

ABSTRACT
ATG5 plays a pivotal role in membrane Atg8ylation, influencing downstream processes encompassing canonical autophagy and noncanonical processes. Remarkably, genetic ablation of ATG5 in myeloid cells leads to an exacerbated pathological state in murine models of tuberculosis, characterized by an early surge in mortality much more severe when compared to the depletion of other components involved in Atg8ylation or canonical autophagy. This study shows that in the absence of ATG5, but not other core canonical autophagy factors, endolysosomal organelles display a lysosomal hypersensitivity phenotype when subjected to damage. This is in part due to a compromised recruitment of ESCRT proteins to lysosomes in need of repair. Mechanistically, in the absence of ATG5, the ESCRT protein PDCD6IP/ALIX is sequestered by the alternative conjugate ATG12–ATG3, contributing to excessive exocytic processes while not being available for lysosomal repair. Specifically, this condition increases secretion of extracellular vesicles and particles, and leads to excessive degranulation in neutrophils. Our findings uncover unique functions of ATG5 outside of the autophagy and Atg8ylation paradigm. This finding is of in vivo relevance for tuberculosis pathogenesis as modeled in mice.
Abbreviations: Atg5: autophagy related 5; ESCRT: endosomal sorting complex required for transport; EVPs: extracellular vesicles and particles; FPR1: formyl peptide receptor 1; LyHYP: lysosomal hypersensitivity phenotype; LysoIP: lysosome immunopurification; Mtb: Mycobacterium tuberculosis; ORF3a: open reading frame 3a protein; PDCD6IP/ALIX: programmed cell death 6 interacting protein; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2, TFEB: transcription factor EB.
Keywords: ATG5, Atg8ylation, autophagy, lysosomal damage, secretion, tuberculosis
The initial studies on the immunological roles of autophagy indicated that this process can control intracellular Mycobacterium tuberculosis (Mtb) in both murine models and human cells. However, in murine models of Mtb infection, genetic knockout (KO) of Atg5 in myeloid-derived cells is associated with more severe pathology and mortality when compared to the loss of other Atg genes. These observations suggest that ATG5 possesses additional functions beyond its well-established role in membrane Atg8ylation and canonical autophagy as one of its outputs.
Mtb infects and parasitizes host macrophages where it inhibits phagosome maturation and partially permeabilizes membranes of endolysosomal and phagosomal compartments avoiding the acidic environment and activity of hydrolases within lysosomes and phagolysosomes. To investigate the impact of ATG5 on lysosomal quality control, we measured the lysosomal damage in diverse cell lines depleted of ATG5 [1]. The ATG5-deficient cells display a lysosomal hypersensitivity phenotype (LyHYP) to a variety of membrane damage agents, including Leu-Leu-OMe/LLOMe, silica crystals, and overexpression of SARS-CoV-2 ORF3a. LyHYP is manifested by increased lysosomal damage marker LGALS3 (galectin 3) and diminished acidification. In contrast to ATG5 inactivation, depleting other autophagy factors, including the key players in membrane Atg8ylation, ATG3 and ATG7, has no significant effects on LyHYP. This indicates a unique function of ATG5 that is independent of its role in canonical autophagy and Atg8ylation. We further demonstrated that LyHYP in atg5 KO cells is rescued by expressing either a conjugation-competent or conjugation-deficient (K130R mutant) form of ATG5. We observed a similar disturbance in lysosome homeostasis in primary cells, such as murine bone marrow-derived macrophages.
Homeostatic response to lysosomal membrane damage occurs in three stages: 1) repair of membranes via ESCRTs and lipid transfer pathways; 2) removal through autophagy coupled with lysosomal membrane protein salvage after irreparable damage; and 3) replacement by TFEB-regulated lysosomal biogenesis. We found that ESCRT components PDCD6IP/ALIX, CHMP4B, and CHMP2A, which are normally recruited to compromised lysosomes, are missing from or are greatly reduced on lysosomes in atg5 KO cells. This was confirmed by unbiased quantitative high-content microscopy, flow cytometry coupled with microscopy imaging, and immune-isolation of lysosomes by the LysoIP procedure. Complementation experiments and comparisons with cells deficient in other autophagy factors further established that the observed absence of ESCRT machinery was linked to the missing ATG5.
We carried out an unbiased search for ATG5 interactors by proximity biotinylation coupled with quantitative LC/MS/MS proteomics using Flp-In cells expressing conjugation-competent (WT) and conjugation-deficient (K130R mutant) ATG5 fused to APEX2. Gene ontology analysis indicated a significant enrichment of proteins involved in secretion, exocytosis, exosomes, and degranulation. We thus addressed effects of ATG5 on secretory processes. We first found that lysosomal damage induced biochemically with L-leucyl-L-leucine methyl ester or upon expression of SARS-CoV-2 ORF3 itself stimulates various exocytic processes. Using nanoparticle tracking analysis and imaging flow cytometry, we observed that lysosomal damage intensifies the release from cells of extracellular vesicles and particles (EVPs). The increased EVPs secretion is also evident in primary macrophages and neutrophils. We detected elevated ITGAM/CD11b on neutrophils’ plasma membrane, an indicator of neutrophil degranulation and their general activation. This is comparable to the effects triggered by conventional inducers of neutrophil degranulation. Because the absence of ATG5 leads to a state of LyHYP, we hypothesized that ATG5 may have a secretory phenotype secondary to heightened lysosomal vulnerability. This was assessed and validated by measuring various exocytosis/degranulation outputs and comparing WT with ATG5-deficient cells including primary murine neutrophils. The atg5 knockout cells display elevated EVPs including exosomes, increased neutrophil cell surface markers (ITGAM/CD11b and FPR1), and enhanced lysosomal exocytosis by detecting LAMP2 on the cell surface. We found that all metrics are increased in ATG5-deficient cells.
The ESCRT protein PDCD6IP/ALIX, which is sequestered away from damaged lysosomes of ATG5-deficient human cells, has been previously linked to exosome secretion through its interaction with the alternative ATG12–ATG3 conjugate. We found that the levels of the ATG12–ATG3 conjugate in human cells and its colocalization with PDCD6IP/ALIX are increased in ATG5 KO cells explaining elevated EVP release from these cells. Consistent with these observations, ATG3 knockout in ATG5-deficient cells reverses the hypersecretory phenotype.
These findings demonstrate that ATG5 acts as a hitherto unappreciated switch between canonical and noncanonical conjugation cascades involving ATG proteins (Figure 1). In this role ATG5 affects endolysosomal quality control beyond its known activities in canonical autophagy and Atg8ylation. When this role is perturbed, the changes in ATG conjugation systems favor lysosomal hypersensitivity leading to lysosomal exocytosis, mirrored by enhanced secretion of exosomes and EVPs. This phenotype is replicated in primary neutrophils and, because excessive activation of neutrophils leads to exacerbated tuberculosis pathogenesis in animal models and in humans, the findings of our study explain at the molecular level the paradoxical dominance of ATG5 effects in vivo relative to other autophagy genes.
Figure 1.
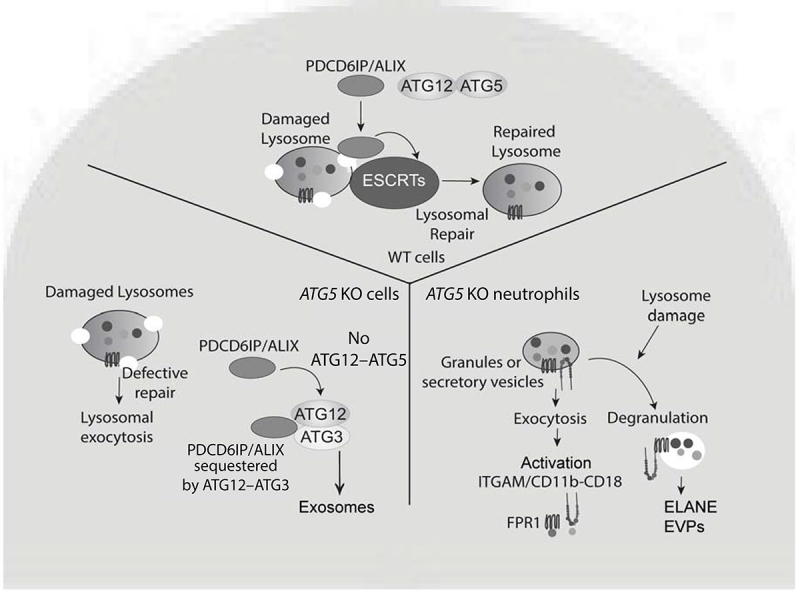
Role of ATG5 in lysosomal quality control and secretion activities. In WT cells, damaged lysosomes are repaired by ESCRT machinery including PDCD6IP/ALIX, which is absent in ATG5-deficient cells. The defective membrane repair leads to increased lysosomal exocytosis. ATG5 absence favors the formation of the alternative conjugate ATG12–ATG3, which sequesters PDCD6IP/ALIX making it unavailable to repair lysosomes and at the same time promotes secretion of exosomes. In neutrophils, ATG5 deficiency leads to increased degranulation, elevated release of EVPs, and a heightened state of activation. In the context of M. tuberculosis infection (not depicted) this contributes to excessive neutrophilic inflammation and exacerbated tuberculosis pathogenesis.
Funding Statement
The work was supported by the National Institute of Allergy and Infectious Diseases [R37AI042999, R01AI111935]; National Institute of General Medical Sciences [P20GM121176]; NIH Office of the Director [S10OD021801].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Wang F, Peters R, Jia J, et al. ATG5 provides host protection acting as a switch in the atg8ylation cascade between autophagy and secretion. Dev Cell. 2023;58(10):866–884 e868. doi: 10.1016/j.devcel.2023.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]