reduction in transcript levels was a consequence of increased cell death induced by TQ administration. For this, the activated splenocytes with and without TQ treatment were treated with a cell viability stain, and the proportion of live cells was analyzed by flow cytometry. We observed no changes in the proportion of live cells between any treatments (Fig. 3B); however, the proportion of inflammatory macrophages and Th17 cells within these live cell populations was reduced by TQ treatment (Figs. 3C and 3D). Thereby, our data show that the reduction of inflammatory cytokine production observed upon TQ treatment is not due to alteration in cell viability or the overall number of macrophages or CD4+ T cells but due to the relative reduction in the abundance of intracellular inflammatory cytokines within these cells.
Fig. 3.
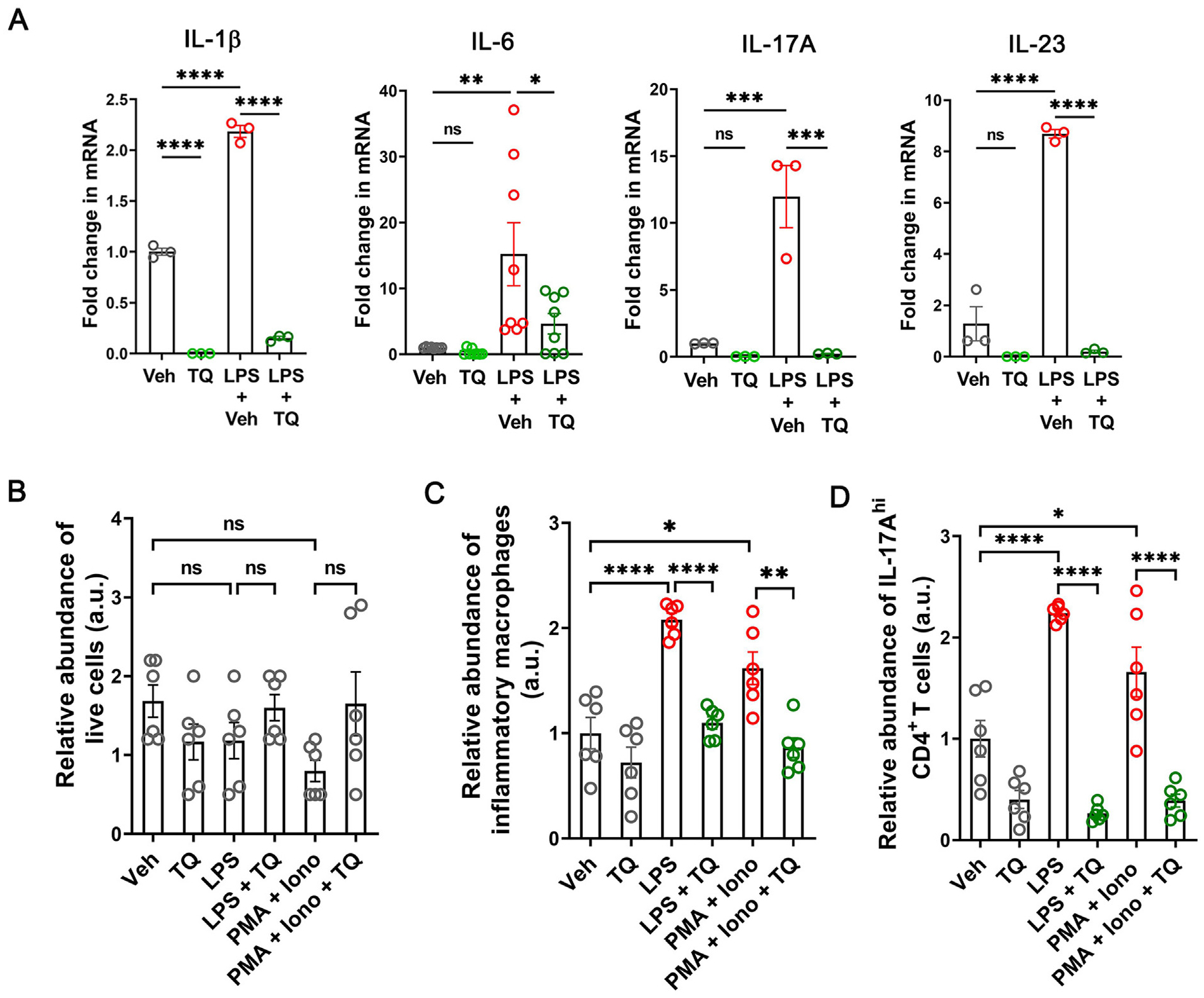
TQ efficiently reduces the effect of inflammatory stimulus in murine splenocytes. (A) TQ (25 μM) reduced the LPS-induced upregulation in the mRNA levels of IL-1β, IL-6, IL-17A, and IL-23 by splenocytes isolated from WT mice. (B) TQ alone or in combination with any of the activators did not affect the survival of the splenocytes. (C) TQ reduced the abundance of IL-6, TNF-α, and IL-1β-enriched inflammatory macrophages within the splenocyte population when they were activated with LPS (10 ng/ml) or a cocktail of PMA and ionomycin (1 μg/ml each). (D) TQ reduced the abundance of IL-17A-enriched RORγt+ CD4+ T cells within the splenocytes when they were activated with LPS or a cocktail of PMA and ionomycin. The flow cytometry data are represented as the ratio of the absolute number of IL-6, and TNF-α -enriched inflammatory macrophages to the total number of macrophages (C) or the ratio of the absolute number of IL-17A-enriched CD4+ T cells to the total number of CD4+ T cells (D), normalized to the average values of the similar ratio of the vehicle-treated control (Veh) cells. Data represented as mean ± SEM, n ≥ 3/group. One-way analysis of variance with Tukey’s post test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant.
TQ reduces the production of inflammatory cytokines by human macrophage U937 cells in an AhR-dependent mechanism
Having observed the anti-inflammatory role of TQ in the murine models of colitis and splenocyte activation, we next investigated the effect of TQ on the human U937 cell line to assess the underlying mechanism. Activation of the human macrophage U937 cells by LPS treatment increased the production of the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α; however, treating cells with LPS in the presence of 25 μM TQ decreased the stimulatory effects of LPS. (Fig. 4A). TQ treatment also reduced the protein levels of IL-6 and IL-1β in cell supernatants as assessed by ELISA (Supplementary Fig. 1). Since TQ was identified as an activator of the AhR by a chemical screen, we examined if TQ activates AhR in immune cells and if its immune-modulatory effect is AhR dependent. To this end, we assessed the nuclear localization of AhR by confocal-immunofluorescence microscopy in the vehicle and TQ-treated U937 cells and observed increased localization of AhR in the nucleus upon TQ treatment (Figs. 4B and 4C). Also, the mRNA level of CYP1A1, the prototypic target of AhR, was elevated in a dose-dependent manner upon TQ treatment, with the maximum effect observed at the 25 μM concentration of TQ (Fig. 4D). Furthermore, in an XRE-luciferase reporter assay, we observed increased luciferase activity in U937 cells treated with TQ (Supplementary Fig. 2), confirming that TQ activates AhR.
Fig. 4.
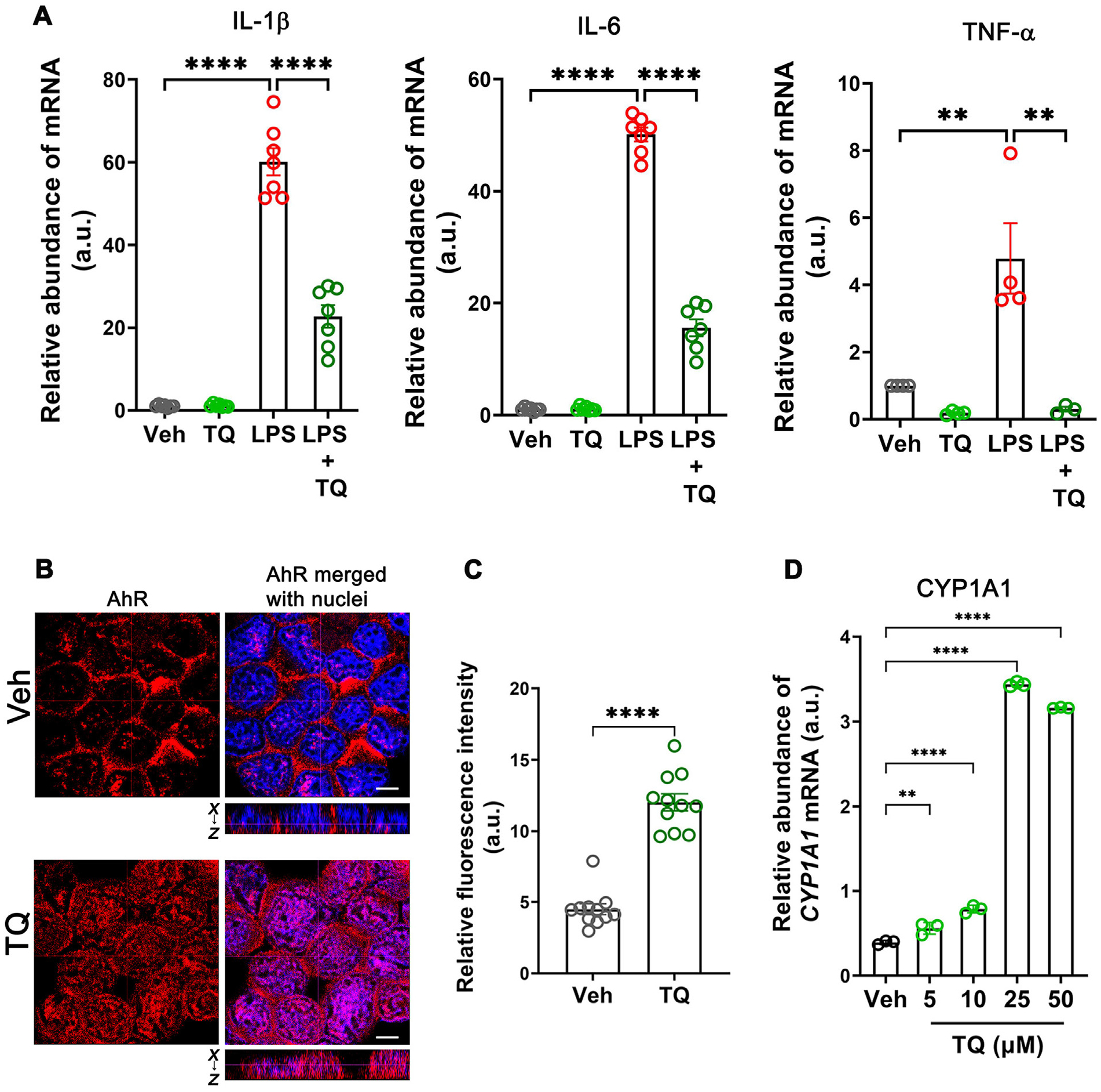
TQ exerts its anti-inflammatory effect by activating the AhR in U937 cells. (A) TQ prevented the LPS-induced (10 ng/ml) increase in the mRNA levels of IL-1β, IL-6, and TNF-α in the human U937 cell line. (B) Confocal images of AhR (red) and nuclei (blue) in U937 cells. The images showed that TQ treatment (4 h) increased AhR levels as well as its nuclear translocation, indicating that TQ activates AhR. The dotted lines represent the optical level for the x-y and x-z planes. White bar: 5 μm. These data are representative of multiple areas from independent samples, n = 5. (C) Quantification of AhR fluorescence in the nucleus from (B). (D) The mRNA levels of CYP1A1, the prototypic transcriptional target of AhR, showed a dose-dependent increase in abundance with increasing concentrations of TQ with the maximum effect observed with 25 μM concentration of TQ. Data represented as mean ± SEM, n > 3 from 3 independent experiments. Student’s t-test (C) and one-way analysis of variance with Tukey’s post test (A and D). **p < 0.01, ****p < 0.0001.
To test if AhR was involved in the TQ-induced regulation of immune responses in the macrophages, we treated U937 cells with LPS and assessed the levels of IL-6 transcripts in the presence or absence of TQ along with the known AhR inhibitors GNF-531(20 μM), SGA-360 (20 μM), or CH-223191(10 μM)27–29. Of the three AhR inhibitors, only GNF-531 treatment successfully inhibited the anti-inflammatory effect of TQ on the production of IL-6 (Fig. 5A). These observations allude to a mechanism where TQ-mediated suppression of inflammation is AhR dependent; however, given that GNF-531 is a cytoplasmic competitive inhibitor of AhR, the anti-inflammatory effect of TQ may be independent of the nuclear translocation of AhR.
Fig. 5.
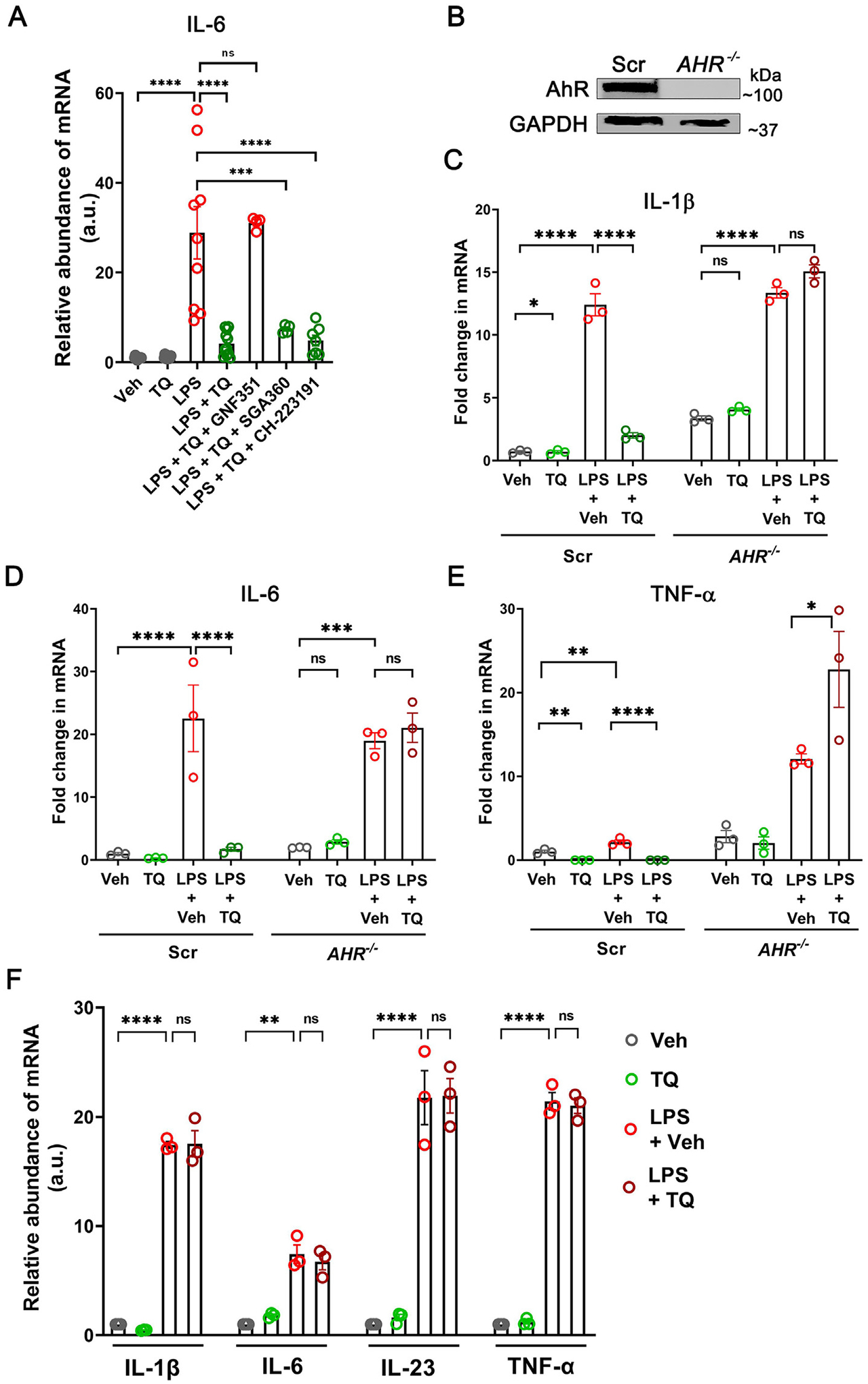
The anti-inflammatory effect of TQ is dependent on AhR. (A) The anti-inflammatory effect of TQ, as measured by the mRNA levels of IL-6 in U937 cells upon activation with LPS, was prevented by the competitive inhibitor of AhR, GNF-531 (20 μM). Other AhR inhibitors SGA-360 (20 μM) and CH-223191 (10 μM) failed to inhibit the anti-inflammatory effect of TQ. (B) CRISPR/Cas9-mediated targeting of the AHR gene produced a complete knockout of AhR in U937 cells. The GAPDH levels are shown as the loading control. (C–E) In the absence of AhR, TQ failed to prevent the LPS-induced upregulation in the mRNA levels of IL-1β (C), IL-6 (D), and TNF-α (E) by U937 cells. Scr- non-target scrambled control. (F) TQ did not prevent the LPS-induced upregulation of IL-1β, IL-6, IL-23, and TNF-α mRNA levels in the Ahr−/− mouse splenocytes (n = 3/group). Data represented as mean ± SEM, n ≥ 3 from 3 independent experiments. One-way analysis of variance with Tukey’s post test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns = not significant.
To account for the non-specific effects of the pharmacological inhibitors and directly assess the anti-inflammatory role of AhR, we employed CRISPR/Cas9 to generate AHR−/− U937 cells. Targeting the AHR locus by sgRNA produced a near-complete deletion of AhR in U937 cells (Fig. 5B). The AHR−/− cells produced high baseline levels of the inflammatory cytokines IL-1β, IL-6, and TNF-α compared to the non-target guide RNA-transfected U937 cells (Scr), which were amplified upon LPS treatment. Additionally, unlike the Scr cells, where TQ treatment successfully quelled the effect of LPS, TQ treatment produced an exacerbated inflammatory effect in the AHR−/− cells, increasing the transcript levels of the cytokines (Figs. 5–E), potentially due to the combination of LPS-mediated inflammatory response and loss of anti-oxidative response to electrophilic stress caused by TQ, in the absence of AhR30. Furthermore, like the AHR−/− U937 cells, TQ did not affect the LPS-induced elevation of IL-1β, IL-6, IL-23, and TNF-α mRNA levels in the splenocytes isolated from Ahr−/− mice (Fig. 5F). Additionally, in acute DSS colitis, unlike the WT mice, TQ did not reduce the abundance of the IL-1βhi, TNF-αhi, and IL-6hi pro-inflammatory macrophages and Th17 cells in Ahr−/− mice colons (data not shown). Taken together, our data show that TQ-mediated dampening of inflammatory cytokine production is AhR-dependent.
TQ uniquely dampens LPS-mediated cytokine production by inhibiting the activation of the NFκB and Stat-3 pathways
Having established the role of AhR in regulating the TQ-mediated modulation of the immune responses, we further examined the mechanism underlying the anti-inflammatory role of TQ. LPS is a bacterial antigen that causes inflammatory cytokine production through the activation of the NFκB pathway31. To this end, we assessed the localization of p65, one of the subunits of the NFκB complex, upon LPS treatment of U937 cells in the presence and absence of TQ. Confocal-immunofluorescence imaging showed that while LPS treatment significantly increased the nuclear localization of p65, TQ significantly reduced the LPS-induced p65 nuclear localization (Figs. 6A and 6B). In a complementary approach, we also assessed the protein levels of p65 and IkBα in the U937 cells and observed increased levels of p65 and a corresponding decrease in IkBα upon LPS treatment, indicating successful activation of the NFκB pathway. The LPS-activated NFκB pathway, however, was inhibited by TQ (Fig. 6C). We next examined the effect of AHR knockdown on the NFĸB pathway. Assessing for the NFκB promoter activity by luciferase reporter showed that TQ treatment significantly reduced the LPS-induced increase in NFκB promoter activity in Scr cells but not AHR−/− cells (Fig. 6D), confirming the AhR dependence of the ability of TQ to inhibit NFκB signaling.
Fig. 6.
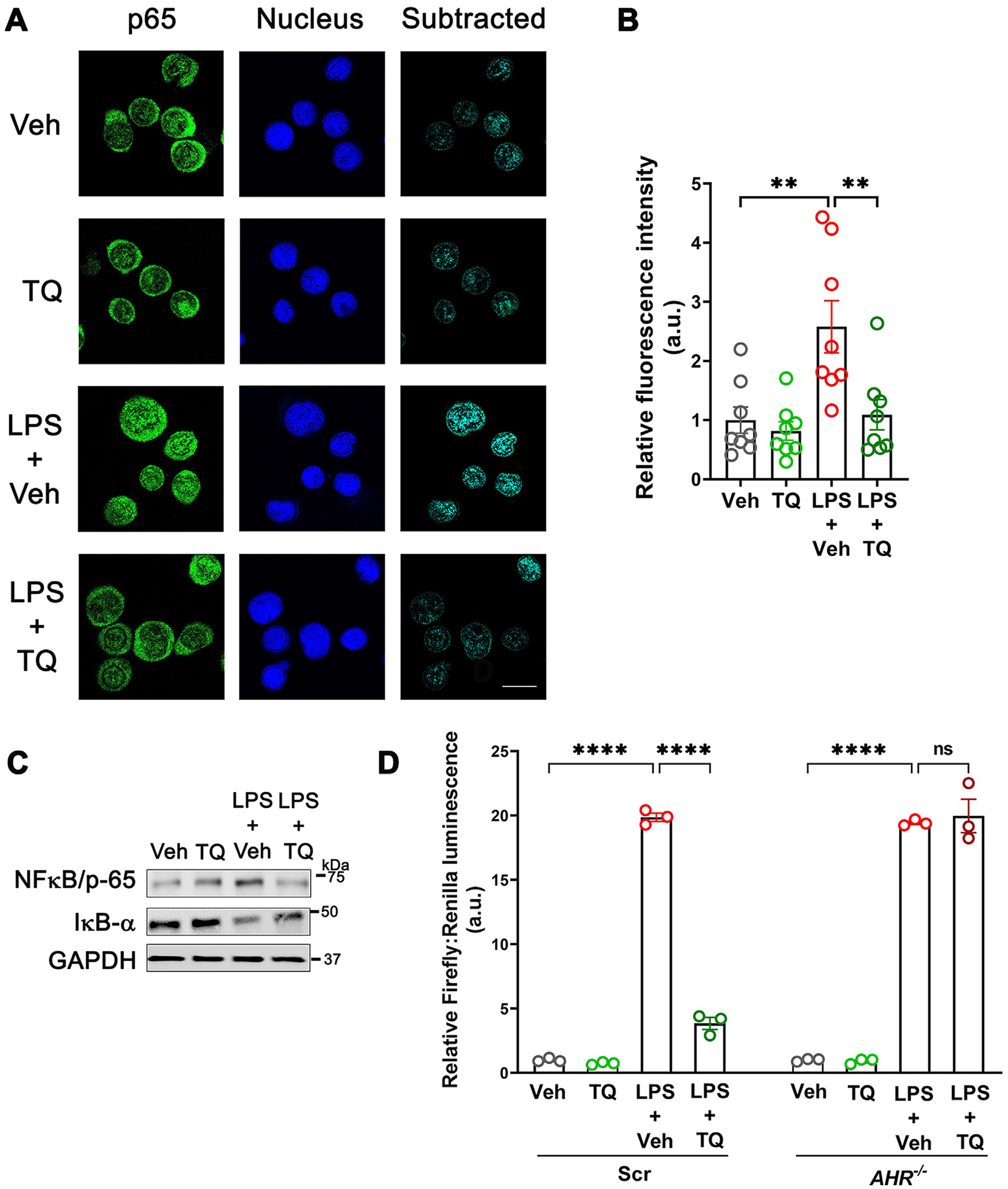
TQ reduces signaling along the NFκB axis. (A) Confocal immunofluorescence imaging showed that TQ reduced the LPS-induced nuclear localization of the NFκB protein p65 (green) in U937 cells. White bar = 10 μM. Image representative 3 independent experiments with > 30 individual fields captured per experimental round. (B) Quantification of the p65 fraction localized to the nucleus from (A). (C) TQ prevented the LPS-induced increase in p65 protein levels and the associated decrease in the levels of IκBα in U937 cells. The GAPDH levels are shown as the loading control. Representation of three independent experiments. (D) In AHR−/− U937 cells, TQ did not prevent the LPS-induced upregulation of luciferase activity where the luciferase reporter was placed downstream of the NFκB binding site. Representation of three independent experiments. Data represented as mean ± SEM. One-way analysis of variance with Tukey’s post test. **p < 0.01, ****p < 0.0001. ns = not significant.
As functional interactions between NFκB and Stat-3 in immune cells control the production of pro-inflammatory cytokines32, and our previous studies also showed inhibition of Stat-3 by TQ in the intestinal epithelium25, we also assessed the activation of the Stat-3 in U937 cells in the presence and absence of TQ. TQ significantly inhibited the LPS-induced phosphorylation and hence activation of Stat-3 (Figs. 7A and 7B) and Stat-3-dependent promoter activity in luciferase assay (Fig. 7E). Furthermore, upon testing the AhR dependence of these anti-inflammatory roles of TQ, we observed that while TQ successfully inhibited the phosphorylation of Stat-3 and the increase in Stat-3 promoter activity in the Scr cells, this effect was nullified in the AHR−/− U937 cells (Figs. 7C–E). Taken together, our data show that TQ mediates its anti-inflammatory action by inhibiting the NFκB and Stat-3 pathways in a process dependent on AhR.
Fig. 7.
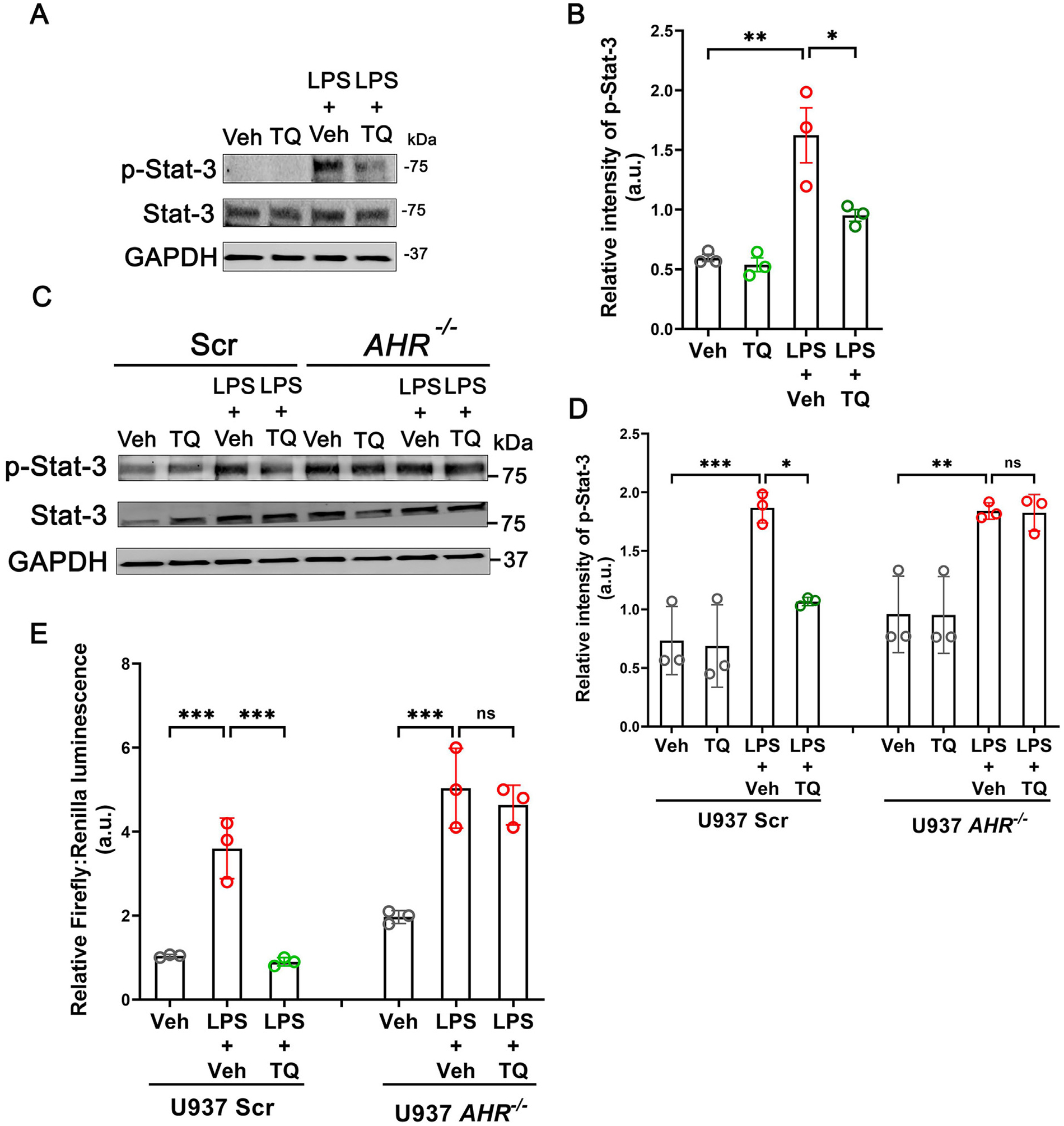
TQ reduces Stat-3 signaling. (A) TQ reduces the LPS-induced phosphorylation and activation of Stat-3 in U937 cells. The GAPDH levels are shown as the loading control. Representation of three independent experiments. (B) Quantification of p-Stat-3 band intensity normalized to total Stat-3 from (A). (C) The inhibitory effect of TQ on Stat-3 activation was dependent on AhR as AHR−/− U937 cells failed to reduce the LPS-induced phosphorylation of Stat-3 upon incubation with TQ. The GAPDH levels are shown as the loading control. Representation of three blots. (D) Densitometric quantification of phospho-Stat-3 bands normalized to total Stat-3 from (C). (E) TQ failed to prevent the LPS-induced upregulation of luciferase activity in the AHR−/− U937 cells where the luciferase reporter was placed downstream of the Stat-3 dimer binding site. Representation of 3 independent experiments. Data represented as mean ± SEM. One-way analysis of variance with Tukey’s post test. * p < 0.05, ** p < 0.01, *** p < 0.001. ns = not significant.
TQ inhibits the IL-6-IL-6R signaling axis in T cells
Considering the specific effect of TQ in the suppression of IL-6 production, an essential cytokine necessary for Th17 cell differ entiation33, and the reduction of IL-17A/F transcript levels in the different models of colitis and activated murine splenocytes, we assessed the effect of TQ on different components of the IL-6 signaling in the human T cell line Jurkat cells. IL-6 signaling is initiated by the binding of IL-6 to the receptor IL-6R34. Treatment of Jurkat cells with TQ significantly reduced the levels of IL-6R (Figs. 8A and 8B). Also, TQ significantly reduced the levels of IL-6R in primary WT mouse-derived CD4+ T cells (Supplementary Figs. 3A and 3B), indicating that this is not a cell-line-specific effect. Furthermore, this effect was unique to TQ as treatment with known AhR agonists 6-Formylindolo-(3,2-b) carbazole (FICZ) (100 nM) or β-naphthoflavone (β-NF) (50 μM) did not affect the levels of IL-6R (Figs. 8C and 8D). We next investigated the effect of TQ on the downstream signaling cascade of the IL-6R. IL-6-IL-6R-mediated activation of the Jak1-Stat-3 pathway is a characteristic hallmark of Th17 differentiation, which upon activation promotes the transcription of RORγt, the signature transcription factor of Th17 cell lineage, and locks in the fate of the naive CD4+ T cell35. Having observed the reduction of IL-6R levels by TQ, and our data showing inhibition of Stat-3 activation by TQ in the macrophages, we also investigated its effect on Stat-3 activation in Jurkat cells. IL-6 treatment of Stat-3-luciferase reporter-transfected Jurkat cells significantly increased the luciferase activity, which was inhibited when combined with TQ. Additionally, only TQ but not FICZ or β-NF inhibited the IL-6-induced increase in Stat-3-luciferase activity (Fig. 8E), further demonstrating the uniqueness of TQ in regulating Th17 development. Taken together, our data highlight the distinctive effect of TQ-mediated AhR activation in blocking the IL-6-IL-6R signaling axis in T cells.
Fig. 8.
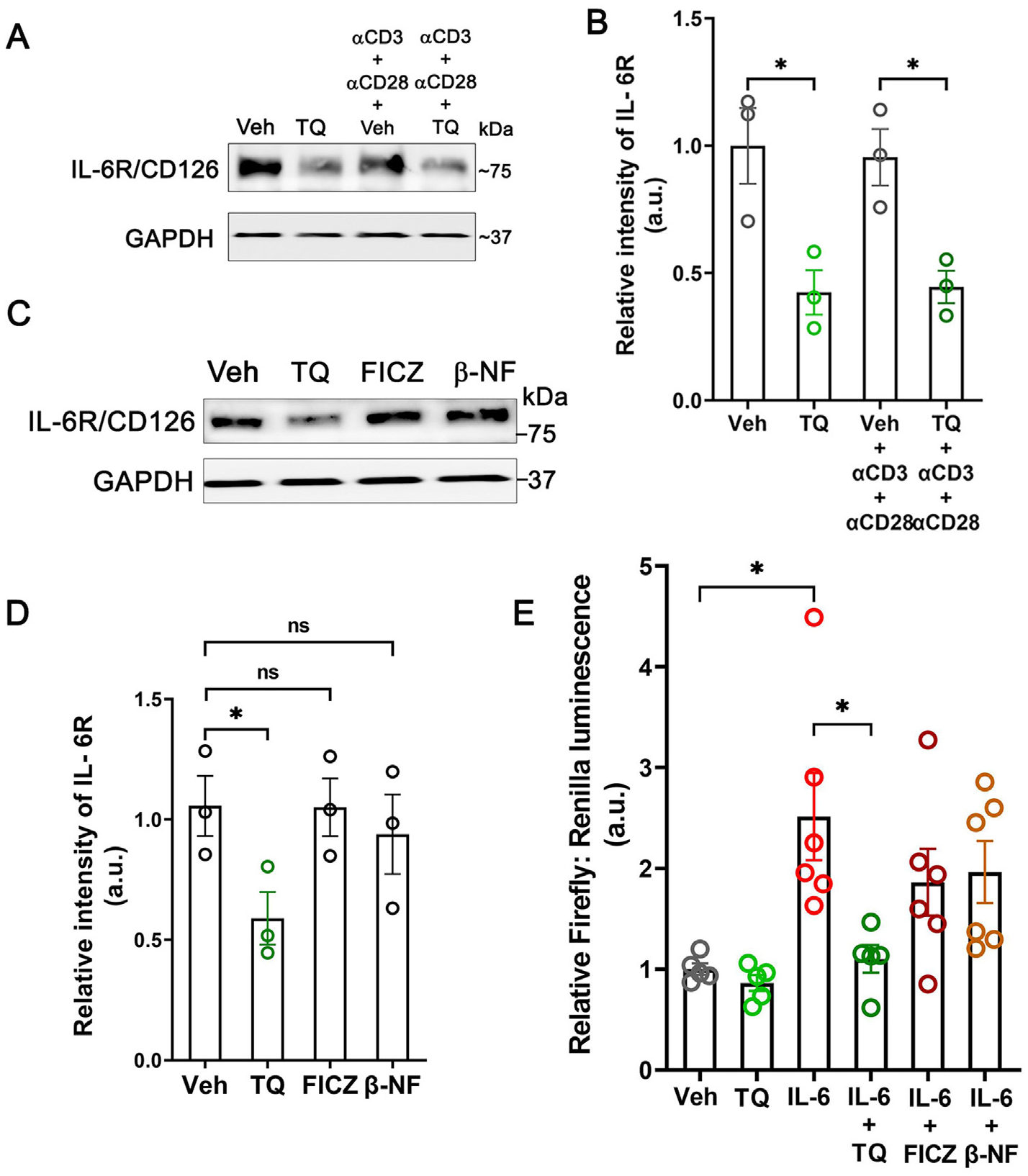
TQ reduces signaling along the IL-6-IL-6R axis in T cells. (A) In the human T cell line, Jurkat cells, TQ reduced the IL-6Rα/CD126 protein levels. The GAPDH levels are shown as the loading control. Representation of 3 blots. (B) Densitometric quantification of IL-6Rα/CD126 protein bands normalized to GAPDH from (A). (C) TQ, but not other AhR agonists FICZ and β-NF, reduced the levels of IL-6Rα/CD126 in Jurkat cells. The GAPDH levels are shown as the loading control. Representation of 3 blots. (D) Densitometric quantification of IL-6Rα/CD126 protein bands normalized to GAPDH from (C). (E) TQ reduced the IL-6-induced upregulation of luciferase activity in Jurkat cells where the luciferase reporter was placed downstream of the Stat-3 dimer binding site. The AhR agonists FICZ and β-NF did not affect the IL-6-induced luciferase activity. Representation of 2 independent experiments with 3 technical replicates each. Data represented as mean ± SEM. One-way analysis of variance with Tukey’s post test. *p < 0.05. ns = not significant.
TQ uniquely activates AhR and reduces Th17 differentiation in vitro
In the intestinal mucosa, CD4+ T lymphocytes are key regulators of immune homeostasis through the production of pro and anti-inflammatory cytokines. De-regulation of this homeostatic state plays a central role both in the induction and in the persistence of chronic inflammation in IBD. Among the different CD4+ T cell differentiated subsets, the Th17 cells have been of particular interest in IBD36. While multiple studies have pointed to the protective role of the IL-17A/F cytokines by the Th17 and γδ T cells in infection and tissue healing, uncontrolled and persistent Th17 responses are implicated in intestinal inflammation and IBD development37,38. Based on the uniqueness of TQ in regulating the IL-6-IL-6R signaling axis, we next investigated whether TQ can regulate the differentiation of naive CD4+ T cells to the Th17 lineage. For this, lymphocytes from the thymus and spleen of WT mice were isolated, and naive CD4+ T cells were sorted as CD4+, CD25−, CD44lo/−, and CD62Lhi/+ cells (Supplementary Fig. 4). The isolated cells were then allowed to differentiate in Th17 differentiation media in the presence of α-CD3 and α-CD28 antibodies in the presence or absence of TQ. We observed successful differentiation of naive T cells into the effector Th17 cell type in the presence of the α-CD3 and α-CD28 antibodies, as evidenced by their expression of CD44, CD28, and increased production of IL-17A/F. However, in combination with TQ, the naive T cells did differentiate to effector CD4+ T cells, as evidenced by high CD44 and CD28 expression, but they produced a significantly lower amount of IL-17A. Furthermore, in the absence of α-CD3 and α-CD28 antibodies, no differentiation was observed, indicating the specificity of the differentiation. To ascertain the involvement of AhR in the process, we next assessed the role of TQ in the differentiation of naive CD4+ isolated from Ahr−/− mice to Th17 cells. Incubation of naive CD4+ T cells in the Th17 differentiation media promoted Th17 differentiation; however, TQ treatment had no effect in inhibiting the production of IL-17A in these Ahr−/− mouse-derived cells (Figs. 9A and 9B and Supplementary Figs. 5 and 6).
Fig. 9.
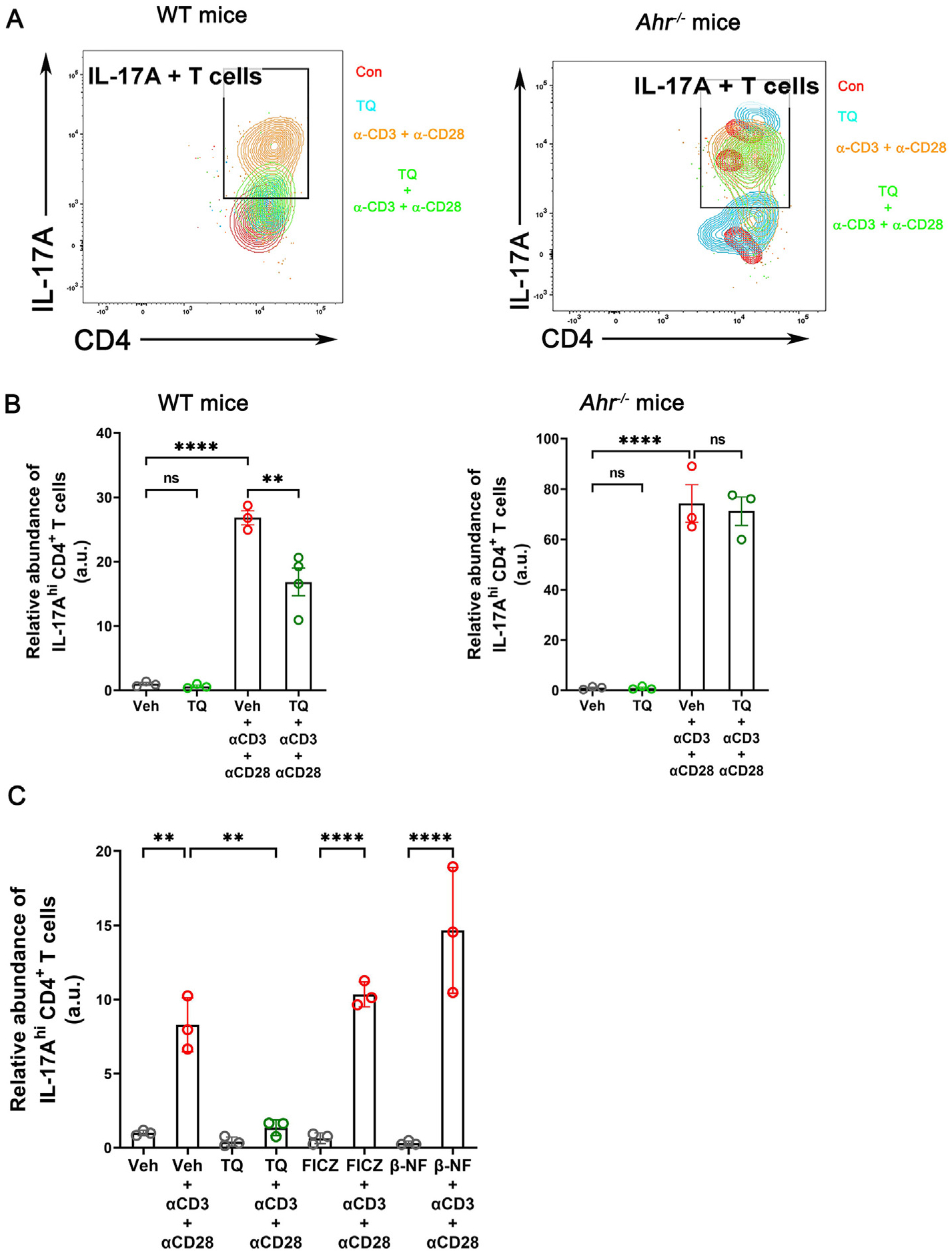
TQ occludes the differentiation of Th17 cells. (A) Flow panel showing the distribution of IL-17hi CD4+ Th17 populations differentiated from naive CD4+ T cells isolated from WT and Ahr−/− mice in Th17 cell differentiating conditions in vitro. (B) Quantification of WT and Ahr−/− naive CD4+ T cells differentiated Th17 cells under each test condition. (C) TQ but not the other AhR agonists FICZ and β-NF reduced the in vitro differentiation of WT mouse-derived naive CD4+ T cells to Th17 cells. Flow cytometry quantification data are represented as the ratio of the absolute number of IL-17A-enriched CD4+ T cells to the total number of CD4+ T cells, normalized to the average values of vehicle-treated control (Veh) without α-CD3 and α-CD28. Data represented as mean ± SEM, n = 3/test group. One-way analysis of variance with Tukey’s post test. **p < 0.01, ****p < 0.0001, ns = not significant.
Previous studies have highlighted the role of AhR activation by known agonists like FICZ and β-NF in promoting the differentiation of naive CD4+ T cells into the Th17 lineage39,40. Since we observed an opposite phenomenon with TQ and established the requirement of AhR for the activity of TQ, we investigated the specificity of TQ in regulating the differentiation of naive T cells to Th17 cells. For this, naive CD4+ T cells from WT mice were allowed to differentiate in the presence or absence of FICZ, β-NF, or TQ. We observed successful differentiation of naive T cells into the effector Th17 cell type in the presence of the α-CD3 and α-CD28 antibodies, as evidenced by their expression of CD44 and CD28 and increased production of IL-17A. As expected, IL-17A responses were heightened by FICZ and β-NF in the presence of CD3 and CD28 antibodies. In the case of TQ, however, while it did not impede the differentiation of the naive T cell to the effector cell type, it specifically impeded the production of IL-17A by these effector CD4+ T cells (Fig. 9C). Taken together, our data show that TQ uniquely inhibits the differentiation of Th17 cells in an AhR-dependent process.
TQ reduces inflammation in human tissues
To demonstrate the clinical relevance of our findings in the in vitro and mouse studies, we next assessed the effect of TQ on human peripheral blood mononuclear cells (PBMCs). For this, we initially incubated PBMCs obtained from healthy human subjects with LPS or PMA + ionomycin in the presence of vehicle or TQ. We observed that activation by both LPS and PMA+ ionomycin significantly increased the mRNA abundance of IL-6, IL-23, and TNF-α, which was prevented by TQ (Fig. 10A). Also, we observed no cytotoxic effect on human PBMCs treated with TQ (Fig. 10B). Next, we examined the effect of TQ on inflammatory macrophages and Th17 responses, specifically in the human colon. For this, surgically resected paired diseased and adjoining normal human colonic tissues (0.5 cm2) from CD patients were incubated with vehicle or TQ for 18 hours. TQ treatment significantly reduced the abundance of inflammatory macrophages and Th17 cells compared to vehicle treatment in both normal tissue (NT) and CD tissues without reducing the overall abundance of macrophages or CD4+ T cells (Figs. 10C and 10D). Taken together, these data indicate the ability of TQ to dampen the inflammatory response in the immune cells in the human gut.
Fig. 10.
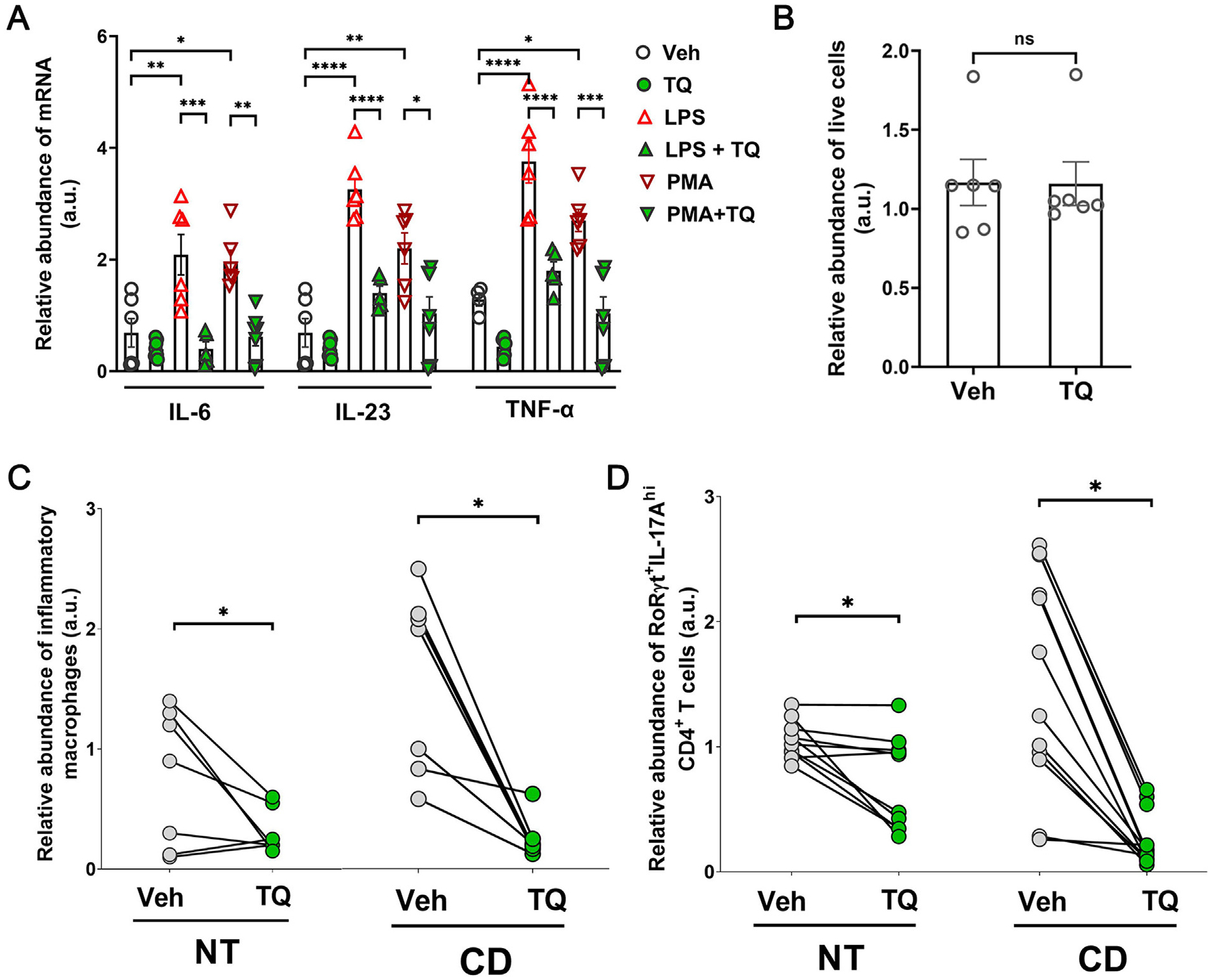
TQ effectively reduces inflammation in human tissues. (A) TQ effectively reduced the upregulation of IL-1β, IL-6, and TNF-α mRNA in human PBMCs when activated with LPS or a cocktail of PMA + ionomycin (PMA). (B) TQ did not affect the cell viability of human PBMCs. (C) In human Crohn’s disease (CD) patient-derived inflamed colonic tissues, TQ treatment reduced the abundance of IL-6, TNF-α, and IL-1β -enriched inflammatory macrophages. Flow cytometry quantification data are represented as the ratio of the absolute number of IL-6, TNF-α, and IL-1β -enriched inflammatory macrophages to the total number of macrophages, pairwise normalized to the average values of three technical replicates for each patient vehicle-treated NT. (D) In human Crohn’s disease (CD) patient-derived inflamed and adjoining normal colonic tissues (NT), TQ treatment reduced the abundance of IL-17A-enriched RORγt+ CD4+ T cells. The flow cytometry quantification data are represented as the ratio of the absolute number of IL-17A-enriched CD4+ T cells to the total number of CD4+ T cells, pairwise normalized to the average values of three technical replicates for each patient vehicle-treated NT. Data represented as mean ± SEM, n ≥ 5/ treatment group. Paired Student’s t-test or one-way analysis of variance with Tukey’s post test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. ns = not significant.
DISCUSSION
It is well established that exaggerated and dysregulated immune response is a key pathogenic factor in IBD, including CD and UC. Dysregulation of both innate and adaptive immune responses is known to contribute to the aberrant intestinal inflammatory response in IBD patients. The cytokines and cytokine-producing immune cells play a crucial role in IBD pathogenesis, as evidenced by the identification of several cytokine and chemokine receptor signaling genes as IBD susceptibility or risk loci41. In this study, we show that TQ acts as a non-cytotoxic immunomodulatory agent that ameliorates colonic inflammation in a mechanism dependent on the AhR. Several studies have established the critical role of AhR in the regulation of the intestinal immune system and immune cell functioning42,43. We show that TQ ameliorated the inflammation-promoting immune responses and reduced the mRNA levels of pro-inflammatory cytokines in the experimental colitis models of acute and chronic DSS administration and T cell transfer-mediated colitis. Furthermore, we also show that TQ reduced the population of inflammatory macrophages, CD4+ T cells, and ILC3 cells enriched in IL-17A/F in the mouse colon in the colitis models. The upregulation of the inflammatory cytokines as a key contributing factor for intestinal inflammation has been established in the different models of colitis44,45. In addition, we also demonstrated the anti-inflammatory role of TQ in murine splenocytes without eliciting cytotoxicity. Therefore, our data suggest that TQ-mediated immune modulation is an important factor in the amelioration of experimental colitis.
As a mechanism for TQ-mediated immunomodulation, we demonstrated that TQ activates AhR signaling based on the nuclear translocation of AhR, XRE reporter assay, and CYP1A1 gene transcription. Beyond the xenobiotic response, AhR has been shown to play several homeostatic roles, including intestinal differentiation, stem cell homeostasis, and maintenance of the gut microbiome46,47. Moreover, AhR activation by various endogenous or exogenous agents has been shown to be protective against experimental colitis via various mechanisms, including inhibition of inflammatory cytokines, induction of regulatory immune responses, microbiome homeostasis, and maintenance of gut barrier integrity9–20. Our data showed that while TQ activated AhR and promoted its nuclear localization, unlike the conventional AhR agonists like FICZ and β-NF, its function was independent of the nuclear translocation of AhR. We showed that TQ prevents the nuclear localization and transcriptional activity of NFκB and the phosphorylation and activation of Stat-3, two key transcription factors involved in the transcription of proinflammatory genes. We also show that the effect of TQ on these two disparately activated pro-inflammatory pathways was dependent on the AhR protein. Pharmacological or genetic inhibition of the AhR completely blocked the effectiveness of TQ in preventing the activation of the NFκB and Stat-3 proteins. This further establishes the AhR dependence of the activity of TQ, pointing to a unified model of the regulation of the NFκB and Stat-3 pathways via the cytoplasmic activity of the activated AhR protein. However, whether this activity is directly mediated by the AhR protein or through intermediaries needs further investigation.
Another key finding of this study is the effect of TQ on the pleiotropic pro-inflammatory cytokine IL-6 and the CD4+ Th17 cells. The inflammatory cytokine IL-6 is an essential component in the immune arsenal to fend off foreign antigens; however, hyperactivation of the IL-6 signaling axis has been implicated in numerous chronic inflammatory diseases, including psoriasis and IBD48. IL-6 signaling in intestinal epithelium also contributes to increased transcription of the pore-forming protein claudin-2, which diminishes the epithelial barrier and is often associated with the leaky gut phenotype associated with IBD49. Furthermore, IL-6 signaling has also been implicated in prolonged T-cell responses by inhibiting T-cell apoptosis, which further contributes to intestinal inflammation in IBD50. In this study, we successfully show that TQ inhibits IL-6 production in mouse intestinal tissue and splenocytes and also by the human macrophage cell line U937 cells. We highlight the role of AhR in regulating this inhibitory effect of TQ and also show the inhibition of the downstream signaling of IL-6 that occurs by Stat-3 activation. We also show the effect of TQ on another key downstream event of IL-6 signaling, that is the development of Th17 cells from naive T cells. The hyperactivation of Th17 cells has been implicated in numerous immune disorders, including IBD51,52. Targeting the Th17 cell response has also been an active area of research for the treatment of IBD; however, the efficacy of said treatment in patient cohorts has not been efficient53,54. Our data show that TQ reduced the mRNA levels of IL-17A, the key cytokine produced by Th17 cells, and also Th17 abundance in the different models of colitis. TQ impeded the differentiation of Th17 cells from naive murine CD4+ T cells in an AhR-dependent manner, as TQ was only able to elicit this response in naive CD4+ T cells isolated from WT but not Ahr−/− mice, which have been shown to have increased susceptibility to experimental colitis13. Moreover, consistent with the ligand and tissue-specific effects of AhR signaling55, we observed that the Th17 suppressive role is unique to TQ-mediated AhR activation. Activation of AhR by other known agonists like FICZ and β-NF was unable to inhibit Th17 development. As an underlying mechanism, we show that TQ uniquely reduced the levels of the IL-6 receptor CD126/IL-6Rα on the human T cell line Jurkat cells and primary murine CD4+ T cells, and as in the case of U937 cells, it also reduced Stat-3 activation and transcriptional activity, two essential factors that contribute to the development of Th17 cells. In this regard, both elevated IL-6R, as well as noncanonical Stat-3 functions, have been implicated in IL-17 production33,56. Besides the above mechanisms, the contradicting regulatory effect of TQ, compared to known AhR agonists such as FICZ and β-NF, on IL-6R and IL-6 expression and the abundance of Th17 cells can be attributed to cell-specific AhR metabolism, coactivators, and corepressors, affinity for AhR binding, and activation of canonical vs. noncanonical AhR pathways. Whereas FICZ and β-NF-dependent AhR activity relies primarily on the nuclear translocation of AhR, TQ-mediated anti-inflammatory effects were dependent on AhR activation and a resultant inhibition of NFκB and Stat-3 signaling, which was accomplished by neither FICZ nor β-NF. The overall reduction of IL-17A production in the presence of TQ may occur due to the reduction of Stat-3 signaling. Stat-3 activation by the IL-6-IL-6R axis is known to promote the transcription of RORγt, the hallmark transcription factor for Th17 cells. The RORγt protein together with active Stat-3 associates with the transcription machinery and binds to conserved non-coding sequence 2 (CNS2) cis-regulatory elements upstream of the IL-17A gene locus to promote the transcription of the cytokine57. We recognize that further investigations are needed into these factors to clarify the differential effect of TQ compared to known AhR ligands. Overall, our data show that TQ employs a unified mode of action, operating via AhR and regulating the pro-inflammatory IL-6, NFκB, and Stat-3 pathways in different cell types, thereby exerting anti-inflammatory responses and protecting against intestinal inflammation.
In addition to the murine and cell line studies, we also showcase the efficacy of TQ on human blood-derived and human colonic tissue-derived immune cells. We show that TQ successfully reduced the mRNA abundance of IL-6, IL-23, and TNF-α by human PBMCs without any cytotoxicity. Finally, we demonstrated the efficacy of TQ in reducing the abundance of pro-inflammatory TNF-α, IL-1β, and IL-6 -enriched inflammatory macrophages and Th17 cells in colonic tissues derived from CD patients, indicating the potential to use TQ in the treatment of IBD.
In summary, we demonstrated the unique immunomodulatory ability of TQ in experimental colitis as well as in healthy and diseased human colonic mucosa. We provided pharmacological and genetic evidence for AhR signaling as an underlying mechanism through which TQ exerts its immune-modulatory effect. Very complex ligand and tissue-specific effects of AhR activation, toxicity considerations, and inadequate potency have precluded the application of AhR modulators against IBD58. Nevertheless, due to the ligand-dependent modulation of AhR activity, an effective, non-toxic AhR modulator is of great scientific interest8. Although only transient prolongation of clotting time without any adverse events was noted in a clinical study59, the anti-coagulative activity of TQ must be considered in its clinical use. Our results indicate that TQ may offer a naturally occurring, non-toxic intervention for the regulation of a broad spectrum of intestinal immune responses and therapeutic for inflammatory conditions.
METHODS
Chemicals and antibodies
D-alpha-tocopheryl quinone (TQ) (T2283) was obtained from TCI (Portland, OR, USA). The primary antibodies used included AhR (MA1–514) and CYP1A1 (PA5–15212) from Life Technologies (Waltham, MA, USA); p65 (AbCam, Boston, MA, USA, AB532536); IkBα (10268-AP), IL-6R (23457–1-AP), Stat-3 (10253–2-AP), and CYP1A1 (13241–1-AP) from ProteinTech (Rosemont, IL, USA); and phospho-Stat-3 (9145) and GAPDH (2118) from Cell Signaling Technologies, Danvers, MA, USA. The anti-rabbit HRP (AB97051) and anti-mouse HRP (AB205719) secondary antibodies were purchased from AbCam (Boston, MA, USA). For confocal-immunofluorescence microscopy, AF488-tagged anti-rabbit antibody (A11008) and Cy3-tagged anti-mouse antibody (APC124C) were purchased from Invitrogen and Sigma-Aldrich, respectively. AhR activators FICZ (SML-1489) and βNF (N3633), Lipopolysaccharide [LPS O127:B8 (L3129)], PMA (79346), and ionomycin (I3909) were purchased from Sigma-Aldrich (St. Louis, MO). Brefeldin A solution 1000X (420601) and all antibodies used for flow cytometry and fluorescence-assisted cell sorting (FACS) sorting were purchased from BioLegend, San Francisco, CA, USA. TaqMan-tagged primers for assaying the expression of all the target genes by qRT-PCR were purchased from Life Technologies, Carlsbad, CA, USA. The AhR inhibitors SGA360 (AOB1971), GNF-531 (182707), and CH223191 (C8124) were obtained from Aobious (Gloucester, MA, USA), EMD Millipore (Burlington, MA, USA), and Sigma-Aldrich, respectively. The untagged cytokines used for T-cell differentiation were purchased from ACROBiosystems (Newark, DE, USA).
Cell culture and in vitro differentiation
Human U937 cells (CRL-1593.2, ATCC) and Jurkat cells (TIB-152, ATCC) were maintained in RPMI-medium (Corning, 10–040CV) supplemented with 10% heat-inactivated FBS (R&D Systems, Minneapolis, MN, USA, S11150H) and antibiotics in a 37 °C incubator with 5% CO2. The U937 cells were activated with 10 ng/ml LPS or a cocktail of 1 μg/ml each of PMA and ionomycin. Mouse T cells were maintained in T cell media (RPMI-1640 + 10% heat-inactivated FBS + antibiotics + 50 μM 2-mercaptoethanol). Jurkat and mouse spleen-derived CD4+ T cells were activated by incubation with 1 μg/ml anti-CD3 antibody (α-CD3) and 1 μg/ml anti-CD28 antibody (α-CD28). Mouse naive CD4+ T cells were isolated and differentiated into Th17 cells by culturing naive cells in T cell media supplemented with 20 ng/ml IL-6, 3 ng/ml tumor growth factor (TGF)-β, 5 ng/ml IL-2, 10 ng/ml IL-23, 5 μg/ml anti-IL-4 antibody (α-IL-4) and 5 μg/ml anti-IFN-γ antibody (α-IFN-γ) in the presence of 2 μg/ml α-CD3 and 1 μg/ml α-CD28.
Isolation of lymphocytes from the murine lymph nodes and spleen
WT and Ahr−/− C57BL.6 mice were anesthetized using 5% isoflurane, and their spleen and mesenteric lymph nodes were harvested in ice-cold PBS supplemented with 2% heat-inactivated FBS. The spleen and lymph nodes were pulverized by gentle rubbing between two frosted slides (Fisher Scientific, 22–034486). Spleen samples were then incubated in 1× RBC lysis buffer (100 mM NH4Cl + 10 mM NaHCO3 + 1 mM EDTA) for 5 minutes at room temperature, followed by the addition of enriched RPMI-1640 medium to stop lysis and centrifugation to remove lysed RBCs. The lymph and spleen cells were then subjected to 40%–80% gradient percoll centrifugation, and the lymphocyte band was isolated for further experimentation. The cells were maintained in RPMI-1640 medium supplemented with 10% FBS and antibiotics.
Cell treatment
TQ treatment of cells in culture was performed as described previously60. Briefly, cells in culture were treated with TQ using bovine serum albumin (BSA) as a carrier for TQ, dissolved first in ethanol, and subsequently added to a 7.5% BSA solution (Sigma-Aldrich, A8412) at 37 °C with mixing, producing a 20-fold concentrated stock. This solution was then added to the standard growth medium at a 5% concentration (i.e. a final con centration of 0.4% BSA) to produce a 25 μM concentration of TQ in the culture medium.
Western blot analysis
Cells were rinsed twice with ice-cold PBS (Corning, Glendale, AZ, 21–040) and lysed using RIPA buffer (Sigma-Aldrich, R0278) containing protease inhibitors (Sigma-Aldrich, 11836170001). The cell lysates were centrifuged at 12,000 rpm for 5 minutes, and the clear supernatant was used for protein quantification. Protein quantification of the extracted aliquots was performed (BCA protein assay kit, Thermo Scientific 23225), and Laemmli gel loading buffer (Invitrogen, Waltham, MA, USA, NP007) was added to the lysate and boiled at 95 °C for 5 minutes. An equal amount of protein was loaded on a 4%–15% SDS-PAGE gel (BioRad, Hercules, CA, USA, 4561086) and transferred to a nitro-cellulose membrane (BioRad, 1620112). The membrane was incubated for 1 hour in the blocking solution (5% non-fat dry milk in TBS-Tween 20 buffer), followed by overnight incubation with the appropriate primary antibody in the blocking solution. After incubation with the primary antibody, the membrane was washed in TBS-0.1% Tween 20 buffer, incubated in the appropriate secondary antibody, and developed using the SuperSignal West Pico PLUS kit (Thermo Scientific, 34580). The densitometry analysis was performed using ImageJ software (NIH, USA).
Quantitative Real-Time PCR
Cells and tissues were suspended in TRIZol (Invitrogen, 15596026), followed by RNA isolation using the Direct-zol RNA Miniprep Plus kit (Zymo Research, Irvine, CA, R2072) by following the manufacturer’s instructions. Transcript levels of the different markers were normalized to GAPDH transcript levels.
Flow cytometry and FACS sorting
Flow cytometry was performed by modifying the previously described protocol44. Briefly, cells were washed with FACS buffer (RPMI-1640 + 1% FBS + 1X DPBS + 5 mM EDTA), followed by staining with fixable viability Dye-eFluor780 (Thermo Scientific, 65-0865-18) for 10 minutes at 4 °C and Fc blocking (Thermo Scientific, 16-0161-82 for mice and BioLegend 422302 for humans) for 10 minutes at room temperatures. To assess intracellular cytokine marker staining, cells were treated with brefeldin A (5 μg/ml) for the last 4 hours of incubation before prepping cells for flow cytometry. Cells were permeabilized using 1× Permeabilization buffer (Thermo Scientific, 00-8333-56) at 37 °C for 30 minutes, followed by staining with cognate fluorescently tagged antibodies (BioLegend). Cells were then either sorted using a FACSAria cell sorter (BD Biosciences, Franklin Lakes, NJ) or were analyzed using an LSR Fortessa flow cytometer (BD Biosciences) using appropriate compensations, isotype, and FMO controls. Flow data were analyzed using Flow Jo V10 software (BD Biosciences). For the subtyping of the murine intestine and spleen-derived immune cells, inflammatory macrophages were identified as the live CD45+, MHC-II+, F4/80+, CD64+, and CXC3CR1+ with TNF-αhi, and IL-6 hi intracellular staining; Th17 cells were identified as the live CD45+, CD4+, CD28+, RORγt+, CCR6+, and CD25− cells with IL-17A and IL-17Fhi intracellular staining; and ILC3 cells were identified as the live CD45+, CD4−, RORγt+, and CCR6+ cells with IL-17A and IL-17Fhi intracellular staining. The gating strategy for each is shown in Supplementary Figs. 7A (macrophages), 7B (Th17 cells), and 7C (ILC3).
Lentiviral-mediated CRISPR/Cas9 knockout of AhR
The plasmid Cas9 nuclease CP-LVC9NU (Genecopoeia, Rockville MD, USA) was used individually with a single guide RNA (sgRNA) targeting the region 5’-CAAGTCGGTCTCTATGCCGCT-3’ of the AHR gene, or scrambled sgRNA for control in pCRISPR- LVSG03 (Genecopoeia), to generate respective lentiviral particles packaged in Lenti-X 293T cells using PMD2.G (12259) and pPAX2 (12260) (Addgene, Watertown MA). The lentiviral particles obtained were used to transduce U937 cells in the presence of polybrene (Sigma-Aldrich, TR-1003) and were selected in their respective antibiotic selection media to generate stable knockout U937 cells. AHR knockout was confirmed using Western blot analysis.
Transient transfection and reporter gene assay
U937 and Jurkat cells were cultured in 12-well tissue culture plates for 24 hours before transfection at 60%–80% confluency. For the measurement of either NFκB or Stat-3 activity in the presence and absence of TQ, the cells were transiently co-transfected with 1 mg/well pLminP_Luc2P_RE1 (NFκB reporter, Addgene 90335) or pLminP_Luc2P_RE5 plasmid (Stat-3 reporter, Addgene 90339) expressing plasmid and 0.1 mg/well Renilla luciferase (hRluc) reporting plasmid (pGL4.74) using Lipofectamine 2000 (Invitrogen, 11668019). After 24 hours of transfection, cells were treated with LPS (for U937 cells) or IL-6 (for Jurkat cells) in the presence or absence of TQ for 48 h, after which the cells were lysed and assayed for luciferase activity using Dual-Luciferase Reporter Assay system (Promega, E1960) on the GloMax 20/20 Luminometer (Promega, Madison, WI, USA). The activity of firefly luciferase was normalized against Renilla reporter activity (control) in the same samples.
Confocal-Immunofluorescence imaging
Confocal Immunofluorescence for AhR and NFκB was performed by standard methods. Cells were washed twice with cold PBS and fixed with 2% paraformaldehyde for 20 minutes. The cells were permeabilized with 0.1% Triton X-100 in PBS at room temperature for 5 minutes. The cells were blocked in normal serum and labeled with primary antibodies in a blocking solution overnight at 4 °C. After PBS washes, the cells were incubated in Alexa Fluor-488 or Cy-3 conjugated secondary antibodies (Invitrogen). ProLong Gold antifade reagent (Invitrogen, P36931) containing DAPI as a nuclear stain was used to mount the sections on glass slides. The slides were examined using a confocal fluorescence microscope, Leica SP8. Images were processed with LAS X software (Leica Microsystems).
Experimental animals
Experimental methodologies used in the study were approved by the Institutional Animal Care and Use Committee of the Pennsylvania State University College of Medicine (approval number 201800202). The mice were housed under standard housing conditions with a 12-h light-dark cycle and ad lib standard chow diet and water supply. The WT (Jackson Laboratories, Bar Harbor, ME, Strain #000664) and Ahr−/− mice with C57BL/6J background of both sexes were randomly allotted to the experimental groups. The experimental murine DSS colitis (acute and chronic DSS) models were established using standard protocols and as described previously44,61,62. In the acute colitis model, 8–10-week-old C57BL/6J mice received 2.5% DSS dissolved in auto-claved water for 7 days. TQ treatment (50 mg/kg/day) was initiated two days prior and continued throughout DSS treatment. In the chronic DSS colitis model, 8–10-week-old C57BL/6J mice received three cycles of 5 days of 1.5% DSS in drinking water followed by 5 days of plain water. Mice were treated with TQ in the last cycle of water and DSS. For T cell transfer colitis, FACS was performed to isolate donor T cells from WT mice. Briefly, the mice’s spleen was harvested and ground in between the rough edges of two frosted slides. The resultant cell slurry was incubated for 5 minutes at room temperature in RBC lysis solution, followed by a wash with RPMI-1640 complete media (RPMI-1640 + 10% FBS). Cells obtained from the lysis step were subjected to an 80%–40% Percoll (Sigma-Aldrich) gradient centrifugation to enrich the leukocyte population and staining for flow cytometry. Effector and memory T cells were sorted as the CD45RB-FITC+ and CD4-APC+ cells. Cells were sorted using a BD-Aria Cell sorter (BD Biosciences, San Jose, CA, USA). Flow cytometry data were analyzed using FlowJo version 10 software (BD Biosciences). Approximately 0.5 × 106 cells were then intraperitoneally injected into recipient Rag−/− mice (Jackson Laboratory, Strain #002216). Isolated T cells from male donor mice were injected into male recipient mice, and cells from female donor mice were administered into female recipient mice. Mice were weighed twice per week and monitored for disease onset for 5 weeks post-injection. Upon the appearance of diarrhea-like symptoms at week 5 postinjection, mice were separated into two groups, with one being gavaged vehicle and the other being gavaged TQ for 7 days. The groups’ allocation, treatments, and outcomes were measured in a blinded fashion. The experimental colitis data represent a minimum of three independent experiments with n ≥ 3 mice per group.
Human samples and treatment
The surgically resected human colon samples were obtained freshly from the Department of Surgery, Division of Colon and Rectal Surgery, as per the protocols approved by the Institutional Review Board (STUDY00010256). Samples were deemed normal or diseased by Anatomic Pathologists. The colonic tissues were washed and incubated in supplemented RPMI-1640 for 18 hours63 in the presence of vehicle or TQ. Tissues were then processed for flow cytometry. For human blood samples, healthy volunteer blood PBMCs were isolated by incubating the samples in 1X RBC lysis solution for 5 minutes at room temperature, followed by percoll gradient centrifugation to obtain PBMCs. Cells were then incubated overnight in supplemented RPMI-1640 culture medium with 10 ng/ml LPS or a cocktail of 1 μg/ml each PMA + ionomycin with or without 25 μM TQ. Cells were then used for RNA isolation and quantitative RT-PCR. Similar to the murine cells, for the human colonic biopsy-derived immune cells, the inflammatory macrophages were identified as the live CD45+, HLA-DR+, CD14+, CD80 +, CD 86+, and CD16+ cells with TNF-α hi, IL-1βhi, and IL-6 hi intracellular staining, and Th17 cells were identified as the live CD45+, CD4+, CD28+, RORγt+, and CD25− cells with IL-17Ahi intracellular staining.
Statistical analysis
Data are reported as means ± SEM. Whenever needed, data were analyzed by using the Student’s t-test or analysis of variance for repeated measures (Sigma Stat, Systat Software, San Jose, CA, USA). Tukey’s test was used for post hoc analysis between treatments following analysis of variance (p < 0.05).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the IBD and Colorectal Diseases Biobank, Flow Cytometry (RRID: SCR_021134), Confocal Microscopy (RRID: SCR_022526), and Animal Facility Cores at the Penn State College of Medicine for their excellent technical assistance.
FUNDING
This research work was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases grant DK100562 (P. N.), the National Institute of Environmental Health Sciences R35ES028244 (G. P.), the Crohn’s & Colitis Foundation Award 694583 (M. N.), and Penn State College of Medicine, Department of Medicine funds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors also acknowledge support from the Peter and Marshia Carlino Fund for IBD Research.
Footnotes
DECLARATION OF COMPETING INTEREST
The authors have no competing interests to declare.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mucimm.2023.09.003.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
REFERENCES
- 1.Netea MG et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol 20, 375–388 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun M, He C, Cong Y & Liu Z Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol 8, 969–978 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo Y & Zheng SG Hall of fame among pro-inflammatory cytokines: interleukin-6 gene and its transcriptional regulation mechanisms. Front. Immunol 7, 604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinrich PC et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J 374, 1–20 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yosef N et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harbour SN, Maynard CL, Zindl CL, Schoeb TR & Weaver CT Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl Acad. Sci. U. S. A 112, 7061–7066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zielinski CE et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484, 514–518 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Mulero-Navarro S & Fernandez-Salguero PM New trends in aryl hydrocarbon receptor biology. Front. Cell Dev. Biol 4, 45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benson JM & Shepherd DM Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol. Sci 120, 68–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goettel JA et al. AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep 17, 1318–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monteleone I et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 141, 237–248.e1 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Singh R et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat. Commun 10, 89 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furumatsu K et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig. Dis. Sci 56, 2532–2544 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Riemschneider S et al. Indol-3-carbinol and quercetin ameliorate chronic DSS-induced colitis in C57BL/6 mice by AhR-mediated anti-inflammatory mechanisms. Int. J. Environ. Res. Public Health 18, 2262 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Y et al. 6-Formylindolo(3,2-b)carbazole induced aryl hydrocarbon receptor activation prevents intestinal barrier dysfunction through regulation of claudin-2 expression. Chem. Biol. Interact 288, 83–90 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Abron JD et al. An endogenous aryl hydrocarbon receptor ligand, ITE, induces regulatory T cells and ameliorates experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol 315, G220–G230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu M et al. Aryl hydrocarbon receptor activation modulates intestinal epithelial barrier function by maintaining tight junction integrity. Int. J. Biol. Sci 14, 69–77 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubbard TD et al. Dietary Broccoli Impacts Microbial Community Structure and Attenuates Chemically Induced Colitis in Mice in an Ah receptor dependent manner. J. Funct. Foods 37, 685–698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Islam J et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J. Nutr. Biochem 42, 43–50 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Ji T et al. Aryl hydrocarbon receptor activation down-regulates IL-7 and reduces inflammation in a mouse model of DSS-induced colitis. Dig. Dis. Sci 60, 1958–1966 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Qiu J et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36, 92–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockwell BR et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niki E Tocopherylquinone and tocopherylhydroquinone. Redox Rep 12, 204–210 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Palan PR, Woodall AL, Anderson PS & Mikhail MS Alpha-tocopherol and alpha-tocopheryl quinone levels in cervical intraepithelial neoplasia and cervical cancer. Am. J. Obstet. Gynecol 190, 1407–1410 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Ganapathy AS et al. Alpha-tocopherylquinone differentially modulates claudins to enhance intestinal epithelial tight junction barrier via AhR and Nrf2 pathways. Cell Rep 42:112705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearson C et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. eLife 5, e10066 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang ZZ et al. In vivo effects of the pure aryl hydrocarbon receptor antagonist GNF-351 after oral administration are limited to the gastrointestinal tract. Br. J. Pharmacol 171, 1735–1746 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray IA et al. Development of a selective modulator of aryl hydrocarbon (Ah) receptor activity that exhibits anti-inflammatory properties. Chem. Res. Toxicol 23, 955–966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao B, Degroot DE, Hayashi A, He G & Denison MS CH223191 is a ligand-selective antagonist of the Ah (dioxin) receptor. Toxicol. Sci 117, 393–403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grishanova AY & Perepechaeva ML Aryl hydrocarbon receptor in oxidative stress as a double agent and its biological and therapeutic significance. Int. J. Mol. Sci 23, 6719 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Zhang L, Joo D & Sun SC NF-κB signaling in inflammation. Signal Transduct. Target. Ther 2, 17023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan Y, Mao R & Yang J NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 4, 176–185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saini C et al. Elevated IL-6R on CD4+ T cells promotes IL-6 driven Th17 cell responses in patients with T1R leprosy reactions. Sci. Rep 10, 15143 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taga T & Kishimoto T Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol 15, 797–819 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Ivanov II et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Weaver CT, Elson CO, Fouser LA & Kolls JK The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu. Rev. Pathol 8, 477–512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujino S et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberg HH, Wesch D, Grüssel S, Rose-John S & Kabelitz D Differential expression of CD126 and CD130 mediates different STAT-3 phosphorylation in CD4+CD25- and CD25high regulatory T cells. Int. Immunol 18, 555–563 (2006). 10.1093/intimm/dxh396. [DOI] [PubMed] [Google Scholar]
- 39.Ho PP & Steinman L The aryl hydrocarbon receptor: a regulator of Th17 and Treg cell development in disease. Cell Res 18, 605–608 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Mohinta S et al. Differential regulation of Th17 and T regulatory cell differentiation by aryl hydrocarbon receptor dependent xenobiotic response element dependent and independent pathways. Toxicol. Sci 145, 233–243 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neurath MF Cytokines in inflammatory bowel disease. Nat. Rev. Immunol 14, 329–342 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Gutiérrez-Vázquez C & Quintana FJ Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 48, 19–33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamas B, Natividad JM & Sokol H Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol 11, 1024–1038 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Nighot M et al. Matrix metalloproteinase MMP-12 promotes macrophage transmigration across intestinal epithelial tight junctions and increases severity of experimental colitis. J. Crohns Colitis 15, 1751–1765 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nighot P et al. Chloride channel ClC-2 is a key factor in the development of DSS-induced murine colitis. Inflamm. Bowel Dis 19, 2867–2877 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metidji A et al. The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 50, 1542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang W et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun 11, 4457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones BE, Maerz MD & Buckner JH IL-6: a cytokine at the crossroads of autoimmunity. Curr. Opin. Immunol 55, 9–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki T, Yoshinaga N & Tanabe S Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J. Biol. Chem 286, 31263–31271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atreya R et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat. Med 6, 583–588 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Choi G et al. A critical role for Th17 cell-derived TGF-β1 in regulating the stability and pathogenicity of autoimmune Th17 cells. Exp. Mol. Med 53, 993–1004 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hou G & Bishu S Th17 cells in inflammatory bowel disease: an update for the clinician. Inflamm. Bowel Dis 26, 653–661 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fauny M et al. Paradoxical gastrointestinal effects of interleukin-17 blockers. Ann. Rheum. Dis 79, 1132–1138 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Burisch J et al. Risk for development of inflammatory bowel disease under inhibition of interleukin 17: a systematic review and meta-analysis. PLoS One 15, e0233781 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanieh H Toward understanding the role of aryl hydrocarbon receptor in the immune system: current progress and future trends. BioMed Res. Int 2014:520763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poholek CH et al. Noncanonical STAT3 activity sustains pathogenic Th17 proliferation and cytokine response to antigen. J. Exp. Med 217, e20191761 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X et al. Transcription of Il17 and Il17f is controlled by conserved noncoding sequence 2. Immunity 36, 23–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stockinger B, Shah K & Wincent E AHR in the intestinal microenvironment: safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol 18, 559–570 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lynch DR et al. A0001 in Friedreich ataxia: biochemical characterization and effects in a clinical trial. Mov. Disord 27, 1026–1033 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Fajardo AM et al. Antioxidants abrogate alpha-tocopherylquinone-mediated down-regulation of the androgen receptor in androgen-responsive prostate cancer cells. PLoS One 11, e0151525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wirtz S, Neufert C, Weigmann B & Neurath MF Chemically induced mouse models of intestinal inflammation. Nat. Protoc 2, 541–546 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Nighot P et al. Matrix metalloproteinase 9-induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am. J. Physiol. Gastrointest. Liver Physiol 309, G988–G997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saha K et al. Autophagy reduces the degradation and promotes membrane localization of occludin to enhance the intestinal epithelial tight junction barrier against paracellular macromolecule flux. J. Crohns Colitis 17, 433–449 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.