

SUMMARY
The phosphoinositide 3-kinase p110α is an essential mediator of insulin signaling and glucose homeostasis. We interrogated the human serine, threonine, and tyrosine kinome to search for novel regulators of p110α and found that the Hippo kinases phosphorylate p110α at T1061, which inhibits its activity. This inhibitory state corresponds to a conformational change of a membrane-binding domain on p110α, which impairs its ability to engage membranes. In human primary hepatocytes, cancer cell lines, and rodent tissues, activation of the Hippo kinases MST1/2 using forskolin or epinephrine is associated with phosphorylation of T1061 and inhibition of p110α, impairment of downstream insulin signaling, and suppression of glycolysis and glycogen synthesis. These changes are abrogated when MST1/2 are genetically deleted or inhibited with small molecules or if the T1061 is mutated to alanine. Our study defines an inhibitory pathway of PI3K signaling and a link between epinephrine and insulin signaling.
Graphical abstract
In brief
Lin et al. describe the finding that p110α, the catalytic subunit of PI3Kα, can be directly inhibited via phosphorylation by MST1, MST2, and other members of the Hippo kinase family. Stimulation of MST1/2 with epinephrine, forskolin, or fasting impedes hepatic PI3K activity and restricts glycogen synthesis.
INTRODUCTION
The binding of insulin to the insulin receptor initiates a surge of tyrosine phosphorylations that promote membrane recruitment of phosphoinositide 3-kinase (PI3Kα), an intracellular lipid kinase.1–3 PI3Kα is composed of a catalytic subunit, p110α, which functions as an obligate heterodimer with the regulatory subunit p85α. Binding of the regulatory subunit to phosphotyrosines relieves its inhibitory contacts on p110α and localizes the complex to the plasma membrane,4 where p110α then engages the lipid bilayer and phosphorylates the D3 positions of the resident phosphoinositide lipids to set off signaling cascades that guide the cellular actions of insulin, including glucose uptake, enhanced glycolysis, and glycogen synthesis.5,6
The glycemic actions of insulin and PI3Kα are opposed by counter-regulatory hormones like glucagon and epinephrine (Epi). Epi is released from the adrenal gland in response to stresses like hypoglycemia. It then binds to β-adrenergic receptors (β-ARs) in the liver to stimulate glycogenolysis and impair the actions of insulin.7–9 The molecular pathways by which Epi acutely blocks insulin action are incompletely understood.8–12
Most of what is known about the regulation of p110α has come from experimental work with accessible cellular model systems, using indirect readouts as a surrogate for PI3Kα activation. We have taken a more direct approach and asked if the production of phosphatidylinositol 3,4,5-triphosphate (PIP3), the product of p110α, can be regulated through phosphorylation by a broad sample of the human protein kinome. This approach has unveiled an inhibitory pathway of PI3K signaling that is used by Epi to block insulin signaling.
RESULTS
The Hippo kinases inhibit p110α through phosphorylation of threonine 1061 at its C terminus
To investigate phospho-regulation of PI3Kα (i.e., the p110α/p85α complex), we developed an in vitro reconstitution assay to screen for the effects of recombinant protein kinases on its catalytic activity. Recombinant, full-length p110α was purified from human suspension cultures as a complex with polyhistidine-tagged p85α.13–15 This complex was then pre-incubated with a panel of 31 functionally diverse human protein kinases from the major branches of the protein kinase evolutionary tree,16 and we evaluated the ability of PI3Kα to phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2), its endogenous substrate (Figures 1A and 1B). Of the 31 kinases, only MST1 (encoded by Stk4) had a major effect on PI3Kα catalytic activity, causing nearly complete inhibition with dose dependence at the level of the protein kinase (Figure 1C).
Figure 1. Kinase screen identifies GCK kinases as negative regulators of PI3Kα activity.
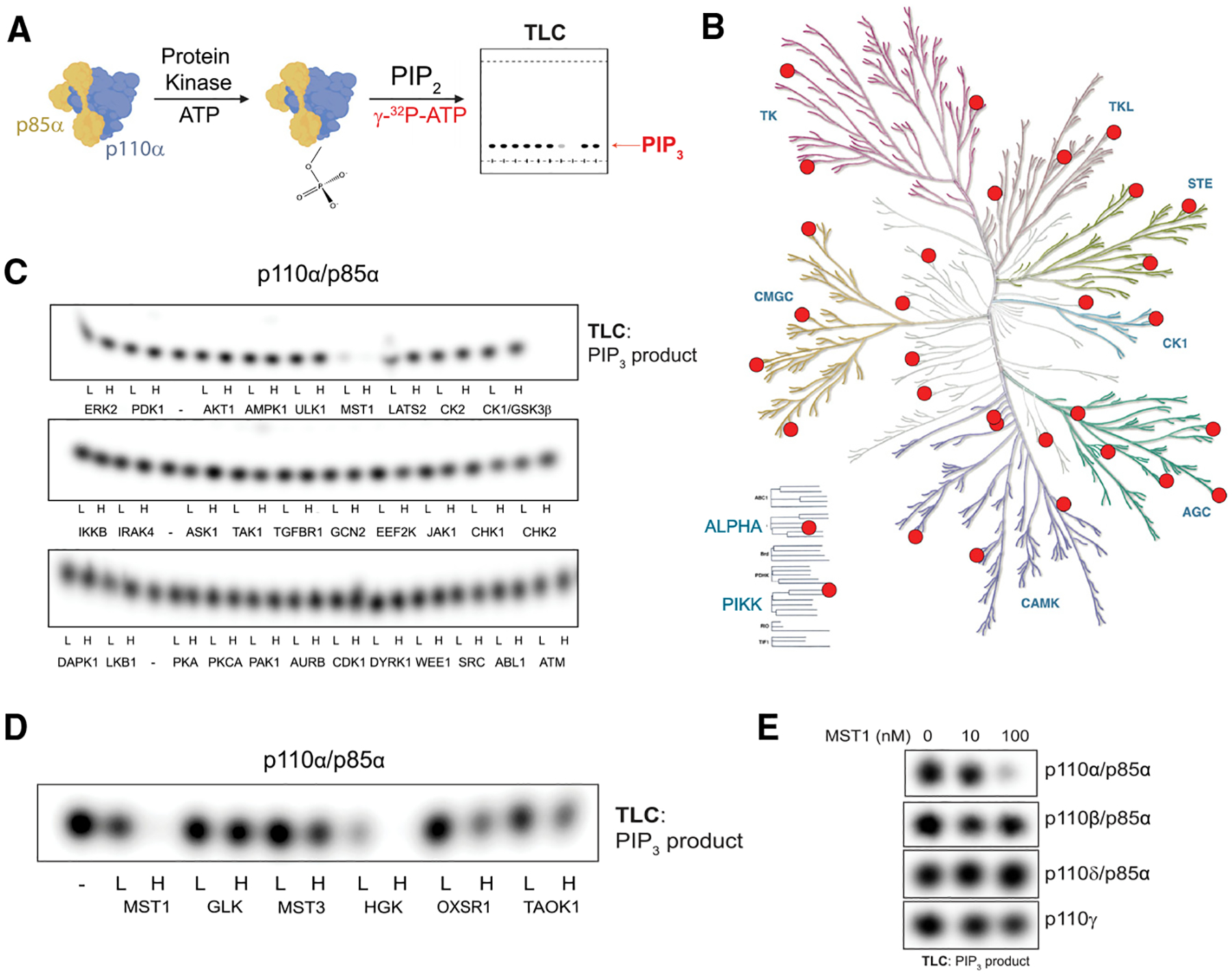
(A) Experimental schema.
(B) Dendrogram of the human protein kinome that highlights the kinases selected for our screen.
(C) Thin-layer chromatography (TLC)-autoradiography of [32P]PIP3 produced by recombinant PI3Kα complexes (p110α/p85α, 500 nM) after incubation with the indicated recombinant protein kinases at 10 (L [low]) or 100 nM (H [high]) concentrations or with enzyme storage buffer (–).
(D) PIP2 kinase assays with PI3Kα after incubation with subset of the GCK family, performed as described in (C).
(E) PIP2 kinase assays with the class I PI3K isoforms (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ, 500 nM) after incubation with MST1.
(D) and (E) are representative of two independent replicates.
MST1, the human ortholog of hippo (hpo) in Drosophila, is a growth-suppressing kinase in the group II germinal center kinase (GCK II) family and a core member of the well-conserved Hippo pathway.17–19 Because the GCK family members have overlapping cellular functions and similar substrate specificities,20,21 we tested whether additional GCK family members (Figure S1) can inhibit PI3Kα’s catalytic activity. We found that HGK (MAP4K4), OXSR1, TAOK1, and MST3 could also inhibit PI3Kα, suggesting that PI3Kα may be under broader regulation by the GCK family (Figure 1D). Next, we investigated MST1’s ability to inhibit other class I PI3K isoforms. MST1 had little effect on the catalytic activities of p110β, p110dδ, and p110γ, indicating a selective regulation of p110α (Figure 1E). Like the p110α preparation, the p110β and p110δ proteins were prepared as complexes with p85α. Given their insensitivity to MST1 inhibition, we inferred that MST1 was directly phosphorylating p110α and not p85α.
To locate the MST1 phosphorylation site(s) on p110α, we profiled MST1’s in vitro substrate specificity using positional scanning peptide arrays (PSPAs). This technique utilizes a combinatorial peptide library that systematically substitutes each of 22 amino acids (20 natural amino acids plus phospho-T and phospho-Y) at nine positions surrounding a central phospho-acceptor containing equivalent amounts of Ser and Thr.22 MST1 preferentially phosphorylated threonine over serine and favored aliphatic amino acids at position +1, aromatic amino acids at position −2, and positively charged amino acids at position +2 (Figure 2A). The PSPA data were converted into a position-specific scoring matrix (PSSM) and used to score all threonine residues in the amino acid sequence of p110α. The most favorable in silico MST1 threonine target was T1061, located in p110α’s C-terminal tail (C-tail), a disordered region spanning residues 1049 to 1068 (Figure 2B). To validate this finding, we incubated recombinant PI3Kα with MST1 and analyzed peptide fragments by mass spectrometry. This approach confirmed that phosphorylation of T1061 increased after MST1 treatment (Figure S2A). Furthermore, we generated the recombinant threonine-substituted phospho-null mutant T1061A of p110α and found its catalytic activity to be completely unaffected by MST1 and HGK (Figures 2C and S2B).
Figure 2. MST1/2 and HGK inhibit catalytic activity of p110α through phosphorylation at T1061.
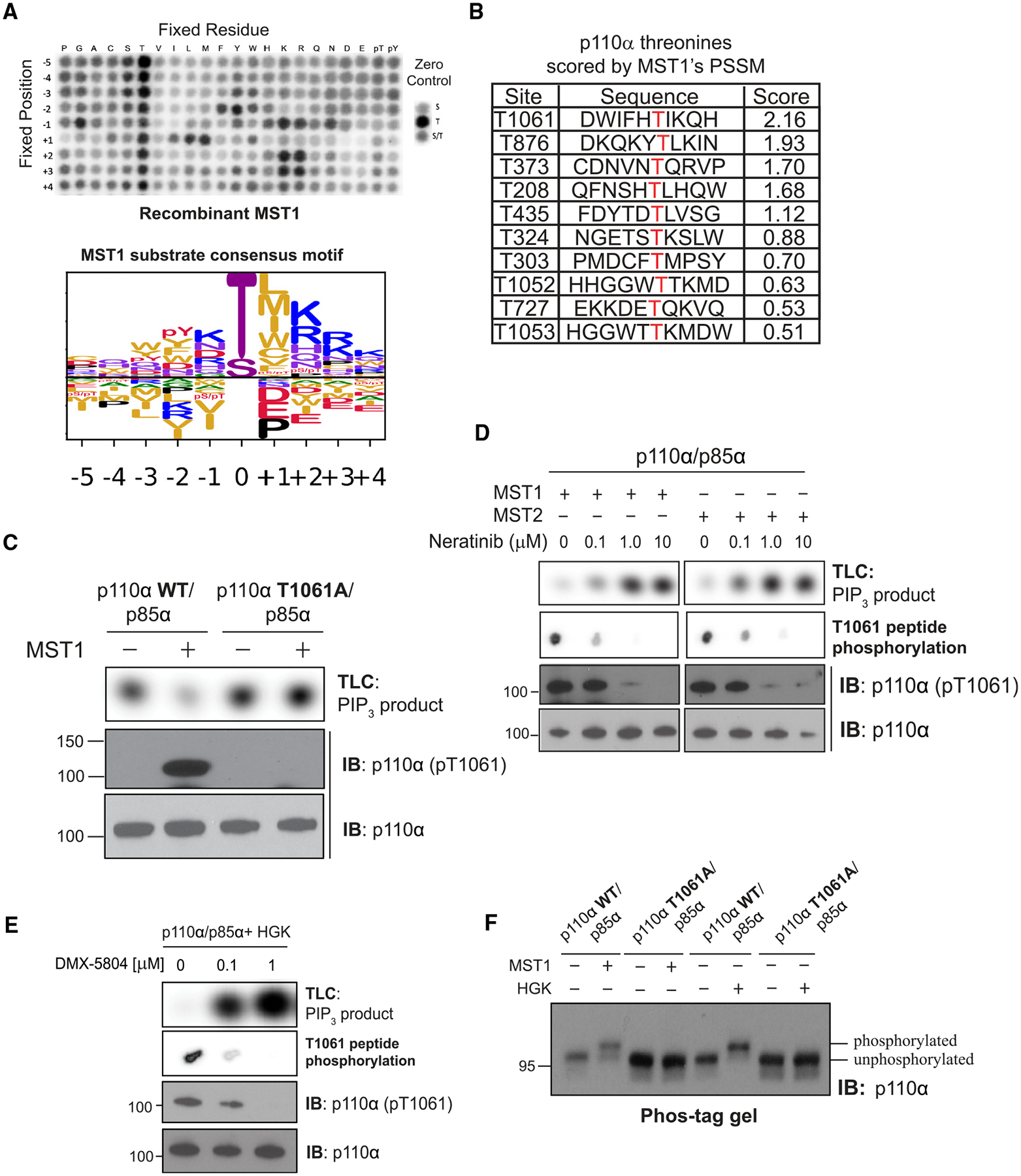
(A) (Top) Peptide phosphorylation by MST1 to characterize its substrate consensus motif. Positional scanning peptide arrays were utilized, where 22 residues (20 amino acids +2 PTM residues) were scanned across nine neighboring positions of the phospho-acceptor. The zero controls (right inset), consisting of serine only, threonine only, or a 1:1 mixture of both, were examined as phospho-acceptors. Phosphorylation was measured by autoradiography. (Bottom) Sequence logo of the substrate consensus motif of MST1 as determined on top. Letter height is proportional to favorability of corresponding amino acid.
(B) p110α’s threonine residues scored by MST1’s position-specific scoring matrix obtained from (A).
(C) Incubation of p110α(WT)/p85α and p110α(T1061A)/p85α with MST1. Top: autoradiography of [32P]PIP3 production by p110α. Bottom: immunoblots of total p110α and pT1061 p110α.
(D) Incubation of MST1 or MST2 with increasing concentrations of neratinib, followed by incubation with PI3Kα. Top: autoradiography of [32P]PIP3 production by p110α. Second from top: autoradiography of T1061-modeled peptide substrate peptide phosphorylation by MST1 or MST2. Bottom: immunoblots of total p110α and pT1061.
(E) Repeat of (D) using HGK and its specific inhibitor, DMX-5804.
(F) Immunoblot of p110α (WT and T1061A) on Phos-tag gel after treatment with MST1 or HGK.
(A)–(F) are representative of two independent replicates.
Next, we generated and validated polyclonal antibodies against a p110α peptide containing phosphothreonine at 1061 (pT1061). The antibodies detected wild-type (WT) p110α only after MST1 treatment and failed to identify MST1-treated T1061A, confirming their utility for readout of pT1061 (Figure 2C). We also showed that blocking MST1/2 or HGK/MAP4K4 activity using neratinib23 or DMX-5804,24 respectively, reversed the inhibition of PI3Kα activity and reduced pT1061 phosphorylation (Figures 2D and 2E). Finally, we performed SDS-PAGE using Phos-tag gels to measure the extent of phosphorylation of p110α by MST1 and HGK.25 Following exposure to MST1 or HGK, the protein band blotted by an anti-p110α antibody exhibited a delay in migration, suggesting stoichiometric modification of p110α (Figure 2F). In contrast, the T1061A protein migrated alongside that of unphosphorylated WT p110α in both control and MST1/HGK-treated samples, indicating that pT1061 is the prominent phosphorylation event. Together, these data indicate that PI3K catalytic activity is inhibited through phosphorylation of T1061 on p110α by the Hippo kinases.
Phosphorylation of T1061 impedes p110α’s interaction with membranes
To gain insight into the inhibitory mechanism of p110a pT1061 phosphorylation, we examined publicly available structures. Only ~15% (8/55) publicly available structures of PI3Kα can resolve T1061, and when resolved, its side chain faces the solvent.26–31 In general, the tail adopts one of two conformations: one that is directed toward the catalytic site in a “frontside conformation” or one where T1061 undergoes a ~42 Å shift to the opposite side of the protein in a “backside conformation” (Figure 3A).32,33 We generated and purified a phosphomimetic mutant of p110α (T1061E) and showed that it partially inhibits PI3K activity (Figures S3A–C). To gain insights into the structural basis for PI3Kα inhibition by T1061 phosphorylation, we solved the crystal structure of the p110α T1061E protein in complex with the nSH2-iSH2 domains of p85α and the catalytic inhibitor GDC-0077 at 2.95Å. The peptide backbone of the C-tail was resolved up to residue 1062 and adopted the backside conformation observed in previous structures of WT p110α (Figures 3A, 3B, S3D, and S3E). The side chain of E1061 was not resolved well, and crystal contacts were observed at the approximate location of this residue and Q205 and K228 from a second complex (Figure S3D); however, we noted multiple intramolecular contact points in this mutant structure that have not been observed in previous structures, including an interaction between a hydrophobic patch of residues on the C-tail, composed of residues 1057–1059, and helix kα11, encompassing residues 1031–1048 (Figure S3E).34 Together, these structural findings are consistent with an allosteric model of inhibition by T1061 phosphorylation where it promotes the backside conformation of the C-tail.
Figure 3. Phosphorylation at T1061 decreases p110α’s association with membranes.
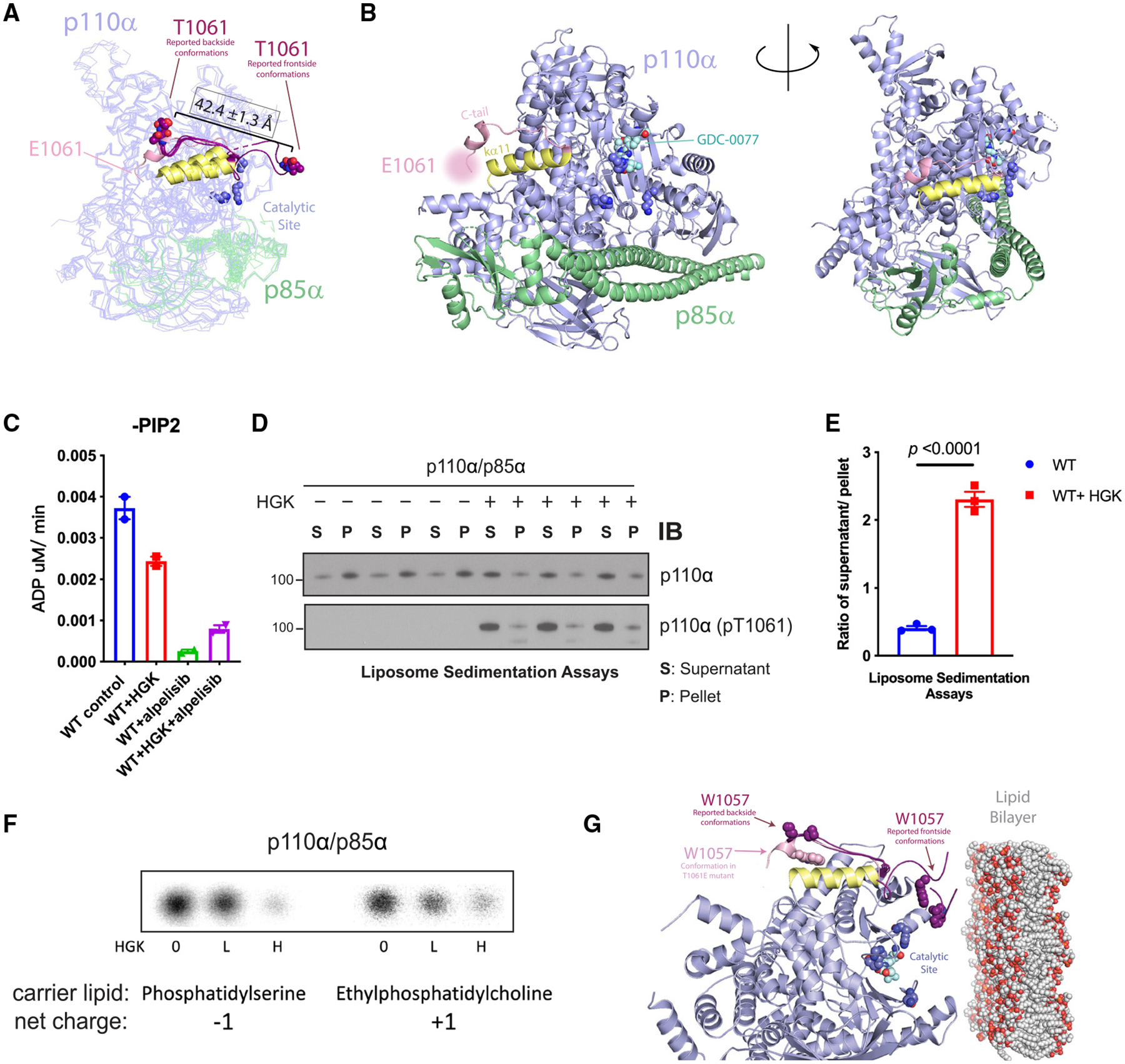
(A) Overlay of the crystal structure of p110α (T1061E) with six reported p110α (WT) structures where threonine 1061 was resolved. C-tails from frontside conformations (PDB: 4A55 and 5DXH) and backside conformations (PDB: 4JPS, 4AWF, 4ZOP, and 5FI4) of reported structures are shown in purple. E1061’s approximate location is indicated. Residues 1032–1048, constituting helix ka11, are displayed in yellow.
(B) Catalytic residue side chains K776, H917, and H93635 are displayed as dark blue spheres.
(C) ADP-Glo measurements of ATPase activity of PI3Kα ± HGK and ± 1 μM alpelisib.
(D) Liposome sedimentation assays of PI3Kα ± HGK, shown as immunoblots of p110α or p110α (pT1061) recovered from membrane-enriched pellet (P) and supernatant (S).
(E) Quantification of densitometries from (D) as ratios of p110α or pT1061 recovery from supernatant over pellet. Data are represented as means ± SEMs. Significance was calculated using Student’s t test (N = 3).
(F) Activity assays of PI3Kα on PI in anionic and cationic liposomes after incubation with HGK. [32P]PIP3 products were resolved by TLC and measured by autoradiography.
(G) Overlay of p110α (T1061E)’s crystal structure (C-tail in pink) with the 6 reported p110α(WT) structures (in purple, C-tails shown only) selected in Figure 2C. The coloring scheme corresponds to Figure 2C. W1057 side chains are represented as spheres. Catalytic site is indicated by residues K776, H917, and H936 (dark blue) and bound GDC-0077 (teal), shown as spheres. The phospholipid membrane model was obtained from RCSB PDB (PDB: 2MLR).
(F) is representative of two independent replicates.
Our crystal structure suggests that phosphorylation of p110α T1061 facilitates a conformation where its C-tail is directed away from the catalytic site, so we investigated how this change causes inhibition. Given that p110α is activated chiefly through enhancement of its catalytic turnover rate and/or membrane binding,13,31,35–42 we considered both of these possibilities for MST1-mediated phosphorylation. In the absence of substrate, p110α exhibits a basal ATPase activity that is a small fraction of the rate of phosphate transfer to PIP2.26 This basal ATPase activity was inhibited by the ATP-competitive, PI3Kα-specific inhibitor alpelisib, indicating that it is intrinsic to p110α.27 Phosphorylation of p110α at T1061 caused a modest reduction in this activity, indicating that the phosphorylation at T1061 does not significantly affect ATP binding and phosphate transfer to water (Figure 3C).
Next, we determined if p110α phosphorylation affects its interaction with membranes using liposomes that were modeled after the inner leaflet of the plasma membrane. The liposomes were incubated with p110α and pelleted by centrifugation. The amounts of p110α recovered from supernatant and pellet were then quantified. Under control conditions, p110α was mostly lipid bound (Figures 3D and 3E). However, in HGK-treated samples, we saw a notable shift into the supernatant, measured as a 6-fold increase in the ratio of p110α recovered from supernatant versus pellet. These data indicate that T1061 phosphorylation impairs membrane binding. Indeed, MST1/HGK treatment of the phosphomimetic, T1061E variant of p110α also reduced membrane binding (Figures S4A and S4B), but these effects were ablated in the T1061A variant (Figures S4C and S4D). Given that the C-tail interacts with membranes, we examined the possibility that membrane dissociation is driven by electrostatic repulsion between the phosphorylated C-tail and the negatively charged membrane surface. To test this theory, cationic liposomes containing phosphatidylinositol (PI) were prepared as substrates. HGK treatment reduced p110α’s conversion of PI to PI3P in both our anionic control liposomes and the cationic liposomes, indicating that charge repulsion is not a significant contributor (Figure 3F). We then asked whether the hydrophobic patch of the C-tail that is occluded in our crystal structure was essential for membrane binding.26 Within this patch, W1057 is universally conserved across all examined p110α orthologs (Figure S4E). In the reported structures of WT p110α, the W1057 side chain only resolves in the frontside conformations of the tail to form part of the predicted membrane-binding interface. Our structure of the T1061E mutant is a novel case where W1057 can be stabilized and resolved in the backside conformation, where it cannot directly contribute to membrane binding (Figure S4F). Therefore, our findings are consistent with a model where T1061 phosphorylation acts by shifting conformational equilibrium of the C terminus such that it is directed away from the catalytic face of p110α, sequestering a membrane-binding domain and thereby reducing its ability to engage membranes and phosphorylate PIP2.
Activation of adenylyl cyclase promotes phosphorylation and inhibition of p110α
We proceeded to explore the biological implications of p110α regulation by the Hippo kinases. Recent work has identified the Hippo pathway as a downstream branch of G protein-coupled receptor (GPCR) signaling.43 Stimulation of the Gαs-coupled receptors or adenylyl cyclase using receptor agonists or forskolin (FSK), respectively, results in activation of the PKA and Hippo kinase signaling pathways.44 To determine if p110α can be regulated by the Hippo pathway under these contexts, we first treated primary mouse hepatocytes with FSK or Epi.43 Both compounds strongly induced phosphorylation of p110α at T1061 as well as PKA activity as denoted by the increase of CREB phosphorylation at S133 (Figure 4A). We confirmed this finding in multiple cell lines (Figures S5A–S5C). To further clarify the effects of pT1061 on PI3K activity, we treated freshly isolated primary human hepatocytes with insulin, followed by FSK. Insulin robustly stimulated p110α activity as determined by AKT phosphorylation at T308 and S473, and the addition of FSK led to increased PKA activity, p110α phosphorylation at T1061, and a reduction of AKT phosphorylation (Figure 4B). This was also demonstrated in MCF10A and AML12 cells (Figures S5D and S5E). The activation of PI3K results in multiple downstream feedback pathways, so we directly examined the catalytic activity of p110α with an immunopurified enzyme taken from cells treated with insulin and FSK (Figures 4C and 4D). In the presence of insulin stimulation, the production of PIP3 was increased over 100-fold, and this was greatly reduced when FSK was added. The phosphorylation and inhibition of p110α by FSK in HEK293A cells also correlated with suppression of the extracellular acidification rate (ECAR), suggesting acute reductions in glycolysis (Figures 4E and 4F). Taken together, these results demonstrate that increased adenylyl cyclase activity leads to phosphorylation and inhibition of p110α and glucose metabolism.
Figure 4. Forskolin and epinephrine exposure phosphorylate and inhibit p110α in cells.
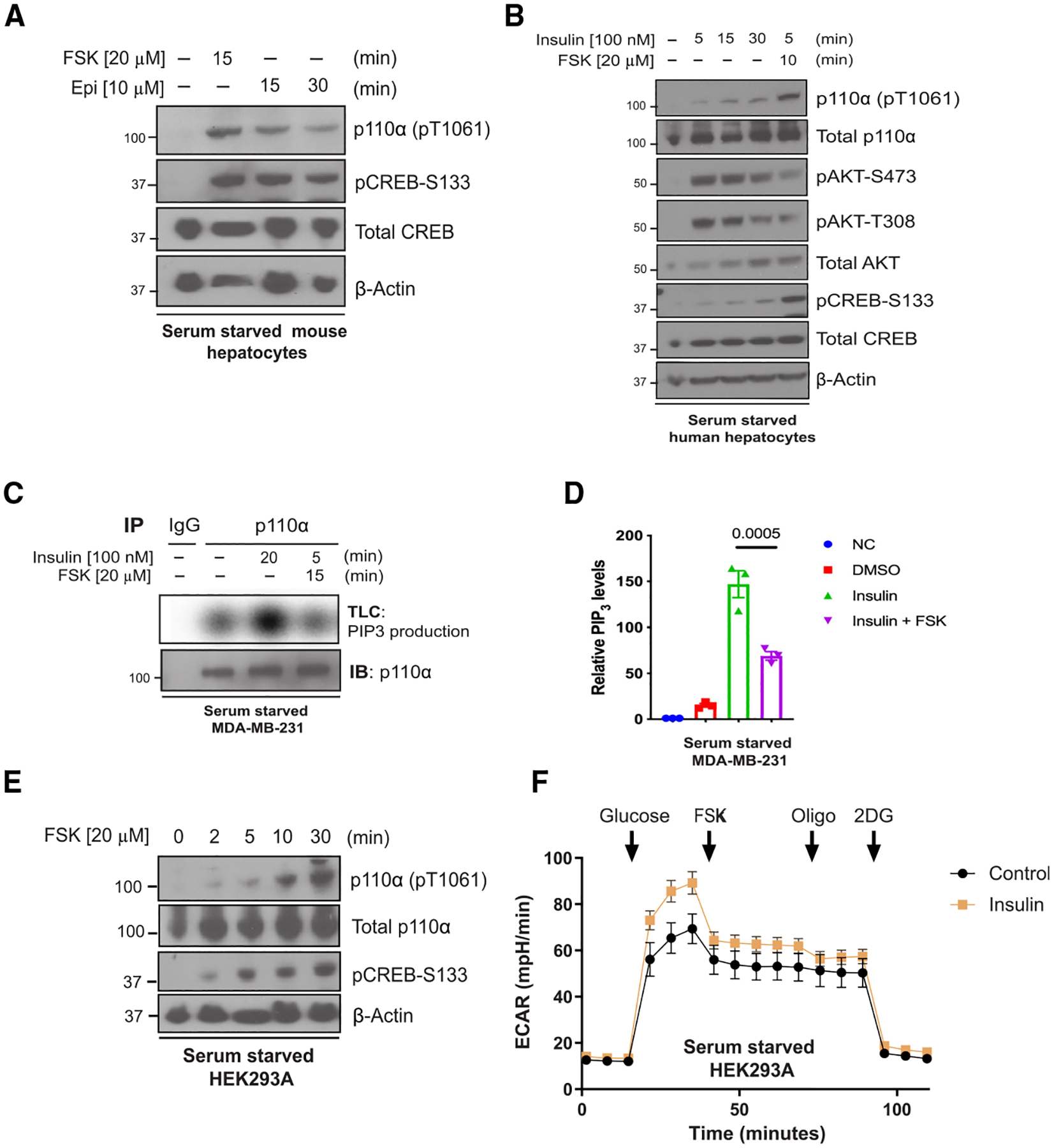
(A) Immunoblot for the indicated proteins using lysates from primary mouse hepatocytes that were serum starved 12 h before stimulation with forskolin (FSK; 20 μM) for 15 min or epinephrine (Epi, 10 μM) for 15 and 30 min.
(B) Immunoblot for the indicated proteins using lysates from primary human hepatocytes that were serum starved for 12 h and treated with insulin (100 nM) for different time points (0, 5, 15, or 30 min) or insulin followed by FSK (20 μM) for 10 min.
(C) Top: radioautograph of a TLC separation demonstrating PIP3 production of endogenous p110α that was immunoprecipitated from serum-starved MDA-MB-231 cells treated with vehicle, insulin (100 nM), or insulin plus FSK (20 μM) for 15 min. Bottom: Corresponding immunoblot for p110α using the same immunoprecipitate lysate.
(D) Quantification of the radioautograph from
(C) averaged over three independent experiments. Means ± SEM. Comparisons made using ANOVA with Tukey’s multiple comparisons post-test (N = 3).
(E) Immunoblot for the indicated proteins using lysates from HEK293A cells that were serum starved for 2 h before being stimulated with FSK 20 μM for 0, 2, 5, 10, and 30 min.
(F) The extracellular acidification rate (ECAR) was monitored in serum-starved HEK293A cells with or without insulin (0.1 mM] pre-treatment for 1 h. Arrows indicate injection of glucose (10 mM), FSK (20 μM), oligomycin (Oligo; 1 μM), and 2-deoxy-D-glucose (2DG; 50 mM). Means ± SEM, N = 19 wells. Results are representative of 3 independent experiments.
Epi stimulates p110α phosphorylation at T1061 in vivo
Epi stimulates adenylyl cyclase activity and regulates the Hippo pathway through GPCRs including the β2-AR, a Gαs-coupled receptor.43,44 Therefore, we hypothesized that Epi could result in phosphorylation and inhibition of p110α in vivo. We injected WT C57BL/6J mice with Epi and collected liver tissue for western blot analysis. In the Epi-treated mice, we observed a significant increase in the phosphorylation of T1061 (Figure 5A) and YAP (Figures S6A and S6B), a marker of Hippo pathway activity, in the liver. This increase was not observed in mice with genetic deletion of the β1- and β2-ARs (Figure 5B), confirming the importance of β-adrenergic signaling in this response. Next, we exposed mice to several physiologic stressors to induce endogenous Epi release and assess the phosphorylation status of p110α. For example, hypoglycemia is known to induce the sympathoadrenal response and cause an acute and robust release of Epi, so we fasted the mice for 18 h. Fasting increased blood Epi levels (Figure S6C) and significantly increased the abundance of p110α phosphorylation at T1061 in the liver as compared to “Fed” mice that were food restricted for 18 h and then given access to food for 4 h (Figures 5C and 5D). The fasted mice also displayed reduced levels of phosphorylated AKT, as expected in conditions of low circulating insulin levels (Figure S6D). Next, we imposed hyperinsulinemic conditions by treating WT mice with insulin until hypoglycemia developed (30 min) and harvested blood and liver tissues. At this time point, Epi levels in the blood were elevated, the abundance of T1061 phosphorylation was significantly increased, and phosphorylation of AKT was reduced, suggesting that Epi blocks PI3K activity in the liver (Figures S6E–S6G). Lastly, we fed mice a high-fat diet (HFD), which is known to increase endogenous Epi levels.45 Following 2 weeks of HFD, systemic Epi levels were elevated, and the abundance of pT1061 was increased as compared to mice fed with normal chow (Figures S6H–S6J).
Figure 5. Epi exposure induces phosphorylation and inhibition of p110α in the liver.
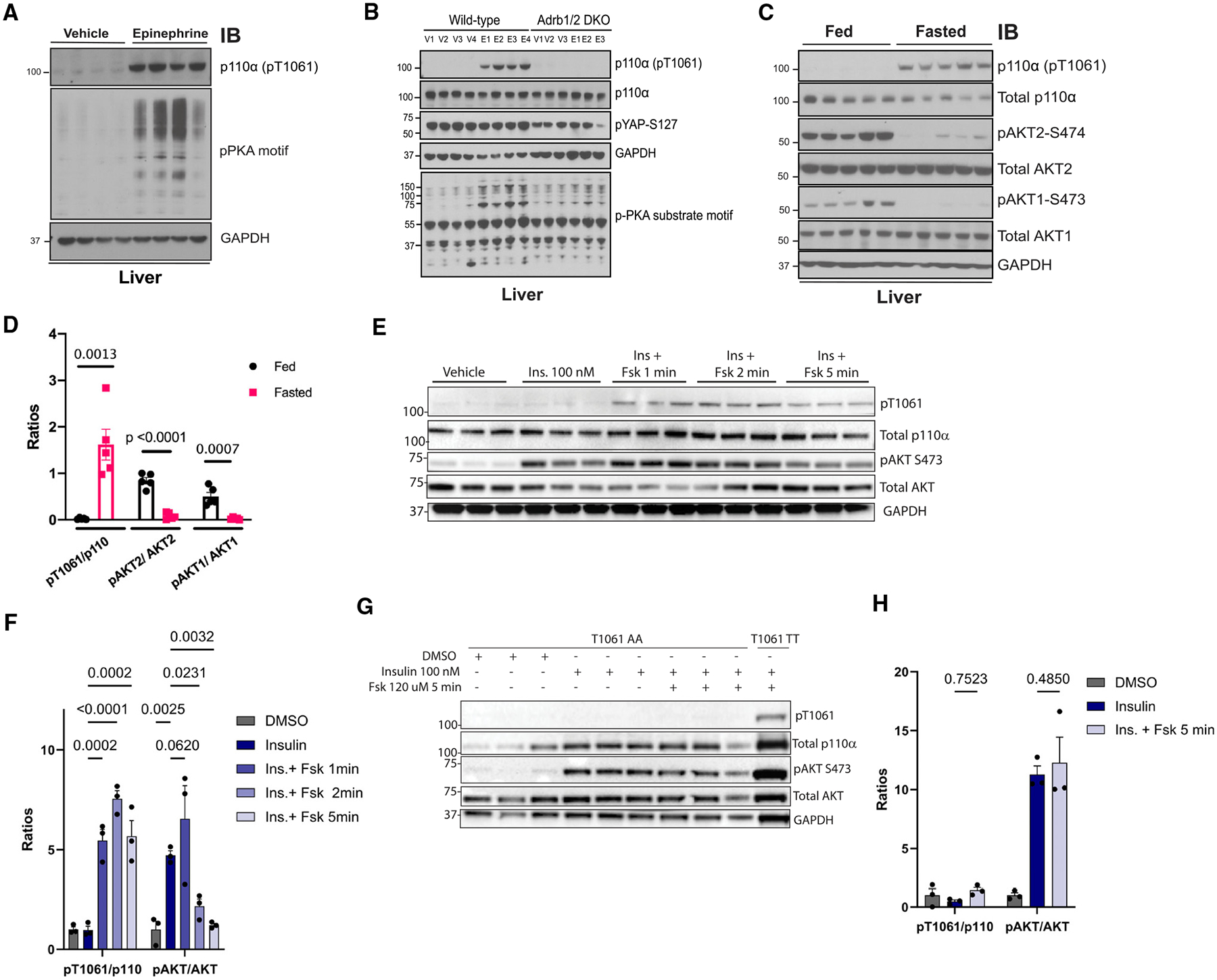
(A) Immunoblot for the indicated proteins using lysates from livers taken from WT mice that were injected with vehicle (normal saline) or Epi (0.3 μg/g) for 10 min (n = 4).
(B) Immunoblot for the indicated proteins using lysates from livers taken from WT and β1/β2 double-knockout mice (Adrb1/2 DKO) treated with vehicle (V; saline) or Epi (E) via i.p. injection for 10 min.
(C) Immunoblot for the indicated proteins using lysates from liver taken from WT mice that were fasted for 18 h (Fasted) or fasted and then exposed for food for 4 h (Fed) (n = 5).
(D) Quantification of the ratios of pT1061 to total p110α, pAKT2 to total AKT2, and pAKT1 to total AKT1 using band intensity from (C). N = 5. Comparisons made via ANOVA with Dunnett’s post-test.
(E) Immunoblot for the indicated proteins using lysates from WT mouse primary hepatocytes exposed to vehicle (DMSO), insulin (Ins.; 100 nM), or insulin plus Fsk for the indicated times.
(F) Quantification of the ratios of pT1061 to total p110α and pAKT to total AKT using band intensity from (E). N = 3. Comparisons made via ANOVA with Dunnett’s post-test comparing to insulin group.
(G) Immunoblot for the indicated proteins using lysates from mouse primary hepatocytes isolated from Pik3caT1061A/T1061A mice (T1061 AA, N = 3) or WT littermates (T1061 TT, N = 1) exposed to insulin and Fsk for the indicated times.
(H) Quantification of the ratios of pT1061 to total p110α and pAKT to total AKT using band intensity from (G). N = 3. Data are presented as mean ± SEM. Comparisons made via ANOVA with Dunnett’s post-test comparing to insulin group.
In addition to the effects on the liver, Epi increases rates of adipocyte lipolysis leading to the release of non-esterified fatty acids (NEFAs) and glycerol, which can then be used by tissues such as the liver. To determine if the lipolytic products from adipose tissue contribute to p110α phosphorylation, we injected Epi into mice with adipocyte-specific deletion of adipose triglyceride lipase (ATGLaKO), an enzyme that catalyzes the hydrolysis of triacylglycerols to diacylglycerols. Epi appropriately activated PKA signaling in the adipose tissue of ATGLaKO; however, there was no rise in NEFAs or glycerol in the blood (Figures S7A–S7D). Nevertheless, Epi increased the abundance of pT1061 in the liver, suggesting that the lipolytic products to do not contribute to this inhibitory signaling event (Figures S7A and S7E).
In order to more stringently assess the effects of Epi on glucose homeostasis, rats were infused with insulin and glucose in order to maintain euglycemic conditions for 90 min prior to the introduction of either vehicle (saline) or Epi (0.75 μg/kg/min). After 30 min, Epi infusion reduced the glucose infusion rate (GIR), a marker of systemic insulin resistance (Figure S8A). The experiment was reproduced with rats who were exposed to Epi for either 10 or 30 min to better understand the timing of this effect. GIR was only suppressed at 30 min, and this suppression correlated with increased pT1061 in hepatic lysates (Figures S8B–S8D). Epi and insulin levels were similar at 10 and 30 min time points, suggesting that there is an intracellular delay in the inhibitory signaling event (Figures S8E and S8F). In line with the effects observed after intraperitoneal Epi injection, we found an increase in markers of MST1/2 activation, including LATS1 phosphorylation, and a reduction in markers of PI3K signaling at the 30 min time point (Figures S8C and S8D).
Next, we modeled the effects of Epi during euglycemic hyperinsulinemic conditions in primary hepatocytes by exposing cells to insulin with or without FSK for different durations of time. FSK treatment led to phosphorylation of T1061 within 1 min; however, the suppression of p110 activity (as determined by the reduction of pAKT) required 5 min (Figures 5E, 5F, and S8G). To test whether T1061 phosphorylation is required for the suppression of p110 activity in this setting, we generated mice with the Thr-to-Ala mutation knocked into position 1061 (T1061A) in the Pik3ca gene. The mice were born in the appropriate Mendelian ratios with equal sex balance (Figures S8H, S8I). The homozygous T1061A (AA) mice weighed about 10% less than the WT littermates (TT) at 8 weeks of age (Figure S8J), with proportional reductions in liver mass (Figure S8K). We isolated primary hepatocytes from the AA mice and exposed them to insulin and FSK for 5 min. Unlike the WT geno-type, FSK did not promote phosphorylation of T1061, and there was no reduction in the pAKT/AKT ratio (Figures 5G, 5H, and S8L).
Taken together, these experiments demonstrate that endogenous or exogenous Epi stimulation results in MST1/2 activation, which is concomitant with p110α phosphorylation at T1061 and reduced PI3K activity.
The Hippo kinases MST1/2 regulate p110α phosphorylation in cells and tissues via PKA
We next sought to examine if the Hippo kinases play a role in the phosphorylation of p110α. Primary mouse hepatocytes were treated with FSK in the absence or presence of a selective MST1/2 inhibitor, XMU-MP-1.46 This agent significantly reduced FSK- and Epi-induced phosphorylation of T1061 (Figures 6A and S9A). The Epi-induced increase in pT1061 was also significantly reduced when the hepatocytes were pre-treated with a non-specific β-adrenergic inhibitor (propranolol HCl) and the PKA inhibitor H89 (Figure S9A). The dependance on PKA was confirmed using another PKA inhibitor, AT13148, and by overexpressing a natural PKA inhibitor (PKIA) in HEK293 cells (Figures S9B and S9C). These results implicate PKA as a critical node connecting Epi and MST1/2 to PI3K.
Figure 6. Loss of MST1/2 activity in cells and liver reduces p110α phosphorylation.
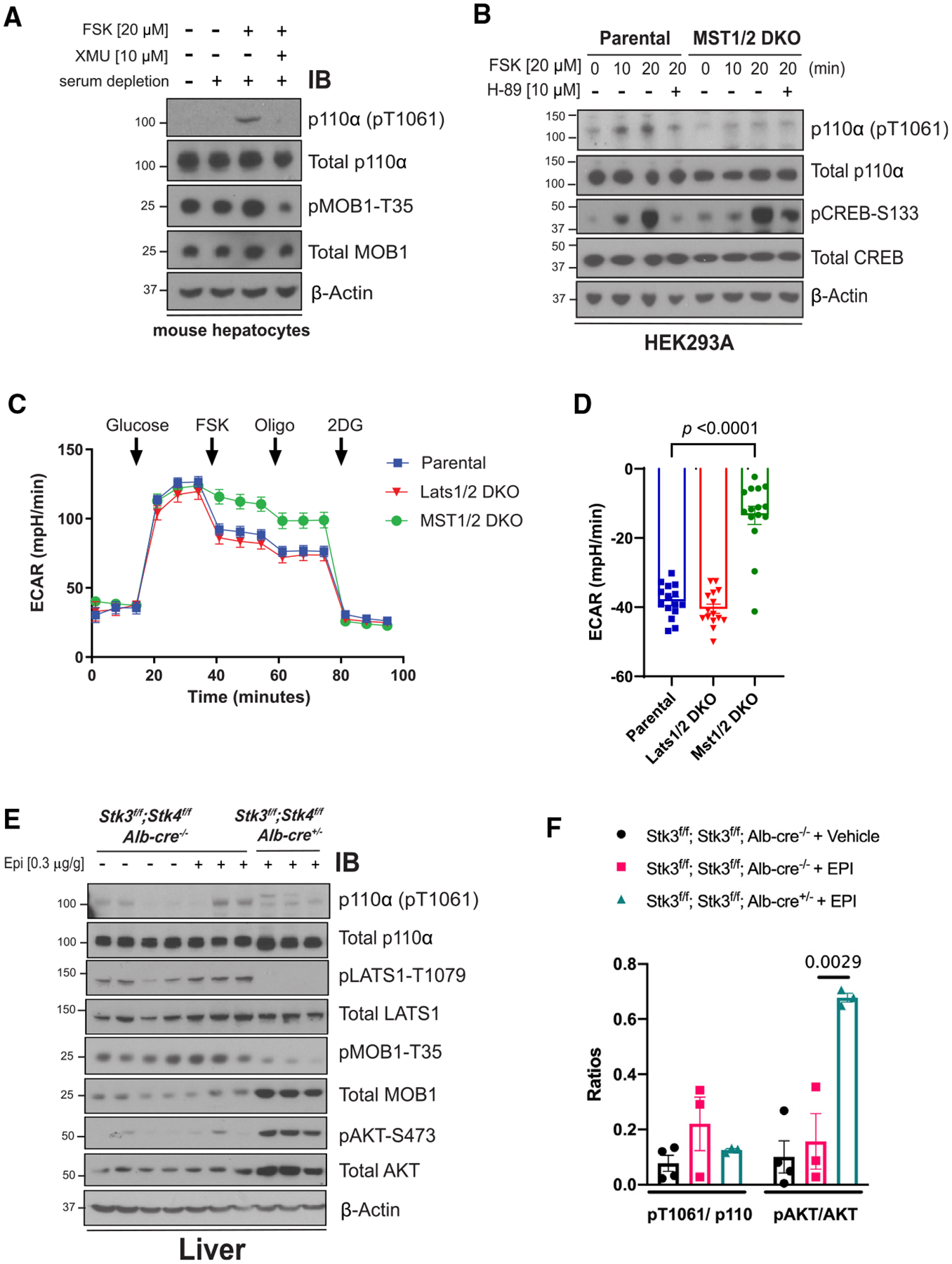
(A) Immunoblot for the indicated proteins using lysates from serum starved primary mouse hepatocytes isolated from WT mice that were stimulated with vehicle (DMSO), FSK (20 μM), or FSK and the MST1/2 inhibitor XMU (10 μM).
(B) Immunoblot for the indicated proteins using lysates from HEK293A parental cells or a line with CRISPR deletion of MST1 and MST2 (MST1/2 DKO) that were serum starved for 2 h and pre-treated with vehicle (DMSO) or the PKA inhibitor H-89 (10 μM) for 1 h before stimulating with FSK (20 μM) for 10 or 20 min.
(C) The ECAR was monitored in HEK293A parental, MST1/2 DKO, and Last1DKO cells for 1 h. Arrows indicate injection of glucose (10 mM), FSK (20 μM), Oligo (1 μM), and 2DG (50 mM). N = 15 wells. Results are representative of 3 independent experiments.
(D) The suppression (%) of ECAR following the addition of FSK (20 μM) using data from (C). N = 15.
(E) Immunoblot for the indicated proteins using lysates from livers taken from animals (Stk3f/f;Stk4f/f, Alb-Cre−/− and Stk3f/f;Stk4f/f, Alb-Cre+/−) that were injected with vehicle (normal saline) or Epi (0.3 μg/g).
(F) Quantification of the ratios of pT1061 to total p110α and pAKT to total AKT using band intensity from (E). In (C), (D), and (F) data are represented as mean ± SEMs. Comparisons made via two-way ANOVA with Sidak’s multiple comparisons post-test (D) and ANOVA with Turkey’s multiple comparisons post-test (F).
We complemented the pharmacologic inhibition of MST1/2 with experiments using cells with CRISPR deletion of MST1/2 (MST1/2 double knockout [DKO]).21 Following treatment with FSK, the MST1/2DKO and parentalcells displayed increased PKA signaling as determined by the phosphorylation of CREB and an increase in phosphorylation of T1061; however, the phosphorylation T1061 was greatly reduced in MST1/2 DKO cells (Figure 6B). We next determined if these changes in signaling correspond to changes in PI3K-mediated glucose metabolism using ECAR as a readout. Both MST1/2 DKO cells and cells deficient in Lats1/2 (Lats1/2 DKO), a key downstream node in the Hippo pathway, displayed similar rates of ECARs as compared to parental cells following glucose exposure (Figure 6C). However, the ability of FSK to suppress ECARs was significantly abrogated in the MST1/2 DKO cells (Figures 6C and 6D), suggesting that MST1/2, and not the remainder of the Hippo pathway, are important in this effect.
To determine whether MST1 and MST2 (encoded by the genes Stk3 and Stk4) are involved in Epi-mediated p110α regulation in vivo, we generated mice with hepatocyte-specific genetic deletion of Stk3 and Stk4 (Alb-Cre+/−; Stk3f/f; Stk4f/f). These mice had significantly larger livers compared to their littermates without the Alb-Cre transgene, consistent with previous reports of hepatocyte hypertrophy following loss of MST1 and MST2 (Figure S9D).47 Loss of MST1 and MST2 in hepatocytes resulted in a significant decrease in the phosphorylation of several target proteins including LATS1 (T1079) and MOB1 (T35) (Figures 6E and S9E). We subjected these mice to acute Epi exposure via intraperitoneal (i.p.) injection to assess the impact of these signaling changes on pT1061. While Epi increased the phosphorylation of T1061 in WT mice, this effect was significantly reduced in liver-specific MST1/2 DKO mice (Figures 6E and 6F). Interestingly, these mice also displayed a paradoxical increase in AKT phosphorylation (pS473) following Epi, consistent with deletion of an endogenous negative regulator of p110α. We confirmed these results by inducing the deletion of MST1 and MST2 in adult mice by injecting an adenovirus containing Cre recombinase (AdCre) into Stk3f/f;Stk4f/f mice (Figure S9F, S9G).
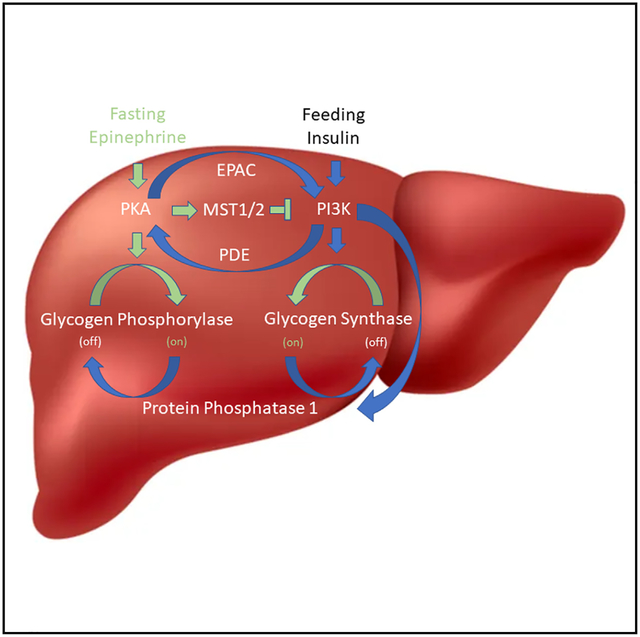
Lastly, we evaluated the role of MST1/2 in the hepatic response to endogenous Epi by fasting the MST1/2 DKO mice and their littermate controls (Stk3f/f;Stk4f/f, referred to as Flox). Fasting increased the phosphorylation of p110α in Flox mice as compared to those that were “Fed”; however, this change did not occur in DKO mice. We also interrogated the abundance of phosphorylated glycogen synthase (pGS) at site S641, an inhibitory modification controlled by PI3K signaling.48 While the Flox mice could appropriately phosphorylate GS and suppress GS activity during fasting, these effects were absent in DKO mice (Figures 7A–7C). Similar trends were observed when mice were directly injected with Epi (Figure S10). The activity of glycogen phosphorylase in the liver during fasting and the rate of glycogenolysis following Epi exposure during a hyperinsulinemic euglycemic clamp were unchanged (Figures 7D and S11). Together, the high glycogen synthase activity and normal glycogen phosphorylase activity associated with more glycogen content in the DKO livers during fasting (Figure 7E). These data suggest that Epi acts via MST1/2 to suppress hepatic glycogen synthesis independent of glycogen phosphorylase activity.49
Figure 7. Loss of MST1/2 alters fasting glycogen metabolism.
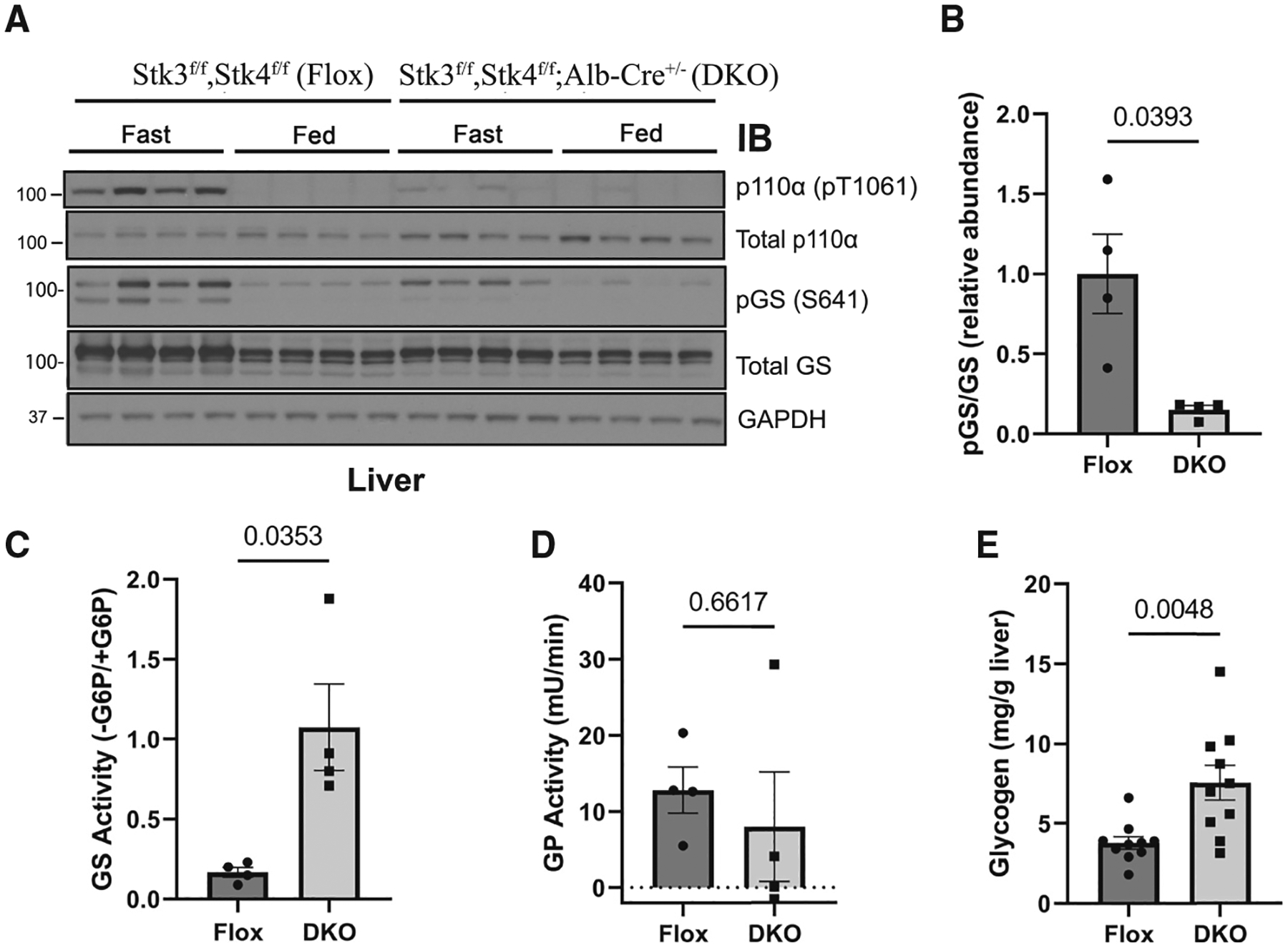
(A) Immunoblot for the indicated proteins using lysates from livers taken from Stk3f/f;Stk4f/f, Alb-Cre−/− (Flox) and Stk3f/f;Stk4f/f, Alb-Cre+/− (DKO) that were fasted for 18 h (Fast) and then euthanized or provided food for 4 h (Fed). N = 4.
(B) Quantification of the ratio of phosphorylated glycogen synthase (pGS) to total GS using band intensity for the Fasted mice in (A). The ratio was normalized to the average of the Fasted Flox mice. (C–E) Hepatic GS activity (C; N = 4), glycogen phosphorylase (GP) activity (D; N = 4), and glycogen content (E; N = 10) were measured from Fasted Flox and DKO mice.
Data are presented as mean ± SEMs. Comparisons made via Student’s t test.
DISCUSSION
We report evidence that a signaling enzyme can inhibit p110α via direct modification and reveal an unexpected connection between the PI3K and Hippo pathways. Following phosphorylation by MST1/2, p110α undergoes a conformational change that impedes membrane binding and inhibits PIP2 phosphorylation. The p110α C terminus is known to be essential for its function and harbors one of the most prevalent oncogenic point mutations of all cancers (H1047R), which, conversely, enhances membrane binding to promote PIP2 phosphorylation. Phosphorylation of p110α by MST1/2 provides cells the opportunity to regulate this process, and like the H1047R mutation, this effect is α-isoform specific. Our work also implicates p110α as an essential mediator and regulatory control point linking cell growth pathways with those regulating cell metabolism.
Several independent laboratories have observed the development of insulin resistance in humans exposed to Epi.9,50 The crosstalk between Epi and PI3K activity is thought to be mediated by phospho-diesterases and guanine nucleotide-exchange factors like exchange protein directly activated by cAMP (EPAC).51–53 Our data add to this body of literature by providing a protein kinase link between the two regulatory pathways that impacts hepatic glycogen metabolism (Figure 7F). These data are in line with those of Liu et al., who demonstrated that hepatic MST1/2 is sequestered in glycogen via an interaction with Laforin.54 Together, our results support a model where counter-regulatory hormones regulate hepatic insulin signaling by activation of PKA-mediated glycogen degradation, MST1/2 release, and direct phosphorylation and inhibition of p110α. In this setting, MST1/2 is also freed to phosphorylate LATS1/2 and reduce the abundance of YAP/TAZ in hepatocytes. Low levels of TAZ can contribute to the activation of gluconeogenesis in the liver via interactions with the glucocorticoid receptor.55
Our study focused on the acute changes in hepatic PI3K signaling following administration of Epi. Glycogen synthesis and the downstream phosphorylation events in the PI3K pathway are the most direct readouts of hepatocellular insulin action.56 In the liver, insulin promotes glycogen synthesis and inhibits glycogenolysis to suppress hepatic glucose production.57 In contrast, Epi increases hepatic glucose release by activating glycogenolysis and inhibiting glycogen synthase.11,58 The loss of MST1/2 in the liver creates a unique scenario where Epi continues to activate glycogenolysis, but the crosstalk to PI3K is lost, and glycogen synthesis continues. These data contribute to the argument that activation of glycogen phosphorylase flux occurs independently from the inhibition of glycogen synthase activity and that the latter may not contribute significantly to EGP.49
We speculate that this crosstalk may also account for Epi-mediated PI3K inhibition in other metabolic tissues, such as skeletal muscle and adipose tissue. Epi is known to reduce glucose uptake into these tissues in both rodents and humans9,59 PI3K inhibition likely plays an important role in this effect. For example, Araiz et al. found that β-adrenergic signaling inactivates p110α in adipose tissue,53 and deletion of the GCK family member HGK (gene: Map4k4), from mouse muscle and that adipocytes improve insulin sensitivity.60,61 In agreement, genetic variants in the MAP4K4 locus are associated with insulin resistance in humans.62 These data suggest that PI3K inhibition via the GCK family may regulate glucose metabolism outside of the liver.
Limitations of the study
The current work is limited by the resolution of the T1061E crystallography, which did not resolve the side chain of the 1061 residue with high confidence. However, we noted multiple novel intramolecular contact points supporting the predicted location, and the totality of our biochemical data suggest an allosteric model of inhibition where T1061 phosphorylation encourages the backside conformation. Our in vivo studies in the DKO mice are limited by the effects of the Hippo pathway on cell growth and proliferation. For example, genetic deletion of Stk3 and Stk4 in the liver promotes hepatomegaly, which can potentially confound our results. We attempted to overcome this confounding by using multiple complementary approaches, including the use of adenovirus to acutely delete MST1/2 from the liver and the use of primary hepatocytes in culture. The Hippo pathway also mediates the control of cell proliferation by contact inhibition, which was not explored in this study. Furthermore, the long-term implications of our work on glucose homeostasis remain unclear. It is interesting to note that other GCK family kinases have been reported as negative regulators of insulin sensitivity. For example, the overexpression of YSK1/STK25 reduces glucose tolerance and insulin sensitivity in mice fed an HFD,63 the deletion of MST3/STK24 improves hyperglycemia and insulin resistance,64 and the deletion of HGK improves insulin response in adipocytes.65 Therefore, the GCK kinases could be an attractive therapeutic target to improve glycemia.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marcus Goncalves (mdg9010@med.cornell.edu).
Materials availability
The Pik3caT1061A mice can be supplied by the lead contact following completion of a material transfer agreement. The p110 (pT1061) polyclonal antibody can be purchased from Cell Signaling Technology.
Data and code availability
All data points used to create the graphs and uncropped western blots are provided in Data S1. Crystallography data have been deposited at Protein DataBank and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Akt (Ser473) Rabbit mAb | Cell Signaling Technology | Cat#3787; RRID:AB_331170 |
| Phospho-Akt (Ser473) Rabbit mAb | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| Phospho-Akt (Thr308) Rabbit mAb | Cell Signaling Technology | Cat#4056; RRID:AB_331163 |
| Phospho-Akt2 (Ser474) Rabbit mAb | Cell Signaling Technology | Cat#8599; RRID:AB_2630347 |
| Akt (pan) (40D4) Mouse mAb | Cell Signaling Technology | Cat#2920; RRID:AB_1147620 |
| AKT1 (C73H10) [#2967] | Cell Signaling Technology | Cat#2938; RRID:AB_915788 |
| Akt2 (D6G4) Rabbit mAb | Cell Signaling Technology | Cat#3063; RRID:AB_2225186 |
| Phospho-CREB (Ser133) Rabbit mAb | Cell Signaling Technology | Cat#9198; RRID:AB_2561044 |
| Phospho-LATS1 (Thr1079) Rabbit mAb | Cell Signaling Technology | Cat#8654; RRID:AB_10971635 |
| Phospho-MOB1 (Thr35) Rabbit mAb | Cell Signaling Technology | Cat#8699; RRID:AB_11139998 |
| Phospho-YAP (Ser127) Rabbit mAb | Cell Signaling Technology | Cat#13008; RRID:AB_2650553 |
| Phospho-PKA Substrate (RRXS*/T*) Rabbit mAb | Cell Signaling Technology | Cat#9624; RRID:AB_331817 |
| PI3 Kinase p110α (C73F8) Rabbit mAb | Cell Signaling Technology | Cat#4249; RRID:AB_2165248 |
| CREB (D76D11) Rabbit mAb | Cell Signaling Technology | Cat#4820; RRID:AB_1903940 |
| MOB1 (E1N9D) Rabbit mAb | Cell Signaling Technology | Cat#13730; RRID:AB_2783010 |
| LATS1 Antibody Rabbit Ab | Cell Signaling Technology | Cat#9153; RRID:AB_2296754 |
| MST1 (D8B9Q) Rabbit mAb | Cell Signaling Technology | Cat#14946; RRID:AB_2798654 |
| GAPDH (D16H11) Rabbit mAb | Cell Signaling Technology | Cat#5174; RRID:AB_10622025 |
| Recombinant Anti-STK3/MST-2 antibody [EP1466Y] | Abcam | Cat#ab52641; RRID:AB_882734 |
| p110 (pT1061) polyclonal antibody | This paper (from CST) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Alpelisib (BYL-719) | MEDCHEM EXPRESS | Cat#HY-15244 |
| Neratinib (HKI-272) | SelleckChem | Cat#S2150 |
| Go6976 | EMD Millipore | Cat#365250 |
| DMX-5804 (HY-111754) | MEDCHEM EXPRESS | Cat#HY-111754 |
| XMU-MP-1 | SelleckChem | Cat#S8334 |
| H 89 dihydrochloride | Tocris Bioscience | Cat#2910 |
| AT13148 | SelleckChem | Cat#S7563 |
| Propranolol hydrochloride | Sigma Aldrich | Cat#P0884 |
| MST1 | SignalChem | S25–10G |
| HGK | SignalChem | M26–11G |
| MST2 | SignalChem | S24–10G |
| MST3 | SignalChem | S42–11G |
| TAOK1 | SignalChem | T24–11G |
| GLK | SignalChem | M25–11G |
| AKT1 | SignalChem | A16–10G |
| AMPK1 | SignalChem | P47–10H |
| ERK2 | SignalChem | M28–10G |
| PDK1 | SignalChem | P14–10H |
| CK1A | SignalChem | C64–10G |
| GSK3A | SignalChem | G08–10G |
| IKKB | SignalChem | I03–10BG |
| IRAK4 | SignalChem | I12–10G |
| ASK1 | SignalChem | M13–11G |
| GCN2 | SignalChem | E12–11G |
| EEF2K | SignalChem | E01–10G |
| JAK1 | SignalChem | J01–11G |
| CHK1 | SignalChem | C47–10G |
| CHK2 | SignalChem | C48–10G |
| DAPK1 | SignalChem | D01–11G |
| PKACA | SignalChem | P51–10G |
| PKCA | SignalChem | P61–18G |
| PAK1 | SignalChem | P02B-10BG |
| AurB | SignalChem | A31–10G |
| CDK1 | SignalChem | C22–10G |
| DYRK1A | SignalChem | D09–10G |
| SRC | SignalChem | S19–18G |
| ABL1 | SignalChem | A03–18H |
| LATS2 | Abnova | P5574 |
| OXSR1 | Abnova | P5614 |
| CK2 | NEB | P6010L |
| TGFBR1 | Proqinase | 0397-0000-1 |
| WEE1 | BPS Biosciences | 40412 |
| PIK3CG was purchased from Thermo Fisher | SignalChem | P27–10CG |
| Alpelisib (BYL-719) | MEDCHEM EXPRESS | Cat#HY-15244 |
| Critical commercial assays | ||
| Transcreener ADP fluorescence intensity assay | Bellbrook Labs | 3013-A |
| Deposited data | ||
| Crystal structure | Protein DataBank | entry D_1292124630 (8a.m.0) |
| Experimental models: Cell lines | ||
| MDA-MB-231 | ATCC | HTB-26 |
| Huh7 | Gift from Lab of Robert Schwartz | N/A |
| HEK293A | ATCC | CRL-1573 |
| MCF10A | ATCC | CRL-10317 |
| Primary mouse hepatocytes | isolated from mouse liver | N/A |
| Experimental models: Organisms/strains | ||
| Stk3f/f,Stk4f/f mice | Jackson laboratories | Stock No: 017635 |
| Liver specific MST1/2 double knockout mice (Stk3f/f,Stk4f/f,Alb-Cre−/+) | This paper | N/A |
| Albumin-Cre mice | Jackson laboratories | Stock No: 003574 |
| Homozygous null mice for the Adrb1 and Adrb2 genes (Adrb1−/−;Adrb2−/−) | Jackson laboratories | Stock No: 003810 |
| Pnpla2f/f mice | Jackson laboratories | Stock No: 024278 |
| Pik3caT1061A mice This paper | N/A | |
| Adipoq-Cre mice | Gift from Lab of James Lo | Stock No: 028020 |
| Oligonucleotides | ||
| PIK3CAV143I to WT isoleucine Forward: GACTTCCGAAGAAATATTCTGAACGTTTGTAAA | This paper | N/A |
| PIK3CAV143Ito WT isoleucine Reverse: TTTACAAACGTTCAGAATATTTCTTCGGAAGTC. | This paper | N/A |
| PIK3CAT1061A mutagenesis primer FW: 5′-caaaaatggattggatcttccacgcaattaaacagcatgcattgaac-3′ | This paper | N/A |
| PIK3CAT1061A mutagenesis primer RV: 5′-gttcaatgcatgctgtttaattgcgtggaagatccaatccatttttg-3 | This paper | N/A |
| PIK3CAT1061E mutagenesis primer FW: 5′-cctcagttcaatgcatgctgtttaatctcgtggaagatccaatccatttttgttg-3 | This paper | N/A |
| PIK3CAT1061E mutagenesis primer Rev: 5′-caacaaaaatggattggatcttccacgagattaaacagcatgcattgaactgagg-3 | This paper | N/A |
| PIK3CAW1057A mutagenesis primer FW: 5′-ctgtttaattgtgtggaagatcgcatccatttttgttgtccagcca-3′ | This paper | N/A |
| PIK3CAW1057A mutagenesis primer RV: 5′-tggctggacaacaaaaatggatgcgatcttccacacaattaaacag-3 | This paper | N/A |
| Recombinant DNA | ||
| pBabe puro HA-PIK3CA | Addgene | plasmid #12522 |
| Software and algorithms | ||
| Prism 7 | GraphPad La Jolla, CA | N/A |
| BioRender | https://biorender.com/ | N/A |
| Geneious | http://www.geneious.com/ | RRID:SCR_010519 |
| Chemdraw | http://www.perkinelmer.co.uk/category/chemdraw | RRID:SCR_016768 |
| Pymol | http://www.pymol.org/ | RRID:SCR_000305 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture and cell lines
All cell lines were cultured at 37°C with 5% CO2. MDA-MB-231, Huh7, HEK293A cell lines were cultured in DMEM (Life Technologies #11965092) supplemented with 10% FBS. MCF10A were cultured in DMEM/F12 (Life Technologies #11330032) containing 5% horse serum, 20 ng/mL EGF, 0.5 μg/mL hydrocortisone, 10 μg/mL insulin, 100 ng/mL cholera toxin, and 50 μg/mL penicillin/streptomycin (P/S). Primary mouse hepatocytes were cultured in William’s E medium (Life Technologies #12551032) containing 10% FBS and 1% P/S. AML12 cells were cultured in DMEM: F12 medium (ATCC #30–2006) supplemented with 10% FBS, Insulin-Transferrin-Selenium (ITS-G) (100X) (Thermo Fisher Scientific #41400045), and 40 ng/mL dexamethasone. For signaling assays, cells were washed 1x in PBS and placed in starvation media (-FBS) for 2–16 h depending upon cell line.
Mouse strain and breeding
All animal studies were conducted following IACUC approved animal protocols (#2013–0116) at Weill Cornell Medicine. Mice were maintained in temperature- and humidity-controlled on a 12-h light/dark cycle and received a normal chow diet with free access to drinking water. Male and female C57/BL6 mice were purchased at 8 weeks of age from Jackson laboratories (Bar Harbor, ME). Male and female Stk3f/f, Stk4f/f mice were purchased from Jackson laboratories (Stock No: 017635). Liver specific MST1/2 double knockout mice (Stk3f/f, Stk4f/f, Alb-Cre−/+) were generated by breeding Stk3f/f, Stk4f/f mice with Albumin-Cre (Stock No: 003574) mice. Both male and female mice were used between 8 and 12 weeks old. Male homozygous null mice for the Adrb1 and Adrb2 genes (Adrb1−/−; Adrb2−/−) were purchased at 8 weeks of age from the Jackson laboratory (Stock No. 003810). Adipocyte-specific deletion of Pnpla2 gene were generated by breeding Pnpla2f/f mice with Adipoq-Cre mice. Pnpla2f/f mice were purchased from the Jackson laboratory (Stock No.024278). Both male and female mice Adipoq-Cre; Pnpla2f/f were used between 8 and 20 weeks old. Pik3caT1061A mice were generated using the guide sequence AGCATGCTGCTTGATGGTG and previously described methods.66 Both male and female mice were used between 8 and 20 weeks old. All mice were fed normal chow (Purina 5053) or a 60% high-fat diet (D12492i, Research Diets, Inc. New Brunswick, NJ, USA), when indicated.
METHOD DETAILS
Mutagenesis and cloning
pBabe puro HA-PIK3CA was purchased from Addgene (plasmid #12522). This construct has an artifactual amino acid change in its coding sequence (I143V) and site-directed mutagenesis (Quikchange II XL, Agilent) was performed to convert it back to WT with the two primers in the Table above. The coding sequence of PIK3CA was then cloned PIK3CA into the pcDNA3.4 vector through in-fusion cloning (Clontech) as an untagged protein or with an N-terminal polyhistidine tag. Sequencing analysis was performed with Snapgene.
Protein purification
Expi293F cells (Thermo Fisher) were cultured in Expi293 Expression Medium (Thermo Fisher) and incubated in 2L spinner flasks on an orbital shaker at 90 rpm at 37°C with 8% CO2. 350 μg of pcDNA 3.4-PIK3CA and 150 μg pcDNA 3.4-His6-flag-TEV-PIK3R1 were combined and diluted in Opti-MEM I Reduced Serum Medium (Thermo Fisher). ExpiFectamine 293 Reagent (Thermo Fisher) was diluted with Opti-MEM separately then combined with diluted plasmid DNA for 10 min at room temperature. The mixture was then transferred to 500 mL EXPI-293F cells (3 ′ 106 cells/mL) and incubated. After 24 h, ExpiFectamine 293 Transfection Enhancer 1 and Enhancer 2 (Thermo Fisher) were added. Two days later, cells were centrifuged at 300 ′ g for 5 min, snap freezed in liquid nitrogen and store at −80°C (3 days post-transfection). All steps of protein purification were performed in the cold room at 4°C. Cell pellets were solubilized in lysis buffer (50 mM Tris pH 8.0, 400 mM NaCl, 2 mM MgCl2, 5% glycerol, 1% Triton X-100, 5 mM β-mercaptoethanol) supplemented with EDTA-free phosphatase and protease inhibitor cocktail (Life technologies) and lysed by Dounce homogenization (20 strokes). Lysates were centrifuged at 100,000 × g for 1 h and the supernatant were affinity purified on Ni-NTA resin (Qiagen) by batch binding for 30 min-1h. Resin was washed with 10 column volumes of base buffer (50 mM Tris pH 8.0, 500 mM NaCl, 2 mM MgCl2, 2% glycerol, 20 mM imidazole) and eluted in 10 column volumes of elution buffer (50 mM Tris pH 8.0, 100 mM NaCl, 2 mM MgCl2, 2% glycerol, 250 mM imidazole). Eluted protein was concentrated using 30 kDa Ultra Centrifugal Filter Units (Amicon) and supplemented with 1 mM DTT and 25% glycerol before snap freezing in liquid nitrogen. The same approach was carried out for purification of untagged p110β: His6-p85 a and untagged p110δ: His6-p85a.
Lipid kinase assay
For phosphorylation reactions, predetermined concentrations of protein kinase and PI3K complexes were added to master mix containing 50 mM of ATP, 0.1 mg/mL of BSA and 1x Assay Buffer I (SignalChem) in 40 mL total volumes. Reactions were carried out at 30°C for 30 min. Next, 0.01 mCi/mL 32P-labeled ATP (PerkinElmer) and 25 μM PIP2 (Thermo Scientific) were included, bringing total volume to 100 mL PIP2 phosphorylation reactions were carried out at 30°C for 20 min 50 μl of 4N HCl was added to quench the reaction and the lipid was extracted with 100 mL of CHCl3: MeOH (1:1) followed by vortexing for 1 min and centrifugation at maximum speed for 2 min. 10 mL of the lower hydrophobic phase was extracted with gel loading pipet tips and spotted onto a silica plate (EMD Millipore #M116487001) for thin-layer chromatography. Plates were placed in a sealed chamber with 1-propanol: 2M acetic acid (65:35). Radioactivity was imaged with a Typhoon FLA 7000 phosphorimager (GE) and quantified by ImageQuant (GE). For cationic liposomes, EPC (#890704C) and PI (840042C) were purchased from Avanti. Liposomes containing 5% PI, 60% EPC, and 35% cholesterol were prepared by extrusion via 20 passes through a 0.1 mm membrane using a Mini-Extruder kit (Avanti). The anionic liposomes in these experiments replaced EPC with PS.
Liposome preparation and liposome sedimentation assays
PS (#840032) and PE (#840026C) were purchased from Avanti and cholesterol (#CH800) was purchased from Nu Chek Prep. Lipid stocks were prepared at 35% PE, 30% PS, and 35% cholesterol. N2 gas was applied for 15 s and tubes were frozen and stored at −20°C. Before the experiments, the lipid stocks were vortexed and 100 μL of chloroform (HPLC-grade) was transferred to a clean glass vial. N2 gas was immediately applied to the stock tube, capped, and stored at −20°C N2 gas was applied to the 100 μL aliquot to evaporate chloroform. Next, 1 mL of 20 mM HEPES/1 mM EGTA was added and lipids were rehydrated at room temperature for 1 h. Liposomes were prepared by extrusion via 7 passes through a 0.8 mm membrane using a Mini-Extruder kit (Avanti). Liposomes were transferred to a clean Eppendorf tube and centrifuged at 10,000 ′ g for 5 min. Supernatant was discarded, and the lipid pellet was resuspended in 50 μL 1′ Assay Buffer I (SignalChem), vigorously until resuspended. In parallel, p110a phosphorylation assays were carried out. 150 μL volume of reaction mix, containing 500 nM p110a/p85a, −/+ 100 nM MST1, 50 mM of ATP, 0.1 mg/mL of BSA and 1′ Assay Buffer I (SignalChem) were added to liposomes and 50 mL volumes were distributed across three Eppendorf tubes. Binding reactions proceeded for 10 min at RT. Samples were centrifuged at 10,000 ′ g for 5 min at RT and 40 mL supernatant was carefully removed without disturbing the pellet and optimal amount of NuPAGE 4′ LDS Sample Buffer (Life Technologies #NP0008) was added. Lipid pellets were resuspended with 40 μL of Buffer I with optimal amount of 4′ LDS Sample Buffer added. The amount of membrane-bound p110α was probed and analyzed by Western blotting. For quantification, densitometry was performed using ImageStudioLite.
X-Ray crystallography
The full length, human, catalytic subunit, p110α, with the phosphomimetic mutation (T1061E, residues 1–1068) was co-expressed and purified as a complex with its regulatory subunit p85α (residues 308–593). An affinity His-tag was attached to the N terminus of p85α. The p110α/p85α heterodimer was expressed in sf21 insect cells, which were infected in 2 million per milliliter in presence of Gentamycin at 300 K. The cells were harvested after an incubation time of 66 h at 300 K and stored at 193 K until lysis. The cells were lysed using Microfluidizer in a buffer containing 50 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 5% glycerol, 10 mM imidazole, 2 mM TCEP, 1 mM MgCl 2, 0.1% NP-40, cOmplete protease inhibitor tablet, 1/100 mL (pH 8.0). Purification was performed by Ni-NTA capture of the His-tagged p85α using the buffer 20 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 5% glycerol, 20 mM imidazole, 1 mM TCEP (pH 8.0) for equilibration. Imidazole was added to this buffer at a concentration of 300 mM for elution. Anion exchange was performed using 20 mM Tris/HCl, 50 mM NaCl, 5% glycerol, 1 mM TCEP (pH 8.5) as buffer, to which 1 M NaCl was added for elution. As final step, size-exclusion chromatography was used to polish and change the buffer into 20 mM Tris/HCl, 150 mM NaCl, 1 mM TCEP (pH 8.0). The protein complexes were then concentrated to 15 mg/mL using Vivaspin Turbo 15 centrifugal concentrators, and stored at 193 K until crystallization. After one freeze-thaw cycle, a small amount of protein was analyzed by HPLC-SEC where one homogeny peak in calculated size was observed. Finally, both fingerprint mass and full intact mass were determined to confirm the formation of the intended protein complex.
Crystals of the p110α (T1061E)/His-tagged p85α heterodimer in complex with GDC-0077 (Inavolisib) were obtained using hanging-drop vapor-diffusion set-ups. The p110α(T1061E)/p85 complex at a concentration of 9 mg/mL (20 mM Tris/HCl, 150 mM NaCl, 1 mM TCEP, pH 8.0) was pre-incubated with 0.5 mM (6.9-fold molar excess) of GDC-0077 (10 mM in DMSO) for 1 h. 1 μL of the protein solution was then mixed with 1 μL of reservoir solution (0.10 M Bis-Tris-Propane pH 8.10, 0.20 M Na 3 -citrate, 10.00%(w/v) PEG 3350) and equilibrated at 20°C over 0.4 mL of reservoir solution. Well diffracting crystals appeared within 4 days and grew to full size over 20 days. Crystals were cryo-protected by the addition of glycerol to a final concentration of 20% to the crystallization drop before mounting. Diffraction data were collected from three crystals at the beamline I04–1 of the Diamond Light Source and the data were integrated, analyzed, merged and scaled by the programs XDS, Pointless and Aimless, respectively. Molecular replacement was performed using the CCP4 program PHASER and a published structure of the heterodimer PI3Kα/p85α (PDB accession code 4L1B) as a starting model. Several rounds of alternating manual re-building and refinement with REFMAC5 resulted in the final model. PI3K structural display and mapping was performed using PyMOL (RRID:SCR_000305; http://www.pymol.org/).
Transcreener assay
Transcreener ADP fluorescence intensity assay (Bellbrook Labs) was applied to determine the ATPase activity of the PI3K complex under different treatment. For preparing the HGK-treated PI3K phosphorylation reaction, 38 μL of PI3K complex (1.3 mg/mL) (EMD Millipore #14–602M) was incubated with 30 μL of HGK, 20 μL of ATP (1 mM) and 412 μL of kinase Buffer to the total volume of 500 μL. Reaction was carried out at 30°C for 1hr. GST-HGK was removed from the reaction using 10 μL of Glutathione Sepharose beads (Thermo Fisher Scientific #45000285) and batched incubation at 4°C for 30 min. The PIK3CA proteins were diluted to 5X stocks in buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 0.5mM EGTA, 0.01% Triton X-100) just before use. 10mM stocks of Alpelisib were serially diluted (3′) in neat DMSO and stored in a dessicator at room temperature. PIK3CA, along with buffer components (except ATP) were incubated with or without compound at 27°C for 1h. After incubation, the reaction was initiated by the addition of 5uL of 125uM ATP. A typical assay mixture (25 μL) contained 40mM HEPES buffer, pH 7.4, 25 mM MgCl2, 0.01% v/v Triton X-100, 5% v/v DMSO, 20 mM NaCl, 1–5 nM Wt or H1047R, 25 μM ATP, and 50 μM PIP2diC8 or PIP2 in membrane. The reaction was allowed to proceed till ~10% conversion (2.5 μM ADP) after which time, 10 μL of reaction mixture was quenched with 25uL of transcreener reagent (transcreener ADP2 FI assay kit, BellBrook labs, Cat. No. 3013). The contents were incubated at RT for 1h and fluorescence was measured using a plate reader (Paradigm, Molecular Devices). The same assay was also run at pH 6.0 or 6.4 using MOPS buffer. A calibration curve was generated under identical buffer conditions with varying ADP amounts. Using that, the observed fluorescence was converted to uM ADP. A plot between [ADP] and log[I] yielded the dose-response curves that enabled the calculation of IC50.
LC-MS/MS-based phospho-peptide detection and analyses
MST1-treated and untreated PI3K phosphorylation reaction were prepared as follows: 2 μL of PI3K complex was incubated−/+ 4.5 mL MST1 and the mastermix containing 2 μL of ATP (1 mM) and 31.5 μL of kinase Buffer to the total volume of 40 μL. Reaction was carried out at 30°C for 1h and stopped by adding 4x sample buffer. Samples were running on the SDS-PAGE and gel was stained using QC Colloidal Coomassie Stain (Bio-Rad Laboratories #1610803). The sample was digested in-gel with trypsin overnight at 37°C following reduction with DTT and alkylation with iodoacetamide. The digest was vacuum centrifuged to near dryness and desalted by C18 stage-tip column. A Thermo Fisher Scientific EASY-nLC 1000 coupled on-line to a Fusion Lumos mass spectrometer (Thermo Fisher Scientific) was used. The raw files were processed using the MaxQuant67 computational proteomics platform version 1.5.5.1 (Max Planck Institute, Munich, Germany) for protein identification. The fragmentation spectra were used to search the UniProt human protein database (downloaded September 21, 2017). Oxidation of methionine, protein N-terminal acetylation and phosphorylation of serine, threonine and tyrosine were used as variable modifications for database searching. The precursor and fragment mass tolerances were set to 7 and 20 ppm, respectively. Both peptide and protein identifications were filtered at 1% false discovery rate based on decoy search using a database with the protein sequences reversed. Identified phosphopeptides were subjected to manual inspection for confirmation of the identification.
Peptide library arrays and computational prediction of phosphorylation
Recombinant MST1 was distributed across 384-well plate, mixed with the peptide substrate library (Anaspec #AS-62017–1 and #AS-62335), kinase assay buffer I (SignalChem) and g-32P-ATP, and incubated for 90 min at 30°C. Each well contains a mixture of peptides with a centralized phosphor-acceptor (serine and threonine at a 1:1 ratio), one fixed amino acid in a randomized background. All 20 amino acids were distributed across the range of −5 to +4, relative to the centralized serine/threonine to determine the individual contributions of amino acids along the substrate peptide, as shown in the heatmap where the x axis represents fixed amino acid and the y axis represents the relative position. Peptides contained C-terminal biotinylations and were spotted on streptavidin-conjugated membranes (Promega #V2861) and imaged with Typhoon FLA 7000 phosphorimager (GE) and quantified by ImageQuant (GE). Intensities were normalized by position to generate a PSSM for MST1 and utilized to score all serines and threonines in p110a′s amino acid sequence.68,69
pT1061 peptide phosphorylation assays
A synthetic peptide modeled after p110a′s C terminus (sequence: [K(LC-biotin)]-K-G-A-M-D-W-I-F-T-I-K-Q-H-A-L-N, where LC = linker chain) were incubated with reaction mixtures, kinase assay buffer I (SignalChem) and g-32P-ATP, for 5 min at 30°C. Reactions were spotted onto streptavidin-conjugated membranes (Promega #V2861) and imaged with Typhoon FLA 7000 phosphorimager (GE).
Illustrations
Experimental schema and illustrative models were generated by BioRender (https://biorender.com/) and Chemdraw (RRID:SCR_016768 http://www.perkinelmer.co.uk/category/chemdraw). Sequence alignments performed with Geneious (RRID:SCR_010519, http://www.geneious.com/). Kinome images generated and modified at http://kinhub.org/kinmap/index.html.
Immunoprecipitation and lipid kinase assay
Cell were lysed using the PI3K buffer [25 mM Tris, pH 7.5, 10 mM EDTA, 10 mM EGTA, 1% Nonidet P-40] with one tablet (per 10 mL) of protease and phosphatase inhibitor (Life Technologies). After lysis on ice for 30 min, cell lysates were centrifuged for 10 min, 4°C and supernatant were used for immunoprecipitation. Lysates were incubated with anti-phospho-Tyr-4G10 (EMD Millipore 05–321), anti-p110a (CST #4249) or anti-p85a antibody (EMD Millipore #ABS233) for 1 h, followed by protein A/G beads (Santa Cruz) for additional 1hr, and then washed twice with PI3K buffer, and three times with TNE buffer [10 mM Tris (pH 7.5), 100 mM NaCl, 1 mM EDTA]. For PI3K activity assays, the beads (50 μL) were incubated in 1′ Assay Buffer I (SignalChem), 0.5 μL ATP (10 mM), 1 μL 32P-labeled ATP (0.01 mCi/mL) and 10 μL of PIP2 (250 μM) in 100 μL total volumes. Reactions were carried out at 30°C for 30 min 50 μl of 4N HCl was added to quench the reaction and the lipid was extracted with 100 mL of CHCl3: MeOH (1:1) followed by vortexing and centrifugation. 20 mL of the lower hydrophobic phase was extracted with gel loading pipet tips and spotted onto a silica plate (EMD Millipore, M116487001) for thin-layer chromatography. Plates were placed in a sealed chamber with 1-propanol: 2M acetic acid (65:35). Radioactivity was visualized and quantified using a Typhoon FLA 7000 phosphorimager.
Immunoblotting
Cell lysates were prepared in the cell lysis buffer [50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA and 1% NP-40 with one tablet (per 10 mL) of protease and phosphatase inhibitor (Life Technologies)]. After incubation on ice for 30 min, lysates were centrifuged at top speed for 10 min and supernatant were collected and proteins were quantified using Protein DC assay (BioRad). Proteins were running and separated on 4%–12% Bis-Tris Pre-cast Gel (Life Technologies) using MOPS buffer and transferred to PVDF membrane at 350 mA for 1.5 h. Membranes were blocked in 5% non-fat milk in TBST and incubated with primary antibody overnight. Primary antibody against pAKT (S473) [#3787, #4060], pAKT (T308) [#4056], pAKT2 (S474) [#8599], pan AKT [#2920], AKT1 [#2967], AKT2 [#3063], pCREB (S133) [#9198], pLATS1 (T1079) [#8654], pMOB1 (T35) [#8699], pYAP (S127) [#13008], pPKA substrate motif antibody [#9624], p110a [#4249], CREB [#4820], MOB1 [#13730], LATS1 [#9153], MST1 [#14946] and GAPDH [#5174] were from Cell Signaling Technology. MST2 [#ab52641] was from Abcam, and p110 (pT1061) polyclonal antibody was developed and purified by Cell Signaling Technology. HRP-conjugated secondary antibody was used at 1:5000 in 5% milk and membrane were developed using ECL solution and exposed to film. 7.5% phos-tag gel was used to resolve phosphor-p110a (T1061) (Wako Diagnostics/Chemicals # 192–18001).
Vectors and compounds
Alpelisib (MEDCHEM EXPRESS LLC #HY-15244), Neratinib (Selleck #S2150), Go6976 (EMD Millipore #365250), DMX-5804 (MEDCHEM EXPRESS LLC# HY-111754), XMU-MP-1 (Selleck #S8334), H-89 (Tocris Bioscience #2910), AT13148 (Selleck #S7563) and Propanolol HCl (Sigma Aldrich #P0884) were purchased and reconstituted using DMSO and stored at −20 before use. cDNA of PKIA were synthesized by IDT (gBlock) and PCR amplified using the two primers: pLenti-PKIA-IF-FW: 5′CGACTCTAGAGGATCCATGACTGATGTGGAAACTACATATG; pLenti-PKIA-IF-Rev: 5′-GAGGTTGATTGTCGACTTAGCTTTCAGATTTTGC TGCTTC and PCR products were purified. Vector pLenti-GFP were double digested with BamHI-HF and SalI-HF and gel purified. Next, PCR product were inserted into the vector backbone using In-fusion HD cloning (Takara) and transformed in Stbl3 competent cells and grown at 37°C overnight.
Mouse injections and metabolic tissue collections
Fed male mice at 10 weeks of age received an intraperitoneal injection of epinephrine (0.3 mg/g) or insulin (0.75 mU/g body weight) for the indicated durations. For assessment of blood glucose, 10 mL of blood was taken from the tail of mice before treatment and at the indicated time points using a handheld glucose meter (OneTouch). Mice were sacrificed using cervical dislocation and the metabolic tissues were harvested and immediately frozen in liquid nitrogen. Samples were stored in −80°C until use. Frozen tissues were homogenized in 1 mL of lysis buffer (50 mM Tris HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% Nonidet P-40, 0.5% Triton X-100) and 1 tablet (per 10 mL) of protease and phosphatase inhibitor (Life Technologies). Homogenates were centrifuged at top speed for 30 min and 50 mg of protein, determined by Protein DC Assay (BioRad) were used for Western blot analysis.
Primary hepatocytes isolation
Mouse hepatocytes were isolated by a 2-step perfusion procedure.70 Briefly, mice were anesthetized with ketamine (100 mg/kg), xylazine (10 mg/kg), and acepromazine (3 mg/kg). Following laparotomy, the liver was perfused in situ through the inferior vena cava with 20 mL of prewarmed liver perfusion medium (Invitrogen) followed by 40 mL of liver digestion medium (Invitrogen). The liver was then placed in ice-cold hepatocyte wash medium (Invitrogen), and the capsule of the liver was then gently disrupted in order to release the hepatocytes. The cell suspension was filtered with a 70-mm cell strainer (Becton, Dickinson), and cells were washed once (30′ g, 4 min, 4°C). Dead cells were removed using Percoll solution (Sigma-Aldrich) as described previously.71 Live cells were washed, pelleted, resuspended in incubation medium, and seeded on 6-well Primaria plates (Becton, Dickinson) at a density of 4 ×105 cells/well. Cells were allowed to adhere to the plates for 3 h, after which the culture medium were replaced by fresh 2 mL medium.
Extracellular acidification rates measurement (ECAR)
Cells were plated in XF96 V3 PET cell culture microplates (Seahorse Biosciences, Cat. #; 101104–004) at a density of 20,000 cells per well in DMEM (Life Technologies #11965092) with 10% FBS. After 24 h, growth media was changed to bicarbonate-free assay media (XF assay medium, Seahorse Biosciences, Cat. #: 102365–100) and incubated at 37°C for 1 h in a CO2-free incubator. Extracellular acidification rate (ECAR) was measured using an XF96 Extracellular Flux Analyzer (Seahorse Biosciences).
Hyperinsulinemic-euglycemic clamps
Rat studies
All procedures for the rat hyperinsulinemic–euglycemic clamp were approved by the University of Pennsylvania Animal Care and Use Committee. Rats were fasted for 5 h prior to the initiation of the clamp and acclimated to the bucket containers. Jugular vein and carotid arterial line were hooked up to the dual swivel 2 h prior to the clamp initiation. During the 90 min clamp period, rats received a constant infusion of insulin (Novolin Regular) at 5 mU/kg/min. Blood glucose levels were clamped at euglycemia with a variable infusion of 50% dextrose. Blood glucose levels were monitored via arterial blood sampling every 10 min and glucose infusion rates adjusted to maintain euglycemia. 87 min after initiation of the clamp, rats received either an IV infusion of vehicle saline for 10 min or epinephrine 0.75 μg/kg/min for 10 or 30 min. Blood samples were collected at the beginning of the clamp (−30 and 0 min, basal period) and during the clamp period (30, 60, 90, 100 or 120 min) for the subsequent assay of catecholamines and insulin levels. At study termination, animals were anesthetized with IV pentobarbital and tissues collected and flash frozen in liquid nitrogen for livers.
Mouse study
All procedures for the hyperinsulinemic–euglycemic clamp were approved by the Vanderbilt University Animal Care and Use Committee. Catheters were implanted into a carotid artery and a jugular vein of mice for sampling and infusions respectively five days before the study as described by Berglund et al.72 Insulin clamps were performed on 5 hr-fasted conscious unhandled mice, using a modification of the method described by Ayala et al.73 After 3h of fast, an arterial blood sample was obtained to determine natural isotopic enrichment of plasma glucose. Immediately following this sample, a quantitative stable isotope delivery to increase isotopic enrichment above natural isotopic labeling was initiated as described previously.74 Briefly, a [6,6-2H2]Glucose-2H2O (99.9%)-saline bolus was infused for 25 min to enrich total body water to 4.5% (t = −120 min to −95 min). A continuous infusion of [6,6-2H2]glucose (t = −95min–0min; 0.8 mg kg−1 min−1) was started following the [6,6-2H2]Glucose-2H2O-saline prime. The insulin clamp was initiated at t = 0min with a continuous insulin infusion (2.5 mU/kg body weight/min). At the same time, a variable infusion of glucose was started (50% dextrose + 2H2O (0.04 MPE) +[6,6-2H2]Glucose (0.08 MPE)) in order to maintain stable euglycemia and stable enrichment of 2H2O and [6,6-2H2]Glucose in plasma. At t = 91min, an epinephrine infusion was initiated (continuous infusion until sacrifice; 4 μg/kg body weight/min, in 0.7 mg/mL ascorbate for stability). Washed red blood cells were also continuously infused during the clamp period to maintain hematocrit. Every infusate was prepared in a 4.5% 2H 2O -enriched saline solution. Arterial glucose levels were monitored every 10 min to provide feedback for adjustment of the glucose infusion rate (GIR). Blood sampling for glucose kinetics was performed at t = −10;−5 (basal), t = 80–90 (insulin) and t = 110–140 min (insulin+epinephrine) of the clamp. Clamp insulin was determined at t = 120 min. At 140 min, 13μCi of 2[14C]deoxyglucose ([14C]2DG) was administered as an intravenous bolus. Blood was taken from t = 142–165min for determination of plasma [14C]2DG. Plasma lactate and catecholamines were determined at t = 165min. At t = 166min, mice were sacrificed by pentobarbital injection, and tissues immediately frozen in liquid nitrogen. Plasma glucose enrichments ([6,6-2H2]Glucose), isotopomer distribution and the enrichment ratio of deuterium on the fifth (C5) and second carbon (C2) of glucose were assessed by GC-MS as described previously.75 Glucose fluxes were assessed using non–steady-state equations (volume of distribution of glucose = 130 mL/kg).76 The contribution of gluconeogenesis was assessed as the ratio of C5 and C2 of plasma glucose.77,78. [14C]2DG in plasma samples, and [14C]2DG-6-phosphate in tissue samples were determined by liquid scintillation counting. The glucose metabolic index (Rg) was calculated as previously described.79 Plasma insulin was determined by RIA. Plasma lactate was determined by colorimetric assay (abcam). Plasma catecholamine levels were determined by HPLC. Stable isotopes were purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA). Radioactive tracers were purchased from PerkinElmer.
Catecholamine measurements
Mice were sacrificed using cervical dislocation and blood were collected immediately using cardiac puncture. EGTA-glutathione solution (pH6.0–7.4) [4.5g EGTA and 3.0g Glutathione/per 50 mL] was added at the time of blood collection (2 mL of solution was added per 100 mL of blood). After centrifuged at 4°C, 10000 rpm for 10 min, supernatants were collected and aliquoted and samples for catecholamine analysis were flash frozen in liquid nitrogen. Plasma for insulin measurements were stored at −20°C. Catecholamine and insulin measurements were performed by the VUMC Hormone Assay and Analytical Services Core.
Glycogen synthase activity, phosphorylase activity, and tissue content
Glycogen synthase activity was measured using the incorporation of glucose from UDP-[6-3H] D-glucose into glycogen using liver homogenates as previously described.80 Activity was calculated as the ratio of activated GS activity (-G6P) versus the total GS activity (+G6P). Glycogen phosphorylase activity was measured from liver homogenates using a calorimetric assay that measures the appearance of G1P in the presence of excess substrate (Abcam ab273271). For glycogen tissue content, frozen liver tissue (30–50 mg) and dilutions of glycogen type III obtained from rabbit liver (Sigma-Aldrich) were homogenized in 0.03 N HCl. An aliquot of the homogenate was mixed with 1.25 N HCl and heated for 1 h at 95°C. Samples were centrifuged at 18,400 × g, and 10 μL of supernatant was mixed with 1 mL of glucose oxidase reagent (Stanbio Laboratory). After a short incubation at 37°C, the absorbance was read at 505 nm.
QUANTIFICATION AND STATISTICAL ANALYSIS
All summary data are expressed as mean ± SEM. One-way, two-way ANOVA or student’s t test were used as indicated followed by correction for multiple comparisons test using Prism 7 (GraphPad La Jolla, CA). Sample size, specific test details, and level of significance is indicated as specific p values in figure legends.
Supplementary Material
Highlights.
The Hippo kinases phosphorylate p110α at T1061, which inhibits its activity
Activation of MST1/2 with epinephrine impairs PI3K signaling and glycogen synthesis
Loss of MST1/2, or mutating T1061A, prevents the effects of epinephrine on glycogen synthesis
ACKNOWLEDGMENTS
We thank Ismail Moarefi and Karine Roewer of CreLux WuXi AppTec for assisting in solving the structure of the p110α(T1061E)/ni-p85α complex. We thank Ed Kesicki and David McElliot of Petra Pharmaceuticals for helping coordinate our work with CreLux. We thank Kun-Liang Guan for providing CRISPR-engineered HEK293 cells. Isolation of primary hepatocytes was performed by Baran Ersoy and the Metabolic Phenotyping Core at Weill Cornell Medicine. Euglycemic clamp studies were performed by the Rodent Metabolic Phenotyping Core supported by Penn Diabetes Research Center grant P30-DK1952 and the Vanderbilt Mouse Metabolic Phenotyping Center (DK059637). The Vanderbilt Hormone Assay and Analytical Core performed the insulin and catecholamine analyses (DK059637 and DK020593). This work was supported in part by NIH CA230318 (M.D.G.), NIH R35 CA197588 (L.C.C.), a grant from the Breast Cancer Research Foundation (L.C.C.), and institutional funds from Weill Cornell Medicine.
DECLARATION OF INTERESTS
N.V. reports consultant and advisory board activities for Novartis, Petra Pharmaceuticals, Reactive Biosciences, and Magnet Biomedicine. E.A.K. is a shareholder of Eli Lilly and Company, and E.A.K. and K.K. are employees of Loxo Oncology at Lilly. L.C.C. is a founder and member of the board of directors of Agios Pharmaceuticals; is a founder and receives research support from Petra Pharmaceuticals; has equity in and consults for Cell Signaling Technologies, Volastra, Larkspur, and 1 Base Pharmaceuticals; and consults for Loxo-Lilly. J.L.J. has received consulting fees from Scorpion Therapeutics and Volastra Therapeutics. J.L.J. reports consultant activities for Scorpion Therapeutics and Volastra Therapeutics. M.D.G. reports personal fees from Novartis AG, Pfizer, Inc., and Scorpion Therapeutics. L.C.C., B.D.H., and M.D.G. are inventors on patents for Combination Therapy for PI3K-associated Disease or Disorder and The Identification of Therapeutic Interventions to Improve Response to PI3K Inhibitors for Cancer Treatment. B.D.H., L.C.C., and M.D.G. are co-founders and shareholders in Faeth Therapeutics. R.E.S. is on the sponsored advisory board for Miromatrix, Inc., and Lime Therapeutics and is a consultant and speaker for Alnylam, Inc. T.M.Y. is a stockholder and on the board of directors of DESTROKE, Inc., an early-stage start-up developing mobile technology for automated clinical stroke detection.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113535.
REFERENCES
- 1.Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, and Harrison SC (1996). Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat. Struct. Biol 3, 364–374. [DOI] [PubMed] [Google Scholar]
- 2.Fritsch R, de Krijger I, Fritsch K, George R, Reason B, Kumar MS, Diefenbacher M, Stamp G, and Downward J (2013). RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153, 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen M, Choi S, Jung O, Wen T, Baum C, Thapa N, Lambert PF, Rapraeger AC, and Anderson RA (2019). The Specificity of EGF-Stimulated IQGAP1 Scaffold Towards the PI3K-Akt Pathway is Defined by the IQ3 motif. Sci. Rep 9, 9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, and Abraham RT (2017). The PI3K Pathway in Human Disease. Cell 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopkins BD, Goncalves MD, and Cantley LC (2020). Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol 16, 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manning BD, and Toker A (2017). AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cori GT, Cori CF, and Buchwald KW (1930). The Mechanism of Epinephrine Action V. Changes in Liver Glycogen and Blood Lactic Acid After Injection of Epinephrine and Insulin. J. Biol. Chem 86, 375–388. [Google Scholar]
- 8.Dufour S, Lebon V, Shulman GI, and Petersen KF (2009). Regulation of net hepatic glycogenolysis and gluconeogenesis by epinephrine in humans. Am. J. Physiol. Endocrinol. Metab 297, E231–E235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deibert DC, and DeFronzo RA (1980). Epinephrine-induced insulin resistance in man. J. Clin. Invest 65, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Battram DS, Graham TE, and Dela F (2007). Caffeine’s impairment of insulin-mediated glucose disposal cannot be solely attributed to adrenaline in humans. J. Physiol 583, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cori CF, and Cori GT (1929). The Influence of Insulin and Epinephrine on Glycogen Formation in the Liver. J. Biol. Chem 85, 275–280. [Google Scholar]
- 12.Rizza RA, Cryer PE, Haymond MW, and Gerich JE (1980). Adrenergic mechanisms for the effects of epinephrine on glucose production and clearance in man. J. Clin. Invest 65, 682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, Ladewig E, Gorelick A, Lin TY, Toska E, et al. (2019). Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science 366, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun M, Hart JR, Hillmann P, Gymnopoulos M, and Vogt PK (2011). Addition of N-terminal peptide sequences activates the oncogenic and signaling potentials of the catalytic subunit p110alpha of phosphoinositide-3-kinase. Cell Cycle 10, 3731–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickson JMJ, Lee WJ, Shepherd PR, and Buchanan CM (2013). Enzyme activity effects of N-terminal His-tag attached to catalytic subunit of phosphoinositide-3-kinase. Biosci. Rep 33, e00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- 17.Harvey KF, Pfleger CM, and Hariharan IK (2003). The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467. [DOI] [PubMed] [Google Scholar]
- 18.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, and Pan D (2007). Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Huang J, Dong J, and Pan D (2003). hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456. [DOI] [PubMed] [Google Scholar]
- 20.Miller CJ, Lou HJ, Simpson C, van de Kooij B, Ha BH, Fisher OS, Pirman NL, Boggon TJ, Rinehart J, Yaffe MB, et al. (2019). Comprehensive profiling of the STE20 kinase family defines features essential for selective substrate targeting and signaling output. PLoS Biol 17, e2006540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo JS, Lu W, Lu S, et al. (2015). MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun 6, 8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, and Turk BE (2004). A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29. [DOI] [PubMed] [Google Scholar]
- 23.Ardestani A, Tremblay MS, Shen W, and Maedler K (2019). Neratinib is an MST1 inhibitor and restores pancreatic β-cells in diabetes. Cell Death Discov 5, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.te Lintel Hekkert M, Newton G, Chapman K, Aqil R, Downham R, Yan R, Merkus D, Whitlock G, Lane CAL, Cawkill D, et al. (2021). Preclinical trial of a MAP4K4 inhibitor to reduce infarct size in the pig: does cardioprotection in human stem cell-derived myocytes predict success in large mammals? Basic Res. Cardiol 116, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, and Koike T (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757. [DOI] [PubMed] [Google Scholar]
- 26.Hon WC, Berndt A, and Williams RL (2012). Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 31, 3655–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, et al. (2013). Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett 23, 3741–3748. [DOI] [PubMed] [Google Scholar]
- 28.Barsanti PA, Pan Y, Lu Y, Jain R, Cox M, Aversa RJ, Dillon MP, Elling R, Hu C, Jin X, et al. (2015). Structure-Based Drug Design of Novel, Potent, and Selective Azabenzimidazoles (ABI) as ATR Inhibitors. ACS Med. Chem. Lett 6, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heffron TP, Heald RA, Ndubaku C, Wei B, Augistin M, Do S, Edgar K, Eigenbrot C, Friedman L, Gancia E, et al. (2016). The Rational Design of Selective Benzoxazepin Inhibitors of the alpha-Isoform of Phosphoinositide 3-Kinase Culminating in the Identification of (S)-2-((2-(1-Isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d] [1, 4]oxazepin-9-yl)oxy)propanamide (GDC-0326). J. Med. Chem 59, 985–1002. [DOI] [PubMed] [Google Scholar]
- 30.Han W, Menezes DL, Xu Y, Knapp MS, Elling R, Burger MT, Ni ZJ, Smith A, Lan J, Williams TE, et al. (2016). Discovery of imidazo [1,2-a]-pyridine inhibitors of pan-PI3 kinases that are efficacious in a mouse xenograft model. Bioorg. Med. Chem. Lett 26, 742–746. [DOI] [PubMed] [Google Scholar]
- 31.Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, and Amzel LM (2009). A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA 106, 16996–17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker EH, Perisic O, Ried C, Stephens L, and Williams RL (1999). Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402, 313–320. [DOI] [PubMed] [Google Scholar]
- 33.Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, and Williams RL (2000). Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6, 909–919. [DOI] [PubMed] [Google Scholar]
- 34.Hol WG, van Duijnen PT, and Berendsen HJ (1978). The alpha-helix dipole and the properties of proteins. Nature 273, 443–446. [DOI] [PubMed] [Google Scholar]
- 35.Gymnopoulos M, Elsliger MA, and Vogt PK (2007). Rare cancer-specific mutations in PIK3CA show gain of function. Proc. Natl. Acad. Sci V 104, 5569–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, and Amzel LM (2007). The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 318, 1744–1748. [DOI] [PubMed] [Google Scholar]
- 37.Shekar SC, Wu H, Fu Z, Yip SC, Nagajyothi, Cahill SM, Cahill SM, Girvin ME, and Backer JM (2005). Mechanism of constitutive phosphoinositide 3-kinase activation by oncogenic mutants of the p85 regulatory subunit. J. Biol. Chem 280, 27850–27855. [DOI] [PubMed] [Google Scholar]
- 38.Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, Dhanak D, Copeland RA, Gontarek RR, Tummino PJ, and Luo L (2008). Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem. J 409, 519–524. [DOI] [PubMed] [Google Scholar]
- 39.Chaussade C, Cho K, Mawson C, Rewcastle GW, and Shepherd PR (2009). Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem. Biophys. Res. Commun 381, 577–581. [DOI] [PubMed] [Google Scholar]
- 40.Gkeka P, Evangelidis T, Pavlaki M, Lazani V, Christoforidis S, Agianian B, and Cournia Z (2014). Investigating the structure and dynamics USAof the PIK3CA wild-type and H1047R oncogenic mutant. PLoS. Comput. Biol 10, e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, and Williams RL (2007). Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317, 239–242. [DOI] [PubMed] [Google Scholar]
- 42.Burke JE (2018). Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 71, 653–673. [DOI] [PubMed] [Google Scholar]
- 43.Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al. (2012). Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y, Pan D, Taylor SS, Lai ZC, and Guan KL (2013). Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev 27, 1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomes A, Soares R, Costa R, Marino F, Cosentino M, Malagon MM, and Ribeiro L (2020). High-fat diet promotes adrenaline production by visceral adipocytes. Eur. J. Nutr 59, 1105–1114. [DOI] [PubMed] [Google Scholar]
- 46.Fan F, He Z, Kong LL, Chen Q, Yuan Q, Zhang S, Ye J, Liu H, Sun X, Geng J, et al. (2016). Pharmacological targeting of kinases MST1 and MST2 augments tissue repair and regeneration. Sci. Transl. Med 8, 352ra108. [DOI] [PubMed] [Google Scholar]
- 47.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, and Bardeesy N (2009). Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakamoto K, Hirshman MF, Aschenbach WG, and Goodyear LJ (2002). Contraction regulation of Akt in rat skeletal muscle. J. Biol. Chem 277, 11910–11917. [DOI] [PubMed] [Google Scholar]
- 49.Petersen KF, Laurent D, Rothman DL, Cline GW, and Shulman GI (1998). Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J. Clin. Invest 101, 1203–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bessey PQ, Brooks DC, Black PR, Aoki TT, and Wilmore DW (1983). Epinephrine acutely mediates skeletal muscle insulin resistance. Surgery 94, 172–179. [PubMed] [Google Scholar]
- 51.De Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, and Bos JL (1998). Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477. [DOI] [PubMed] [Google Scholar]
- 52.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, and Graybiel AM (1998). A Family of cAMP-Binding Proteins That Directly Activate Rap1. Science 282, 2275–2279. [DOI] [PubMed] [Google Scholar]
- 53.Araiz C, Yan A, Bettedi L, Samuelson I, Virtue S, McGavigan AK, Dani C, Vidal-Puig A, and Foukas LC (2019). Enhanced β-adrenergic signalling underlies an age-dependent beneficial metabolic effect of PI3K p110α inactivation in adipose tissue. Nat. Commun 10, 1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Q, Li J, Zhang W, Xiao C, Zhang S, Nian C, Li J, Su D, Chen L, Zhao Q, et al. (2021). Glycogen accumulation and phase separation drives liver tumor initiation. Cell 184, 559–5576.e19. [DOI] [PubMed] [Google Scholar]
- 55.Xu S, Liu Y, Hu R, Wang M, Stöhr O, Xiong Y, Chen L, Kang H, Zheng L, Cai S, et al. (2021). TAZ inhibits glucocorticoid receptor and coordinates hepatic glucose homeostasis in normal physiological states. Elife 10, e57462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petersen MC, and Shulman GI (2018). Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev 98, 2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis GF, Carpentier AC, Pereira S, Hahn M, and Giacca A (2021). Direct and indirect control of hepatic glucose production by insulin. Cell Metab 33, 709–720. [DOI] [PubMed] [Google Scholar]
- 58.Jensen J, Ruge T, Lai Y-C, Svensson MK, and Eriksson JW (2011). Effects of adrenaline on whole-body glucose metabolism and insulin-mediated regulation of glycogen synthase and PKB phosphorylation in human skeletal muscle. Metabolism 60, 215–226. [DOI] [PubMed] [Google Scholar]
- 59.Raje V, Ahern KW, Martinez BA, Howell NL, Oenarto V, Granade ME, Kim JW, Tundup S, Bottermann K, Gödecke A, et al. (2020). Adipocyte lipolysis drives acute stress-induced insulin resistance. Sci. Rep 10, 18166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danai LV, Flach RJR, Virbasius JV, Menendez LG, Jung DY, Kim JH, Kim JK, and Czech MP (2015). Inducible Deletion of Protein Kinase Map4k4 in Obese Mice Improves Insulin Sensitivity in Liver and Adipose Tissues. Mol. Cell Biol 35, 2356–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang X, Guilherme A, Chakladar A, Powelka AM, Konda S, Virbasius JV, Nicoloro SMC, Straubhaar J, and Czech MP (2006). An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARγ, adipogenesis, and insulin-responsive hexose transport. Proc. Natl. Acad. Sci. USA 103, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sartorius T, Staiger H, Ketterer C, Heni M, Machicao F, Guilherme A, Grallert H, Schulze MB, Boeing H, Stefan N, et al. (2012). Association of common genetic variants in the MAP4K4 locus with prediabetic traits in humans. PLoS One 7, e47647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cansby E, Amrutkar M, Mannerås Holm L, Nerstedt A, Reyahi A, Stenfeldt E, Borén J, Carlsson P, Smith U, Zierath JR, and Mahlapuu M (2013). Increased expression of STK25 leads to impaired glucose utilization and insulin sensitivity in mice challenged with a high-fat diet. FASEB J 27, 3660–3671. [DOI] [PubMed] [Google Scholar]
- 64.Iglesias C, Floridia E, Sartages M, Porteiro B, Fraile M, Guerrero A, Santos D, Cuñarro J, Tovar S, Nogueiras R, et al. (2017). The MST3/STK24 kinase mediates impaired fasting blood glucose after a high-fat diet. Diabetologia 60, 2453–2462. [DOI] [PubMed] [Google Scholar]
- 65.Virbasius JV, and Czech MP (2016). Map4k4 Signaling Nodes in Metabolic and Cardiovascular Diseases. Trends Endocrinol. Metab 27, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Romanienko PJ, Giacalone J, Ingenito J, Wang Y, Isaka M, Johnson T, You Y, and Mark WH (2016). A Vector with a Single Promoter for In Vitro Transcription and Mammalian Cell Expression of CRISPR gRNAs. PLoS One 11, e0148362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 68.Johnson JL, Yaron TM, Huntsman EM, Kerelsky A, Song J, Regev A, Lin TY, Liberatore K, Cizin DM, Cohen BM, et al. (2023). An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yaron TM, Heaton BE, Levy TM, Johnson JL, Jordan TX, Cohen BM, Kerelsky A, Lin TY, Liberatore KM, Bulaon DK, et al. (2022). Host protein kinases required for SARS-CoV-2 nucleocapsid phosphorylation and viral replication. Sci. Signal 15, eabm0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scapa EF, Pocai A, Wu MK, Gutierrez-Juarez R, Glenz L, Kanno K, Li H, Biddinger S, Jelicks LA, Rossetti L, and Cohen DE (2008). Regulation of energy substrate utilization and hepatic insulin sensitivity by phosphatidylcholine transfer protein/StarD2. FASEB J 22, 2579–2590. [DOI] [PubMed] [Google Scholar]
- 71.Lee P, Peng H, Gelbart T, and Beutler E (2004). The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-transferrin receptor 2-and beta 2-microglobulin-deficient hepatocytes. Proc. Natl. Acad. Sci. USA 101, 9263–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berglund ED, Li CY, Poffenberger G, Ayala JE, Fueger PT, Willis SE, Jewell MM, Powers AC, and Wasserman DH (2008). Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 57, 1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ayala JE, Bracy DP, McGuinness OP, and Wasserman DH (2006). Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55, 390–397. [DOI] [PubMed] [Google Scholar]
- 74.Hasenour CM, Wall ML, Ridley DE, Hughey CC, James FD, Wasserman DH, and Young JD (2015). Mass spectrometry-based micro-assay of (2)H and (13)C plasma glucose labeling to quantify liver metabolic fluxes in vivo. Am. J. Physiol. Endocrinol. Metab 309, E191–E203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hughey CC, James FD, Bracy DP, Donahue EP, Young JD, Viollet B, Foretz M, and Wasserman DH (2017). Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice. J. Biol. Chem 292, 20125–20140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steele R, Wall JS, De Bodo RC, and Altszuler N (1956). Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am. J. Physiol 187, 15–24. [DOI] [PubMed] [Google Scholar]
- 77.Burgess SC, Hausler N, Merritt M, Jeffrey FMH, Storey C, Milde A, Koshy S, Lindner J, Magnuson MA, Malloy CR, and Sherry AD (2004). Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J. Biol. Chem 279, 48941–48949. [DOI] [PubMed] [Google Scholar]
- 78.Antoniewicz MR, Kelleher JK, and Stephanopoulos G (2011). Measuring deuterium enrichment of glucose hydrogen atoms by gas chromatography/mass spectrometry. Anal. Chem 83, 3211–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kraegen EW, James DE, Jenkins AB, and Chisholm DJ (1985). Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am. J. Physiol 248, E353–E362. [DOI] [PubMed] [Google Scholar]
- 80.Li Q, Zhao Q, Zhang J, Zhou L, Zhang W, Chua B, Chen Y, Xu L, and Li P (2019). The Protein Phosphatase 1 Complex Is a Direct Target of AKT that Links Insulin Signaling to Hepatic Glycogen Deposition. Cell Rep 28, 3406–3422.e7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data points used to create the graphs and uncropped western blots are provided in Data S1. Crystallography data have been deposited at Protein DataBank and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Akt (Ser473) Rabbit mAb | Cell Signaling Technology | Cat#3787; RRID:AB_331170 |
| Phospho-Akt (Ser473) Rabbit mAb | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| Phospho-Akt (Thr308) Rabbit mAb | Cell Signaling Technology | Cat#4056; RRID:AB_331163 |
| Phospho-Akt2 (Ser474) Rabbit mAb | Cell Signaling Technology | Cat#8599; RRID:AB_2630347 |
| Akt (pan) (40D4) Mouse mAb | Cell Signaling Technology | Cat#2920; RRID:AB_1147620 |
| AKT1 (C73H10) [#2967] | Cell Signaling Technology | Cat#2938; RRID:AB_915788 |
| Akt2 (D6G4) Rabbit mAb | Cell Signaling Technology | Cat#3063; RRID:AB_2225186 |
| Phospho-CREB (Ser133) Rabbit mAb | Cell Signaling Technology | Cat#9198; RRID:AB_2561044 |
| Phospho-LATS1 (Thr1079) Rabbit mAb | Cell Signaling Technology | Cat#8654; RRID:AB_10971635 |
| Phospho-MOB1 (Thr35) Rabbit mAb | Cell Signaling Technology | Cat#8699; RRID:AB_11139998 |
| Phospho-YAP (Ser127) Rabbit mAb | Cell Signaling Technology | Cat#13008; RRID:AB_2650553 |
| Phospho-PKA Substrate (RRXS*/T*) Rabbit mAb | Cell Signaling Technology | Cat#9624; RRID:AB_331817 |
| PI3 Kinase p110α (C73F8) Rabbit mAb | Cell Signaling Technology | Cat#4249; RRID:AB_2165248 |
| CREB (D76D11) Rabbit mAb | Cell Signaling Technology | Cat#4820; RRID:AB_1903940 |
| MOB1 (E1N9D) Rabbit mAb | Cell Signaling Technology | Cat#13730; RRID:AB_2783010 |
| LATS1 Antibody Rabbit Ab | Cell Signaling Technology | Cat#9153; RRID:AB_2296754 |
| MST1 (D8B9Q) Rabbit mAb | Cell Signaling Technology | Cat#14946; RRID:AB_2798654 |
| GAPDH (D16H11) Rabbit mAb | Cell Signaling Technology | Cat#5174; RRID:AB_10622025 |
| Recombinant Anti-STK3/MST-2 antibody [EP1466Y] | Abcam | Cat#ab52641; RRID:AB_882734 |
| p110 (pT1061) polyclonal antibody | This paper (from CST) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Alpelisib (BYL-719) | MEDCHEM EXPRESS | Cat#HY-15244 |
| Neratinib (HKI-272) | SelleckChem | Cat#S2150 |
| Go6976 | EMD Millipore | Cat#365250 |
| DMX-5804 (HY-111754) | MEDCHEM EXPRESS | Cat#HY-111754 |
| XMU-MP-1 | SelleckChem | Cat#S8334 |
| H 89 dihydrochloride | Tocris Bioscience | Cat#2910 |
| AT13148 | SelleckChem | Cat#S7563 |
| Propranolol hydrochloride | Sigma Aldrich | Cat#P0884 |
| MST1 | SignalChem | S25–10G |
| HGK | SignalChem | M26–11G |
| MST2 | SignalChem | S24–10G |
| MST3 | SignalChem | S42–11G |
| TAOK1 | SignalChem | T24–11G |
| GLK | SignalChem | M25–11G |
| AKT1 | SignalChem | A16–10G |
| AMPK1 | SignalChem | P47–10H |
| ERK2 | SignalChem | M28–10G |
| PDK1 | SignalChem | P14–10H |
| CK1A | SignalChem | C64–10G |
| GSK3A | SignalChem | G08–10G |
| IKKB | SignalChem | I03–10BG |
| IRAK4 | SignalChem | I12–10G |
| ASK1 | SignalChem | M13–11G |
| GCN2 | SignalChem | E12–11G |
| EEF2K | SignalChem | E01–10G |
| JAK1 | SignalChem | J01–11G |
| CHK1 | SignalChem | C47–10G |
| CHK2 | SignalChem | C48–10G |
| DAPK1 | SignalChem | D01–11G |
| PKACA | SignalChem | P51–10G |
| PKCA | SignalChem | P61–18G |
| PAK1 | SignalChem | P02B-10BG |
| AurB | SignalChem | A31–10G |
| CDK1 | SignalChem | C22–10G |
| DYRK1A | SignalChem | D09–10G |
| SRC | SignalChem | S19–18G |
| ABL1 | SignalChem | A03–18H |
| LATS2 | Abnova | P5574 |
| OXSR1 | Abnova | P5614 |
| CK2 | NEB | P6010L |
| TGFBR1 | Proqinase | 0397-0000-1 |
| WEE1 | BPS Biosciences | 40412 |
| PIK3CG was purchased from Thermo Fisher | SignalChem | P27–10CG |
| Alpelisib (BYL-719) | MEDCHEM EXPRESS | Cat#HY-15244 |
| Critical commercial assays | ||
| Transcreener ADP fluorescence intensity assay | Bellbrook Labs | 3013-A |
| Deposited data | ||
| Crystal structure | Protein DataBank | entry D_1292124630 (8a.m.0) |
| Experimental models: Cell lines | ||
| MDA-MB-231 | ATCC | HTB-26 |
| Huh7 | Gift from Lab of Robert Schwartz | N/A |
| HEK293A | ATCC | CRL-1573 |
| MCF10A | ATCC | CRL-10317 |
| Primary mouse hepatocytes | isolated from mouse liver | N/A |
| Experimental models: Organisms/strains | ||
| Stk3f/f,Stk4f/f mice | Jackson laboratories | Stock No: 017635 |
| Liver specific MST1/2 double knockout mice (Stk3f/f,Stk4f/f,Alb-Cre−/+) | This paper | N/A |
| Albumin-Cre mice | Jackson laboratories | Stock No: 003574 |
| Homozygous null mice for the Adrb1 and Adrb2 genes (Adrb1−/−;Adrb2−/−) | Jackson laboratories | Stock No: 003810 |
| Pnpla2f/f mice | Jackson laboratories | Stock No: 024278 |
| Pik3caT1061A mice This paper | N/A | |
| Adipoq-Cre mice | Gift from Lab of James Lo | Stock No: 028020 |
| Oligonucleotides | ||
| PIK3CAV143I to WT isoleucine Forward: GACTTCCGAAGAAATATTCTGAACGTTTGTAAA | This paper | N/A |
| PIK3CAV143Ito WT isoleucine Reverse: TTTACAAACGTTCAGAATATTTCTTCGGAAGTC. | This paper | N/A |
| PIK3CAT1061A mutagenesis primer FW: 5′-caaaaatggattggatcttccacgcaattaaacagcatgcattgaac-3′ | This paper | N/A |
| PIK3CAT1061A mutagenesis primer RV: 5′-gttcaatgcatgctgtttaattgcgtggaagatccaatccatttttg-3 | This paper | N/A |
| PIK3CAT1061E mutagenesis primer FW: 5′-cctcagttcaatgcatgctgtttaatctcgtggaagatccaatccatttttgttg-3 | This paper | N/A |
| PIK3CAT1061E mutagenesis primer Rev: 5′-caacaaaaatggattggatcttccacgagattaaacagcatgcattgaactgagg-3 | This paper | N/A |
| PIK3CAW1057A mutagenesis primer FW: 5′-ctgtttaattgtgtggaagatcgcatccatttttgttgtccagcca-3′ | This paper | N/A |
| PIK3CAW1057A mutagenesis primer RV: 5′-tggctggacaacaaaaatggatgcgatcttccacacaattaaacag-3 | This paper | N/A |
| Recombinant DNA | ||
| pBabe puro HA-PIK3CA | Addgene | plasmid #12522 |
| Software and algorithms | ||
| Prism 7 | GraphPad La Jolla, CA | N/A |
| BioRender | https://biorender.com/ | N/A |
| Geneious | http://www.geneious.com/ | RRID:SCR_010519 |
| Chemdraw | http://www.perkinelmer.co.uk/category/chemdraw | RRID:SCR_016768 |
| Pymol | http://www.pymol.org/ | RRID:SCR_000305 |