

Abstract
It has been reported that CDK8 plays a key role in acute myeloid leukaemia. Here, a total of 40 compounds were rational designed and synthesised based on the previous SAR. Among them, compound 12 (3-(3-(furan-3-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzamide) showed the most potent inhibiting activity against CDK8 with an IC50 value of 39.2 ± 6.3 nM and anti AML cell proliferation activity (molm-13 GC50 = 0.02 ± 0.01 μM, MV4-11 GC50 = 0.03 ± 0.01 μM). Mechanistic studies revealed that this compound 12 could inhibit the phosphorylation of STAT-1 and STAT-5. Importantly, compound 12 showed relative good bioavailability (F = 38.80%) and low toxicity in vivo. This study has great significance for the discovery of more efficient CDK8 inhibitors and the development of drugs for treating AML in the future.
Keywords: CDK8 inhibitor, STAT-1, STAT-5, AML
Introduction
Acute myeloid leukaemia (AML), a highly heterogeneous disease derived from the malignant clonal proliferation of abnormally differentiated myeloid lineage cells, is very difficult to be cured in young adults1,2. In the recent 40 years, daunorubicin or idarubicin and cytarabine as the standard induction (initial) chemotherapy were selected for the treatment of AML with only 70-80% of the complete response due to the intrinsic disease resistance1. In recent years, more and more targeted inhibitors have been used to treat AML with better therapeutic effects, such as IDH1/IDH2 inhibitors3,4, FLT3 inhibitors3,5, BCL-2 inhibitors3,4 etc. Therefore, to discover novel targeted inhibitors for treatment of AML is of great significance, especially for patients who have been resistant to drugs.
Signal transducer and activator of transcription 5 (STAT-5) had been found to exist in AML cells and exhibited sustained high activity. Importantly, STAT-5 also played a key role in mediating the relationship between several malignant diseases in AML cells5–9. In the recent ten years, many CDK8 inhibitors have been reported to exhibited good anti-tumour activity, such as cortistainA10, AU1-10011, MK-25612, SEL120-34A7, CCT25154513, CCT25159114, MSC253081815, BI-134716,17, etc. It has been reported compound SEL120-34A (Figure 1) as the potent CDK8 inhibitor could inhibit the phosphorylation of STAT1 S727 and STAT5 S726 in AML cells7. More importantly, in 2019, SEL120-34A was approved for clinical trials for treatment of advanced AML (NCT04021368)18. In addition, many CDK8 inhibitors were also reported to have good anti proliferative activity on AML cells10–12,19–22, such as cortistatinA10, AU1-10011, MK-25612, etc (Figure 1). Although, three CDK8 inhibitors, such as BCD-115 (NCT03065010), SEL120-34A (NCT04021368, NCT05052255) and TSN-084 (NCT05300438) have been approved for clinical trials, however, due to toxicity and drug PK efficacy, there are no commercially available drugs on the market. So, we think it is great significance to discover novel potent CDK8 inhibitors for treatment of AML through inhibiting the phosphorylation of STAT1 S727 and STAT5 S726.
Figure 1.

Chemical structures of some CDK8 inhibitors.
In this work, based on rational design and structure-activity relationship discussion, compound 12 (3–(3-(furan-3-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzamide) as a potent CDK8 inhibitor was found. Further mechanistic studies showed that compound 12 could inhibit the phosphorylation of STAT5 S726 and showed good antiproliferative activity for acute myeloid leukaemia cell lines (molm-13 GC50 = 0.02 ± 0.01 μM, MV4-11 GC50 = 0.03 ± 0.01 μM). Importantly, compound 12 showed relatively good bioavailability (F = 38.80%, which is better than our previous report) and low toxicity in vivo. It is of great significance for the discovery of more efficient CDK8 inhibitors and the development of drugs for treating AML.
Results and discussions
Design and optimisation
In order to further optimise on the hit compound C43, we analysed the docking model of it with CDK8. As shown in Figure 2, we noticed that the azaindole of compound C43 forms hydrogen-bonding interactions with amino acid residues Asp98 and Ala100. The benzamide fragment forms cation-pi stacking with amino acid residue Arg356 and hydrogen-bonding interaction with the hydroxyl group of Tyr32. Importantly, there is a small cavity near the 3-position of the azaindole ring, which consists of the phenyl ring of the amino acid residue Phe97 and the amino group of Lys52 (Figure 2). In addition, based on the previous SAR21, we found that the activity of compounds that were introduced into other groups in the small cavity was significantly reduced, only compounds C5 and C8 which with 3-furyl and phenyl still had good activity, with IC50 values of 85.1 ± 5.0 nM and 60.3 ± 1.2 nM, respectively. Therefore, we believe that the introduction of aromatic heterocycle or benzene ring derivatives at the 3-position of the azaindole ring can enhance the activity by forming hydrophobic interactions, pi-pi stacking interactions or hydrogen bonding interactions.
Figure 2.

Binding mode of compound C43 with active site of CDK8 (PDB: 5IDN). CDK8 is shown in gray ribbons with selected residues coloured green. Hydrogen bonds are drawn as yellow dashed lines, and pi-pi stacking is drawn as magenta dashed lines. Compound 43 is shown with blue stick. The illustration was generated using PyMOL.
2.2. Chemistry
The preparation of title compounds 1-40 was described in Schemes 1–2. As shown in Scheme 1, compounds 1-11 were obtained through Suzuki Reaction. As shown in Scheme 2, compounds 12-40 were obtained through 5-step reaction. (1) The nitrogen atom of 5-bromo-7-azaindole was protected by p-methylbenzenesulfonyl to obtain compound M1; (2) The key intermediate M2 was synthesised by Suzuki Reaction; (3) Compound M2 went through Halogenated Reaction to obtain compound M3; (4) The key intermediate M4 was prepared through Suzuki-Miyaura reaction and the synthesis method of compounds M5-M32 was the same as compound M4; (5) p-methylbenzenesulfonyl of compound M4 was removed under the condition of sodium hydroxide at 75 °C to obtain compounds 12, the synthesis method of compounds 13-40 was the same as compound 12.
Scheme 1.

Synthesis of compounds 1-11a
aReagents and conditions: A. K2CO3, Pd(dppf)Cl2, 1,4-dioxne, H2O, 85 °C, 14 h.
Scheme 2.

Synthesis of compounds 12-40a
aReagents and conditions: A. K2CO3, Pd(dppf)Cl2, 1,4-dioxne, H2O, 85 °C, 14 h; B. NaOH, THF, H2O, 35 °C, 6 h; C. DMF, NIS, 85 °C, 12 h; D. NaOH, CH3CH2OH, H2O, 75 °C, 2 h.
SAR study
A total of 40 compounds were designed and synthesised, and their CDK8 inhibition rates at 200 nM were screened. Some of them with high inhibition rates were further measured for their IC50 values. Compound SEL-120 34 A was selected as a positive control.
First, in order to determine whether the previous 7-azaindole is still a good skeleton after the introduction of the amide group, compounds 1-10 were designed and synthesised. As shown in Table 1, their activity was significantly reduced, except for compound 6, with the IC50 value of 64.5 ± 3.8 nM, showing that 7-azaindole is still a good skeleton.
Table 1.
The evaluation of the activity of compounds 1-10 on CDK8.
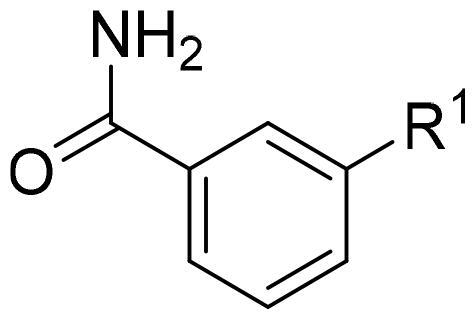
| |||
|---|---|---|---|
| Compounds | R1 | Inhibition rate @ 200 nMa (%)a | CDK8 IC50 (nM)b |
| 1 |
|
63.0 ± 4.5 | 105.9 ± 4.6 |
| 2 |
|
35.2 ± 2.8 | NTc |
| 3 |
|
50.8 ± 2.3 | 176.5 ± 7.2 |
| 4 |
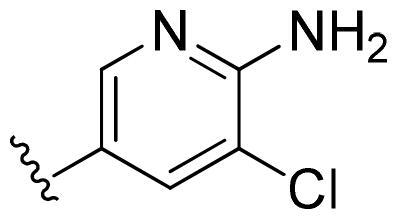
|
45.6 ± 4.2 | NT |
| 5 |
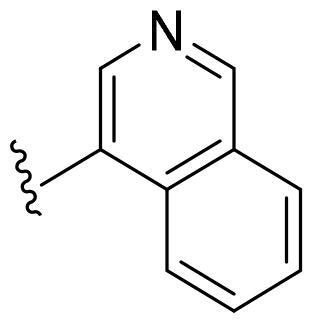
|
55.7 ± 3.8 | 186.3 ± 4.5 |
| 6 |
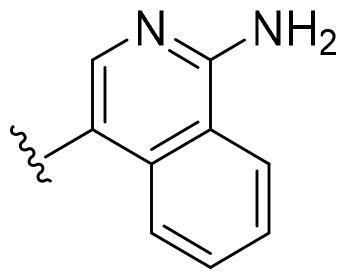
|
75.8 ± 5.9 | 64.5 ± 3.8 |
| 7 |
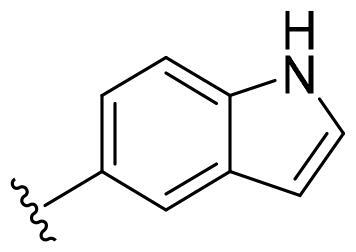
|
59.1 ± 2.1 | 212.3 ± 6.1 |
| 8 |
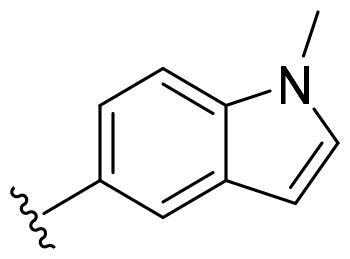
|
51.7 ± 5.4 | 255.3 ± 4.5 |
| 9 |
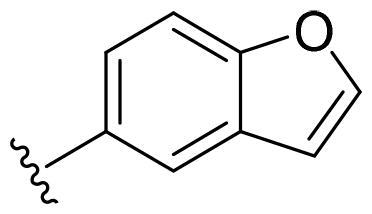
|
60.4 ± 4.3 | 189.6 ± 4.9 |
| 10 |
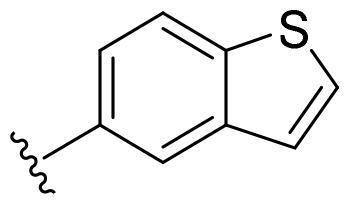
|
38.3 ± 2.2 | NT |
| SEL-120-34A | / | 82.5 ± 5.8 | 37.2 ± 2.5 |
| C43 | / | 73.2 ± 4.3 | 52.6 ± 3.5 |
CDK8 inhibition rate at 200 nM was determined by CDK8 enzyme activity assay. Values from two experiments and results were shown as means ± SD.
IC50 values were determined by CDK8 enzyme activity assay. Values from three dependent experiments and results were shown as means ± SD. cNT: Not Test.
Based on these, compounds 11-41 were designed and synthesised through introducing chlorine atom, aromatic heterocycle or benzene ring derivatives. As shown in Table 2, the activity of compound 12 with 3-furyl is the most active and significantly higher than that of C43, with an IC50 value of 39.2 ± 6.3 nM. When 3-furyl was replaced by 3-thienyl to give compound 13, its activity decreased a little, with an IC50 value of 55.7 ± 3.0 nM. The activity of other compounds decreased, especially compound 17, its activity decreased significantly, due to the introduction of 3-methylimidazolyl group, it has a large steric hindrance, which is the reason for the decreased activity. There is a significant difference in activity between compounds 20-41, showing that the size of the functional group and the electron cloud density of the aromatic ring have a significant impact on the activity.
Table 2.
The evaluation of the activity of compounds 11-40 on CDK8.
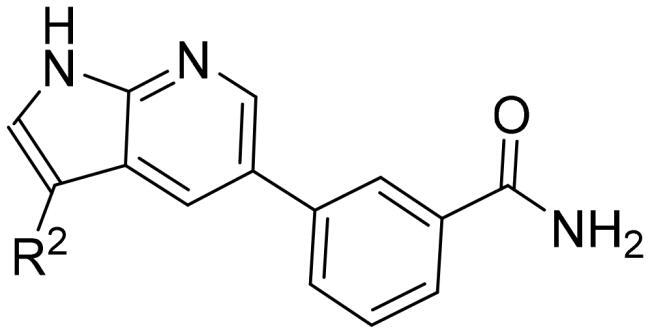
| |||
|---|---|---|---|
| Compounds | R2 | Inhibition rate @ 200 nM (%)a |
CDK8 IC50 (nM)b |
| 11 | Cl | 51.8 ± 4.3 | NTc |
| 12 |
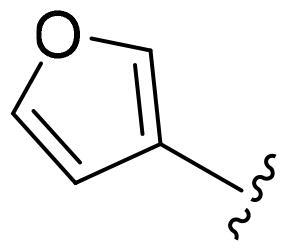
|
80.9 ± 3.5 | 39.2 ± 6.3 |
| 13 |
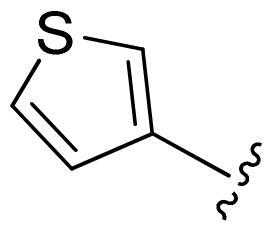
|
70.4 ± 3.6 | 65.7 ± 3.1 |
| 14 |
|
52.5 ± 2.8 | 185.7 ± 4.6 |
| 15 |
|
49.1 ± 3.4 | NT |
| 16 |
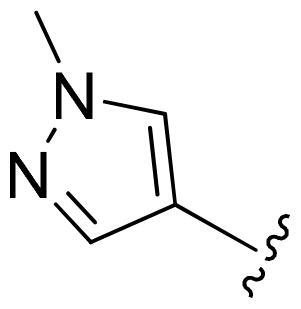
|
29.5 ± 2.3 | NT |
| 17 |
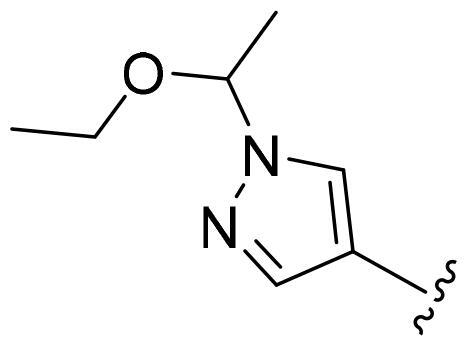
|
46.5 ± 1.8 | NT |
| 18 |
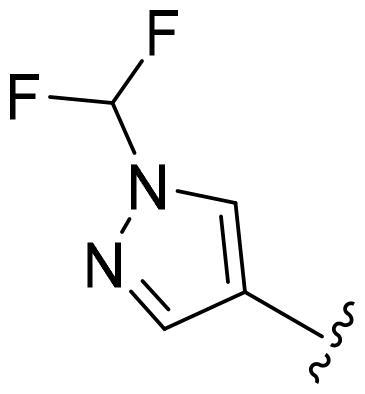
|
32.5 ± 4.9 | NT |
| 19 |
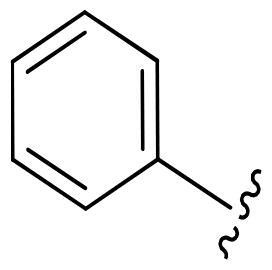
|
57.4 ± 1.4 | 157.7 ± 4.3 |
| 20 |
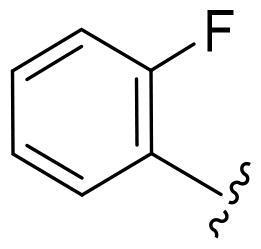
|
58.7 ± 3.3 | 160.5 ± 3.1 |
| 21 |
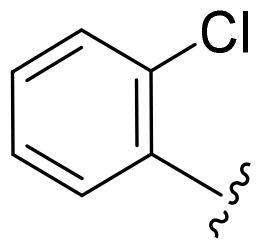
|
23.6 ± 2.6 | NT |
| 22 |
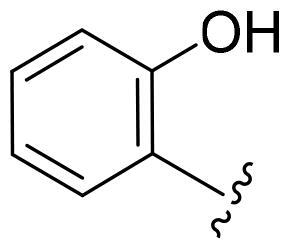
|
36.2 ± 2.7 | NT |
| 23 |
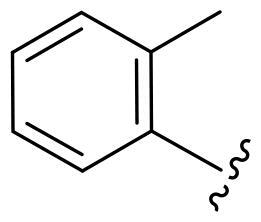
|
51.6 ± 2.6 | 192.6 ± 6.6 |
| 24 |
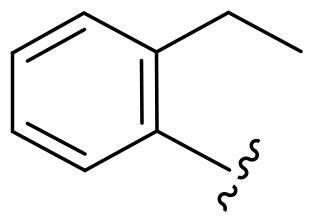
|
24.6 ± 2.4 | NT |
| 25 |
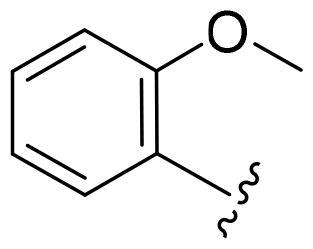
|
61.98 ± 4.6 | 135.7 ± 5.3 |
| 26 |
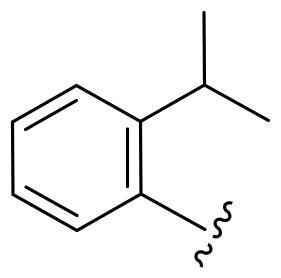
|
33.1 ± 4.2 | NT |
| 27 |
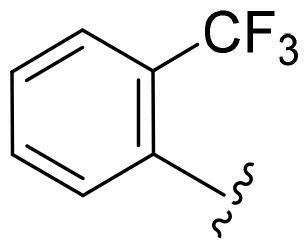
|
15.9 ± 5.2 | NT |
| 28 |
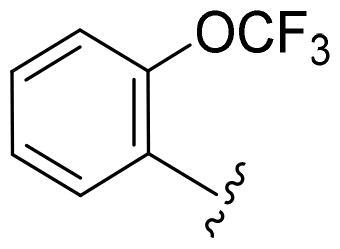
|
16.9 ± 2.2 | NT |
| 29 |
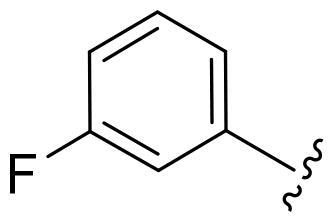
|
23.1 ± 2.5 | NT |
| 30 |
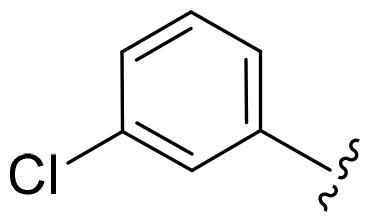
|
65.5 ± 3.8 | 86.7 ± 6.5 |
| 31 |
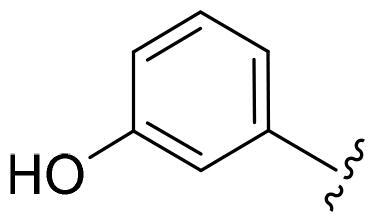
|
30.5 ± 2.3 | NT |
| 32 |
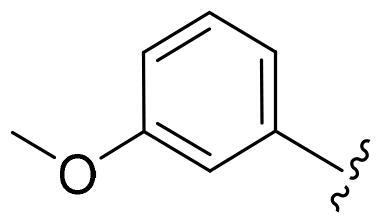
|
43.97 ± 3.8 | NT |
| 33 |
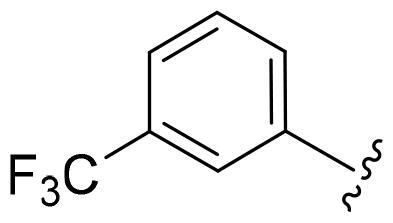
|
34.6 ± 4.1 | NT |
| 34 |
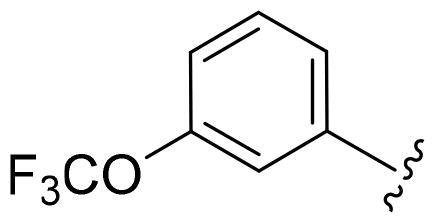
|
23.4 ± 3.3 | NT |
| 35 |
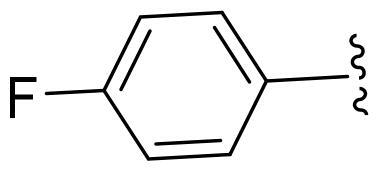
|
55.8 ± 4.6 | NT |
| 36 |
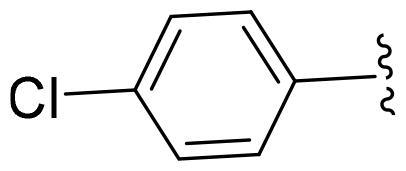
|
61.6 ± 5.4 | 127.9 ± 3.8 |
| 37 |
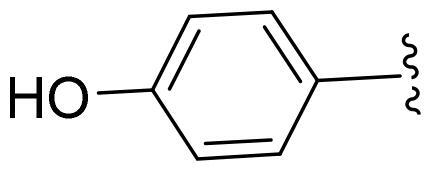
|
28.3 ± 3.1 | NT |
| 38 |
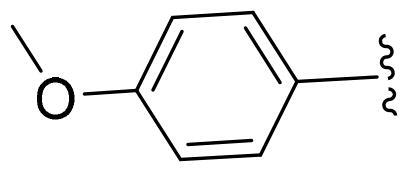
|
26.42 ± 1.6 | NT |
| 39 |
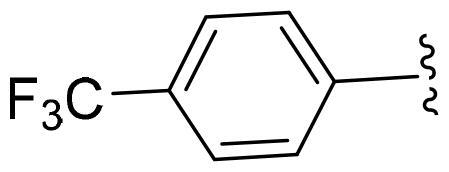
|
22.3 ± 3.7 | NT |
| 40 |
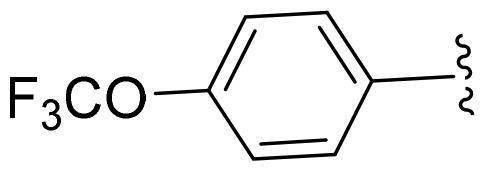
|
46.3 ± 1.9 | NT |
| SEL-120-34A | / | 82.5 ± 5.8 | 37.2 ± 2.5 |
| C43 | / | 73.2 ± 4.3 | 52.6 ± 3.5 |
CDK8 inhibition rate at 200 nM was determined by CDK8 enzyme activity assay. Values from two experiments and results were shown as means ± SD.
IC50 values were determined by CDK8 enzyme activity assay. Values from three dependent experiments and results were shown as means ± SD. cNT = Not Test.
Finally, for a better overall structure-activity relationship discussion, we analyse and compare the molecular docking of compounds C43 and 12 (Figure 3B). It is obvious that compound 12 forms more pi-pi stacking interactions and one hydrogen bond interaction than compound C43, which is also the reason why compound 12 (CDK8 IC50 =39.2 ± 6.3 nM) is more active than compound C43 (CDK8 IC50 =52.6 ± 3.5 nM), but the activity is not greatly improved. The reason was analysed: on the one hand, it may be that the furan ring has a certain steric hindrance, which is not conducive to improving the activity; The significant difference in activity between compounds 17 and 20-41 indirectly proved large spatial hindrance could weaken the activity. On the other hand, it is important that the furan ring is connected to 7-azaindole, and the two aromatic systems may form a large delocalised π bond due to coplanarity, especially affecting the charge distribution of the pyrrole ring results in decreased activity. The significantly lower activity of compound 2 than that of C43 may be due to the charge change of the pyrrole ring. Therefore, these results are instructive to design novel and highly active CDK8 inhibitors in the future.
Figure 3.

Molecular docking of target compounds. (A) The docking model of compound 12 with CDK8 (B) Superposition of spatial structures of compound C43 and 12 within active site of CDK8 (PDB: 5IDN). CDK8 is shown in gray ribbons with selected residues coloured green. Hydrogen bonds are drawn as yellow dashed lines, and pi-pi stacking is drawn as magenta dashed lines. Compound 12 is shown with yellow stick and compound C43 is shown with blue stick. The illustration was generated using PyMOL.
The evaluation of anti-proliferation activity and cytotoxicity of compounds
Compounds12, 13 and 30 were selected for further cell activity assessments based on the enzymatic activity assessment results. molm-13 and MV4-11(acute myeloid lineage leukaemia cells), MGC-803 (gastric cancer cell), MDA-MB-231(breast cancer cell), A375 (melanoma cell), A549 (lung cancer cell), HCT-116, SW-480 and HT-29 (colorectal cancer cells) were selected to evaluate antitumor activity in vitro, GES-1 (gastric mucosal epithelial cell) was selected to evaluate toxicity in vitro. As shown in Table 3, compound 12 and 13 exhibited potent antiproliferative activity on selected tumour cells, especially on AML cells. However, compound 30 did not exhibited potent antiproliferative activity on selected tumour cells.
Table 3.
Antiproliferative activity and preliminary safety of selected compounds.
| Compounds | 12 | 13 | 30 | SEL120-34A | |
|---|---|---|---|---|---|
| CDK8 IC50 (nM) a | / | 39.2 ± 6.3 | 65.7 ± 3.1 | 86.7 ± 6.5 | 37.2 ± 2.5 |
| GC50 (μM) b | molm-13 | 0.02 ± 0.01 | 0.16 ± 0.02 | >50 | 0.02 ± 0.01 |
| MV4-11 | 0.03 ± 0.01 | 0.36 ± 0.12 | >50 | 0.007 ± 0.001 | |
| MGC-803 | 11.72 ± 1.93 | 9.19 ± 2.42 | >50 | 16.26 ± 1.86 | |
| MDA-MB-231 | >50 | 31.47 ± 1.27 | >50 | 9.60 ± 0.81 | |
| A375 | 1.00 ± 0.23 | 0.95 ± 0.52 | >50 | 5.90 ± 1.43 | |
| A549 | 15.49 ± 2.78 | 21.75 ± 1.66 | >50 | 12.06 ± 0.37 | |
| HCT-116 | 3.66 ± 1.94 | 2.48 ± 0.99 | >50 | 11.3 ± 1.9 | |
| SW480 | 6.05 ± 2.26 | 5.45 ± 2.82 | >50 | 17.7 ± 2.1 | |
| HT-29 | 8.26 ± 0.65 | 9.31 ± 1.67 | >50 | 33.8 ± 1.4 | |
| GES-1 | 25.70 ± 4.97 | 24.15 ± 3.62 | >100 | 55.3 ± 3.2 | |
IC50 values were determined by CDK8 enzyme activity assay. Values from three dependent experiments and results were shown as means ± SD.
GI50 values were determined by CCK-8 assay. Values from three dependent experiments and results were shown as means ± SD.
CDKs Selectivity of compound 12
CDK7 and CDK9 with the same transcription function and CDK2 and CDK6 were selected to evaluate the selectivity of compound 12. As shown in Table 4, compound 12 showed good family protein selectivity.
Table 4.
CDKs Selectivity of Compound 12.
| Compound | CDK2 (nM) | CDK6 (nM) | CDK7 (nM) | CDK9 (nM) | CDK8 (nM) |
|---|---|---|---|---|---|
| 12 | >1000 | 351.9 ± 36.5 | 301.5 ± 27.8 | 473.6 ± 25.3 | 39.2 ± 6.3 |
Cellular thermal shift assay
Cellular thermal shift assay has been recognised as a means of demonstrating the binding of small molecules to target proteins. Here, in order to identify compound 12 could bind CDK8, HCT-116 cells, a CDK8 high expression cell line, were treated with 5 μM compound 12 and DMSO for 6 h, respectively. Cell suspension was divided into 11 PCR tubes equally, then the samples were heated from 37 to 67 °C at intervals of 3 °C and the results were analysed through western blot. As shown in Figure 4, CDK8 was degraded almost completely at 49 °C after treated with DMSO. However, CDK8 was degraded almost completely at 55 °C after treated with compound 12, which indicated compound 12 enhanced the thermal stability of CDK8 and compound 12 could bind CDK8.
Figure 4.

Compound 12 enhanced the thermal stability of intracellular CDK8 protein. (A-B)The CDK8 protein intensity at different temperature.
Compound 12 and biotinylated compound bind to CDK8 competitively
In previous study, a biotinylated compound with favourable CDK8 inhibition activity was designed and synthesized23. Here, the pulldown assay was performed to determine compound 12 could bind CDK8. HCT-116 cells or HEK293T cells transfected with CDK8 plasmid labelled were treated with 2 μM biotinylated compound and compounds 12 with different concentrations of 1 μM, 2 μM, 4 μM separately. As shown in Figure 5, the amount of protein pulled down by biotinylated compound decreased as the concentration of compound 12 increased, which indicated that compound 12 could inhibit the binding of biotinylated compound to CDK8, indirectly proving that the compound 12 could bind CDK8.
Figure 5.

Compound 12 and biotin bind to CDK8 protein competitively. (A) Compound 12 binds to CDK8 protein in HCT-116 cells. (B)Compound 12 binds to CDK8 protein in HEK293T cells.
Effect of compound 12 on phosphorylation of STAT1 S727 and STAT5 S726
It has been reported that CDK8 could specifically target the transactivation domain of STAT1 at Ser727 phosphorylation24. HCT-116 cells were treated with compounds 12 with different concentrations of 1 μM, 2 μM, 4 μM separately to verify compound 12 could inhibit the phosphorylation of STAT1 Ser727. As shown in Figure 6(A), after treating cells with compound 12, the phosphorylation of STAT1 S727 was significantly inhibited in a dose-dependent manner, while JAK-mediated the phosphorylation of STAT1 Tyr701 was not inhibited. Above results indicated that compound 12 could affect the biological function of CDK8. It’s reported that CDK8 was also involved in S726 phosphorylation of STAT5 in AML cells7. As shown in Figure 6(B), compound 12 could inhibit phosphorylation of STAT5 S726 in a dose-dependent manner.
Figure 6.

Compound 12 inhibited phosphorylation of STAT1 S727 and STAT5 S726. (A) Compound 12 suppressed the phosphorylation of STAT1 S727 in a dose-dependent manner. HCT-116 cells were treated with compound for 12 h. (B) Compound 12 suppressed the phosphorylation of STAT5 S726 in a dose dependent manner. HL-60 cells were treated with or without compound for 12 h. The samples were analysed by Western blot.
In vivo pharmacokinetic evaluation
Pharmacokinetic data is an important parameter for small molecule drugs. Here, the pharmacokinetic data of compound 12 was performed. As shown in Table 5, after oral administration of compound 12 at 10 mg/kg, there was a good blood drug concentration (AUC0−∞ = 1611.6 μg/L × h). In addition, we noticed that the Cmax was 821.8 μg/L at 0.88 h and t1/2 was 0.95 h, which indicated that compound 12 exhibietd acceptable pharmacokinetic properties. After iv administration of compound 12 at 2 mg/kg, the Cmax was 647.7 μg/L, the half-time (t1/2) was about 1.12 h and the area under the concentration time curve (AUC0−∞) was 829.7 μg/L × h. Importantly, compound 12 exhibited relatively satisfied bioavailability (F%= 38.8%). In addition, compound 12 showed high permeability and no obvious inhibition activity against five cytochrome p450 isoenzymes (Tables 6 and 7), which indicated that compound 12 may be suitable for co-administering with other drugs. Based on the above data analysis, we believed it is possible that compound 12 could be used as an oral medication (Figure 7).
Table 5.
In vivo PK properties of compound 12.
| Dose/routes | t1/2 (h) | Tmax (h) | MRT (h) | Cmax (μg/L) | AUC 0-∞ (μg/L × h) |
CL(L/h/kg) | F(%) |
|---|---|---|---|---|---|---|---|
| 10 mg/kg (po) | 0.95 | 0.88 | 2.24 | 821.82 | 1611.67 | 6.21 | 38.80 |
| 2 mg/kg (iv) | 1.12 | 0.33 | 1.64 | 647.79 | 829.75 | 2.45 | / |
Table 6.
Caco-2 permeability determination.
| Compound | Papp (10 −6 cm/s) |
Efflux ratio | Recovery (%) |
||
|---|---|---|---|---|---|
| A→B | B→A | A→B | B→A | ||
| 12 | 27.0 | 35.1 | 1.3 | 107.0 | 108.1 |
| Nadolol | 0.2 | 0.5 | – | 99.0 | 110.9 |
| Propranolol | 40.2 | 34.1 | – | 99.9 | 107.0 |
| Digoxin | 0.9 | 5.6 | 6.2 | 92.5 | 99.2 |
Table 7.
CYP inhibition activity.
| Compound | IC50 (μM) |
||||
|---|---|---|---|---|---|
| 1A2 | 2C9 | 2C19 | 2D6 | 3A4/5 (M) | |
| 12 | >50 | >50 | >50 | >50 | >50 |
| Positive control | 0.209 | 0.569 | 9.30 | 0.0356 | 0.0118 |
Figure 7.

(A-B) The concentrations versus time curve of compound 12.
Acute toxicity study
The safety of compound 12 in vivo was evaluated through an acute toxicity study on C57BL/6 mice. The compound 12 was administered by gastric tube at a dose of 1000 mg/kg. The normal group was given the same amount of normal saline. During the whole 7 days, there is no obvious change in physiological phenomena such as anorexia, drowsiness, seizures and hyperactivity. Finally, the mice were anaesthetised and sacrificed, and the main organs such as heart, liver, spleen, lung and kidney were detected through pathological section. As shown in Figure 8, there were no diseased tissue, indicating good safety of compound 12 in vivo.
Figure 8.

Compound 12 had low toxicity in vivo. The HE staining of tissues after mice were treated with 1000 mg/kg compound 12. The scan bar: 100 μM.
Conclusion
STAT1, STAT3 and STAT5 are constitutively activated in AML cell lines. CDK8 can positively regulate phosphorylation of S727 in STAT1 and S726 in STAT5 in AML cells. SEL120-34A (SEL120), a clinical trial phase I CDK8 inhibitor, has been shown to downregulate phosphorylation level of STAT1 and STAT5 in AML cells lines. Thus, the synergistic effects of STAT inhibitor with CDK8 inhibitor could provide new therapeutic opportunities for AML, rather than reverse the effects of the CDK8 inhibitor. Based on the previous work, a total of 40 compounds were designed and synthesised through rational design and discussion on structure-activity relationships. Through comprehensive evaluation, compound 12 (3–(3-(furan-3-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzamide) was selected as potent CDK8 inhibitor (CDK8 IC50 = 39.2 ± 6.3 nM) for further research. The studies showed that compound 12 could bind the CDK8 and inhibit the phosphorylation of the STAT 1 Ser 727 and STAT 5 Ser 726. compound 12 showed good antiproliferative activity against AML cells at nanomolar concentrations. Importantly, compound 12 exhibited acceptable pharmacokinetic properties that could be taken orally. This study has guiding significance for the development of novel and efficient CDK8 inhibitors.
Supporting information
1H NMR,13C NMR and HR-MS spectra.
Supplementary Material
Funding Statement
These studies were supported by the National Natural Science Funding of China (21977001).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Yogarajah M, Stone RM.. A concise review of BCL-2 inhibition in acute myeloid leukemia. Expert Rev Hematol. 2018;11(2):145–154. [DOI] [PubMed] [Google Scholar]
- 2.Cao Z, Shu Y, Wang J, Wang C, Feng T, Yang L, Shao J, Zou L.. Super enhancers: Pathogenic roles and potential therapeutic targets for acute myeloid leukemia (AML). Genes Dis. 2022;9(6):1466–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kayser S, Levis MJ.. Updates on targeted therapies for acute myeloid leukaemia. Br J Haematol. 2022;196(2):316–328. [DOI] [PubMed] [Google Scholar]
- 4.Thol F, Ganser A.. Treatment of relapsed acute myeloid leukemia. Curr Treat Options Oncol. 2020;21(8):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosono N, Yokoyama H, Aotsuka N, Ando K, Iida H, Ishikawa T, Usuki K, Onozawa M, Kizaki M, Kubo K, et al. Gilteritinib versus chemotherapy in Japanese patients with FLT3-mutated relapsed/refractory acute myeloid leukemia. Int J Clin Oncol. 2021;26(11):2131–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brachet-Botineau M, Deynoux M, Vallet N, Polomski M, Juen L, Hérault O, Mazurier F, Viaud-Massuard MC, Prié G, Gouilleux F.. A novel inhibitor of STAT5 signaling overcomes chemotherapy resistance in myeloid leukemia cells. Cancers. 2019;11(12):2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rzymski T, Mikula M, Żyłkiewicz E, Dreas A, Wiklik K, Gołas A, Wójcik K, Masiejczyk M, Wróbel A, Dolata I, et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget. 2017;8(20):33779–33795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wingelhofer B, Maurer B, Heyes EC, Cumaraswamy AA, Berger-Becvar A, de Araujo ED, Orlova A, Freund P, Ruge F, Park J, et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. 2018;32(5):1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, Iino T, Rocnik JL, Kikushige Y, Mori Y, Shima T, Iwasaki H, Takenaka K, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood. 2009;114(24):5034–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cee VJ, Chen DY, Lee MR, Nicolaou KC.. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew Chem Int Ed Engl. 2009;48(47):8952–8957. [DOI] [PubMed] [Google Scholar]
- 11.Yu M, Long Y, Yang Y, Li M, Teo T, Noll B, Philip S, Wang S.. Discovery of a potent, highly selective, and orally bioavailable inhibitor of CDK8 through a structure-based optimisation. Eur J Med Chem. 2021;218:113391. [DOI] [PubMed] [Google Scholar]
- 12.Lee JC, Liu S, Wang Y, Liang Y, Jablons DM.. MK256 is a novel CDK8 inhibitor with potent antitumor activity in AML through downregulation of the STAT pathway. Oncotarget. 2022;13(1):1217–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dale T, Clarke PA, Esdar C, Waalboer D, Adeniji-Popoola O, Ortiz-Ruiz MJ, Mallinger A, Samant RS, Czodrowski P, Musil D, et al. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat Chem Biol. 2015;11(12):973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallinger A, Schiemann K, Rink C, Stieber F, Calderini M, Crumpler S, Stubbs M, Adeniji-Popoola O, Poeschke O, Busch M, et al. Discovery of potent, selective, and orally bioavailable small-molecule modulators of the mediator complex-associated kinases CDK8 and CDK19. J Med Chem. 2016;59(3):1078–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czodrowski P, Mallinger A, Wienke D, Esdar C, Pöschke O, Busch M, Rohdich F, Eccles SA, Ortiz-Ruiz MJ, Schneider R, et al. Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J Med Chem. 2016;59(20):9337–9349. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann MH, Mani R, Engelhardt H, Impagnatiello MA, Carotta S, Kerenyi M, Lorenzo-Herrero S, Böttcher J, Scharn D, Arnhof H, et al. Selective and potent CDK8/19 inhibitors enhance NK-cell activity and promote tumor surveillance. Mol Cancer Ther. 2020;19(4):1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boehringer Ingelheim GmbH . New phenyl pyrazolyl acetamide compounds and derivatives as CDK8/CDK19 inhibitors [P]. WO2017202719A1, 2017.11.30. [Google Scholar]
- 18.NIH. U.S. National Library of Medicine. https://clinicaltrials.gov. (accessed May 25, 2023).
- 19.Yu M, Teo T, Yang Y, Li M, Long Y, Philip S, Noll B, Heinemann GK, Diab S, Eldi P, et al. Potent and orally bioavailable CDK8 inhibitors: design, synthesis, structure-activity relationship analysis and biological evaluation. Eur J Med Chem. 2021;214:113248. [DOI] [PubMed] [Google Scholar]
- 20.Solum E, Hansen TV, Aesoy R, Herfindal L.. New CDK8 inhibitors as potential anti-leukemic agents - design, synthesis and biological evaluation. Bioorg Med Chem. 2020;28(10):115461. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XX, Yan YY, Ma X, Xiao Y, Lei CC, Wang YM, Liu C, Wang Q, Zhang XT, Cheng WD, et al. Discovery of a novel oral type I CDK8 inhibitor against acute myeloid leukemia. Eur J Med Chem. 2023;251:115214. [DOI] [PubMed] [Google Scholar]
- 22.Xie Z, Hou S, Yang X, Duan Y, Han J, Wang Q, Liao C.. Lessons learned from past cyclin-dependent kinase drug discovery efforts. J Med Chem. 2022;65(9):6356–6389. [DOI] [PubMed] [Google Scholar]
- 23.Yan YY, Zhang XX, Xiao Y, Shen XB, Jian YJ, Wang YM, She ZH, Liu MM, Liu XH.. Design and synthesis of a 2-amino-pyridine derivative as a potent CDK8 inhibitor for anti-colorectal cancer therapy. J Med Chem. 2022;65(19):13216–13239. [DOI] [PubMed] [Google Scholar]
- 24.Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, Dölken L, Strobl B, Müller M, Taatjes DJ, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013;38(2):250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.